

Abstract
Idiopathic pulmonary fibrosis is a fatal interstitial lung disease characterized by the TGF-β (transforming growth factor-β)–dependent differentiation of lung fibroblasts into myofibroblasts, which leads to excessive deposition of collagen proteins and progressive scarring. We have previously shown that synthesis of collagen by myofibroblasts requires de novo synthesis of glycine, the most abundant amino acid found in collagen protein. TGF-β upregulates the expression of the enzymes of the de novo serine–glycine synthesis pathway in lung fibroblasts; however, the transcriptional and signaling regulators of this pathway remain incompletely understood. Here, we demonstrate that TGF-β promotes accumulation of ATF4 (activating transcription factor 4), which is required for increased expression of the serine–glycine synthesis pathway enzymes in response to TGF-β. We found that induction of the integrated stress response (ISR) contributes to TGF-β–induced ATF4 activity; however, the primary driver of ATF4 downstream of TGF-β is activation of mTORC1 (mTOR Complex 1). TGF-β activates the PI3K-Akt-mTOR pathway, and inhibition of PI3K prevents activation of downstream signaling and induction of ATF4. Using a panel of mTOR inhibitors, we found that ATF4 activation is dependent on mTORC1, independent of mTORC2. Rapamycin, which incompletely and allosterically inhibits mTORC1, had no effect on TGF-β–mediated induction of ATF4; however, Rapalink-1, which specifically targets the kinase domain of mTORC1, completely inhibited ATF4 induction and metabolic reprogramming downstream of TGF-β. Our results provide insight into the mechanisms of metabolic reprogramming in myofibroblasts and clarify contradictory published findings on the role of mTOR inhibition in myofibroblast differentiation.
Keywords: fibrosis, metabolism, glycolysis, mitochondria
Clinical Relevance
Metabolic reprogramming in lung fibroblasts is required for their differentiation into myofibroblasts and for matrix protein production. We have identified signaling and transcriptional regulators of this metabolic reprogramming that could be therapeutically targeted to treat lung fibrosis.
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease with a median survival of 3.5 years, affecting ∼89,000 people in the United States (1, 2). IPF is associated with the TGF-β (transforming growth factor-β)–dependent differentiation of fibroblasts into myofibroblasts, contractile cells that secrete excessive amounts of extracellular matrix, including collagen (3–6). Myofibroblasts are the primary cells responsible for the structural remodeling and impairment of lung function characteristic of IPF and thus represent a key therapeutic target for the treatment of the disease (6).
Metabolic reprogramming is increasingly understood to be a key regulator of myofibroblast biology. Treatment of fibroblasts with TGF-β increases both glycolytic rate and mitochondrial respiration, inhibition of which prevents their differentiation into myofibroblasts as well as matrix production (7–9). We have previously shown that myofibroblasts utilize carbon derived from glucose to support de novo synthesis of glycine, an amino acid that constitutes over 30% of the primary structure of collagen protein (9–11). This requires TGF-β–mediated induction of the enzymes of the de novo serine–glycine synthesis pathway, which catalyze the conversion of the glycolytic intermediate 3-phosphoglycerate into the amino acids serine and glycine (12, 13). Inhibition of this pathway reduces TGF-β–induced collagen protein production in vitro and ameliorates bleomycin-induced lung fibrosis in vivo (9–11).
Although a role for metabolic reprogramming in the promotion of fibrogenesis is increasingly accepted, the signaling and transcriptional regulators of myofibroblast metabolism are incompletely understood. ATF4 (activating transcription factor 4) is a basic leucine zipper transcription factor best known for its role in regulation of stress responses, including endoplasmic reticulum stress (14). Among the transcriptional targets of ATF4 are amino acid transporters and biosynthetic enzymes, including those of the serine–glycine synthesis pathway (15–17). ATF4 is also induced downstream of growth factor signaling through activation of mTORC1 (mTOR Complex 1), allowing cells to coordinate biosynthetic capacity with growth signals (18–21).
Here, we demonstrate that TGF-β–induced expression of the serine–glycine synthesis pathway enzymes requires ATF4. Though the integrated stress response (ISR) contributes to ATF4 accumulation, it is activation of the PI3K-Akt-mTORC1 signaling axis that is the major regulator of ATF4 and metabolic reprogramming downstream of TGF-β. Moreover, we show that ATF4 induction and metabolic reprogramming are refractory to mTOR inhibition by rapamycin, an allosteric inhibitor that incompletely inhibits mTORC1. By contrast, ATP-competitive mTOR kinase inhibitors (TORKi) such as TORIN1, which inhibits both mTORC1 and mTORC2, and Rapalink-1, which selectively inhibits mTORC1, abolished TGF-β–induced ATF4 accumulation and expression of the enzymes of the serine–glycine synthesis pathway. Furthermore, Rapalink-1, but not rapamycin, prevented TGF-β–mediated increases in glycolytic rate and mitochondrial respiration. Our results demonstrate that mTORC1 is a critical regulator of myofibroblast metabolism and highlights the divergent effects of various mTOR inhibitors on myofibroblasts.
Methods
Fibroblast Culture
Normal human lung fibroblasts (NHLFs, Lonza) were cultured as previously described (9). For experiments, cells were cultured in the absence of serum in Dulbecco’s modified Eagle medium (Corning) containing 0.1% BSA, 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate for 24 hours prior to treatment with TGF-β (1 ng/ml; Millipore-Calbiochem). ISRIB (Sigma), SB431542 (R&D Systems), LY294002 (Cell Signaling), BKM120 (Cayman), rapamycin (R&D Systems), TORIN1 (R&D Systems), and Rapalink-1 (ApexBio) were added 30 minutes prior to TGF-β. Tunicamycin was from Millipore.
siRNA Knockdowns
For knockdowns using siRNA, 1 × 106 NHLFs were transfected with 250 pmol ON-TARGETplus siRNA (Dharmacon) using a Nucleofector 2b (Lonza) with program T-016. Twenty-four hours after transfection, cells were replated for experiments as above. For a list of siRNAs used, see data supplement.
Western Blotting
Cells were lysed in urea sample buffer, and electrophoresis was performed as we have described previously (22). A list of primary antibodies used is included in the data supplement. Western blots were quantified using ImageJ software (National Institutes of Health, https://imagej.nih.gov/ij/).
Quantitative PCR
RNA was isolated using Direct-zol RNA MiniPrep Plus (Zymo). Reverse transcription was performed using iScript Reverse Transcription Supermix (Bio-Rad). Quantitative real-time RT-PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad). A list of primers used is included in the data supplement.
