

Abstract
There is an urgent need to develop new efficacious antimalarials to address the emerging drug-resistant clinical cases. Our previous phenotypic screening identified styrylquinoline UCF501 as a promising antimalarial compound. To optimize UCF501, we herein report a detailed structure–activity relationship study of 2-arylvinylquinolines, leading to the discovery of potent, low nanomolar antiplasmodial compounds against a Plasmodium falciparum CQ-resistant Dd2 strain, with excellent selectivity profiles (resistance index < 1 and selectivity index > 200). Several metabolically stable 2-arylvinylquinolines are identified as fast-acting agents that kill asexual blood-stage parasites at the trophozoite phase, and the most promising compound 24 also demonstrates transmission blocking potential. Additionally, the monophosphate salt of 24 exhibits excellent in vivo antimalarial efficacy in the murine model without noticeable toxicity. Thus, the 2-arylvinylquinolines represent a promising class of antimalarial drug leads.
Graphical Abstract
INTRODUCTION
Malaria afflicts almost half of the world’s population, causing an estimated 228 million clinical cases and 405,000 deaths in 2018. Predominant mortality was among children below the age of five and pregnant women in Africa.1 Significant progress has been made, over the last decade, to reduce the global malaria burden using artemisinin-based combination therapies (ACTs), long-lasting insecticide-treated nets, and indoor residual spraying for vector control. However, the loss of efficacy of ACTs in the chloroquine (CQ)-resistant malaria strains underscores the fragility of gains accomplished in the global effort for malaria eradication. To mitigate this alarming situation and achieve the eventual elimination of malaria, it is important to develop inexpensive chemical entities against drug-resistant malaria parasite strains, ideally those that possess transmission-blocking properties.
Important strategies to develop new antimalarial compounds involve structural re-engineering of exiting drugs such as CQ a landmark 4-aminoquinoline compound, because of its efficacy against all types of human malaria parasites, long half-life, low cost, and favorable safety profiles.2 However, CQ has lost it efficacy because of mutations in the gene encoding the digestive vacuole (DV) membrane protein Pf CRT, leading to reduced drug accumulation in its site of action and ultimately CQ resistance.3 Based on this premise, numerous CQ analogues with conformational rigid nitrogen-containing side chains4,5,6 and CQ hybrids (CQ-resistance reverse agents,7–9 CQ-artemisinin,10–12 CQ-synthetic peroxide,13–15 CQ-ferrocene,16,17 CQ-chalcone,17–19 CQ-N-contained heterocyclic compound,9,20–22 etc.) have been developed to overcome the resistance and improve the antimalarial activity. Piperaquine and ferroquine are examples of re-engineered CQ analogues. The former containing two 7-chloro-aminoquinoline moieties is extensively used in Southeast Asia for prophylaxis and treatment.23 The latter, a 7-chloroaminoquinoline covalently linked to an aminoferrocenyl group, is in phase II pilot clinical trials.24,25 Thus, the quinoline scaffold is a privileged structure that can be re-engineered to design new antimalarial candidates2,9,26–28.
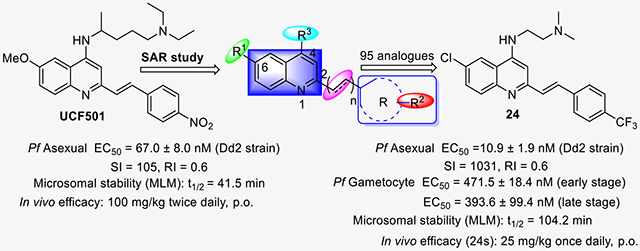
Within the quinoline class, styrylquinoline derivatives display a wide spectrum of pharmacological properties, such as antileishmanial,29 anticancer,30 anti-HIV,31 and anti-Alzheimer’s activity.32 Recently, we have reported the discovery of styrylquinoline UCF501 with promising in vitro and in vivo antiplasmodial activity.33 In particular, it exhibited a more potent inhibitory activity against CQ-resistant strain than that against CQ-sensitive strain. From a mechanistic perspective, UCF501 may in fact act differently from CQ, while its exact molecular target remained elusive. Although a number of analogues were reported in the previous investigation, a detailed account is highly desirable to elucidate the structure–activity relationship (SAR) of this chemotype. Notably, all of them were less active than the lead compound UCF501. Herein, we describe a comprehensive medicinal chemistry study on arylvinylquinolines (Figure 1), report the key structural determinants for the antimalarial activity, and identify new compounds that show improved microsomal stability over UCF501. In addition, we disclose the details of the specific stage action, the rate of killing, and an in vivo evaluation of the frontrunner compounds.
