

ABSTRACT
Background
Cerebellar atrophy is a nonspecific imaging finding observed in a number of neurological disorders. Genetic ataxias associated with cerebellar atrophy are a heterogeneous group of conditions, rendering the approach to diagnosis challenging.
Objectives
To define the spectrum of genetic ataxias associated with cerebellar atrophy in a Canadian cohort and the diagnostic yield of exome sequencing for this group of conditions.
Methods
A total of 92 participants from 66 families with cerebellar atrophy were recruited for this multicenter prospective cohort study. Exome sequencing was performed for all participants between 2011 and 2017 as part of 1 of 2 national research programs, Finding of Rare Genetic Disease Genes or Enhanced Care for Rare Genetic Diseases in Canada.
Results
A genetic diagnosis was established in 53% of families (35/66). Pathogenic variants were found in 21 known genes, providing a diagnosis for 31/35 families (89%), and in 4 novel genes, accounting for 4/35 families (11%). Of the families, 31/66 (47%) remained without a genetic diagnosis. The most common diagnoses were channelopathies, which were established in 9/35 families (26%). Additional clinical findings provided useful clues to specific diagnoses.
Conclusions
We report on the high frequency of channelopathies as a cause of genetic ataxias associated with cerebellar atrophy and the utility of exome sequencing for this group of conditions.
Keywords: ataxia, cerebellar atrophy, channelopathies, exome sequencing
Cerebellar atrophy is a nonspecific imaging finding observed in a number of acquired and genetic neurological disorders in both pediatric and adult populations. 1 , 2 , 3 It is characterized by a loss of cerebellar tissue, with evidence on brain imaging of enlarged interfolial spaces compared to the foliae, in a posterior fossa of normal size. 2 , 4 , 5 , 6
Genetic ataxias associated with cerebellar atrophy are a clinically and genetically heterogeneous group of conditions, which makes the approach to diagnosis challenging. 1 , 2 , 3 Possible causes include chromosomal abnormalities; repeat expansions; inborn errors of metabolism, in particular mitochondrial disorders; and other single gene disorders. 1 , 2 , 7 , 8 , 9 Genetic ataxias were historically classified based on their mode of inheritance, with early‐onset autosomal recessive conditions being the most common. 10 , 11 Additional clinical and neuroimaging findings are essential to selecting appropriate genetic investigations. 1 , 3 , 12 , 13 Specific etiologies for genetic ataxias associated with cerebellar atrophy are distinct from those for genetic ataxias without this imaging finding.
The goal of this study was to define the spectrum of genetic ataxias associated with cerebellar atrophy in a Canadian cohort and the diagnostic yield of exome sequencing (WES) for this group of conditions.
Methods
A total of 92 participants from 66 families with ataxia and cerebellar atrophy of suspected genetic etiology were recruited from 6 centers across Canada between 2011 and 2017 in the context of a prospective cohort study. Patients of all ages were included in the study. Atrophy of the cerebellar vermis and/or hemispheres on brain imaging was assessed by the referring clinicians. Not all patients had a family history of ataxia, and participants were not selected based on suspected mode of inheritance.
WES was performed as part of 1 of 2 national research programs, Finding of Rare Genetic Disease Genes or Enhanced Care for Rare Genetic Diseases in Canada. Genetic investigations obtained by the referring clinicians prior to enrollment in the study included biochemical, cytogenetic, and molecular investigations (including for expansion‐related ataxias), depending on provincially funded clinical testing. Diagnostic tests prior to recruitment were based on clinical indication. Acquired causes of ataxia were also excluded.
Patients from 15 families were previously published as part of a brief report on utility of WES in pediatric‐onset ataxia and in separate publications reporting novel genes or expanding the phenotypic spectrum associated with known genes. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23
WES was performed on total genomic DNA from the 92 participants. DNA was extracted from peripheral blood following standard procedures. Target enrichment was performed using the Agilent SureSelect 50 Mb (V3, V4, or V5) All Exon Kit (Agilent, Santa Clara, CA). Sequencing was performed on Illumina Hiseq 2000 and generated 6 Gbp of 100 bp paired‐end reads per sample (Illumina, San Diego, CA). Read alignment, variant calling, and annotations were performed as previously described. 24 The mean sequencing coverage depth was 80 to 120x.
A systematic approach was used for data interpretation. Rare (allele frequency of <1% in Exome Variant Server and 1000 Genomes Project) and nonsynonymous variant splicing as well as coding indels were examined first. Pathogenicity and conservation of affected residues were predicted using tools such as SIFT, Polyphen‐2, Mutation Taster, and GERP. 25 , 26 , 27 , 28 Genes relevant to the phenotype were examined based on a Human Phenotype Ontology–driven gene list. All known disease‐causing genes included in Online Mendelian Inheritance in Man (approximately 4500) were reviewed. Finally, all rare variants were examined to identify possible novel disease‐causing genes. Candidate variants were manually visualized using the Integrative Genomics Viewer. Variants in candidate genes were confirmed by Sanger sequencing, and segregation was documented when parental samples were available. The American College of Medical Genetics and Genomics variant classification guidelines were used to determine pathogenicity of the variants. 29
Clinical, radiologic, and molecular information for all 92 participants was provided by the referring clinicians. The available information was collected and analyzed using Phenotips. 30 This study was approved by the ethics committees of all participating centers. Informed consent was obtained from the patients or their legal representatives to participate in this study.