Metabolic Assays
Mitochondrial and glycolytic stress tests were performed as we have previously shown (9, 11, 22–24). NHLFs were treated with TGF-β for 48 hours as above. Cells were replated onto Seahorse XFe24 plates at 3 × 104/well, and media was replaced with serum-, glucose-, glutamine-, and bicarbonate-free Dulbecco’s modified Eagle medium (Agilent). For mitochondria stress tests, media was supplemented with 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate. For glycolysis stress tests, media was supplemented with 2 mM glutamine. Injections for mitochondria stress tests included oligomycin A (1 μM), FCCP (carbonyl cyanide-4-trifluoromethoxyphenylhydrazone, 2 μM), and antimycin A and rotenone (1 μM each). Injections for glycolysis stress tests included glucose (10 mM), oligomycin A (1 μM), and 2-DG (100 mM). All drugs were purchased from Sigma.
Immunohistochemistry
The collection and use of human IPF lung specimens was approved by the University of Chicago Institutional Review Board. After deparaffinization and rehydration, antigens were retrieved using citrate buffer (S1699; DAKO) in a steamer for 20 minutes. Tissue sections were incubated with anti-ATF4 antibody (1:200; Proteintech) for 1 hour at room temperature in a humidity chamber. Tissue sections were washed in TBS and incubated with biotinylated antirabbit IgG (1:200, BA-1000; Vector Laboratories) for 30 minutes at room temperature. Antigen-antibody binding was detected by Elite kit (PK-6100; Vector Laboratories) and DAB (K3468; DAKO). Tissue sections were counterstained with hematoxylin and covered with cover glasses.
Statistical Analysis
Statistical analysis was done using Prism 8 (GraphPad Software, Inc). All data presented in bar charts are mean ± SEM. Statistical significance was determined by using unpaired two-tailed Student’s t test (comparisons between two samples) or by using one-way ANOVA with Bonferroni’s correction (multiple comparisons).
Results
ATF4 Is Required for TGF-β–dependent Induction of serine–glycine Synthesis Pathway Enzymes and Collagen Protein Synthesis
We have previously demonstrated that TGF-β induces the expression of all enzymes of the serine–glycine synthesis pathway, which converts the glycolytic intermediate 3-phosphoglycerate to glycine, the most abundant amino acid in collagen protein (9). As shown in Figure 1A, concomitant with TGF-β–induced accumulation of collagen and α-SMA (α-smooth muscle actin) proteins, the expression of serine–glycine synthesis enzymes (PHGDH [phosphoglycerate dehydrogenase], PSAT1 [phosphoserine aminotransferase 1], PSPH [phosphoserine phosphatase], and SHMT2 [serine hydroxymethyltransferase 2]) was also induced. As ATF4 has been shown to be a key regulator of amino acid biosynthesis enzymes (15–17), we examined expression of ATF4 after TGF-β treatment. As shown in Figure 1A, ATF4 protein accumulated prior to induction of serine–glycine synthesis enzymes.
Figure 1.

ATF4 (activating transcription factor 4) is required for induction of serine–glycine synthesis enzymes and collagen protein downstream of TGF-β (transforming growth factor-β). (A) Protein levels of collagen I, α-SMA (α-smooth muscle actin), ATF4, and the serine–glycine synthesis enzymes PHGDH (phosphoglycerate dehydrogenase), PSAT1 (phosphoserine aminotransferase 1), PSPH (phosphoserine phosphatase), and SHMT2 (serine hydroxymethyltransferase 2) in normal human lung fibroblasts (NHLFs) treated with TGF-β (1 ng/ml) for the indicated intervals. (B) Collagen I, α-SMA, serine–glycine synthesis enzyme, and P5CS (pyrroline-5-carboxylate synthase) protein levels in control and ATF4 knockdown NHLFs treated with TGF-β for the indicated intervals. (C) mRNA expression of ATF4 and serine–glycine synthesis enzymes in control and ATF4 knockdown NHLFs measured by qRT-PCR. Cells were treated with TGF-β for 24 hours or left untreated (mean ± SEM, n = 3). (D) mRNA expression of COL1A1 (collagen type I α 1), ACTA2 (α-SMA), CTGF (connective tissue growth factor), and SERPINE1 (serpin family E member 1) measured by qRT-PCR. NHLFs were treated with TGF-β for 24 hours or left untreated (mean ± SEM, n = 3). (E) Immunohistochemistry analysis of ATF4 protein expression in fibrotic and nonfibrotic areas of paraffin-embedded lung sections from a patient with idiopathic pulmonary fibrosis. Scale bar, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001. Cont siRNA = control nontargeting siRNA; NS = not significant (P ≥ 0.05).
To determine whether ATF4 is required for induction of serine–glycine synthesis enzymes downstream of TGF-β, we knocked down the expression of ATF4 using two independent siRNAs. As shown in Figure 1B, knockdown of ATF4 prevented the TGF-β–mediated induction of all of the serine–glycine synthesis enzymes and prevented myofibroblast differentiation and collagen protein production. Although ATF4 knockdown prevented induction of serine–glycine synthesis enzymes at the mRNA level (Figures 1C and E1 in the data supplement), induction of collagen (COL1A1), α-SMA (ACTA2), and other SMAD target genes were not affected (Figure 1D), consistent with our previous reports that serine–glycine synthesis pathway supports collagen protein synthesis at the post-transcriptional level (9–11).
Proline is the second most abundant amino acid found in collagen protein, and we and others have shown that glutamine-dependent proline synthesis also supports collagen protein synthesis downstream of TGF-β (11, 25). ATF4 has been shown to regulate the expression of proline biosynthesis enzymes, including P5CS (pyrroline-5-carboxylate synthase, encoded by the ALDH18A1 gene) and PYCR1 (P5C reductase 1) (26–28). We thus examined the effect ATF4 knockdown on proline biosynthetic enzymes. In contrast to serine–glycine synthesis enzymes, ATF4 knockdown had no effect on proline biosynthetic enzymes (Figures 1B and E2).
Activation of the ISR in alveolar epithelial cells and downstream effectors such as ATF4 has been demonstrated in familial interstitial pneumonia as well as in IPF, suggesting that stress responses in epithelial cells may promote disease pathogenesis (29, 30). Little is known about how these ISR effectors may contribute to myofibroblast biology. Consistent with previous studies, we found that ATF4 was highly enriched in the alveolar epithelium overlaying fibroblastic foci in lung samples from patients with IPF (Figure 1E). Though staining was most prominent in the epithelium, myofibroblasts stained positively for ATF4 expression as well. This was in contrast to nonfibrotic areas of the same lung, which did not stain positive for ATF4 (Figure 1E).
