

Abstract
Rationale: Animal models have been highly informative for understanding the characteristics, onset, and progression of cystic fibrosis (CF) lung disease. In particular, the CFTR−/− rat has revealed insights into the airway mucus defect characteristic of CF but does not replicate a human-relevant CFTR (cystic fibrosis transmembrane conductance regulator) variant.
Objectives: We hypothesized that a rat expressing a humanized version of CFTR and harboring the ivacaftor-sensitive variant G551D could be used to test the impact of CFTR modulators on pathophysiologic development and correction.
Methods: In this study, we describe a humanized-CFTR rat expressing the G551D variant obtained by zinc finger nuclease editing of a human complementary DNA superexon, spanning exon 2–27, with a 5′ insertion site into the rat gene just beyond intron 1. This targeted insertion takes advantage of the endogenous rat promoter, resulting in appropriate expression compared with wild-type animals.
Measurements and Main Results: The bioelectric phenotype of the epithelia recapitulates the expected absence of CFTR activity, which was restored with ivacaftor. Large airway defects, including depleted airway surface liquid and periciliary layers, delayed mucus transport rates, and increased mucus viscosity, were normalized after the administration of ivacaftor.
Conclusions: This model is useful to understand the mechanisms of disease and the extent of pathology reversal with CFTR modulators.
Keywords: cystic fibrosis, mucus, ivacaftor, rat model, G551D
At a Glance Commentary
Scientific Knowledge on the Subject
New CFTR (cystic fibrosis transmembrane conductance regulator) modulators have reached patients with cystic fibrosis, improving lung function and quality of life. Effects of these drugs, including ivacaftor, on more complicated mechanisms of lung disease are still unclear and require new animal models to study these effects in mechanistic detail.
What This Study Adds to the Field
We have generated a new rat model of cystic fibrosis to address the mechanistic role of CFTR modulator therapy by inserting the human Cftr complementary DNA sequence harboring a G551D mutation into the rat genome downstream of the endogenous Cftr promoter. The resultant CFTR is appropriately expressed and responds to the modulator ivacaftor with increased tracheal currents, restored mucociliary transport, and normalized airway mucus viscosity.
Cystic fibrosis (CF) is a genetic disorder caused by disruption of the Cftr (cystic fibrosis transmembrane conductance regulator) gene, which is expressed in multiple organ systems (1). The absence of CFTR-dependent chloride and bicarbonate transport (2) causes widespread pathology, but lung disease remains the most common cause of morbidity and mortality (3). Although in vitro model systems of CF have provided valuable mechanistic data regarding CF pathophysiology (4, 5) and supported the development of new therapeutics (6–8), the complicated nature of this disease necessitates animal models to fully investigate the underlying mechanisms.
Since the first mouse with disruption to the Cftr gene was created (9), the CF research community has continued to generate new animal models using higher-order species with increased gene homology and similar airway anatomy (10–12), new murine models with patient-relevant mutations (13), or humanized wild-type (WT) Cftr (14). The rat offers many advantages and has been used for evaluation of complex disease phenotypes (15–18). The CF rat lung steadily develops decreased mucus transport (MCT) and increased mucus viscosity (15, 18). The progressive nature allows for mechanistic investigation into the development of mucus abnormalities.
Recent advances include the approval of CFTR modulators (small molecules that rescue the function of the CFTR protein). Ivacaftor (VX-770) was the first to be approved, targeting patients with G551D and other gating variants (6, 19, 20). Ivacaftor acts as a potentiator, augmenting surface-localized CFTR channel activity (6). Despite clinical efficacy, questions remain about the long-term effects of these drugs on CF pathophysiology (21, 22). An animal model that exhibits CF pathophysiology and is responsive to ivacaftor is needed to investigate the chronic effects of modulator therapy and to predict how patients might respond to novel CFTR modulators.
In the present study, we have addressed an unmet need by creating a new CF rat model that expresses humanized G551D-Cftr. By inserting a human complementary DNA (cDNA) Cftr sequence that encompassed exons 2–27 into the rat gene after intron 1, we take advantage of the endogenous promotor to achieve appropriate protein and tissue expression, which was determined by analysis of the lung and tracheal tissues. We have generated a novel model that expresses the same phenotypic characteristics as the original knockout (KO) rat and responds to ivacaftor (VX-770), providing a framework to reveal key elements of the effects of CFTR modulation on CF pathophysiology. We also have established the activity of a large human cDNA Cftr as a means to apply gene editing and correct a diversity of disease-causing variants.
Methods
Full experimental details are provided in the online supplement.
Model Generation
The hCFTR G551D rat model was designed and generated by Horizon Discovery (formerly SAGE Labs; now part of Envigo) using ZFN (zinc finger nuclease)-based technology. Full details are in the online supplement. The donor DNA plasmid, together with the selected pair of Cftr ZFN mRNA, was microinjected into pronuclei of fertilized one cell–stage eggs isolated from superovulated Sprague-Dawley donor female rats. After microinjection, 25–30 eggs were transferred into each pseudopregnant female rat, leading to the birth of the founder.
Cell Culture, Breeding Scheme, Husbandry, Gene Expression, Western Blot, and Immunohistochemistry
See online supplement.
Treatment with Ivacaftor
Rats received ivacaftor, here referred to as VX-770 (Selleckchem). For the experiments analyzing mucus parameters using micro–optical coherence tomography (μOCT), the VX-770 was obtained from Vertex Pharmaceuticals under a material transfer agreement. Rats were dosed for 14 days with VX-770 at 30 mg/kg/day or 3% methylcellulose vehicle by oral gavage.
Tracheal Short-Circuit Current
WT, hG551D, and KO rats between 3 and 6 weeks of age were killed, and short-circuit current (Isc) measurements were performed on excised tracheas (15). Full details are in the online supplement. The administration of amiloride at 100 μM (Sigma-Aldrich) to inhibit the epithelial sodium channel (ENaC) was followed by forskolin 10 at μM (Sigma-Aldrich); VX-770 at 1 nM, 10 nM, 100 nM, 1 μM, and 10 μM; and CFTRInh-172 at 10 μM to block CFTR-dependent current.
Nasal Potential Difference
Rats were anesthetized with ketamine (200 mg/kg), acepromazine (0.6 mg/kg), and xylazine (30 mg/kg). Potential difference was measured using AgCl electrodes and electronic data capture (AD Instruments) (15). Traces were interpreted in a blinded fashion.
μOCT Image Acquisition and Analysis
Functional microanatomic measurements of ex vivo tissue were performed using μOCT (15, 18, 23, 24). The MCT rate was determined using the time elapsed and distance traveled of native particulates in the mucus over multiple frames. Images were acquired at standard distances along the ventral surface, with the optical beam scanned along the longitudinal axis.
Particle-Tracking Microrheology
Particle-tracking microrheological techniques were used to measure the viscosity of mucus on rat tracheae in situ, as previously described. Dynamic viscosity was derived from mean squared displacement by the application of the generalized Stokes-Einstein relation (25).
