

Abstract
OBJECTIVE:
The risk of thrombosis in myeloproliferative neoplasms (MPN), such as primary myelofibrosis (PMF) varies depending on the type of key driving mutation (JAK2, CALR and MPL) and the accompanying mutations in other genes. In the current study, we sought to examine the propensity for thrombosis, as well as platelet activation properties in a mouse model of PMF induced by JAK2V617F mutation.
APPROACH:
Vav1-hJAK2V617F transgenic mice show hallmarks of PMF, including significant megakaryocytosis and bone marrow fibrosis, with a moderate increase in red blood cells and platelet number. This mouse model was used to study responses to two models of vascular injury and to investigate platelet properties.
RESULTS:
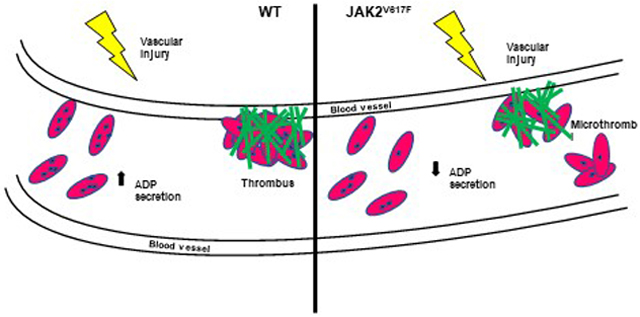
Platelets derived from the mutated mice have reduced aggregation in response to collagen, reduced thrombus formation and thrombus size, as demonstrated using laser-induced or FeCl3-induced vascular injury models, and increased bleeding time. Strikingly, the mutated platelets had a significantly reduced number of dense granules, which could explain impaired adenosine diphosphate (ADP) secretion upon platelet activation, and a diminished second wave of activation.
CONCLUSIONS:
Together, our study highlights for the first time the influence of a hyperactive JAK2 on platelet activation-induced ADP secretion and dense granule homeostasis, with consequent effects on platelet activation properties.
Keywords: Platelets, aggregation, thrombosis, activation, granule, secretion, hemostasis
Subject terms: Animal Models of Human Disease, Fibrosis, Translational Studies, Thrombosis
Graphical Abstract
INTRODUCTION
Philadelphia chromosome negative myeloproliferative neoplasms (MPN) include polycythemia vera (PV), essential thrombocytosis (ET) and primary myelofibrosis (PMF). The latter three are typically classified according to somatic mutations mainly in three genes (JAK2, CALR and MPL) combined with abnormalities in the myeloid lineage. PMF consists of augmented proliferation of megakaryocytes, fibrosis in the bone marrow and presence of a clonal marker1, 2. The classification by the World Health Organization makes a distinction between pre-PMF and overt PMF, the main difference being the presence of fibrosis in overt PMF3.
Several mouse models of Philadelphia chromosome-negative MPN have been developed4. Most recent focus has been on incorporating the JAK2V617F mutation in the myeloid lineages through knock-in (KI). Differences in expression of the JAK2V617F mutation appear to correlate with the phenotypic manifestations4. Differentiating between PV, ET and PMF is pivotal as the clinical presentation, the prognosis and propensity for thrombosis are heterogeneous in these patient populations5. Expression of JAK2V617F in KI murine models at similar levels or higher than wildtype JAK2 is associated with a PV-like phenotype, whereas expression levels of JAK2V617F lower than wildtype JAK2 results in increased platelet levels consistent with an ET-like phenotype6, 7.
All Philadelphia chromosome negative MPN have an increased thrombogenic tendency8. Overall, arterial or venous thrombosis in patients with PMF has been shown to be approximately 20%9. All MPN, including PMF, have an increased tendency to develop Budd-Chiari syndrome, involving vein occlusion10. Unlike PV and ET, hemorrhagic events are more common in PMF (approximately 10% vs 3%)11. Mimicking thrombogenic phenotypes in mouse models has uncovered heterogeneity which is also observed in patients with MPN. Several JAK2V617F models exist and exhibit different characteristics on the spectrum of MPN. For example, one human JAK2V617F KI model exhibited a more ET-like phenotype with observed megakaryocyte and platelet hyperactivation7. In this instance the mice were heterozygous for the JAK2V617F mutation. In later studies, the same group showed that homozygous JAK2V617F mutation resulted in a more profound PV-like phenotype with marked erythrocytosis and increased platelet turnover12. On the other hand, a JAK2V617F KI mouse model showed hyporesponsive platelets, a mild GPVI deficiency in vitro and rapid occlusive, but unstable clots in vivo6. These examples illustrate that the heterogenicity of hemostatic disorders observed in mouse models likely depends on the level and site of JAK2V617F mutation. Indeed, new evidence indicates that endothelial cells13 and neutrophils are contributing to the increased thrombosis in MPN through neutrophil extracellular traps (NETs)14. Endothelial cells contribute in the ET phenotype to an acquired von Willebrand deficiency, resulting in increased bleeding tendency13. In the current study, we sought to examine propensity for thrombosis and platelet activation properties in a transgenic mouse model in which human JAK2V617F is expressed in megakaryocytes. This mouse line shows hallmarks of PMF including significant megakaryocytosis and bone marrow fibrosis, with moderate increase in red blood cells and platelet number15.
MATERIALS AND METHODS
The authors declare that all supporting data are available within the article [and its online supplementary files].
JAK2V617F transgenic mice
Vav1-hJAK2V617F (JAK2V617F) mice were gift from Dr. Zhizhuang Joe Zhao (University of Oklahoma). The mouse line A, harboring 13 copies of the transgene, has the hallmarks of PMF, including expansion of the megakaryocyte lineage, a fibrotic bone marrow and splenomegaly (Supplemental Figure 1A)15, 16. Expansion of the mouse colonies was performed at Boston University School of Medicine and all studies involving mice were approved by the Boston University Institutional Animal Care and Use Committee. Animal housing conditions and treatment protocols were approved by the Institutional Animal Care and Use Committee of Boston University School of Medicine.
Peripheral blood analysis
Peripheral blood collection was performed via retro-orbital plexus bleeding using a heparinized capillary tube in animals under isoflurane anesthesia. Blood parameters were measured in blood collected in EDTA tubes (Sarstedt, Germany) using a Hemavet HV950FS hematology counter (Drew Scientific, Waterbury, CT).
Vascular injury models
The carotid artery injury model of vascular thrombosis was performed as previously described17 Mice were anesthetized by 5% isoflurane gas and placed on a temperature-regulated pad to maintain body temperature at 37°C. The right carotid artery was exposed and basal blood flow recorded using a 0.5PSB S-Series flowprobe connected to a TS420 perivascular transit-time flow meter (Transonic, Ithaca, NY). The probe was removed and a piece of Whatman filter paper (1mm × 3 mm) soaked in 7.5% ferric chloride (FeCl3) (Sigma-Aldrich, St. Louis, MO) was placed in the artery for 1 min. The probe was then placed on the carotid artery downstream of the injury site and the flow of blood was measured for a maximum of 25 min starting from the placement of the filter. The mean, maximum, and minimum carotid flow was recorded using Powerlab Chart5 version 5.2 software in 1 second intervals. Time to occlusion (tOc) was determined as the first measurement ≤ 0.299 mL/min.
For laser-injury thrombosis experiments, animal care and experimental procedures were performed in accordance with and under the approval of the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee. Mice were anaesthetized with intraperitoneal injection of a ketamine (125 mg/kg) and xylazine (12.5 mg/kg) mixture in sterile saline. Anesthesia was maintained with pentobarbital (5 mg/kg) through a jugular vein cannula. The intravital fluorescence microscopy system has previously been described18. Modifications of the imaging system that were used for these studies include an Orca-Flash 4.0 sCMOS camera (Hamamatsu-Hamamatsu City, Japan) to capture digital video images, a LED-based SpectraX light engine (Lumencor-Beaverton, OR) using six solid state light sources, and a LED-based white light source (Prior Scientific-Rockland, MA). Cremaster arterioles were injured using a MicroPoint Laser system (Photonics Instruments). We use a cremaster preparation since this surgical window provides access to a microvasculature that is mostly free of connective tissue and can be accessed with minimal trauma to the vessel. Other preparations are associated with more connective tissue, which can interfere with the path of the laser, and trauma, which can activate leukocyte rolling and affect thrombus formation. Since only male mice have a cremaster muscle, only male mice are used for these studies. Ablation injuries were observable by white-light trans illumination of the cremaster muscle, presenting as a distortion of the vessel wall in the region adjacent to the injury site. This distortion was quantified by measuring the length of the disrupted region in micrometers. All ablation injury measurements were obtained from the frame immediately following injury for consistency. Data from wild-type (WT) mice (35 thrombi, 4 mice) and Vav1-hJAK2V617F mice (31 thrombi, 3 mice) were used to determine the median value of the integrated fluorescence intensity to account for the variability of thrombus formation at any given set of experimental conditions. Data were analyzed using Slidebook 6.0 (Intelligent Imaging Innovations).
