

Abstract
Pulmonary hypertension (PH) and right ventricular (RV) hypertrophy frequently develop in patients with hypoxic lung disease. Chronic alveolar hypoxia (CH) promotes sustained pulmonary vasoconstriction and pulmonary artery (PA) remodeling by acting on lung cells, resulting in the development of PH. RV hypertrophy develops in response to PH, but coronary arterial hypoxemia in CH may influence that response by activating HIF-1α (hypoxia-inducible factor 1α) and/or HIF-2α in cardiomyocytes. Indeed, other studies show that the attenuation of PH in CH fails to prevent RV remodeling, suggesting that PH-independent factors regulate RV hypertrophy. Therefore, we examined the role of HIFs in RV remodeling in CH-induced PH. We deleted HIF-1α and/or HIF-2α in hearts of adult mice that were then housed under normoxia or CH (10% O2) for 4 weeks. RNA-sequencing analysis of the RV revealed that HIF-1α and HIF-2α regulate the transcription of largely distinct gene sets during CH. RV systolic pressure increased, and RV hypertrophy developed in CH. The deletion of HIF-1α in smooth muscle attenuated the CH-induced increases in RV systolic pressure but did not decrease hypertrophy. The deletion of HIF-1α in cardiomyocytes amplified RV remodeling; this was abrogated by the simultaneous loss of HIF-2α. CH decreased stroke volume and cardiac output in wild-type but not in HIF-1α–deficient hearts, suggesting that CH may cause cardiac dysfunction via HIF-dependent signaling. Collectively, these data reveal that HIF-1 and HIF-2 act together in RV cardiomyocytes to orchestrate RV remodeling in CH, with HIF-1 playing a protective role rather than driving hypertrophy.
Keywords: hypoxia, right ventricular hypertrophy, hypoxia-inducible factors
Clinical Relevance
These studies reveal that alveolar hypoxia leads to the development of pulmonary hypertension through a HIF-1–dependent mechanism in smooth muscle cells of the pulmonary artery. The associated systemic hypoxemia also leads to the activation of HIF-1 and HIF-2 in the right ventricle, leading to transcriptional responses that affect the remodeling response.
Pulmonary hypertension (PH) occurs in a variety of clinical disorders and affects patients with highly prevalent diseases, including chronic obstructive pulmonary disease, interstitial lung disease, and obstructive sleep apnea (1). Chronic alveolar hypoxia in these conditions promotes sustained pulmonary vasoconstriction, pulmonary artery (PA) wall thickening, and the development of PH classified by the World Health Organization as group 3. Right ventricular (RV) hypertrophy occurs in these patients, presumably in response to the increase in RV afterload associated with PH. Many large cohort studies have sought to identify biomarkers for predicting outcomes in patients with PH (2–7); these consistently point to RV function as a key predictor of survival.
HIF-1 and HIF-2 (hypoxia-inducible factors 1 and 2) play dominant roles in regulating transcriptional responses to hypoxia. HIF-dependent genes regulate a large number of processes related to PH, including vascular endothelial growth factor–induced angiogenesis, cellular proliferation and migration, and cellular energy production and metabolism (8–10). The loss of HIF-1α in PA smooth muscle cells leads to a blunting of chronic hypoxia–induced PH (11), and mice with HIF-1α haploinsufficiency show a decrease in HIF-1α protein expression and an attenuation of hypoxia-induced RV hypertrophy (12, 13). Relatively less is known about the role of HIF-2 in chronic hypoxia, but it does regulate EPO expression and thereby affects the hematocrit (14). HIF-2α+/− mice have been reported to show protection against PH development in hypoxia (15), and endothelial HIF-2α has been reported to contribute to severe pulmonary hypertension (16). A recent study using systemic antisense oligonucleotides and small molecule inhibitors found that HIF-2, but not HIF-1, inhibition prevented hypoxia-induced PH in mice (17). Finally, the genetic deletion of PHD2, a negative regulator of HIF-1/2α protein stability, in the vascular endothelium during development led to a severe form of HIF-2–dependent PH in the mice (18). Collectively these studies demonstrate that HIF-1 and HIF-2 in the pulmonary vasculature contribute importantly to the development of PH in chronic hypoxia.
However, the roles of HIF-1 and HIF-2 in the heart have not been extensively studied in the context of PH. We had previously found that PH was attenuated by the deletion of HIF-1α in PA smooth muscle, but this failed to attenuate RV hypertrophy during chronic hypoxia (11). We therefore hypothesized (Figure E1 in the data supplement) that coronary (systemic) arterial hypoxemia during chronic hypoxia might contribute to cardiac remodeling by activating HIF-1 and/or HIF-2 in the RV itself, thereby augmenting the hypertrophy caused by the increase in pulmonary arterial pressure (19). In the present study, we therefore sought to clarify the roles of HIF-1 and HIF-2 in RV remodeling and function in chronic hypoxia–induced PH by inducing tissue-specific inducible deletions in adult animals in a model of hypoxia-induced PH associated with RV hypertrophy.
Methods
Expanded Methods are contained in the data supplement.
Animal Models
All animal protocols were performed after review and approval by the Northwestern University Institutional Animal Care and Use Committee. To generate mice lacking HIF-1α in cardiomyocytes, mice with LoxP sites flanking exon 2 of the HIF-1α gene (HIF-1αfl/fl) were bred with mice containing a tamoxifen-inducible Cre recombinase under the control of a cardiomyocyte-specific promoter (α myosin heavy chain 6; Myh6Cre) (20) to generate Myh6Cre-HIF-1αfl/fl (Myh6Cre-HIF-1α) mice. A similar procedure was used to generate mice with an inducible deletion of HIF-2α in the heart (Myh6Cre-HIF-2α). To generate mice lacking HIF-1α in smooth muscle cells, HIF-1αfl/fl mice were bred with a tamoxifen-inducible Cre recombinase under the control of a smooth muscle–specific promoter (smooth muscle myosin heavy chain 11; SMCCre) (21) to generate SMCCre-HIF-1αfl/fl (SMCCre-HIF-1α) mice. To generate mice lacking HIF-1α in both cardiomyocytes and smooth muscle cells, Myh6Cre-HIF-1α mice were bred with SMCCre-HIF-1α mice to generate SMCCre-Myh6Cre-HIF-1α mice. To generate mice with an inducible cardiac deletion of HIF-1α and HIF-2α, the corresponding floxed mice were bred to homozygosity with Myh6Cre mice to generate Mhy6Cre-HIF-1/2α mice. To activate Cre-mediated excision of the floxed gene(s), tamoxifen (20 mg/kg body weight dissolved in corn oil) was administered via intraperitoneal injection at 4–6 weeks of age for 5 consecutive days. Mice were used for experiments 2 weeks after the final tamoxifen administration. To confirm the efficiency of Cre recombination, SMCCre and Myh6Cre mice were crossed with mT/mG mice (22). All mice were in the C57Bl/6 genetic background and were genotyped by tail-snip analysis (Transnetyx).
Real Time RT-PCR
To confirm the deletion of HIF-α in cardiomyocytes and smooth muscle cells, total RNA was extracted and column-purified from RV and abdominal aortas of experimental mice and their littermate control mice using the RNeasy kit (Qiagen).
Chronic Hypoxia
Mice were housed in normoxia (room air; 21% O2) or hypoxia (10% O2) in the same room for 4 weeks (range: 28–31 d) beginning 2 weeks after tamoxifen administration (Coy Laboratory Products, Inc.).
Echocardiography
Cardiac function was assessed by echocardiography using a VisualSonics Vevo-770 echo system (VisualSonics).
Hemodynamics
Hypoxia-induced changes in RV systolic pressure (RVSP) were assessed in mechanically ventilated mice by performing a thoracotomy and inserting a microtip pressure transducer catheter (Millar) into the right ventricle via an apical puncture.
Systemic Arterial Blood Pressure
The mean arterial blood pressure was measured noninvasively using a tail-cuff system (CODA System; Kent Scientific).
Vascular Remodeling
Pulmonary vascular remodeling was assessed in the lungs, which were inflation-fixed with 4% formaldehyde and embedded in paraffin.