Data Availability Statement
All data presented in this study are available upon reasonable request.
Results
Diagnostic Yield of WES
A total of 92 individuals from 66 families were included in this study. A genetic diagnosis was established in 52/92 patients (57%) from 35 families (35/66 families, 53%). Pathogenic variants were found in 21 known genes, providing a diagnosis for 45 participants from 31 families (31/35 families, 89%), and in 4 novel genes (LAMA1, PUM1, SLC39A8, XRCC1), accounting for 7 participants from 4 families (4/35 families, 11%). Of the patients, 40 from 31 families (31/66 families, 47%) remained without a genetic diagnosis.
Inheritance was autosomal recessive for the majority of the diagnoses for which a genetic cause was established, accounting for 34 individuals from 21 families (21/35 families, 60%). Autosomal dominant inheritance was documented in 18 individuals from 14 families (14/35 families, 40%). There were 9/14 families (64%) with de novo and 3/14 families (21%) with inherited dominant variants. Segregation information was not available for 2/14 families (14%) with autosomal dominant disorders.
The most common diagnoses were channelopathies in 12 patients from 9 families (9/35 families, 26%). There were 9 patients from 5 families (5/35 families, 14%) with autosomal recessive mitochondrial disorders, 8 patients from 5 families (5/35 families, 14%) with metabolic disorders (congenital disorders of glycosylation and peroxisomal fatty acid beta‐oxidation defects), and 4 patients from 4 families (4/35 families, 12%) with classic autosomal recessive ataxias caused by pathogenic variants in genes well known to cause cerebellar ataxia (SACS, SYNE1, or SETX). 12
In the remaining 19 patients from 12 families (12/35 families, 34%), pathogenic variants in 10 different genes were identified. These conditions included rare spinocerebellar ataxias, lamininopathy disorders, pontocerebellar hypoplasia subtypes, Gordon Holmes syndrome, and Warburg Micro syndrome. We classified these disorders based on prominent ancillary clinical findings in addition to cerebellar ataxia; 11 patients from 6 families (6/12 families, 50%) had a syndromic presentation with significant systemic manifestations and/or extracerebellar symptoms (RAB3GAP1, RNF216, EXOSC3, LAMA1, KIF1A), 3 patients from 2 families (2/12 families, 17%) had ataxia with ocular motor apraxia (BRAT1, XRCC1), 3 patients from 2 families (2/12 families, 17%) had a relatively pure spinocerebellar ataxia (PUM1, PRKCG), and 2 unrelated patients (2/12 families, 17%) had prominent seizures (SPTAN1). Diagnostic yield, mode of inheritance, and diagnostic categories are summarized in Figure 1 and Table 1.
FIG 1.
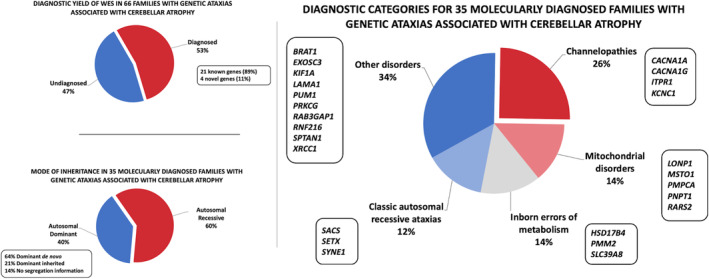
Diagnostic yield of exome sequencing (WES) in genetic ataxias associated with cerebellar atrophy. Diagnostic yield of WES in 66 families with genetic ataxias associated with cerebellar atrophy (A), mode of inheritance (B), and diagnostic categories (C) for 35 molecularly diagnosed families.
TABLE 1.
Molecular characteristics of 35 families with confirmed diagnoses of genetic ataxias associated with cerebellar atrophy, classified by diagnostic categories
| Identification | Diagnostic Category | Gene | Pathogenic Variant(s) | Age(s) at Last Examination, yr |
|---|---|---|---|---|
| 254b | Channelopathies | ITPR1 | Heterozygous, de novoc.7748T>C, p.Ile2583Thr | 11 |
| 423 a | Channelopathies | ITPR1 | Heterozygous, de novo c.800C>T, p.Thr267Met | 10 |
| 521 | Channelopathies | CACNA1A | Heterozygousc.593G>A, p.Arg198Gln | 61 |
| 552 | Channelopathies | ITPR1 | Heterozygous, de novoc.7727A>C, p.Asn2576Thr | 14 |
| 614 | Channelopathies | KCNC1 | Heterozygous, de novo c.959G>A, p.Arg320His | 17 |
| 620 | Channelopathies | CACNA1A | Heterozygous, de novo c.1999G>A, p.Glu667Lys | 20 |
| 671 | Channelopathies | CACNA1G | Heterozygousc.5144G>A, p.Arg1715His | 33, 53, 65 |
| 800 | Channelopathies | CACNA1A | Heterozygous, de novo c.4055G>A, p.Arg1352Gln | 8 |
| 895 | Channelopathies | CACNA1A | Deletion of exons 23 to 47 | 29, 32 |
| 59 a | Mitochondrial | MSTO1 |
c.706G>C, p.Asp236His c.836G>A, p.Arg279His |
7, 8, 10 |