Tail bleeding time
Mice induced and maintained by isoflurane anesthesia were placed on a heat pad and 3 mm of tail tip was cut with a scalpel and immersed immediately in saline (0.9% NaCl) at 37°C in 50 ml conical tube. Time to cessation of blood stream was measured and recorded. Absence of bleeding for 1 minute was considered complete cessation. Bleeding is observed for a total of 20 minutes from the cutting of the tail, including partial cessations that resume within 1 minute19.
Isolation of washed platelets and platelet aggregation assay
Blood was collected via retro-orbital plexus in 10% final volume of acid-citrate-dextrose anti-coagulant solution (85 mM trisodium citrate dihydrate, 66.6 mM citric acid monohydrate, 111 mM dextrose, 450 mOsm/L, pH 4.5) and diluted with modified Tyrode’s buffer (137 mM NaCl, 11.9 mM NaHCO3, 0.4 mM Na2HPO4, 2.7 mM KCl, 1.1 mM MgCl2, 5.6 mM glucose, pH 7.3) and centrifuged at 100g for 30 minutes. Platelet-rich plasma and buffy coat were transferred to a new tube and centrifuged at 100g for 20 minutes. Prostacyclin (Sigma Aldrich, St. Louis, MO) was added at 1 μg/mL and apyrase (Sigma Aldrich, St. Louis, MO) was added at 2 U/mL. Platelets were then pelleted at 900g for 7 minutes and resuspended in modified Tyrode’s buffer for quantitation on the Hemavet HV950FS hematology counter and diluted to the appropriate concentration. Platelet aggregation was monitored in a PAP-4 aggregometer equipped with a micro-volume adaptor (Bio/Data Corporation, Horsham, PA) at 37°C for 6 minutes, at 1200 rpm stirring speed, as previously described17. Aggregation was performed with 180 μl of washed platelets at 1.0 × 108 platelets/mL, added with 20 μL of murine thrombin (Enzyme Research Laboratories, South Bend, IN), acid-soluble calf skin type I collagen (Bio/Data Corp, Horsham), and adenosine diphosphate (ADP) (Bio/Data) with doses indicated in Figures.
Flow cytometry
Flow cytometry analysis of washed platelets was completed as previously described17. For analysis of GPVI, α2 integrin subunit, and β1 integrin subunit expression, washed platelets were incubated for 30 minutes in modified Tyrode’s buffer with the following antibodies: PE conjugated anti-mouse CD41a (eBioscience, catalog # 12-0411-83); FITC conjugated rat anti-mouse GPVI (Emfret Analytics, catalog # M011–1); Anti-mouse CD49b FITC (α2 subunit) (eBioscience, catalog # 11-0491-82); Anti-Rat CD29 Alexa Fluor 647 (β1 subunit) (BD Pharmingen, catalog # 562153). For analysis of P-selectin expression in agonist-stimulated platelets, platelets were resuspended at 1 × 108 platelets/ml, aliquoted into tubes and stained with FITC Rat anti-mouse CD62p (P-selectin) (BD Pharmingen, catalog # 553744) and APC Rat anti-mouse CD41a (eBioscience, catalog # 17-0411-82). Acid-soluble calf skin type I collagen (Bio/Data Corp, Horsham) were added with doses indicated in Figures. Samples were analyzed after 15 minutes of incubation at room temperature. Cells were analyzed on the LSRII flow cytometer using FACS Diva software (BD Biosciences) and FlowJo (Becton, Dickinson and Company).
Platelet granule ADP and adenosine triphosphate (ATP) measurement
Platelets were isolated from whole blood as described above. 50 μl of 108 platelets/mL per reaction were prepared. Agonist was added at the indicated doses and incubated with platelets for the indicated time, followed by centrifugation at 900g for 1 min. The supernatant was used to measure ADP and ATP secretion using the Abcam ADP/ATP Ratio Assay Kit (Abcam ab65313, Cambridge, MA) per manufacturer’s instructions. Technical duplicates for each biological replicate were performed. Phorbol-myristate-acetate (PMA) (Sigma) was dissolved as per manufacture instructions.
Electron microscopy (EM)
Platelets were isolated from whole blood as described above. Platelets from the same mouse line were pooled and centrifuged at 900g for 15 min. A volume of approximately 50–100 μl of platelet pellets was supplemented with about 200 μl of Modified Tyrode’s buffer (137mM NaCl, 11.9mM NaHCO3, 0.4mM Na2HPO4, 2.7mM KCl, 1.1mM MgCl2, 5.6mM glucose, pH7.3). Then, a volume of 1.3 mL of Formaldehyde/Glutaraldehyde, 2.5% each in 0.1 M Sodium Cacodylate Buffer (fixative) pH 7.4 at 37°C was added to the solution of platelets. Pellets were allowed to incubate in fixative for 5 min and then pelleted and resuspended in fixative again and allowed to fix overnight. Pellets were stored in cacodylate buffer (Electron Microscopy Sciences 11652, Hatfield, PA) until ready for electron microscopy analysis. For thin-section EM, platelets were fixed with 1.25% paraformaldehyde, 0.03% picric acid, 2.5% glutaraldehyde in 0.1–M cacodylate buffer (pH 7.4) for 1 h, post-fixed with 1% osmium tetroxide, dehydrated through a series of alcohols, infiltrated with propylene oxide, and embedded in epoxy resin. Ultrathin sections were stained and examined with a Tecnai G2 Spirit BioTwin electron microscope (Hillsboro, OR) at an accelerating voltage of 80 kV. Images were recorded with an Advanced Microscopy Techniques (AMT) 2-K charged coupled device camera, using AMT digital acquisition and analysis software (Advanced Microscopy Techniques, Danvers, MA).
Measurement of blood fibrinogen and prothrombin time
Plasma was collected per IDEXX Laboratories (Westbrook, Maine) instructions. Briefly, whole blood was collected in 3.2% citrate at a 1:9 ratio of citrate: whole blood and centrifuged at 1500 rpm, 15 minutes. Plasma was collected, frozen, and shipped to IDEXX for analysis.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism. Student’s t test and Mann-Whitney U test with an α of at least 0.05 were considered significant. Normality and variance were not tested. As described in a previous publication20, the platelet accumulation and fibrin formation data do not demonstrate a normal distribution. For this reason, the non-parametric analysis Mann-Whitney test was used for these data. In the Mann-Whitney nonparametric U test, the values are ranked from low to high and the P-values were calculated based on the ranks of the two groups. Area Under the Curves were extrapolated from Igor Pro using the trapezoid method based on the equation:
Please see the Major Resources Table in Supplemental Materials.
RESULTS
JAK2V617F mice show decreased tOc in vascular injury models of thrombosis
The Vav1-hJAK2V617F mouse model of PMF was subjected to FeCl3-induced vascular injury. Both young and older JAK2V617F mice showed prolonged tOc (Figure 1A) and prolonged tail bleeding time (Figure 1B), compared to matching controls. Since older JAK2V617F mice have significant bone marrow fibrosis, we conclude that myelofibrosis per se is not a significant determinant of the thrombotic response in these mice, compared to younger JAK2V617F mice. Analysis of plasma isolated from the experimental groups indicated that fibrinogen level as well as prothrombin time (indicative of plasma coagulation properties) were similar in JAK2V617F and matching WT control mice (Table 1). Of note, platelet counts were slightly higher in JAK2V617F mice compared to controls, as typical of PMF (blood cell count is shown in Table 2).