Statistics
Inferential statistics (mean, SD, and SE) were computed using ANOVA, with an unpaired or paired t test as appropriate. P values of 0.05 were considered significant. Statistics are presented as mean ± SE, except as indicated.
Study Approvals
Animal procedures were approved by the institutional animal care and use committees at University of Alabama at Birmingham and Horizon Discovery.
Results
Validation of a Rat-Human Chimera Gene to Express Cftr
A construct was designed to insert a humanized copy of Cftr cDNA into the rat genome, harboring a G551D variant, targeted to the second exon of the rat sequence. This strategy preserved exon 1 and intron 1 and allowed the human cDNA to be controlled by the endogenous rat Cftr promoter (Figure 1A). Before animal generation, constructs consisting of either human Cftr cDNA or the human cDNA sequence combined with rat exon 1 and intron 1 were stably transfected into Fischer rat thyroid cells. Fischer rat thyroid cells expressing each construct were analyzed for Isc measurements after the addition of the CFTR agonist forskolin, the CFTR potentiator VX-770, and the CFTR inhibitor CFTRInh-172. Responses were identical between the human sequence and the rat + human sequence (Figure 1B) and gave the expected response to VX-770, indicating that the hybrid sequence was likely to result in a functional protein in vivo. Tracings from cells expressing WT Cftr (see Figure E1 in the online supplement) indicate that the G551D construct, in both human and rat + human cells, express minimal CFTR activity at baseline but are correctable to ∼42% of the WT activity.
Figure 1.
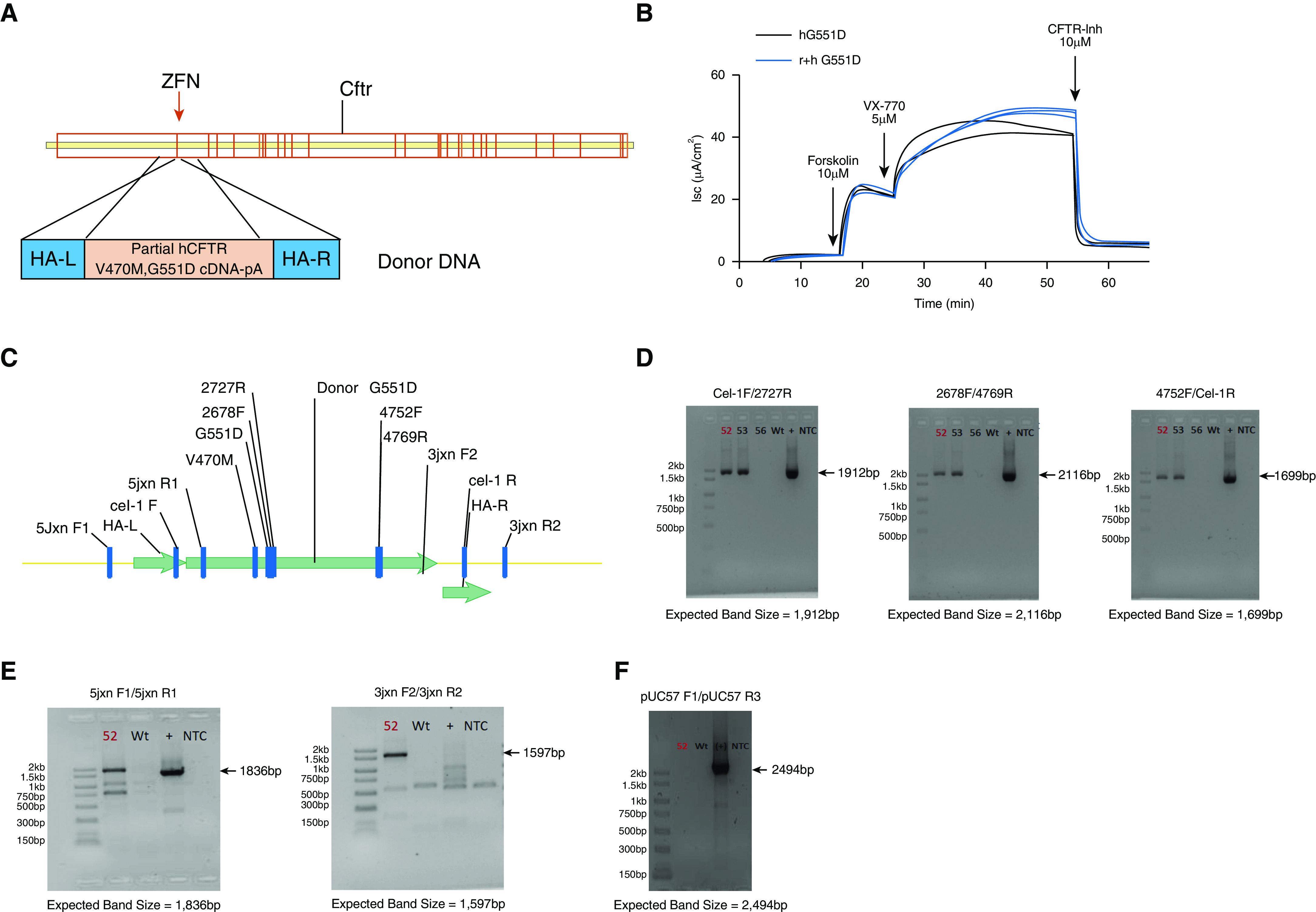
Generation of the model was achieved using a zinc finger nuclease approach. The targeting strategy included zinc finger nuclease–paired targets of the rat Cftr (cystic fibrosis transmembrane conductance regulator) exon 2. (A) Donor DNA contains the human Cftr V470M, G551D complementary DNA without the first exon. (B) This human-rat hybrid (r+h G551D, blue) product was tested in Fischer rat thyroid cells to ensure that the response to forskolin, VX-770, and CFTR-Inh 172 were similar to the human sequence (h G551D, black). (C) Once the construct was inserted into the rat sequence, several sets of primers were designed to test for presence of the complete sequence. (D) Rats that were positive for insertion were identified with primer sets specific for the internal sequence. (E) Targeted insertion was identified using primers specific for the 5′ and 3′ junctions. (F) Primers identifying the vector were used to ensure that none of the vector sequence was inserted as well. Donor number 52 (labeled in each gel) was selected for propagation to F1. Donor number 56 was negative for the targeted insertion, whereas donor number 53 was positive for both targeted and random insertion and was therefore not selected for propagation. cDNA = complementary DNA; HA-L = left homology arm; HA-R = right homology arm; Isc = short-circuit current; NTC = no template control; WT = wild-type; ZFN = zinc finger nuclease.