The ISR Is Not a Major Contributor to TGF-β–induced ATF4 Activation
ATF4 is best known as a downstream effector of the ISR (14). The ISR is activated by various cellular stresses that lead to phosphorylation of eIF2α (eukaryotic initiation factor 2α), resulting in inhibition of bulk protein translation, whereas translation of several transcripts, including that of ATF4, is increased. We found that treatment of NHLFs with the endoplasmic reticulum stressor tunicamycin was sufficient to induce ATF4 accumulation to a similar extent as TGF-β treatment (Figures 2A and E3). TGF-β induced accumulation of the spliced form of the transcription factor XBP-1 to a lesser extent than tunicamycin, and induction of the ISR effector CHOP (C/EBP homologous protein) was not seen with TGF-β, suggesting an overall lower level of stress induced by TGF-β than by tunicamycin (Figures 2A and E3). Although tunicamycin increased ATF4 levels, ATF4 accumulation itself was not sufficient to cause significant upregulation of protein levels of serine-glycine synthesis enzymes (PHGDH, PSAT1, PSPH, SHMT2) (Figures 2A and E3) despite an increase in their transcript levels (Figure 2B), suggesting that these enzymes themselves may be the subject of translational regulation. To determine whether activation of the ISR was required for TGF-β–induced ATF4 activation, we treated cells with TGF-β in the presence or absence of the ISR inhibitor ISRIB (31) and assessed the protein (Figures 2C and E3) and mRNA (Figures 2D and E4) expression of ATF4, serine–glycine synthesis pathway enzymes, and ISR effectors. Treatment with ISRIB reduced TGF-β–induced accumulation of ATF4, although its expression was still higher than baseline (Figures 2C and E3). Similarly, ISRIB reduced the TGF-β–induced increase in the mRNA expression of serine–glycine synthesis pathway enzymes, although their TGF-β–induced expression was still higher than baseline (Figure 2D). Despite the reduction in mRNA levels, ISRIB failed to prevent the protein induction of serine–glycine synthesis pathway enzymes (Figures 2C and 2D and E3). Interestingly, ISRIB treatment, across a range of concentrations, resulted in increased accumulation of collagen protein downstream of TGF-β (Figures 2C and E3). This suggests that basal levels of cellular stress result in translational repression and decreased levels of collagen protein production downstream of TGF-β. Relieving this translational repression with ISRIB results in increased protein translation, including collagen protein production. Though ISRIB inhibited induction of CHOP (DDIT3) mRNA downstream of TGF-β, ISRIB did not affect TGF-β–induced expression of XBP-1 unspliced mRNA (a transcriptional target of ATF6) or spliced XBP-1 (a target of IRE-1 endonuclease), suggesting that the effects of ISRIB are specific to stress responses downstream of eIF2α (Figure E4). No compensatory increase in the protein levels of eIF2α or the ISRIB binders eIF2Bβ or eIF2Bδ was seen after ISRIB treatment (Figure E5).
Figure 2.

The Integrated Stress Response is not required for induction of serine–glycine synthesis enzymes and collagen protein downstream of TGF-β. (A) Protein levels of collagen I, α-SMA, ATF4, serine–glycine synthesis enzymes, XBP-1s (X-box-binding protein 1, spliced isoform), and CHOP (C/EBP homologous protein) in NHLFs treated with TGF-β (1 ng/ml) or tunicamycin (2 μg/ml) for the indicated intervals. (B) mRNA expression of serine–glycine synthesis enzymes measured by qRT-PCR. NHLFs were treated with tunicamycin for 24 hours or left untreated (mean ± SEM, n = 4). (C) Western blot analysis of collagen I, α-SMA, ATF4, and serine–glycine synthesis enzyme protein levels in NHLFs treated with TGF-β in the presence or absence of ISRIB (1 μM) for the indicated intervals. (D) mRNA expression of serine–glycine synthesis enzymes measured by qRT-PCR. NHLFs were treated with TGF-β in the presence or absence of ISRIB (mean ± SEM, n = 4). (E and F) Western blot analysis of collagen I, α-SMA, ATF4, and serine–glycine synthesis enzyme protein levels in control and either (E) PERK or (F) GCN2 knockdown NHLFs treated with TGF-β for the indicated intervals. **P < 0.01 and ***P < 0.001. GCN2 = general control nonderepressible 2; ISRIB = integrated stress response inhibitor; PERK = protein kinase R-like endoplasmic reticulum kinase; Tun = tunicamycin.
We hypothesized that myofibroblastic differentiation may result in increased levels of endoplasmic reticulum stress and amino acid stress; thus, we used siRNA to knock down PERK and GCN2, two eIF2α kinases that respond to these stresses, respectively (Figures 2E and 2F). Knockdown of PERK resulted in increased collagen protein production, similar to ISRIB treatment, suggesting that basal levels of endoplasmic reticulum stress act to repress collagen protein synthesis in lung fibroblasts. Collectively, our results suggest that ISR activation contributes to ATF4 activation downstream of TGF-β but is not required for serine–glycine synthesis enzyme expression or collagen protein production. Furthermore, the effects of baseline ISR activation on protein translation act to repress collagen protein accumulation downstream of TGF-β.
TGF-β Activates the PI3K-Akt-mTOR Pathway, Which Is Required for Downstream Activation of ATF4
ATF4 has more recently been shown to be activated by growth factor signaling independent of stress responses, allowing cells to coordinate growth signals with biosynthetic capacity (18–21). TGF-β initiates signaling through a receptor complex composed of type I and type II receptors, leading to phosphorylation and nuclear localization of SMAD transcription factors, which promote downstream gene expression (32). TGF-β also activates other signaling pathways, including the PI3K-Akt-mTOR pathway, through less well-established mechanisms (Figure 3A) (32, 33). We found that TGF-β promotes phosphorylation of Akt at the PDK1 (phosphoinositide dependent kinase 1)-dependent T308 site (Figures 3B and 3C and E6A). Downstream phosphorylation of TSC2, mTOR, and the major mTORC1 targets S6 kinase and 4EBP1 (eukaryotic initiation factor 4E binding protein 1) were also induced by TGF-β. All of these signaling events were inhibited by cotreatment with the TGF-β receptor kinase inhibitor SB431542 (Figure 3B) or with the PI3K inhibitors LY294002 or BKM120 (Figures 3C and E6A). Pretreatment of lung fibroblasts with LY294002 did not prevent phosphorylation of SMAD3 downstream of TGF-β (Figure 3D), demonstrating that the canonical signaling pathway of TGF-β receptor complex to SMAD occurs independent of PI3K. In contrast to treatment with ISRIB, cotreatment of cells with LY294002 or BKM120 abolished ATF4 accumulation and expression of serine–glycine synthesis enzymes downstream of TGF-β (Figures 3E and 3F and E6B). Furthermore, PI3K inhibition prevented TGF-β–induced collagen protein production (Figures 3E and E6B). Collectively, these results suggest that TGF-β induces ATF4 via activation of PI3K-Akt-mTOR pathway.
