

Crystal structures of the C-domain of P140 from M. genitalium adopt an alternative conformation to that recently found in the intact extracellular region of P140. The plasticity of the C-domain is related to the functioning of the mycoplasma adhesion complex.
Keywords: Mycoplasma genitalium, adhesins, Nap, gliding motility, infection
Abstract
The human pathogen Mycoplasma genitalium is responsible for urethritis in men, and for cervicitis and pelvic inflammatory disease in women. The adherence of M. genitalium to host target epithelial cells is mediated through an adhesion complex called Nap, which is essential for infectivity. Nap is a transmembrane dimer of heterodimers of the immunodominant proteins P110 and P140. The M. genitalium genome contains multiple copies of portions that share homology with the extracellular regions of P140 and P110 encoded by the genes mg191 and mg192, respectively. Homologous recombination between the genes and the copies allows the generation of a large diversity of P140 and P110 variants to overcome surveillance by the host immune system. Interestingly, the C-terminal domain (C-domain) of the extracellular region of P140, which is essential for the function of Nap by acting as a flexible stalk anchoring the protein to the mycoplasma membrane, presents a low degree of sequence variability. In the present work, the X-ray crystal structures of two crystal forms of a construct of the P140 C-domain are reported. In both crystal forms, the construct forms a compact octamer with D4 point-group symmetry. The structure of the C-domain determined in this work presents significant differences with respect to the structure of the C-domain found recently in intact P140. The structural plasticity of the C-domain appears to be a possible mechanism that may help in the functioning of the mycoplasma adhesion complex.
1. Introduction
Mycoplasmas are microorganisms that are characterized by their reduced genomes and their lack of a cell wall, presenting only a single phospholipid bilayer, the plasma membrane (Razin, 1992 ▸; Razin et al., 1998 ▸). These features make mycoplasmas unique among bacteria, explaining why they are used as model organisms in systems and synthetic biology (Xavier et al., 2014 ▸; Gibson et al., 2008 ▸; Glass et al., 2006 ▸). Many mycoplasmas live in close association with eukaryotic cells, either intracellularly or at the cell surface (Rottem, 2003 ▸). Mycoplasma genitalium, the causative agent of urethritis in men and cervicitis and pelvic inflammatory disease in women, presents arguably the smallest genome amongst organisms that are capable of self-replication and axenic growth (McGowin & Anderson-Smits, 2011 ▸; Glass et al., 2006 ▸). The adherence of M. genitalium to host target epithelial cells is mediated by an ∼0.5 MDa transmembrane complex called Nap, which is composed of a dimer of the heterodimer formed by the P110 and P140 proteins (Aparicio et al., 2018 ▸, 2020 ▸; Burgos et al., 2006 ▸; Mernaugh et al., 1993 ▸; Scheffer et al., 2017 ▸). In M. genitalium and in other members of the pneumonia cluster of mycoplasmas, such as the important human pathogen M. pneumoniae, the Nap adhesion complex also participates in a unique type of gliding cell motility that is essential for infectivity (Krause, 1996 ▸; Krause & Baseman, 1982 ▸; Radestock & Bredt, 1977 ▸; Burgos et al., 2006 ▸; García-Morales et al., 2016 ▸; Nakane et al., 2011 ▸; Seto et al., 2005 ▸). Besides their roles in adhesion and gliding motility, the P110 and P140 proteins are probably the most immunodominant proteins in M. genitalium (Svenstrup et al., 2002 ▸; Morrison-Plummer et al., 1987 ▸). The genome of M. genitalium contains several non-identical copies of portions of the extracellular regions of P110 and P140, named MgPar sequences (Ma et al., 2010 ▸; Peterson et al., 1995 ▸). By orthologous recombination with the corresponding genes, the copies allow the generation of a large diversity of variants of P110 and P140 in order to overcome surveillance by the host immune system (Iverson-Cabral et al., 2007 ▸; Ma et al., 2007 ▸). The only genetically invariable continuous stretches in the extracellular regions of P110 and P140 correspond to the C-terminal domain (C-domain) and the hinge connecting the N-domain and C-domain (Ma et al., 2010 ▸). The C-domain of P140 is even highly conserved when compared with the corresponding domain of the orthologous protein P1 from M. pneumoniae, suggesting a critical functional role that, together with its low genetic variability, makes this domain a promising therapeutic target. A better understanding of the cytoadherence and gliding mechanisms could facilitate the implementation of more effective antimicrobial approaches based on anti-adhesion therapies.