| 287 a | Mitochondrial(pontocerebellar hypoplasia type 6) | RARS2 | c.1432G>A, p.Gly478Arg c.997C>G, p.Arg333Gly | 0.5, 1 |
| 301 a | Mitochondrial | PNPT1 | c.2234T>C, p.Met745Thr c.1748dupA, p.Glu584Glyfs*17 | 60, 62 |
| 483 | Mitochondrial | PMPCA | Homozygousc.766G>A, p.Val256Met | 34 |
| 517 | Mitochondrial | LONP1 | Homozygousc.1387G>A, p.Asp463Asn | 20 |
| 78 a | IEM(congenital disorder of glycosylation, type Ia) | PMM2 | c.422G>A, p.Arg141Hisc.722G>C, p.Cys241Ser | 18 |
| 171 a | IEM(Perrault syndrome) | HSD17B4 | c.101C>T, p.Ala34Val c.1547T>C, p.Ile516Thr | 18, 22 |
| 324 a | IEM(congenital disorder of glycosylation, type IIn) | SLC39A8 b | Homozygousc.112G>C, p.Gly38Arg | 13 |
| 330 | IEM(Perrault syndrome) | HSD17B4 | c.1537C>A, p.Pro513Thr c.1628G>C, p.Arg543Pro | 13, 16, 21 |
| 611 | IEM(congenital disorder of glycosylation, type Ia) | PMM2 | c.430T>C, p.Phe144Leu c.459A>G, p.Ile153Met | 22 |
| 109 a | Classic AR ataxias | SACS | Homozygousc.9284dupC, p.Ala3096Cysfs*2 | 42 |
| 254a | Classic AR ataxias | SETX | Homozygousc.6139G>T, p.Gly2047Cys | 24 |
| 326 | Classic AR ataxias(autosomal recessive spinocerebellar ataxia 8) | SYNE1 | c.17407A>G, p.Lys5803Glu c.12511A>G, p.Met4171Val | 16 |
| C1026 a | Classic AR ataxias(Charlevoix‐Saguenay spastic ataxia) | SACS | Homozygousc.5151dupA, p.Ser1718Ilefs*20 | 9 |
| 242d | Other, syndromic presentation | KIF1A | Heterozygous, de novo c.650C>T, p.Ser217Phe | 12 |
| 545 a | Other, syndromic presentation | LAMA b | Homozygousc.588+2T>G | 3, 5 |
| 615 a | Other, ataxia with ocular motor apraxia | XRCC1 b | c.1293G>C, p.Lys431Asn c.1393C>T, p.Gln465* | 47 |
| 843a | Other, syndromic(pontocerebellar hypoplasia type 1B) | EXOSC3 | Homozygousc.395A>C, p.Asp132Ala | 6, 8 |
| 843b | Other, syndromic(pontocerebellar hypoplasia type 1B) | EXOSC3 | Homozygousc.395A>C, p.Asp132Ala | 4, 10 |
| 619 | Other, ataxia with ocular motor apraxia | BRAT1 | Homozygous c.185T>A, p.Val62Glu | 7, 24 |
| 661d | Other, with seizures | SPTAN1 | Heterozygous, de novo c.6899A>T, p.Asp2300Val | 11 |
| 661e | Other, with seizures | SPTAN1 | Heterozygous, de novo c.6605_6607delAGC, p.Gln2202del | 10 |
| 108 a | Other, syndromic(Gordon Holmes Syndrome) | RNF216 | Homozygousc.2056C>T, p.Arg686* | 26, 30 |
| 242b a | Other, syndromic(Warburg Micro syndrome) | RAB3GAP1 | Homozygousc.363G>A, p.Trp121Ter | 10 |
| 407l a | Other, pure spinocerebellar ataxia (spinocerebellar ataxia 47) | PUM1 b | Heterozygousg.31414862T>A, p.Thr1035Ser | 52, 58, 59 |
| 407 m | Other, pure spinocerebellar ataxia (spinocerebellar ataxia 14) | PRKCG | Heterozygousc.220C>A, p.His74Asn | 53 |
Genetically Diagnosed Subgroup: Clinical Characteristics
Age at onset of ataxia ranged from the neonatal period to adulthood (middle age). Overall, onset of ataxia was reported to occur in the neonatal period, infancy, or childhood in 29/52 patients (56%) and in adulthood in 11/52 individuals (21%). This information was not available for 12/52 participants (23%). Patients with dominant de novo variants tended to have earlier onset of symptoms, ranging from infancy to 6 years of age. Early age at onset was also observed in patients with autosomal recessive conditions, with ataxia presenting in infancy or childhood in 21/34 participants (62%) and in adulthood in 3/34 (9%) patients with autosomal recessive disorders.
Ataxia was reported by the referring clinicians to be progressive over time in 19/52 patients (36%) and nonprogressive in 32/52 (62%). This information was not available for 1/52 patients (2%). The clinicians reported that 26 patients (26/52, 50%) had cognitive impairment, 24/52 (48%) had normal cognition, and cognitive status was not documented for 1/52 patients (2%). Cognitive impairment appeared to correlate with earlier onset of symptoms; 20/25 patients (80%) with cognitive impairment had onset of ataxia in infancy or childhood, and information on age of onset was not available for the other 5 patients with cognitive impairment. For patients who were reported to have normal cognition, onset of ataxia was in infancy or childhood in 9/24 (38%), in adulthood in 8/24 (33%), and was not reported in 7/24 (29%).
