

Abstract
Fibrosis is characterized by fibroblast activation, leading to matrix remodeling culminating in a stiff, type I collagen–rich fibrotic matrix. Alveolar epithelial cell (AEC) apoptosis is also a major feature of fibrogenesis, and AEC apoptosis is sufficient to initiate a robust lung fibrotic response. TGF-β (transforming growth factor-β) is a major driver of fibrosis and can induce both AEC apoptosis and fibroblast activation. We and others have previously shown that changes in extracellular matrix stiffness and composition can regulate the cellular response to TGF-β. In the present study, we find that type I collagen signaling promotes TGF-β–mediated fibroblast activation and inhibits TGF-β–induced AEC death. Fibroblasts cultured on type I collagen or fibrotic decellularized lung matrix had augmented activation in response to TGF-β, whereas AECs on cultured on type I collagen or fibrotic lung matrix were more resistant to TGF-β–induced apoptosis. Both of these responses were mediated by integrin α2β1, a major collagen receptor. AECs treated with an α2 integrin inhibitor or with deletion of α2 integrin had loss of collagen-mediated protection from apoptosis. We found that mice with fibroblast-specific deletion of α2 integrin were protected from fibrosis whereas mice with AEC-specific deletion of α2 integrin had more lung injury and a greater fibrotic response to bleomycin. Intrapulmonary delivery of an α2 integrin–activating collagen peptide inhibited AEC apoptosis in vitro and in vivo and attenuated the fibrotic response. These studies underscore the need for a thorough understanding of the divergent response to matrix signaling.
Keywords: collagen, apoptosis, integrin
Progressive fibrosis is characterized by an accumulation of activated myofibroblasts, which produce a collagen-rich scar (1, 2). Thus, much of the prior fibrosis research has focused on the activation and function of myofibroblasts (3). However, many drugs that were found to block fibroblast activation and collagen production by myofibroblasts have resulted in negative clinical trials (4), and the therapies that have shown effective results in clinical trials only modestly slow the progression of disease (5–7). There is increasing recognition that fibrogenesis is a multicellular process with important contributions from other profibrotic cell types including macrophages and alveolar epithelial cells (AECs) (8–14). Mounting evidence suggests that AEC death is not merely a feature or bystander of the fibrotic process but rather that AEC dysfunction and apoptosis are critical in initiating and propagating fibrogenesis (15). Mutations in AEC-specific genes, such as surfactant proteins, leads to AEC dysfunction and familial lung fibrosis (16–20). In animal models, inhibiting prominent apoptosis pathways blocks fibrosis (21, 22), and we have shown that induction of AEC-specific apoptosis is sufficient to initiate lung scarring (23, 24). TGF-β (transforming growth factor-β) is among the most well-studied profibrotic cytokines and is a key mediator of both fibroblast activation and AEC apoptosis (1). However, we and others have shown that several intracellular signaling pathways that augment the fibroblast activation response to TGF-β are also involved in inhibiting TGF-β–induced AEC apoptosis, potentially yielding a mixed overall effect on fibrosis and limiting the effectiveness of targeting these pathways for fibrosis therapy (25–27).
The extracellular matrix (ECM) strongly influences cell behavior and the response to TGF-β (1).
Fibrosis is characterized by progressive matrix remodeling with destruction of the laminin-rich basement membrane and initial replacement by a temporary provisional matrix made up of plasma proteins (8). Changes in matrix composition are accompanied by increased stiffness or rigidity. Matrix composition and stiffness regulates cell behavior through cell surface matrix receptors, such as integrins, which activate intracellular adaptor proteins such as FAK and Rho (28). For fibrotic diseases, this pathway has primarily been studied in the context of TGFβ-induced fibroblast activation, and a fibrotic matrix has been shown to promote fibroblast activation in a positive feedback fashion (29, 30). However, this pathway is involved in both normal wound healing and pathologic fibrosis. This is important because inhibitors for matrix signaling including specific antagonist to integrins, FAK, and Rho have all been proposed as potential targets for fibrosis therapy (26, 31, 32). However, nonspecific inhibition of these pathways may concurrently inhibit signaling that suppresses ongoing AEC apoptosis or activation and may further activate fibrogenesis (25).
As injury progresses into a more chronic phase, the provisional matrix is degraded and replaced by a fibrotic matrix primarily made up of fibrillar type I collagen. Although collagen I is often regarded as an end product of fibrosis, it is also a key signaling molecule with a number of cell surface receptors that can influence cell behavior (33, 34). The role of collagen I signaling during fibrosis has not been well studied. Early collagen deposition may therefore have an important role in further collagen production and may help determine the extent of fibrosis.
In this report, we find that type I collagen signaling through integrin α2β1 is critical for promoting fibroblast activation and inhibiting AEC apoptosis in response to TGF-β, leading to conflicting effects on the overall fibrotic outcome. We further find that delivery of an α2β1-activating collagen-mimetic peptide into the pulmonary airway lumenal space inhibits pulmonary fibrosis.
Methods
Reagents
Purified TGF-β1, TC-I15, and obtustatin were purchased from R&D Systems. Antibody against integrin α2 is from Abcam. Type I collagen antibody is from ThermoFisher. Caspase-Glo 3/7 is from Promega. Tissue culture hydrogels of different matrix stiffness are from Matrigen Life Technologies. Small airway growth media (SAGM) is from Lonza. All other reagents are from Sigma. Triple helical collagen activating peptide, GFOGER, or control peptide GPP was purchased from CambCol (Cambridge University).
Mice
All mice were housed under specific pathogen–free conditions. The Institutional Animal Care and Use Committee at The University of Michigan approved all the experiments. CMV/β-actin promoter-CreERT (CAG-CreERT), Col1a2-CreERT, and floxed integrin α2 in a C57b6 background were purchased from Jackson Laboratories. Floxed col1a1 mice, SFTPC-reverse tetracycline transactivator (SPC-rtTA) mice, and CMV promoter tetO-Cre (CMV-tetO-Cre) mice were previously described (25, 35). Mice were used at 6–8 weeks old. For mice carrying the SPC-rtTA/CMV-tetO-Cre transgenes, recombination of the floxed allele was achieved by feeding mice chow supplemented with doxycycline (200 mg/kg) for the times indicated. For mice carrying CAG-CreERT or Col1a2-CreERT transgenes, recombination of the floxed allele was achieved by daily intraperitoneal injections of tamoxifen (1 mg) for the times indicated (36). Littermate control mice lacking at least one component of the multitransgene system were also treated with doxycycline or tamoxifen and were used as controls.