This work presents X-ray crystal structures of a construct from the P140 C-domain from M. genitalium in two different crystal forms. The structure of the isolated domain determined here shows major differences with respect to the recently reported structure of the C-domain when forming part of the intact Nap. Possible roles for the structural plasticity of the C-domain in the functioning of Nap are discussed.
2. Experimental procedures
2.1. Cloning, expression and protein purification
The C-domain of P140 was amplified from a synthetic clone of M. genitalium gene mg191 (GenScript) using the primers P140cF and P140cR (Supplementary Table S1). The PCR fragment was cloned to generate a construct comprising residues 1220–1351 with a His tag at the N-terminal end containing an upstream proteolytic sequence for protease 3C (Supplementary Fig. S1). Recombinant protein was obtained after 4 h of expression at 37°C in Escherichia coli B834 (DE3) cells (Merck) upon induction with 0.5 mM isopropyl β-d-1-thiogalactopyranoside. The cells were harvested and lysed by sonication in 1× PBS and 40 mM imidazole (binding buffer) followed by centrifugation at 20 000g at 4°C. The supernatant was charged onto a pre-equilibrated HisTrap 5 ml column (GE Healthcare) with binding buffer and was eluted with binding buffer containing 400 mM imidazole. Soluble aliquots were treated overnight with protease 3C in a 1:100 molar ratio (protease 3C:P140 C-domain) and subsequently passed through a reverse HisTrap column. Finally, the aliquots were pooled and loaded onto a Superdex 200 GL 10/300 column (GE Healthcare) pre-equilibrated with 20 mM Tris pH 7.4, 150 mM NaCl. To prepare a selenomethionine (SeMet) derivative of the C-domain, bacteria were grown in SelenoMethionine Medium Complete (Molecular Dimensions) using the protocol provided with the kit. Purification of the SeMet-labeled C-domain was performed following the same steps as described for the native protein.
2.2. Crystallization, data collection and structure determination
Crystallization screening was performed using either a Cartesian or a Crystal Phoenix robot by mixing 150 nl crystallization condition with 150 nl protein solution at 6 mg ml−1 in 96-well plates. The best diffracting crystals belonged to the monoclinic space group C2 (unit-cell parameters a = 114.5, b = 83.0, c = 118.2 Å, β = 117.5°) and the tetragonal space group I422 (unit-cell parameters a = 90.1, b = 90.1, c = 58.0 Å). The monoclinic crystals were obtained in crystallization buffer consisting of 0.15 M dl-malic acid, 0.1 M bis-Tris propane pH 7.0 and the tetragonal crystals were obtained in crystallization buffer consisting of 20% PEG 3350, 0.02 M MgCl2·6H2O, 0.1 M Tris pH 8.5. All crystals used for data collection were flash-cooled in liquid nitrogen with 15% glycerol as a cryoprotectant.