Other neurological manifestations and systemic findings, such as neuromuscular symptoms and ophthalmological abnormalities, provided useful clues to some of the diagnoses. There were 5 patients from 2 families with Perrault syndrome caused by biallelic variants in HSD17B4, encoding a peroxisomal fatty acid beta‐oxidation enzyme (MIM number 233400). Of the patients, 2 had sensorineural hearing loss, retinal abnormalities, and evidence of peripheral neuropathy. The other 3 patients were reported to have ophthalmoparesis.
The patient with Warburg Micro syndrome also had evidence of peripheral neuropathy, as did the patient with autosomal recessive ataxia with ocular motor apraxia type 5 (AOA5, AOA‐XRCC1; MIM number 617633). Myopathy was described in autosomal recessive MSTO1‐related disorder (3 individuals from 1 family, MIM number 617675) and in 1 patient with autosomal recessive spinocerebellar ataxia 8 (Beauce ataxia, MIM number 610743) caused by biallelic variants in SYNE1.
Patients with pontocerebellar hypoplasia type 1B caused by biallelic variants in EXOSC3 (4 patients from 2 families, MIM number 614678) had unique clinical characteristics. In addition to severe developmental delay, they showed a combination of upper and lower motor neuron signs with appendicular spasticity, hyperreflexia, extensor plantar response, and tongue fasciculations.
Retinal changes were also seen in 1 of the 2 unrelated patients with congenital disorder of glycosylation type Ia (PMM2‐CDG, caused by biallelic variants in PMM2, MIM number 212065). Optic atrophy was identified in the 2 siblings with mitochondrial PNPT1‐related disorder (MIM number 614932). Cataracts were present in Warburg Micro syndrome caused by biallelic variants in RAB3GAP1 (1 patient, MIM number 600118).
Seizures were reported in the 2 siblings with pontocerebellar hypoplasia type 6 caused by biallelic variants in RARS2 (MIM number 611523), in the single patient with KCNC1‐related disorder (MIM number 616187), in 1 of the patients with CACNA1G‐related disorder (MIM number 616795), and in 1 of the patients with SPTAN1‐related disorder (MIM number 613477). SPTAN1‐related disorder was also associated with prominent spasticity and upper motor neuron signs. The patient with autosomal dominant KIF1A‐related disorder (MIM number 614255) had a history of febrile seizures.
Genetically Diagnosed Subgroup: Neuroimaging Characteristics
Brain imaging revealed isolated cerebellar atrophy in the majority of patients with an established genetic diagnosis (44/52 patients, 85%). Additional imaging findings were reported in a few patients (8/52, 15%). Hyperintensities of the cerebellar cortex on T2‐weighted and fluid‐attenuated inversion recovery sequences were observed in 1 patient with BRAT1‐related disorder and the 2 unrelated patients with SPTAN1‐related disorder. There were nonspecific supratentorial white matter signal changes on brain magnetic resonance imaging of the patient with KCNC1‐related disorder and in 2 unrelated patients with pontocerebellar hypoplasia type 1B caused by biallelic variants in EXOSC3. Another patient with pontocerebellar hypoplasia type 1B had cortical and corpus callosum anomalies. Thinning of the corpus callosum was noted in the patient with Warburg Micro syndrome caused by biallelic variants in RAB3GAP1.
Genetically Diagnosed Subgroup: Channelopathies
A total of 12 patients from 9 families were diagnosed with channelopathies, which was the most common diagnosis in this cohort (9/36 families, 25%). Their clinical, radiologic, and molecular characteristics are summarized in Table 2.
TABLE 2.
Clinical radiologic and molecular characteristics of 12 patients diagnosed with channelopathies
| Identification | 521 | 620 | 800 | 895_MT0181 | 895_MT0182 | 671 | 6712 | 6713 | 254b | 423 | 552 | 614 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | CACNA1A | CACNA1A | CACNA1A | CACNA1A | CACNA1A | CACNA1G | CACNA1G | CACNA1G | ITPR1 | ITPR1 | ITPR1 | KCNC1 |
| Pathogenic variant | c.593G>A (p. Arg198Gln) | c.1999G>A (p. Glu667Lys) | c.4055G>A (p. Arg1352Gln) | Deletion of exons 23 to 47 | Deletion of exons 23 to 47 | c.5144G>A (p. Arg1715His) | c.5144G>A (p. Arg1715His) | c.5144G>A (p. Arg1715His) | c.7748 T>C (p. Ile2583Thr) | c.800C>T (p. Thr267Met) | c.7727A>C (p. Asn2576Thr) | c.959G>A (p. Arg320His) |
| De novo | N/A | + | + | − | − | − | − | − | + | + | + | + |
| Sex/age, yr | F/61 | M/20 | F/8 | M/29 | F/32 | F/65 | M/33 | F/53 | F/11 | F/10 | M/14 | F/17 |
| Age at onset of ataxia, yr | 35 | 1 | 1 | Teens | Teens | 30 | 28 | 20s | 1.5 | 1 | 2 | 4 |
| Presenting symptoms | Ataxia | GDD,Hypotonia | GDD,Hypotonia | Ataxia | Ataxia | Ataxia | Ataxia | Ataxia | Hypotonia | GDD | Hypotonia | Ataxia |
| Gait ataxia/limb ataxia | +/‐ | +/+ | +/+ | +/+ | +/+ | +/+ | +/‐ | +/+ | +/+ | +/+ | +/+ | +/+ |
| Progressive ataxia | + | − | − | + | + | + | + | + | + | − | + | − |
| Walking aid required | + | − | + | + | − | − | − | − | + | + | + | − |
| Developmental delay | − | + | + | − | − | − | − | − | + | + | + | − |
| Cognitive impairment | − | + | + | + | − | − | − | + | + | + | + | + |
| Nystagmus | + | + | + | + | + | − | − | + | + | − | + | + |
| Dysarthria | + | + | + | + | − | + | + | + | + | + | + | + |
| Bulbar symptoms | + | − | − | − | − | + | − | − | − | − | − | − |
| Episodic symptoms | − | − | − | + | + | − | − | − | + | − | − | − |
| Headache | + | + | − | − | + | − | − | − | − | − | − | + |
| Muscle weakness | + | − | + | − | − | − | − | + | + | − | + | − |
| Upper motor neuron signs | − | − | + | − | − | − | − | − | − | − | − | − |
| Seizures | − |
+ FS |
− | − | − | − | − | + GTC | − | − | − | + M, GTC |
| Isolated cerebellar atrophy on neuroimaging | + | + | + | + | − | + | N/A | + | + | + | + | + |
N/A, not available; F, female; M, male; GDD, global developmental delay; FS, febrile seizures; GTC, generalized tonic–clonic seizures; M, myoclonic seizures.