Fulton’s Index
Hearts were removed from mice, and the RV was dissected from the left ventricle and septum. The Fulton’s index was calculated as a ratio of the weight of the RV to the weight of the left ventricle plus septum.
Cardiomyocyte Diameter
To measure the cardiomyocyte diameter, formalin-fixed, paraffin-embedded hearts were transversely sectioned and stained with Wheat Germ Agglutinin (Molecular Probes). Cardiomyocyte diameter in the RV was measured using Image J (National Institutes of Health).
Statistics
Data were analyzed using one-way or two-way ANOVA as appropriate, with post hoc testing using Sidak’s multiple comparisons test (Prism 8; GraphPad Software). Statistical significance was set at P < 0.05.
Results
To investigate the role of HIF in regulating the cardiovascular response to chronic hypoxia, we generated mice with tissue-specific Cre-inducible deletion of HIF-1α and/or HIF-2α. For all studies, tamoxifen was administered daily for 5 days to induce Cre activation in a specific tissue; tamoxifen-administered littermate Cre-negative mice served as control animals. Two weeks were allotted for the specific floxed gene and protein deletion before the start of the experiment intervention. To confirm the cell specificity of the inducible smooth muscle Cre (SMCCre) (21) and cardiac muscle Cre (Myh6Cre) (20) transgenes, mice were bred with mTmG reporter mice that constitutively express membrane-targeted red fluorescent protein (22). The activation of Cre causes a genetic recombination resulting in a shift in the expression of the fluorescent protein from red to green. In the absence of Cre, mice exhibited the expected widespread red fluorescence in both the lungs and the heart (Figure 1A; wild-type [WT]). The activation of SMCCre led to the appearance of green fluorescence in the PA walls; a residual layer of red fluorescence was retained in the vessel lumen, confirming that genetic recombination had not occurred in the endothelium (Figure 1A; SMCCre). Green fluorescence was also evident in coronary arteries within the heart, indicating that the SMCCre affected both systemic and pulmonary smooth muscle cells. Some green fluorescence was also visible in blood vessels in the lungs after Myh6Cre activation (Figure 1A; Myh6Cre), which are most likely pulmonary veins containing cardiomyocytes, as previously described (23).
Figure 1.
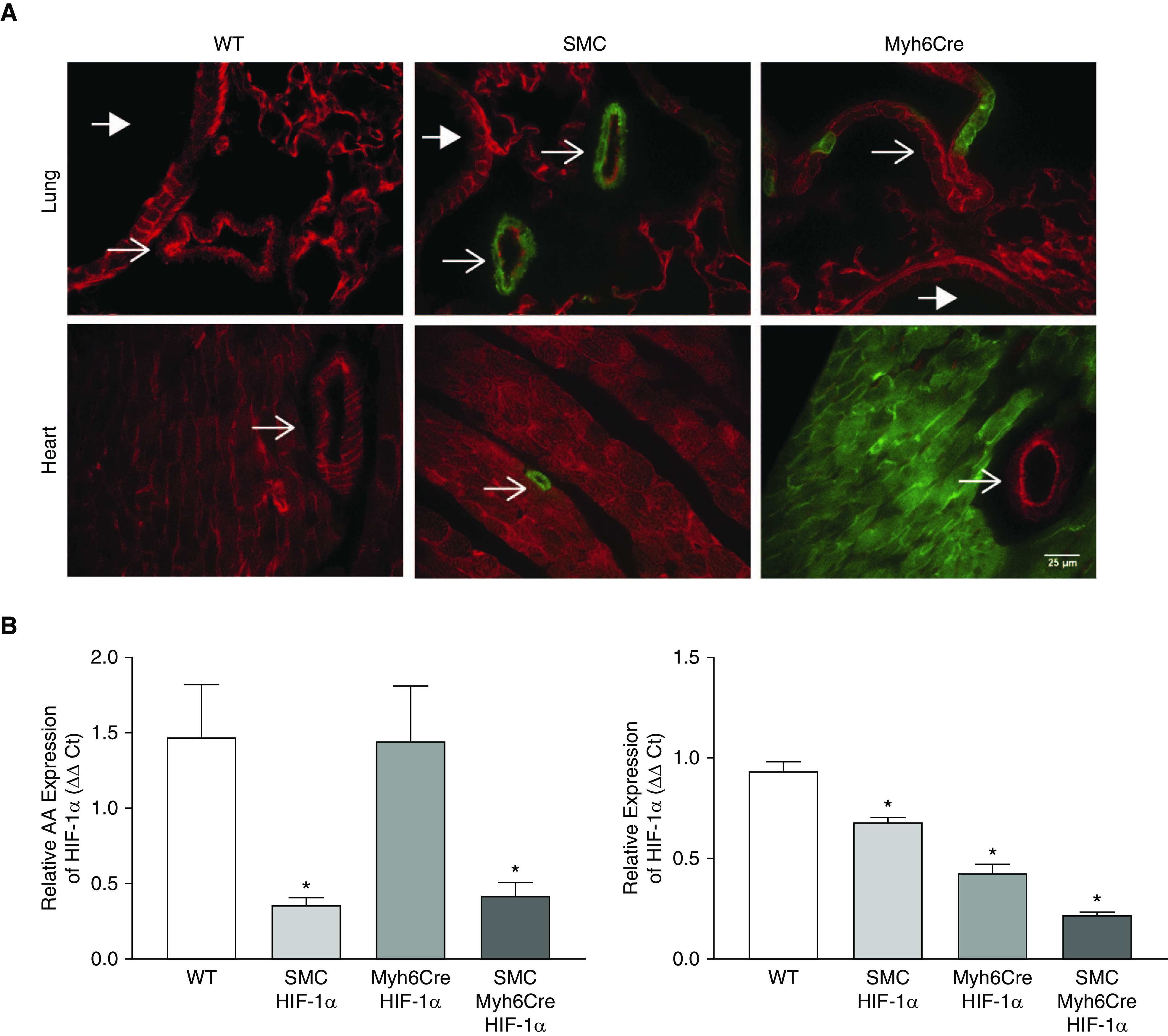
Cre-mediated deletion of HIF-1α from smooth muscle cells and cardiomyocytes is highly efficient. (A) Fluorescence images of mouse lung (top) and heart (bottom) in wild-type, SMCCre-mTmG, and Myh6Cre-mTmG mice. Green fluorescence indicates the activation of Cre recombinase. Thin arrows indicate vessels, and thick arrows indicate airways. Scale bar, 25 μm. (B) Relative expression of HIF-1α in abdominal aorta (left) and right ventricle (right) measured by RT-PCR. n = 4. *P < 0.05 versus wild-type. Values are means ± SEM. Ct = cycle threshold; HIF-1α = hypoxia-inducible factor 1α; Myh6Cre HIF-1α = deletion of HIF-1α in cardiomyocytes; SMC = smooth muscle cell; SMC HIF-1α = deletion of HIF-1a in smooth muscle; SMC Myh6Cre HIF-1α = deletion of HIF-1α in smooth muscle and cardiomyocytes; WT = wild-type.
To confirm the deletion of HIF-1α, mRNA levels of HIF-1α were measured in the descending aorta and RV of WT, SMCCre-HIF-1α, Myh6Cre-HIF-1α, and SMCCre-Myh6Cre-HIF-1α mice. In the descending aorta, HIF-1α mRNA levels were significantly decreased in SMCCre-HIF-1α and SMCCre-Myh6Cre-HIF-1α mice (36% and 42%, respectively, Figure 1B). In the RV, HIF-1α mRNA was significantly reduced in Myh6Cre-HIF-1α and SMCCre-Myh6Cre-HIF-1α mice (43% and 22%, respectively, Figure 1B). In addition, HIF-1α mRNA levels were decreased in the RV of SMCCre-HIF-1α mice (68%, Figure 1B) compared with those in the WT mice, likely reflecting a contribution from vascular smooth muscle. Collectively, these data demonstrate that Cre activation led to significant decreases in HIF-1α message in smooth muscle cells and cardiomyocytes.
