

Abstract
Hsp90 is a molecular chaperone and important driver of stabilization and activation of several oncogenic proteins that are involved in the malignant transformation of tumor cells. Therefore, it is not surprising that Hsp90 has been reported to be a promising target for the treatment of several neoplasias, such as non-small-cell lung cancer and HER2-positive breast cancer. Hsp90 chaperone function depends on its ability to bind and hydrolyze ATP and Hsp90 inhibitors have been shown to compete with nucleotides for binding to Hsp90. Multiple factors, such as co-chaperones and post-translational modification, are involved in regulating Hsp90 ATPase activity. Here, the impact of post-translational modifications and co-chaperones on the efficacy of Hsp90 inhibitors are reviewed.
Hsp90 structure & function
Hsp90 is an evolutionarily conserved molecular chaperone and has two sides to its cellular function [1–3]. On one side, it is involved in the normal development and proliferation of cells [4–6], but conversely, it is an important driver of malignant transformation, growth, survival and possibly invasiveness of cancer cells (Figure 1) [7–9]. Therefore, it can be argued that cancer cells are ‘addicted’ to Hsp90 [10,11]. There are two isoforms of Hsp90 encoded by two separate genes in eukaryotes. These include the constitutively expressed human Hsp90β (yeast Hsc82), and the stress-induced human Hsp90α (hHsp90α) or yeast Hsp82 [12,13]. This molecular chaperone belongs to the ATPase/kinase GHKL (DNA Gyrase, Hsp90, Histidine Kinase, MutL) superfamily [14], sharing the unifying feature of an ATP-binding site. Each protomer of the Hsp90 dimer contains three domains: the N-domain that contains an ATP- and drug-binding site, and co-chaperone-interacting motifs; a middle domain that harbors sites for clients and cochaperones; and a carboxy-terminal domain that contains a dimerization motif, a second drug-binding region and interaction region for other co-chaperones (Figure 2) [15–21]. Driven by ATP, Hsp90 has the ability to undergo conformational changes, known as the chaperone cycle, allowing it to interact with other distinct co-chaperones (Figure 2). The cycle involves several conformational states that bind and release client proteins, ultimately altering their stability. An updated list of Hsp90 clients can be found online [18,19,22,23,201]. Hsp90 inhibitors interfere with this cycle by replacing ATP at the nucleotide-binding site and, consequently, leading to ubiquitination and proteasome degradation of the majority of client proteins (Figure 2) [24,25]. Several studies have assessed the effects of Hsp90 inhibitors on different tumor cells. A relatively recent and less developed area of investigation is the regulatory factors that affect drug sensitivity or resistance. This article will review the evidence assessing post-translational modifications and other regulatory mechanisms, such as co-chaperones, that affect and influence cells sensitivity and resistance to various Hsp90 inhibitors.
Figure 1. Two sides to Hsp90 function.
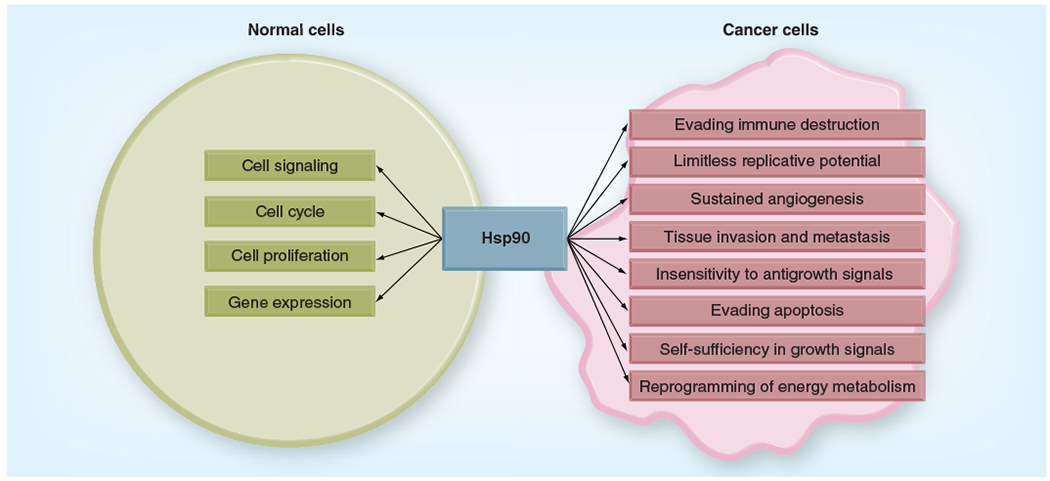
Hsp90 looks after proteins that are involved in normal cellular function. Hsp90 also chaperones clients that are crucial for the maintenance of each of the proposed hallmarks of cancer.
Figure 2. Hsp90 chaperone function.
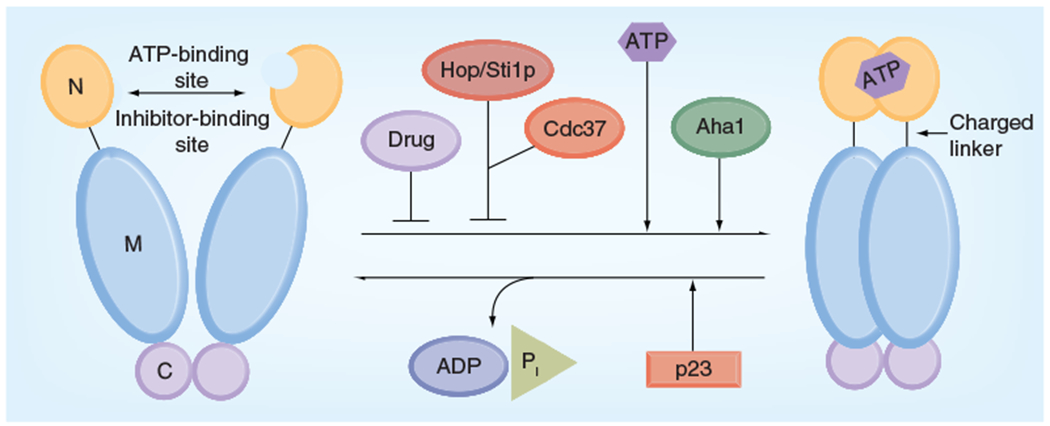
ATP binding to the N-terminal domain of Hsp90 promotes transient dimerization of the N-domains. The co-chaperone Aha1 enhances Hsp90 ATPase activity by promoting various conformational changes, while Hop/Sti1 and Hsp90 inhibitors such as geldanamycin or radicicol exert the opposite effect by inhibiting N-domain dimerization. p23 slows ATP hydrolysis at a late stage of the chaperone cycle.
Hsp90 inhibitors
Hsp90 inhibitors and their clinical development are reviewed in depth elsewhere [2,26]. This section contains a brief summary of this area to provide background for the sections on sensitivity and resistance to Hsp90 inhibitors.