X-ray data sets for the monoclinic and tetragonal crystal forms were collected to resolutions of 1.98 and 1.43 Å, respectively. X-ray data collection was carried out on the XALOC beamline (Juanhuix et al., 2014 ▸) at the ALBA Synchrotron, Spain. SAD data were collected from an SeMet-derivatized tetragonal crystal at the Se K-edge energy. Data were processed in xia2 (Winter, 2010 ▸) using XDS (Kabsch, 2010 ▸), and AIMLESS and POINTLESS (Evans, 2006 ▸) from the CCP4 suite of programs (Winn et al., 2011 ▸). The structure of the C-terminal domain construct was solved by SAD from a tetragonal SeMet-derivatized crystal at 1.65 Å resolution using SHELX/E (Sheldrick, 2015 ▸). The structures of the native tetragonal and monoclinic crystals were then solved by molecular replacement using Phaser, and contained one and eight subunits in the asymmetric unit, respectively. The structures were refined by alternating interactive and automatic cycles with Coot (Emsley et al., 2010 ▸) and REFMAC (Murshudov et al., 2011 ▸), giving agreement factors R and R free of 18.3% and 20.0%, respectively, for the I422 structure, and 17.6% and 20.6%, respectively, for the C2 structure (Table 1 ▸). The final refined structures have been deposited in the PDB as entry 6rcc for the I422 structure and 6rcd for the C2 structure.
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| P140 C-domain, SeMet derivative | P140 C-domain | P140 C-domain | |
|---|---|---|---|
| Data-collection statistics | |||
| Space group | I422 | I422 | C2 |
| a, b, c (Å) | 90.21, 90.21, 58.17 | 90.10, 90.10, 57.98 | 114.51, 83.02, 118.16 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 117.50, 90.00 |
| Unique reflections | 14807 (2121) | 20651 (3175) | 67417 (9766) |
| Resolution (Å) | 48.89–1.65 (1.74–1.65) | 45.04–1.43 (1.51–1.43) | 52.41–1.98 (2.09–1.98) |
| Wavelength (Å) | 0.97911 | 1.07216 | 1.07216 |
| R merge † (%) | 0.104 (1.565) | 0.09 (1.73) | 0.08 (0.90) |
| 〈I/σ(I)〉 | 16.3 (2.0) | 11.50 (1.5) | 10.50 (1.9) |
| CC1/2 | 0.997 (0.644) | 0.998 (0.71) | 0.998 (0.64) |
| CCano | 0.289 (0.011) | ||
| Completness (%) | 100.00 (99.90) | 92.41 (99.8) | 98.30 (99.5) |
| Multiplicity | 6.4 (4.1) | 6.3 (6.3) | 3.40 (3.4) |
| Refinement statistics | |||
| Resolution | 45.04–1.43 (1.51–1.43) | 82.41–1.98 (1.99–1.98) | |
| No. of reflections | 20650 (3012) | 67416 (1284) | |
| R cryst ‡ (%) | 18.30 (23.84) | 17.80 (22.00) | |
| R free § (%) | 19.90 (26.87) | 20.60 (27.24) | |
| No. of residues (in unit cell) | 99 | 782 | |
| No. of ligands | 2 | 0 | |
| Solvent content (%) | 51.80 | 54.43 | |
| Average B factor (Å2) | 29.94 | 46.22 | |
| Coordinate error¶ (Å) | 0.20 | 0.23 | |
| R.m.s. deviation, bond lengths (Å) | 0.01 | 0.01 | |
R
merge = 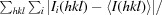
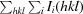 , where Ii(hkl) is the intensity of an observation and 〈I(hkl)〉 is the mean value of observations for an unique reflection.
, where Ii(hkl) is the intensity of an observation and 〈I(hkl)〉 is the mean value of observations for an unique reflection.
R
cryst = 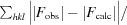
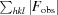 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated with 5% of the data, which were excluded from refinement.
Based on maximum likelihood.
2.3. SEC-MALS analysis
The molecular weight and oligomerization of the construct in solution were measured using a Superdex 75 10/300 GL column (GE Healthcare) in a Prominence liquid-chromatography system (Shimadzu) connected to a DAWN HELEOS II multi-angle light-scattering (MALS) detector and an Optilab T-rEX refractive-index (dRI) detector (Wyatt Technology). The ASTRA 7 software (Wyatt Technology) was used for data processing and analysis of the results. A dn/dc value of 0.185 ml g−1 (typical of proteins) was assumed for calculations.