Pathogenic variants were identified in CACNA1A (3 unrelated patients with different missense variants and 2 patients from 1 family with an intragenic deletion), CACNA1G (3 patients from 1 family with a missense variant), ITPR1 (3 unrelated individuals with different missense variants), and KCNC1 (1 participant with a missense variant). In half of the patients, variants were de novo.
Overall, age of onset ranged from 1 to 35 years, with a mean of 12 years. Patients with de novo variants tended to have earlier onset of symptoms (between 1 and 4 years of age), often with global developmental delay and/or hypotonia as their first clinical manifestations. The other patients presented with ataxia. Half of the patients required a walking aid, and clinicians reported that ataxia progressed over time in 8/12 patients (67%). Episodic symptoms were reported in 3/12 (25%) patients. Additional clinical features included muscle weakness on physical examination (5/12, 42%), headaches (4/12, 33%), and seizures (3/12, 25%). Of the patients with channelopathies, 8 (8/12, 67%) had cognitive impairment and had early onset of symptoms (between age 1 and 20 years).
The 5 patients with CACNA1A‐related disorder had highly variable phenotypes. Age of onset ranged from 1 to 35 years. Those with early onset (at age 1 year) had developmental delay and hypotonia. Three patients had cognitive impairment. Only the 2 siblings with intragenic deletions were reported to have episodic symptoms. Two patients exhibited muscle weakness.
The 3 patients with CACNA1G‐related disorder were from a single family. All 3 had an onset of symptoms in early adulthood. Of the 3 patients, 2 had a relatively pure, slowly progressive cerebellar syndrome, whereas the third also had cognitive impairment, muscle weakness, and seizures.
The 3 unrelated patients with ITPR1‐related disorder presented between 1 and 2 years of age. All 3 had developmental delay and cognitive impairment. They required a walking aid before adolescence. Two of them exhibited muscle weakness.
The patient with KCNC1‐related disorder presented at age 4 years with ataxia. This patient also had myoclonic and generalized tonic–clonic seizures.
Neuroimaging was available for 11/12 patients (92%) with channelopathies and was characterized by isolated cerebellar atrophy in 10 patients (10/11, 91%). There were no additional imaging findings suggestive of the specific diagnoses. One patient with CACNA1A‐related disorder did not have significant cerebellar atrophy. Brain magnetic resonance imaging characteristics of patients with channelopathies are illustrated in Figure 2.
FIG 2.
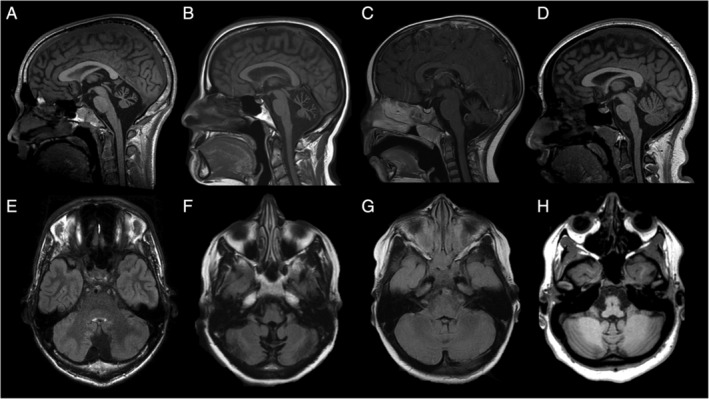
Neuroimaging characteristics of 4 patients diagnosed with channelopathies. Brain magnetic resonance imaging of patients 620 (A,E; CACNA1A), 671 (B,F; CACNA1G), 423 (C,G; ITPR1), and 614 (D,H; KCNC1). Sagittal T1 (A–D) and axial fluid‐attenuated inversion recovery (E–H) images show atrophy of the cerebellar vermis and hemispheres.
Undiagnosed Subgroup
Limited clinical information was available for the 40 patients from 31 families for whom a genetic diagnosis was not established (31/66 families, 47%). Symptom onset was in childhood in 13/40 patients (33%) and in adulthood in 9/40 patients (23%). Information on age of onset was not available for 18/40 patients (45%). Referring clinicians reported the ataxia to be progressive in 10/40 patients (25%) and nonprogressive in 28/40 (70%), and this information was not available for 2/40 patients (5%).