Figure 1.
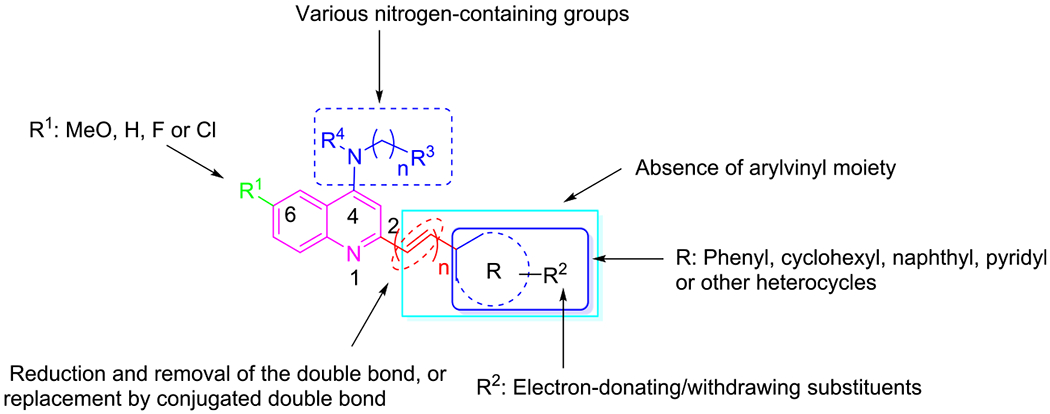
SAR strategy around the quinoline scaffold.
RESULTS AND DISCUSSION
Chemistry.
The synthesis of compounds 8–37 (Scheme 1 and Table 1) started from commercially available anilines 1a–d. The reaction of aniline 1a with ethyl acetoacetate 2 in the presence of acetic acid afforded an imine intermediate, which was converted to hydroxyquinoline 3a at elevated temperature.34 Alternatively, hydroxyquinolines 3a–d were synthesized by mixing anilines 1a–d with 2 in the presence of polyphosphoric acid (PPA).35 The chlorination of hydroxyquinolines 3a–d with phosphorus oxychloride gave 4-chloroquinolines 4a–d in quantitative yields, which were then reacted with neat N,N-dimethylaminoalkylamines 5a–b via nucleophilic substitution to produce the aminoquinolines 6a–e in excellent yields. Subsequent olefination of 6a–e with appropriate aromatic aldehydes 7a–l using p-toluenesulfonamide (p-TsNH2) as a catalyst was carried out in xylene to afford (E)-styrylquinolines 8–37.33
Scheme 1. Synthesis of 6-Substituted 2-Styrylquinolines 8–37a.
