

Crystal structures of a bacterial nonhydrolyzing UDP-GlcNAc 2-epimerase with and without substrates are described. The structures, when compared with homologous enzymes, shed additional light on the mechanism and understanding of the non-allosteric nature of the enzyme.
Keywords: UDP-GlcNAc, UDP-ManNAc, epimerases, epimerization, Rossmann fold, X-ray crystallography, Neisseria meningitidis
Abstract
Bacterial nonhydrolyzing UDP-N-acetylglucosamine 2-epimerases catalyze the reversible interconversion of UDP-N-acetylglucosamine (UDP-GlcNAc) and UDP-N-acetylmannosamine (UDP-ManNAc). UDP-ManNAc is an important intermediate in the biosynthesis of certain cell-surface polysaccharides, including those in some pathogenic bacteria, such as Neisseria meningitidis and Streptococcus pneumoniae. Many of these epimerases are allosterically regulated by UDP-GlcNAc, which binds adjacent to the active site and is required to initiate UDP-ManNAc epimerization. Here, two crystal structures of UDP-N-acetylglucosamine 2-epimerase from Neisseria meningitidis serogroup A (NmSacA) are presented. One crystal structure is of the substrate-free enzyme, while the other structure contains UDP-GlcNAc substrate bound to the active site. Both structures form dimers as seen in similar epimerases, and substrate binding to the active site induces a large conformational change in which two Rossmann-like domains clamp down on the substrate. Unlike other epimerases, NmSacA does not require UDP-GlcNAc to instigate the epimerization of UDP-ManNAc, although UDP-GlcNAc was found to enhance the rate of epimerization. In spite of the conservation of residues involved in binding the allosteric UDP-GlcNAc observed in similar UDP-GlcNAc 2-epimerases, the structures presented here do not contain UDP-GlcNAc bound in the allosteric site. These structural results provide additional insight into the mechanism and regulation of this critical enzyme and improve the structural understanding of the ability of NmSacA to epimerize modified substrates.
1. Introduction
Bacterial capsular polysaccharides (CPSs) are distinct, organized structures that are found on the surface of a wide range of bacterial species and are important virulence factors that provide protection from various circumstances, ranging from harsh environmental conditions to the immune system of a host (Roberts, 1996 ▸; Cress et al., 2014 ▸).
Neisseria meningitidis is a Gram-negative bacterium with at least 12 known serogroups which are classified based on their CPS structures. N. meningitidis serogroup A has historically been responsible for large epidemics of meningitis and septicemia in the meningitis-belt countries and still causes life-threatening invasive meningococcal diseases in some countries (Aye et al., 2020 ▸; Dutta et al., 2020 ▸; Novak et al., 2019 ▸). The CPS of N. meningitidis serogroup A is a homopolymer of (–6ManNAcα1-PO4–) and is unique compared with other disease-causing N. meningitidis serogroups, including serogroup X, which has a CPS consisting of a (–4GlcNAcα1-PO4–) homopolymer (Xie et al., 2012 ▸), and serogroups B, C, W-135 and Y, which all contain sialic acid in their CPSs (Fiebig et al., 2014 ▸; Jennings et al., 1977 ▸). The cps operon of N. meningitidis serogroup A contains four open reading frames, with the first being sacA, which encodes an enzyme (NmSacA) that catalyzes the first step in the N. meningitidis serogroup A CPS biosynthetic pathway (Swartley et al., 1998 ▸). This enzyme (EC 5.1.3.14) catalyzes the interconversion of uridine 5′-diphosphate N-acetylglucosamine (UDP-GlcNAc) to its C2′′-epimer UDP-N-acetylmannosamine (UDP-ManNAc) (Zhang et al., 2016 ▸; Swartley et al., 1998 ▸).
NmSacA has been verified to be a nonhydrolyzing UDP-GlcNAc 2-epimerase (Zhang et al., 2016 ▸). The mechanism of this type of bacterial epimerase is believed to involve anti elimination of the C2′′ proton and UDP from UDP-GlcNAc, generating enzyme-bound intermediates of UDP and 2-acetamidoglucal, followed by the subsequent syn addition of a proton to C2′′ and UDP to the same face of the double bond, producing the UDP-ManNAc product (Fig. 1 ▸; Morgan et al., 1997 ▸; Tanner, 2002 ▸). NmSacA was shown to release 2-acetamidoglucal and UDP, which were easily observed when the reactions were carried out for an extended period of time (Zhang et al., 2016 ▸).
Figure 1.

The reaction catalyzed by NmSacA.
Bacterial nonhydrolyzing UDP-GlcNAc 2-epimerases are found in both Gram-negative and Gram-positive strains and vary widely in their regulation. Those from the Gram-positive bacteria Bacillus anthracis (Velloso et al., 2008 ▸), B. cereus (Kawamura et al., 1978 ▸) and Staphylococcus aureus (Mann et al., 2016 ▸), as well as those from the Gram-negative bacterium Escherichia coli O14:K7:H (Morgan et al., 1997 ▸) and the archaeon Methanococcus jannaschii (Chen et al., 2014 ▸), have been shown to be allosterically regulated, requiring UDP-GlcNAc to catalyze the reversible epimerization of UDP-ManNAc to UDP-GlcNAc (Kawamura et al., 1979 ▸; Morgan et al., 1997 ▸). In the absence of UDP-GlcNAc, the enzymes were shown not to epimerize UDP-ManNAc, but UDP-GlcNAc alone can be epimerized readily to form UDP-ManNAc until equilibrium is reached. The crystal structures of these enzymes reveal that the allosteric UDP-GlcNAc binds in a conserved site adjacent to the active site, which contains a bound UDP after the GlcNAc is hydrolyzed from UDP-GlcNAc during an extended incubation time for crystal growth (Chen et al., 2014 ▸; Velloso et al., 2008 ▸). The GlcNAc moiety of the UDP-GlcNAc allosteric effector makes extensive interactions with the pyrophosphate of the UDP in the active site. The allosteric UDP-GlcNAc binding is presumed to optimize the conformation of the active site and to exclude solvent.