Lung injury and fibrosis was initiated with bleomycin as described before (8, 37). Briefly, mice were given 40 μl of saline or bleomycin (1.5–3 U/kg as indicated) dissolved in saline via oropharyngeal aspiration (38). At the indicated time points after bleomycin, mice were killed for collection of samples.
In some experiments, mice were also treated with daily doses of GFOGER or GPP (20 μg) by oropharyngeal aspiration for the time points indicated.
Masson’s Trichrome Staining
Lung sections were analyzed as described previously (39). Briefly, mice were killed and lungs were inflated with formaldehyde to a pressure of 25 cm H2O. Tissues were embedded in paraffin, sectioned, and stained with Masson’s trichrome by the McClinchey Histology Laboratory.
Hydroxyproline Assay
The hydroxyproline assay was performed as described previously (24, 40). Briefly, entire lungs were isolated and homogenized 3 weeks after bleomycin administration. Tissue was heated in 12N HCl overnight at 120°C. Aliquots of tissue samples were neutralized with citrate buffer and incubated with chloramine T solution at room temperature for 20 minutes. The samples were then mixed with Ehrlich’s solution and incubated at 65°C for 15 minutes. Absorption at 540 nm was determined. Hydroxyproline concentration was quantified by comparison against a standard curve.
Active Caspase 3/7 Assay
Levels of active caspase 3/7 was measured in equal protein amounts from cell or lung lysate using the Caspase-Glo 3/7 Assay, per the manufacturer’s protocol. Briefly, cultured cells were washed with PBS and then lysed in RIPA. For lung lysate, lungs tissue was removed from killed mice and immediately frozen in liquid nitrogen. Frozen lung tissue was then pulverized using a Cryogrinder Kit (OPS Diagnostics) and then lysed RIPA. Samples mixed with the Caspase-Go reagent and luminescence quantified using a Veritas Microplate Luminometer. Values are expressed as fold differences in relative luminescence units.
Immunoblot
Immunoblot was performed on lung tissue or cells lysed in RIPA as previously described (41). Immunoblots are representative of at least three independent experiments.
Decellularized Lung Tissue
IPF lung tissue was obtained at the time of lung biopsy or lung resection of transplantation and normal lung tissue was obtained from lung deemed unsuitable for lung transplantation following procedures approved by the University of Michigan Institutional Review Board. Lung tissue was decellularized as previously described (42, 43). Briefly, human or murine lung tissues were washed with sterile, deionized distilled water. Lung tissue was then incubated at 4°C under gentle agitation with the following sequence of solutions: 0.1% Triton X-100, Na deoxycholate, 1 M NaCl, DNase I in 1.3 mM MgSO4 (30 μg/ml), and 0.18% peracetic acid in 4.8% ethanol for ∼24 hours for each solution. Lungs were then embedded in 3% ultra–low-melting-temperature agarose and sliced on a vibratome. Low-melt agarose was removed by incubating lung slices in water at 50°C and lungs stored in 0.18% peracetic acid in 4.8% ethanol at 4°C until used for cell culture. Prior to cell culture, decellularized lung tissue was washed with sterile water and placed in a sterile multiwell plate and covered with appropriate cell culture media.
Isolation and Culture of Type II AECs and Fibroblasts
Primary murine type II AECs were harvested as described previously (9, 25, 40, 44). Briefly, mice were killed with CO2. Lungs were perfused and lavaged with PBS and then filled with dispase. After incubation at room temperature for 45 minutes, lungs were teased apart and then incubated with DNase for 10 minutes. The digested lungs were then sequentially passed through 70-μm, 40-μm, and 20-μm filters. The crude cell suspension was incubated with biotin-conjugated antibodies against CD16/32 and CD45 for 30 minutes at 37°C followed by incubation with streptavidin magnetic beads. CD16/32- and CD45-positive cells were then removed using a magnetic separator. The remaining cells were further negatively selected by plating on a plastic petri dish for 2 hours at 37°C. AECs were then cultured in a 37°C, 5% CO2 incubator in SAGM (Lonzo) supplemented with KGF (10 ng/ml).
Some AECs were treated with an adenovirus expressing Cre recombinase (50 pfu/cell) or control adenovirus expressing GFP for 1 week before reseeding under the conditions indicated.
Primary murine fibroblasts were isolated as previously described (33). Briefly, mice were killed with CO2. Lungs were perfused and lavaged with PBS and then removed. Lungs were minced and cultured in Dulbecco’s modified Eagle medium with 10% FBS in a 37°C, 5% CO2 incubator to allow fibroblast outgrowth. Fibroblasts were trypsinized and filtered as above to isolate fibroblasts from remaining lung tissue and seeded under the conditions described.
Equal numbers of AECs or fibroblasts were cultured on tissue culture hydrogels of defined matrix stiffness (2 kPa or 50 kPa) coated with laminin or collagen (10 mg/ml). Twenty-four hours after seeding, cells were washed with PBS and the media replaced with serum-free media (Dulbecco’s modified Eagle medium for fibroblasts and SAGM for AECs). After another 24 hours, cells were treated with TGFβ (4 ng/ml) for 24 hours before analysis. In some experiments, AECs were pretreated with GFOGER (10 μg/ml), GPP (10 μg/ml), obtustatin (2.5 μM), TC-I15 (2.5 μM), or vehicle control for 1 hour before treatment with TGF-β. This dose of TGF-β is consistent with prior recent reports (11, 45–50).