Figure 3.
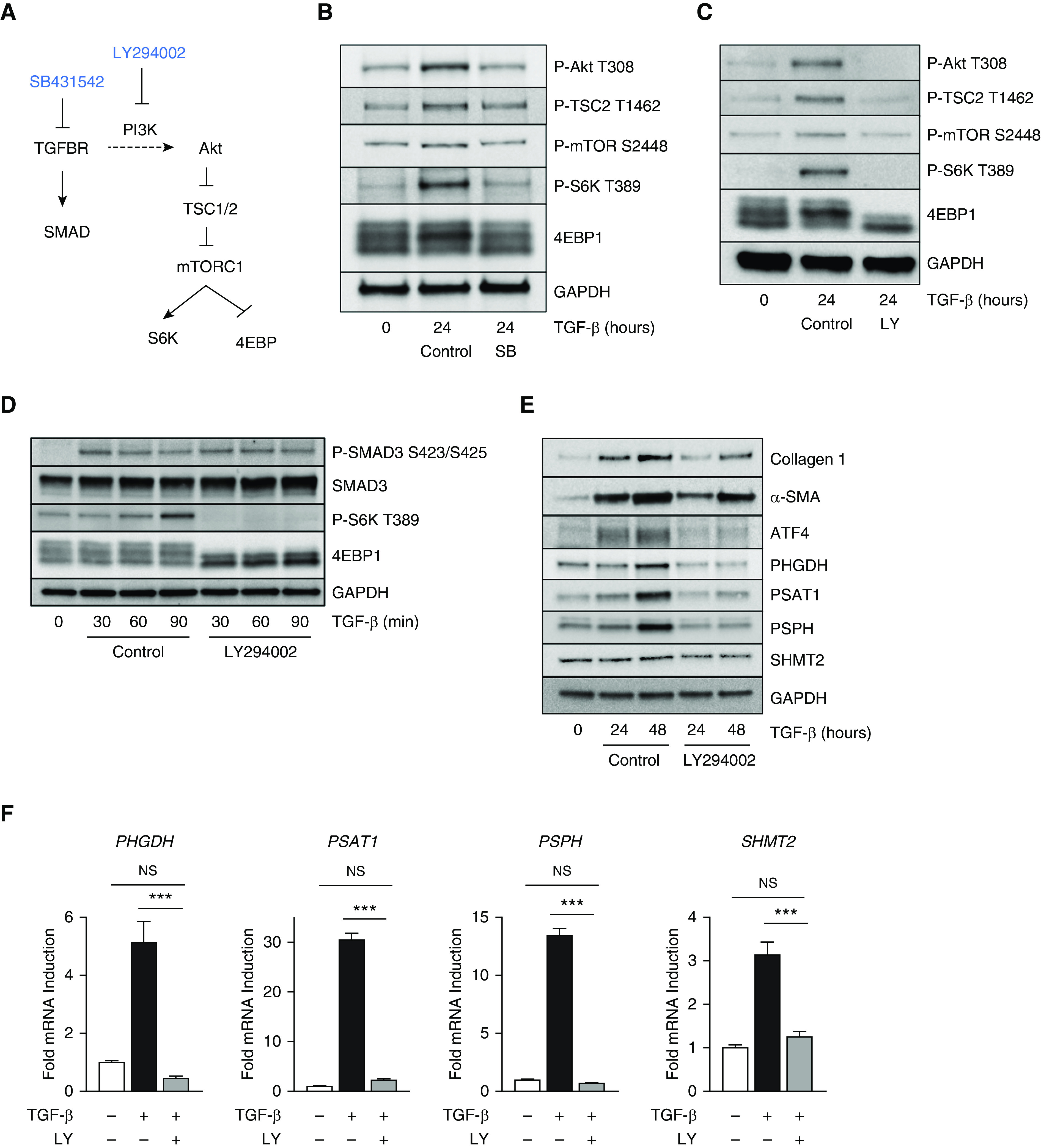
The PI3K-Akt-mTOR pathway is activated by TGF-β and is required for ATF4 activation and collagen protein synthesis. (A) Schematic representation of signaling downstream of TGF-β through the canonical (SMAD) pathway and noncanonical (PI3K) pathway. (B and C) Western blot analysis of signaling downstream of PI3K in NHLFs treated with TGF-β (1 ng/ml) for 24 hours and cotreated with either (B) the TGF-β receptor kinase inhibitor SB431542 (SB; 10 μM) or (C) the PI3K inhibitor LY294002 (LY; 25 μM). (D) Western blot analysis of signaling downstream of the TGF-β receptor in NHLFs treated with TGF-β for the indicated intervals. Cells were pretreated with LY294002 for 30 minutes or left untreated. (E) Western blot analysis of collagen I, α-SMA, ATF4, and serine–glycine synthesis enzyme protein levels in NHLFs treated with TGF-β in the presence or absence of LY294002 for the indicated intervals. (F) mRNA expression of serine–glycine synthesis enzymes measured by qRT-PCR. NHLFs were treated with TGF-β in the presence or absence of LY294002 (mean ± SEM, n = 4). ***P < 0.001. 4EBP = eukaryotic translation initiation factor 4E-binding protein.
mTORC1 Regulates ATF4 and serine–glycine Synthesis Pathway Enzyme Expression in Human Lung Fibroblasts Downstream of TGF-β
The downstream effector of the PI3K-Akt pathway, mTORC1, is a major regulator of cellular growth and anabolic metabolism (34–36). Through promotion of ATF4 activation, mTORC1 has been shown to promote amino acid and nucleotide synthesis (18–21). We thus cotreated NHLFs with TGF-β and rapamycin, an allosteric inhibitor specific to mTORC1. Interestingly, we observed only a partial inhibitory effect of rapamycin on ATF4 protein accumulation and target gene induction downstream of TGF-β (Figures 4A and 4B). Furthermore, we did not detect any reduction in TGF-β–induced collagen production in cells treated with rapamycin (Figure 4A).