UDP-GlcNAc was not required for the activity of NmSacA in epimerizing UDP-ManNAc to UDP-GlcNAc, but UDP-GlcNAc did appear to increase the initial rate of epimerization (Zhang et al., 2016 ▸). To better understand the epimerization reaction mechanism of NmSacA and its apparent lack of a requirement for UDP-GlcNAc for reactivity, and to determine the structural basis of its activity on modified substrates, we solved two crystal structures of NmSacA: a ligand-free structure and a structure bound to the ligand UDP-GlcNAc.
2. Materials and methods
2.1. Cloning, expression and purification
As reported previously (Zhang et al., 2016 ▸), the gene that encodes NmSacA was amplified from the genomic DNA of N. meningitidis serogroup A strain M1027 and cloned into pET-22b(+) vector (Novagen) with a C-terminal His6 tag using the NdeI and XhoI restriction sites. Plasmids were sequenced to verify the correct ligation and were transformed into E. coli BL21 (DE3) cells (Invitrogen) for expression.
Plasmid-bearing strains were grown in LB-rich medium with ampicillin (100 µg ml−1) to an OD at 600 nm of 0.8. Overexpression was induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside and the cultures were incubated at 293 K for 20 h with shaking.
After 20 h of incubation, the cells were pelleted from the culture by centrifugation at 3400g for 2 h. The cell pellet was resuspended in lysis buffer consisting of 100 mM Tris–HCl buffer pH 8.0, 0.1% Triton X-100. The cells were lysed by treatment with 50 µg ml−1 lysozyme and 3 µg ml−1 DNase I for 60 min at 310 K with vigorous shaking. The cell lysate was cleared by centrifugation at 14 905g for 30 min. The His6-tagged protein was purified from the cell lysate using a nickel–nitrilotriacetic acid (Ni2+–NTA) column. The column was equilibrated with ten column volumes (CV) of binding buffer consisting of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 5 mM imidazole. The cell lysate was loaded onto the column, which was then washed with 8 CV of binding buffer followed by 10 CV of washing buffer consisting of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 40 mM imidazole. The protein was eluted from the column using an elution buffer consisting of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 200 mM imidazole. Elution fractions containing the purified protein were collected, dialyzed and stored at 277 K.
2.2. Crystallization
For the ligand-free structure, purified NmSacA at 10 mg ml−1 was crystallized by hanging-drop vapor diffusion using a reservoir solution consisting of 50%(v/v) PEG 200, 100 mM phosphate–citrate pH 4.2, 200 mM NaCl (condition H12 of the Wizard Cryo screen, Rigaku Reagents, USA). The crystals were flash-cooled in liquid nitrogen prior to data collection.
For the substrate-bound structure, NmSacA (13 mg ml−1) was crystallized by sitting-drop vapor diffusion against a solution consisting of 22% PEG 5000, 100 mM sodium citrate/citric acid pH 5.5, 10 mM UDP-GlcNAc. The crystals were briefly soaked in a solution of 30% ethylene glycol with 10 mM UDP-GlcNAc in mother liquor and flash-cooled in liquid nitrogen.
2.3. Data collection and structure determination
X-ray diffraction data for the ligand-free structure were collected on beamline 7-1 at SSRL with a wavelength of 1.12709 Å and a 100 µm beam size using an ADSC Q315 detector at a distance of 200 mm. Diffraction data were integrated and scaled with XDS and XSCALE (Kabsch, 2010a ▸,b ▸). The phases were determined by molecular replacement with Phaser (McCoy et al., 2007 ▸) using the structure of a nonhydrolyzing UDP-GlcNAc 2-epimerase from B. subtilis (PDB entry 4fkz; C.-S. Yang, S.-C. Chen, S.-M. Kuan, Y.-R. Chen, Y.-H. Liu & Y. Chen, unpublished work) as a phasing model. X-ray diffraction data for the UDP-GlcNAc-bound structure were collected on beamline 7-1 at SSRL with a wavelength of 1.03317 Å and a beam size of 150 × 100 µm using an ADSC Q315 detector at a distance of 300 mm. Data were collected using an oscillation angle of 0.2° with a 7 s exposure time and were integrated and scaled with XDS and XSCALE (Kabsch, 2010a ▸,b ▸). The initial phases were determined by molecular replacement (Phaser) using the two individual domains of the ligand-free structure as search models. The structures were refined using the Phenix package (Liebschner et al., 2019 ▸). The data-collection and refinement statistics are summarized in Table 1 ▸. Less than 1% of the residues reside in the disallowed region of the Ramachandran plot. The majority of these outliers fall in the β2–α2 and β3–α3 loops, which are juxtaposed and have weakly defined electron density.