Gene Expression Analysis
Differences in gene expression were measured by qRT-PCR as described before (10, 51). Briefly, RNA was isolated from cells or lung tissues with TRIzol Reagent (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized with SuperScript III first-stand synthesis kit (Invitrogen). RT-PCR was quantified using POWER SYBR Green PCR MasterMix kit (Applied Biosystems) and the Applied Biosystems 7000 sequence detection system. The fold change in gene expression was normalized to the housekeeping genes β-actin and GAPDH. Primer sequences were as follows: β-actin forward: 5′-CCGTGAAAAGATGACCCAGATC-3′, β-actin reverse: 5′-CACAGCCTGGATGGCTACGT-3′; GAPDH forward: 5′-AACTTTGGCATTGTGGAAGG-3′, GAPDH reverse: 5′-ACACATTGGGGGTAGGAACA-3′; COL1A1 forward: 5′-GCCAAGAAGACAAACTTT-3′, COL1A1 reverse: 5′-GGCCTTGGAAACCTTGTGGAC-3′; ACTA2 forward: 5′AGAGACTCTCTTCCAGCCATC-3′, ACTA2 reverse: 5′-GACGTTGTTAGCATAGAGATC-3′.
Statistical Analysis
Data are shown as individual values plots ± SD. For comparison of differences, the two-tailed Student’s t test was used for experiments between two groups, assuming equal variance, and a one-way ANOVA was used to determine differences for experiments with more than two groups. A P value of less than 0.05 was accepted as significant.
Results
Newly Synthesized Type I Collagen Regulates Fibroblast Activation and AEC Apoptosis
Fibrosis is characterized by AEC death and fibroblast activation leading to changes in ECM composition and stiffness. To determine the role of collagen I on AEC death and fibroblast activation, primary murine cells (AECs or lung fibroblasts) were cultured on compliant (2 kPa) and stiff (50 kPa) hydrogels coated with laminin, a major component of the alveolar basement membrane, or type I collagen, a major component of the fibrotic matrix. Cells were treated with TGF-β1 (4 ng/ml). After 24 hours, profibrotic activation of lung fibroblasts in response to TGF-β was determined by qPCR for makers of activation, ACTA2 (acta) and collagen 1a1 (col1a1). Fibroblasts cultured on a stiff collagen I matrix had greater myofibroblast activation in response to TGF-β compared with fibroblasts cultured on a softer laminin matrix (Figures 1A and 1B). Fibroblasts did not demonstrate significant changes in caspase activation under these conditions (not shown). The apoptotic response of AECs was determined by measuring the levels of active caspase 3/7, a canonical final common pathway for apoptosis. AECs cultured on stiff type I collagen were more resistant to TGF-β–induced apoptosis compared with AECs cultured on a softer laminin matrix (Figure 1C).
Figure 1.

Collagen augments TGF-β (transforming growth factor-β)–induced fibroblast activation and inhibits TGF-β–induced alveolar epithelial cell apoptosis in vitro. (A and B) Primary lung fibroblasts were cultured on hydrogels of defined stiffness (Matrigen Life Technologies) coated with laminin (Lam) or collagen I (Col) (10 μg/ml), and relative expression of ACTA2 (Acta) (A) and collagen I (Col1a1) (B) in response to TGF-β was determined. n = 6. *P < 0.05 compared with fibroblasts cultured on 2 kPa Lam with TGF-β. **P < 0.05 compared with fibroblasts on cultured on 50 kPa Lam with TGF-β and compared with fibroblasts cultured on 2 kPa Col with TGF-β. (C) Alveolar epithelial cells (AECs) were cultured on Lam- and Col-coated hydrogels and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs on Lam 2 kPa without TGF-β and AECs on 2 kPa Col with TGF-β. **P < 0.05 TGF-β–treated AECs on 2 kPa Lam.
To extend these results to a more complex lung matrix, we used decellularized lung matrix from mice 3 weeks after treatment with saline or bleomycin (3 U/kg) to induce lung fibrosis. Fibroblasts cultured on fibrotic lung matrix had more robust activation in response to TGF-β compared with fibroblasts cultured on normal lung matrix (Figures 2A and 2B). Similarly, fibroblasts cultured on decellularized IPF lung matrix had greater activation in response to TGF-β compared with fibroblasts cultured on normal human lung matrix (Figures 2C and 2D), suggesting a feed-forward, profibrotic effect of fibrotic lung, consistent with prior reports (30). In contrast, AECs cultured on uninjured mouse or human lung matrix had a robust apoptotic response to TGF-β compared with an attenuated apoptotic response by AECs cultured on fibrotic lung matrix (Figures 2E and 2F).
Figure 2.

Fibrotic lung matrix augments TGF-β–induced fibroblast activation and inhibits TGF-β–induced alveolar epithelial cell apoptosis in vitro. (A and B) Primary lung fibroblasts were cultured on decellularized lung matrix from saline- or bleomycin-treated mice, and relative expression of Acta (A) and Col1a1 (B) in response to TGF-β was determined. n = 6. *P < 0.05 compared with fibroblasts cultured on decellularized lung matrix from saline-treated mice and TGF-β–treated fibroblasts cultured on decellularized lung matrix from bleomycin-injured mice. (C and D) Primary lung fibroblasts were cultured on normal human decellularized lung matrix or decellularized lung matrix from patients with idiopathic pulmonary fibrosis (IPF) and relative expression of Acta (C) and Col1a1 (D) in response to TGF-β was determined. n = 6. *P < 0.05 compared with fibroblasts cultured on normal human lung matrix and TGF-β–treated fibroblasts cultured on IPF lung matrix treated with TGF-β. (E) AECs were cultured on decellularized lung matrix from saline- or bleomycin-treated mice, and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs on uninjured lung matrix and TGF-β–treated AECs cultured on bleomycin-injured lung matrix. (F) AECs were cultured on normal human decellularized lung matrix or on decellularized lung matrix from patients with IPF, and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs on normal human lung matrix and TGF-β–treated AECs cultured on IPF lung matrix.