3. Results
3.1. Structure determination of the C-domain from P140
Tetragonal and monoclinic crystals were obtained from a construct of the C-domain of P140 from M. genitalium spanning residues Lys1220–Asp1351 (Supplementary Fig. S1) with a two-residue extension (Gly-Pro) at the N-terminal end (remaining after the removal of the 6×His tag). The sequence contained two methionines, which allowed the preparation of an SeMet derivative in order to collect an anomalous data set (at 1.65 Å resolution) from the tetragonal crystals that was solved by SAD using SHELXD and SHELXE (Sheldrick, 2015 ▸; Table 1 ▸). These tetragonal crystals, which belonged to space group I422, contained one subunit in the crystal asymmetric unit, with a solvent content of 52% by volume. A complete model was built in the experimental map (Fig. 1 ▸ a), which was then refined with REFMAC (Murshudov et al., 2011 ▸) using a diffraction data set at 1.4 Å resolution obtained from a native protein crystal, giving final agreement factors R and R free of 18.3% and 20.0%, respectively (Table 1 ▸). The monoclinic crystals, which belonged to space group C2, were then solved by molecular replacement with Phaser (McCoy, 2007 ▸) using the structure determined from the tetragonal crystals as a search model. The monoclinic crystals contained eight subunits in the asymmetric unit, with a solvent-content volume of 54%, and the structure was refined using REFMAC to a resolution of 1.98 Å using noncrystallographic symmetry restraints, giving final agreement factors R and R free of 17.6% and 20.6%, respectively (Fig. 1 ▸ b and Table 1 ▸).
Figure 1.

Structure of the isolated C-domain from P140. (a) Two views (90° apart from each other) of the structure of one subunit of the C-domain from P140 as found in the asymmetric unit of the tetragonal crystals. The three strands in the β-sheet are indicated. The third strand is interrupted by an insertion of ten residues that is partially disordered. The positions of an Na+ ion and a Cl− ion are also depicted. (b) Two views, 90° apart, down and perpendicular to the fourfold axis, of the octamer with D4 symmetry found in the asymmetric unit of the monoclinic crystals. (c) In spite of the differences in the unit-cell content, the structures of the subunits and the packing interactions are similar in both the tetragonal (I422, upper panels, with one subunit in the asymmetric unit shown in red) and the monoclinic (C2, lower panels, with a whole octamer shown in color) crystals.
3.2. Overall structure of the isolated C-domain
The C-domain construct presents a structure that is essentially identical in the tetragonal and monoclinic crystal forms. In both crystals, the closest neighboring subunits are organized as compact octamers with D4 point-group symmetry (Fig. 1 ▸ c). In the tetragonal crystal form, with only one subunit in the asymmetric unit, the fourfold and the twofold axes of the octamer are coincident with crystallographic symmetry axes, implying that all of the subunits in the octamer are identical. In the monoclinic crystal, a whole octamer is found in the asymmetric unit, with the eight subunits related by accurate fourfold and twofold noncrystallographic symmetries. The packing of the octamers is very similar in both crystal forms, despite the differing space groups (Fig. 1 ▸ c).
The structure determined contains a few disordered residues at the N-terminal end, which were not included in the model. These are followed by an extended stretch of about 20 residues that protrudes away from the subunit (Fig. 1 ▸ a) and interacts extensively with other subunits in the octamer (Figs. 2 ▸ a and 2 ▸ b). There is then a zeta-like motif, approximately from Pro1268 to Ala1298, with two loosely antiparallel helices and an irregular stretch that reaches the first β-strand of a three-stranded antiparallel β-sheet. The third β-strand, which is the last strand in the β-sheet, is interrupted by a ten-residue β-bulge (Pro1330–Pro1339) that is exposed to the solvent and contains a few disordered residues in its central part, with the coordinates corresponding to residues Asn1333–Ser1336 missing in most of the subunits. The interaction between the zeta-like motif and the β-sheet defines a hydrophobic core of the domain, in which not even a single solvent (water) molecule has been found despite the large number of solvent molecules that were modeled in the tetragonal crystal, with a ratio of 1.3 waters per residue, owing to the quality and the resolution available.