Table 1. Data-collection and refinement statistics for NmSacA.
Values in parentheses are for the highest resolution shell.
| Structure | No substrate | With UDP-GlcNAc |
|---|---|---|
| PDB code | 6vlb | 6vlc |
| Data-collection statistics | ||
| X-ray source | Beamline 7-1, SSRL | Beamline 7-1, SSRL |
| Wavelength (Å) | 1.12709 | 1.03317 |
| Temperature (K) | 100 | 100 |
| Detector | ADSC Q315 | ADSC Q315 |
| Crystal-to-detector distance (mm) | 200 | 300 |
| Rotation range per image (°) | 0.2 | 0.2 |
| Exposure time per image (s) | 10 | 7 |
| Space group | C2 | C2221 |
| a, b, c (Å) | 211.55, 49.51, 81.22 | 124.88, 129.74, 213.39 |
| α, β, γ (°) | 90, 90.3, 90 | 90, 90, 90 |
| Resolution (Å) | 105.76–1.85 (1.89–1.85) | 38.98–2.15 (2.20–2.15) |
| R merge † (%) | 7.7 (49.8) | 6.3 (56.4) |
| 〈I/σ(I)〉 | 10.80 (2.65) | 18.68 (2.45) |
| CC1/2 (%) | 99.7 (89.0) | 99.9 (73.6) |
| No. of reflections | 254006 (19336) | 348834 (24367) |
| No. of unique reflections | 71340 (5160) | 92958 (6787) |
| Completeness (%) | 98.7 (97.0) | 98.8 (98.6) |
| Multiplicity | 3.56 (3.75) | 3.75 (3.59) |
| Refinement statistics | ||
| Resolution (Å) | 105.76–1.85 (1.89–1.85) | 38.98–2.15 (2.20–2.15) |
| No. of reflections (F > 0) used in refinement | 67746 (4873) | 94030 (2719) |
| R factor‡ (%) | 18.59 | 16.79 |
| R free ‡ (%) | 21.56 | 21.17 |
| R.m.s.d., bond lengths (Å) | 0.011 | 0.012 |
| R.m.s.d., bond angles (°) | 1.22 | 1.492 |
| Overall B value (Å2) | 36.0 | 43.3 |
| Ramachandran plot statistics§ | ||
| No. of residues | 371 | 371 |
| Most favored region (%) | 97.6 | 96.5 |
| Allowed region (%) | 2.3 | 2.8 |
| Disallowed (%) | 0.1 | 0.7 |
R
merge = 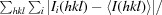
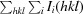 , where 〈I(hkl)〉 is the mean of i observations of reflection I(hkl).
, where 〈I(hkl)〉 is the mean of i observations of reflection I(hkl).
R factor and R
free = 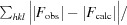
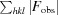 for 95% of recorded data (R factor) or 5% of data that were not used in refinement (R
free).
for 95% of recorded data (R factor) or 5% of data that were not used in refinement (R
free).
From MolProbity (Chen et al., 2010 ▸).
3. Results and discussion
3.1. Crystal structure of NmSacA
The NmSacA monomeric structure consists of two domains, each folding into the three-layer (αβα) sandwich architecture of a Rossmann fold (Fig. 2 ▸ a). Domain 1 consists of residues 1–170 and is augmented by an additional C-terminal helix, residues 356–371. Domain 2 spans residues 171–355. Domain 1 is made up of a central seven-stranded parallel β-sheet flanked by seven α-helices with the topology of a dinucleotide-binding Rossmann domain. Domain 2 is composed of a central six-stranded β-sheet with nine α-helices. Helices α9–α15 pack against the central β-sheet, forming the dinucleotide-binding domain, while helices α7 and α8 form a connecting segment that packs against domain 2 (Fig. 2 ▸ a). The cleft between the two domains defines the substrate-binding pocket.
Figure 2.

Structure of NmSacA. (a) Overall structure of the NmSacA monomer bound to the substrate UDP-GlcNAc. The protein is color-coded with a rainbow spectrum from blue at the N-terminus to red at the C-terminus. Secondary-structure elements mentioned in the manuscript are labeled. The UDP-GlcNAc substrate is shown in ball-and-stick representation with gray-colored C atoms. (b) Dimeric structure of one dimer in the crystallographic asymmetric unit of the substrate-bound NmSacA structure. Each dimer only displays UDP-GlcNAc substrate binding (space-filling spheres) in one monomer of each dimer (chain A, green). The substrate-free monomer is in an open conformation (chain B, sand). For clarity, the other dimer (chains C and D) is not shown. (c) Dimer interaction is mediated by three helices (α3, α4 and α5) of each monomer, forming a six-helix bundle at the dimer interface. The UDP-GlcNAc substrate is shown as space-filling spheres with green C atoms (bound to chain A). Similar dimeric interactions are observed in the substrate-free structure.
The ligand-free NmSacA structure contains one dimer in the crystallographic asymmetric unit, with both monomers displaying nearly identical conformations (r.m.s.d. of 1.00 Å for 350 equivalent Cα atoms). The substrate-bound structure contains two dimers in the asymmetric unit, with the UDP-GlcNAc substrate binding to only one monomer of each dimer (chains A and C; Fig. 2 ▸ b). UDP-GlcNAc epimerases have been shown to be dimeric in solution (Kawamura et al., 1979 ▸). The dimer interface is very similar between the two types of dimers: substrate-bound and substrate-free. Three α-helices (α3, α4 and α5) from each monomer of domain 1 form a six-helix bundle mediating the dimer interaction, which buries surface areas of 1401 Å2 and 1368 Å2 for the structures without and with substrate, respectively (Fig. 2 ▸ c).