Figure 4.
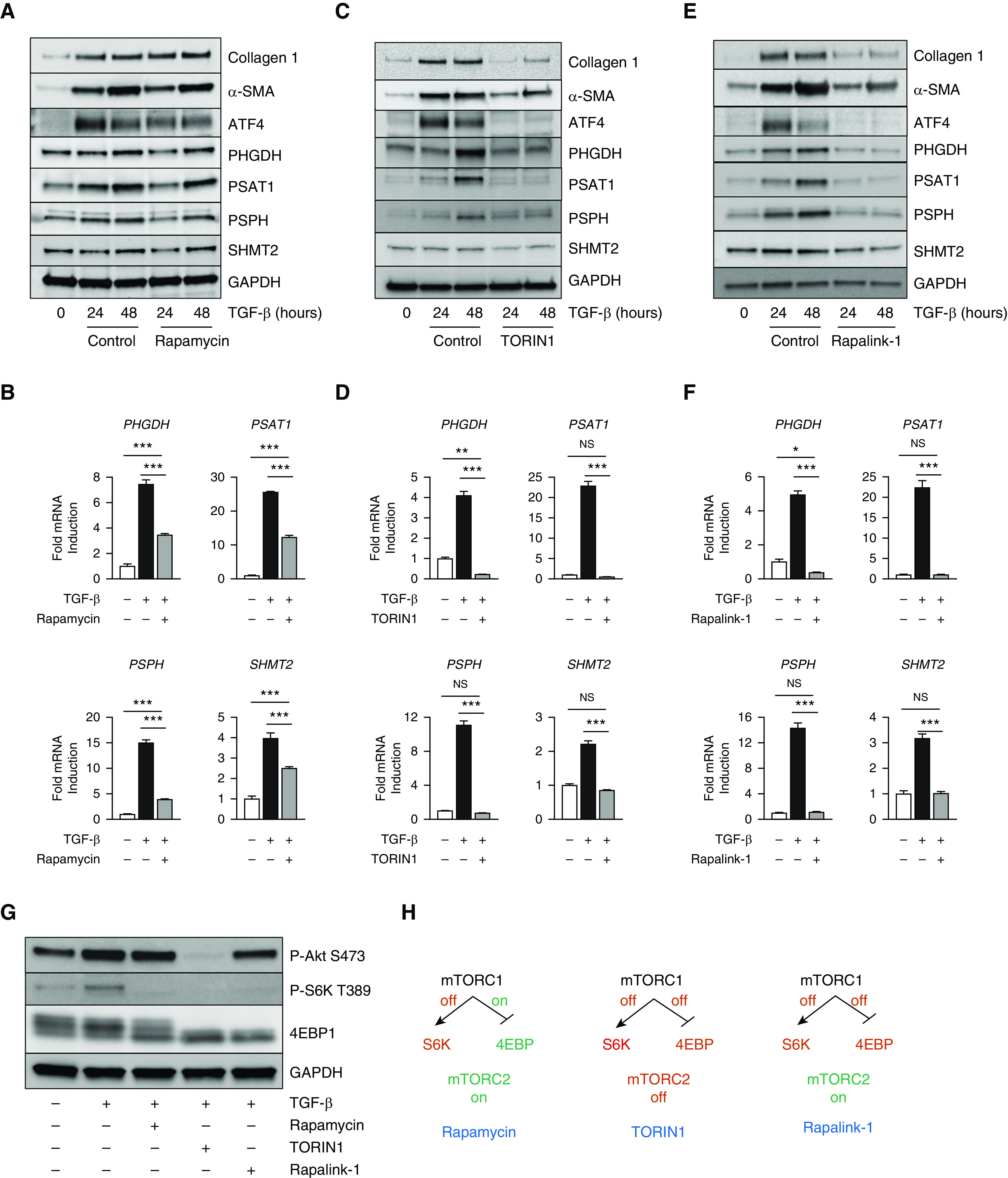
mTORC1 (mTOR complex 1) promotes ATF4 activation and collagen protein synthesis downstream of TGF-β. (A, C, and E) Western blot analysis of collagen I, α-SMA, ATF4, and serine–glycine synthesis enzyme protein levels or (B, D, and F) mRNA expression of serine–glycine synthesis enzymes measured by qRT-PCR (mean ± SEM, n = 4). NHLFs were treated with TGF-β (1 ng/ml) in the presence or absence of the indicated mTOR inhibitors for the indicated intervals. (A and B) Rapamycin (30 nM). (C and D) TORIN1 (125 nM). (E and F) Rapalink-1 (2 nM). (G) Western blot analysis of signaling downstream of PI3K in NHLFs treated with TGF-β for 24 hours and cotreated with either rapamycin, TORIN1, or Rapalink-1. (H) Schematic representation of the bioactivities of rapamycin, TORIN1, and Rapalink-1 on mTORC1 and mTORC2 signaling targets. *P < 0.05, **P < 0.01, and ***P < 0.001. S6K = ribosomal protein S6 kinase.
Rapamycin forms a complex with FKBP12 (FK506-binding protein 12), which then interacts with mTOR and allosterically inhibits its activity. Certain targets of mTORC1, such as S6-kinase, are acutely sensitive to rapamycin, whereas others such as 4EBP1 are less sensitive (37–39). The development of ATP-competitive mTOR inhibitors, which directly inhibit the kinase domain of mTOR, has highlighted the rapamycin-insensitive targets of mTORC1 (40, 41). We thus cotreated NHLFs with TGF-β and TORIN1, a TORKi. In contrast to rapamycin, cotreatment with TORIN1 abolished ATF4 protein accumulation and target gene induction downstream of TGF-β (Figures 4C and 4D). TGF-β–dependent collagen induction was also inhibited by TORIN1 (Figure 4C).
As mTORC1 and mTORC2 share the same ATP-binding site, it is not possible to determine whether the effects of TORIN1 are dependent on mTORC1, mTORC2, or both. Recently, a bitopic mTOR inhibitor was developed, allowing for the specific and complete inhibition of mTORC1. Rapalink-1 consists of rapamycin connected to the TORKi MLN0128 with a chemical linker (42, 43). When cotreated with Rapalink-1, TGF-β–induced ATF4 protein accumulation and target gene induction were abolished similar to treatment with TORIN1 (Figures 4E and 4F). Collagen protein production downstream of TGF-β was also inhibited by treatment with Rapalink-1 (Figure 4E).