The first identified Hsp90 inhibitors were the natural products, radicicol (RD; macrocyclic antifungal antibiotic) and geldanamycin (GA; benzaquinoid ansamycin antibiotic) [2,27]. They work by mimicking the unusual structure ATP adopts when binding to the N-terminal nucleotide binding pocket, therefore blocking ATP binding and hydrolysis, and consequently interaction with Hsp90 client proteins, leading to their degradation. Both GA and RD are poorly soluble, unstable and highly toxic, minimizing their clinical value. However, they provided a chemical foundation to build clinically suitable, better tolerated drugs. An example is 17-allylamino-demothoxygeladanamycin (17-AAG: tanespimysin), a geldanamycin derivative with low toxicity and significant clinical response in HER2-positive breast cancer, used in combination with bortezomib in relapsed/refractory multiple myeloma (Figure 3) [28,29]. The water-soluble 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG, alvespimycin) and the soluble stabilized hydroquinone form of 17-AAG, IPI-504 (retaspimycin), have improved pharmokinetic properties, which circumvent the hepatotoxicity problems of 17-AAG in clinical trials [30,31]. RD has inhibitory effects in vitro but not in vivo [5,32,33]. Derivatives of resorcinals, such as ganetespib (formerly STA-9090 developed by Synta Pharmaeuticals, MA, USA), AUY922 [34–36], KW-2478 [37] and AT13387 [38], have been found to be more effective in multiple clinical trials (Figure 3).
Figure 3.
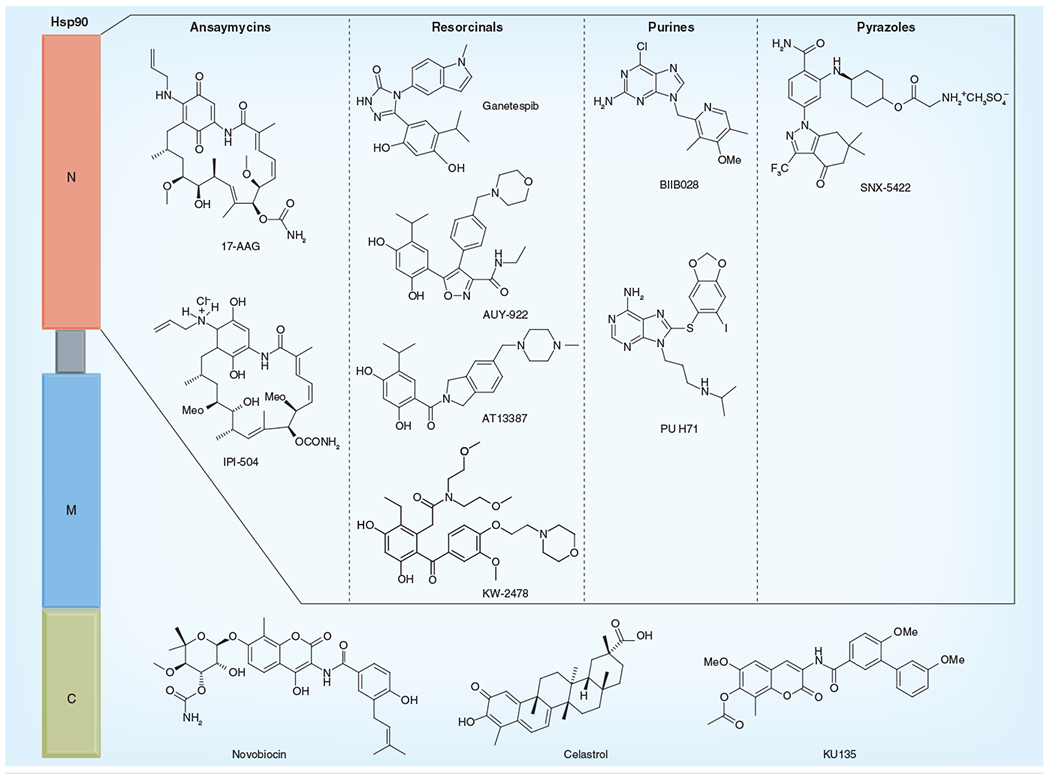
Hsp90 N- and C-domain inhibitors.
The purine scaffold series were the first synthetic small molecules to inhibit Hsp90. They mimicked the shape adopted by ADP when bound to the nucleotide-binding site [27]. These inhibitors were based initially on the prototype PU3 [39,40], and led to the clinical candidates BIIB021 and BIIB028, as well as PU-H71, currently in Phase I clinical trials [41].
The resorcylic pyrazoles and isoxazoles (based on CCT018159 [42]) led to the development of NVP-AUY922 (Novartis)/VER52296 [43], the resorcylic dihydroxybenza-mide AT13387 [38], and the structurally related KW-2478 [44]. These purine derivatives proved to be more beneficial than 17-AAG because they were not affected by NAD(P)H:NQO1 or P-gp as discussed below [45].
Hsp90 also possess a second drug-binding site in the C-domain [46,47]. Coumarin antibiotics, such as novobiocin, target this site and recent works have made significant advances in improving the affinity of these compounds for Hsp90 (Figure 3). For example, KU135, a novel novobiocin derivative has the ability to induce apoptosis in cancer cells, in some cases with superior efficacy compared with tanespimycin (Figure 3) [48]. Another potential benefit that is associated with some of the Hsp90 C-domain inhibitors is their inability to activate HSF1. This is unlike the effect connected to the N-terminal inhibitors [49]. Existing data strongly support further medicinal chemistry optimization and preclinical evaluation of C-terminal Hsp90 inhibitors [48,50,51].
Impact of co-chaperones on Hsp90 drug binding
Co-chaperones are a group of proteins that regulate the function of chaperones and tend to interact with distinct conformations of Hsp90, therefore, forming a multi-protein complex also known as ‘the molecular chaperone machine’ [52]. Some co-chaperones such as Hop/Sti1, CHIP (E3 ubiquitin ligase), FKBP-51 and -52 (immunophilins-peptidyl-prolyl isomerases), PP5/Ppt1 (serine/threonine protein phosphatase 5) rely on their tetratricopeptide repeat (TPR) domains to interact with Hsp90. These co-chaperones have additional domains that catalyze reactions such as ubiquitin ligation, dephosphorylation and peptidylprolyl isomerization. Non-TPR co-chaperones include Aha1/Hch1, p23 (PGES3)/Sba1, Sgt1 and p50/Cdc37 [53–69]. A number of co-chaperones (e.g., p50/Cdc37, Hop/Sti1, p23 and Aha1) have the ability to modulate the rate of Hsp90 ATPase activity, thereby tailoring the chaperone machine for specific clients. Most Hsp90 co-chaperones are conserved in the single cell eukaryote budding yeast, Saccharomyces cerevisiae. Therefore, yeast has proven to be a valuable model organism to dissect the function of both Hsp90 and its co-chaperones in maintaining cellular homeostasis [70–75].
Cdc37
The co-chaperone Cdc37 recruits protein kinases to the Hsp90 chaperone complex [58,59,76]. This is supported by a 3D structure of Hsp90–Cdc37–Cdk4 obtained using single-particle electron microscopy [22]. Cdc37 also has an inhibitory effect on Hsp90 ATPase activity, thus, allowing kinase clients to be loaded on to the chaperone complex. [77]. Consequently, disruption of Hsp90–Cdc37 complex not only can affect the chaperoning of kinase clients but can also impact drug binding.