Figure 2.

Interactions between subunits in the octamer. Two views, 90° apart from each other, showing the intersubunit interactions within the octamer. The insets show details of the residues involved in the interactions, with hydrogen bonds depicted as dashed lines. (a) View down the fourfold axis, showing interactions between subunits related by fourfold symmetry. (b) View down a twofold axis perpendicular to that in (a). (c) View down a twofold axis, showing interactions between the N- and C-terminal tails of the subunits related by this symmetry.
In the octamer, interactions between subunits cover a large surface across the fourfold axis, with an averaged contact area between each neighboring pair of subunits of about 1200 Å2 and an estimated free energy of −14 kcal mol−1 according to PISA (Krissinel & Henrick, 2007 ▸; Figs. 2 ▸ a and 2 ▸ b). Interactions between pairs of subunits cover a smaller surface across the twofold axis, but each subunit interacts with three other subunits through the twofold symmetries (with contact areas of about 500, 250 and 30 Å2, respectively), which add up in the octamer (Figs. 2 ▸ a and 2 ▸ c). Therefore, although the tendency to form dimers between subunits might be weak, dimerization of the tetramers is expected to be favorable, with a contacting surface between tetramers of about 2750 Å2 and with the observed D4 octamers as the most stable oligomer. However, SEC-MALS analysis indicates that at the concentrations used (see Section 2) the C-domain construct remains monomeric in solution (Fig. 3 ▸). The interface between the two tetramers exclusively involves the N- and C-terminal ends of the eight subunits that are sandwiched at both sides by the four domain cores and contains a large number of solvent (water) molecules, reflecting that it is a hydrophilic interface. The entropic cost of fixing the many solvent molecules and all of the subunit ends together with the hydrophilic character of the interface might explain the presence of monomers in solution at the concentrations tested despite the large intersubunit contacting surface in the octamers.
Figure 3.

SEC-MALS analysis of the C-domain of P140. SEC-MALS analysis of purified samples of the C-domain from P140 detected two species. The most abundant (peak 1, 98.86%), with a molecular weight of 16.71 kDa, corresponds to the monomeric species. A second species is observed (peak 2), with a molecular weight of 42.49 kDa, that could correspond to either unstable oligomers or to some contamination.
3.3. Comparison of the C-domain structures
Superposition of the structures of the C-domain construct solved in this work with the C-domain in the recently reported structure of P140 gives an r.m.s.d. of 1.0 Å for 55 equivalent residues (Fig. 4 ▸ a). Thus, only about half (53%) of the residues are structurally equivalent between the two C-domain structures, while the other half of the residues show large structural differences, although the sequence is the same in both structures. The hydrophobic core, which includes the zeta-like motif and the three antiparallel strands of the β-sheet described above, is well conserved. In contrast, the N-terminal end region and the β-bulge (Pro1330–Pro1339), which emerges from the middle of the third β-strand in the three-stranded β-sheet, adopt different structures (Fig. 4 ▸ a). There are also some structural differences for the residues at the very C-terminal end.
Figure 4.

Comparison of the P140 C-domain structures. (a) Two views (90° apart from each other) of the superposed C-domain structures as found in this work (green) and in the structure of the whole P140 ectodomain (PDB entry 6s3u; red; Aparicio et al., 2020 ▸). The largest differences are found in the N- and C-terminal tails and in the β-bulge. (b) The presence of the P140 N-domain would clash with the C-domain β-bulge as seen in this work, suggesting that movement of the β-bulge can be associated with movement of the N- and C-domains with respect to one another. (c) The insets show details of the interactions found in the whole P140 ectodomain structure that occur in the N-terminal end with either residues Leu1290 and Asn1291 (left) or with the β-bulge (right). (d) Scheme of the possible transition between the C-domain structures found in this work and in the whole P140 ectodomain. Disturbing the interactions of the N-terminus from the whole P140 ectodomain structure would destabilize this region, which in turn would eliminate the interactions with the β-bulge.