HIFs Regulate Some, but Not All, of the RV Transcriptional Response to Chronic Hypoxia
To identify the RV transcriptional response to chronic hypoxia, mice aged 4–6 weeks were housed in an environmental chamber (10% O2) for 4 wk, whereas normoxic control mice were kept under room air in the same room. Parallel studies were conducted in mice with homozygous floxed alleles for HIF-1α, HIF-2α, and HIF-1/2α with or without the Myh6Cre transgene. After 4 weeks in hypoxia, RV tissue was harvested and RNA-sequencing (RNA-seq) analysis was performed using total RNA. Principal component analysis revealed distinct signatures in comparing WT normoxic hearts with WT hypoxia, as well as differences between WT hypoxia and HIF-1 α, HIF-2 α, and HIF1/2 α cardiac knockout hypoxic responses (Figure E2). Figure 2 shows that chronic hypoxia in WT mice was associated with a significant upregulation of some genes in the RV and a downregulation of others. As expected, the deletion of HIF-1α in the heart led to a decrease in the number of genes whose expression was altered by chronic hypoxia. Similarly, HIF-2α deletion decreased the number of genes affected by chronic hypoxia, as did deletion of both HIF-1α and HIF-2α. Venn diagram comparisons of the gene sets affected by hypoxia show that, for example, many genes affected by hypoxia in the WT RV were distinct from the genes altered by hypoxia in the HIF-knockout groups (Figure E3). This is consistent with the known effects of HIF-1 and HIF-2 in regulating gene expression during hypoxia. However, a subset of genes in the RV were affected by hypoxia but unaffected by HIF deletions (shown in the overlap regions), indicating that some hypoxia-responsive genes in the heart are not regulated by HIF. Finally, a comparison of the transcriptomic responses to hypoxia in the HIF-1α–deficient hearts versus the HIF-2α–deficient hearts indicates that these transcription factors regulate distinct gene sets in the heart with an overlap of a relatively small number of transcripts.
Figure 2.

Chronic hypoxia alters gene expression in the right ventricle; inactivation of HIF-1 and/or HIF-2 significantly alters that response, as detected by RNA-sequencing analysis. Chronic hypoxia altered the expression of nearly 400 genes in wild-type hearts, with upregulation of some and downregulation of others. HIF-1α deletion decreased the number of genes altered by hypoxia, as did the deletion of HIF-2α and HIF-1/2α, compared with wild-type hypoxic mice. Genes shown exhibited a log2-fold change of ≥1.5. (n = 4 in each group). HIF-1α KO hypoxia = Myh6Cre-HIF-1α KO in chronic hypoxia; HIF-2α KO hypoxia = Myh6Cre-HIF-2α KO in chronic hypoxia; HIF-1/2α KO hypoxia = Myh6Cre-HIF-1/2α KO in chronic hypoxia; KO = knockout.
Gene ontology analysis of the transcriptomic response identified a number of processes and functions that were significantly altered in the RV of WT mice during chronic hypoxia, including signaling, development, and ion transport (Table 1). The deletion of HIF-1α in the heart led to a decrease in the number of affected processes and functions, although signaling, development, and electrophysiology were still affected (Table 2). HIF-2α deletion in the RV affected fewer processes during chronic hypoxia, although inflammation, cell migration, and immune cell functions were significantly altered (Table 3). The deletion of HIF-1/2α in the chronically hypoxic heart also led to the alteration of processes related to lymphoid and myeloid cell migration, chemotaxis, immune cell function, ion transport, and calcium regulation (Table 4).
Table 1.
Wild-Type Normoxia versus Chronic Hypoxia Right Ventricle
| GO Term | Description | P Value | FDR q Value | Enrichment |
|---|---|---|---|---|
| GO process (log fold change >1.5) | ||||
| GO:0007186 | G-protein–coupled receptor signaling pathway | 3.47 × 10−5 | 4.90 × 10−1 | 2.79 |
| GO:0007154 | Cell communication | 1.03 × 10−4 | 7.24 × 10−1 | 2.45 |
| GO:0007218 | Neuropeptide signaling pathway | 1.39 × 10−4 | 6.54 × 10−1 | 13.86 |
| GO:0060428 | Lung epithelium development | 2.61 × 10−4 | 9.19 × 10−1 | 20.8 |
| GO:0003008 | System process | 2.71 × 10−4 | 7.63 × 10−1 | 1.91 |
| GO:0043269 | Regulation of ion transport | 2.90 × 10−4 | 6.82 × 10−1 | 2.22 |
| GO:0002684 | Positive regulation of immune system process | 4.59 × 10−4 | 9.25 × 10−1 | 2.01 |
| GO:1903814 | Regulation of collecting lymphatic vessel constriction | 5.76 × 10−4 | 1.00 × 1 | 41.59 |
| GO: 0021941 | Negative regulation of cerebellar granule cell precursor proliferation | 5.76 × 10−4 | 9.03 × 10−1 | 41.59 |
| GO:0009786 | Regulation of asymmetric cell division | 5.76 × 10−4 | 8.13 × 10−1 | 41.59 |
| GO:0051610 | Serotonin uptake | 5.76 × 10−4 | 7.39 × 10−1 | 41.59 |
| GO:0060481 | Lobar bronchus epithelium development | 5.76 × 10−4 | 6.77 × 10−1 | 41.59 |
| GO:0023052 | Signaling | 5.89 × 10−4 | 6.39 × 10−1 | 2.64 |
| GO:0007189 | Adenylate cyclase-activating G-protein–coupled receptor signaling pathway | 6.79 × 10−4 | 6.84 × 10−1 | 5.55 |
| GO:0060638 | Mesenchymal-epithelial cell signaling | 7.04 × 10−4 | 6.62 × 10−1 | 15.6 |
| GO:0007267 | Cell–cell signaling | 8.71 × 10−4 | 7.68 × 10−1 | 2.77 |
| GO:0006811 | Ion transport | 8.75 × 10−4 | 7.26 × 10−1 | 1.85 |
| GO:0007155 | Cell adhesion | 8.79 × 10−4 | 6.89 × 10−1 | 2.02 |
| GO:0007187 | G-protein–coupled receptor signaling pathway, coupled to cyclic nucleotide second messenger | 9.09 × 10−4 | 6.75 × 10−1 | 3.96 |
| GO function (log fold change >1.5) | ||||
| GO:0048018 | Receptor ligand activity | 2.53 × 10−8 | 1.03 × 10−4 | 4.42 |
| GO:0030545 | Receptor regulator activity | 8.48 × 10−8 | 1.72 × 10−4 | 4.12 |
| GO:0060089 | Molecular transducer activity | 7.11 × 10−7 | 9.63 × 10−4 | 2.6 |
| GO:0038023 | Signaling receptor activity | 8.01 × 10−7 | 8.14 × 10−4 | 2.69 |
| GO:0004888 | Transmembrane signaling receptor activity | 1.51 × 10−6 | 1.23 × 10−3 | 2.93 |
| GO:0022836 | Gated channel activity | 3.20 × 10−6 | 2.17 × 10−3 | 4.16 |
| GO:0022839 | Ion gated channel activity | 8.05 × 10−6 | 4.67 × 10−3 | 4.1 |
| GO:0005216 | Ion channel activity | 2.32 × 10−5 | 1.18 × 10−2 | 3.36 |
| GO:0022838 | Substrate-specific channel activity | 2.97 × 10−5 | 1.34 × 10−2 | 3.29 |
| GO:0022803 | Passive transmembrane transporter activity | 5.98 × 10−5 | 2.43 × 10−2 | 3.11 |
| GO:0015267 | Channel activity | 5.98 × 10−5 | 2.21 × 10−2 | 3.11 |
| GO:0005261 | Cation channel activity | 6.63 × 10−5 | 2.25 × 10−2 | 3.6 |
| GO:0070492 | Oligosaccharide binding | 9.46 × 10−5 | 2.96 × 10−2 | 15.12 |
| GO:0022832 | Voltage-gated channel activity | 1.10 × 10−4 | 3.20 × 10−2 | 4.29 |
| GO:0005244 | Voltage-gated ion channel activity | 1.10 × 10−4 | 2.98 × 10−2 | 4.29 |
| GO:0050840 | Extracellular matrix binding | 1.56 × 10−4 | 3.97 × 10−2 | 5.94 |
| GO:0046873 | Metal ion transmembrane transporter activity | 2.54 × 10−4 | 6.07 × 10−2 | 2.86 |
| GO:0015318 | Inorganic molecular entity transmembrane transporter activity | 3.00 × 10−4 | 6.77 × 10−2 | 2.22 |
| GO:0015075 | Ion transmembrane transporter activity | 4.10 × 10−4 | 8.77 × 10−2 | 2.13 |
| GO:0005125 | Cytokine activity | 4.24 × 10−4 | 8.61 × 10−2 | 4.44 |
| GO:0022890 | Inorganic cation transmembrane transporter activity | 4.73 × 10−4 | 9.15 × 10−2 | 2.43 |
| GO:0004930 | G-protein–coupled receptor activity | 4.95 × 10−4 | 9.14 × 10−2 | 3.32 |
| GO:0008324 | Cation transmembrane transporter activity | 5.54 × 10−4 | 9.80 × 10−2 | 2.33 |
| GO:0005184 | Neuropeptide hormone activity | 5.76 × 10−4 | 9.76 × 10−2 | 41.59 |
| GO:0005139 | IL-7 receptor binding | 5.76 × 10−4 | 9.37 × 10−2 | 41.59 |
| GO:0003858 | 3-hydroxybutyrate dehydrogenase activity | 5.76 × 10−4 | 9.01 × 10−2 | 41.59 |
| GO:0022857 | Transmembrane transporter activity | 6.53 × 10−4 | 9.83 × 10−2 | 1.96 |
Definition of abbreviations: FDR = false discovery rate; GO = gene ontology.