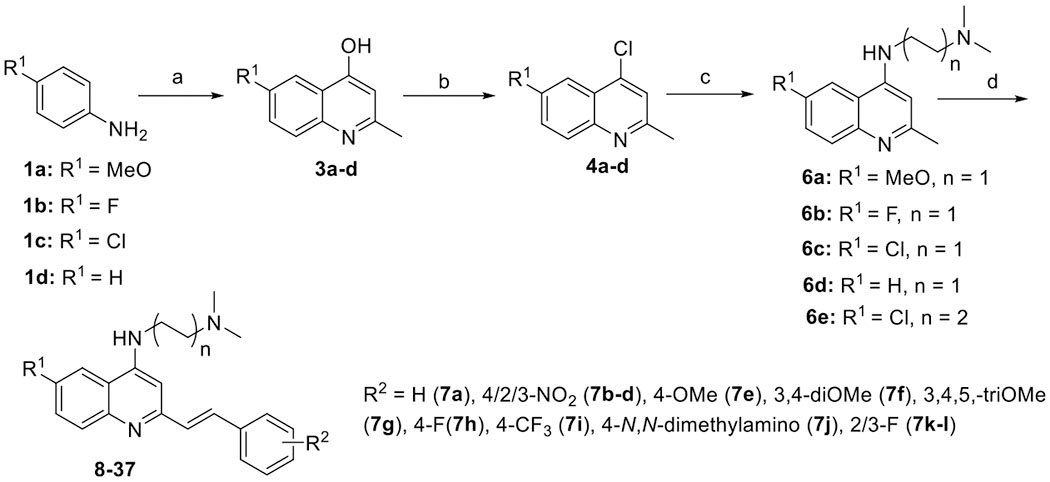
aReagents and conditions: (a) method A for 3a: (i) p-anisidine 1a, ethyl acetoacetate 2, acetic acid, anhydrous magnesium sulfate, and ethanol, 90 °C, 6 h; (ii) Dowtherm, 270 °C, 30 min, 43% for two steps; method B for 3a–d: anilines 1a–d, ethyl acetoacetate 2, PPA, 150 °C, 2 h, 57–68%; (b) phosphorus oxychloride, 105 °C, 2 h, 85–90%; (c) N,N-dimethylaminoalkylamines 5a–b, 130 °C, 24 h, 87–93%; and (d) aromatic aldehydes 7a–l, p-TsNH2, xylene, 130 °C, 12 h, 60–89%.
Table 1.
Antiplasmodial Activity of SAR1 Targets against the Dd2 Strain
 | ||||
|---|---|---|---|---|
| compd | R1 | n | R2 | EC50 (nM) |
| 8 | MeO | 1 | H | 41.2 ± 5.3 |
| 9 | MeO | 1 | 4-NO2 | 28.6 ± 0.9 |
| 10 | MeO | 1 | 2-NO2 | 56.3 ± 8.1 |
| 11 | MeO | 1 | 3-NO2 | 49.5 ± 4.0 |
| 12 | MeO | 1 | 4-OMe | 43.6 ± 2.0 |
| 13 | MeO | 1 | 3,4-diOMe | 187.3 ± 13.6 |
| 14 | MeO | 1 | 3,4,5-triOMe | 74.2 ± 8.8 |
| 15 | MeO | 1 | 4-F | 32.9 ± 5.1 |
| 16 | F | 1 | H | 21.0 ± 2.1 |
| 17 | F | 1 | 4-NO2 | 30.9 ± 5.9 |
| 18 | F | 1 | 4-F | 30.9 ± 5.5 |
| 19 | F | 1 | 4-OMe | 37.8 ± 8.7 |
| 20 | F | 1 | 3,4-diOMe | 41.1 ± 0.6 |
| 21 | F | 1 | 3,4,5-triOMe | 38.6 ± 1.8 |
| 22 | Cl | 1 | H | 22.4 ± 2.0 |
| 23 | Cl | 1 | 4-NO2 | 28.7 ± 3.8 |
| 24 | Cl | 1 | 4-CF3 | 10.9 ± 1.9 |
| 25 | Cl | 1 | 4-OMe | 34.8 ± 7.9 |
| 26 | Cl | 1 | 3,4-diOMe | 38.9 ± 7.8 |
| 27 | Cl | 1 | 3,4,5-triOMe | 44.4 ± 3.6 |
| 28 | Cl | 1 | 4-N,N-dimethylamino | 88.3 ± 2.2 |
| 29 | Cl | 1 | 4-F | 4.8 ± 2.0 |
| 30 | Cl | 1 | 2-F | 26.0 ± 0.9 |
| 31 | Cl | 1 | 3-F | 5.9 ± 1.4 |
| 32 | H | 1 | H | 80.7 ± 22.4 |
| 33 | H | 1 | 4-NO2 | 47.9 ± 9.5 |
| 34 | H | 1 | 4-F | 25.5 ± 7.1 |
| 35 | Cl | 2 | H | 68.0 ± 9.0 |
| 36 | Cl | 2 | 4-NO2 | 52.5 ± 2.0 |
| 37 | Cl | 2 | 4-F | 51.2 ± 1.6 |
| UCF501 | 67.0 ± 8.0 | |||
| CQ | 174.0 ± 15.5 | |||
To study the effect of heterocycles and carbocycles other than benzenoid on the antiplasmodial potency, 2-arylvinylquinolines 39–57 (Scheme 2 and Table 2) were synthesized from 2-methylquinolines 6a–c, following the same synthetic sequence as shown for styrylquinolines 8–37.