3.2. Substrate UDP-GlcNAc bound in the active site
The closed conformation had UDP-GlcNAc bound in the active site. While the electron density strongly defines the UDP moiety, the density was weaker for the sugar GlcNAc and was refined to have ∼60% occupancy, suggesting that some hydrolysis occurred in the crystal (Fig. 3 ▸ a). At 100% occupancy, the sugar is surrounded by negative electron density in the F o − F c map. With only UDP modeled in the structure, strong positive electron density extends from the end of the β-phosphate, suggesting that GlcNAc is present. Decreasing the occupancy of the GlcNAc to 60% provided continuous electron density in the 2F o − F c map and no density in the F o − F c map.
Figure 3.

The UDP-GlcNAc substrate binds at the interface between the two domains of NmSacA and is bound by both aromatic interactions and hydrogen bonds. (a) Domain 1 is shown in salmon and domain 2 is shown in green. The electron-density map (2F o − F c) around the UDP-GlcNAc is shown as a blue mesh and is contoured at 1σ. (b) Interactions between UDP-GlcNAc (gray C atoms) and NmSacA are shown. Hydrogen bonds to the UDP-GlcNAc substrate are shown as yellow dashed lines. New inter-domain ionic interactions formed by substrate-induced enzyme closure are shown as gray dashed lines. Ordered water molecules that interact with UDP-GlcNAc are shown as small red spheres. The residues involved in binding are labeled and the catalytic residues are highlighted with underlined text. (c) Superposition of substrate-bound (green) and ligand-free (brown) NmSacA structures. Domain 1 is aligned (r.m.s.d. of 0.799 Å), showing the closure of domain 2 onto the substrate. Domain 2 rotates by 29°.
UDP-GlcNAc binds at the interface between the two domains. The uracil ring π-stacks with Phe275. The main-chain carbonyl of Gln270 hydrogen-bonds to N3 of the uracil ring (2.8 Å) and the side-chain amide hydrogen-bonds to O4 (3.0 Å). Both hydroxyls on the ribose sugar hydrogen-bond to the side chain of Glu295 (2.8 Å for both O atoms). The ring O atom in the ribose sugar is hydrogen-bonded by Arg10 (2.8 Å), which also binds to the α-phosphate O atom (3.0 Å). The side-chain hydroxyl of Ser289 hydrogen-bonds to both the α- and β-phosphates (2.7 and 3.1 Å, respectively). The main-chain amide N atoms of Gly290 and Gly291 both interact with the β-phosphate (both at 2.9 Å). These two glycine residues are part of the conserved sequence DSGG and initiate the N-terminus of helix α12, suggesting that the helix dipole contributes to anchoring the UDP. The closed conformation also orders a loop from His212 to Glu219, which has sparse electron density in the substrate-free open-conformation monomer in the crystal. This loop is in close proximity to the substrate-binding site, with His212 hydrogen-bonding to the α- and β-phosphate O atoms (3.3 and 3.0 Å, respectively).
The GlcNAc sugar moiety is held in place by a series of hydrogen-bonding interactions. Lys15 reaches into the binding pocket and binds to the C4 hydroxyl of GlcNAc (3.2 Å). The C4 hydroxyl is also ligated by the carboxyl group of Glu117 (2.5 Å). The C3 hydroxyl is bound by the carboxyl group of Asp95 (2.8 Å) and the guanidinium group of Arg312 (3.0 Å). Arg135 makes a long hydrogen bond to the acetyl-group carbonyl O atom of the carbohydrate (3.4 Å; Fig. 3 ▸ b). In summary, most of the contacts to the UDP moiety come from domain 2, while most of the interactions with the GlcNAc sugar originate from domain 1.
The annotated catalytic residues Asp95, Glu117, Glu131 and His212 (Samuel & Tanner, 2004 ▸) are all in close proximity to the sugar (Fig. 3 ▸ b). The carboxyl group of Asp95 is 3.3 Å from the GlcNAc C2′′ atom, suggesting that it serves as the general base to generate the 2-acetamidoglucal intermediate. However, it is uncertain which residue can serve as the general acid for the syn-addition protonation of C2′′. Nevertheless, it is interesting to note that His212 hydrogen-bonds to a β-phosphate O atom, which in turn is 4.0 Å from the syn face of the C2′′ atom, suggesting that His212 may be a general acid to which the proton is shuttled by the β-phosphate in a substrate-assisted fashion.
3.3. UDP-GlcNAc substrate binding triggers a closed conformation
As seen in other epimerases of this family (Chen et al., 2014 ▸; de Azevedo & Nascimento, 2019 ▸; Velloso et al., 2008 ▸), substrate binding induces a conformational change in which each domain closes down upon the UDP-GlcNAc substrate, which binds at the cleft between the two Rossmann domains. The ligand-free monomers of each dimer (chains B and D) reside in the open conformation, very similar to both monomers of the completely ligand-free structure (r.m.s.d.s range from 0.374 to 1.16 Å). Comparing the two conformations, each domain superimposes well between the substrate-free open structure and the closed UDP-GlcNAc-bound structure, with an r.m.s.d. of 0.799 Å for domain 1 and 2.077 Å for domain 2 (184 and 182 equivalent Cα atoms, respectively). However, aligning domain 1 from each structure reveals that domain 2 rotates by about 29°, clamping down on the substrate (as calculated by DynDom; Hayward & Lee, 2002 ▸; Fig. 3 ▸ b). The interdomain connecting helix α7 serves as the pivot point or hinge upon closure, which results in some Cα-atom movements over 11 Å distant from the hinge in helix α10.