Gray et al. have demonstrated the role of Cdc37 in the proliferation of prostate cancer cells by using RNAi to knockdown CDC37 expression. This leads to induced growth arrest both in androgen receptor-negative and -positive prostate carcinoma cells [78]. Furthermore, they reported that knockdown of Cdc37 was able to sensitize PC3 and DU145 cells to 17-AAG and proved to be highly effective in inhibiting cell growth in these cell lines. Of notice, they describe that in spite of this effect, targeting Cdc37 does not significantly deplete intracellular levels of other Hsp90 clients [78]. Similarly, Smith et al. reported that silencing of Cdc37 in human colon carcinoma-derived HCT116 cells led to an increased sensitization to 17-AAG (threefold) and to VER49009 (twofold). Silencing of Cdc37 interfered with Hsp90 association with kinase clients and induced proteasome dependent degradation of clients such as ErbB2, c-Raf, CDK4 and CDK6. The levels of non-kinase clients were unaffected [79]. It has been reported that celastrol binds to the C-domain of Hsp90 and affects chaperone activity by disrupting the Hsp90–Cdc37 complex [80]. It would be of interest to know if celastrol or novobiocin-derived C-terminal inhibitors of Hsp90 (i.e., KU compounds) can provide synergistic effects when combined with N-domain inhibitors [50].
Post-translational modifications of co-chaperones add another layer of regulation in controlling Hsp90 chaperone function both in normal and cancer cells. Using yeast, Bandhakavi et al. have elegantly shown that CK2 targets two conserved serines, S14/17 in yeast Cdc37 (S13 in human Cdc37). This phosphorylation is necessary for Cdc37 interaction with Hsp90 and also for chaperoning of numerous kinase clients including CK2 itself, Cdc28Cdc2, Ste11RAF, Kin28 and Mps1 [81]. Lack of phosphorylation hypersensitizes cells to the Hsp90 inhibitor GA. Later studies confirmed that a similar process also exists for mammalian Cdc37 [82,83]. Finally, Vaughan et al. have reported that Cdc37 and PP5/Ppt1 form complexes with Hsp90 in yeast and in human tumor cells. The protein phosphatase PP5/Ppt1 dephosphorylates the S13 on Cdc37 in vivo, therefore, affecting its interaction with Hsp90 and negatively impacting the chaperoning of numerous kinase clients by the Hsp90–Cdc37 complex [60]. Recent work by Xu and colleagues demonstrated that the nonreceptor tyrosine kinase c-Yes can target Y4 and Y298 on Cdc37. It would be interesting to know if the phosphorylation status of these residues can contribute to Hsp90 inhibitor sensitivity [84].
Aha1
The co-chaperone Aha1 is an activator of Hsp90 ATPase activity [65,66]. Recent work by Retzlaff and co-workers demonstrated that both N- and C-domains of Aha1 interact in a cooperative manner with both the N-terminal and middle domains of Hsp90 via an asymmetric activation mechanism [85]. Aha1 is involved in various cellular processes, including the quality control of the cystic fibrosis transmembrane conductance regulator [86], as well as activation of glucocorticoid receptor and p60v-src [64,66,87]. Interestingly, Holmes et al. demonstrated that siRNA knockdown of AHA1 in human colon carcinoma HCT116 cells and in the ovarian cancer cell line A2780 resulted in a three- to six-fold increase of sensitivity to 17-AAG after 48 h of treatment [87]. It would be of interest to know if downregulation of Aha1 also sensitizes cancer cells to the new generation of Hsp90 inhibitors.
p23/Sba1
p23 (Sba1 in yeast), is known to interact with ATP-bound Hsp90, slowing the chaperone cycle and stabilizing the conformation required for client–protein activation [61,88]. Hsp90 inhibitors such as GA and RD are known to interfere with the binding of p23/Sba1 by competing with ATP binding [89]. Forafonov et al. reported that when p23/Sba1p is absent, both yeast and mammalian cells become hypersensitive to GA and RD. Furthermore, based on the fact that Hsp90 inhibitors induce apoptosis in several cell types, the group observed that low doses of GA inhibited cell growth in p23-null cells and induced cell death, as opposed to wild-type p23 cells, which continued to grow for 2 days and had greater survival. This suggests that p23 may have a protective role against Hsp90 inhibitors. In addition, p23 mutants A13S, R106A and K113A interfered with GA binding by at least twofold in yeast [74,90].
Hop/Sti1 & Cyp40/Cpr6–Cpr7
Hop is a TPR-containing co-chaperone with an important role in facilitating interactions between Hsp70 and Hsp90 [91–93]. Hop has the ability to bind to the MEEVD peptide in the C-domain of Hsp90, as well as to additional sites in the C-terminal and middle domain. Deletion of Hop/Sti1 makes yeast cells hypersensitive to GA and RD [94]. The observed drug sensitivity was further enhanced in the absence of Hsc82 (constitutively expressed yeast Hsp90) compared with Hsp82 (stress-induced yeast Hsp90). These data highlight the functional differences between Hsp90 isoforms [95].
The immunophilin Cyp40 is another TPR-containing co-chaperone that binds solely to the C-domain of Hsp90 and is involved in the chaperoning of steroid hormone receptors [66,96,97]. Displacement of Hsp90–Hop/Sti1 complexes by Cpr6, (Cyp40 homolog in yeast), including formation of an Hsp90–Sti1–Cpr6 ternary complex has been proposed to be one of the key components of the Hsp90 cycle [7,97]. Work by Zuehlke et al. indicated that an Hsp90–Cpr6–Cpr7 ternary complex also exists [98]. The authors have proposed a model where Hsp90–Sti1–Cpr6 complex is followed by formation of an Hsp90–Cpr6–Cpr7 complex that is required for Cpr6 and Cpr7 dissociation. Li and colleagues recently reported that Cpr6 enhances the affinity between Aha1 and Hsp90 and further stimulates the Hsp90 ATPase activity [99]. Finally, single or double deletion of CPR6, CPR7in yeast hyposensitizes these mutants to GA [100]. Interestingly, Cyp40 interacts with Wee1 kinase [101]. This kinase directly targets a conserved tyrosine residue in the Hsp90 N-domain and regulates its chaperone function (see later discussion) [102]. Inhibition of Wee1 function hypersensitizes cells to Hsp90 inhibitors [103]. Whether deregulated or destabilized, Wee1 contributes to the hypersensitivity of CPR7 knock out cells to Hsp90 inhibitors and requires further investigation.
Post-translational modifications that enhance sensitivity to Hsp90 inhibitors
Hsp90 is the target of various post-translational modifications that affect its chaperone function [8,104]. These include phosphorylation, acetylation, S-nitrosylation, oxidation and ubiquitination. Multiple studies have demonstrated the influence of post-translational modifications on Hsp90 function and their impact on drug sensitivity [102,105–107].
Phosphorylation
Hsp90 is a phosphoprotein and early work reported that treating NIH 3T3 cells with the serine/threonine phosphatase inhibitor okadaic acid led to hyper-phosphorylation of Hsp90 and compromised chaperoning of its client kinase p60v-src [108]. This was the first evidence linking Hsp90 phosphorylation to its ability to chaperone client proteins [108]. These data were supported by the fact that the co-chaperone PP5/Ppt1 has the ability to dephosphorylate Hsp90 in vitro and positively regulate its chaperone activity in vivo [109,110]. Recently, it was observed that phosphorylation of threonine and tyrosine sites on Hsp90 can influence its sensitivity to Hsp90 inhibitors [8].