Scheme 2. Synthesis of 6-Substituted 2-Arylvinylquinolines 39–57a.
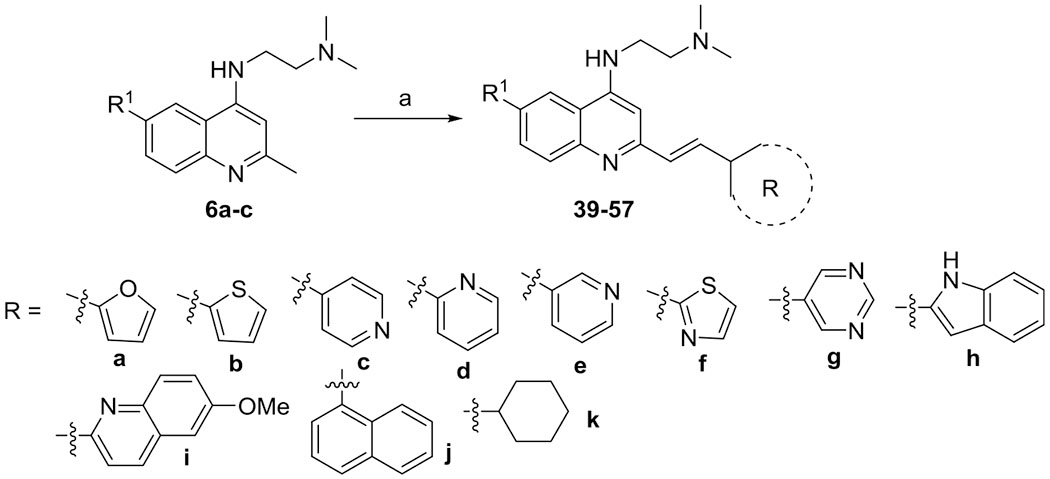
aReagents and conditions: (a) p-TsNH2, aldehydes 38a–k, xylene, 130 °C, 12 h, 48–86%.
Table 2.
Antiplasmodial Activity of SAR 2 Targets against the Dd2 Strain
 |
To investigate the influence of the double bond on the antimalarial activity, 2-pyridylethylquinolines 58 and 59 were prepared in good yields (Scheme 3 and Table 3) through the reduction of 2-pyridylvinylquinolines (41 and 50) with hydrazine hydrate at 80 °C.36
Scheme 3. Synthesis of 6-Substituted 2-Alkylquinolines 58–59a.
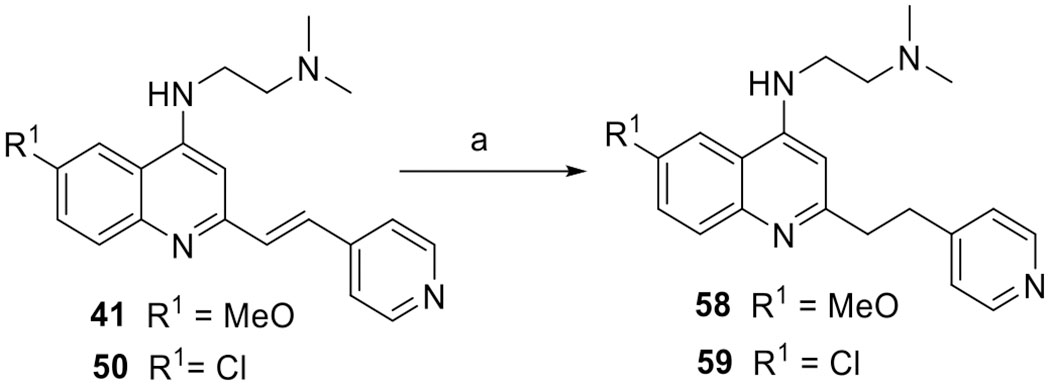
aReagents and conditions: (a) hydrazine hydrate, EtOH, 80 °C, 36 h, 81–83%.
