

Abstract
Rationale: Exogenous angiotensin II increases mean arterial pressure in patients with catecholamine-resistant vasodilatory shock (CRVS). We hypothesized that renin concentrations may identify patients most likely to benefit from such therapy.
Objectives: To test the kinetic changes in renin concentrations and their prognostic value in patients with CRVS.
Methods: We analyzed serum samples from patients enrolled in the ATHOS-3 (Angiotensin II for the Treatment of High-Output Shock) trial for renin, angiotensin I, and angiotensin II concentrations before the start of administration of angiotensin II or placebo and after 3 hours.
Measurements and Main Results: Baseline serum renin concentration (normal range, 2.13–58.78 pg/ml) was above the upper limits of normal in 194 of 255 (76%) study patients with a median renin concentration of 172.7 pg/ml (interquartile range [IQR], 60.7 to 440.6 pg/ml), approximately threefold higher than the upper limit of normal. Renin concentrations correlated positively with angiotensin I/II ratios (r = 0.39; P < 0.001). At 3 hours after initiation of angiotensin II therapy, there was a 54.3% reduction (IQR, 37.9% to 66.5% reduction) in renin concentration compared with a 14.1% reduction (IQR, 37.6% reduction to 5.1% increase) with placebo (P < 0.0001). In patients with renin concentrations above the study population median, angiotensin II significantly reduced 28-day mortality to 28 of 55 (50.9%) patients compared with 51 of 73 patients (69.9%) treated with placebo (unstratified hazard ratio, 0.56; 95% confidence interval, 0.35 to 0.88; P = 0.012) (P = 0.048 for the interaction).
Conclusions: The serum renin concentration is markedly elevated in CRVS and may identify patients for whom treatment with angiotensin II has a beneficial effect on clinical outcomes.
Clinical trial registered with www.clinicaltrials.gov (NCT 02338843).
Keywords: angiotensin I, renin–angiotensin–aldosterone system, distributive shock, angiotensin-converting enzyme defect
At a Glance Commentary
Scientific Knowledge on the Subject
The renin–angiotensin–aldosterone system is a powerful regulator of blood pressure. Renin drives the formation of angiotensin I, which is the precursor of angiotensin II, a powerful vasoconstrictor. Angitenin II is generated from angiotenin I by ACE (angiotensin-converting enzyme). High angiotensin II levels inhibit renin activity and generation. In patients with severe vasodilatory shock, ACE activity is typically decreased, leading to angioteninsin I/II ratios and logically to high renin levels.
What This Study Adds to the Field
This study shows that renin levels correlate with angiotensin I/II ratios and decrease after initiation of angiotensin II infusion compared with placebo. In patients with renin levels >173 pg/ml, angiotensin II infusion was associated with a marked decrease in mortality compared with placebo.
Vasodilatory shock requiring vasopressor therapy is associated with poor outcomes (1, 2). In particular, catecholamine-resistant vasodilatory shock (CRVS), in which hypotension persists despite the use of high-dose vasopressors, carries a 50–80% mortality risk (3). In these patients, first-line vasopressor therapy, which includes norepinephrine followed by epinephrine or vasopressin, may be inadequate to achieve target mean arterial pressure (MAP) (4). Accordingly, and recently, synthetic human angiotensin II has been approved for the treatment of CRVS in the United States and Europe (5, 6).
Angiotensin II is an integral part of the renin–angiotensin–aldosterone system (RAAS) (7, 8) (Figure 1). Angiotensin I, the precursor of angiotensin II, is cleaved from angiotensinogen by renin, a proteolytic enzyme released by juxtaglomerular cells in response to sympathetic nerve activation, hypotension, or decreased sodium delivery to the distal tubule. Renin release is inhibited by high angiotensin II generation and promoted by low angiotensin II generation via a biofeedback loop involving the angiotensin type 1 receptor (9, 10). As such, renin concentrations are increased when there is insufficient activation of the angiotensin II type 1 receptor. This can be caused by decreased angiotensin II generation or angiotensin II–receptor blockade (11, 12). In this regard, the primary pathway for angiotensin II generation is via ACE (angiotensin-converting enzyme), an endothelial membrane–bound enzyme that cleaves angiotensin I to angiotensin II. Thus, it is logical to suppose that the endothelial injury accompanying CRVS should decrease ACE function, increase angiotensin I/II ratios, and promote renin release. If this were true, high renin concentrations would potentially identify patients most likely to benefit from angiotensin II therapy (13–15). In support of this theory, high ratios of angiotensin I to angiotensin II correlate with negative outcomes in such patients (16). Moreover, in a study of patients with sepsis, reduced angiotensin II and ACE concentrations outperformed established illness-severity scores in predicting outcomes (17).
Figure 1.