Figure 1.
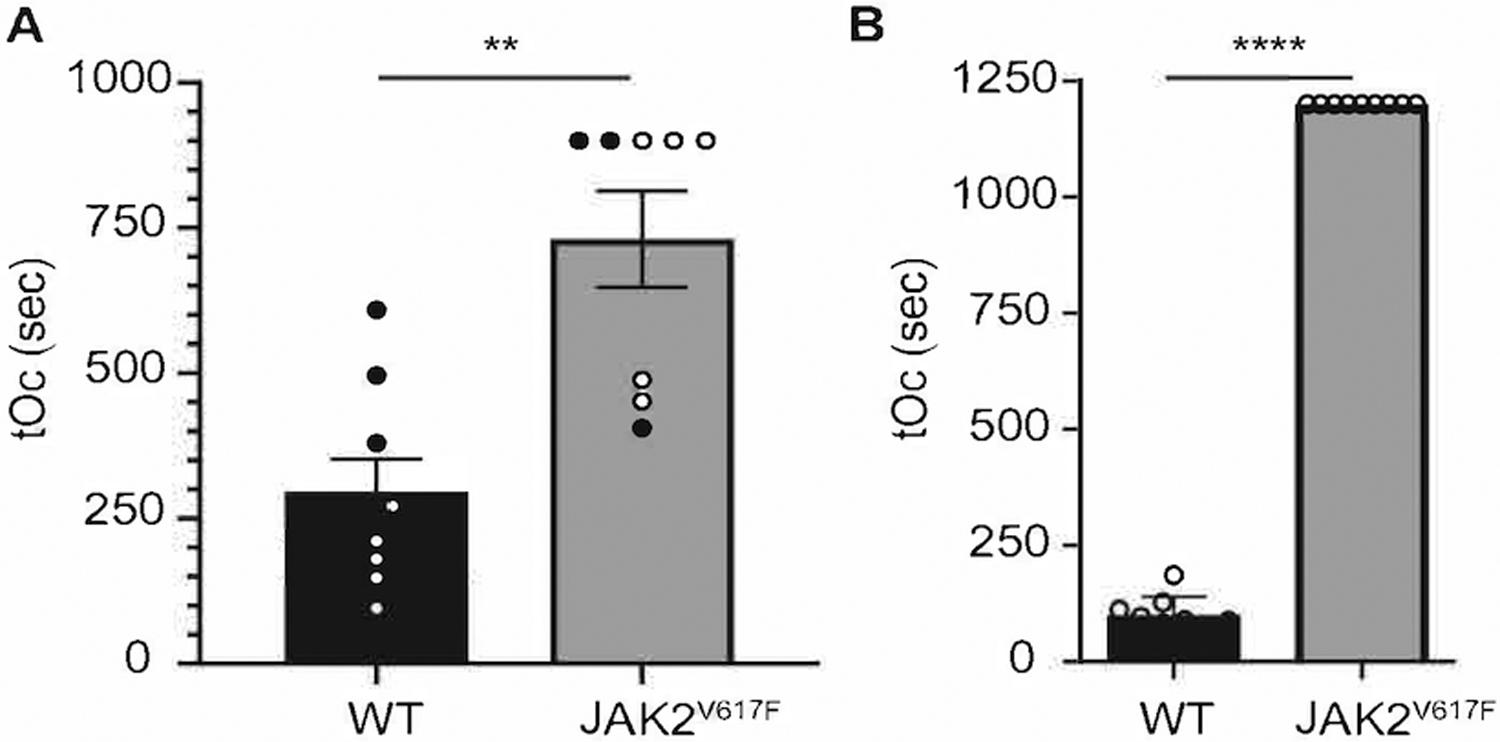
JAK2V617F mice show prolonged time to occlusion in a FeCl3 model of vascular injury, as well as prolonged bleeding. A. Carotid artery injury was performed on WT (n = 8) and JAK2V617F (n= 9) male mice. tOc for each mouse in shown. Black circles represent mice 12 weeks old, open circles represent mice age 30 weeks old. Data are averages +/− SD. B. Tail bleeding assay of WT (n = 9) and JAK2V617F (n = 9) male mice 14–20 weeks old. ** p < 0.01, **** p < 0.0001, unpaired two-tailed t-test.
Table 1.
Analysis of blood fibrinogen and prothrombin time in WT and JAK2V617F plasma.
| Prothrombin Time (sec)* | Fibrinogen (mg/dL)* | |
|---|---|---|
| WT† | 12.2 ± 0.7 (n=5) | 222.6 ± 77.0 (n=5) |
| JAK2V617F† | 11.7 ± 0.9 (n=4) | 281.5 ± 67.7 (n=4) |
| P ‡ | NS | NS |
Data are mean values +/− standard deviation (SD).
4–5 WT and JAK2V617F female mice, approximately 30 weeks old.
Unpaired two-tailed t-test.
Table 2.
Peripheral blood complete blood count.
| WBC (103/μl) * | RBC (M/μl) * | Hb (g/dL) * | HCT (%)* | PLT (K/μl) * | |
|---|---|---|---|---|---|
| WT† | 8.1 ± 2.6 | 6.2 ± 0.3 | 7.5 ± 0.83 | 27.8± 2.2 | 709.3 ± 77.1 |
| JAK2V617F† | 17.0 ± 6.9 | 6.9 ± 0.74 | 6.1 ± 1.0 | 25.4 ± 3.4 | 1010.7 ± 558.6 |
| P‡ | NS | NS | NS | NS | 0.021 |
WBC, white blood cell count; RBC, red blood cell count; Hb, hemoglobin; HCT, hematocrit; PLT, platelet count.
Data are mean values +/− SD, 4 biological replicates.
Male mice, approximately 30 weeks old.
Unpaired two-tailed t-test.
We used intravital microscopy to monitor platelet accumulation and fibrin formation in real-time following laser-induced injury of cremaster arterioles. Platelets and fibrin were visualized using Dylight 647-labeled anti-platelet antibody (CD42b) and Dylight 488-labeled anti-fibrin antibody (59D8). Both platelet accumulation and fibrin formation were impaired in Vav1-hJAK2V617F mice compared with controls (Figure 2A–E). Vav1-hJAK2V617F mice showed decreased platelet accumulation over time compared to control mice (Figure 2B). Total platelet accumulation as determined by area under the curve (AUC) analysis indicated a median value of 9.92×108 in Vav1-hJAK2V617F mice whereas control mice showed a median AUC value of 1.44×1010 (p < 0.01; Figure 2C), indicating a >14-fold reduction in platelet accumulation. The kinetics of fibrin formation were also decreased in Vav1-hJAK2V617F mice (Figure 2D), with median fibrin AUC measurements indicating 1.34×108 in Vav1-hJAK2V617F mice and 1.74×109 in control mice (p < 0.01; Figure 2E). Differences in thrombus formation can occur from differences in injury severity. To determine whether reduced thrombus formation in the Vav1-hJAK2V617F mice resulted from decreased injury sizes, we measured ablation injury length in Vav1-hJAK2V617F and control mice. No significant differences in injury size were detected (Figure 2F). Furthermore, when platelet accumulation and fibrin formation for individual thrombi were normalized on the basis of injury size, differences in the median normalized platelet accumulation (2.0×107 AUC/injury length in Vav1-hJAK2V617F mice vs 2.4×108 AUC/injury length in controls) and fibrin formation (2.80×106 AUC/injury length in Vav1-hJAK2V617F mice vs 2.99×107 AUC/injury length in controls) persisted (p < 0.01: Figure 2G). These data indicate that platelet accumulation and fibrin formation are defective in Vav1-hJAK2V617F mice despite demonstrating similar injury sizes following laser ablation of arterioles.
Figure 2.