The referring clinicians reported that 17 patients (17/40, 43%) had cognitive impairment and 22 (55%) had normal cognition, and this was not reported for 1 patient (1/40, 3%). About one third of the undiagnosed patients (14/40, 35%) were reported to have at least 1 additional clinical feature, including headaches, hearing loss, high myopia, myopathy, neuropathy, or dystonia. Information on the presence or absence of additional clinical features was not provided for 22/40 participants (55%).
A total of 8 patients (8/40, 20%) had other neuroimaging findings in addition to cerebellar atrophy: T2/fluid‐attenuated inversion recovery hyperintensities in the cerebellar cortex, cerebellar calcifications, supratentorial white matter signal changes or volume loss, abnormalities of the corpus callosum, or atrophy of the basal ganglia. Information on the presence or absence of additional neuroimaging features was not available for 27/40 patients (68%).
Discussion
Our results are consistent with previously published diagnostic rates in patients with genetic ataxias associated with cerebellar atrophy. 1 , 6 , 8 , 13 , 31 , 32 , 33 , 34 Channelopathies were more frequent in our cohort compared with previously published studies and likely represent an under‐recognized cause of cerebellar atrophy in both adults and children. One large cohort study recently reported the frequency of channelopathies in patients with autosomal dominant cerebellar ataxias, in particular CACNA1A‐related disorder. 7 , 35 , 36 Our cohort was not limited to patients with autosomal dominant conditions, and our study focused on cerebellar atrophy as a specific clinical feature, which supports the applicability of our results to this population. Our results highlight cerebellar atrophy as being an early sign of CACNA1A‐related disorder and other channelopathies.
No chromosomal abnormalities were identified in our cohort, as these disorders were excluded prior to enrollment in the study. The classic autosomal recessive ataxias were also less common, which could be attributed to the fact that some of these disorders, for example, ataxia telangiectasia, are clinically recognizable and were identified prior to considering WES. Of note, patient selection appears to significantly impact the diagnostic yield in many studies of genetic ataxias associated with cerebellar atrophy. For example, in a retrospective study of 300 patients with cerebellar atrophy in a pediatric tertiary care center in Canada, mitochondrial disorders were found to be most common, followed by neuronal ceroid lipofuscinoses. 1 This was likely due to the exclusive inclusion of pediatric patients. In our study, patients of all ages were recruited, and they were not selected based on suspected mode of inheritance. This reflects a realistic approach to clinical practice, as patients do not always have a family history of ataxia that is suggestive of a specific inheritance pattern.
In our study, pathogenic variants in 25 different genes were identified in 35 families, highlighting the genetic heterogeneity in this group of conditions. Clinical features were also highly variable; as our study included both pediatric and adult patients, onset of symptoms ranged from the neonatal period to adulthood (middle age). The majority of patients in our cohort had pediatric‐onset disorders, often presenting with global developmental delay and/or hypotonia as the initial clinical manifestations. Patients with pediatric‐onset disorders had higher frequencies of cognitive impairment and additional neurological and/or systemic features. Conversely, adults with later onset disorders tended to have gait difficulties as the predominant symptom.
Younger age of onset in patients with autosomal dominant de novo variants was striking. For example, among patients diagnosed with channelopathies, symptom onset in patients with de novo variants ranged from age 1 to age 4 years, whereas in patients with inherited autosomal dominant variants, onset was in adolescence or adulthood. Clinical heterogeneity was also particularly notable in patients with channelopathies. The most severely affected individuals presented with childhood‐onset gait and limb ataxia, with additional clinical features such as developmental delay, cognitive impairment, and seizures. At the milder end of the spectrum, other patients with channelopathies presented in adulthood with isolated gait difficulties. The patients with CACNA1A‐related disorder illustrated this broad phenotypic range, with age of onset ranging between 1 and 35 years in those 5 patients alone.
Even within the same family, clinical features were variable. For example, 2 of the 3 patients with CACNA1G‐related disorder had a relatively pure cerebellar syndrome, whereas the third also had cognitive impairment, muscle weakness, and seizures. We cannot exclude that this patient could have a second, distinct condition, however. Episodic symptoms, classically associated in the literature with channelopathies, were reported in only a few participants (3/12, 25%). Of them, 2 were siblings with intragenic deletions in CACNA1A. Deletions in this gene have previously been associated with the episodic ataxia type 2 phenotype. 37
Channelopathies accounted for the majority of the autosomal dominant conditions. The diagnostic yield of WES is typically thought to be better for de novo variants compared with inherited dominant variants, as filtering the proband's variants against those of their unaffected parents facilitates interpretation. In our study, 64% of families with autosomal dominant disorders carried de novo variants and 21% had inherited dominant variants.
Several of the 25 different genes identified in our cohort are not considered classical ataxia genes. It is therefore probable that WES was an important diagnostic tool for a subset of patients, as these genes would not have been included in commercially available ataxia gene panels. For example, variants in KCNC1 are typically associated with a progressive myoclonic epilepsy phenotype, with cerebellar atrophy as an additional manifestation. 38 Our patient with KCNC1‐related disorder, however, first presented with ataxia and later developed myoclonic and generalized tonic–clonic seizures. The BRAT1 gene is another example; it is typically associated with a lethal neonatal disorder, but our patients' phenotype was characterized by nonprogressive cerebellar ataxia with ocular motor apraxia. 23
Our results also reveal that not all patients with genetic ataxias present with gait or limb ataxia initially, especially when there is early onset of symptoms. The first clinical manifestations can be nonspecific, such as global developmental delay and hypotonia. Clinicians may not choose an ataxia gene panel in this context.