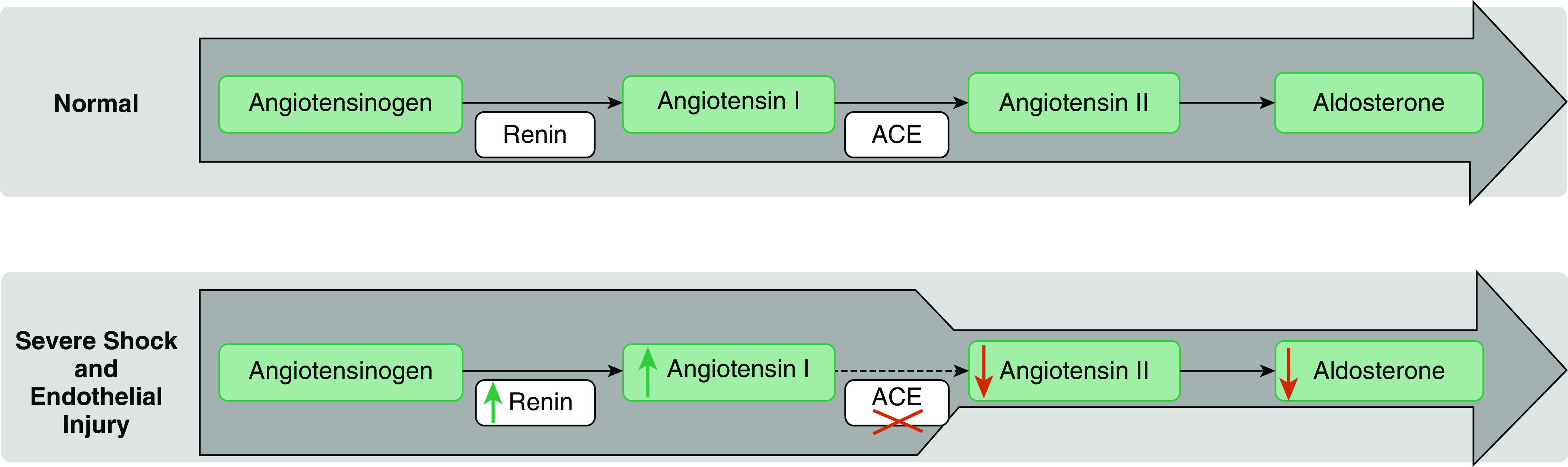
Renin–angiotensin–aldosterone system disturbance hypothesis. The green arrows indicate an increase, the red arrows a decrease, and the red cross marks loss of function. ACE = angiotensin-converting enzyme.
Unfortunately, assessing ACE activity is not amenable to simple plasma measurement, and measurement of the angiotensin I/II ratio is difficult and not available in a timely manner. Renin concentrations, on the other hand, which are expected to increase in patients with CRVS with ACE dysfunction and high angiotensin I to angiotensin II concentrations, could potentially be used to identify such high-risk patients. Laboratory assays that measure renin concentrations are inexpensive, are U.S. Food and Drug Administration– and European Medicines Agency–approved, have demonstrated established performance, and can be easily set up by most laboratories; importantly, measurement of renin concentrations outperforms that of maximum serum lactate concentrations as a predictor of mortality in the ICU (18, 19). Accordingly, we hypothesized that plasma renin concentrations would be high in patients with CRVS, would correlate with angiotensin I/II ratios, and, when elevated, would help to identify those patients most likely to benefit from intravenous angiotensin II therapy.
Methods
Study Design and Patients
The ATHOS-3 (Angiotensin II for the Treatment of High-Output Shock 3) study has been previously described (clinicaltrials.gov identifier NCT 02338843) (5, 20). In brief, adults with vasodilatory shock despite volume resuscitation with ≥25 ml/kg and high-dose vasopressors (defined as a norepinephrine equivalent dose [NED] >0.2 μg/kg/min) were randomly assigned 1:1 to receive synthetic human angiotensin II (La Jolla Pharmaceutical Co.) or saline placebo plus standard vasopressors. Randomization was stratified according to MAP at screening and the Acute Physiology and Chronic Health Evaluation II (APACHE II) score.
Study-drug infusion started at 20 ng/kg/min and was adjusted during the first 3 hours to increase the MAP to ≥75 mm Hg while keeping other vasopressors constant. Thereafter, the study drug and other vasopressors were adjusted at the discretion of the treating ICU team to maintain a MAP between 65 and 75 mm Hg. At 48 hours, the infusion was discontinued according to a protocol-specified tapering process. However, continuation was allowed for up to 7 days at the discretion of the ICU team.
Ethics Approval and Consent to Participate
The study, inclusive of the sample collection, was conducted in accordance with Good Clinical Practice guidelines, applicable local regulations, and the ethical principles described in the Declaration of Helsinki. The protocol, informed-consent form, and all other documents were reviewed and approved by the respective independent institutional review boards before study initiation. The current analysis was conducted on the deidentified data set from ATHOS-3.
Serum Renin, Angiotensin I, and Angiotensin II Measurement
Serum concentrations of renin, angiotensin I, and angiotensin II were measured after randomization but before administration of the study drug and at 3 hours after initiation of the study drug (see online supplement).
Statistical Analysis
We used descriptive statistics with 95% confidence intervals (CIs) to summarize data according to treatment group. We analyzed differences between treatment groups using the Wilcoxon rank-sum test or ANOVA for continuous or ordinal variables and the chi-square or Fisher’s exact test for discrete variables. We summarized time-to-event data, including survival, using Kaplan-Meier estimates and compared them by log-rank test. We estimated hazard ratios (HRs) from a proportional hazards model. A two-sided α amount of 0.05 was used to test for differences in treatment outcomes without adjustments for multiplicity. Correlation coefficients were calculated for renin versus angiotensin I, and the angiotensin I/II ratio. Correlation coefficients were also calculated for renin versus angiotensin I for Hour 0 (baseline) and Hour 3.