Table 2.
Wild-Type Chronic Hypoxia versus Chronic Hypoxia HIF-1α KO Right Ventricle
| GO Term | Description | P Value | FDR q Value | Enrichment |
|---|---|---|---|---|
| GO Process (log fold change >1.5) | ||||
| GO:0007186 | G-protein–coupled receptor signaling pathway | 1.13 × 10−4 | 1.00 × 1 | 3.92 |
| GO:1902533 | Positive regulation of intracellular signal transduction | 1.46 × 10−4 | 1.00 × 1 | 2.73 |
| GO:0032642 | Regulation of chemokine production | 1.60 × 10−4 | 7.53 × 10−1 | 9.69 |
| GO:0050731 | Positive regulation of peptidyl-tyrosine phosphorylation | 1.66 × 10−4 | 5.88 × 10−1 | 6 |
| GO:0050917 | Sensory perception of umami taste | 2.42 × 10−4 | 6.84 × 10−1 | 73.66 |
| GO:0021569 | Rhombomere 3 development | 2.42 × 10−4 | 5.70 × 10−1 | 73.66 |
| GO:0035284 | Brain segmentation | 2.42 × 10−4 | 4.89 × 10−1 | 73.66 |
| GO:0042327 | Positive regulation of phosphorylation | 4.10 × 10−4 | 7.24 × 10−1 | 2.5 |
| GO:0051897 | Positive regulation of protein kinase B signaling | 4.21 × 10−4 | 6.61 × 10−1 | 7.89 |
| GO:0006032 | Chitin catabolic process | 4.81 × 10−4 | 6.80 × 10−1 | 55.24 |
| GO:0006030 | Chitin metabolic process | 4.81 × 10−4 | 6.18 × 10−1 | 55.24 |
| GO:0050916 | Sensory perception of sweet taste | 4.81 × 10−4 | 5.67 × 10−1 | 55.24 |
| GO:0032722 | Positive regulation of chemokine production | 4.98 × 10−4 | 5.41 × 10−1 | 10.78 |
| GO:0050906 | Detection of stimulus involved in sensory perception | 5.46 × 10−4 | 5.51 × 10−1 | 10.52 |
| GO:0051896 | Regulation of protein kinase B signaling | 5.81 × 10−4 | 5.47 × 10−1 | 5.82 |
| GO:1904894 | Positive regulation of STAT cascade | 6.53 × 10−4 | 5.77 × 10−1 | 10.04 |
| GO:0001934 | Positive regulation of protein phosphorylation | 6.67 × 10−4 | 5.54 × 10−1 | 2.48 |
| GO:0098870 | Action potential propagation | 7.98 × 10−4 | 6.26 × 10−1 | 44.19 |
| GO:0019227 | Neuronal action potential propagation | 7.98 × 10−4 | 5.93 × 10−1 | 44.19 |
| GO:0021546 | Rhombomere development | 7.98 × 10−4 | 5.64 × 10−1 | 44.19 |
| GO:0010562 | Positive regulation of phosphorus metabolic process | 8.73 × 10−4 | 5.87 × 10−1 | 2.34 |
| GO:0045937 | Positive regulation of phosphate metabolic process | 8.73 × 10−4 | 5.61 × 10−1 | 2.34 |
| GO function (log fold change >1.5) | ||||
| GO:0048018 | Receptor ligand activity | 1.26 × 10−7 | 5.12 × 10−4 | 7.05 |
| GO:0030545 | Receptor regulator activity | 2.89 × 10−7 | 5.90 × 10−4 | 6.53 |
| GO:0008083 | Growth factor activity | 1.14 × 10−5 | 1.55 × 10−2 | 9.1 |
| GO:0038023 | signaling receptor activity | 2.69 × 10−5 | 2.74 × 10−2 | 3.45 |
| GO:0008201 | Heparin binding | 3.76 × 10−5 | 3.06 × 10−2 | 7.58 |
| GO:0004930 | G-protein–coupled receptor activity | 5.78 × 10−5 | 3.93 × 10−2 | 5.97 |
| GO:0005125 | Cytokine activity | 6.28 × 10−5 | 3.66 × 10−2 | 8.72 |
| GO:0060089 | Molecular transducer activity | 8.34 × 10−5 | 4.25 × 10−2 | 3.13 |
| GO:0004888 | Transmembrane signaling receptor activity | 1.22 × 10−4 | 5.53 × 10−2 | 3.6 |
| GO:0005539 | Glycosaminoglycan binding | 1.66 × 10−4 | 6.78 × 10−2 | 6 |
| GO:0071837 | HMG box domain binding | 2.44 × 10−4 | 9.06 × 10−2 | 23.68 |
| GO:0004568 | Chitinase activity | 4.81 × 10−4 | 1.64 × 10−1 | 55.24 |
| GO:0008061 | Chitin binding | 4.81 × 10−4 | 1.51 × 10−1 | 55.24 |
| GO:0016709 | Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen NAD(P)H as one donor, and incorporation of one atom of oxygen | 7.36 × 10−4 | 2.14 × 10−1 | 16.57 |
| GO:1901681 | Sulfur compound binding | 7.68 × 10−4 | 2.09 × 10−1 | 4.66 |
Definition of abbreviations: HIF-1α = hypoxia-inducible factor-1α; HMG = high mobility group; KO = knockout; NAD(P)H = nicotinamine adenine dinucleotide phosphate (reduced); STAT = signal transducers and activators of transcription.
Table 3.
GO Process* Wild-Type Chronic Hypoxia versus Chronic Hypoxia HIF-2α KO Right Ventricle (Log Fold Change >1.5)
| GO Term | Description | P Value | FDR q Value | Enrichment |
|---|---|---|---|---|
| GO:0030595 | Leukocyte chemotaxis | 4.84 × 10−5 | 6.85 × 10−1 | 12.47 |
| GO:1901700 | Response to oxygen-containing compound | 1.63 × 10−4 | 1.00 × 1 | 3.19 |
| GO:0050900 | Leukocyte migration | 3.23 × 10−4 | 1.00 × 1 | 8.35 |
| GO:0060326 | Cell chemotaxis | 3.64 × 10−4 | 1.00 × 1 | 8.14 |
| GO:0097529 | Myeloid leukocyte migration | 3.79 × 10−4 | 1.00 × 1 | 11.64 |
| GO:0031349 | Positive regulation of defense response | 5.60 × 10−4 | 1.00 × 1 | 5.82 |
| GO:0006954 | Inflammatory response | 7.25 × 10−4 | 1.00 × 1 | 5.54 |
| GO:0033993 | Response to lipid | 7.45 × 10−4 | 1.00 × 1 | 4.05 |
| GO:0006952 | Defense response | 7.81 × 10−4 | 1.00 × 1 | 3.31 |
| GO:0050727 | Regulation of inflammatory response | 8.21 × 10−4 | 1.00 × 1 | 5.41 |
| GO:0050729 | Positive regulation of inflammatory response | 9.88 × 10−4 | 1.00 × 1 | 9.04 |
*No pathways were significantly altered for GO function.