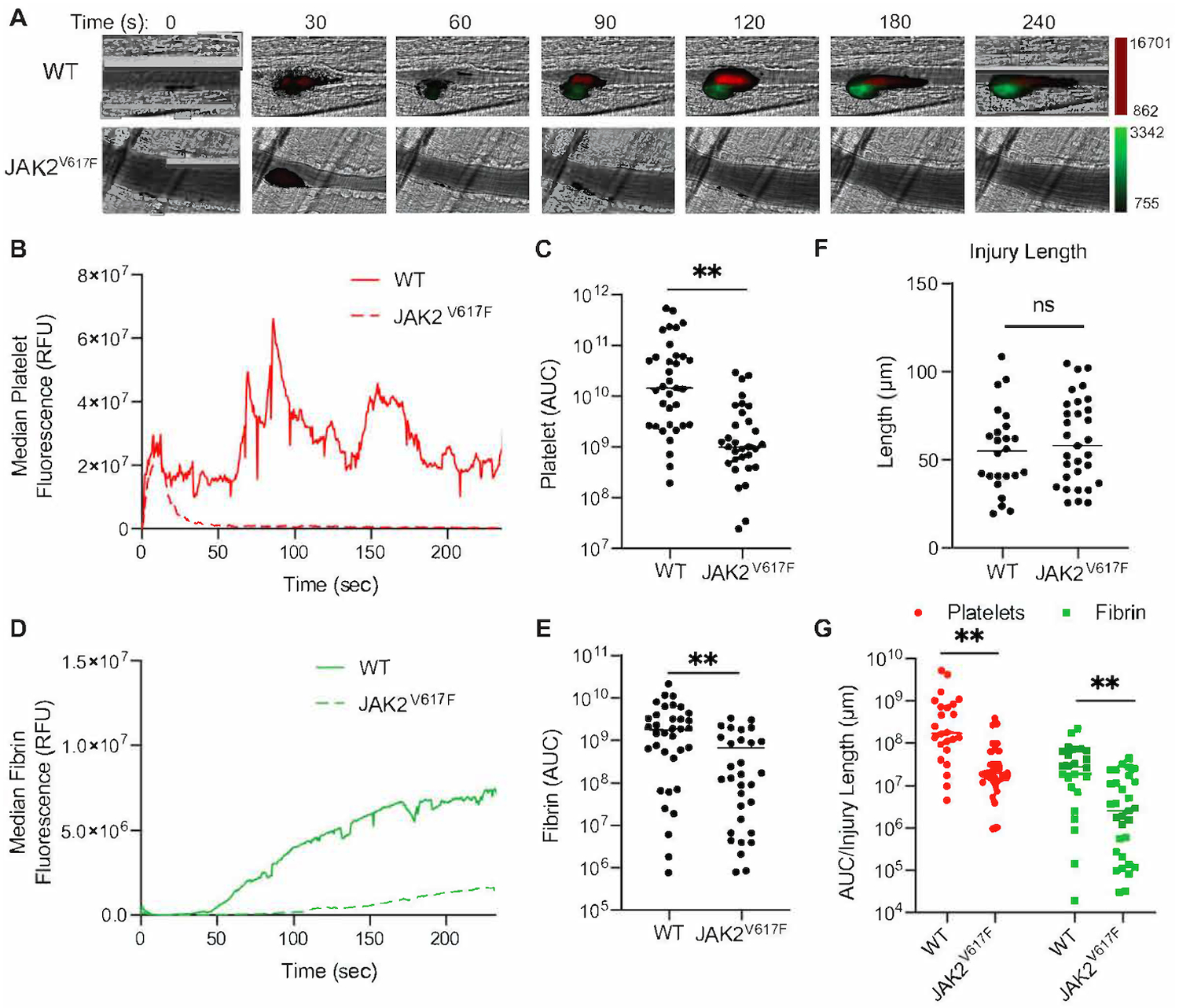
JAK2V617F mice demonstrate decreased platelet accumulation and fibrin formation. (A) 28.4 weeks old males (n=5 each) WT and JAK2V617F mice were injected with Dylight 647-labeled anti-platelet antibody (CD42b) and Dylight 488-labeled anti-fibrin antibody (59D8). Representative images obtained at the indicated times following laser-induced injury are shown. Fluorescence intensity in each channel was normalized to background fluorescence obtained from images prior to laser-induced injury. (B) Median integrated platelet intensities following laser injury were calculated and plotted at 0.5 sec intervals for all thrombi in WT (n=35 thrombi) and Vav1-hJAK2V617F mice (n=31 thrombi). (C) Platelet AUCs were calculated and plotted for each thrombus formed in WT and Vav1-hJAK2V617F mice. Median values are indicated. (D) Median integrated fibrin intensities following laser injury were calculated and plotted at 0.5 sec intervals for all thrombi in vehicle and Vav1-hJAK2V617F mice. (E) Fibrin AUCs were calculated and plotted for each thrombus formed in WT and Vav1-hJAK2V617F mice. Median values are indicated. (F) Ablation injury lengths were measured in WT and Vav1-hJAK2V617F mice as a marker of injury severity. Median values were plotted. (G) Platelet accumulation and fibrin formation data were normalized for injury severity by dividing the AUC value for each thrombus by its corresponding ablation injury length. ns: not significant, **p < 0.01, Mann Whitney U test.
Platelet aggregation response to collagen is compromised in JAK2V617F mice
Platelets derived from age- and sex-matched control and JAK2V617F mice were tested for their response to different agonists. Of the ones tested, collagen seemed to induce less platelet aggregation in the mutated mice, compared to controls (Figure 3A–C). Activation by ADP was comparable in the experimental groups at higher doses, but at lower doses a tendency for impaired aggregation was observed in JAK2V617F platelets (Figure 3D), and one concentration of thrombin had a slightly reduced effect on JAK2V617F platelets compared to controls (Figure 3E). Myelofibrosis did not seem to affect platelet response to agonists as the defect was consistently observed between young (non-myelofibrotic) and old (myelofibrotic) mice (Supplemental Figure 1B–C). The major receptors for collagen are α2β1 and GPVI. Flow cytometry analysis of the level of these receptors on platelet surface indicated a reduction in the level of α2 and GPVI, but not of β1 in JAK2V617F mice, compared to matching controls (Figure 4). This JAK2 mutation-induced changes in the above integrins could explain a compromised platelet aggregation response to collagen in the mutated mice.
Figure 3.