A Cftr Superexon Insertion Generated a Humanized G551D-CFTR–Expressing Rat Model
A pair of ZFNs were designed to insert the human cDNA sequence (Figure 1C). After the microinjection and embryo transfer, pups were screened using custom primer sets (Figure 1C and Table 1) to identify donor-positive pups and to ensure complete insertion. Additional junction PCR primer sets (Figure 1C and Table 2) were used to confirm the targeted insertion of the superexon. A last set of primers (Table 3) were used to screen for random insertion. A total of 69 pups were screened, and pup 52 (red) and pup 53 were confirmed to be donor positive (Figure 1D) in contrast to a donor-negative pup (pup 56). These numbers of positive pups were expected based on the large size of the genomic insert. Pup 53 was found to contain random insertion; however, pup 52 showed evidence of targeted insertion of the superexon (Figure 1E) without random insertion (Figure 1F) and was selected for propagation. This F0 male generated 11 pups that were positive for the insertion.
Table 1.
Donor-Positive Primer Sets
| Cel-1F |
5′-GCAAAGGCATCGGTTTAGAG |
| 2727R | 5′-GCCATCAGTTTACAGACACAGC |
| Expected band size | 1,912 bp |
| 2678F | 5′-GACTCTCCTTTTGGATACCTAGATG |
| 4769R | 5′-TTGAATCCCAAGACACACCA |
| Expected band size | 2116 bp |
| 4752F | 5′-GGAGAAATCCAGATCGATGG |
| Cel-1R | 5′-GGATTTTCCCTACTGCTGGA |
| Expected band size | 1,699 bp |
Table 2.
Targeted Integration Primer Sets
| 5′ Junction |
|
| 5jxnF1 | TCCTTTTCTTCTCTGCTTTCTTCC |
| 5jxnR1 | AAATGGCTGGGTGTAGGAGC |
| Expected band size | 1,836 bp |
| 3′ Junction | |
| 3jxnF2 | 5′-CCCCCTGAACCTGAAACATA |
| 3jxnR2 | 5′-GGAGCTGAATGGGTTTTCAA |
| Expected band size | 1,597 bp |
Table 3.
Random Integration Primer Sets
| pUC57 F1 |
5′-TATCCGCTCACAATTCCACA |
| pUC57 R3 | 5′-CTGGCGTAATAGCGAAGAGG |
| Expected band size | 2,494 bp |
hG551D Rats Recapitulate the Phenotype Seen in the CFTR-KO Rat
Eleven (four male and seven female) F1 heterozygous rats were used to establish the colony (see breeding scheme, Figure E2). Data are reported from rats that are homozygous for the hG551D insert, termed “hG551D rats.” Litters generated from hG551D heterozygotes were found to have a genotype distribution of 28.3% WT, 46.4% hG551D heterozygous, and 25.3% hG551D homozygous, corresponding closely with Mendelian expectation. Homozygous hG551D pups had reduced survival, beginning at approximately 21 days of life (Figure 2A), even when raised with GoLTYELY to reduce the incidence of gastrointestinal obstruction. However, once hG551D rats reach 40 days of life, the GoLYTELY supplement is sufficient to prevent obstruction, and 100% of surviving hG551D rats reach 120 days of life. Corresponding weights showed that the hG551D rats, similar to the KO, were significantly smaller than the WT rats in both male (Figure 2B) and female (Figure 2C) pups. Rats that did not survive past the first 40 days of life even with Go-LTYELY treatment underwent necropsy. Gross analysis of hG551D rats at death revealed distended, occluded intestines (Figure 2D) compared with those of their WT littermates. Intestinal impaction initiated at the ileocecal junction was the cause of death, a finding that replicates that of the KO rat (15) and is a phenotype seen in the other animal models of CF (9–12). In addition, the hG551D pups replicated the white incisor phenotype (Figure 2E) compared with the normal yellow color seen in the WT pups. This is a finding previously observed (15) and is a rodent-specific manifestation of absent functional CFTR (26).
Figure 2.
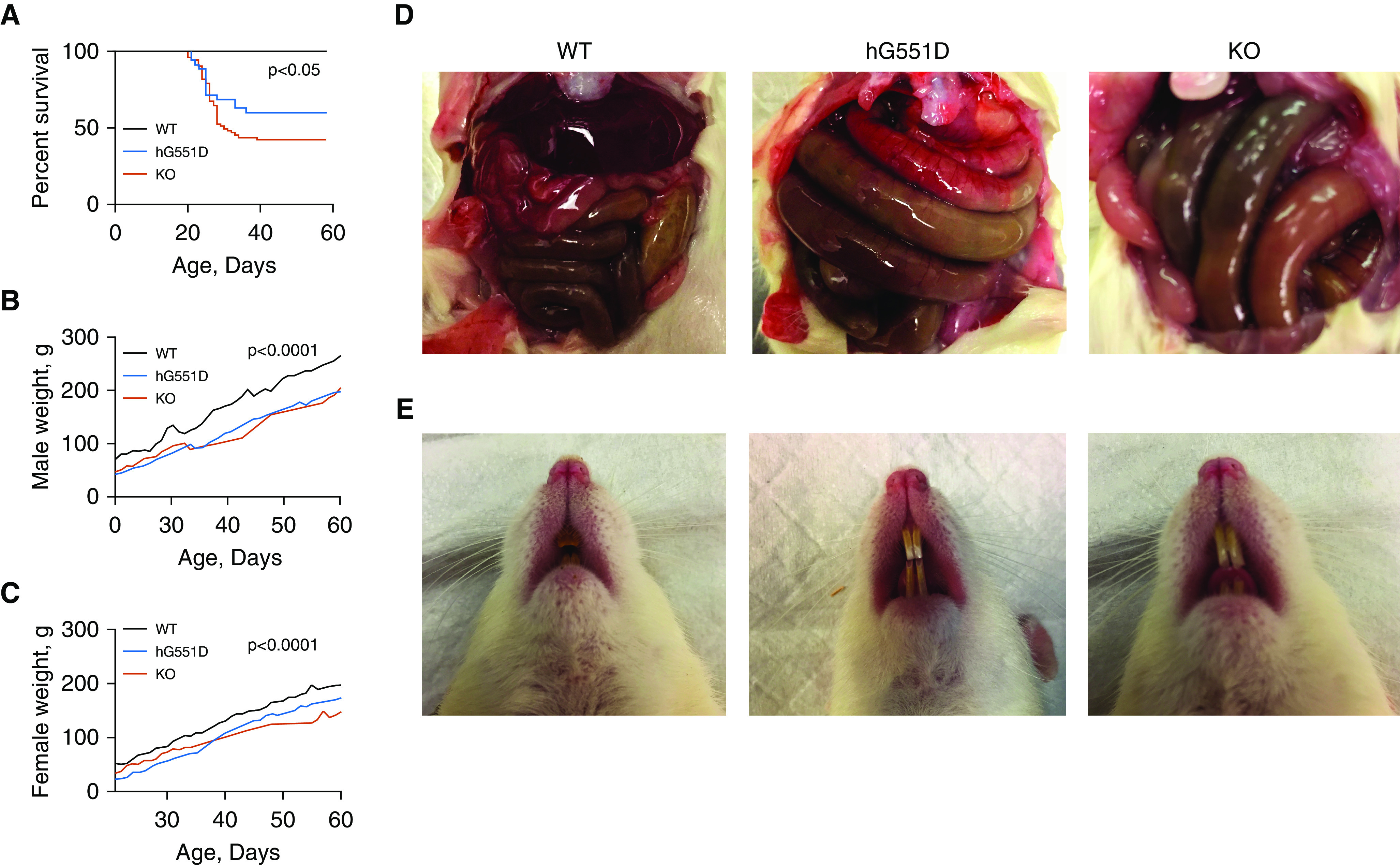
The hG551D homozygous rat closely resembles the CFTR (cystic fibrosis transmembrane conductance regulator)-knockout (KO) intestinal phenotype. (A) Survival analysis of wild-type (WT), hG551D, and CFTR-KO rats to postnatal Day 60 demonstrates increased mortality of both hG551D and CFTR-KO rats, with 47% survival at 60 days in the KO group and 60% survival in the hG551D group. P < 0.05; WT compared with hG551D and WT compared with KO. (B) Weight was tracked from weaning at 21–60 days of age in male pups. (C) Weight was tracked from weaning at 21–60 days of age in female pups. (D) Death in the hG551D rats was largely due to gastrointestinal obstruction, as occurs in the KO rat. (E) Teeth enamel in the hG551D rat appears as the characteristic white overgrown teeth present in the KO rat and dramatically different from the normal WT phenotype. All genotypes received Go-LYTELY supplement from weaning. n = 6/group. Data are presented as means.