Table 4.
Wild-Type Chronic Hypoxia versus Chronic Hypoxia HIF-1α/HIF-2α KO Right Ventricle
| GO Term | Description | P Value | FDR q Value | Enrichment |
|---|---|---|---|---|
| GO process (log fold change >1.5) | ||||
| GO:0030595 | Leukocyte chemotaxis | 2.26 × 10−6 | 3.20 × 10−2 | 9.25 |
| GO:0050900 | Leukocyte migration | 6.30 × 10−6 | 4.45 × 10−2 | 6.87 |
| GO:0097529 | Myeloid leukocyte migration | 8.28 × 10−6 | 3.90 × 10−2 | 9.5 |
| GO:0010959 | Regulation of metal ion transport | 2.62 × 10−5 | 9.25 × 10−2 | 3.94 |
| GO:0043269 | Regulation of ion transport | 4.72 × 10−5 | 1.33 × 10−1 | 3.15 |
| GO:0060326 | Cell chemotaxis | 5.24 × 10−5 | 1.23 × 10−1 | 6.05 |
| GO:0030593 | Neutrophil chemotaxis | 7.51 × 10−5 | 1.52 × 10−1 | 11.25 |
| GO:0006952 | Defense response | 1.03 × 10−4 | 1.83 × 10−1 | 2.72 |
| GO:0007204 | Positive regulation of cytosolic calcium ion concentration | 1.13 × 10−4 | 1.77 × 10−1 | 4.78 |
| GO:0071621 | Granulocyte chemotaxis | 1.23 × 10−4 | 1.73 × 10−1 | 10.18 |
| GO:0070488 | Neutrophil aggregation | 1.36 × 10−4 | 1.74 × 10−1 | 85.52 |
| GO:1990266 | Neutrophil migration | 1.37 × 10−4 | 1.62 × 10−1 | 9.94 |
| GO:0097530 | Granulocyte migration | 2.11 × 10−4 | 2.29 × 10−1 | 9.1 |
| GO:1904062 | Regulation of cation transmembrane transport | 2.14 × 10−4 | 2.16 × 10−1 | 3.98 |
| GO:0060340 | Positive regulation of type I IFN-mediated signaling pathway | 2.41 × 10−4 | 2.27 × 10−1 | 23.32 |
| GO:0006935 | Chemotaxis | 2.51 × 10−4 | 2.22 × 10−1 | 4.3 |
| GO:0042330 | Taxis | 2.72 × 10−4 | 2.26 × 10−1 | 4.25 |
| GO:0051480 | Regulation of cytosolic calcium ion concentration | 2.84 × 10−4 | 2.23 × 10−1 | 4.23 |
| GO:0002684 | Positive regulation of immune system process | 3.13 × 10−4 | 2.33 × 10−1 | 2.58 |
| GO:0002523 | Leukocyte migration involved in inflammatory response | 3.19 × 10−4 | 2.25 × 10−1 | 21.38 |
| GO:0031349 | Positive regulation of defense response | 3.75 × 10−4 | 2.52 × 10−1 | 4.07 |
| GO:0023052 | Signaling | 4.66 × 10−4 | 2.99 × 10−1 | 3.61 |
| GO:0007267 | Cell–cell signaling | 4.71 × 10−4 | 2.89 × 10−1 | 3.95 |
| GO:0048705 | Skeletal system morphogenesis | 5.68 × 10−4 | 3.35 × 10−1 | 5.83 |
| GO:0051049 | Regulation of transport | 5.82 × 10−4 | 3.29 × 10−1 | 1.91 |
| GO:0034765 | Regulation of ion transmembrane transport | 6.32 × 10−4 | 3.44 × 10−1 | 3.22 |
| GO:0007589 | Body fluid secretion | 6.33 × 10−4 | 3.31 × 10−1 | 10.06 |
| GO:0050801 | Ion homeostasis | 6.38 × 10−4 | 3.22 × 10−1 | 2.61 |
| GO:0050878 | Regulation of body fluid levels | 6.78 × 10−4 | 3.31 × 10−1 | 4.17 |
| GO:0051707 | Response to other organism | 6.93 × 10−4 | 3.26 × 10−1 | 2.99 |
| GO:0032101 | Regulation of response to external stimulus | 6.93 × 10−4 | 3.16 × 10−1 | 2.59 |
| GO:0018119 | Peptidyl-cysteine S-nitrosylation | 8.02 × 10−4 | 3.54 × 10−1 | 42.76 |
| GO:0051459 | Regulation of corticotropin secretion | 8.02 × 10−4 | 3.44 × 10−1 | 42.76 |
| GO:0051461 | Positive regulation of corticotropin secretion | 8.02 × 10−4 | 3.34 × 10−1 | 42.76 |
| GO:0010524 | Positive regulation of calcium ion transport into cytosol | 8.78 × 10−4 | 3.54 × 10−1 | 9.25 |
| GO:0140013 | Meiotic nuclear division | 8.78 × 10−4 | 3.45 × 10−1 | 9.25 |
| GO:0060338 | Regulation of type I IFN-mediated signaling pathway | 9.44 × 10−4 | 3.61 × 10−1 | 15.09 |
| GO:0050678 | Regulation of epithelial cell proliferation | 9.53 × 10−4 | 3.55 × 10−1 | 3.29 |
| GO:0006874 | Cellular calcium ion homeostasis | 9.82 × 10−4 | 3.56 × 10−1 | 3.28 |
| GO:0051924 | Regulation of calcium ion transport | 9.98 × 10−4 | 3.53 × 10−1 | 3.93 |
| GO function (log fold change >1.5) | ||||
| GO:0005509 | Calcium ion binding | 4.36 × 10−6 | 1.78 × 10−2 | 3.82 |
| GO:0035662 | Toll-like receptor 4 binding | 4.04 × 10−4 | 8.24 × 10−1 | 57.01 |
Deletion of HIF-1α in Smooth Muscle, but Not Cardiomyocytes, Attenuates Chronic Hypoxia–induced PH
We previously found that deletion of HIF-1α in smooth muscle attenuated the pulmonary hypertensive and vascular remodeling responses to chronic hypoxia, yet the degree of RV hypertrophy was not diminished (11). That suggested that systemic hypoxemia might contribute to RV hypertrophy by activating HIF-dependent gene expression in the heart (Figure E1). To identify a possible role of cardiac HIF-1 and/or HIF-2 in the chronic hypoxia–induced pulmonary hypertensive response, we compared WT, SMCCre-HIF-1α, Myh6Cre-HIF-1α, SMCCre-Myh6Cre-HIF-1α, Myh6Cre-HIF-2α, and Myh6Cre-HIF-1/2α mice housed in normoxia or chronic hypoxia. After hypoxic exposure, RVSP was measured during mechanical ventilation with normoxic (room air; normoxic mice) or hypoxic (10% O2; chronic hypoxia mice) gas. No differences in RVSP were detected among the groups housed under normoxia (Figure 3A). Chronic hypoxia increased RVSP in all groups compared with the corresponding group housed in normoxia except for the Myh6Cre-HIF-2α mice (P = 0.067). In the hypoxic groups, smooth muscle HIF-1α loss attenuated the increase in RVSP compared with WT control mice, as did SMCCre-Myh6Cre-HIF-1α deletion compared with Myh6Cre-HIF-1α deletion. The deletion of HIF-1α in cardiomyocytes did not attenuate hypoxia-induced increases in RVSP compared those in with WT hypoxic control mice. These findings confirm that HIF-1α in smooth muscle contributes to the increase in RVSP in response to hypoxia, and they demonstrate that HIF-1α deletion in the heart did not affect the development of PH.