Only partial clinical information was available for several participants in our cohort, which represents a limitation of the study. This was most striking for the patients in the undiagnosed subgroup and may have contributed to the inability to define a genetic diagnosis. Missing information included age of onset (45%), the presence or absence of additional clinical manifestations (55%), and the presence or absence of additional neuroimaging features (68%). We suggest that these are key features in the evaluation of patients with genetic ataxias associated with cerebellar atrophy. We observed that more detailed clinical information correlated with a higher diagnostic yield of WES.
It is well established that WES remains of limited use for large deletions and duplications, repeat disorders, deep intronic variants, other forms of noncoding variants, and mosaicism. Some of the patients could have also had nongenetic causes of ataxia and cerebellar atrophy. In our study, investigations obtained by the referring clinicians prior to WES were based on clinical indication, which reflects a realistic diagnostic approach to genetic ataxias associated with cerebellar atrophy, and a practical clinical use of WES for this population. Given the genetic heterogeneity in this group of conditions and the contribution of chromosomal abnormalities and repeat disorders as causative mechanisms, we suggest that a multimodal approach is best, with a focus on detailed clinical evaluation and neuroimaging.
In summary, we report a high frequency of channelopathies in our large cohort of pediatric and adult patients with cerebellar atrophy. We suggest that WES is an important tool in the diagnostic approach for patients with genetic ataxias associated with cerebellar atrophy given the clinical and genetic heterogeneity in this group of conditions, especially when isolated cerebellar atrophy is the only imaging finding identified. WES is most useful in conjunction with a detailed clinical assessment and targeted investigations to identify additional neurological and systemic manifestations. Channelopathies are more common than previously reported in genetic ataxias associated with cerebellar atrophy, and likely represent an under‐recognized cause of these disorders.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
L.G.: 1C, 2A, 2B, 3A
T.H.: 1B, 1C, 2B, 2C, 3B
M. Tarnopolsky: 1C, 2C, 3B
D.A.D.: 1C, 2C, 3B
B.B.: 1C, 2C, 3B
M.T.G.: 1C, 2C, 3B
M. Tétreault 1C, 2B, 2C, 3B
S.A.: 1C, 2C, 3B
S.R.: 1C, 2C, 3B
K.C.: 1C, 2C, 3B
J.M.: 1C, 2B, 2C, 3B
F.B.: 1C, 2C, 3B
A.M.I.: 1C, 2C, 3B
G.R.: 1C, 2C, 3B
O.S.: 1C, 2C, 3B
K.M.B.: 1A, 1B, 1C, 2A, 2B, 2C, 3B
G.Y.: 1A, 1B, 1C, 2A, 2B, 2C, 3B
Disclosures
Ethical Compliance Statement
The study was approved by research ethics boards from all participating sites under research ethics board Protocol CHEOREB#11/04E—Enhanced Care for Rare Genetic Diseases in Canada. All patients and family members were enrolled following informed consent. The authors confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
This work was performed by the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI‐147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, Children's Hospital of Eastern Ontario Foundation, and The Hospital for Sick Children. The authors have no conflicts of interest to declare.
Financial Disclosures from the Previous 12 Months
L.G. has received grants from the Canadian Gene Cure Advanced Therapies for Rare Disease (Can‐GARD) and from the R.S. McLaughlin and Teva Canada Innovation funds from the Faculty of Medicine, Université Laval. The other authors have nothing to disclose.