To determine the role of newly synthetized type I collagen on AEC apoptosis, we generated mice with conditional generalized deletion of the col1a1 gene. Previously reported floxed col1a1 mice (10) were crossed with mice carrying a gene for tamoxifen-inducible cre regulated by the CMV-β-actin promoter (CAG-CreERT). Six-week-old CAG-CreERT/floxed col1a1 mice (vs. littermate controls lacking either transgene) were treated with tamoxifen (1 mg daily i.p. injections) for 1 week to eliminate new collagen I synthesis in all cell types (Figure 3A). After an additional recovery week, mice were injured with bleomycin or saline control. Fourteen days after bleomycin treatment, lungs were lysed and analyzed by immunoblot (Figure 3B). As expected, control mice treated with bleomycin had an increase in lung type I collagen accumulation whereas CAG-CreERT/floxed col1a1 mice did not have an increase in lung type I collagen. Seven and 14 days after bleomycin, total lung active caspase 3/7 was determined as a measure of apoptosis (Figure 3C). Both control mice and CAG-CreERT/floxed col1a1 mice demonstrated an increase in active caspase 3/7 7 days after bleomycin, indicating robust bleomycin-induced apoptosis. However, 14 days after bleomycin, CAG-CreERT/floxed col1a1 mice had persistent levels of apoptosis whereas levels of active caspase 3/7 in control mice were significantly reduced. Furthermore, CAG-CreERT/floxed col1a1 mice demonstrated a non–statistically significant trend toward less survival after bleomycin compared with littermate control mice with preserved collagen expression (Figure 3D). Finally, lungs from CAG-CreERT/floxed col1a1 and littermate control mice were removed, decellularized, and used to culture wild-type (WT) primary cells. As expected, AECs cultured on control fibrotic lungs were protected from TGF-β–induced apoptosis. Lung matrix from CAG-CreERT/floxed collagen mice treated with bleomycin demonstrated a dramatically reduced ability to inhibit AEC apoptosis (Figure 3E). Fibroblasts cultured on fibrotic lungs demonstrated enhanced upregulation of acta and col1a1, but fibroblasts cultured on lung matrix from mice with deletion of collagen and bleomycin had reduced levels of acta and col1a1 (Figure E1 in the data supplement). Collectively these results indicate that type I collagen signaling promotes fibroblast activation but inhibits AEC apoptosis in response to TGF-β.
Figure 3.

Type I collagen regulates in vivo lung cell apoptosis. (A) Schematic of mice with postnatal deletion of col1a1. Postnatal deletion of col1a1 is achieved in transgenic mice carrying a gene for tamoxifen-inducible cre regulated by the CMV-β-actin promoter (CAG-CreERT) and homozygous for floxed col1a1 (Δcol1a1). Six- to 8-week-old mice are treated with intraperitoneal tamoxifen (1 mg) for 1 week. (B) Immunoblot of lung lysate from CAG-CreERT/floxed col1a1 or littermate control lacking one of the transgenes 2 weeks after treatment with saline or bleomycin. (C) Seven and 14 days after bleomycin or saline, whole lung active caspase 3/7 was measured in Δcol1a1 and littermate control mice. n = 6. *P < 0.05 compared with bleomycin control mice. (D) Percent survival of control and Δcol1a1 mice after bleomycin (n = 18). (E) AECs were cultured on decellularized lung matrix from saline- or bleomycin-treated control or Δcol1a1 mice, and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs on lung matrix from control mice treated with bleomycin. bleo = bleomycin; CMV = cytomegalovirus; sal = saline.
α2 Integrin Is a Critical Regulator of AEC Apoptosis In Vitro
Whereas the role of ECM receptors in regulating fibroblast activation has been studied fairly extensively, less is known about the function of collagen receptors in regulating AEC apoptosis. We recently reported that ECM/integrin signaling is a critical regulator of AEC apoptosis (8, 25). We therefore investigated the role of two major collagen-binding integrins, α1β2 and α2β1, on apoptosis of AECs cultured on collagen-coated hydrogels. AECs treated with an α2 integrin inhibitor, TC-I15 (2.5 μM) (33), demonstrated marked upregulation of TGF-β–induced apoptosis, whereas an inhibitor of α1 integrin, obtustatin (2.5 μM) (52), had a reduced effect (Figure 4A). We next confirmed an important role for α2 integrin in regulating AEC apoptosis using mice with conditional deletion of α2 integrin (floxed α2). Primary AECs from floxed α2 mice were treated with adenovirus expressing Cre or GFP control, and loss of α2 integrin expression after 5 days was confirmed by immunoblot (Figure 4B). AECs were then cultured on stiff (50 kPa) collagen-coated hydrogels and treated with TGF-β as before. Similar to AECs treated with the α2 inhibitor, AECs with loss of α2 expression demonstrated marked increase in apoptosis in response to TGF-β (Figure 4C). Finally, AECs with or without deletion of α2 integrin were cultured on uninjured or fibrotic decellularized lung matrix (from bleomycin-injured WT mice) and treated with TGF-β. As expected, AECs with loss of α2 integrin had a robust increase in apoptosis in response to TGF-β (Figure 4D). Collectively, these results suggest that collagen deposition during fibrosis inhibits AEC apoptosis through collagen I/α2 integrin signaling. Furthermore, we recently reported that fibroblasts treated with an α2 integrin inhibitor had decreased activation in response to TGF-β in vitro (33), suggesting that α2 integrin is a major regulator of the cellular response to collagen I signaling during fibrosis.
Figure 4.

Integrin α2β1 regulates alveolar epithelial cell apoptosis in vitro. (A) Primary AECs were cultured on collagen I–coated 50-kPa hydrogels and treated with an α1β1 integrin inhibitor, obtustatin (2.5 μM), or an α2β1 inhibitor, TC-I15 (2.5 μM), and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with all other conditions. (B) Immunoblot for α2 integrin from primary AECs from floxed α2 mice after treatment with adenovirus expressing Cre recombinase (AdCre, 50 pfu/cell) compared with cells treated with control adenovirus. (C) Floxed α2 AECs were treated with AdCre or control adenovirus and cultured on collagen I–coated 50-kPa hydrogels, and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with all other conditions. (D) Floxed α2 AECs were treated with AdCre or control adenovirus and cultured on decellularized lung from bleomycin-injured mice and treated with TC-I15 or vehicle control, and activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with control AECs treated with TGF-β. pfu = plaque-forming unit.