Figure 3.

Chronic hypoxia induces pulmonary hypertension and right ventricle (RV) remodeling. (A) In normoxia, no differences in RV systolic pressure (RVSP) were detected among groups (P = not significant [ns]). In chronic hypoxia, RVSP increased compared with corresponding normoxic control animals (*P < 0.05). Compared with WT mice, SMCCre-HIF-1α mice exhibited lower RVSP (*P < 0.05). Compared with Myh6Cre-HIF-1α mice, SMCCre-HIF-1α and SMCCre-Myh6Cre-HIF-1α mice exhibited lower RVSP (*P < 0.05). (B) In normoxia, no differences in Fulton’s index were detected among groups (P = ns). In chronic hypoxia, Fulton’s index increased compared with corresponding normoxic control animals (*P < 0.05). Compared with hypoxic WT mice, Myh6Cre-HIF-1α mice exhibited increased Fulton’s index (**P < 0.01). Compared with Myh6Cre-HIF-1α mice, Myh6Cre-HIF-1/2α mice exhibited lower Fulton’s index (**P < 0.01). (C) In normoxia, no differences in RV weight were detected among groups (P = ns). In chronic hypoxia, RV weight increased compared with that in corresponding normoxic control mice (*P < 0.05). In hypoxia, Myh6Cre-HIF-1α mice exhibited increased RV weight compared with WT mice (**P < 0.01). For all panels, WT: n = 23–25; SMCCre-HIF-1α: n = 12; Myh6Cre-HIF-1α: n = 8–12; SMCCre-Myh6Cre-HIF-1α: n = 10–12; Myh6Cre-HIF-1/2α: n = 10–11; and Myh6Cre-HIF-2α: n = 5–11. Values are means ± SEM. LV = left ventricle; S = septum.
Chronic Hypoxia Induces RV Hypertrophy, and Loss of HIF-1 in the RV Augments That Response
To assess the role of HIF in chronic hypoxia–induced RV remodeling, we measured Fulton’s index [RV weight/(LV+septum weight)] in the hearts of mice exposed to normoxia or chronic hypoxia. No differences in Fulton’s index were detected among the experimental groups housed in normoxia (Figure 3B). Chronic hypoxia significantly increased Fulton’s index in all experimental groups compared with their respective normoxic control animals. In chronic hypoxia, the cardiac deletion of HIF-1α produced a significant increase in Fulton’s index (P < 0.01) compared with WT hearts, whereas the cardiac deletion of HIF-1/2α caused a significant attenuation of that response (P < 0.01). These findings indicate that HIF-1 in the heart protects the RV against hypertrophic remodeling, whereas simultaneous loss of HIF-2 abrogates the detrimental effect of HIF-1α deletion, indicating that both HIF-1 and HIF-2 in the RV act in concert to regulate RV remodeling during chronic hypoxia.
Direct measurements of RV mass did not differ among groups housed under normoxia (Figure 3C). Chronic hypoxia increased RV mass in all groups compared with their respective normoxic control animals. RV mass in the hypoxic cardiac HIF-1α–knockout mice was significantly greater than that in WT hypoxic control mice (P < 0.01). To determine whether chronic hypoxia induces cardiomyocyte hypertrophy, we measured the cross-sectional area of cardiomyocytes in mice exposed to normoxia or chronic hypoxia. Although cell area was increased by chronic hypoxia, no differences were detected among the chronic hypoxia groups (Figure 4A).
Figure 4.

Chronic hypoxia induces cardiomyocyte hypertrophy in the RV and pulmonary hypertension, but these responses were not altered by HIF-1 and/or HIF-2 knockout. (A) Cardiomyocyte area as a measure of hypertrophic remodeling. In chronic hypoxia, WT, SMCCre-HIF-1α, and SMCCre-Myh6Cre-HIF-1α heart cells exhibited increased cross-sectional area compared with normoxic WT hearts (*P < 0.05) (n = 4–5 in each group). (B) PAT/ET as an estimate of pulmonary hypertension. In normoxia, no differences among groups were detected (P = ns). Chronic hypoxia WT mice exhibited a decrease in pulmonary acceleration time/ejection time compared with normoxic WT mice, indicating pulmonary hypertension (*P < 0.05). However, no differences among hypoxia groups were detected (P = ns). (WT: n = 21–22; SMCCre-HIF-1α: n = 10–12; Myh6Cre-HIF-1α: n = 10–11; SMCCre-Myh6Cre-HIF-1α: n = 11–12; Myh6Cre-HIF-1α-2α: n = 12; Myh6Cre-HIF-2α: n = 3–9). (C) Mean arterial pressures. In normoxia, Myh6Cre-HIF2α blood pressures were decreased compared with those of WT mice (*P < 0.05). In chronic hypoxia, blood pressures were decreased compared with corresponding normoxic control mice (*P < 0.05). No differences among chronic hypoxia groups were detected (P = ns). (WT: n = 18–19; SMCCre-HIF-1α: n = 8–10; Myh6Cre-HIF-1α: n = 6–7; SMCCre-Myh6Cre-HIF-1α: n = 10–11; Myh6Cre-HIF-1α-2α: n = 7–13). Values are means ± SEM. ET = ejection time; PAT = pulmonary acceleration time.
To further evaluate the degree of PH, pulmonary acceleration time (PAT) and ejection time (ET) were measured by echocardiography. A decrease in the PAT/ET is an indication of PH. No differences in PAT/ET were detected between groups housed in normoxia (Figure 4B). Chronic hypoxia significantly decreased the PAT/ET in WT mice compared with that in normoxic control mice. However, no differences in the PAT/ET were detected across groups housed in chronic hypoxia, perhaps reflecting the larger measurement variability among animals compared with the direct micromanometer measurements.
We also measured the effect of chronic hypoxia on systemic arterial pressure. In normoxia, HIF-1/2α deletion in the heart decreased mean arterial pressure compared with WT. Hypoxic WT, Myh6Cre-HIF-1α, and SMCCre-Myh6Cre-HIF-1/2α mice showed a decrease in mean arterial pressure compared with normoxic WT control animals (Figure 4C). These data show that chronic hypoxia tends to lower blood pressure (measured during acute return to normoxia).
PA Wall Thickness Increased during Chronic Hypoxia, but Deletion of HIFs Did Not Alter That Response
We next examined the medial wall thickness of PAs in mice exposed to normoxia or chronic hypoxia (Figure 5A). No measured differences in PA wall thickness were detected among groups housed under normoxia (Figure 5B). Chronic hypoxia caused a significant increase in the medial wall thickness (PA cross-sectional area normalized to PA diameter) of WT, SMCCre-HIF-1α, and Myh6Cre-HIF-1/2α mice compared with the corresponding normoxic control mice. However, no differences in cross-sectional area were seen across groups housed under chronic hypoxia. These findings confirm that hypoxia elicits remodeling of pulmonary arteries, but the lack of differences among the hypoxic groups tends to downplay the role of cardiomyocyte HIF-1 and HIF-2 in the extent of that remodeling.
Figure 5.

Chronic hypoxia induces pulmonary vascular remodeling, but that response was not altered by HIF-1 and/or HIF-2 knockout. (A) Representative hematoxylin and eosin–stained mouse lung tissue sections. Thin arrows indicate vessels and thick arrows indicate airways. Scale bar, 25 μm. (B) Quantification of pulmonary artery wall cross-sectional area. No differences in area were detected among normoxic groups (P = ns). Chronic hypoxia increased wall thickness compared with wild-type normoxic control mice (*P < 0.05). No differences among chronic hypoxia groups were detected (P = ns). WT: n = 8–13; SMCCre-HIF-1α: n = 7–8; Myh6Cre-HIF-1α: n = 5; SMCCre-Myh6Cre-HIF-1α: n = 9; Myh6Cre-HIF-1α-2α: n = 3. Values are means ± SEM. PA = pulmonary artery.