Recent work has shown that CK2 phosphorylates a conserved threonine residue (T22) in the N-domain of yeast Hsp82 both in vitro and in vivo (Figure 4) [111]. Thr22, and adjacent amino acids, participate in a vital hydrophobic interaction with the catalytic loop in the middle domain of Hsp90 [15,112–115]. It was also reported that non-phosphorylatable T22A mutant did not compromise Hsp90 ATPase activity, while the phosphomimetic mutant (T22E) had only approximately 40% of wild-type ATPase activity. These mutants and their equivalent in hHsp90α (T36A and T36E), affected Hsp90-dependent chaperoning of kinase and non-kinase clients [111]. In addition, the phosphomimetic mutant (T22E) resulted in yeast sensitivity to Hsp90 inhibitors GA and ganetespib (formerly STA-9090, Synta Pharmaceuticals), and SNX-2112 (Serenex/Pfizer [NY, USA]) [116]. These findings identify T22 as an important determinant of Hsp90 drug sensitivity in yeast and suggest that hyperphosphorylation of T22 (hHsp90α-T36) may contribute to drug sensitivity in vivo [116]. Previous work has shown that members of the Src family kinases, c-Src and c-Yes, can specifically target hHsp90 [84,117]. Recent work has shown that Wee1 kinase, a key regulator of the cell cycle check-point and also an Hsp90 client, is responsible for phosphorylating a highly conserved tyrosine residue in the N-domain of yeast Hsp40 (Y24) and hHsp90 (Y38) (Figure 4) [102]. This phosphorylation occurs in the nucleus and eventually leads to ubiquitination and degradation of Hsp90 in the proteasome. Furthermore, regulation of Hsp90 chaperone function also depends on tyrosine phosphorylation. It was observed that lack of phosphorylation as a result of either SWE1 deletion (Saccharomyces cerevisiae WEE1) in yeast or knocking down WEE1 in prostate or cervical cancer cell lines led to hypersensitization of these cells to Hsp90 inhibitors (GA, RD, ganetespib and SNX-2112). This phenotype is explained by the increased binding affinity of non-phosphorylatable Y24 or Y38 mutants to Hsp90 inhibitors [107,118]. Our recent study has demonstrated that pharmacological inhibition of Wee1 made prostate cancer cells sensitive to Hsp90 inhibitors both in vivo and in vitro, and this led to the activation of the intrinsic apoptotic pathway [103]. This downstream effect also coincided with the transcriptional downregulation of survivin and Wee1. These findings provide a novel therapeutic strategy using combined Wee1/Hsp90 inhibitors, and also support a mechanistic rationale for enhancing the pro-apoptotic activity of Hsp90 inhibitors.
Figure 4. Post-translational modification residues on human Hsp90α.
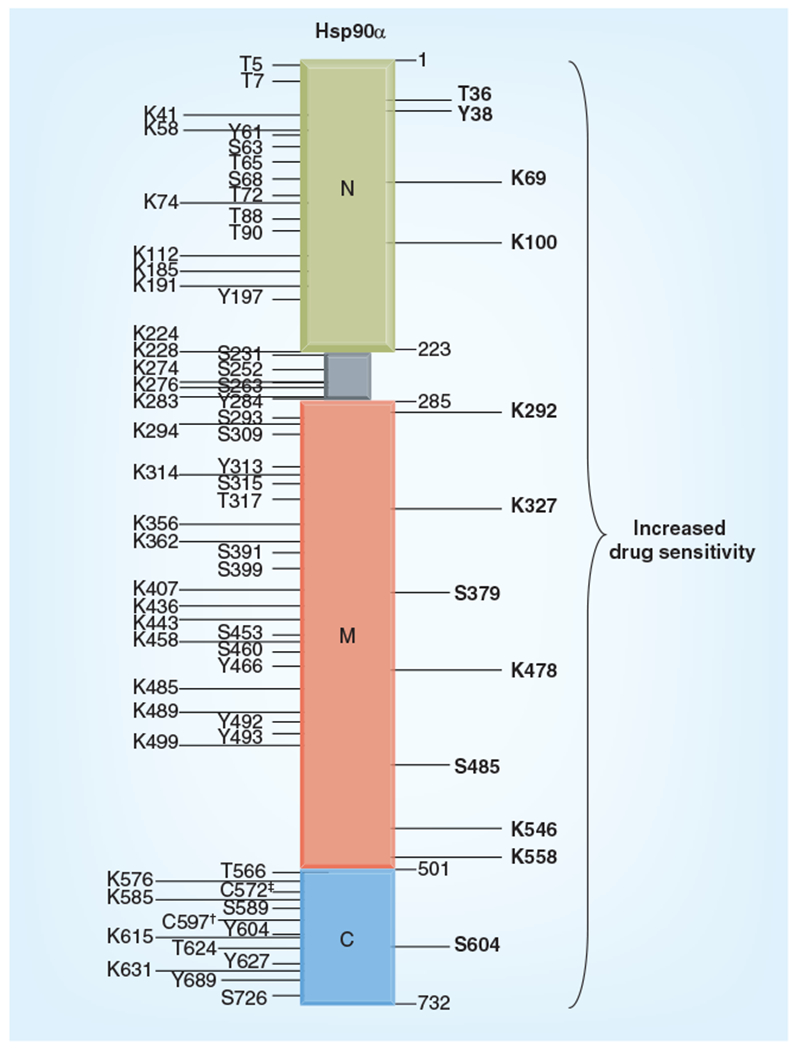
Domain location of phosphorylated serine (S), theronine (T) and tyrosine (Y) sites for which kinases are known. Acetylated lysine (K) residues, S-nitrosylated cysteine (†) and cysteine oxidation sites (‡) on human Hsp90α. Phosphorylation and acetylation sites that enhance sensitivity to Hsp90 inhibitors are also marked in bold.
Acetylation–deacetylation
Acetylation is another post-translational modification that impacts Hsp90 chaperone function [8,104,119,120]. Enzymes that catalyze the addition/removal of acetyl groups on lysine residues were initially discovered in histones, hence they were termed histone acetylases and HDAC. Initial work by Yu et al. reported that the HDACi depsipeptide (romidepsin) caused hyperacetylation of Hsp90 and decreased ATP binding. This post-translational modification also simultaneously abrogated Hsp90 interaction with several client proteins, including ErbB2, Raf-1 and mutant p53 [121]. Subsequent work demonstrated that p300 promotes acetylation, and HDAC1, 6 and 10 cause deacetylation, of Hsp90, affecting chaperone function [122–127]. Rao et al. have specifically shown that silencing HDAC6 by siRNA led to hyperacetylation of Hsp90 and also increased binding to the Hsp90 inhibitor 17-AAG [106]. Work by Yang et al. identified acetylation of seven lysine residues in Hsp90 when HEK293 cells were treated with the pan-HDACi panobinostat (LBH589) [105]. Acetylation of all seven lysines enhanced the binding of hHsp90α to 17-AAG [105]. Interestingly, recent work by Robbins et al. has shown that mutation of one of those seven lysines (K290) to arginine or glutamine conferred sensitivity to GA [105]. These observations suggest an evolutionarily conserved mechanism, where acetylation–deacetylation of Hsp90 impacts chaperone function and also influences its ability to bind to its inhibitors.