α2 Integrin Is a Critical Regulator of Apoptosis and Fibrosis
To explore thein vivo importance of these findings, we generated mice with conditional deletion of α2 integrin. Floxed α2 integrin mice were crossed with mice expressing Cre in all cells (CAG-CreERT), fibroblasts (Col1a2-CreERT), and within AECs (SPC-rtTA/tetO-Cre). Each system enables deletion of α2 integrin after full development by treating mice (or controls) with tamoxifen or doxycycline. As before, 6-week-old mice were treated with tamoxifen or doxycycline for 1 week. After an additional week, mice were injured with saline or bleomycin. Mice with fibroblast-specific deletion of α2 integrin (Col1a2-CreERT) demonstrated a robust reduction in fibrosis after bleomycin injury compared with littermate control mice with preserved α2 expression, indicating an important role of collagen I/α2 signaling within fibroblasts in promoting fibrosis (Figure 5A), consistent with our prior report demonstrating an important role for α2 integrin in promoting fibroblast activation in vitro (33). Surprisingly, mice with total deletion of α2 integrin had fairly preserved levels of fibrosis after bleomycin compared with littermate control mice with intact α2 integrin. In contrast, mice with AEC-specific deletion of α2 integrin demonstrated greater injury and increased fibrosis (Figure 5). Seven days after bleomycin injury, lungs from SPC-rtTA/tetO-Cre/floxed α2 (SCα2) mice had increased levels of active caspase 3/7 compared with littermate control mice (Figure 5B). SCα2 mice also demonstrated increased amounts of protein in the BAL fluid, indicating greater loss of barrier integrity (Figure 5C). In preliminary experiments, there was a trend toward greater death in SCα2 mice by 21 days after bleomycin precluding an assessment of the fibrotic response (not shown). Therefore, a reduced level of bleomycin (1.5 U/kg) was used. Whereas littermate control mice demonstrated a weak fibrotic response to this lower dose of bleomycin, SCα2 mice demonstrated robust fibrosis, indicating an inhibitor effect of AEC α2 on the fibrotic response (Figures 5D–5F).
Figure 5.

Integrin α2β1 regulates profibrotic and antifibrotic responses. (A) Three weeks after intrapulmonary bleomycin (3 U/kg) or saline, collagen content of littermate control mice, mice with deletion of α2 in fibroblasts (col1a2-CreERT), and mice with deletion of α2 in all cell types (CAG-CreERT) was determined by hydroxyproline. n = 7–10. *P < 0.05 compared with littermate control mice treated with bleomycin. (B) Seven days after bleomycin or saline, whole lung active caspase 3/7 was measured in mice with AEC-specific deletion of α2 integrin (SC α2) and littermate control mice. n = 6–9. *P < 0.05 compared with bleomycin control mice. (C) SC α2 mice have greater BAL protein 7 days after bleomycin compared with littermate control mice treated with bleomycin. (D) Three weeks after intrapulmonary low-dose bleomycin or saline, collagen content of littermate control mice and SC α2 mice was assessed by hydroxyproline. n = 6–10. *P < 0.05 compared with littermate control mice treated with bleomycin. (E and F) Three weeks after low-dose bleomycin (1.5 U/kg), the extent of fibrosis was visualized from trichrome-stained lung sections (200×) of littermate control mice (E) and SC α2 mice (F).
Intratracheal Delivery of an α2 Agonist Inhibits Fibrosis
A triple helical peptide based on the collagen I α2 integrin binding site (GFOGER) has previously been shown to promote integrin activation (53). To determine if α2 activation by GFOGER could attenuate TGF-β–induced apoptosis, primary murine AECs were cultured on laminin-coated hydrogels. AECs were treated with GFOGER (10 μg/ml) or a control triple helical collagen peptide GPP. After 2 hours, AECs were then stimulated with or without TGF-β. AECs treated with GFOGER demonstrated reduced apoptosis, similar to levels of apoptosis by AECs cultured on type I collagen–coated wells (Figure 6A). We next cultured AECs on uninjured murine or human lung matrix and treated cells with GFOGER or GPP. Similar to AECs cultured on laminin, AECs treated with GFOGER demonstrated marked inhibition in TGF-β–induced apoptosis similar to AECs cultured on fibrotic lung matrix (Figures 6B and 6C). Finally, WT mice were injured with bleomycin and treated with intrapulmonary delivery of GFOGER (20 μg) or GPP. After 7 days, lung caspase 3/7 levels were measured. As before, bleomycin induced significant apoptosis; however, mice treated with GFOGER demonstrated a significant reduction in apoptosis compared with mice treated with control collagen peptide (Figure 6D). The fibrotic response was determined 21 days after bleomycin and collagen peptide treatment by hydroxyproline and trichrome staining. Whereas mice treated with bleomycin and GPP had a robust fibrotic response, mice treated with GFOGER demonstrated marked reduction in fibrosis (Figures 6E–6G).
Figure 6.