Absence of CFTR Activity in the hG551D Airway Is Restored With VX-770
To determine that the inserted human cDNA superexon was expressed appropriately under the endogenous rat promoter, we analyzed lung tissues for Cftr mRNA. Rat Cftr is approximately 75% homologous to the human sequence (27), allowing the use of primer sets that were either species-specific or crossreactive. Comparing the hG551D rats to the WT and KO rats indicated that both models of CF had minimally detectable rat Cftr expression (Figure 3A). The hG551D lung had robust expression of the human mRNA sequence (Figure 3B), and quantitation using the crossreactive primers showed that the overall expression of Cftr in the hG551D rats was similar to that detected in WT rats (Figure 3C). Protein expression using Western blot analysis (Figure 3D) of homogenized lung tissues indicated similar expression of the fully glycosylated CFTR C-band protein in both WT and hG551D rats. No difference in C-band density was observed, reflecting normal steady-state CFTR expression (Figure 3E). KO rats showed a faint band at the molecular weight of the C-band on Western blot, as previously described (15). Density measurements, coupled with functional data, indicate that this does not produce surface-active CFTR protein. To confirm that cell-surface expression matched that of the WT rats, hG551D tracheae were stained for CFTR and cilia. Immunohistochemistry indicated that CFTR was present at the cell surface, similar to the WT expression, but was absent from the KO animals (Figure 3F).
Figure 3.
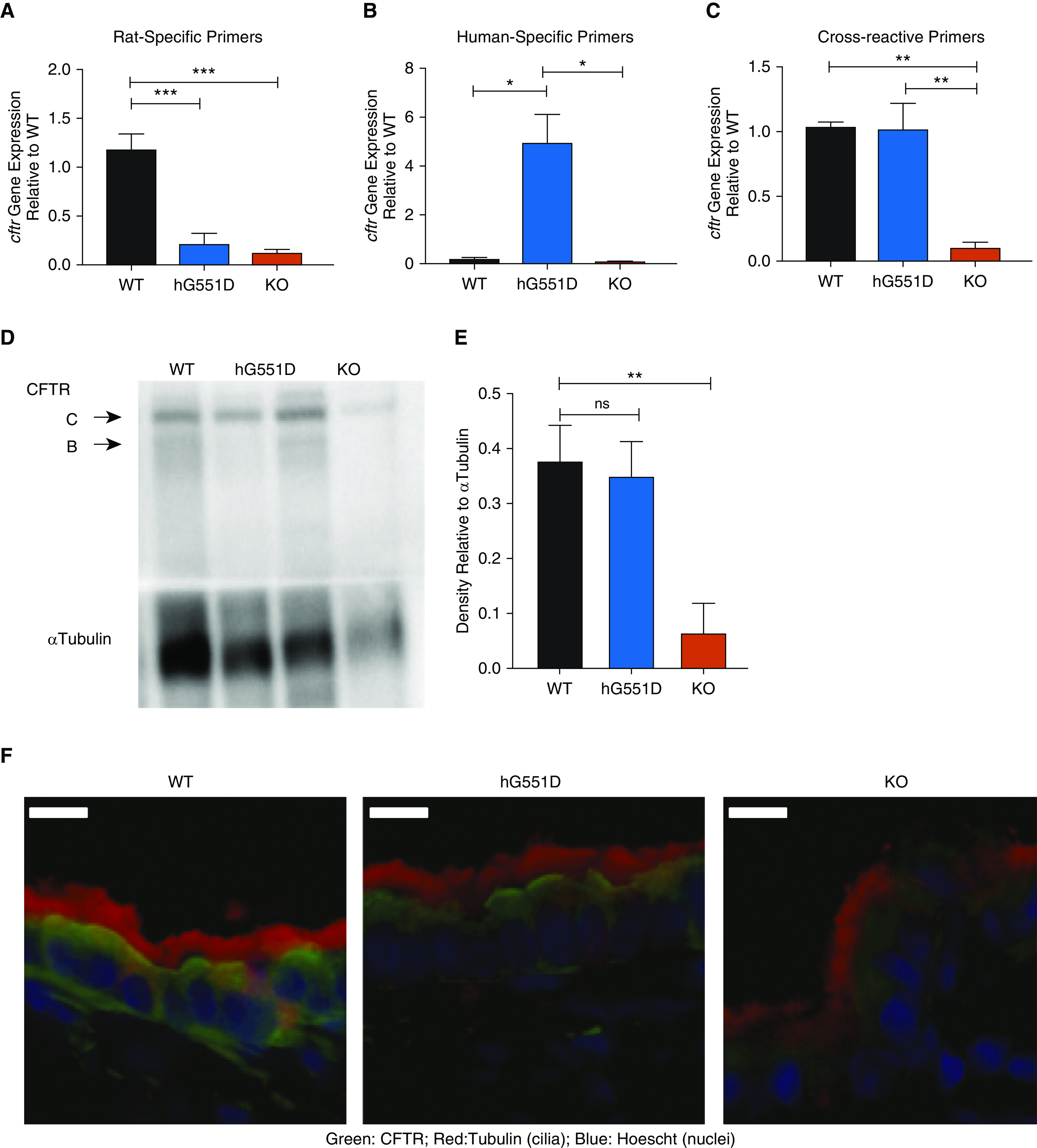
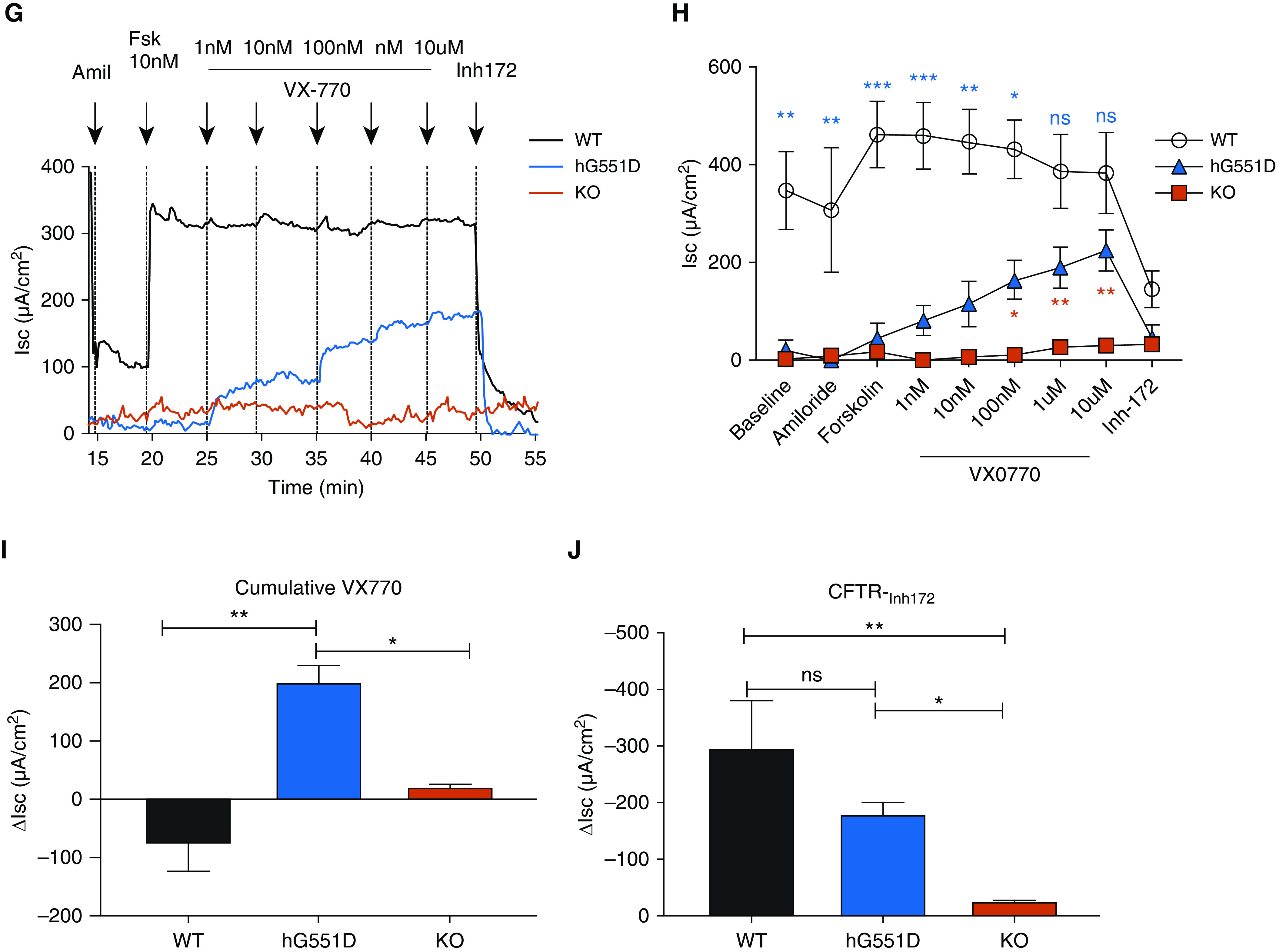
The hG551D-CFTR (cystic fibrosis transmembrane conductance regulator) is expressed and functional in the airway. (A–C) Gene expression analysis of lungs from wild-type (WT), hG551D, and knockout (KO) rat using primers specific to rats (A), humans (B), and both (C) indicate human specificity of the sequence and appropriate expression. (D) Western blot indicates that the protein concentration in the lung is also similar to that of WT rats. (E) Blots were analyzed for density of CFTR bands compared with loading control (α-tubulin). (F) To confirm that hG551D-CFTR localized to the surface of the epithelium, tracheal sections from WT, hG551D, and KO rats were stained with antibodies against CFTR (green), cilia (red), and nuclei (blue). White bar = 10 μm. (G) Short-circuit current (Isc) analysis was performed on trachea, with representative tracings from each genotype. (H) Combined summary data indicate that the hG551D rat has a lower baseline current than the WT rat but responds specifically to increasing doses of VX-770. Statistical comparison between WT and hG551D rats is in blue; comparison between hG551D and KO rats is in red. (I) Δ currents 10 μM VX770 demonstrate a selective response to ivacaftor in the hG551D animal. (J) Δ currents after CFTR-Inh172 indicate different responses between genotypes. The CFTR-Inh172 response indicates that hG551D rats can be potentiated to approximately 50% of WT activity. *P < 0.05, **P < 0.01, and ***P < 0.001. n = 6/group. Amil = amiloride; Fsk = forskolin; ns = not significant.
Isc measurements were performed on tracheae excised from WT, hG551D, and KO rats to assess the functional activity of CFTR. After baseline measurements and amiloride (100 μM) to inhibit the ENaC, the trachea were exposed to the CFTR agonist forskolin (10 nM, the known half maximal effective concentration [EC50] in rats) (15, 28) followed by increasing concentrations of the CFTR potentiator VX-770. Representative tracings (Figure 3G) show that baseline Isc was significantly lower in the hG551D rats compared with WT rats (Figure 3H) but was not statistically different than that of the KO rats. The difference in baseline persisted after the addition of amiloride because of the preactivation of CFTR (15, 28). The Isc change in response to forskolin in the hG551D rats was substantially reduced compared with that in the WT rats and was similar to the Isc change of the KO rats. However, the hG551D rats responded robustly to increasing concentrations of ivacaftor, with a total change of 202.33 ± 28.6 μA/cm2 attributable to VX-770 (Figure 3I). The addition of the CFTR inhibitor-172 indicated that differences in Isc were CFTR dependent and showed that VX-770 increased CFTR activity in the hG551D rats to approximately 50% of WT rats (Figure 3J). These data confirm that the humanized G551D protein is correctly expressed in the airway, exhibits low baseline activity, and can be potentiated by VX-770 to concentrations that resemble the response in primary human bronchial epithelial cells, in which 10 μM VX-770 increased CFTR activity by 48% (6, 29), and in humans (19, 30).