The contacting surface between the N- and C-domains in P140 is only about 650 Å2 (according to PISA; Krissinel & Henrick, 2007 ▸; Fig. 4 ▸ b). In the C-domain these interactions only involve the β-bulge and five other residues from the first and second β-strands of the β-sheet (Lys1303, Ile1305, Ser1306 and Val1316-Arg1317). As the conformations of these residues and of the corresponding strands remain essentially unchanged in the C-domain structures, only the absence of the interactions between the N-domain and the β-bulge appears to be responsible for the changes that appear in the isolated C-domain. The absence of these interactions can directly explain the conformational change of the β-bulge, which is extended in the structure solved in this work and bent in P140 (Fig. 4 ▸ b). In turn, the extended β-bulge in the isolated C-domain lacks interactions with the N-terminal end region of the domain in P140, which changes the conformation of this N-terminal end region (Figs. 4 ▸ c and 4 ▸ d). Therefore, it appears that the β-bulge acts like a trigger that can be activated, one way or another, by just a few interactions with the N-domain of P140. Finally, it is worth mentioning that there are no interactions similar to those found between the subunits in the octamer that forms the isolated C-domain either in P140 or in the whole Nap (the structure of which has also recently been reported at ∼15 Å resolution from cryo-electron tomography and subtomogram averaging studies; Aparicio et al., 2020 ▸).
Superposition of the C-domains from the Nap proteins P140 and P110 (Aparicio et al., 2018 ▸), which present a unique topology (according to DALI; Holm & Rosenström, 2010 ▸), gives an r.m.s.d. of 2.2 Å for 70 equivalent residues (Fig. 5 ▸ a). A β-bulge is also present in the corresponding β-strand from P110 and interacts with the N-domain (Fig. 5 ▸ b). However, in P110 the β-bulge is extended and oriented in a direction that is not like that in P140, but is close to that found in the structure of the isolated C-domain. In P110 the N-end region of the C-domain appears to be stabilized by an extra hairpin with respect to P140 (Fig. 5 ▸ a), which suggests that the C-domain of P110 does not have a triggering mechanism like that in P140.
Figure 5.

Comparison of P140 and P110 C-domain structures. (a) Two views (90° apart from each other) of the superposed C-domain structures as found in this work (green) and in the structure of the whole P110 ectodomain (PDB entry 6r3t; purple; Aparicio et al., 2018 ▸). Both structures present a β-bulge, with a similar extended conformation, inserted at the same position in the structurally equivalent β-strand. (b) The relative position of the N- and C-domains differs between the P140 and P110 structures determined. The inset shows details of the interactions between the extended β-bulge and the P110 N-domain. Similar interactions might take place in P140 when the β-bulge adopts the extended conformation.
4. Discussion
The extracellular region of P140, which is probably the most immunodominant protein from M. genitalium, was predicted to contain a large N-domain and a small C-domain inmediately followed by a transmembrane helix. Here, we present the structure of the isolated C-domain. Initially, the reasons for undertaking structure determination of the isolated C-domain were mainly methodological as a possible means of contributing to the structure determination of the whole extracellular region of P140, for which crystals were available but were difficult to solve. However, the structure of the isolated C-domain, determined in this work by SAD using an SeMet derivative and refined at high resolution, did not provide any match on molecular replacement with the P140 crystals, indicating important differences between the structures of the isolated domain and of the domain within P140. Recently, the structure of P140 has become available (Aparicio et al., 2020 ▸), confirming the important structural differences between the two C-domain structures, with only about half of the residues retaining structurally equivalent positions.