The closed substrate-bound NmSacA conformation is stabilized by interactions between UDP-GlcNAc and residues from both domains. The closed state is further reinforced through the creation of new inter-domain ionic interactions. Arg213 from domain 2 forms a new ionic interaction with Glu131 from domain 1. Glu311 of domain 2 also forms a new salt bridge with Arg135 of domain 1. Finally, Glu294 from domain 2 forms an ion pair with Lys15 of domain 1. This substrate-induced closure is also seen in other nonhydrolyzing UDP-GlcNAc 2-epimerases (Chen et al., 2014 ▸).
3.4. Na+ ion ligated in the open conformation
The ligand-free open-conformation structure revealed electron density for a potential metal ion with short contacts to the main-chain carbonyl O atoms of Pro297, Ala349 and Ile351 as well as two water molecules in a trigonal bipyramidal geometry. Since the crystals were grown in 200 mM NaCl, we modeled the cation as Na+ (Fig. 4 ▸ a). The geometry and ligation distances are consistent with an Na+ ion, as confirmed using the CheckMyMetal web server (Zheng et al., 2017 ▸). The Na+ ion was observed in both subunits in the asymmetric unit of the completely ligand-free structure as well as the two monomers in the open conformation that do not contain UDP-GlcNAc (chains B and D) of the substrate-bound crystal form.
Figure 4.

An Na+ ion is coordinated in the open conformation. (a) The open conformation coordinates what is assumed to be an Na+ ion through three main-chain carbonyls (Pro297, Ala349 and Ile351) and two waters. It is coordinated in a trigonal bipyramidal geometry. The ligation distances are shown. (b) The Na+ ion is not found in the closed conformation. Upon substrate-induced conformational change, the loop between helices α15 and α16 shifts, breaking the planar geometry of the main-chain coordinating residues and displacing a water and consequently the Na+ ion.
This cation-binding site is distant from the active site (>20 Å). It is located at a place where domain 2 transitions back into domain 1 and fastens the loop between helices α15 and α16 to helix α12. Interestingly, the Na+ ion is absent in the ligand-bound closed conformation. Upon closing, the end of helix α15 unravels a partial turn, shifting the loop connecting helices α15 to α16. Ile351 in the loop swings over and occludes the sodium site. This results in the flipping of Ile351 and the movement of Ala349, breaking the trigonal planar geometry of the main-chain ligations and displacing a water ligand (Fig. 4 ▸ b). The cation does not appear to be involved in catalysis or conformational flexibility, as treatment with ethylenediaminetetraacetic acid (EDTA) had little to no effect on catalytic activity (Zhang et al., 2016 ▸).
3.5. Comparison with other epimerase structures
NmSacA shares high sequence and structural homology with many epimerase structures in the PDB (Fig. 5 ▸ a; Badger et al., 2005 ▸; Campbell et al., 2000 ▸; Chen et al., 2014 ▸; Mann et al., 2016 ▸; Velloso et al., 2008 ▸). The most similar protein is the UDP-GlcNAc 2-epimerase from E. coli (UniProtKB P27828; PDB entry 1vgv; Badger et al., 2005 ▸), which shares 56% identity with NmSacA. The E. coli structure also crystallized with two dimers in the asymmetric unit, where UDP-GlcNAc is bound to only one monomer of each dimer. It should be noted that this structure, which is the result of a high-throughput structural genomics consortium, modeled the UDP-ManNAc epimer in the structure, but mistakenly labeled it UDP-GlcNAc.
Figure 5.


Comparison of bacterial nonhydrolyzing UDP-GlcNAc 2-epimerases. (a) Sequence alignment of all known bacterial UDP-GlcNAc 2-epimerase structures in the PDB. The secondary-structure elements defined by the NmSacA structure are drawn above the sequence alignment. Red boxes indicate residues that are conserved in all epimerases. The putative catalytic residues are designated by blue asterisks below the sequence, residues that bind UDP-GlcNAc substrate are designated by magenta triangles and residues that are observed to bind the UDP-GlcNAc allosteric effector in other epimerase structures are represented by orange crosses. (b) Superposition of substrate-bound NmSacA (green) on substrate-bound E. coli epimerase (salmon; PDB entry 1vgv). UDP-GlcNAc is shown in stick representation. The r.m.s.d. ranges between 0.655 and 0.949 Å (300 Cα atoms) on comparing the two substrate-bound monomers from each structure. (c) Close-up view of the active site, revealing the major structural difference between the two epimerases: a shift of helix α10 and the β9–α9 loop. Residues are labeled in their respective colors for each epimerase. Yellow dashed lines represent interactions.
The two UDP-GlcNAc-bound chains of NmSacA align with the two substrate-bound chains of E. coli UDP-GlcNAc 2-epimerase (PDB entry 1vgv) with r.m.s.d.s that range from 0.655 to 0.949 Å for 300 Cα atoms (Fig. 5 ▸ b). However, the E. coli structure exhibits slightly more of a domain closure upon binding substrate, resulting in higher r.m.s.d.s for ligand-free superposition comparisons between the E. coli enzyme and NmSacA, which range between 1.15 and 1.44 Å for 300 Cα atoms.