To confirm the differential effects of the various mTOR inhibitors on signaling downstream of mTORC1 and mTORC2, we observed phosphorylation of mTORC1 and mTORC2 targets after treatment with TGF-β in the presence or absence of the inhibitors. Though cotreatment with any of the mTOR inhibitors (rapamycin, TORIN-1, Rapalink-1) abolished the TGF-β–induced phosphorylation of mTORC1 target S6-kinase, only TORIN1 resulted in complete loss of phosphorylation of the mTORC2 target S473 of Akt (Figures 4G and E7). Phosphorylation of 4EBP1 was abolished by both TORIN1 and Rapalink-1; however, significant phosphorylation of 4EBP1 remained in the presence of rapamycin, consistent with a role of 4EBP1-dependent regulation of ATF4 (Figures 4G and E7). Together, our results suggest that mTORC1 is the major regulator of ATF4, serine–glycine synthesis, and collagen protein production downstream of TGF-β. Furthermore, the differential effects of the various mTOR inhibitors on the downstream effects of TGF-β can be explained by their ability to block signaling to specific targets downstream of mTORC1 (Figure 4H).
TGF-β–induced Glycolysis and Mitochondrial Oxygen Consumption Require mTORC1 but Not ATF4
We and others have shown that TGF-β induces metabolic reprogramming in NHLFs characterized by increased rates of glycolysis and mitochondrial oxygen consumption (7–9, 11). Although increased glycolysis and mitochondrial activity has been shown to be required for myofibroblast differentiation, little is known about how this metabolic reprogramming is regulated. We thus sought to determine whether ATF4 and mTORC1 regulate the glycolytic and mitochondrial reprogramming observed in NHLFs after exposure to TGF-β.
Control and ATF4 knockdown NHLFs were treated with TGF-β for 48 hours or left untreated and then subjected to a glycolysis stress test, measuring extracellular acidification rate as a surrogate for glycolytic lactate production. As we have previously shown, TGF-β increased both basal (after injection of glucose) and maximal (after inhibition of mitochondrial ATP production with oligomycin A) extracellular acidification rate in NHLFs (Figures 5A and 5B and E8A and E8B). TGF-β also increased mitochondrial oxygen consumption rate (OCR) in NHLFs (Figures 5C and E8C). ATF4 knockdown did not affect either the increase in glycolytic rate or mitochondrial oxygen consumption downstream of TGF-β (Figures 5A–5C and E8).
Figure 5.

mTORC1 regulates glycolytic and mitochondrial reprogramming downstream of TGF-β. (A) Analysis of extracellular acidification rate (ECAR) in control and ATF4 knockdown NHLFs treated with TGF-β (1 ng/ml) for 48 hours or left untreated. ECAR was measured at baseline and after injection of glucose (glycolytic rate), oligomycin A (maximal glycolytic rate), and 2-deoxyglucose (2-DG; nonglycolytic acidification) as indicated. (B) Average basal and maximal ECAR values from A (mean ± SEM, n = 4). (C) Oxygen consumption rate (OCR) in control and ATF4 knockdown NHLFs treated with TGF-β for 48 hours or left untreated (mean ± SEM, n = 4). (D) Analysis of ECAR in NHLFs treated with TGF-β for 48 hours or left untreated. Cells were cotreated with either rapamycin (30 nM) or Rapalink-1 (2 nM) or left untreated. (E) Average basal and maximal ECAR values from D (mean ± SEM, n = 4). (F) OCR in NHLFs treated with TGF-β for 48 hours or left untreated (mean ± SEM, n = 4). Cells were cotreated with either rapamycin or Rapalink-1 or left untreated. (G) Analysis of cellular OCR in NHLFs treated with TGF-β for 48 hours or left untreated. Cells were cotreated with either rapamycin or Rapalink-1 or left untreated. OCR was measured at baseline and after injection of oligomycin A (coupled respiration), FCCP (maximal respiration), and antimycin and rotenone (nonmitochondrial OCR) as indicated. (H) Average basal and maximal OCR values from G (mean ± SEM, n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001. FCCP = carbonyl cyanide-4-trifluoromethoxyphenylhydrazone.
To measure the effect of mTORC1 inhibition on TGF-β–induced glycolytic flux, we treated NHLFs with TGF-β in the presence or absence of rapamycin or Rapalink-1 for 48 hours and then performed a glycolysis stress test. Cotreatment of cells with rapamycin reduced TGF-β–dependent glycolysis (Figures 5D and 5E); however, this reduction in glycolytic rate did not differ significantly from cells treated with TGF-β alone (Figure 5E). By contrast, cotreatment of cells with Rapalink-1 resulted in complete inhibition of TGF-β–induced glycolytic reprogramming (Figures 5D and 5E). Treatment with Rapalink-1 also prevented the TGF-β–induced increase in mitochondrial oxygen consumption (Figure 5F).
Because Rapalink-1 prevented the increase in mitochondrial oxygen consumption downstream of TGF-β, we subjected rapamycin and Rapalink-1–treated cells to a mitochondrial stress test to gain a fuller understanding of the effect of mTORC1 on mitochondrial function in NHLFs. OCR was measured at baseline, after injection of oligomycin A (to measure ATP-coupled respiration), and after uncoupling with FCCP (carbonyl cyanide-4-trifluoromethoxyphenylhydrazone, to measure maximal oxygen consumption). As we previously showed, TGF-β increases basal OCR in NHLFs but does not affect maximal OCR. Cotreatment of cells with rapamycin had no effect on either basal or maximal OCR; however, treatment with Rapalink-1 abolished TGF-β–induced basal OCR (Figures 5G and 5H). Rapalink-1 also significantly decreased maximal OCR in NHLFs. Together, our results demonstrate that mTORC1 promotes glycine synthesis in NHLFs through ATF4 and regulates cellular glycolytic and mitochondrial function through ATF4-independent mechanisms (Figure 6).
Figure 6.

TGF-β activates mTORC1-dependent metabolic reprogramming in lung fibroblasts. TGF-β activates mTORC1, which promotes cellular glucose usage in lung fibroblasts. mTORC1 also promotes biosynthetic reprogramming by upregulating the enzymes of the serine–glycine synthesis pathway, which convert the glycolytic intermediate 3-phosphoglycerate (3-PG) to glycine, the most abundant amino acid in collagen protein. mTORC1 is required for ATF4 induction downstream of TGF-β. Glycolytic reprogramming occurs independent of ATF4.