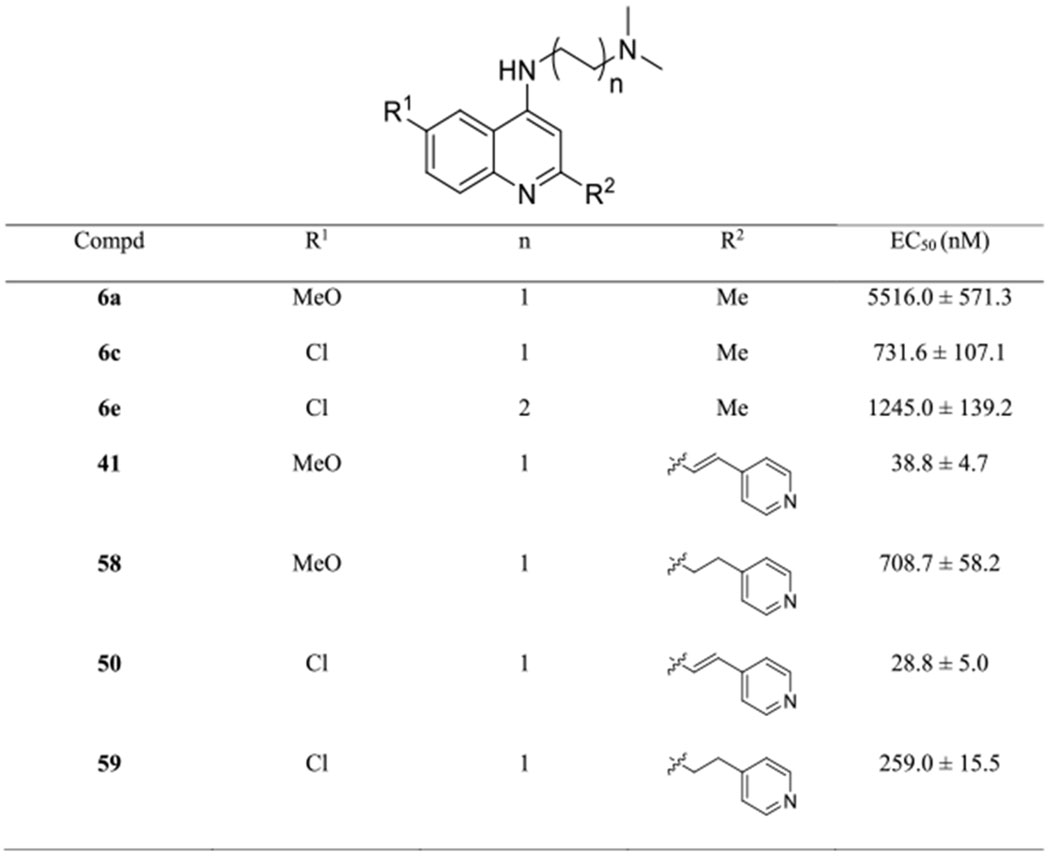
Table 3.