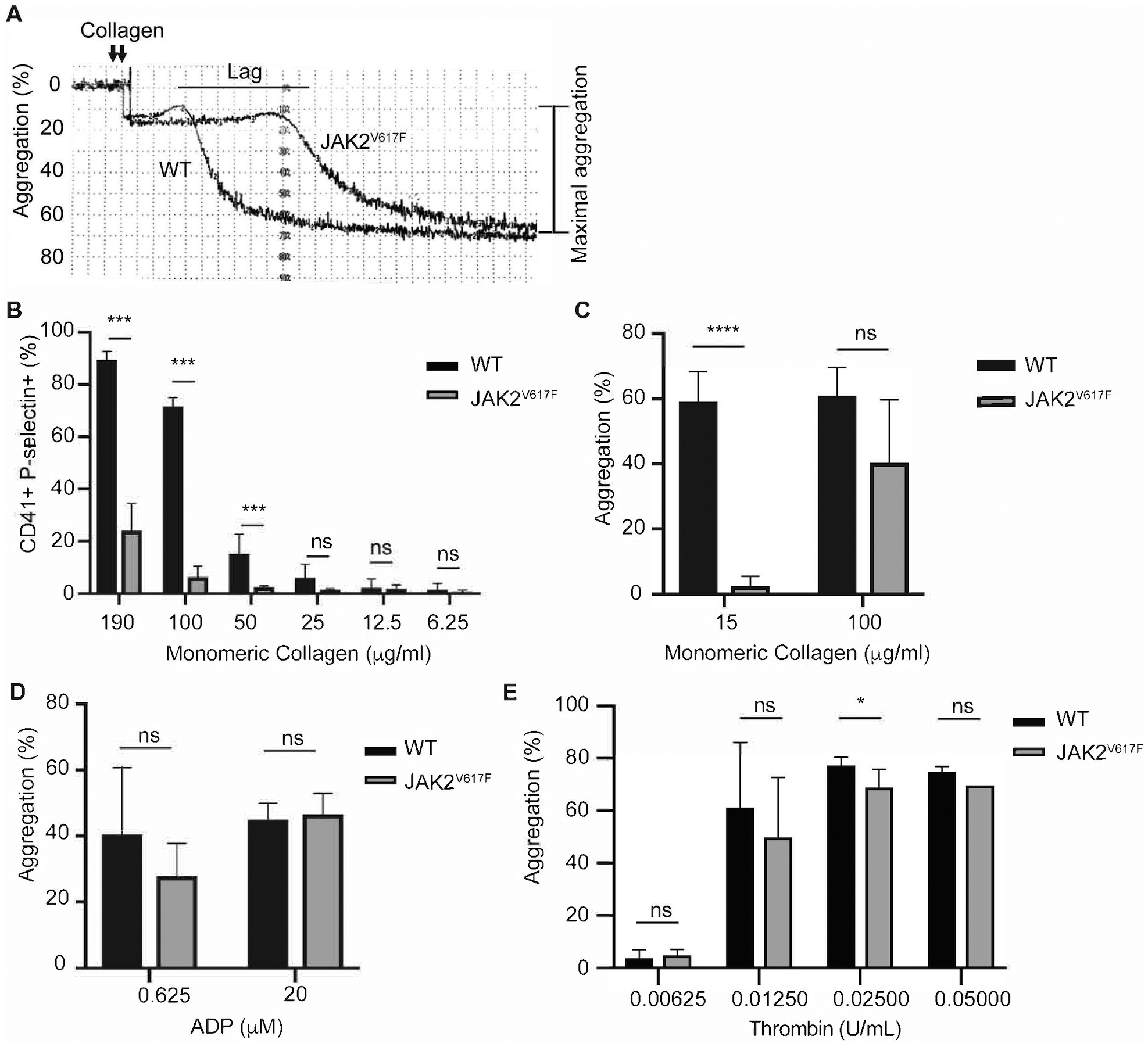
JAK2V617F platelets exhibit decreased platelet aggregation response to collagen. A. Representative aggregation trace of platelets derived from 15 weeks old males JAK2V617F and WT mice, activated by 10 μg/mL monomeric collagen. The tracing is representative of at least three biological replicates. B. Flow cytometry of 10 weeks old females WT (n = 3) and JAK2V617F (n = 4) washed platelets labeled with CD41 (platelet marker) and p-selectin after activation with monomeric collagen at the indicated concentrations. Platelet aggregation as measured in response to indicated concentrations of collagen (C) ADP (D) and thrombin (E), using five to six 15 weeks old male mice in each case. Data are averages +/− SD. ns: not significant, *p <0.05; *** p <0.001, **** p <0.0001.
Figure 4.
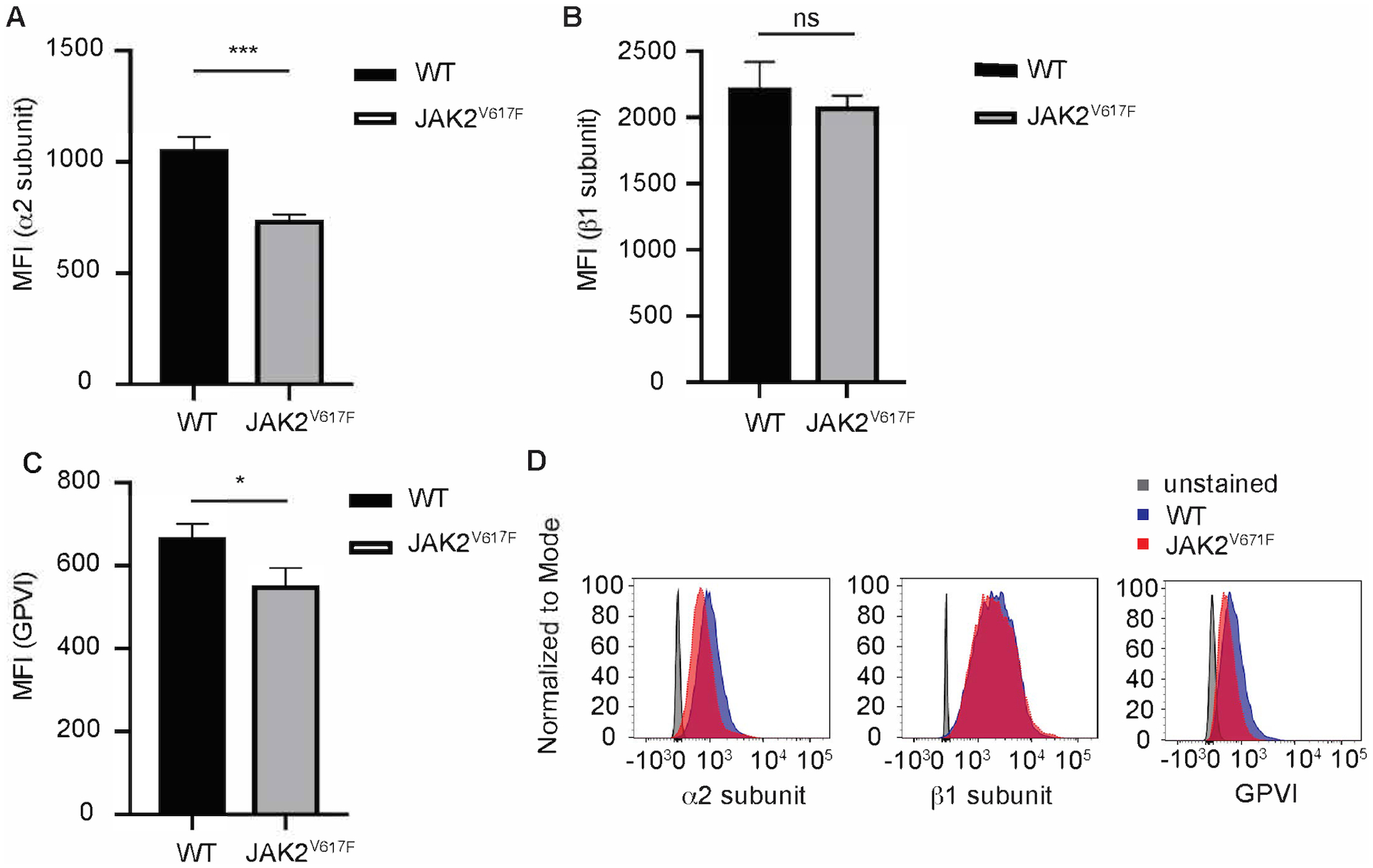
JAK2V617F platelets show decreased cell surface levels of α2 integrin subunit and GPVI. Platelets derived from JAK2V617F and WT mice (as in Figure 3) were subjected to flow cytometry analysis to measure cell surface α2 integrin subunit (A), β1 integrin subunit (B), and GPVI (C), using antibodies as described under Methods. A representative flow cytometry tracing is shown in panel D. Data are averages +/−SD. 15 weeks old male WT and JAK2V617F mice (n=3 each). ns: not significant, *p<0.05; ***p< 0.001.
Platelet ADP and ATP secretion
Tracing aggregation kinetics revealed that while WT platelets have a sharp aggregation response, typically mediated by a second wave of activation induced by intracellularly released ATP/ADP, the response was shallower in platelets of the JAK2V617F mice, before reaching a plateau (Figure 3A). This, plus the appearance of smaller thrombi in JAK2V617F mice subjected to vascular injury (Figure 2B) led us to suspect that ATP/ADP secretion is compromised in JAK2V617F platelets. To examine this possibility, the release of ATP and ADP was measured following collagen-induced platelet activation. Interestingly, ATP and ADP release by JAK2V617F platelets was significantly reduced compared to controls (Figure 5A–C). Platelet activation by PMA, a calcium ionophore known to induce platelet aggregation and secretion18, also resulted in diminished extracellular ADP and ATP in JAK2V617F samples compared to controls (Figure 5D). Considering that ADP/ATP is stored in and released from platelet dense granules, we sought to examine the platelet ultrastructure in the experimental samples. EM analysis showed a significantly reduced number of dense granules in platelets of JAK2V617F mice compared to controls, suggesting that JAK2 hyperactivating mutation affects the development and/or assembly of these granules (Figure 6).