Molecular mechanisms of resistance to Hsp90 inhibitors
As more Hsp90 inhibitors enter clinical trials, the question of how to increase sensitivity to these drugs is of great interest. However, equally important, but a far less explored area, is the development of possible resistance to Hsp90 inhibitors and understanding the underlying mechanisms of this process.
Hsp90 inhibitors used in the clinic target the nucleotide-binding pocket in the N-domain of Hsp90. Therefore, mutations to this region that weaken drug binding can potentially give rise to drug resistance. Furthermore, most mutations within this pocket can potentially compromise the essential chaperone function of Hsp90. Recent work by Millson et al. demonstrated a partial resistance to GA or RD by engineering mutations to yeast Hsp90 [128,129]. Those mutations were based on unusual features of the N-domain ADP-/ATP-binding site of Hsp90 from drug producing organisms; Streptomyces hygroscopicus (GA) and Humicola fuscoatra (RD) [128,129]. Other work have shown that single amino acid substitution I123T in hHsp90β, and the corresponding hHsp90α-I128T, can make yeast and HEK293 cells resistant to Hsp90 inhibitors [130]. This mutation did not diminish drug binding in vitro, but it had strong Aha1 binding and increased ATPase activity, therefore, protecting Hsp90 from its inhibitors [130]. Although these data complement an earlier report that decreased AHA1 expression increases cellular sensitivity to Hsp90 inhibitors [87], it does not explain that tumor cells have highly active Hsp90 with increased binding affinity to 17-AAG [131]. Holmes et al., have also reported that over-expression of AHA1 in human cancer cell lines did not significantly affect their sensitivity to 17-AAG [87]. Finally, recent work by Armstrong et al. has demonstrated that deletion of HCH1, related to co-chaperone AHA1 in yeast, confers resistance to Hsp90 inhibitor NVP-AUT922 [132]. Although Hch1 does not exist in higher eukaryotes, it does, however, suggest a distinct role in the regulation of Hsp90 that may be independent of its ability to stimulate ATPase activity [132].
As mentioned in previous sections, NQO1, also known as DT-diaphorase, plays an important role in drug sensitivity or resistance. Cancer cells with high levels of NQO1 are particularly sensitive to 17-AAG, probably due to reduction of the quinone, which favors binding of more potent Hsp90 inhibitors [133]. Gaspar and colleagues reported an acquired resistance to 17-AAG in four glioblastoma cell lines in vitro. They proposed that decreased NQO1 expression and activity is the mechanism of resistance in these cells [134]. They obtained similar findings in melanoma cells (WM266.4). Benchekroun et al. observed that human breast cancer cells (MCF7/ADRR) are resistant to Hsp90 inhibitors such as GA and herbimycin A (a benzoquinonoid ansamycin antibiotic). They used photo-affinity labeling methods to show GA interaction with the P-gp efflux pump and that cells expressing this pump have lower concentrations of GA [135]. However, McCollum et al. demonstrated that even though P-gp plays a role in resistance to 17-AAG, Hsp70 and Hsp27 were the more important players in determining resistance to Hsp90 inhibitors [136]. A number of studies have linked the upregulation of these two chaperones to an increase in the cellular capacity for evasion of apoptosis [137,138]. It has been reported that downregulation of HSP27 or treating the cells with buthionine sulfoximine, an inhibitor of GSH synthesis, sensitizes cells to 17-AAG [139]. Conversely, 17-AAG can upregulate Hsp27 and make cell lines resistant to 17-AAG. There are few molecular mechanisms that can contribute towards this drug resistance. First, 17-AAG can directly bind to GSH and, therefore, may reduce binding to Hsp90 in vivo, and could, therefore, explain protection of tumor cells by increased GSH levels. Second, ansamycins can produce reactive oxygen species in cells and the formation of free radicals. GSH can eliminate this by either direct binding to 17-AAG, binding to oxygen species formed during 17-AAG treatment, such as superoxide, or binding oxidative species from mitochondrial leakage observed during 17-AAG-mediated apoptosis. Third, GSH may play an important role in oxidative protein folding that is disrupted as a result of 17-AAG inhibiting Hsp90 chaperone functions. Increased levels of GSH in tumor cells could prevent cell damage and possibly death resulting from these oxidative species. In any of these events, GSH plays an important role in cellular homeostasis and inhibits cell death as a result of 17-AAG treatment. These data suggest an explanation for disparate sensitivity and also drug resistance to ansamycin derivatives of Hsp90 inhibitors. However, mechanisms that may lead to tumor resistance to other Hsp90 inhibitors, such as the synthetic purine analog PU-H71 (Samus Therapeutics, NY, USA), or the resorcinol analogs ganetespib and NVP-AUY922, remain to be examined. These drugs are currently being extensively evaluated in clinical trials.
Conclusion
Hsp90 is an essential and evolutionarily conserved molecular chaperone in eukaryotes. Cancer cells hijack Hsp90 chaperone machinery in order to protect an array of mutated and overexpressed oncoproteins from misfolding and degradation. Therefore, Hsp90 plays a crucial role as a facilitator of oncogenic addiction and cancer cell survival. Thus, it is not surprising that 17 Hsp90 inhibitors have entered into clinical trials during the past two decades. Hence, it is important to evaluate the factors that can influence cell susceptibility to Hsp90 inhibitors. Co-chaperones and post-translational modifications play a major role in fine-tuning Hsp90 chaperone function. Targeting and preventing the interaction of certain co-chaperones, including Aha1, Cdc37, p23, Hop/Sti1 and Cyp40/Cpr6–Cpr7 with Hsp90 has made cancer cells, or the model eukaryotic organism yeast, hypersensitive to Hsp90 inhibitors. Although Hsp90 is subject to numerous post-translational modifications, only a few studies have linked the contributions of phosphorylation or acetylation of certain amino acids to Hsp90 drug binding. Remarkably, the vast majority of these post-translational modifications are catalyzed by Hsp90 clients and, therefore, targeting these enzymes may provide novel strategies to improve drug sensitivity and/or avoid drug resistance.
Future perspective
During the past two decades significant progress has been made in understanding the complex regulation of the Hsp90 machinery. Post-translational modifications of Hsp90 and co-chaperones have been recently the focus of intense scrutiny. A large number of studies have mapped various post-translational modifications on Hsp90 and their impact on chaperone activity (Figure 4). However, the contributions of these individual modifications to drug sensitivity or resistance remain to be explored. Other important areas of Hsp90 research that require further investigation are:
Identification of the enzymes (e.g., kinases, phosphatases, histone acetylases and HDACs) that target Hsp90 and co-chaperones;
Development of post-translational site-specific drugs to Hsp90 and co-chaperones to understand the impact of individual Hsp90 inhibitors or environmental stimuli on sub-cellular localization of the chaperone complex;
Unraveling the cross-talk between several post-translational modifications of the Hsp90 chaperone machine (Figure 5).
Figure 5. Post-translational modifications and co-chaperones fine-tune Hsp90 chaperone machinery.