For multivariate analyses, covariates that were used for study stratification (model A), covariates that were different between groups as defined by P < 0.10 (model B), and statistically significant covariates from models A and B were included in a multivariate logistic regression (model C). The dichotomized baseline covariates that were used for study stratification were MAP < 65 mm Hg and APACHE II score > 30 (20).
Safety was evaluated by assessment of treatment-emergent adverse events, serious adverse events, and adverse event–related drug discontinuations. Data were analyzed with the use of SAS software version 9.4 (SAS Institute) and R version 3.5.3 (R Foundation).
Results
Of the 321 patients with CRVS studied in ATHOS-3, 255 (79.4%) had suitable serum samples available for analysis of renin concentrations at baseline (Hour 0) (see Tables E8–E10 in the online supplement). In these patients, baseline serum renin concentrations were comparable between the angiotensin II and placebo arms and were elevated to concentrations above the upper limit of normal in 194 (76%) patients. The median serum renin concentration of the entire population was 172.7 pg/ml (interquartile range [IQR], 60.7–440.6 pg/ml); this was approximately three times the upper limit of normal (58.78 pg/ml). Figure E7 and Table E4 further show baseline renin concentrations in patient subsets. As expected, baseline serum renin concentrations were positively correlated with the baseline angiotensin I/II ratio (P < 0.001) and with angiotensin I (P < 0.001) (Figures E3 and E4). In addition, renin and angiotensin I were also positively correlated at 3 hours of treatment (P < 0.001) (Figure E4).
Table E1 shows patient demographic and clinical data dichotomized by median serum renin concentrations, and Table 1 shows these data further dichotomized by treatment group within each renin-concentration stratum. At baseline, patients with serum renin concentrations above the study population median were similar to those with concentrations below the study population median, except for baseline concentrations of angiotensin I, angiotensin II, angiotensin I/II ratio, and NED, all of which were significantly higher in the former group.
Table 1.
Demographics and Clinical Characteristics of Study Patients
| Study Population below the Median Renin |
P Value | Study Population above the Median Renin |
P Value | |||
|---|---|---|---|---|---|---|
| Treatment with Placebo (n = 63) | Treatment with Angiotensin II (n = 64) | Treatment with Placebo (n = 73) | Treatment with Angiotensin II (n = 55) | |||
| Age, yr, median (IQR) | 66 (53–75) | 61.5 (51–74) | 0.51* | 63 (51–75) | 62 (50–72) | 0.66* |
| Sex, n (%) | 0.20† | 0.21† | ||||
| F | 20 (31.7) | 28 (43.8) | 26 (35.6) | 26 (47.3) | ||
| M | 43 (68.3) | 36 (56.3) | 47 (64.4) | 29 (52.7) | ||
| Race, n (%) | 1.00† | 0.20† | ||||
| White | 52 (82.5) | 53 (82.8) | 54 (74.0) | 46 (83.6) | ||
| Nonwhite | 11 (17.5) | 11 (17.2) | 19 (26.0) | 9 (16.4) | ||
| Baseline albumin (g/dl), median (IQR) | 2.3 (1.8–2.8) | 2.2 (1.6–2.6) | 0.21* | 2.3 (1.9–2.7) | 2.3 (1.9–2.7) | 0.75* |
| Baseline MELD, median (IQR) | 22 (15–25) | 20 (14–25) | 0.26* | 23 (20–28) | 22 (18–26) | 0.09* |
| Baseline MAP, mm Hg, median (IQR) | 66.7 (64.0–68.7) | 66.0 (63.7–67.9) | 0.40* | 66.3 (62.3–68.0) | 66.7 (63.3–69.7) | 0.28* |
| Baseline APACHE II score, median (IQR) | 29 (20–34) | 27 (22–33) | 0.70* | 31 (25–36) | 28 (22–34) | 0.14* |
| ARDS,‡n (%) | 17 (27.0) | 17 (27.0) | 1.00 | 28 (38.4) | 13 (23.6) | 0.088 |
| Medical history, sepsis, n (%) | 56 (88.9) | 50 (78.1) | 0.15† | 57 (78.1) | 42 (76.4) | 0.83† |
| Recent ARB exposure, n (%) | 2 (3.2) | 4 (6.3) | 0.68† | 7 (9.6) | 3 (5.5) | 0.51† |
| Recent ACEi exposure, n (%) | 5 (7.9) | 2 (3.1) | 0.27† | 8 (11.0) | 10 (18.2) | 0.31† |
| Severe AKI, n (%) | 14 (22.2) | 18 (28.1) | 0.54† | 41 (56.2) | 21 (38.2) | 0.05† |
| ESRD, n (%) | 2 (3.2) | 4 (6.3) | 0.68† | 1 (1.4) | 2 (3.6) | 0.58† |
| Baseline NED, μg/kg/min, median (IQR) | 0.29 (0.22–0.49) | 0.32 (0.22–0.54) | 0.40* | 0.40 (0.29–0.69) | 0.36 (0.23–0.50) | 0.06* |
| Angiotensin I, pg/ml, median (IQR) | 85.6 (36.70–173.00) | 133.0 (44.10–356.00) | 0.15* | 602 (238–1,110) | 655 (304.5–1,375) | 0.45* |
| Angiotensin II, pg/ml, median (IQR) | 52.4 (24.60–137.00) | 98.2 (24.50–168.00) | 0.35* | 108 (16.7–523) | 151 (41.4–439) | 0.46* |
| Baseline angiotensin I/II ratio, median (IQR) | 1.25 (0.79–2.39) | 1.34 (0.91–2.59) | 0.70* | 3.41 (1.17–10.39) | 3.01 (1.17–12.43) | 0.96* |
Definition of abbreviations: ACEi = angiotensin-converting enzyme inhibitor; AKI = acute kidney injury; APACHE II = Acute Physiology and Chronic Health Evaluation II; ARB = angiotensin receptor blocker; ARDS = acute respiratory distress syndrome; ESRD = end-stage renal disease; IQR = interquartile range; MAP = mean arterial pressure; MELD = Model for End-Stage Liver Disease; NED = norepinephrine equivalent dose.