Discussion
Metabolic reprogramming in myofibroblasts is an emerging mechanism that promotes myofibroblast differentiation and matrix production, contributing to fibrotic disease. We have previously demonstrated that TGF-β induces metabolic reprogramming in lung fibroblasts characterized by increased levels of glycolysis and mitochondrial oxygen consumption (9, 11). Increased glycolytic flux promotes de novo production of glycine, the most abundant amino acid found in collagen protein (9, 10). TGF-β induces the expression of the enzymes of serine–glycine synthesis pathway, which converts the glycolytic intermediate 3-phosphoglycerate into glycine. Inhibition of this pathway prevents collagen protein synthesis downstream of TGF-β (9–11). Here, we sought to better understand the mechanisms by which metabolic reprogramming and the de novo serine–glycine synthesis pathway is regulated in lung fibroblasts.
ATF4 is a basic leucine zipper transcription factor best known to be induced by stress responses. Among the targets of ATF4 are amino acid transporters and biosynthetic enzymes, including those of the de novo serine–glycine synthesis pathway (15–17). Our results demonstrate that ATF4 is induced in lung fibroblasts early after TGF-β exposure, preceding the induction of serine–glycine synthesis pathway enzymes. We show that ATF4 is required for TGF-β–induced expression of serine–glycine synthesis enzymes and that ATF4 knockdown prevents collagen protein accumulation downstream of TGF-β. We previously reported that in addition to de novo glycine synthesis, TGF-β also induces glutamine-dependent proline synthesis (11). Though enzymes of this pathway have been shown to be regulated by ATF4 (26–28), we found that induction of these enzymes downstream of TGF-β occurred independent of ATF4.
Because ATF4 is known to be regulated by the ISR, we examined the effect of ISR activation on ATF4 induction downstream of TGF-β. We found that ISR inhibition reduced ATF4 induction and mRNA expression of serine–glycine synthesis enzymes downstream of TGF-β, suggesting that ISR activation does contribute to TGF-β–induced ATF4 activation; however, gene expression in the presence of ISRIB remained significantly induced compared with baseline, and induction of serine–glycine synthesis enzyme protein expression was maintained. This was consistent with previous work demonstrating that activation of ATF4 downstream of insulin is independent of eIF2α phosphorylation (19).
ISR activation was sufficient to induce ATF4 activation but not to increase protein levels of serine–glycine synthesis enzymes or collagen protein production. This is consistent with in vivo work by Lawson and colleagues, which showed that intratracheal tunicamycin instillation was insufficient to induce fibrosis in mice but promoted bleomycin-induced fibrosis (44). Interestingly, we found that ISR inhibition resulted in increased TGF-β–induced collagen protein production. It is possible that baseline levels of stress act to repress collagen protein synthesis during myofibroblast differentiation and that relieving ISR-associated translational repression acts to increase collagen production. Alternatively, it is possible that inhibition of this homeostatic pathway leads to increased levels of cellular stress, leading to increased myofibroblastic differentiation downstream of TGF-β. IPF is associated with activation of the ISR in multiple cells of the lung, including fibroblasts, epithelial cells, and macrophages (45, 46). Our results suggest that therapeutic attempts to target the effects of ISR activation in other cell types may have the unintended effect of promoting collagen protein production in myofibroblasts.
Recently, mTORC1-mediated activation of ATF4 has been demonstrated in cancer cells, providing a mechanism by which growth signals coordinate biosynthetic pathways to promote proliferation (18, 19). mTOR signaling has been implicated in myofibroblast differentiation; however, there have been conflicting reports on the relative contributions of mTORC1 and mTORC2 in driving fibrogenesis (47, 48). Furthermore, a recent clinical trial using the rapamycin analog Everolimus failed, calling into question the role of mTOR in promoting fibrosis (49). Using a panel of mTOR inhibitors, we dissected the relative contributions of mTOR signaling targets to myofibroblast metabolic reprogramming downstream of TGF-β. The activity of rapamycin depends on the formation of a rapamycin-FKBP12 complex, which allosterically inhibits mTORC1 with differential activities toward the various mTORC1 targets. Despite the fact that rapamycin completely inhibited phosphorylation of S6-kinase downstream of TGF-β, phosphorylation of 4EBP1 was only partially inhibited, and induction of ATF4 and of the serine–glycine synthesis enzymes was not inhibited by rapamycin. The TORKi TORIN1 abolished TGF-β–induced phosphorylation of S6-kinase and 4EBP1 as well as the mTORC2 target S147 of Akt. TORIN1 treatment inhibited ATF4 activation downstream of TGF-β as well as induction of serine–glycine synthesis enzymes and collagen protein production. Because TORIN1 inhibits both mTORC1 and mTORC2, we utilized a new bitopic mTOR inhibitor that uses a chemical linker to deliver a TORKi specifically to mTORC1 (42, 43). Our results using Rapalink-1 demonstrate that mTORC1 inhibition, independent of effects on mTORC2 activity, is sufficient to prevent the TGF-β–induced activation of ATF4 and expression of serine–glycine synthesis enzymes. This finding is in agreement with previous studies that demonstrated that knockdown of Raptor prevents ATF4 activation downstream of growth factors (20, 21). The effects of Rapalink-1 on collagen protein synthesis were similar to the effects of TORIN1 treatment, suggesting that the effects of TORIN1 on mTORC2 are dispensable for its inhibitory effect on myofibroblast differentiation.
Selvarajah and colleagues recently published a manuscript demonstrating that the nonspecific TORKi AZD8055 prevents ATF4 induction and serine–glycine synthesis enzyme expression downstream of TGF-β in lung fibroblasts (20). Our findings agree with those of Selvarajah and colleagues and demonstrate that mTORC1 is the primary mTOR complex driving metabolic reprogramming during myofibroblast differentiation. We demonstrate that Rapalink-1 treatment not only prevents TGF-β–induced ATF4 activation and expression of the serine–glycine synthesis pathway enzymes, it also completely abolished TGF-β–induced increases in glycolytic rate and mitochondrial oxygen consumption in lung fibroblasts. In addition to regulating ATF4, mTORC1 has been shown to regulate other metabolism-regulating transcription factors, including HIF-1α, SREBPs, and PGC-1α (50–54). Our results suggest that ATF4 does not regulate glycolysis and mitochondrial respiration in NHLFs. Future work will be required to determine which downstream targets of mTORC1 are required to regulate these pathways. Our finding that rapamycin does not affect glycolysis or mitochondrial function suggest that a possible explanation for the failure of the rapamycin analog Everolimus in an IPF clinical trial (49) may be due to its inability to target metabolic reprogramming in myofibroblasts.