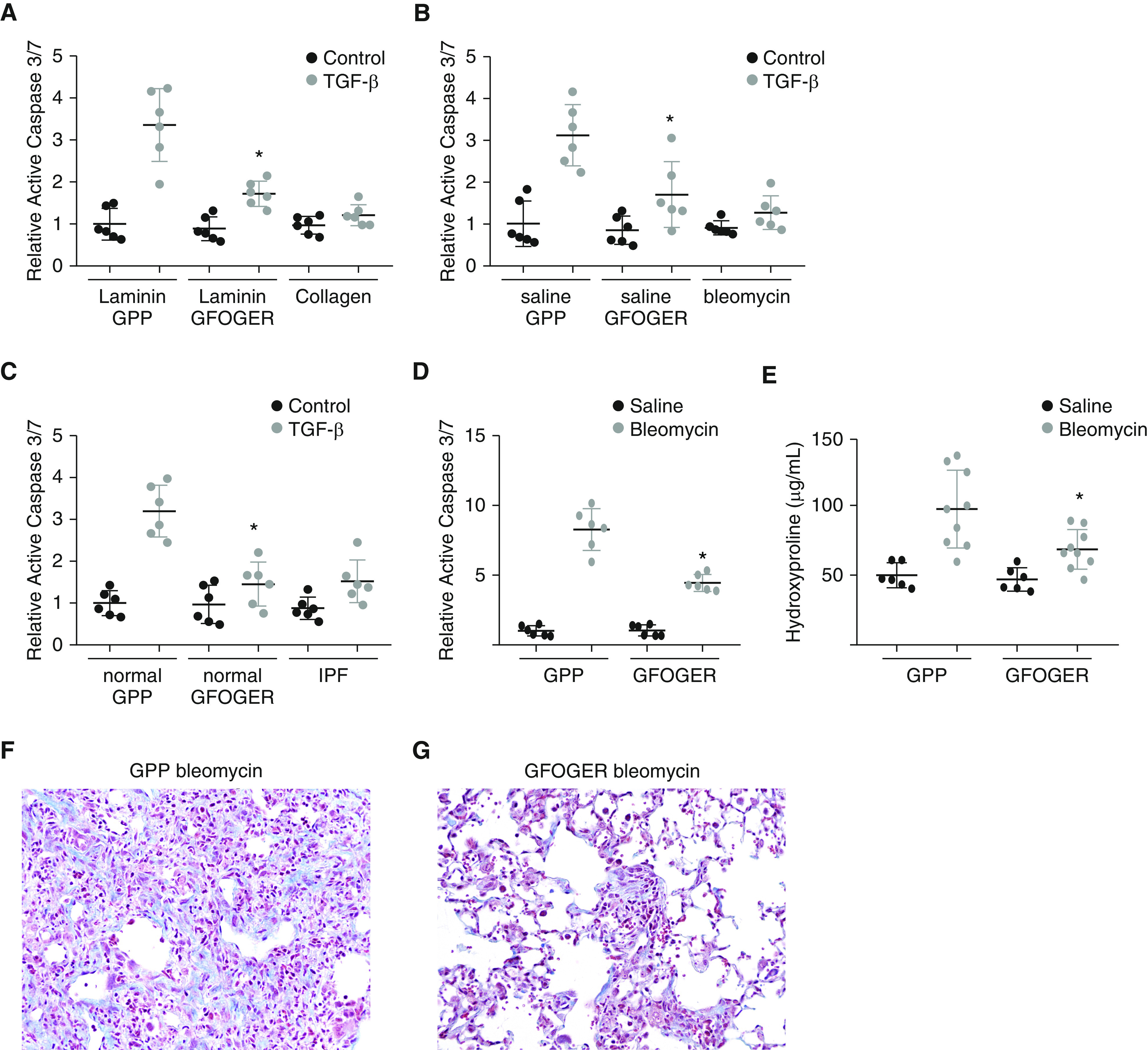
An integrin α2β1–activating collagen peptide blocks alveolar epithelial cell apoptosis and attenuates fibrosis. (A) Primary AECs were cultured on laminin-coated 2-kPa hydrogels and treated with an α2β1-activating collagen triple helical peptide (GFOGER) or a control collagen triple helical peptide (GPP), and the activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs cultured on laminin and treated with GPP and TGF-β. Response by AECs cultured on collagen-coated hydrogels is shown as a reference. (B) AECs were cultured on normal (saline) mouse lung matrix and treated with GFOGER or GPP, and the activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs cultured on normal lung matrix and treated with GPP and TGF-β. Caspase activation by AECs cultured on fibrotic (bleomycin) lung matrix is shown as a reference. (C) AECs were cultured on normal human lung matrix and treated with GFOGER or GPP (10 μg/ml), and the activation of caspase 3/7 in response to TGF-β (4 ng/ml) was determined. n = 6. *P < 0.05 compared with AECs cultured on normal lung matrix and treated with GPP and TGF-β. Caspase activation by AECs cultured on IPF lung matrix is shown as a reference. (D) Seven days after bleomycin or saline, whole lung active caspase 3/7 was measured in mice treated with daily intrapulmonary doses of GFOGER or GPP (20 μg). n = 6. *P < 0.05 compared with bleomycin GPP mice. (E) Three weeks after intrapulmonary bleomycin (3 U/kg) or saline, collagen content of mice treated with GPP or GFOGER was assessed by hydroxyproline. n = 6–9. *P < 0.05 compared with bleomycin GPP-treated mice. (F and G) Three weeks after bleomycin (3 U/kg), the extent of fibrosis was visualized from trichrome-stained lung sections (200×) of mice treated with GPP (F) or GFOGER (G).
Discussion
Collagen I is often regarded as an end product of fibrosis, but we and others have found that collagen I expression increases early after injury (10, 54–56) and leads to activation of collagen receptors (10), potentially influencing cell behavior during progressive fibrosis. Thus, type I collagen deposition itself may be important in determining both the resolution of injury and the progression of fibrosis. In this report, we find that type I collagen signaling regulates AEC apoptosis. AEC injury and apoptosis are believed to be critical for the initiation of scarring and progressive fibrosis (15, 23, 24). Indeed, some degree of fibrillar collagen scarring after injury may be a necessary physiologic response that limits ongoing injury and AEC death and ultimately brings closure to the injury and fibrosis cycle. In fibrotic diseases, this scarring process is aberrant or excessive. Attempts to block fibrosis without understanding how scar formation could limit ongoing injury may be problematic. Consistent with this notion, we found that mice with generalized deletion of new collagen synthesis had greater AEC apoptosis and greater death after bleomycin injury.
We previously found that provisional matrix proteins such as vitronectin and fibronectin likely signaling through integrins such as αvβ3 regulate AEC resistance to TGF-β–mediated apoptosis (8). Downstream integrin signaling molecules such as FAK and Rho are critical to this survival effect. This is problematic because an ECM/integrin/FAK/Rho pathway is also known to be a strong driver of fibroblast activation in response to TGF-β, and there has been some consideration in using inhibitors to this pathway as a therapeutic strategy. In the current report, we extend this argument to collagen I itself creating a potential dilemma in which inhibition of collagen deposition, which is a focus of many fibrosis studies, itself may lead to greater AEC injury and apoptosis, which ultimately may cause continued activation of downstream profibrotic pathways.
A number of cell surface collagen receptors have been identified (34, 57). We previously found that discoidin domain receptor 2, a receptor tyrosine kinase activated by collagen, regulates fibroblast survival and fibrosis. Integrin such as α1β1 and α2β1 are activated by collagens, but the importance of collagen integrin signaling on fibrosis has not been well studied. One report found that α2β1 integrin regulates fibroblast proliferation (58).