ACEi exposure was determined by the presence of an ACEi in the medical chart within 7 days before study enrollment.
Wilcoxon rank-sum test.
Fisher’s exact test.
ARDS was determined by chest X-ray reading. Patients with ARDS noted on their chest X-ray during screening were denoted as having ARDS.
Figure 2 shows serum concentrations of angiotensin I and renin in patients who had samples available at baseline and at 3 hours. Median baseline renin concentrations were similar among patients in the placebo group (193.7 [IQR, 58.1 to 489.8] pg/ml) and patients in the angiotensin II group (146.1 [IQR, 62.4 to 412.2] pg/ml) (P = 0.4277). Patients treated with synthetic angiotensin II experienced a median reduction of 54.3% (IQR, 37.9 to 66.5% reduction) in renin concentration compared with a median reduction of 14.1% (IQR, 37.6% reduction to 5.1% increase) in patients treated with placebo (P < 0.0001) at Hour 3. Median baseline concentrations of angiotensin I were also similar between treatment groups, and as was seen with serum renin concentrations, treatment with synthetic angiotensin II led to a significantly greater reduction in angiotensin I concentrations at Hour 3 (39.7% median reduction [IQR, 11.8% to 56.1% reduction]) compared with treatment with placebo (7.0% reduction [IQR, 26.7% reduction to 20.0% increase]) (P < 0.0001).
Figure 2.

Serum angiotensin I and renin concentrations. (A) Serum angiotensin I concentrations. (B) Serum renin concentrations. All values are presented as the median (interquartile range).
The MAP response at 3 hours was not affected by baseline renin concentrations. MAP response in patients with renin concentrations below the population median treated with placebo was 30.2% compared with 78.1% in patients treated with angiotensin II, whereas in patients with renin concentrations above the population median, MAP response in patients treated with placebo was 20.5% compared with 61.8% in patients treated with angiotensin II (data not shown). Thus, renin was not identified as significantly modifying the effect of study group assignment (treatment arm by renin interaction odds ratio, 0.76 [95% CI, 0.25–2.32]; P = 0.6266).
Survival by Baseline Serum Renin Concentrations
For patients with renin concentrations above the study population median, baseline characteristics were well balanced between the placebo and angiotensin II treatment arms (Table 1). Despite this balance, patients treated with placebo had a 28-day mortality rate of 69.9% compared with 51.1% in patients treated with angiotensin II (unstratified HR, 0.556 [95% CI, 0.35–0.88]; P = 0.0115) (Table 2 and Figure 3; see also Figures E1, E2, and E5). In contrast, there was no statistically significant difference in 28-day mortality for those with serum renin concentrations below the study population median (Table 2 and Figure 3).
Table 2.
Outcomes and Renin Concentration
| Patients below the Study Population Median Renin [Median (IQR)] |
P Value | Patients above the Study Population Median Renin [Median (IQR)] |
P Value | |||
|---|---|---|---|---|---|---|
| Treatment with Placebo (n = 63) | Treatment with Angiotensin II (n = 64) | Treatment with Placebo (n = 73) | Treatment with Angiotensin II (n = 55) | |||
| 28-d mortality, % | 44 (33–58) | 45 (34–58) | 0.70 | 70 (59–80) | 51 (39–65) | 0.01 |
| Ventilator liberation by Day 7 (alive and vent-free), % | 38 (27–52) | 27 (18–40) | 0.20 | 14 (8–25) | 28 (18–43) | 0.07 |
| RRT liberation by Day 7 (alive and RRT free), % | 14 (4–46) | 28 (13–54) | 0.33 | 12 (5–27) | 43 (25–66) | 0.01 |
| ICU discharge by Day 28, % | 52 (41–65) | 39 (28–52) | 0.13 | 22 (14–33) | 44 (32–58) | 0.02 |
Definition of abbreviations: IQR = interquartile range; RRT = renal replacement therapy.
Figure 3.

Kaplan-Meier survival plot according to renin concentrations and treatment with angiotensin II or placebo. (A) Day 28 survival: renin concentration below population median. (B) Day 28 survival: renin concentration above population median. CI = confidence interval; HR = hazard ratio.