Figure 5.
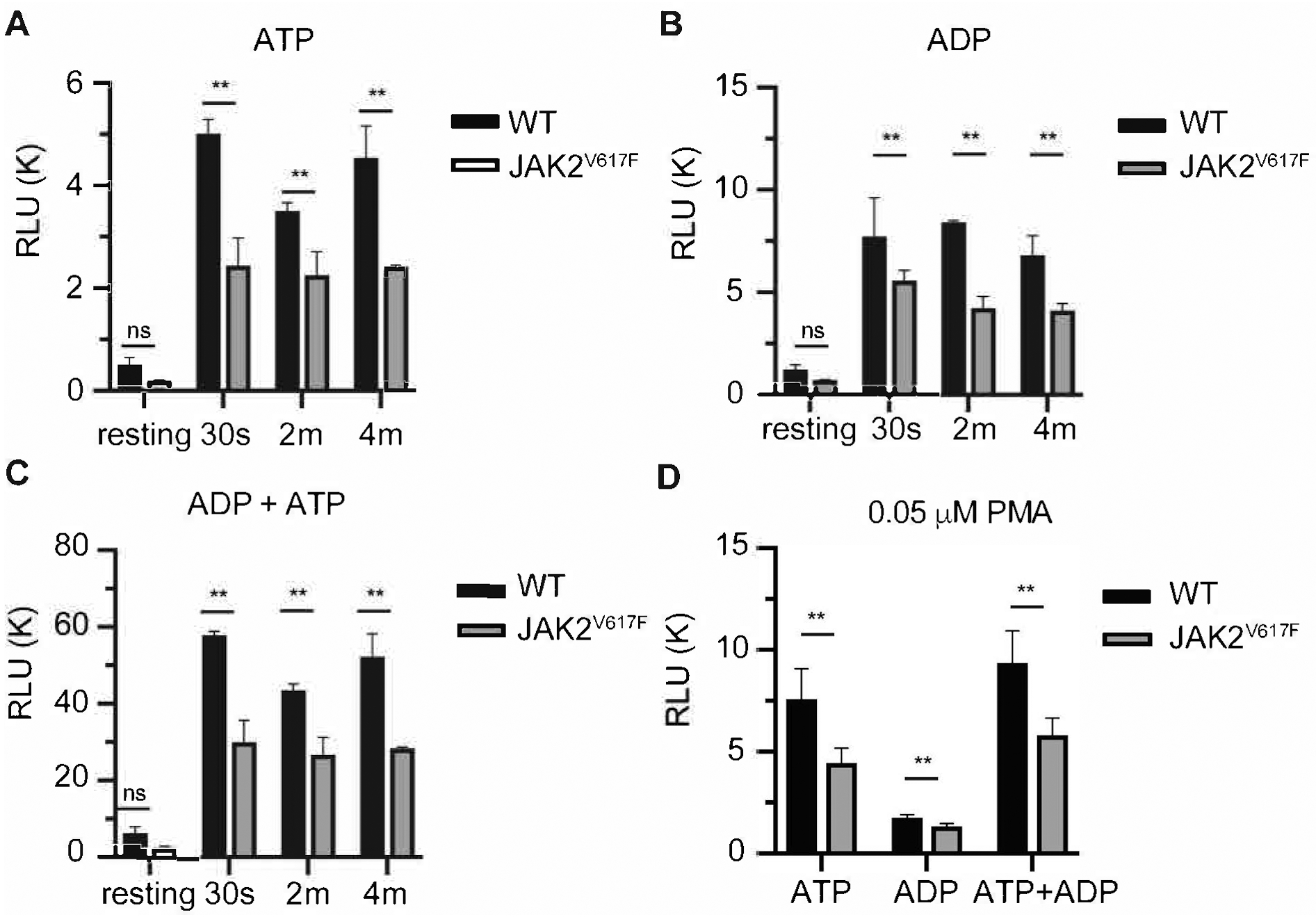
JAK2V617F platelets show decreased secretion of ADP and ATP in response to collagen activation. Extracellular A. ADP, B. ATP and C. ADP+ATP were measured in response to 10 μg/mL monomeric collagen (see Methods). For this set of analysis 3 separate experiments were performed with a total of 7 male mice; 4 mice 26–30 weeks of age, and 3 mice 15 weeks of age. Data are averages+/−SD. Results were collectively averaged as the trend was similar in the young and older mice. D. Platelets were activated by a concentration of PMA (0.05 μM) found following preliminary exploration to activate platelets and to induce ATP and ADP release. Values of secreted nucleotides are shown. RLU: relative luminescence units. Data are averages+/−SD of n=4 male mice, 14 weeks of age. ns: not significant, **p< 0.01
Figure 6.
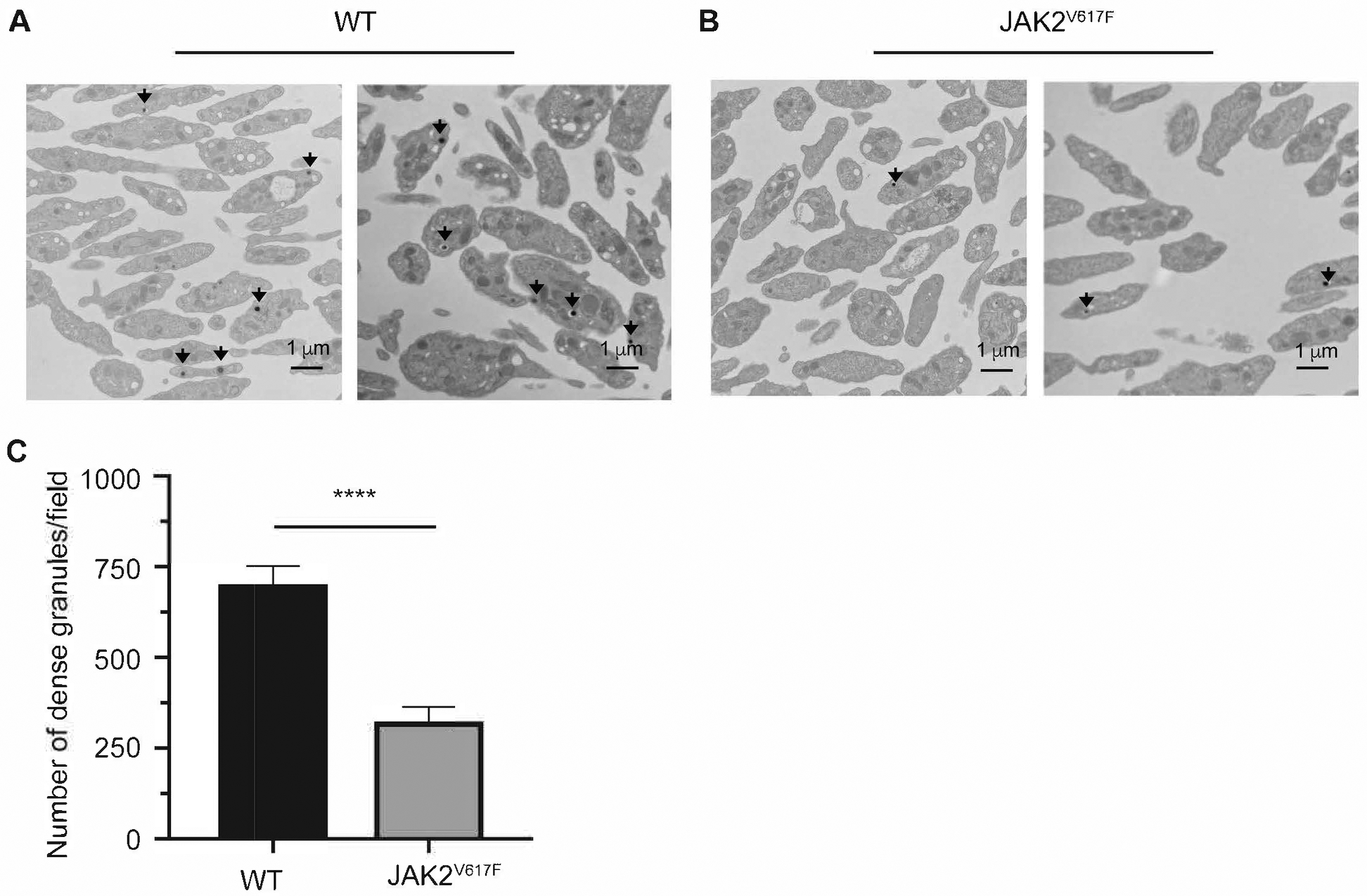
EM reveals a dense granule deficiency in JAK2V617F mice. Representative images of EM analysis of platelets derived from two WT (A) or JAK2V617F (B) male mice (about 30 weeks old). Black arrows represent dense granules found in platelets. C. Quantification of dense granules. Shown are averages+/−SD of 41 fields and 27 fields analyzed for WT and JAK2V617F mice, respectively. Mann Whitney was performed as a two tailed test using the Prism software. **** p <0.0001.