In Vivo Potentiation of hG551D-CFTR Restores Transepithelial Nasal Potential Difference
To assess the utility of hG551D rats to model patients with CF receiving highly effective modulators, we treated the rats with VX-770 systemically in vivo, dosing hG551D rats and KO control rats with 30 mg/kg/d by oral gavage for 14 days beginning at postnatal day 21, which is when the pups were weaned. The 14-day treatment would simultaneously ensure that the drug was at steady-state concentrations, mimic the initial 30-day treatment studies in the human patients (accounting for shortened rodent pharmacokinetics), and be long enough to allow for changes in weight or body condition. Additional hG551D rats and WT littermates were treated with 3% methylcellulose as a vehicle control. After 14 days, rats underwent nasal potential difference (NPD) measurements to assess CFTR activity. Representative tracings of untreated (Figure 4A) and VX-770–treated (Figure 4B) hG551D rats are depicted. The weights of VX-770–treated hG551D rats diverged from those of untreated hG551D rats (Figures 4C and 4D) and approximated the curve of WT rats by the end of the study, indicating that VX-770 prevented the expected decline in growth rate. No differences were detected in mortality or intestinal phenotype, although the change in weight suggests that these effects may be present in a longer-term study. In contrast, VX-770 had no effect in KO rats. NPD measurements were minimally different at baseline between the groups and exhibited no difference in amiloride-sensitive transepithelial potential difference (Figure 4E), as seen previously (15). In contrast, hG551D rats treated with VX-770 exhibited a strong response to CFTR stimulus (chloride-free Ringers + forskolin) compared with those treated with the vehicle (Figure 4F). Systemic CFTR potentiation by ivacaftor restored CFTR activity to 63% of WT in the hG551D rat, which was similar to the effect observed in NPD studies of patients with the G551D variant (19, 30, 31).
Figure 4.

The hG551D rat responds to in vivo treatment with VX-770. VX-770 was dosed by oral gavage to hG551D and knockout rats for 14 days, beginning at weaning, and nasal potential difference was measured. Representative tracings of (A) hG551D + vehicle and (B) hG551D + VX-770 are depicted. (C) Weights of these groups, compared with untreated hG551D and wild-type (WT) rats, indicate increased weight in the last few days of therapy for hG551D + VX-770 rats compared with untreated hG551D rats. (D) The final weight change per group indicates that hG551D rats were able to gain as much weight as WT rats after 14 days of treatment. (E) Summary data of nasal potential difference measurements of WT, hG551D, hG551D + VX-770, and knockout + VX-770 rats at baseline and after amiloride, with Cl− free and Cl− free + 20 μM forskolin. (F) Untreated hG551D rats had a smaller change in potential difference after the Cl− free + forskolin addition compared with WT rats; this was increased in hG551D rats + VX-770. ns = P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. n = 4–6/condition. FSK = forskolin; KO = knockout.
Airway Mucus Defects in the hG551D Rat Are Normalized by VX-770 Treatment
After confirmation of the in vivo effect of VX-770, μOCT imaging was performed to assess the effect on the airway epithelial surface. Previously, we have shown that the KO rat develops abnormal airway surface liquid (ASL) depth, compressed periciliary layer (PCL), delayed MCT rates, and hyperviscous mucus in the large airways (18, 32). The administration of VX-770 restores these parameters in a human bronchial epithelial cell model of CF (29). hG551D rats at 3 months of age, when the rats are past the risk of fatal intestinal obstruction and the mucus abnormalities emerge (18), were treated with VX-770 or vehicle control for 14 days to allow for the drug to reach steady state and the mucus parameters to normalize, and their tracheae were excised and treated with acetylcholine to augment mucus secretion (18). Representative μOCT images of untreated WT rats, hG551D rats treated with vehicle, and hG551D rats treated with VX-770 (Figure 5A) depict the epithelium and the overlying mucus layer. Quantitation demonstrated that ASL depths in the vehicle-treated hG551D rats were significantly depleted but increased with VX-770 administration (Figure 5B) to levels that approached those observed in WT rats. PCL depths were similarly decreased in the vehicle-treated hG551D rats but were restored to normal levels with VX-770 treatment (Figure 5C). MCT rates were delayed in hG551D rats and were augmented in response to VX-770 (Figure 5D). Improvement in MCT suggested the potential for improved viscoelastic properties like those we observed in cells derived from patients with the G551D variant (29). To test this, in situ particle-tracking microrheology was performed in the absence of ciliary movement. Representative particle tracings (Figure 5E) showed that the VX-770 treatment of hG551D rats augmented the Brownian motion of particles in airway mucus compared with that of the vehicle-treated hG551D rats, which was severely restricted compared with that of WT rats. Fourier transforms were used to calculate effective viscosity, which is markedly elevated in untreated hG551D rats but normalized in response to VX-770 (Figure 5F). These data established that VX-770 treatment induced profound improvements in the mucociliary transport apparatus of hG551D rats and that the functional microanatomy approached that observed in untreated WT control rats.
Figure 5.
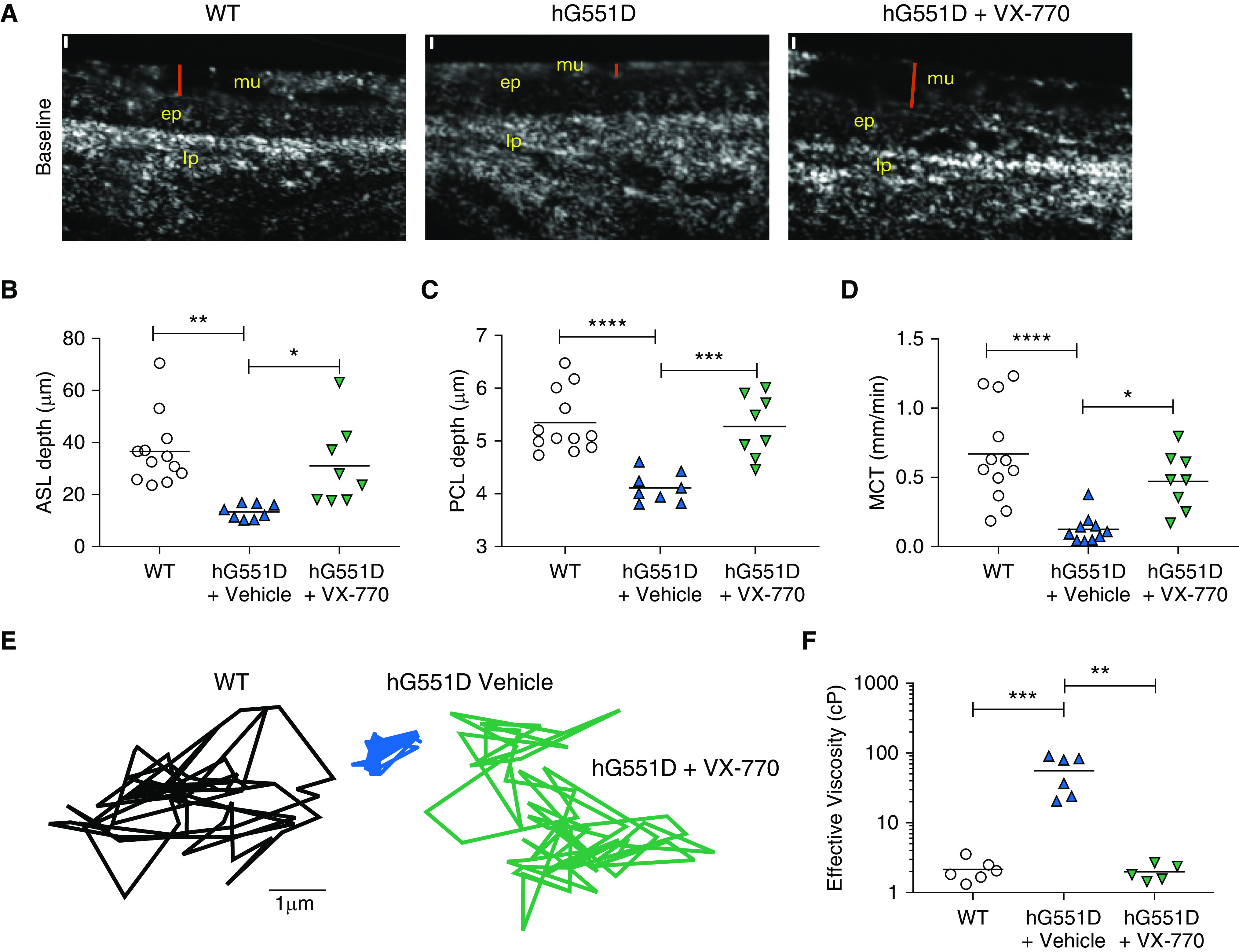
The hG551D airway defect is rescued by VX-770 administration. Tracheae from 3-month-old wild-type and hG551D rats were imaged with μOCT. (A) Representative images of wild-type rats, hG551D rats, and hG551D rats treated with VX-770 depict the surface analyzed (red bar = airway surface liquid). Scale bars (white), 10 μm. Quantitative analysis of the images yields (B) airway surface liquid depth, (C) periciliary layer depth, and (D) mucociliary transport rates. After the ciliary beating was arrested, particles were added to the surface and Brownian motion was tracked. (E) Representative tracings depict freedom of movement in the mucus from each trachea. (F) Conversion of mean squared displacement yielded effective viscosities. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ASL = airway surface liquid; ep = epithelial layer; lp = lamina propria layer; MCT = mucociliary transport; mu = mucus layer; PCL = periciliary layer; WT = wild-type.