Multivariate logistic regression within the placebo-treated arm alone showed that, after adjusting for age, sex, APACHE II score, MAP, and NED, elevated serum renin concentrations were independently associated with an increased risk of death (HR, 2.15 [95% CI, 1.35–3.42]; P = 0.0013) (Table E5). Moreover, in patients with a renin concentration above the study population median, multivariable analysis identified treatment with angiotensin II as associated with decreased risk of mortality (HR, 0.62 [95% CI, 0.39–0.98]; P = 0.0423) (Table 3). See also Tables E11 and E12 in the online supplement for sensitivity analyses of these multivariable analyses. Figure 4 displays survival by serum renin concentration as a continuous variable. Although the P value for the interaction term including the dichotomized renin concentration was 0.048 (HR, 0.50 [95% CI, 0.25–0.99]), the P value for the interaction term including continuous renin (log2) was 0.28 (95% CI, 0.78–1.08). The difference reflects the apparent nonlinear HR provided in Figure 4.
Table 3.
Multivariable Analysis for the Prediction of Mortality: Patients with Baseline Renin Above Study Median Only
| Hazard Ratio (95% CI) | P Value | |
|---|---|---|
| Model A | ||
| Treatment arm, angiotensin II | 0.58 (0.36–0.93) | 0.02 |
| Baseline APACHE II score > 30 | 2.02 (1.29–3.18) | 0.002* |
| Baseline MAP < 65 mm Hg | 1.76 (1.12–2.77) | 0.01 |
| Model B | ||
| Treatment arm, angiotensin II | 0.62 (0.38–0.99) | 0.04 |
| Baseline NED ≥ 0.5 μg/kg/min | 1.88 (1.19–2.98) | 0.007* |
| Baseline MELD ≥ 30 | 1.43 (0.81–2.50) | 0.21 |
| Model C | ||
| Treatment arm, angiotensin II | 0.62 (0.39–0.98) | 0.04 |
| Baseline APACHE II score > 30 | 2.03 (1.29–3.19) | 0.002* |
| Baseline MAP < 65 mm Hg | 1.66 (1.06–2.62) | 0.03 |
| Baseline NED ≥ 0.5 μg/kg/min | 1.78 (1.13–2.83) | 0.01 |
Definition of abbreviations: APACHE II = Acute Physiology and Chronic Health Evaluation II; CI = confidence interval; MAP = mean arterial pressure; MELD = Model for End-Stage Liver Disease; NED = norepinephrine equivalent dose.
Model A includes treatment assignment and stratification variables. Model B includes covariates that were imbalanced by P < 0.10 in the population clinical characteristics. Model C includes the statistically significant variables from model A and model B.
P < 0.01.
Figure 4.

Cubic splines of mortality and baseline serum renin. Baseline renin is shown with log scale. LJPC501 = angiotensin II.
Other Outcomes of Interest
Table 2 shows rates of ventilator and renal replacement therapy liberation, as well as ICU discharge according to treatment group in patients dichotomized by serum renin concentrations. Whereas there were no differences in these outcomes between treatment groups in patients with renin concentrations below the study population median, in patients with renin concentrations above the study population median, the rate of renal replacement therapy liberation by Day 7 was significantly greater in patients treated with angiotensin II (43%) compared with those treated with placebo (12%; P = 0.01) (Table 2 and Figure E6 and Table E3), as was the rate of ICU discharge by Day 28 (angiotensin II, 44%; placebo, 22%; P = 0.02). The rate of ventilator liberation by Day 7, however, did not differ significantly between treatment groups (angiotensin II, 28%; placebo, 14%; P = 0.07).
Table E7 shows the changes in cardiovascular and total Sequential Organ Failure Assessment (SOFA) scores between baseline and Hour 48 according to renin status and treatment group. In the total ATHOS-3 population, the change in cardiovascular score was significantly greater in the angiotensin II compared with the placebo group, whereas the change in the total SOFA score was not (5). As shown in Table E7, neither the change in cardiovascular SOFA nor the total SOFA score differed significantly between treatment groups in patients with serum renin concentrations below the study-population median. In patients with serum renin concentrations above the study population median, the mean ± SD change in cardiovascular SOFA score at 48 hours was significantly greater in the angiotensin II group (−1.56 ± 1.793) compared with the placebo group (−0.78 ± 1.387), whereas the change in total SOFA score was not (angiotensin II, 1.35 ± 6.032; placebo, 2.86 ± 5.611; P = 0.07).
Adverse Events
In patients with elevated renin concentrations at baseline, there was no significant difference in adverse events or serious adverse events between the angiotensin II and placebo groups (Table 4).
Table 4.
Summary of Adverse Events
| Placebo | Angiotensin II | Total | |
|---|---|---|---|
| Patients with shock and renin above the median | |||
| Patients, n | 73 | 55 | 128 |
| TEAE | 69 (94.5) | 47 (85.5) | 116 (90.6) |
| Grade 3/4 TEAE | 61 (83.6) | 39 (70.9) | 100 (78.1) |
| Serious TEAE | 61 (83.6) | 39 (70.9) | 100 (78.1) |
| Serious related TEAE | 5 (6.8) | 4 (7.3) | 9 (7.0) |
| Patients with shock and renin below the median | |||
| Patients, n | 63 | 64 | 127 |
| TEAE | 56 (88.9) | 58 (90.6) | 114 (89.8) |
| Grade 3/4 TEAE | 40 (63.5) | 40 (62.5) | 80 (63.0) |
| Serious TEAE | 36 (57.1) | 38 (59.4) | 74 (58.3) |
| Serious related TEAE | 1 (1.6) | 2 (3.1) | 3 (2.4) |
Definition of abbreviation: TEAE = treatment-emergent adverse event.