Deletion of HIFs in the Heart Does Not Significantly Affect Cardiac Function in Normoxia or Chronic Hypoxia
To determine whether deletion of HIF in the heart alters cardiac function, we examined the hearts of normoxic and chronically hypoxic WT, SMCCre-HIF-1α, Myh6Cre HIF-1α, Myh6Cre HIF-1α, SMCCre-Myh6Cre HIF-1α, Myh6Cre-HIF-1/2α, and Myh6Cre-HIF-2α mice by echocardiography during isoflurane breathing in 100% O2. Heart rate, stroke volume, ejection fraction, cardiac output, end-systolic volume, and end-diastolic volume were not affected by gene knockout in normoxia (Figure 6). Chronic hypoxia led to a decrease in stroke volume in WT mice, but no differences among the hypoxic groups were detected. Cardiac output decreased in WT and Myh6Cre-HIF-1/2α relative to normoxic control mice and was higher in SMCCre-HIF-1α mice than in WT hypoxic animals. These data suggest that HIF-1α and HIF-2α in the heart have only minor effects on cardiac function in normoxic animals or in mice housed in chronic hypoxia.
Figure 6.
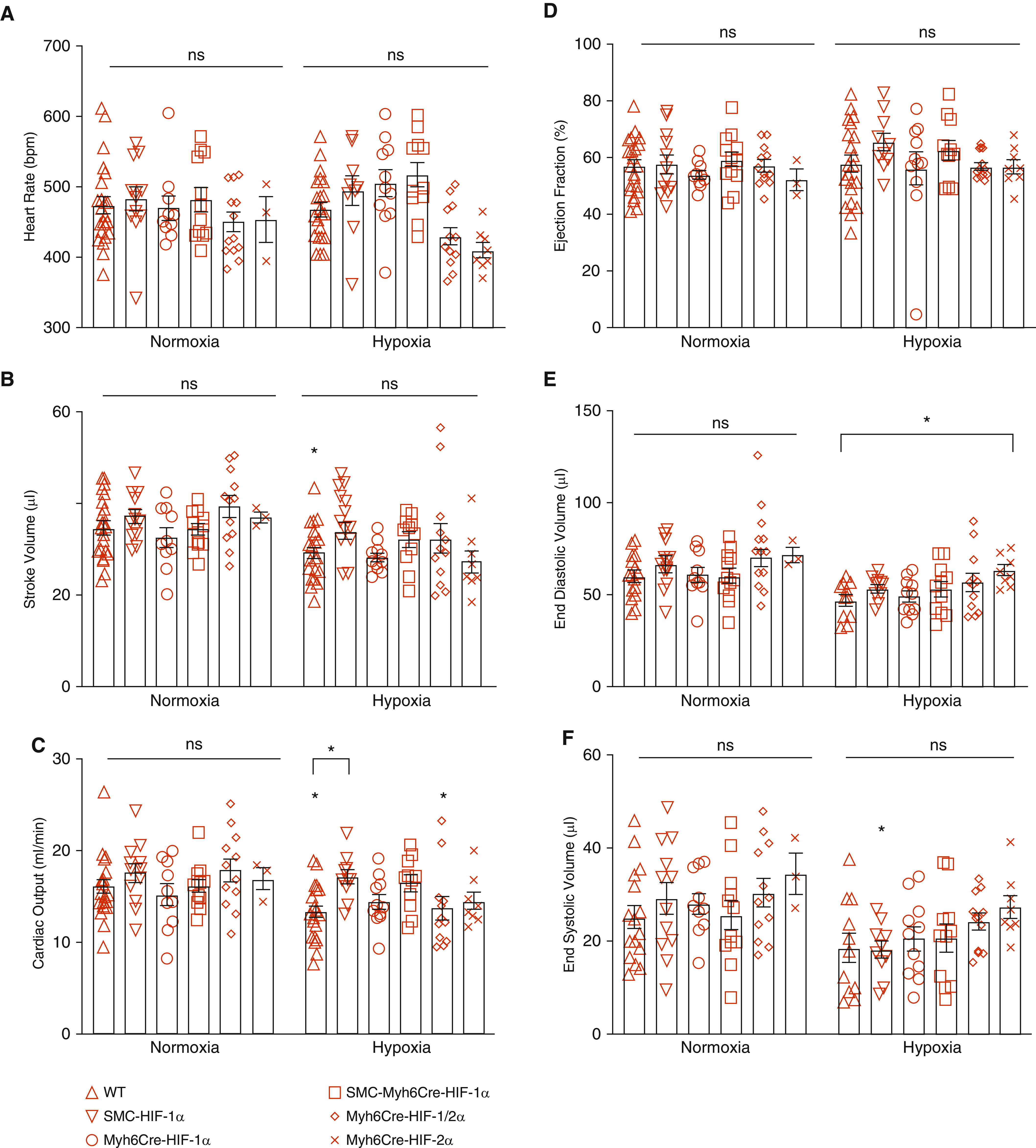
Chronic hypoxia induces minimal changes in cardiac function; these responses are not significantly altered by HIF-1 and/or HIF-2 knockout. (A) Heart rate was not different among groups in normoxia (P = ns) or in chronic hypoxia (P = ns), nor was it different between normoxia and corresponding hypoxic groups (P = ns). (B) Stroke volume was not different among groups in normoxia (P = ns) or in chronic hypoxia (P = ns). In WT mice, stroke volume was lower in chronic hypoxia compared with normoxia (*P < 0.05). (C) Cardiac output was not different among groups in normoxia (P = ns) but was lower in hypoxia in WT and Myh6Cre-HIF-1α-2α mice compared with corresponding normoxic groups (*P < 0.05). Also, cardiac output was greater in SMCCre-Myh6Cre-HIF-1α mice compared with hypoxic WT mice (*P < 0.05). (D) Ejection fraction was not different among groups in normoxia (P = ns) or chronic hypoxia (P = ns). No differences were detected in between normoxic groups and corresponding chronic hypoxia groups. (E) End-diastolic volume was not different among groups in normoxia (P = ns). No differences were detected between normoxia and corresponding hypoxic groups. In chronic hypoxia, end-diastolic volume was greater in Myh6Cre-HIF-2α mice compared with hypoxic WT mice (*P < 0.05). (F) End-systolic volume was not different among normoxic groups or chronic hypoxia groups (P = ns). In chronic hypoxia, SMCCre-Cre-HIF-α end-systolic volume was lower compared with that in corresponding normoxic control animals (signified by asterisk above the column). WT: n = 22; SMCCre-HIF-1α: n = 10–12; Myh6Cre-HIF-1α: n = 10–11; SMCCre-Myh6Cre-HIF-1α: n = 10–12; Myh6Cre-HIF-1α-2α: n = 12; Myh6Cre-HIF-2α: n = 3–8. Values are means ± SEM.
Discussion
Chronic hypoxia acts on the lung to cause HIF-dependent vascular remodeling that leads to PH. Alveolar hypoxia also causes coronary arterial hypoxemia, which can activate HIF-dependent transcriptional responses in the heart and conceivably affect RV function and hypertrophic remodeling. Understanding how HIF-1 and HIF-2 regulate hypertrophic remodeling in response to chronic hypoxia is complicated by the fact that systemic interventions to inhibit HIF activity (e.g., HIF inhibitors) tend to lower PA pressure, the primary stimulus driving RV hypertrophy. We sought to address this by inducing conditional, cardiac-specific deletion of HIF-1α and/or HIF-2α in adult mice and then evaluating the consequences in terms of the effects on the development of chronic hypoxia–induced PH and RV hypertrophy.
We found that chronic hypoxia modified the expression of nearly 400 genes in the RV of WT animals. Diverse biological processes were associated with gene pathways broadly related to cardiac growth, development, and cell division, which likely contributed to the RV hypertrophic remodeling we observed. Those transcriptional responses reflected both the effects of cardiac tissue hypoxia and the PH-induced increase in RVSP, which is a major driver of RV remodeling.