In the structure of P140, the interface between the N- and C-domains is small, involving only a few residues from the C-domain, all of which belong to a three-stranded β-sheet or to a β-bulge (Pro1330–Pro1339) that emerges from the middle of the third β-strand and is where the strongest interactions occur. The β-strands remain essentially unchanged between the two C-domain structures, while the β-bulge undergoes a complete rearrangement. The absence of interactions between the N-domain and the β-bulge might be the main trigger for the changes that are observed between the C-domain structures. It is interesting that these few interactions could be critical for the structural stability of about half of the residues in the C-domain. It is also surprising to find that in the second half of the domain the residues remain essentially unchanged.
The C-domain of P140 appears to be designed to contain a malleable region that is very sensible to interactions with the N-domain, and a second region where the transmembrane helix is connected that is not affected by interactions with the N-domain. From the experimental information on P140, it is clear that the N- and C-domains can move with respect to each another (Aparicio et al., 2020 ▸). These movements may be required by the conformational transitions that Nap experiences during the cycle of attachment to and release from the sialic acid oligosaccharide cell host receptors. Therefore, even subtle signals from the N-domain, possibly initiated by the binding/release of Nap to the cell receptors, that alter the interaction with the C-domain β-bulge are expected to trigger structural changes in the C-domain that allow a global conformational transition of Nap. The residues involved in this mechanism, in particular the presence of the β-bulge in the C-domain, are well preserved in P1, the orthologue of P140 from M. pneumoniae, suggesting that this might be a general characteristic of the adhesion/release process in mycoplasmas from the pneumoniae cluster.
5. Related literature
The following reference is cited in the supporting information for this article: McGuffin et al. (2000 ▸).
Supplementary Material
PDB reference: P140 C-domain, space group C2, 6rcd
PDB reference: space group I422, 6rcc
Schematic representation of the regions in P140 and the construct used in this study. DOI: 10.1107/S2053230X20012297/jb5021sup1.pdf
Acknowledgments
Many thanks are given to the Crystallization Platform at the PCB and particularly to Dr Roman Bonet. The authors also acknowledge the beamtime granted through proposal 2017062252 at BL13-XALOC, ALBA Synchrotron, Spain.
Funding Statement
This work was funded by Ministerio de Economía, Industria y Competitividad, Gobierno de España grant BFU2018-101265-B-100.
References
- Aparicio, D., Scheffer, M. P., Marcos-Silva, M., Vizarraga, D., Sprankel, L., Ratera, M., Weber, M. S., Seybert, A., Torres-Puig, S., Gonzalez-Gonzalez, L., Reitz, J., Querol, E., Piñol, J., Pich, O. Q., Fita, I. & Frangakis, A. S. (2020). Nat. Commun. 11, 2877. [DOI] [PMC free article] [PubMed]
- Aparicio, D., Torres-Puig, S., Ratera, M., Querol, E., Piñol, J., Pich, O. Q. & Fita, I. (2018). Nat. Commun. 9, 4471. [DOI] [PMC free article] [PubMed]
- Burgos, R., Pich, O. Q., Ferrer-Navarro, M., Baseman, J. B., Querol, E. & Piñol, J. (2006). J. Bacteriol. 188, 8627–8637. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- García-Morales, L., González-González, L., Querol, E. & Piñol, J. (2016). Mol. Microbiol. 100, 125–138. [DOI] [PubMed]