Data are n (%), unless otherwise indicated.
Discussion
In this post hoc analysis of patients enrolled in the ATHOS-3 study, we tested the hypothesis that there is a disturbance in the RAAS, likely resulting from insufficient ACE activity in CRVS. We reasoned that such ACE insufficiency could be identified through the analysis of serum renin concentrations and that increased serum renin concentrations would predict worse outcomes. We found that serum renin concentrations were significantly elevated in most patients with CRVS and that they were positively and significantly associated with angiotensin I concentrations and angiotensin I/II ratios. This ratio, a surrogate for ACE activity, has previously been reported to be associated with increased mortality risk in CRVS (16). As with those patients having elevated angiotensin I/II ratios, patients with serum renin concentrations above the study population median had a significantly increased risk of mortality. However, treatment with synthetic angiotensin II was associated with a significant reduction in this risk. These data suggest that renin has the potential to be used to identify patients with CRVS at high risk for poor outcomes and who may benefit from treatment with synthetic angiotensin II.
Our observations are consistent with those of previous studies demonstrating that patients with septic shock can develop decreased ACE concentrations, which can lead to impaired ability to convert angiotensin I to angiotensin II (7, 17). This may reflect endothelial injury or ACE-gene SNPs, both of which are factors that correlate with increased 28-day mortality (21, 22). Our findings also confirm previous reports that elevated renin is associated with worse outcomes in patients with septic shock and other critical illness (23–25). Specifically, previous reports in this disease population show that plasma renin activity is elevated while aldosterone concentrations are inappropriately low, suggesting a defect in the RAAS pathway (Figure 1) (23, 24). Finally, our findings in the CRVS population are consistent with observations in patients treated with ACE inhibitors. Specifically, patients receiving ACE inhibitors have substantial increases in both serum angiotensin I and renin concentrations, both of which were effectively suppressed after the administration of exogenous angiotensin II (11, 26, 27).
The mechanistic implications of ACE dysfunction for patients with CRVS are potentially important. ACE inhibition induced by drugs (e.g., captopril or enalapril) increases the concentrations of not only angiotensin I but of vasodilatory angiotensin I metabolites, such as angiotensin 1–7 (27). As the absolute concentrations of angiotensin II do not decrease significantly during ACE-inhibitor therapy, and may actually increase, the hypotensive effects of ACE inhibitors may occur as a consequence of the activity of these vasodilatory angiotensin metabolites (27). The similar impact of shock on angiotensin I and II concentrations in the present analysis suggests that ACE dysfunction resulting from endothelial damage may be a significant contributor to the pathophysiology in some patients with CRVS.
We believe that renin concentrations may modify the effect of treatment group on outcomes independent of blood pressure because renin has other important clinical effects related to its neurohormonal activity. Renin binds the (pro)renin receptor, which has been described on many cell and tissue types, including leukocytes. One study showed that incubation of leukocytes with renin activated the (pro)renin receptor and elicited the production of proinflammatory cytokines independently of downstream angiotensin II effects (28). Preclinical studies of sepsis demonstrate that (pro)renin receptor blockade improves survival and is associated with lower concentrations of proinflammatory cytokines (29). Thus, our finding that synthetic angiotensin II rapidly reduces renin concentrations compared with standard-of-care vasopressors suggests that synthetic angiotensin II potentially modulates the inflammatory response caused by excess renin and that this mechanism may explain our findings of enhanced survival. In addition, normalization of serum renin concentrations with angiotensin II may also indicate adequate angiotensin II type 1 receptor activation in end organs such as the kidney. Activation of this receptor by synthetic angiotensin II can increase the glomerular filtration rate and urine output in sepsis (30). This notion is supported by previous findings that patients with severe acute kidney injury and shock treated with synthetic angiotensin II experienced enhanced renal recovery and improved survival (31).
Prospective validation of these data is warranted, and if confirmed, these findings may influence patient care, as renin assays are widely available and inexpensive. Thus, use of this biomarker for vasopressor-targeted therapy is logistically feasible. In this regard, our findings imply that using angiotensin II as a vasopressor, which also lowers renin concentrations, may offer a combined biological and clinical target during angiotensin II administration. Finally, the findings that both renin and angiotensin I are suppressible by exogenous angiotensin II suggests that patients with elevated renin may have insufficient activation of their angiotensin type I receptor.
This study has several strengths. First, the theory that decreased ACE activity may be a key mechanism in CRVS that is sensitive to angiotensin II was proposed before the start of the ATHOS-3 trial, and this analysis is mechanistically linked to such a RAAS-disturbance hypothesis (Figure 1) (7, 20). Second, our findings are consistent with the well-described physiology of the RAAS and the effects of ACE inhibition (26, 27). The clear reduction in both renin and angiotensin I with exogenous angiotensin II administration compared with minimal changes in the placebo arm gives credence to the concept of decreased activity of the RAAS pathway and relative preservation of a biofeedback response in CRVS. Third, ATHOS-3 was an international, prospective randomized placebo-controlled trial in which adequate resuscitation per international consensus guidelines was a prerequisite for trial entry, thus increasing the generalizability to our findings.