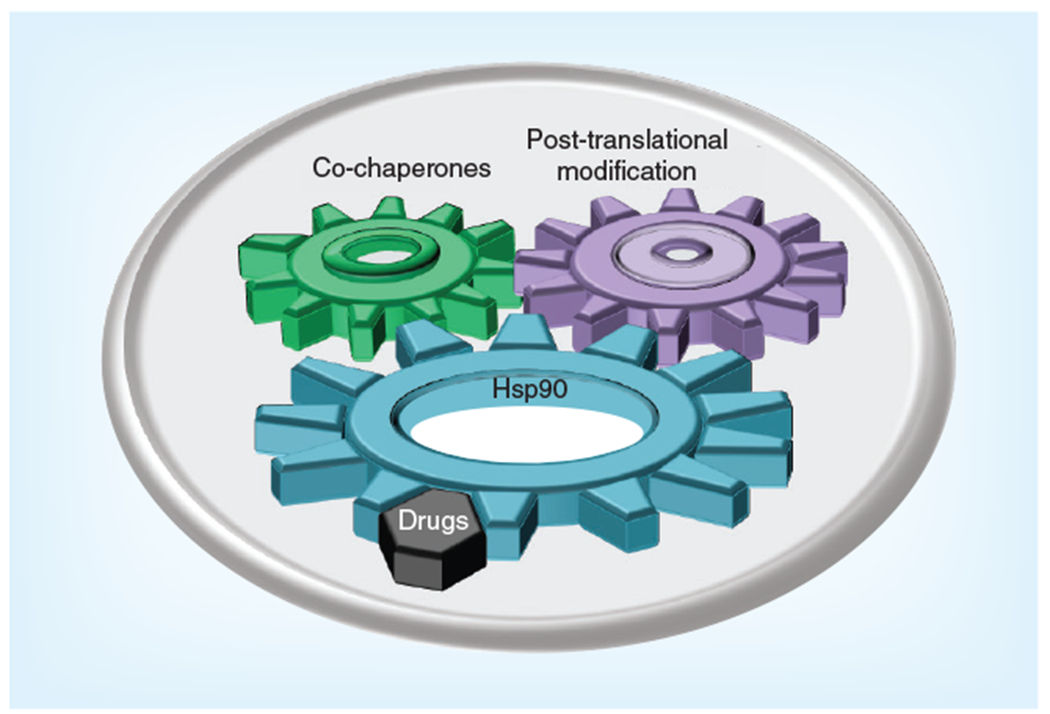
The Hsp90 chaperone cycle is regulated by the interplay between ATP binding to Hsp90 and the regulated association/dissociation of various co-chaperones. The Hsp90 chaperone machine is also regulated by a number of diverse post-translational modifications of Hsp90 and co-chaperones, including phosphorylation, S-nitrosylation, oxidation, acetylation and ubiquitination. Hsp90 inhibitors effectively displace ATP from its binding pocket in the N-domain of Hsp90, disrupt this complex mechanism and prevent chaperone cycling.
These data will expand our knowledge of the multilayered control mechanisms used by both normal and cancer cells to fine-tune Hsp90 function. They will also provide information relevant to improving drug sensitivity and inhibiting drug resistance.
Key Terms.
Molecular chaperone: Group of proteins that assist in the folding or unfolding and the assembly or disassembly of other macromolecular structures.
Co-chaperones: Proteins that are involved in regulating chaperone function.
Post-translational modification: Covalent addition of functional groups or protein.
Excutive Summary.
Hsp90 is a promising target in cancer therapy and Hsp90 inhibitors have demonstrated efficacy in clinical trials in patients with non-small-cell lung cancer and HER2-positive breast cancer.
Impact of co-chaperones on Hsp90 drug binding are discussed.
Post-translational modifications regulate Hsp90 chaperone function.
Targeting the regulators of Hsp90 function may provide a strategy to increase the potency of Hsp90 inhibitors and to prevent/minimize drug resistance.
Acknowledgments
The authors are grateful to their colleagues S Tsutsumi, W Xu, K Beebe, and collaborators C Prodromou, LH Pearl, C Vaughan, PW Piper, J Buchner, M Mayer, B Blagg, WG Stetler-Stevenson, G Colombo, B Panaretou, M-J Lee, G Morra, S Lee and A Truman for their scientific contributions.
Financial & competing interests disclosure
This work was supported by the Intramural Research Program of the National Cancer Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the Hsp90/Hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228(2), 111–133 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res 18(1), 64–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prodromou C, Pearl LH. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 3(5), 301–323 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89(2), 239–250 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem 42(2), 260–266 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Richter K, Hendershot LM, Freeman BC. The cellular world according to Hsp90. Nat. Struct. Mol. Biol 14(2), 90–94 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823(3), 624–635 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823(3), 648–655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JL. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 1823(3), 607–613 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10(8), 537–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5(10), 761–772 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Grad I, Cederroth CR, Walicki J et al. The molecular chaperone Hsp90alpha is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS ONE 5(12), el5770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol 11(7), 515–528 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Picard D Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci 59(10), 1640–1648 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali MM, Roe SM, Vaughan CK et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440(7087), 1013–1017 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94–nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 28(1), 41–56 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 28(5), 602–613 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hessling M, Richter K, Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol 16(3), 287–293 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol 16(3), 281–286 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Neckers L, Mollapour M, Tsutsumi S. The complex dance of the molecular chaperone Hsp90. Trends Biochem. Sci 34(5), 223–226 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural analysis of E. coli Hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 127(2), 329–340 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Vaughan CK, Gohlke U, Sobott F et al. Structure of an Hsp90–Cdc37–Cdk4 complex. Mol. Cell 23(5), 697–707 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SE. Hsp90: structure and function. Top. Curr. Chem 328, 155–240 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem 75, 271–294 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Neckers L Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb. Exp. Pharmacol (172), 259–277 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Whitesell L, Lin NU. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim. Biophys. Acta 1823(3), 756–766 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Alarcon SV, Mollapour M, Lee MJ et al. Tumor-intrinsic and tumor-extrinsic factors impacting Hsp90-targeted therapy. Curr. Mol. Med 12(9), 1125–1141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modi S, Stopeck A, Linden H et al. Hsp90 inhibition is effective in breast cancer: a Phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res 17(15), 5132–5139 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC. Inhibition of heat shock protein 90 (Hsp90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol 152(4), 367–379 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Roue G, Perez-Galan P, Mozos A et al. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by downregulation of the prosurvival ER chaperone BiP/Grp78. Blood 117(4), 1270–1279 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Scaltriti M, Serra V, Normant E et al. Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol. Cancer Ther 10(5), 817–824 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Schulte TW, Akinaga S, Soga S et al. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 3(2), 100–108 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, Hsp90, by the transformation suppressing agent, radicicol. Oncogene 16(20), 2639–2645 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Jensen MR, Schoepfer J, Radimerski T et al. NVP-AUY922: a small molecule Hsp90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 10(2), R33 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moser C, Lang SA, Hackl C et al. Targeting Hsp90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 32(7), 2551–2561 (2012). [PubMed] [Google Scholar]
- 36.Nagengast WB, De Korte MA, Oude Munnink TH et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with Hsp90 inhibitor NVP-AUY922. J. Nucl. Med 51(5), 761–767 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Ishii T, Seike T, Nakashima T et al. Antitumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J. 2(4), e68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodhead AJ, Angove H, Carr MG et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl] methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem 53(16), 5956–5969 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Chiosis G, Timaul MN, Lucas B et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes HER2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol 8(3), 289–299 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Wright L, Barril X, Dymock B et al. Structure–activity relationships in purine-based inhibitor binding to Hsp90 isoforms. Chem. Biol 11(6), 775–785 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Caldas-Lopes E, Cerchietti L, Ahn JH et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl Acad. Sci. USA 106(20), 8368–8373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheung KM, Matthews TP, James K et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett 15(14), 3338–3343 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Eccles SA, Massey A, Raynaud FI et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68(8), 2850–2860 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Nakashima T, Ishii T, Tagaya H et al. New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin. Cancer Res 16(10), 2792–2802 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Mccleese