Acknowledgments
We thank the patients and families for participating in this study; without them this work would not be possible. This work was performed under the Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Génome Québec, and Children's Hospital of Eastern Ontario Foundation.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Al‐Maawali A, Blaser S, Yoon G. Diagnostic approach to childhood‐onset cerebellar atrophy: a 10‐year retrospective study of 300 patients. J Child Neurol 2012;27(9):1121–1132. 10.1177/0883073812448680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poretti A, Wolf NI, Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur J Paediatric Neurol 2008;12(3):155–617. 10.1016/j.ejpn.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 3. Poretti A, Wolf NI, Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood: an update. Neuropediatrics 2015;46(6):359–370. 10.1055/s-0035-1564620. [DOI] [PubMed] [Google Scholar]
- 4. Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. Am J Neuroradiol 2002;23(7):1074–1087. [PMC free article] [PubMed] [Google Scholar]
- 5. Boltshauser E. Cerebellum‐small brain but large confusion: a review of selected cerebellar malformations and disruptions. Am J Med Genet A 2004;126a(4):376–385. 10.1002/ajmg.a.20662. [DOI] [PubMed] [Google Scholar]
- 6. D'Arrigo S, Vigano L, Grazia Bruzzone M, et al. Diagnostic approach to cerebellar disease in children. J Child Neurol 2005;20(11):859–866. 10.1177/08830738050200110101. [DOI] [PubMed] [Google Scholar]
- 7. Coutelier M, Coarelli G, Monin ML, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 2017;140(6):1579–1594. 10.1093/brain/awx081. [DOI] [PubMed] [Google Scholar]
- 8. Coutelier M, Hammer MB, Stevanin G, et al. Efficacy of exome‐targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 2018;75(5):591–599. 10.1001/jamaneurol.2017.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hadjivassiliou M, Martindale J, Shanmugarajah P, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry 2017;88(4):301–309. 10.1136/jnnp-2016-314863. [DOI] [PubMed] [Google Scholar]
- 10. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet 1983;1(8334):1151–1155. 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 11. Tirada N, Levy LM. Genetics of ataxias: hereditary forms. Am J Neuroradiol 2014;35(9):1681–1682. 10.3174/ajnr.A3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beaudin M, Klein CJ, Rouleau GA, Dupre N. Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias 2017;4:3. 10.1186/s40673-017-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steinlin M, Blaser S, Boltshauser E. Cerebellar involvement in metabolic disorders: a pattern‐recognition approach. Neuroradiology 1998;40(6):347–354. [DOI] [PubMed] [Google Scholar]
- 14. Sawyer SL, Schwartzentruber J, Beaulieu CL, et al. Exome sequencing as a diagnostic tool for pediatric‐onset ataxia. Hum Mutat 2014;35(1):45–49. 10.1002/humu.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zambonin JL, Bellomo A, Ben‐Pazi H, et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: a case series and review of this emerging congenital ataxia. Orphanet J Rare Dis 2017;12(1):121 10.1186/s13023-017-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gennarino VA, Palmer EE, McDonell LM, et al. A mild PUM1 mutation is associated with adult‐onset ataxia, whereas haploinsufficiency causes developmental delay and seizures. Cell 2018;172(5):924–36.e11. 10.1016/j.cell.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aldinger KA, Mosca SJ, Tetreault M, et al. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am J Hum Genet 2014;95(2):227–234. 10.1016/j.ajhg.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoch NC, Hanzlikova H, Rulten SL, et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017;541(7635):87–91. 10.1038/nature20790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boycott KM, Beaulieu CL, Kernohan KD, et al. Autosomal‐recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet 2015;97(6):886–893. 10.1016/j.ajhg.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eaton A, Bernier FP, Goedhart C, et al. Is PNPT1‐related hearing loss ever non‐syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1‐related disorders. Am J Med Genet A 2018;176(11):2487–2493. 10.1002/ajmg.a.40516. [DOI] [PubMed] [Google Scholar]
- 21. Joseph JT, Innes AM, Smith AC, et al. Neuropathologic features of pontocerebellar hypoplasia type 6. J Neuropathol Exp Neurol 2014;73(11):1009–1025. 10.1097/nen.0000000000000123. [DOI] [PubMed] [Google Scholar]
- 22. Donkervoort S, Sabouny R, Yun P, et al. MSTO1 mutations cause mtDNA depletion, manifesting as muscular dystrophy with cerebellar involvement. Acta Neuropathol 2019;138(6):1013–1031. 10.1007/s00401-019-02059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahjoub A, Cihlarova Z, Tétreault M, et al. Homozygous pathogenic variant in BRAT1 associated with nonprogressive cerebellar ataxia. Neurol Genet 2019;5(5):e359 10.1212/nxg.0000000000000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beaulieu CL, Majewski J, Schwartzentruber J, et al. FORGE Canada consortium: outcomes of a 2‐year national rare‐disease gene‐discovery project. Am J Hum Genet 2014;94(6):809–817. 10.1016/j.ajhg.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng PC, Henikoff S. SIFT predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31(13):3812–3814. 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7(4):248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 2010;7(8):575–576. 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 28. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15(7):901–913. 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Girdea M, Dumitriu S, Fiume M, et al. PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat 2013;34(8):1057–1065. 10.1002/humu.22347. [DOI] [PubMed] [Google Scholar]
- 31. Boddaert N, Desguerre I, Bahi‐Buisson N, et al. Posterior fossa imaging in 158 children with ataxia. J Neuroradiol 2010;37(4):220–230. 10.1016/j.neurad.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 32. Ohba C, Osaka H, Iai M, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013;14(3–4):225–232. 10.1007/s10048-013-0375-8. [DOI] [PubMed] [Google Scholar]
- 33. Kashimada A, Hasegawa S, Nomura T, et al. Genetic analysis of undiagnosed ataxia‐telangiectasia‐like disorders. Brain Dev 2019;41(2):150–157. 10.1016/j.braindev.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 34. Ramaekers VT, Heimann G, Reul J, Thron A, Jaeken J. Genetic disorders and cerebellar structural abnormalities in childhood. Brain 1997;120(Pt 10):1739–1751. 10.1093/brain/120.10.1739. [DOI] [PubMed] [Google Scholar]
- 35. Chen Z, Wang P, Wang C, et al. Updated frequency analysis of spinocerebellar ataxia in China. Brain 2018;141(4):e22 10.1093/brain/awy016. [DOI] [PubMed] [Google Scholar]
- 36. Coutelier M, Brice A, Stevanin G, Durr A. Reply: updated frequency analysis of spinocerebellar ataxia in China. Brain 2018;141(4):e23 10.1093/brain/awy018. [DOI] [PubMed] [Google Scholar]
- 37. Wan J, Mamsa H, Johnston JL, et al. Large genomic deletions in CACNA1A cause episodic ataxia type 2. Front Neurol 2011;2:51. 10.3389/fneur.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47(1):39–46. 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data presented in this study are available upon reasonable request.