We acknowledge several limitations. The current analysis is post hoc, and we did not have enough remaining samples to measure concurrent angiotensin 1–7, bradykinin, or aldosterone concentrations, which might have provided more indirect information on ACE activity and on the activity of nonclassical angiotensin I and II metabolism by ACE2. We also did not have renin samples for all patients in the cohort; however, the patient characteristics of the patients with missing samples were not different from those with samples available. In addition, we were not able to rigorously assess for the presence or severity of preexisting hypertension, renovascular disease, or chronic kidney disease, all of which can affect background renin concentrations. Finally, we conducted many sensitivity analyses, and the treatment effect of angiotensin II in the high-renin arm did not achieve statistical significance in all assessments. Furthermore, the effect modification of renin on treatment group, present when treating renin as a dichotomous variable, was not evident when treating it as a continuous variable and adjusting for baseline covariates. Therefore, the observed findings may represent chance imbalances between groups in the baseline severity of illness.
Conclusions
We show that in most people with CRVS, there is significant disturbance in the RAAS, likely resulting from impairment of ACE function. RAAS disturbance may play a significant role in the pathophysiology of vasodilatory shock in a subset of patients, suggesting that such patients may benefit from alternative management approaches and specifically from intervention targeting the RAAS. Importantly, we show that patients with RAAS disturbance can be readily identified through simple laboratory assessment of serum renin concentrations and that their outcomes may be improved when receiving synthetic angiotensin II. Our data further suggest that renin assessment could be used to identify patients without RAAS disturbance, in whom treatment with angiotensin II would likely be futile. Although additional data are needed to confirm our findings, this study suggests that a personalized approach to the management of vasodilatory shock may soon be feasible and could potentially improve outcomes in vasodilatory shock. In a setting where mortality rates can reach or even exceed 50%, such an approach is urgently needed. Prospective confirmation of these findings is warranted.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank the patients and their families for entrusting them to conduct this study. They also thank the study investigators, study coordinators, and support staff across all sites for their compassion, dedication, and diligence in the conduct of this trial. Medical editorial assistance was provided by Robert J. Schoen, PharmD, of ApotheCom (Yardley, Pennsylvania) and funded by La Jolla Pharmaceutical Company (San Diego, California).
Footnotes
Supported by La Jolla Pharmaceutical Co. (primary study and this post hoc analysis).
Author Contributions: Acquisition of data: R.B., L.W.B., M.T.M., K.R.H., D.W.B., J.H., A.K.K., T.E.A., J.T., K.S., L.S.C., and M.O. Conception and design: R.B. and L.S.C. Analysis and interpretation: R.B., L.G.F., L.W.B., M.T.M., T.E.A., J.T., D.H., G.F.T., L.S.C., and M.O. Wrote the manuscript: R.B., L.G.F., L.S.C., and M.O. Approved and edited the manuscript: R.B., L.G.F., L.W.B., M.T.M., K.R.H., J.H., T.E.A., and L.S.C. All authors approved the final manuscript.
A complete list of ATHOS-3 investigators may be found in the online supplement.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.201911-2172OC on July 1, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
Collaborators: on behalf of the ATHOS-3 Investigators
References
- 1.Lambden S, Creagh-Brown BC, Hunt J, Summers C, Forni LG. Definitions and pathophysiology of vasoplegic shock. Crit Care. 2018;22:174. doi: 10.1186/s13054-018-2102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sterling SA, Puskarich MA, Shapiro NI, Trzeciak S, Kline JA, Summers RL, et al. Emergency Medicine Shock Research Network (EMSHOCKNET) Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock. 2013;40:11–14. doi: 10.1097/SHK.0b013e318298836d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown SM, Lanspa MJ, Jones JP, Kuttler KG, Li Y, Carlson R, et al. Survival after shock requiring high-dose vasopressor therapy. Chest. 2013;143:664–671. doi: 10.1378/chest.12-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock. 2016. Crit Care Med. 2017;45:486–552. doi: 10.1097/CCM.0000000000002255. [DOI] [PubMed] [Google Scholar]
- 5.Khanna A, English SW, Wang XS, Ham K, Tumlin J, Szerlip H, et al. ATHOS-3 Investigators. Angiotensin II for the treatment of vasodilatory shock. N Engl J Med. 2017;377:419–430. doi: 10.1056/NEJMoa1704154. [DOI] [PubMed] [Google Scholar]