JK, Bear MD, Fossey SL et al. The novel Hsp90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int. J. Cancer 125(12), 2792–2801 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem 275(47), 37181–37186 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl Cancer Inst 92(3), 242–248 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem 15(26), 2702–2717 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conde R, Belak ZR, Nair M, O’Carroll RF, Ovsenek N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem. Cell Biol 87(6), 845–851 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Matts RL, Brandt GE, Lu Y et al. A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem 19(1), 684–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shelton SN, Shawgo ME, Matthews SB et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol 76(6), 1314–1322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caplan AJ. What is a co-chaperone? Cell Stress Chaperones 8(2), 105–107 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abbas-Terki T, Briand PA, Donze O, Picard D. The Hsp90 co-chaperones Cdc37 and Sti1 interact physically and genetically. Biol. Chem 383(9), 1335–1342 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Chang HC, Nathan DF, Lindquist S. In vivo analysis of the Hsp90 cochaperone Sti1 (p60). Mol. Cell. Biol 17(1), 318–325 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richter K, Muschler P, Hainzl O, Reinstein J, Buchner J. Sti1 is a non-competitive inhibitor of the Hsp90 ATPase. Binding prevents the N-terminal dimerization reaction during the atpase cycle. J. Biol. Chem 278(12), 10328–10333 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Song Y, Masison DC. Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein Sti1 (Hop1). J. Biol. Chem 280(40), 34178–34185 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee P, Shabbir A, Cardozo C, Caplan AJ. Sti1 and Cdc37 can stabilize Hsp90 in chaperone complexes with a protein kinase. Mol. Biol. Cell 15(4), 1785–1792 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maclean M, Picard D. Cdc37 goes beyond Hsp90 and kinases. Cell Stress Chaperones 8(2), 114–119 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siligardi G, Panaretou B, Meyer P et al. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem 277(23), 20151–20159 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Vaughan CK, Mollapour M, Smith JR et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 31(6), 886–895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mclaughlin SH, Sobott F, Yao ZP et al. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J. Mol. Biol 356(3), 746–758 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Picard D Intracellular dynamics of the Hsp90 co-chaperone p23 is dictated by Hsp90. Exp. Cell. Res 312(2), 198–204 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Sullivan WP, Owen BA, Toft DO. The influence of ATP and p23 on the conformation of Hsp90. J. Biol. Chem 277(48), 45942–45948 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Lotz GP, Lin H, Harst A, Obermann WM. Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem 278(19), 17228–17235 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Meyer P, Prodromou C, Liao C et al. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23(3), 511–519 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panaretou B, Siligardi G, Meyer P et al. Activation of the ATPase activity of Hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10(6), 1307–1318 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Johnson JL, Halas A, Flom G. Nucleotide-dependent interaction of Saccharomyces cerevisiae Hsp90 with the cochaperone proteins Sti1, Cpr6, and Sba1. Mol. Cell. Biol 27(2), 768–776 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayr C, Richter K, Lilie H, Buchner J. Cpr6 and Cpr7, two closely related Hsp90-associated immunophilins from Saccharomyces cerevisiae, differ in their functional properties. J. Biol. Chem 275(44), 34140–34146 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Catlett MG, Kaplan KB. Sgt1p is a unique co-chaperone that acts as a client adaptor to link Hsp90 to Skp1p. J. Biol. Chem 281(44), 33739–33748 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell. Biol 17(2), 87–92 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Boter M, Li K et al. Structural and functional coupling of Hsp90- and Sgt1-centred multi-protein complexes. EMBO J. 27(20), 2789–2798 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Millson SH, Nuttall JM, Mollapour M, Piper PW. The Hsp90/Cdc37p chaperone system is a determinant of molybdate resistance in Saccharomyces cerevisiae. Yeast 26(6), 339–347 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Vaughan CK, Piper PW, Pearl LH, Prodromou C. A common conformationally coupled ATPase mechanism for yeast and human cytoplasmic Hsp90s. FEBS J. 276(1), 199–209 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forafonov F, Toogun OA, Grad I, Suslova E, Freeman BC, Picard D. p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol. Cell. Biol 28(10), 3446–3456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson JL, Brown C. Plasticity of the Hsp90 chaperone machine in divergent eukaryotic organisms. Cell Stress Chaperones 14(1), 83–94 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roe SM, Ali MM, Meyer P et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116(1), 87–98 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Siligardi G, Hu B, Panaretou B, Piper PW, Pearl LH, Prodromou C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem 279(50), 51989–51998 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Gray PJ Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 67(24), 11942–11950 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Smith JR, Clarke PA, de Billy E, Workman P. Silencing the cochaperone Cdc37 destabilizes kinase clients and sensitizes cancer cells to Hsp90 inhibitors. Oncogene 28(2), 157–169 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit Hsp90 and Cdc37 interaction. J. Biol. Chem 284(51), 35381–35389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bandhakavi S, Mccann RO, Hanna DE, Glover CV. A positive feedback loop between protein kinase CKII and Cdc37 promotes the activity of multiple protein kinases. J. Biol. Chem 278(5), 2829–2836 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Miyata Y Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell. Mol. Life Sci 66(11–12), 1840–1849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shao J, Prince T, Hartson SD, Matts RL. Phosphorylation of serine 13 is required for the proper function of the Hsp90 cochaperone, Cdc37. J. Biol. Chem 278(40), 38117–38120 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Xu W, Mollapour M, Prodromou C et al. Dynamic tyrosine phosphorylation modulates cycling of the Hsp90-P50(Cdc37)-AHA1 chaperone machine. Mol. Cell 47(3), 434–443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Retzlaff M, Hagn F, Mitschke L et al. Asymmetric activation of the Hsp90 dimer by its cochaperone aha1. Mol. Cell 37(3), 344–354 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Venable J, Lapointe P et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127(4), 803–815 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of Hsp90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 68(4), 1188–1197 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Richter K, Walter S, Buchner J. The cochaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J Mol Biol 342(5), 1403–1413 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Johnson JL, Toft DO. Binding of p23 and Hsp90 during assembly with the progesterone receptor. Mol. Endocrinol 9(6), 670–678 (1995). [DOI] [PubMed] [Google Scholar]