Discussion
This report describes the generation of a novel animal model of CF, constructed by inserting a humanized-Cftr superexon into the rat genome, producing an in vivo model that expresses hG551D-CFTR in native context and responds to a CFTR potentiator in a clinically relevant manner (19, 20, 29, 31). Airway tissue from hG551D homozygous rats indicate that gene and protein expression are similar to that seen in WT rats, and functional assessments demonstrate the expected lack of CFTR-dependent activity at baseline. Assessments of the functional microanatomy of the airway surface indicated that the ASL in the hG551D rat is depleted, accompanied by decreased MCT rates and increased viscosity, which is consistent with the fundamental abnormalities of the CF defect (18, 33–35). Each of these parameters was restored when the hG551D rats received ivacaftor. These results provide the first evidence of normalization of MCT and viscosity with CFTR modulators in an animal model.
The most relevant animal models for understanding human physiology are those that most closely resemble the characteristics of disease observed in the human population (5). Here, we have reconstructed both the electrophysiologic phenotype of patients with CF and the phenotype of the epithelial layer. Periciliary layer depletion and viscoelastic abnormalities of the associated mucus layer have long been postulated to be underlying mechanisms of a host of downstream lung sequelae, including inadequate host defense (36) and increased susceptibility to bacterial infections (37), and may be markers for predicting the clinical response to therapeutics (29). Although recent studies have demonstrated the ability of CFTR modulators to prevent disease (38), we show that mucus abnormalities characteristic of CF lung disease, such as delayed MCT and increased mucus viscosity, can be corrected with modulator treatment. This allows new therapeutics, such as mutation-agnostic therapies, to be directly compared with the efficacy seen with highly effective modulators to help predict how these newer drugs might benefit those patients who currently are in the “unmet need” category. Importantly, because the CF patient population will predominantly include patients with highly effective CFTR modulator therapy, animal models that mimic this population will be essential to correctly understand disease progression and potential perturbation moving forward.
The rodent models, although not as close to human physiology as some of the larger animals (39), allow some benefits over the pig and ferret models, such as greater accessibility, better immunologic reagents, and shorter gestation and maturation times, enabling more experiments with higher numbers. Similar to the KO rats previously reported, the hG551D rat is not susceptible to spontaneous infection or overt mucus plugging while kept in specific pathogen-free housing unlike the phenotype seen in the larger animals (40, 41). The lack of elevated ENaC activity noted in the NPD experiments replicates studies seen previously (15) and studies in other animals (42) but contradicts results collected from human cell models (43). Future and ongoing studies will determine whether this difference is relevant to the human airway disease, whether it impacts disease expression in the rat airway, or whether this phenotype offers lessons to therapeutic development. Additional benefits of the rodent model will allow administration of ivacaftor to the hG551D rats from birth to enable studies to understand the long-term effects on organs beyond the lung; because CFTR modulators are approved in infants, these studies may be of interest to help predict the nature of organ pathophysiology after years or decades of modulator treatment. Although CF pathology that is ordinarily present at birth has been prevented with in utero ivacaftor treatment in a G551D-expressing ferret (38), it is still unknown whether life-long modulator therapy will preserve lung function or merely delay the decline or whether these patients will experience altered susceptibilities to bacteria and viruses. Early intervention has long been understood to be critical to this patient population (44), but the long-term sequelae of a corrected severe variant, such as G551D, is still poorly understood. This model will support investigations to predict the more nuanced responses patients might have to modulators, including the long-term effects on infection and inflammation, which have so far been difficult to discern from patient populations (21, 22).
The approach taken here to create the first humanized-Cftr expression rat by the insertion of a superexon into the genome behind the endogenous rat promoter is the first of its kind in CF. The benefit of this approach is that the use of the rat Cftr regulatory elements ensures that the gene retains the correct expression profile in the lung and trachea, as evidenced by the restoration of CFTR-dependent ion channel activity after ivacaftor administration. The insertion of a superexon of Cftr has been explored as a potential therapeutic approach in gene editing in CF because it would enable a mutation-agnostic means of Cftr replacement that could be accomplished in the genome and would result in a functional protein (45). Although this approach has been accomplished in cell lines (46), there is evidence to show that intronic regions are important for the regulation of Cftr expression in different tissues (47). An animal model expressing a superexon would be an ideal setting to test how these tissues regulate Cftr expression in the absence of those intronic regions and may help shed light on eventual efficacy of this approach to gene editing in patients with CF.
Supplementary Material
Acknowledgments
Acknowledgment
The authors acknowledge the assistance of Dezhi Wang and the Histomorphometry and Molecular Analysis Core in the Center for Metabolic Bone Disease.
Footnotes
This work was supported by the NHLBI (1K08HL131867 [S.E.B.]); the National Institute of Diabetes, Digestive and Kidney Diseases (DK072482 [E.J.S. and S.M.R.]); the Cystic Fibrosis Foundation (R464-CF and ROWE19R0); and a compound-only material transfer agreement from Vertex Pharmaceuticals. Funders had no role in the content of the manuscript or decision to publish.