- Gibson, D. G., Benders, G. A., Andrews-Pfannkoch, C., Denisova, E. A., Baden-Tillson, H., Zaveri, J., Stockwell, T. B., Brownley, A., Thomas, D. W., Algire, M. A., Merryman, C., Young, L., Noskov, V. N., Glass, J. I., Venter, J. C., Hutchison, C. A. & Smith, H. O. (2008). Science, 319, 1215–1220. [DOI] [PubMed]
- Glass, J. I., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M. R., Maruf, M., Hutchison, C. A., Smith, H. O. & Venter, J. C. (2006). Proc. Natl Acad. Sci. USA, 103, 425–430. [DOI] [PMC free article] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Iverson-Cabral, S. L., Astete, S. G., Cohen, C. R. & Totten, P. A. (2007). Mol. Microbiol. 66, 55–73. [DOI] [PubMed]
- Juanhuix, J., Gil-Ortiz, F., Cuní, G., Colldelram, C., Nicolás, J., Lidón, J., Boter, E., Ruget, C., Ferrer, S. & Benach, J. (2014). J. Synchrotron Rad. 21, 679–689. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Krause, D. C. (1996). Mol. Microbiol. 20, 247–253. [DOI] [PubMed]
- Krause, D. C. & Baseman, J. B. (1982). Infect. Immun. 37, 382–386. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Ma, L., Jensen, J. S., Mancuso, M., Hamasuna, R., Jia, Q., McGowin, C. L. & Martin, D. H. (2010). PLoS One, 5, e15660. [DOI] [PMC free article] [PubMed]
- Ma, L., Jensen, J. S., Myers, L., Burnett, J., Welch, M., Jia, Q. & Martin, D. H. (2007). Mol. Microbiol. 66, 220–236. [DOI] [PMC free article] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- McGowin, C. L. & Anderson-Smits, C. (2011). PLoS Pathog. 7, e1001324. [DOI] [PMC free article] [PubMed]
- McGuffin, L. J., Bryson, K. & Jones, D. T. (2000). Bioinformatics, 16, 404–405. [DOI] [PubMed]
- Mernaugh, G. R., Dallo, S. F., Holt, S. C. & Baseman, J. B. (1993). Clin. Infect. Dis. 17, S69–S78. [DOI] [PubMed]
- Morrison-Plummer, J., Jones, D. H., Daly, K., Tully, J. G., Taylor-Robinson, D. & Baseman, J. B. (1987). Isr. J. Med. Sci. 23, 453–457. [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nakane, D., Adan-Kubo, J., Kenri, T. & Miyata, M. (2011). J. Bacteriol. 193, 715–722. [DOI] [PMC free article] [PubMed]
- Peterson, S. N., Bailey, C. C., Jensen, J. S., Borre, M. B., King, E. S., Bott, K. F. & Hutchison, C. A. (1995). Proc. Natl Acad. Sci. USA, 92, 11829–11833. [DOI] [PMC free article] [PubMed]
- Radestock, U. & Bredt, W. (1977). J. Bacteriol. 129, 1495–1501. [DOI] [PMC free article] [PubMed]
- Razin, S. (1992). FEMS Microbiol. Lett. 100, 423–431. [DOI] [PubMed]
- Razin, S., Yogev, D. & Naot, Y. (1998). Microbiol. Mol. Biol. Rev. 62, 1094–1156. [DOI] [PMC free article] [PubMed]
- Rottem, S. (2003). Physiol. Rev. 83, 417–432. [DOI] [PubMed]
- Scheffer, M. P., Gonzalez-Gonzalez, L., Seybert, A., Ratera, M., Kunz, M., Valpuesta, J. M., Fita, I., Querol, E., Piñol, J., Martín-Benito, J. & Frangakis, A. S. (2017). Mol. Microbiol. 105, 869–879. [DOI] [PubMed]
- Seto, S., Kenri, T., Tomiyama, T. & Miyata, M. (2005). J. Bacteriol. 187, 1875–1877. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Svenstrup, H. F., Nielsen, P. K., Drasbek, M., Birkelund, S. & Christiansen, G. (2002). J. Med. Microbiol. 51, 361–373. [DOI] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Winter, G. (2010). J. Appl. Cryst. 43, 186–190.
- Xavier, J. C., Patil, K. R. & Rocha, I. (2014). Microbiol. Mol. Biol. Rev. 78, 487–509. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: P140 C-domain, space group C2, 6rcd
PDB reference: space group I422, 6rcc
Schematic representation of the regions in P140 and the construct used in this study. DOI: 10.1107/S2053230X20012297/jb5021sup1.pdf