The only major difference between the substrate-bound structures is the disposition of helix α10 and the loop between β9 and α9 above the UDP moiety in domain 2. In the E. coli enzyme, helix α10 shifts away from helix α9 owing to the position of the β9–α9 loop and the substitution of Phe222 (E. coli) at the helix interface, which is Ile221 in NmSacA (Fig. 5 ▸ c). The β9–α9 loop in NmSacA contains Arg213, which makes a new inter-domain ionic bond with Glu131 upon binding substrate. In the E. coli structure, even though Glu121 is conserved, this loop shifts greatly (Fig. 5 ▸ c). While the electron density is poorly defined in the E. coli structure, weak electron density defines the placement of the main chain. The electron density in NmSacA is well defined, which is likely to be owing to the salt bridge between Arg213 and Glu131, which stabilizes the loop. The β9–α9 loop in NmSacA also contains a 310-helix, which is not observed in the E. coli structure.
The structures of other epimerases with ligands bound in the active site, including those from Burkholderia vietnamiensis (PDB entry 5dld; 48.7% identity; Seattle Structural Genomics Center for Infectious Disease, unpublished work), Bacillus subtilis (PDB entry 4fkz; 51.3% identity), B. anthracis (PDB entry 3beo; 50.6% identity; Velloso et al., 2008 ▸), S. aureus (PDB entry 5enz; 47.2% identity; Mann et al., 2016 ▸) and M. jannaschii (PDB entry 4nes; 36.4% identity; Chen et al., 2014 ▸), all have a similar conformation of α10 and the β9–α9 loop to that observed in NmSacA. This suggests that the E. coli enzyme may be unique in displaying a different conformation near the active site when binding the substrates. However, the residues implicated in catalysis are in similar positions and orientations to all other epimerases.
3.6. Allosteric site
Most of the nonhydrolyzing UDP-GlcNAc 2-epimerases have been shown to require UDP-GlcNAc to initiate the epimerization of UDP-ManNAc (Kawamura et al., 1978 ▸, 1979 ▸), suggesting that the enzyme possesses a distinct allosteric regulatory site. It has also been demonstrated that UDP-GlcNAc stimulates the enzyme activity, resulting in sigmoidal velocity curves with a Hill coefficient of over 2 (Kawamura et al., 1979 ▸). However, it appears that NmSacA may be a unique nonhydrolyzing epimerase in that it does not require UDP-GlcNAc to initiate the epimerization of UDP-ManNAc, although UDP-GlcNAc does stimulate the epimerase activity, resulting in sigmoidal kinetic curves (Zhang et al., 2016 ▸).
Four nonhydrolyzing UDP-GlcNAc 2-epimerase structures have been determined with UDP-GlcNAc bound in the allosteric site, all with UDP also bound in the adjacent active site. These epimerases are from B. vietnamiensis (PDB entry 5dld), B. subtilis (PDB entry 4fkz), B. anthracis (PDB entry 3beo) and M. jannaschii (PDB entry 4nes). Overlaying the NmSacA structure with the epimerase from B. anthracis reveals that while the overall structures are very similar (r.m.s.d. of 1.01 Å), the β2–α2 and β3–α3 loops are in a different conformation in the B. anthracis enzyme compared with NmSacA because these loops help to shape the allosteric UDP-GlcNAc binding pocket (Fig. 6 ▸). These loops in the B. anthracis enzyme are in a similar conformation in the three other epimerase structures with a bound allosteric effector. Interestingly, all of the residues involved in binding allosteric UDP-GlcNAc are conserved in all epimerases, with the exception of Glu41 in NmSacA, which is Gln in many other epimerases, including that from B. subtilis (Fig. 5 ▸ a). It is unknown why UDP-GlcNAc is not observed binding to the allosteric site in NmSacA. It could be that the allosteric effector may have a weaker binding affinity for NmSacA since this enzyme does not require UDP-GlcNAc binding to initiate UDP-ManNAc epimerization. Alternatively, it could be a consequence of the low pH of crystal growth, which has been hypothesized to prevent allosteric binding in the E. coli epimerase (Velloso et al., 2008 ▸). The E. coli enzyme was crystallized at pH 5.2 and the NmSacA crystals presented here were grown at pH 5.5, which is well below the optimal enzyme activity pH of 8.5 (Zhang et al., 2016 ▸).
Figure 6.

Allosteric UDP-GlcNAc binding site. A superposition is shown of NmSacA (green) on UDP-GlcNAc 2-epimerase from B. anthracis (PDB entry 3beo; sand), the structure of which was determined with UDP-GlcNAc bound in the allosteric site (brown C atoms) and UDP bound in the active site. Shown are side chains that interact with the allosteric UDP-GlcNAc in the B. anthracis structure, with the corresponding NmSacA residues labeled in green. Yellow dashed lines show interactions with allosteric UDP-GlcNAc in B. anthracis. All residues are conserved in binding UDP-GlcNAc except for Glu41, which is Gln in the B. anthracis epimerase. The UDP-GlcNAc bound in the NmSacA active site is also shown (light green C atoms).