Author Contributions: S.E.B., G.Z., E.J.S., and S.M.R. conceived the experiments. S.E.B., J.M.D., C.M.F.-P., A.G.H., A.M.O., L.T., H.W., J.H., L.F., A.C., and A.F. performed the experiments. S.E.B., C.M.F.-P., J.H., and G.Z. analyzed the data. S.E.B., G.J.T., E.J.S., and S.M.R. contributed reagents, materials, and/or analysis tools. S.E.B., G.Z., E.J.S., and S.M.R. wrote the manuscript. S.E.B., E.J.S., and S.M.R. supervised the project.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202002-0369OC on June 25, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA. 1994;91:5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Boeck K, Vermeulen F, Dupont L. The diagnosis of cystic fibrosis. Presse Med. 2017;46:e97–e108. doi: 10.1016/j.lpm.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Mou H, Brazauskas K, Rajagopal J. Personalized medicine for cystic fibrosis: establishing human model systems. Pediatr Pulmonol. 2015;50:S14–S23. doi: 10.1002/ppul.23233. [DOI] [PubMed] [Google Scholar]
- 5.Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, et al. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. 2018;17:S28–S34. doi: 10.1016/j.jcf.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadida S, Van Goor F, Zhou J, Arumugam V, McCartney J, Hazlewood A, et al. Discovery of N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, ivacaftor), a potent and orally bioavailable CFTR potentiator. J Med Chem. 2014;57:9776–9795. doi: 10.1021/jm5012808. [DOI] [PubMed] [Google Scholar]
- 9.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 10.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Z, Perisse IV, Cotton CU, Regouski M, Meng Q, Domb C, et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight. 2018;3:e123529. doi: 10.1172/jci.insight.123529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHugh DR, Steele MS, Valerio DM, Miron A, Mann RJ, LePage DF, et al. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS One. 2018;13:e0199573. doi: 10.1371/journal.pone.0199573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gawenis LR, Hodges CA, McHugh DR, Valerio DM, Miron A, Cotton CU, et al. A BAC transgene expressing human CFTR under control of its regulatory elements rescues Cftr knockout mice. Sci Rep. 2019;9:11828. doi: 10.1038/s41598-019-48105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS One. 2014;9:e91253. doi: 10.1371/journal.pone.0091253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stalvey MS, Havasi V, Tuggle KL, Wang D, Birket S, Rowe SM, et al. Reduced bone length, growth plate thickness, bone content, and IGF-I as a model for poor growth in the CFTR-deficient rat. PLoS One. 2017;12:e0188497. doi: 10.1371/journal.pone.0188497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plyler ZE, Birket SE, Schultz BD, Hong JS, Rowe SM, Petty CF, et al. Non-obstructive vas deferens and epididymis loss in cystic fibrosis rats. Mech Dev. 2019;155:15–26. doi: 10.1016/j.mod.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birket SE, Davis JM, Fernandez CM, Tuggle KL, Oden AM, Chu KK, et al. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight. 2018;3:e97199. doi: 10.1172/jci.insight.97199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, et al. GOAL (the G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60:703–712. doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195:1617–1628. doi: 10.1164/rccm.201609-1954OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L, Chu KK, Houser GH, Diephuis BJ, Li Y, Wilsterman EJ, et al. Method for quantitative study of airway functional microanatomy using micro-optical coherence tomography. PLoS One. 2013;8:e54473. doi: 10.1371/journal.pone.0054473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, et al. A functional anatomic defect of the cystic fibrosis airway. Am J Respir Crit Care Med. 2014;190:421–432. doi: 10.1164/rccm.201404-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu KK, Mojahed D, Fernandez CM, Li Y, Liu L, Wilsterman EJ, et al. Particle-tracking microrheology using micro-optical coherence tomography. Biophys J. 2016;111:1053–1063. doi: 10.1016/j.bpj.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright JT, Kiefer CL, Hall KI, Grubb BR. Abnormal enamel development in a cystic fibrosis transgenic mouse model. J Dent Res. 1996;75:966–973. doi: 10.1177/00220345960750041101. [DOI] [PubMed] [Google Scholar]
- 27.Semaniakou A, Croll RP, Chappe V. Animal models in the pathophysiology of cystic fibrosis. Front Pharmacol. 2019;9:1475. doi: 10.3389/fphar.2018.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCormick LL, Phillips SE, Kaza N, Tang LP, Rasmussen L, Byzek SA, et al. Maternal smoking induces acquired CFTR dysfunction in neonatal rats. Am J Respir Crit Care Med. 2018;198:672–674. doi: 10.1164/rccm.201805-0827LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birket SE, Chu KK, Houser GH, Liu L, Fernandez CM, Solomon GM, et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am J Physiol Lung Cell Mol Physiol. 2016;310:L928–L939. doi: 10.1152/ajplung.00395.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowe SM, Liu B, Hill A, Hathorne H, Cohen M, Beamer JR, et al. VX06-770-101 Study Group. Optimizing nasal potential difference analysis for CFTR modulator development: assessment of ivacaftor in CF subjects with the G551D-CFTR mutation. PLoS One. 2013;8:e66955. doi: 10.1371/journal.pone.0066955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Petty CM, Hughes GW, Bowers HL, Watson JD, Rosen BH, Townsend SM, et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight. 2019;4:e125954. doi: 10.1172/jci.insight.125954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoegger MJ, Awadalla M, Namati E, Itani OA, Fischer AJ, Tucker AJ, et al. Assessing mucociliary transport of single particles in vivo shows variable speed and preference for the ventral trachea in newborn pigs. Proc Natl Acad Sci USA. 2014;111:2355–2360. doi: 10.1073/pnas.1323633111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345:818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126:879–891. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA. 2006;103:18131–18136. doi: 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med. 2019;11:eaau7531. doi: 10.1126/scitranslmed.aau7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med. 2011;17:478–483. doi: 10.1097/MCP.0b013e32834b14c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosen BH, Evans TIA, Moll SR, Gray JS, Liang B, Sun X, et al. Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. Am J Respir Crit Care Med. 2018;197:1308–1318. doi: 10.1164/rccm.201708-1616OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chinet TC, Fullton JM, Yankaskas JR, Boucher RC, Stutts MJ. Mechanism of sodium hyperabsorption in cultured cystic fibrosis nasal epithelium: a patch-clamp study. Am J Physiol. 1994;266:C1061–C1068. doi: 10.1152/ajpcell.1994.266.4.C1061. [DOI] [PubMed] [Google Scholar]
- 44.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol. 2001;32:356–366. doi: 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 45.Harrison PT, Hoppe N, Martin U. Gene editing & stem cells. J Cyst Fibros. 2018;17:10–16. doi: 10.1016/j.jcf.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 46.Bednarski C, Tomczak K, Vom Hövel B, Weber WM, Cathomen T. Targeted integration of a super-exon into the CFTR locus leads to functional correction of a cystic fibrosis cell line model. PLoS One. 2016;11:e0161072. doi: 10.1371/journal.pone.0161072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerschner JL, Harris A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem J. 2012;446:203–212. doi: 10.1042/BJ20120693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.