- 90.Cullinan SB, Whitesell L. Heat shock protein 90: a unique chemotherapeutic target. Semin. Oncol 33(4), 457–465 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Onuoha SC, Coulstock ET, Grossmann JG, Jackson SE. Structural studies on the cochaperone Hop and its complexes with Hsp90. J. Mol. Biol 379(4), 732–744 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Schmid AB, Lagleder S, Grawert MA et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31(6), 1506–1517 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li J, Richter K, Buchner J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Bio/. 18(1), 61–66 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Piper PW, Panaretou B, Millson SH et al. Yeast is selectively hypersensitised to heat shock protein 90 (Hsp90)-targetting drugs with heterologous expression of the human Hsp90beta, a property that can be exploited in screens for new Hsp90 chaperone inhibitors. Gene 302(1–2), 165–170 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol 9(9), 3919–3930 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Millson SH, Vaughan CK, Zhai C et al. Chaperone ligand-discrimination by the TPR-domain protein Tah1. Biochem. J 413(2), 261–268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prodromou C, Siligardi G, O’Brien R et al. Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 18(3), 754–762 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zuehlke AD, Johnson JL. Chaperoning the chaperone: a role for the co-chaperone Cpr7 in modulating Hsp90 function in Saccharomyces cerevisiae. Genetics 191(3), 805–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li J, Richter K, Reinstein J, Buchner J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol 20(3), 326–331 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Dolinski KJ, Cardenas ME, Heitman J. CNS1 encodes an essential p60/Sti1 homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol. Cell. Biol 18(12), 7344–7352 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goes FS, Martin J. Hsp90 chaperone complexes are required for the activity and stability of yeast protein kinases Mik1, Wee1 and Swe1. Eur. J. Biochem 268(8), 2281–2289 (2001). [DOI] [PubMed] [Google Scholar]
- 102.Mollapour M, Tsutsumi S, Donnelly AC et al. Swe1 Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 37(3), 333–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iwai A, Bourboulia D, Mollapour M et al. Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 11(19), 3649–3655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scroggins BT, Neckers L. Post-translational modification of heat shock protein 90: impact on chaperone function. Expert Opin. Drug Discov 2(10), 1403–1414 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Yang Y, Rao R, Shen J et al. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 68(12), 4833–4842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rao R, Fiskus W, Yang Y et al. HDAC6 inhibition enhances 17-AAG-mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 112(5), 1886–1893 (2008). [DOI] [PubMed] [Google Scholar]
- 107.Mollapour M, Tsutsumi S, Kim YS, Trepel J, Neckers L. Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity. Oncotarget 2(5), 407–417 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the Hsp90 stress protein and the p60v-src tyrosine kinase. J. Biol. Chem 270(48), 28654–28659 (1995). [DOI] [PubMed] [Google Scholar]
- 109.Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 25(2), 367–376 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang S, Pashtan I, Tsutsumi S, Xu W, Neckers L. Cancer cells harboring MET gene amplification activate alternative signaling pathways to escape MET inhibition but remain sensitive to Hsp90 inhibitors. Cell Cycle 8(13), 2050–2056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mollapour M, Tsutsumi S, Truman AW et al. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell 41(6), 672–681 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Prodromou C, Panaretou B, Chohan S et al. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 19(16), 4383–4392 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hawle P, Siepmann M, Harst A, Siderius M, Reusch HP, Obermann WM. The middle domain of Hsp90 acts as a discriminator between different types of client proteins. Mol. Cell. Biol 26(22), 8385–8395 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol 15(7), 3917–3925 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cunningham CN, Krukenberg KA, Agard DA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J. Biol. Chem 283(30), 21170–21178 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mollapour M, Neckers L. Detecting Hsp90 phosphorylation. Methods Mol. Biol 787, 67–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol. Biol. Cell 18(11), 4659–4668 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9(12), 2310–2316 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scroggins BT, Robzyk K, Wang D et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25(1), 151–159 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robbins N, Leach MD, Cowen LE. Lysine deacetylases Hda1 and Rpd3 regulate Hsp90 function thereby governing fungal drug resistance. Cell Rep. 2(4), 878–888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu X, Guo ZS, Marcu MG et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl Cancer Inst 94(7), 504–513 (2002). [DOI] [PubMed] [Google Scholar]
- 122.Park JH, Kim SH, Choi MC et al. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun 368(2), 318–322 (2008). [DOI] [PubMed] [Google Scholar]
- 123.Bali P, Pranpat M, Bradner J et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem 280(29), 26729–26734 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Kovacs JJ, Murphy PJ, Gaillard S et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18(5), 601–607 (2005). [DOI] [PubMed] [Google Scholar]
- 125.Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem 284(12), 7436–7445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of Hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem 280(40), 33792–33799 (2005). [DOI] [PubMed] [Google Scholar]
- 127.Ai J, Wang Y, Dar JA et al. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol. Endocrinol 23(12), 1963–1972 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Millson SH, Chua CS, Roe SM et al. Features of the Streptomyces hygroscopicus HtpG reveal how partial geldanamycin resistance can arise with mutation to the ATP binding pocket of a eukaryotic Hsp90. FASEB J. 25(11), 3828–3837 (2011). [DOI] [PubMed] [Google Scholar]
- 129.Millson SH, Prodromou C, Piper PW. A simple yeast-based system for analyzing inhibitor resistance in the human cancer drug targets Hsp90alpha/beta. Biochem. Pharmacol 79(11), 1581–1588 (2010). [DOI] [PubMed] [Google Scholar]
- 130.Zurawska A, Urbanski J, Matuliene J et al. Mutations that increase both Hsp90 ATPase activity in vitro and Hsp90 drug resistance in vivo. Biochim. Biophys. Acta 1803(5), 575–583 (2010). [DOI] [PubMed] [Google Scholar]
- 131.Kamal A, Thao L, Sensintaffar J et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425(6956), 407–410 (2003). [DOI] [PubMed] [Google Scholar]
- 132.Armstrong H, Wolmarans A, Mercier R, Mai B, Lapointe P. The co-chaperone Hch1 regulates Hsp90 function differently than its homologue Aha1 and confers sensitivity to yeast to the Hsp90 inhibitor NVP-AUY922. PLoS ONE 7(11), e49322 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J. Natl Cancer Inst 91(22), 1940–1949 (1999). [DOI] [PubMed] [Google Scholar]
- 134.Gaspar N, Sharp SY, Pacey S et al. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 69(5), 1966–1975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Benchekroun MN, Schneider E, Safa AR, Townsend AJ, Sinha BK. Mechanisms of resistance to ansamycin antibiotics in human breast cancer cell lines. Mol. Pharmacol 46(4), 677–684 (1994). [PubMed] [Google Scholar]
- 136.Mccollum AK, Teneyck CJ, Stensgard B et al. P-glycoprotein-mediated resistance to Hsp90-directed therapy is eclipsed by the heat shock response. Cancer Res. 68(18), 7419–7427 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Guo F, Rocha K, Bali P et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylaminodemethoxy geldanamycin. Cancer Res. 65(22), 10536–10544 (2005). [DOI] [PubMed] [Google Scholar]
- 138.Powers MV, Clarke PA, Workman P. Dual targeting of Hsc70 and Hsp72 inhibits Hsp90 function and induces tumor-specific apoptosis. Cancer Cell 14(3), 250–262 (2008). [DOI] [PubMed] [Google Scholar]
- 139.Cysyk RL, Parker RJ, Barchi JJ Jr, Steeg PS, Hartman NR, Strong JM. Reaction of geldanamycin and C17-substituted analogues with glutathione: product identifications and pharmacological implications. Chem. Res. Toxicol 19(3), 376–381 (2006). [DOI] [PubMed] [Google Scholar]
Website
- 201. Updated list of Hsp90 clients. www.picard.ch/downloads/downloads.html.