We initially hypothesized that HIF-1 and/or HIF-2 in the heart might be contributing to the development of RV hypertrophy in chronic hypoxia, but the data clearly indicate that this is not the case. The genetic pathways related to cardiac growth that were observed in the WT chronic hypoxia mice were not downregulated in the HIF-1α–knockout hearts, so it is not surprising that hypertrophic remodeling did not decrease. In fact, hypertrophy was increased in the HIF-1–knockout animals relative to WT control animals in association with the upregulation of other pathways, including G-coupled protein receptor signaling, action potential propagation, tyrosine and protein phosphorylation, and signal transducer and activator of transcription (STAT) signaling, which has been linked to hypertrophic remodeling (24). On this basis, we infer that HIF-1 acts to suppress some genes that contribute to hypertrophy such that its inactivation leads to an upregulation of genes that augmented the cardiac remodeling response.
By contrast, the loss of HIF-2α in chronic hypoxia had no effect on Fulton’s index. Why would loss of HIF-1 augment hypertrophy yet loss of HIF-2α does not? One possible explanation relates to the distinct gene sets regulated by these two transcription factors. Biological pathways upregulated in the HIF-2–knockout mice were heavily related to inflammation, immune cell activation/migration, and host defense. This is consistent with the inflammatory phenotype previously seen in mice that survived global HIF-2α deletion (25). Because these pathways differed from those that were activated in the HIF-1–knockout animals, it is reasonable to conclude that HIF-2 acts primarily to modify inflammation and immune cell function in chronic hypoxia and that this neither augments nor suppresses RV hypertrophic remodeling. Distinct and sometimes opposing roles for HIF-1 and HIF-2 have been demonstrated previously in cancer cells (26), so the concept of disparate roles for these transcription factors is not new.
Does HIF-1 and HIF-2 activation play any role in the heart in relation to cardiac hypertrophy? Insight comes from the study by Xie and colleagues (27), who genetically deleted prolyl hydroxylases (PHDs) 2 and 3 in mouse hearts. Because PHDs are negative regulators of HIFs, their deletion led to the stable activation of these transcription factors, which in turn produced cardiac hypertrophy at the cellular and tissue level. Although major changes in cardiac function were not observed, these hearts were more susceptible to failure in response to chronic isoproterenol. The development of hypertrophy in the PHD2/3 knockout seemingly conflicts with our finding that HIF-1 deletion augmented hypertrophy. However, the deletion of PHDs produces an extreme and sustained activation of HIFs compared with their modest activation during chronic hypoxia. Hence, the comparison between chronic hypoxia and PHD-knockout models may not be meaningful.
Increased stabilization of HIF-1α has been found in heart samples of patients with acute myocardial ischemia resulting from infarction (28), and constitutive overexpression of HIF-1α decreased infarct size in a mouse model (29). These findings suggest that HIF-1 plays a beneficial role of in maintaining O2 delivery to cardiomyocytes (30). However, overexpression of HIF-1α in the heart has been reported to cause cardiomyopathy (31) and may contribute to disease progression in failing hearts (32). Collectively, these observations suggest that the physiological activation of HIF by tissue hypoxia is beneficial, whereas excessive activation results in supraphysiological activation of HIF-1 target genes that may contribute to the development of cardiomyopathy. HIF-1α has also been reported to induce cellular hypertrophy in cultured cardiomyocytes by inducing expression of transient receptor potential channel (TRPC) (33), and increased TRPC6 expression has been shown to contribute to nuclear factor of activated T cells (NFAT)-mediated pathologic remodeling in pressure-overloaded hearts (34). We found no evidence of altered TRPC expression in WT mice during chronic hypoxia or any effect of HIF-1α deletion on TRPC expression in hypoxia; indeed, the loss of HIF-1 activity worsened the cardiac remodeling. Therefore, it seems unlikely that TRPC channels contributed to the RV remodeling in our mice.
Finally, the inactivation of HIF-1 and/or HIF-2 in the heart did not affect the increase in RVSP that developed during chronic hypoxia. Hence, the HIF-dependent expression of secreted factors in the heart does not contribute to the pulmonary vascular remodeling response that leads to PH.
In summary, our data show that HIF-1 and HIF-2 regulate largely distinct sets of genes in the RV during chronic hypoxia. Loss of HIF-1α in the heart worsens chronic hypoxia–induced RV remodeling, without significantly altering RVSP. Simultaneous loss of HIF-2α in the heart mitigates that effect, suggesting that the gene sets regulated by HIF-1 and HIF-2 play interacting roles in controlling RV remodeling.
Supplementary Material
Acknowledgments
Acknowledgment
Histology services were provided by the Northwestern University Mouse Histology and Phenotyping Laboratory, which is supported by NCI P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. RNA-Seq analysis was performed in the High-Throughput RNA-Seq Lab, Division of Pulmonary and Critical Care Medicine, Northwestern University. Transcriptomic data analysis was carried out using the Northwestern University Genomics Computing Cluster, which is jointly supported by the Feinberg School of Medicine, the Center for Genetic Medicine, and Feinberg’s Department of Biochemistry and Molecular Genetics, the Office of the Provost, the Office for Research, and Northwestern Information Technology; it is maintained by the Feinberg IT and Research Computing Group.
Footnotes
Supported by U.S. National Institutes of Health grants HL35440, HL109478, and HL122062 (P.T.S.) and a Parker B. Francis Fellowship (K.A.S.).
Author Contributions: Conception and design: K.A.S., G.B.W., G.R.S.B., and P.T.S. Collection and assembly of data: K.A.S., G.B.W., V.J.D., H.A.-V., and P.T.S. Experimental work: K.A.S., G.B.W., V.J.D., and H.A.-V. Data analysis and interpretation: K.A.S., G.B.W., V.J.D., H.A.-V., E.B., and P.T.S. Manuscript writing: K.A.S., G.B.W., G.R.S.B., and P.T.S.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0023OC on July 21, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(Suppl):D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 3.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 4.Sachdev A, Villarraga HR, Frantz RP, McGoon MD, Hsiao JF, Maalouf JF, et al. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest. 2011;139:1299–1309. doi: 10.1378/chest.10-2015. [DOI] [PubMed] [Google Scholar]
- 5.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. doi: 10.1183/09031936.00072709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 7.Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62(Suppl):D22–D33. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis Nature 1998394485–490.[Published erratum appears in Nature 395:525.] [DOI] [PubMed] [Google Scholar]
- 9.Gleadle JM, Ratcliffe PJ. Induction of hypoxia-inducible factor-1, erythropoietin, vascular endothelial growth factor, and glucose transporter-1 by hypoxia: evidence against a regulatory role for Src kinase. Blood. 1997;89:503–509. [PubMed] [Google Scholar]
- 10.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med. 2014;189:314–324. doi: 10.1164/rccm.201302-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291:L941–L949. doi: 10.1152/ajplung.00528.2005. [DOI] [PubMed] [Google Scholar]
- 13.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2018;314:L256–L275. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CJ, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J. 2019;54:1900378. doi: 10.1183/13993003.00378-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation. 2016;133:2447–2458. doi: 10.1161/CIRCULATIONAHA.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 20.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 21.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 22.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 23.Folmsbee SS, Morales-Nebreda L, Van Hengel J, Tyberghein K, Van Roy F, Budinger GRS, et al. The cardiac protein αT-catenin contributes to chemical-induced asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308:L253–L258. doi: 10.1152/ajplung.00331.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 25.Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 26.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie L, Pi X, Townley-Tilson WH, Li N, Wehrens XH, Entman ML, et al. PHD2/3-dependent hydroxylation tunes cardiac response to β-adrenergic stress via phospholamban. J Clin Invest. 2015;125:2759–2771. doi: 10.1172/JCI80369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 29.Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 30.Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–690. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- 31.Bekeredjian R, Walton CB, MacCannell KA, Ecker J, Kruse F, Outten JT, et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS One. 2010;5:e11693. doi: 10.1371/journal.pone.0011693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hölscher M, Schäfer K, Krull S, Farhat K, Hesse A, Silter M, et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc Res. 2012;94:77–86. doi: 10.1093/cvr/cvs014. [DOI] [PubMed] [Google Scholar]
- 33.Chu W, Wan L, Zhao D, Qu X, Cai F, Huo R, et al. Mild hypoxia-induced cardiomyocyte hypertrophy via up-regulation of HIF-1α-mediated TRPC signalling. J Cell Mol Med. 2012;16:2022–2034. doi: 10.1111/j.1582-4934.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.