DISCUSSION
A recent population-based study21 of 9,429 patients with different forms of MPNs and 35,820 matched control participants found that the calculated hazard ratio for venous thrombosis was about three-fold greater than for arterial thrombosis across all age groups and among all MPN subtypes, compared to controls, although the degree of venous thromboembolism (VTE) seems to vary between ET, PV and PMF. MPN patients with high JAK2V617F allele burden are prone to developing complications associated with hemorrhages and/or thrombosis22, 23. Development of pathological thrombosis is controlled by an array of factors, such as platelet activation, changes in hyperviscosity and shear stress affected by an increase in red blood cell level, changes in inflammatory cytokines and white blood cells activation, the integrity of the endothelium, and coagulation factors. A combination of some or all of these conditions in PMF patients will be affected by the type of MPN and could influence which patients develop venous and/or arterial thrombosis or hemorrhages.
The complexity of the human MPN condition is amplified by secondary accessory mutations that often accompany the JAK2V617F allele burden. To study discrete mechanisms associated with MPN development, several mouse models bearing a single JAK2V617F mutation have been generated (reviewed in24). Such models were reported to have either reduced or increased thrombosis, depending on the mouse studied and whether the MPN phenotype is associated with bone marrow fibrosis, or an increased level of specific blood cells. In the current study, we used the Vav1-hJAK2V617F mice (bearing human JAK2V617F), which display hallmarks of PMF, including expansion of the megakaryocyte lineage and a fibrotic bone marrow15. The mice have a mild PV phenotype, with increased platelet counts and, anticipated, elevated phosphorylated Stat5 level at baseline (data not shown). This model has allowed us to focus on platelet properties in the context of an isolated, single mutation (JAK2V617F) that was engineered to impact primarily the megakaryocytic lineage and to lead to a fibrotic phenotype. Under these conditions, we made the striking observation that platelets bearing JAK2V617F have reduced aggregation response to collagen, and a significantly reduced number of dense granules, which could explain an observed compromised ability to secret ADP upon platelet activation, and a diminished second wave of activation. The role of JAK2 signaling in controlling platelet dense granule number has never been reported before. Prior studies involving human cohorts identified dense granule storage defects. In one study of 9 patients with myeloproliferative disorders of myelofibrosis platelets were found to have significant storage pool depletion (measured by ADP/ATP ratio and 14C-serotonin platelet disappearance patterns)25. Another study reported a defect in mepacrine uptake in platelet dense granules in 71% of the PV subjects and in 48% of the ET subjects26. Dense granule formation has been reported to be controlled by various factors, such as pallidin (HPS9) transcription27. Although there was no significant change in the level of HPS9 between control and JAK2V617F platelets (our unpublished data), future investigations could focus on mechanisms leading to dysregulated dense granule numbers.
P-selectin expression in response to collagen stimulation was decreased in platelets derived from JAK2V617F mice, which could be reflecting the defect in collagen receptor expression and/or alpha granules. However, further analysis revealed that at baseline there is a tendency for elevated P-selectin expression in JAK2V617F platelets (Supplemental Figure 1D). Interestingly, prior studies that looked at alpha granules in platelets of patients with MPN or myelofibrosis identified a less profound effect in cargo depletion in these granules when compared to dense granules. However, the plasma concentration of platelet factor 4 and beta-thromboglobulin in patients with MPN / myelofibrosis in this study were elevated, when compared to control samples25.
In two vascular injury models used in our study, thrombus formation was significantly delayed in the Vav1-hJAK2V617F mice. The size of the thrombi in the mutated mice was smaller, compared to controls, suggesting an impact on platelet aggregation properties. At the same time, Vav1-hJAK2V617F mice showed reduced platelet aggregation response to collagen that is associated with diminished levels of platelet cell surface expression of components of the collagen receptors, integrins GPVI and α2. A clear prolonged bleeding time in the PMF mice suggest that this overall PMF phenotype depicts the hemorrhagic events associated with MPN.
The platelet phenotype of the PMF mice we analyzed is reminiscent of a report using the Hasan et al. model28 in which the mouse Jak2V617F was knocked-in leading to a clear PV phenotype. In this case too tail bleeding was prolonged, in vitro thrombosis on collagen was reduced in association with diminished levels of the collagen receptor GPVI, and rapidly forming platelet aggregates in a FeCl3-induced thrombosis model were unstable6. The mechanism for this observation was not fully explored. Similarly, inducible transgenic expression of human JAK2V617F 29 in hematopoietic and endothelial cells led to thrombocytosis, yet, these mice showed reduced thrombosis following vascular injury associated with compromised Willebrand factor function13. On the other hand, using the ET mouse model generated by Li et al30 in which human JAK2V617F was knocked-into the genome, the group reported enhanced platelet reactivity and aggregation in vitro and a reduced duration of bleeding in vivo7. Moreover, Zhao et al31 demonstrated that animals that express mouse Jak2V617F at physiologic levels, generated by Mullally et al32, die of thrombotic events constituting of large vascular occlusions most prominent in lungs and kidneys. Those authors also demonstrated the contribution of pleckstrin-2 (Plek2) in the thrombotic phenotype, which they mostly attributed to elevated red cell mass. Further, a recent study reported that neutrophils from MPN patients with JAK2V617F mutation tend to form NETs and mice with conditional KI of JAK2V617F have an augmented capacity to form NETs and increased thrombosis14. These opposing results in the different MPN mouse models, albeit induced by JAK2V617F, are likely consistent with the variability of the disorders observed in MPN patients vis-à-vis a balance between augmented incidence of thrombosis vs. hemorrhagic events.
Taken together, our study identified a link between JAK2 hyperactivity and a compromised second wave of platelet activation, associated with reduced number of platelet dense granules. Increased tendency for hemorrhages and smaller thrombi in the JAK2V617F mice could at least partially explain the VTE and bleeding phenotypes in human PMF.
Supplementary Material
HIGHLIGHTS.
The Vav1-hJAK2V617F transgenic mice show prolonged bleeding time and reduced thrombus formation and thrombus size using two in vivo models of vascular injury.
Washed platelets from Vav1-hJAK2V617F mice have reduced aggregation in response to collagen, and reduced expression of α2 integrin subunit and GPVI.
ADP secretion is reduced in activated mouse JAK2V617F platelets
Dense granule number is reduced in JAK2V617F platelets
Acknowledgments:
KR, SM and CRT generated hypotheses, designed experiments, and analyzed data, and CRT and SM performed most of the experiments with the help of AP. KR wrote the manuscript with input from CRT, SM, RHB, and RF. OL, MEB, VCC, GMS, MY and RHB performed and analyzed the in vivo vascular injury models. JI and AR assisted in EM studies.
SOURCES OF FUNDING: This work was supported by NIHLBI grant R01HL136363 to KR and grant R35HL135775 to RF. KR is an established investigator with the American Heart Association. CRT was supported by NHLBI Cardiovascular Research Training grant T32 HL007224 and MY is supported by T32HL007917. SM was supported by the NIH ORIP SERCA K01 award K01OD025290.