- 6.Chawla LS, Busse L, Brasha-Mitchell E, Davison D, Honiq J, Alotaibi Z, et al. Intravenous angiotensin II for the treatment of high-output shock (ATHOS trial): a pilot study. Crit Care. 2014;18:534. doi: 10.1186/s13054-014-0534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla LS, Busse LW, Brasha-Mitchell E, Alotaibi Z. The use of angiotensin II in distributive shock. Crit Care. 2016;20:137. doi: 10.1186/s13054-016-1306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 9.Corrêa TD, Takala J, Jakob SM. Angiotensin II in septic shock. Crit Care. 2015;19:98. doi: 10.1186/s13054-015-0802-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res. 2012;318:1049–1056. doi: 10.1016/j.yexcr.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kono T, Ikeda F, Oseko F, Imura H, Endo J. Effects of angiotensin I-converting enzyme inhibitor, SQ 14225, in normal men. Endocrinol Jpn. 1979;26:411–418. doi: 10.1507/endocrj1954.26.411. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg MR, Tanaka W, Barchowsky A, Bradstreet TE, McCrea J, Lo MW, et al. Effects of losartan on blood pressure, plasma renin activity, and angiotensin II in volunteers. Hypertension. 1993;21:704–713. doi: 10.1161/01.hyp.21.5.704. [DOI] [PubMed] [Google Scholar]
- 13.Orfanos SE, Armaganidis A, Glynos C, Psevdi E, Kaltsas P, Sarafidou P, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation. 2000;102:2011–2018. doi: 10.1161/01.cir.102.16.2011. [DOI] [PubMed] [Google Scholar]
- 14.Deitz DM, Swartz KR, Wright M, Murphy E, Connell RS, Harrison MW. Effects of E. coli endotoxin on rat plasma angiotensin converting enzyme activity in vitro and in vivo. Circ Shock. 1987;21:23–29. [PubMed] [Google Scholar]
- 15.Levi M, van der Poll T. Endothelial injury in sepsis. Intensive Care Med. 2013;39:1839–1842. doi: 10.1007/s00134-013-3054-1. [DOI] [PubMed] [Google Scholar]
- 16.Bellomo R, Wunderink RG, Szerlip H, English SW, Busse LW, Deane AM, et al. Angiotensin I and angiotensin II concentrations and their ratio in catecholamine-resistant vasodilatory shock. Crit Care. 2020;24:43. doi: 10.1186/s13054-020-2733-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Chen X, Huang L, Lu N, Zhou L, Wu G, et al. Severe sepsis: low expression of the renin-angiotensin system is associated with poor prognosis. Exp Ther Med. 2014;7:1342–1348. doi: 10.3892/etm.2014.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleeson PJ, Crippa IA, Mongkolpun W, Cavicchi FZ, Van Meerhaeghe T, Brimioulle S, et al. Renin as a marker of tissue-perfusion and prognosis in critically ill patients. Crit Care Med. 2019;47:152–158. doi: 10.1097/CCM.0000000000003544. [DOI] [PubMed] [Google Scholar]
- 19.Khanna AK. Tissue perfusion and prognosis in the critically ill-is renin the new lactate? Crit Care Med. 2019;47:288–290. doi: 10.1097/CCM.0000000000003582. [DOI] [PubMed] [Google Scholar]
- 20.Chawla LS, Russell JA, Bagshaw SM, Shaw AD, Goldstein SL, Fink MP, et al. Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3): protocol for a phase III, double-blind, randomised controlled trial. Crit Care Resusc. 2017;19:43–49. [PubMed] [Google Scholar]
- 21.Hannula-Jouppi K, Massinen S, Siljander T, Mäkelä S, Kivinen K, Leinonen R, et al. Genetic susceptibility to non-necrotizing erysipelas/cellulitis. PLoS One. 2013;8:e56225. doi: 10.1371/journal.pone.0056225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dou XM, Cheng HJ, Meng L, Zhou LL, Ke YH, Liu LP, et al. Correlations between ACE single nucleotide polymorphisms and prognosis of patients with septic shock. Biosci Rep. 2017;37:BSR20170145. doi: 10.1042/BSR20170145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davenport MW, Zipser RD. Association of hypotension with hyperreninemic hypoaldosteronism in the critically ill patient. Arch Intern Med. 1983;143:735–737. [PubMed] [Google Scholar]
- 24.du Cheyron D, Lesage A, Daubin C, Ramakers M, Charbonneau P. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med. 2003;29:1703–1709. doi: 10.1007/s00134-003-1986-6. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen M, Denimal D, Dargent A, Guinot PG, Duvillard L, Quenot JP, et al. Plasma renin concentration is associated with hemodynamic deficiency and adverse renal outcome in septic shock. Shock. 2019;52:e22–e30. doi: 10.1097/SHK.0000000000001285. [DOI] [PubMed] [Google Scholar]
- 26.Azizi M, Guyene TT, Chatellier G, Wargon M, Ménard J. Additive effects of losartan and enalapril on blood pressure and plasma active renin. Hypertension. 1997;29:634–640. doi: 10.1161/01.hyp.29.2.634. [DOI] [PubMed] [Google Scholar]
- 27.Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14:799–805. doi: 10.1097/00004872-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Narumi K, Hirose T, Sato E, Mori T, Kisu K, Ishikawa M, et al. A functional (pro)renin receptor is expressed in human lymphocytes and monocytes. Am J Physiol Renal Physiol. 2015;308:F487–F499. doi: 10.1152/ajprenal.00206.2014. [DOI] [PubMed] [Google Scholar]
- 29.Hirano Y, Takeuchi H, Suda K, Hagiwara T, Miyasho T, Kawamura Y, et al. (Pro)renin receptor blocker improves survival of rats with sepsis. J Surg Res. 2014;186:269–277. doi: 10.1016/j.jss.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Wan L, Langenberg C, Bellomo R, May CN. Angiotensin II in experimental hyperdynamic sepsis. Crit Care. 2009;13:R190. doi: 10.1186/cc8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tumlin JA, Murugan R, Deane AM, Ostermann M, Busse LW, Ham KR, et al. Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) Investigators. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med. 2018;46:949–957. doi: 10.1097/CCM.0000000000003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.