3.7. Structural basis for the tolerance of modified UDP-ManNAc substrates
NmSacA has been shown to epimerize UDP-ManNAc substrates with various modifications at the N-acetyl position of carbon 2 (the carbon that is epimerized; Zhang et al., 2016 ▸). The mannosamine sugar must still contain an N-acyl group, but some variations of the acyl group can be tolerated. UDP-sugars containing a sugar without the N-acyl group, such as UDP-mannose or its derivatives in which the C2′′-hydroxyl group is replaced by a fluorine, amine or azide group, do not serve as substrates. Only small structural additions to the N-acetyl group in UDP-ManNAc such as N-propyl (an additional methyl) and N-glycolyl (an additional hydroxyl) groups can be tolerated, while UDP-ManNAc derivatives with larger N-acyl groups such as N-butyl, N-azidoacetyl and N-phenylacetyl are not epimerized by NmSacA (Zhang et al., 2016 ▸).
The structure of NmSacA complexed with UDP-GlcNAc reported here helps to explain its substrate specificity and the tolerance of certain sugar modifications. The O atom of the acetyl group forms a weak hydrogen bond (3.4 Å) to the conserved Arg135 (Fig. 3 ▸ b). This explains the requirement for the N-acyl group, or at least a hydrogen-bonding accepting atom, three atoms from the C2′′ sugar ring. Sugars without the acetyl group or other small acyl groups are incapable of hydrogen bonding to Arg135, and therefore may not properly position the sugar for the initial C2′′ proton abstraction. For the tolerance of small additions to the acetyl group, the structure revealed that the methyl moiety of the N-acetyl group points towards a small pocket defined by the loop between β11 and α12 and the loop between β12 and α13. It is interesting to note that the methyl group points towards Gly290 at the start of α12, which is strictly conserved. Inspection of this pocket explains why NmSacA can only tolerate a sugar with the addition of a single hydroxyl or methyl group to the acetyl methyl, because anything larger would clash with the protein (Fig. 7 ▸).
Figure 7.

Slice through a space-filling representation of the NmSacA structure with UDP-GlcNAc bound in the active site. The protein is shown as a green surface with the UDP-GlcNAc shown in stick representation with gray C atoms. The C2′′ atom is labeled along with the small pocket near the N-acetyl group (red circle). The structure explains how NmSacA can accept small modifications of the N-acetyl methyl group in UDP-GlcNAc analogs.
4. Conclusions
Two crystal structures of NmSacA were determined: one in the absence of any ligands and one with UDP-GlcNAc bound in the active site. The substrate-bound structure contains two dimers in the asymmetric unit. Each dimer consists of a monomer without a bound substrate and another with the substrate UDP-GlcNAc bound in the active site. The structure reveals a common fold among nonhydrolyzing UDP-GlcNAc 2-epimerases, consisting of two domains each with the three-layer αβα sandwich of a Rossmann fold. As observed in similar epimerases, substrate binding triggers closure of the two domains, which adds important information on the potential mechanism of epimerization. The reaction has been shown to be nonhydrolyzing (Zhang et al., 2016 ▸) and the mechanism is likely to proceed through Asp95, which would act as the general base to deprotonate the sugar moiety, but the general acid cannot be discerned from the structure. Nevertheless, the conserved His212 may serve as a general acid to which the proton is relayed by the β-phosphate in a substrate-assisted fashion.
Orthogonal enzymes share a very similar structures and active-site pockets, and the conserved residues are nearly identical in NmSacA. The allosteric site of the nonhydrolyzing UDP-GlcNAc 2-epimerases is largely conserved in NmSacA, yet nothing was observed to bind to the allosteric site. Structural differences in the β2–α2 and β3–α3 loops explain why no allosteric effector was bound, despite all but one of the allosteric binding residues being conserved in NmSacA. Further studies are required to determine whether NmSacA does indeed bind UDP-GlcNAc in an allosteric site. Overall, the structure of NmSacA and the comparative analysis with structurally homologous enzymes shed additional light on the enzymatic mechanism and provide an initial understanding of the non-allosteric nature of the enzyme.
Supplementary Material
PDB reference: UDP-GlcNAc 2-epimerase, ligand-free, 6vlb
PDB reference: substrate-bound, 6vlc
Acknowledgments
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the NIH National Institute of General Medical Sciences (NIGMS) (P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH or NIGMS.
Funding Statement
This work was funded by National Institutes of Health grants R01GM094523 , U01GM125288 , and T32GM007377. National Institute of Food and Agriculture grant CA-D-MCB-2310-H to Andrew Fisher.
References
- Aye, A. M. M., Bai, X., Borrow, R., Bory, S., Carlos, J., Caugant, D. A., Chiou, C.-S., Dai, V. T. T., Dinleyici, E. C., Ghimire, P., Handryastuti, S., Heo, J. Y., Jennison, A., Kamiya, H., Tonnii Sia, L., Lucidarme, J., Marshall, H., Putri, N. D., Saha, S., Shao, Z., Sim, J. H. C., Smith, V., Taha, M.-K., Van Thanh, P., Thisyakorn, U., Tshering, K., Vazquez, J., Veeraraghavan, B., Yezli, S. & Zhu, B. (2020). J. Infect., https://doi.org/10.1016/j.jinf.2020.07.025.