ABBREVIATIONS
- JAK2V617F
janus kinase 2 with valine to phenylalanine substitution on codon 617
- PMF
primary myelofibrosis
- MPN
myeloproliferative neoplasms
- CALR
calreticulin
- MPL
myeloproliferative leukemia protein or thrombopoietin receptor
- ADP
adenosine diphosphate
- PV
polycythemia vera
- ET
essential thrombocythemia
- KI
knock-in
- GPVI
platelet glycoprotein VI
- NETs
neutrophil extracellular traps
- tOc
time to occlusion
- WT
wild-type
- ATP
adenosine triphosphate
- EM
electron microscopy
- AUC
area under the curve
- PMA
phorbol-myristate-acetate
- RLU
relative luminescence units
- VTE
venous thromboembolism
- SD
standard deviation
Footnotes
DISCLOSURES: authors declare no conflict of interest.
REFERENCES
- 1.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129:667–679 [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1262–1271 [DOI] [PubMed] [Google Scholar]
- 3.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405 [DOI] [PubMed] [Google Scholar]
- 4.Li J, Kent DG, Chen E, Green AR. Mouse models of myeloproliferative neoplasms: Jak of all grades. Dis Model Mech. 2011;4:311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tefferi A Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am J Hematol. 2016;91:50–58 [DOI] [PubMed] [Google Scholar]
- 6.Lamrani L, Lacout C, Ollivier V, Denis CV, Gardiner E, Ho Tin Noe B, Vainchenker W, Villeval JL, Jandrot-Perrus M. Hemostatic disorders in a jak2v617f-driven mouse model of myeloproliferative neoplasm. Blood. 2014;124:1136–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobbs CM, Manning H, Bennett C, Vasquez L, Severin S, Brain L, Mazharian A, Guerrero JA, Li J, Soranzo N, Green AR, Watson SP, Ghevaert C. Jak2v617f leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood. 2013;122:3787–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbui T, Carobbio A, Cervantes F, Vannucchi AM, Guglielmelli P, Antonioli E, Alvarez-Larran A, Rambaldi A, Finazzi G, Barosi G. Thrombosis in primary myelofibrosis: Incidence and risk factors. Blood. 2010;115:778–782 [DOI] [PubMed] [Google Scholar]
- 9.Rungjirajittranon T, Owattanapanich W, Ungprasert P, Siritanaratkul N, Ruchutrakool T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of philadelphia-negative myeloproliferative neoplasms. BMC Cancer. 2019;19:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rupoli S, Goteri G, Picardi P, Micucci G, Canafoglia L, Scortechini AR, Federici I, Giantomassi F, Da Lio L, Zizzi A, Honorati E, Leoni P. Thrombosis in essential thrombocytemia and early/prefibrotic primary myelofibrosis: The role of the who histological diagnosis. Diagn Pathol. 2015;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bleeding Landolfi R. and thrombosis in myeloproliferative disorders. Curr Opin Hematol. 1998;5:327–331 [DOI] [PubMed] [Google Scholar]
- 12.Li J, Kent DG, Godfrey AL, Manning H, Nangalia J, Aziz A, Chen E, Saeb-Parsy K, Fink J, Sneade R, Hamilton TL, Pask DC, Silber Y, Zhao X, Ghevaert C, Liu P, Green AR. Jak2v617f homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood. 2014;123:3139–3151 [DOI] [PubMed] [Google Scholar]
- 13.Etheridge SL, Roh ME, Cosgrove ME, Sangkhae V, Fox NE, Chen J, Lopez JA, Kaushansky K, Hitchcock IS. Jak2v617f-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111:2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D, Castellano CA, Schneider RK, Padera RF, DeAngelo DJ, Wadleigh M, Steensma DP, Galinsky I, Stone RM, Genovese G, McCarroll SA, Iliadou B, Hultman C, Neuberg D, Mullally A, Wagner DD, Ebert BL. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X, Li Q, Fu X, Xu M, Zhao ZJ. Transgenic expression of jak2v617f causes myeloproliferative disorders in mice. Blood. 2008;111:5109–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leiva O, Ng SK, Matsuura S, Chitalia V, Lucero H, Findlay A, Turner C, Jarolimek W, Ravid K. Novel lysyl oxidase inhibitors attenuate hallmarks of primary myelofibrosis in mice. Int J Hematol. 2019;110:699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuura S, Mi R, Koupenova M, Eliades A, Patterson S, Toselli P, Thon J, Italiano JE Jr., Trackman PC, Papadantonakis N, Ravid K. Lysyl oxidase is associated with increased thrombosis and platelet reactivity. Blood. 2016;127:1493–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaumenhaft R, Dilks JR, Rozenvayn N, Monahan-Earley RA, Feng D, Dvorak AM. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. 2005;105:3879–3887 [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Jennings NL, Dart AM, Du XJ. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J Exp Med. 2012;2:30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, Schulman S, Libermann TA, Lu S, Shapiro NI, Christiani DC, Flaumenhaft R, Parikh SM. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest. 2018;128:1471–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hultcrantz M, Bjorkholm M, Dickman PW, Landgren O, Derolf AR, Kristinsson SY, Andersson TML. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: A population-based cohort study. Ann Intern Med. 2018;168:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertozzi I, Bogoni G, Biagetti G, Duner E, Lombardi AM, Fabris F, Randi ML. Thromboses and hemorrhages are common in mpn patients with high jak2v617f allele burden. Ann Hematol. 2017;96:1297–1302 [DOI] [PubMed] [Google Scholar]
- 23.Ball S, Thein KZ, Maiti A, Nugent K. Thrombosis in philadelphia negative classical myeloproliferative neoplasms: A narrative review on epidemiology, risk assessment, and pathophysiologic mechanisms. J Thromb Thrombolysis. 2018;45:516–528 [DOI] [PubMed] [Google Scholar]
- 24.Dunbar A, Nazir A, Levine R. Overview of transgenic mouse models of myeloproliferative neoplasms (mpns). Curr Protoc Pharmacol. 2017;77:14 40 11–14 40 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malpass TW, Savage B, Hanson SR, Slichter SJ, Harker LA. Correlation between prolonged bleeding time and depletion of platelet dense granule adp in patients with myelodysplastic and myeloproliferative disorders. J Lab Clin Med. 1984;103:894–904 [PubMed] [Google Scholar]
- 26.Coucelo M, Caetano G, Sevivas T, Almeida Santos S, Fidalgo T, Bento C, Fortuna M, Duarte M, Menezes C, Ribeiro ML. Jak2v617f allele burden is associated with thrombotic mechanisms activation in polycythemia vera and essential thrombocythemia patients. Int J Hematol. 2014;99:32–40 [DOI] [PubMed] [Google Scholar]
- 27.Mao GF, Goldfinger LE, Fan DC, Lambert MP, Jalagadugula G, Freishtat R, Rao AK. Dysregulation of pldn (pallidin) is a mechanism for platelet dense granule deficiency in runx1 haplodeficiency. J Thromb Haemost. 2017;15:792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasan S, Lacout C, Marty C, Cuingnet M, Solary E, Vainchenker W, Villeval JL. Jak2v617f expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by ifnalpha. Blood. 2013;122:1464–1477 [DOI] [PubMed] [Google Scholar]
- 29.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, Skoda RC. Ratio of mutant jak2-v617f to wild-type jak2 determines the mpd phenotypes in transgenic mice. Blood. 2008;111:3931–3940 [DOI] [PubMed] [Google Scholar]
- 30.Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C, Chen E, Forrai A, Scott LM, Ferreira R, Campbell PJ, Watson SP, Liu P, Erber WN, Huntly BJ, Ottersbach K, Green AR. Jak2 v617f impairs hematopoietic stem cell function in a conditional knock-in mouse model of jak2 v617f-positive essential thrombocythemia. Blood. 2010;116:1528–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, Mei Y, Cao L, Zhang J, Sumagin R, Yang J, Gao J, Schipma MJ, Wang Y, Thorsheim C, Zhao L, Stalker T, Stein B, Wen QJ, Crispino JD, Abrams CS, Ji P. Loss of pleckstrin-2 reverts lethality and vascular occlusions in jak2v617f-positive myeloproliferative neoplasms. J Clin Invest. 2018;128:125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, Paktinat M, Haydu JE, Housman E, Lord AM, Wernig G, Kharas MG, Mercher T, Kutok JL, Gilliland DG, Ebert BL. Physiological jak2v617f expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.