The problems with the pleiotropic effects of cytokines and intracellular signaling pathways on different cell types as being problematic for targeting fibrosis are also well described (59). For example, attempts to inhibit TGF-β itself can lead to increased inflammation because of the known effects of TGF-β in suppressing inflammation (60). Apoptosis itself can be pro- or antifibrotic depending on the cell type. For example, we have also shown that enhanced apoptosis of fibroblasts can be targeted to attenuate fibrosis in animal models (37, 61, 62). An emerging concept is that cell type–specific targeting may be required to more effectively inhibit fibrosis. Many intracellular signaling pathways, such as FAK and abl kinase, are shared among different cell types and may have opposing effects on fibrosis in different cell types (25, 27). The lung provides fairly easy access to the intralumenal space, which is lined by epithelial cells. This may offer a means to primarily target epithelial cells.
Collectively, we found that collagen signaling through integrin α2β1 augments fibroblast activation and inhibits AEC apoptosis, which may have opposing effects on fibrosis. We found that airway delivery of an α2 integrin–activating peptide blocks AEC apoptosis and attenuates fibrosis. The potential for airway delivery of therapeutic compounds as a way to favor an effect on epithelial cells requires further investigation.
Supplementary Material
Footnotes
Supported by the U.S. National Institutes of Health grant R01 HL108904 (K.K.K.).
Author Contributions: Conception and design: M.A., M.G., S.J., E.S.W., and K.K.K. Analysis and interpretation: M.A., M.G., S.L., and K.K.K. Drafting of the manuscript: M.A., M.G., and K.K.K.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0150OC on July 21, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Kim KK, Sheppard D, Chapman HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. 2018;10:a022293. doi: 10.1101/cshperspect.a022293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thannickal VJ. Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase. Fibrogenesis Tissue Repair. 2012;5:S23. doi: 10.1186/1755-1536-5-S1-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 2010;65:537–553. doi: 10.1111/j.1398-9995.2009.02305.x. [DOI] [PubMed] [Google Scholar]
- 5.Ogura T, Taniguchi H, Azuma A, Inoue Y, Kondoh Y, Hasegawa Y, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–1392. doi: 10.1183/09031936.00198013. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 7.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 8.Wheaton AK, Velikoff M, Agarwal M, Loo TT, Horowitz JC, Sisson TH, et al. The vitronectin RGD motif regulates TGF-β-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1206–L1217. doi: 10.1152/ajplung.00424.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol. 2014;306:L786–L796. doi: 10.1152/ajplung.00243.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Wheeler SE, Velikoff M, Kleaveland KR, LaFemina MJ, Frank JA, et al. Activated alveolar epithelial cells initiate fibrosis through secretion of mesenchymal proteins. Am J Pathol. 2013;183:1559–1570. doi: 10.1016/j.ajpath.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Agarwal M, Ling S, Teitz-Tennenbaum S, Zemans RL, Osterholzer JJ, et al. Diverse injury pathways induce alveolar epithelial cell CCL2/12, which promotes lung fibrosis. Am J Respir Cell Mol Biol. 2020;62:622–632. doi: 10.1165/rcmb.2019-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cipolla E, Fisher AJ, Gu H, Mickler EA, Agarwal M, Wilke CA, et al. IL-17A deficiency mitigates bleomycin-induced complement activation during lung fibrosis. FASEB J. 2017;31:5543–5556. doi: 10.1096/fj.201700289R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L, Hult EM, Cornell TT, Kim KK, Shanley TP, Wilke CA, et al. Loss of myeloid-specific protein phosphatase 2A enhances lung injury and fibrosis and results in IL-10-dependent sensitization of epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2019;316:L1035–L1048. doi: 10.1152/ajplung.00299.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleaveland KR, Moore BB, Kim KK. Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev Respir Med. 2014;8:163–172. doi: 10.1586/17476348.2014.862154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N, et al. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018;9:1056. doi: 10.1038/s41419-018-1074-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borie R, Kannengiesser C, Nathan N, Tabèze L, Pradère P, Crestani B. Familial pulmonary fibrosis. Rev Mal Respir. 2015;32:413–434. doi: 10.1016/j.rmr.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 17.Cottin V, Cordier JF.SFTPC mutations in patients with familial pulmonary fibrosis: combined with emphysema? Am J Respir Crit Care Med 20111831113author reply 1113–1114. [DOI] [PubMed] [Google Scholar]
- 18.Maitra M, Cano CA, Garcia CK. Mutant surfactant A2 proteins associated with familial pulmonary fibrosis and lung cancer induce TGF-β1 secretion. Proc Natl Acad Sci USA. 2012;109:21064–21069. doi: 10.1073/pnas.1217069110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Marshall H. Researchers identify mutations in familial pulmonary fibrosis. Lancet Respir Med. 2015;3:428. doi: 10.1016/S2213-2600(15)00195-2. [DOI] [PubMed] [Google Scholar]
- 20.Ono S, Tanaka T, Ishida M, Kinoshita A, Fukuoka J, Takaki M, et al. Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur Respir J. 2011;38:861–869. doi: 10.1183/09031936.00143610. [DOI] [PubMed] [Google Scholar]
- 21.Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem. 2007;282:7723–7732. doi: 10.1074/jbc.M610764200. [DOI] [PubMed] [Google Scholar]
- 22.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courey AJ, Horowitz JC, Kim KK, Koh TJ, Novak ML, Subbotina N, et al. The vitronectin-binding function of PAI-1 exacerbates lung fibrosis in mice. Blood. 2011;118:2313–2321. doi: 10.1182/blood-2010-12-324574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheaton AK, Agarwal M, Jia S, Kim KK. Lung epithelial cell focal adhesion kinase signaling inhibits lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2017;312:L722–L730. doi: 10.1152/ajplung.00478.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagares D, Busnadiego O, García-Fernández RA, Kapoor M, Liu S, Carter DE, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vittal R, Zhang H, Han MK, Moore BB, Horowitz JC, Thannickal VJ. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther. 2007;321:35–44. doi: 10.1124/jpet.106.113407. [DOI] [PubMed] [Google Scholar]