- Azevedo, E. C. de & Nascimento, A. S. (2019). J. Struct. Biol. 207, 158–168. [DOI] [PubMed]
- Badger, J., Sauder, J. M., Adams, J. M., Antonysamy, S., Bain, K., Bergseid, M. G., Buchanan, S. G., Buchanan, M. D., Batiyenko, Y., Christopher, J. A., Emtage, S., Eroshkina, A., Feil, I., Furlong, E. B., Gajiwala, K. S., Gao, X., He, D., Hendle, J., Huber, A., Hoda, K., Kearins, P., Kissinger, C., Laubert, B., Lewis, H. A., Lin, J., Loomis, K., Lorimer, D., Louie, G., Maletic, M., Marsh, C. D., Miller, I., Molinari, J., Muller-Dieckmann, H. J., Newman, J. M., Noland, B. W., Pagarigan, B., Park, F., Peat, T. S., Post, K. W., Radojicic, S., Ramos, A., Romero, R., Rutter, M. E., Sanderson, W. E., Schwinn, K. D., Tresser, J., Winhoven, J., Wright, T. A., Wu, L., Xu, J. & Harris, T. J. (2005). Proteins, 60, 787–796. [DOI] [PubMed]
- Campbell, R. E., Mosimann, S. C., Tanner, M. E. & Strynadka, N. C. J. (2000). Biochemistry, 39, 14993–15001. [DOI] [PubMed]
- Chen, S.-C., Huang, C.-H., Yang, C. S., Liu, J.-S., Kuan, S.-M. & Chen, Y. (2014). Proteins, 82, 1519–1526. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cress, B. F., Englaender, J. A., He, W., Kasper, D., Linhardt, R. J. & Koffas, M. A. (2014). FEMS Microbiol. Rev. 38, 660–697. [DOI] [PMC free article] [PubMed]
- Dutta, A. K., Swaminathan, S., Abitbol, V., Kolhapure, S. & Sathyanarayanan, S. (2020). Infect. Dis. Ther. 9, 537–559. [DOI] [PMC free article] [PubMed]
- Fiebig, T., Freiberger, F., Pinto, V., Romano, M. R., Black, A., Litschko, C., Bethe, A., Yashunsky, D., Adamo, R., Nikolaev, A., Berti, F. & Gerardy-Schahn, R. (2014). J. Biol. Chem. 289, 19395–19407. [DOI] [PMC free article] [PubMed]
- Hayward, S. & Lee, R. A. (2002). J. Mol. Graph. Model. 21, 181–183. [DOI] [PubMed]
- Jennings, H. J., Bhauacharjee, A. K., Bundle, D. R., Kenny, C. P., Martin, A. & Smith, I. C. (1977). J. Infect. Dis. 136, S78–S83. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kawamura, T., Ishimoto, N. & Ito, E. (1979). J. Biol. Chem. 254, 8457–8465. [PubMed]
- Kawamura, T., Kimura, M., Yamamori, S. & Ito, E. (1978). J. Biol. Chem. 253, 3595–3601. [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Mann, P. A., Müller, A., Wolff, K. A., Fischmann, T., Wang, H., Reed, P., Hou, Y., Li, W., Müller, C. E., Xiao, J., Murgolo, N., Sher, X., Mayhood, T., Sheth, P. R., Mirza, A., Labroli, M., Xiao, L., McCoy, M., Gill, C. J., Pinho, M. G., Schneider, T. & Roemer, T. (2016). PLoS Pathog. 12, e1005585. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Morgan, P. M., Sala, R. F. & Tanner, M. E. (1997). J. Am. Chem. Soc. 119, 10269–10277.
- Novak, R. T., Ronveaux, O., Bita, A. F., Aké, H. F., Lessa, F. C., Wang, X., Bwaka, A. M. & Fox, L. M. (2019). J. Infect. Dis. 220, S279–S285. [DOI] [PMC free article] [PubMed]
- Roberts, I. S. (1996). Annu. Rev. Microbiol. 50, 285–315. [DOI] [PubMed]
- Samuel, J. & Tanner, M. E. (2004). Biochim. Biophys. Acta, 1700, 85–91. [DOI] [PubMed]
- Swartley, J. S., Liu, L. J., Miller, Y. K., Martin, L. E., Edupuganti, S. & Stephens, D. S. (1998). J. Bacteriol. 180, 1533–1539. [DOI] [PMC free article] [PubMed]
- Tanner, M. E. (2002). Acc. Chem. Res. 35, 237–246. [DOI] [PubMed]
- Velloso, L. M., Bhaskaran, S. S., Schuch, R., Fischetti, V. A. & Stebbins, C. E. (2008). EMBO Rep. 9, 199–205. [DOI] [PMC free article] [PubMed]
- Xie, O., Bolgiano, B., Gao, F., Lockyer, K., Swann, C., Jones, C., Delrieu, I., Njanpop-Lafourcade, B. M., Tamekloe, T. A., Pollard, A. J. & Norheim, G. (2012). Vaccine, 30, 5812–5823. [DOI] [PubMed]
- Zhang, L., Muthana, M. M., Yu, H., McArthur, J. B., Qu, J. & Chen, X. (2016). Carbohydr. Res. 419, 18–28. [DOI] [PMC free article] [PubMed]
- Zheng, H., Cooper, D. R., Porebski, P. J., Shabalin, I. G., Handing, K. B. & Minor, W. (2017). Acta Cryst. D73, 223–233. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: UDP-GlcNAc 2-epimerase, ligand-free, 6vlb
PDB reference: substrate-bound, 6vlc