Myofibroblasts are the primary effector cell in the pathogenesis of IPF, responsible for deposition of extracellular matrix that replaces alveoli with scar tissue and resulting in impairment of gas exchange (1, 2). The altered matrix environment of fibrotic lungs has bioactive and mechanical properties that potentiates further fibroblast activation in a feed-forward cycle (55–57). Thus, targeting of the metabolic reprogramming in myofibroblasts that allows for exaggerated matrix production has the potential to inhibit disease progression and possibly lead to resolution (47). We find that mTORC1 and ATF4 are important regulators of metabolic reprogramming in myofibroblasts and suggest that therapies that target this signaling axis may be effective treatments for IPF.
Supplementary Material
Footnotes
Supported by U.S. National Institutes of Health grants R01ES010524, U01ES026718, P01HL144454 (G.M.M.), and R01HL151680 (R.B.H.) as well as Department of Defense grant W81XWH-16-1-0711 (G.M.M.), ATS Foundation Unrestricted Grant, and Respiratory Health Association grant RHA2018-01-IPF (R.B.H.).
Author Contributions: Conception and design: L.J.W., A.Y.M., P.S.W., L.M.K., K.A.S., G.M.M., and R.B.H. Acquisition of data: E.M.O’L., Y.T., R.N., L.J.W., G.A.G., and R.B.H. Analysis and interpretation of data: E.M.O’L., R.N., R.C.-A., A.Y.M., P.S.W., L.M.K., K.A.S., G.M.M., and R.B.H. Manuscript writing: E.M.O’L., G.M.M., and R.B.H. Final approval of manuscript: E.M.O’L., Y.T., R.N., L.J.W., R.C.-A., A.Y.M., P.S.W., L.M.K., K.A.S., G.M.M., and R.B.H.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0143OC on July 15, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 2.Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21:355–361. doi: 10.1183/09059180.00002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–417. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 7.Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem. 2015;290:25427–25438. doi: 10.1074/jbc.M115.646984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. 2015;192:1462–1474. doi: 10.1164/rccm.201504-0780OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nigdelioglu R, Hamanaka RB, Meliton AY, O’Leary E, Witt LJ, Cho T, et al. Transforming growth factor (TGF)-β promotes de novo serine synthesis for collagen production. J Biol Chem. 2016;291:27239–27251. doi: 10.1074/jbc.M116.756247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamanaka RB, Nigdelioglu R, Meliton AY, Tian Y, Witt LJ, O’Leary E, et al. Inhibition of phosphoglycerate dehydrogenase attenuates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2018;58:585–593. doi: 10.1165/rcmb.2017-0186OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamanaka RB, O’Leary EM, Witt LJ, Tian Y, Gökalp GA, Meliton AY, et al. Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am J Respir Cell Mol Biol. 2019;61:597–606. doi: 10.1165/rcmb.2019-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28:794–806. doi: 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 16.Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA. 2012;109:6904–6909. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep. 2017;19:1083–1090. doi: 10.1016/j.celrep.2017.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G, et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β1-induced collagen biosynthesis. Sci Signal. 2019;12:eaav3048. doi: 10.1126/scisignal.aav3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, et al. Increased serine and one-carbon pathway metabolism by PKCλ/ι deficiency promotes neuroendocrine prostate cancer. Cancer Cell. 2019;35:385–400, e9. doi: 10.1016/j.ccell.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamanaka RB, Mutlu GM. PFKFB3, a direct target of p63, is required for proliferation and inhibits differentiation in epidermal keratinocytes. J Invest Dermatol. 2017;137:1267–1276. doi: 10.1016/j.jid.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun KA, Li Y, Meliton AY, Woods PS, Kimmig LM, Cetin-Atalay R, et al. Endogenous itaconate is not required for particulate matter-induced NRF2 expression or inflammatory response. Elife. 2020;9:e54877. doi: 10.7554/eLife.54877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O’Leary EM, et al. Tissue-resident alveolar macrophages do not rely on glycolysis for LPS-induced inflammation. Am J Respir Cell Mol Biol. 2020;62:243–255. doi: 10.1165/rcmb.2019-0244OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwörer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, et al. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J. 2020;39:e103334. doi: 10.15252/embj.2019103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Aniello C, Fico A, Casalino L, Guardiola O, Di Napoli G, Cermola F, et al. A novel autoregulatory loop between the Gcn2-Atf4 pathway and (L)-Proline [corrected] metabolism controls stem cell identity. Cell Death Differ. 2015;22:1094–1105. doi: 10.1038/cdd.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams CM. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem. 2007;282:16744–16753. doi: 10.1074/jbc.M610510200. [DOI] [PubMed] [Google Scholar]
- 28.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 31.Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, et al. Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science. 2018;359:eaaq0939. doi: 10.1126/science.aaq0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal. 2019;12:eaav5183. doi: 10.1126/scisignal.aav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans. 2013;41:906–912. doi: 10.1042/BST20130041. [DOI] [PubMed] [Google Scholar]
- 35.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 2011;71:2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 38.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan Q, Aksoy O, Wong RA, Ilkhanizadeh S, Novotny CJ, Gustafson WC, et al. A kinase inhibitor targeted to mTORC1 drives regression in glioblastoma. Cancer Cell. 2017;31:424–435. doi: 10.1016/j.ccell.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534:272–276. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, Wang Y, Pandupuspitasari NS, Wu G, Xiang X, Gong Q, et al. Endoplasmic reticulum stress, a new wrestler, in the pathogenesis of idiopathic pulmonary fibrosis. Am J Transl Res. 2017;9:722–735. [PMC free article] [PubMed] [Google Scholar]
- 46.Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68-69:355–365. doi: 10.1016/j.matbio.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep. 2014;6:16. doi: 10.12703/P6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang W, Wei K, Ho L, Berry GJ, Jacobs SS, Chang CH, et al. A critical role for the mTORC2 pathway in lung fibrosis. PLoS One. 2014;9:e106155. doi: 10.1371/journal.pone.0106155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malouf MA, Hopkins P, Snell G, Glanville AR Everolimus in IPF Study Investigators. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology. 2011;16:776–783. doi: 10.1111/j.1440-1843.2011.01955.x. [DOI] [PubMed] [Google Scholar]
- 50.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [Published erratum appears in Science 346:aaa1503.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 55.Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marinković A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol. 2013;48:422–430. doi: 10.1165/rcmb.2012-0335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.