- 28.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 29.Hinz B. Matrix mechanics and regulation of the fibroblast phenotype. Periodontol 2000. 2013;63:14–28. doi: 10.1111/prd.12030. [DOI] [PubMed] [Google Scholar]
- 30.Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia S, Agarwal M, Yang J, Horowitz JC, White ES, Kim KK. Discoidin domain receptor 2 signaling regulates fibroblast apoptosis through PDK1/Akt. Am J Respir Cell Mol Biol. 2018;59:295–305. doi: 10.1165/rcmb.2017-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol. 2007;26:146–155. doi: 10.1016/j.matbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Liu T, Gonzalez De Los Santos F, Zhao Y, Wu Z, Rinke AE, Kim KK, et al. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J Biol Chem. 2019;294:8861–8871. doi: 10.1074/jbc.RA118.006615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu B, Wu Z, Nakashima T, Phan SH. Mesenchymal-specific deletion of C/EBPβ suppresses pulmonary fibrosis. Am J Pathol. 2012;180:2257–2267. doi: 10.1016/j.ajpath.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashley SL, Sisson TH, Wheaton AK, Kim KK, Wilke CA, Ajayi IO, et al. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol. 2016;54:482–492. doi: 10.1165/rcmb.2015-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakatos HF, Burgess HA, Thatcher TH, Redonnet MR, Hernady E, Williams JP, et al. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse when compared to intratracheal instillation. Exp Lung Res. 2006;32:181–199. doi: 10.1080/01902140600817465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, et al. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol. 2014;193:5229–5239. doi: 10.4049/jimmunol.1400753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horowitz JC, Tschumperlin DJ, Kim KK, Osterholzer JJ, Subbotina N, Ajayi IO, et al. Urokinase plasminogen activator overexpression reverses established lung fibrosis. Thromb Haemost. 2019;119:1968–1980. doi: 10.1055/s-0039-1697953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghaedi M, Calle EA, Mendez JJ, Gard AL, Balestrini J, Booth A, et al. Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J Clin Invest. 2013;123:4950–4962. doi: 10.1172/JCI68793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warsinske HC, Wheaton AK, Kim KK, Linderman JJ, Moore BB, Kirschner DE. Computational modeling predicts simultaneous targeting of fibroblasts and epithelial cells is necessary for treatment of pulmonary fibrosis. Front Pharmacol. 2016;7:183. doi: 10.3389/fphar.2016.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Liang Y, Zeng X, Yang X, Xu D, Gou X, et al. Long noncoding RNA FENDRR exhibits antifibrotic activity in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2020;62:440–453. doi: 10.1165/rcmb.2018-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santos DM, Pantano L, Pronzati G, Grasberger P, Probst CK, Black KE, et al. Screening for YAP inhibitors identifies statins as modulators of fibrosis. Am J Respir Cell Mol Biol. 2020;62:479–492. doi: 10.1165/rcmb.2019-0296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rana T, Jiang C, Liu G, Miyata T, Antony V, Thannickal VJ, et al. PAI-1 regulation of TGF-β1-induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am J Respir Cell Mol Biol. 2020;62:319–330. doi: 10.1165/rcmb.2019-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scruggs AM, Grabauskas G, Huang SK. The role of KCNMB1 and BK channels in myofibroblast differentiation and pulmonary fibrosis. Am J Respir Cell Mol Biol. 2020;62:191–203. doi: 10.1165/rcmb.2019-0163OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato K, Logsdon NJ, Shin YJ, Palumbo S, Knox A, Irish JD, et al. Impaired myofibroblast dedifferentiation contributes to nonresolving fibrosis in aging. Am J Respir Cell Mol Biol. 2020;62:633–644. doi: 10.1165/rcmb.2019-0092OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nanri Y, Nunomura S, Terasaki Y, Yoshihara T, Hirano Y, Yokosaki Y, et al. Cross-talk between transforming growth factor-β and periostin can be targeted for pulmonary fibrosis. Am J Respir Cell Mol Biol. 2020;62:204–216. doi: 10.1165/rcmb.2019-0245OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Velikoff M, Agarwal M, Disayabutr S, Wolters PJ, Kim KK. Overexpression of inhibitor of DNA-binding 2 attenuates pulmonary fibrosis through regulation of c-Abl and Twist. Am J Pathol. 2015;185:1001–1011. doi: 10.1016/j.ajpath.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown MC, Staniszewska I, Del Valle L, Tuszynski GP, Marcinkiewicz C. Angiostatic activity of obtustatin as alpha1beta1 integrin inhibitor in experimental melanoma growth. Int J Cancer. 2008;123:2195–2203. doi: 10.1002/ijc.23777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farndale RW. Collagen-binding proteins: insights from the Collagen Toolkits. Essays Biochem. 2019;63:337–348. doi: 10.1042/EBC20180070. [DOI] [PubMed] [Google Scholar]
- 54.Deheinzelin D, Jatene FB, Saldiva PH, Brentani RR. Upregulation of collagen messenger RNA expression occurs immediately after lung damage. Chest. 1997;112:1184–1188. doi: 10.1378/chest.112.5.1184. [DOI] [PubMed] [Google Scholar]
- 55.Clark JG, Milberg JA, Steinberg KP, Hudson LD. Type III procollagen peptide in the adult respiratory distress syndrome: association of increased peptide levels in bronchoalveolar lavage fluid with increased risk for death. Ann Intern Med. 1995;122:17–23. doi: 10.7326/0003-4819-122-1-199501010-00003. [DOI] [PubMed] [Google Scholar]
- 56.Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury: pathogenetic and prognostic significance. Am J Respir Crit Care Med. 1997;156:840–845. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- 57.Leitinger B. Transmembrane collagen receptors. Annu Rev Cell Dev Biol. 2011;27:265–290. doi: 10.1146/annurev-cellbio-092910-154013. [DOI] [PubMed] [Google Scholar]
- 58.Xia H, Seeman J, Hong J, Hergert P, Bodem V, Jessurun J, et al. Low α(2)β(1) integrin function enhances the proliferation of fibroblasts from patients with idiopathic pulmonary fibrosis by activation of the β-catenin pathway. Am J Pathol. 2012;181:222–233. doi: 10.1016/j.ajpath.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim KK, Sisson TH, Horowitz JC. Fibroblast growth factors and pulmonary fibrosis: it’s more complex than it sounds. J Pathol. 2017;241:6–9. doi: 10.1002/path.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 61.Ajayi IO, Sisson TH, Higgins PD, Booth AJ, Sagana RL, Huang SK, et al. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol. 2013;49:86–95. doi: 10.1165/rcmb.2012-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. 2015;185:969–986. doi: 10.1016/j.ajpath.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.