Antiplasmodial Activity of SAR 3 Targets against the Dd2 Strain
 |
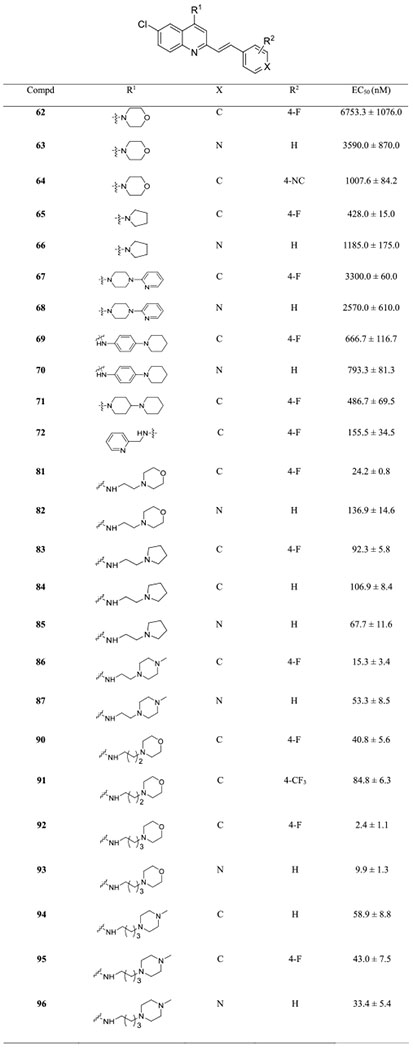
To understand the effect of the C4-amino group on the antimalarial potency, arylvinylquinoline derivatives 62–72, 81–87, and 90–96 were synthesized (Table 4). The synthetic route of 62–72 is depicted in Scheme 4, using a similar chemistry as described in Scheme 1. A growing number of studies demonstrated that the isonitrile group displays good antimalarial activity,37–40 and thus, we prepared compound 64 for antimalarial evaluation. Initially, we employed the direct olefination reaction to construct the arylvinylquinoline motif, but no discernible amount of 64 was detected. Therefore, an alternative synthetic route was adopted to prepare isonitrile 64 (Scheme 5). In detail, benzyl bromide 73 was reacted with a stoichiometric amount of triethylphosphite to give phosphonate 74 via an Arbuzov reaction,41 which was then converted to amine 75 by the reduction of the nitro group. Isonitrile 76 was then obtained by treatment of amine 75 with chloroform in the presence of a base. Subsequently, aldehyde 77, derived from selenium dioxide oxidation of 2-methylquinoline 61a,42 was reacted with phosphonate 76 to afford isonitrile 64 in moderate yield via a Horner–Wadsworth–Emmons reaction.43
Table 4.
Antiplasmodial Activity of SAR 4 Targets against the Dd2 Strain
 |
Scheme 4. Synthesis of 4-Substituted-arylvinylquinolines 62–72a.
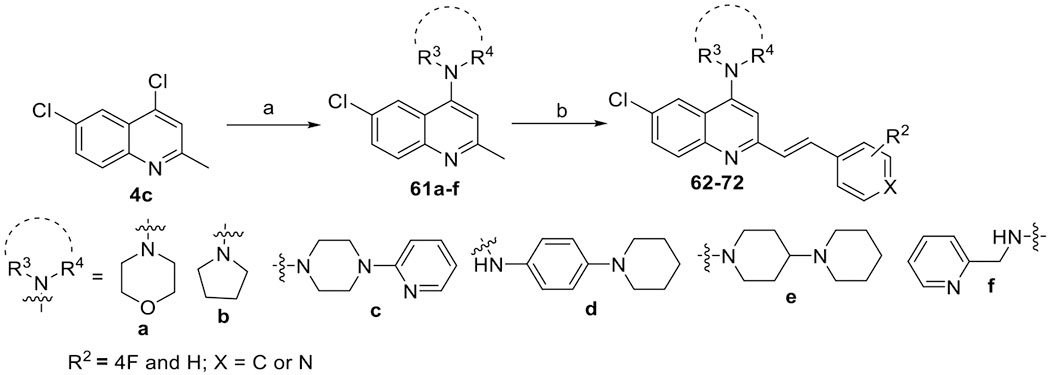
aReagents and conditions: (a) amines 60a–f, 130 °C, EtOH, 12 h, 73–92% and (b) aldehydes 7a, 7h, or 38c, p-TsNH2, xylene, 130 °C, 12 h, 48–89%.
Scheme 5. Synthesis of Isonitrile Styrylquinoline 64a.
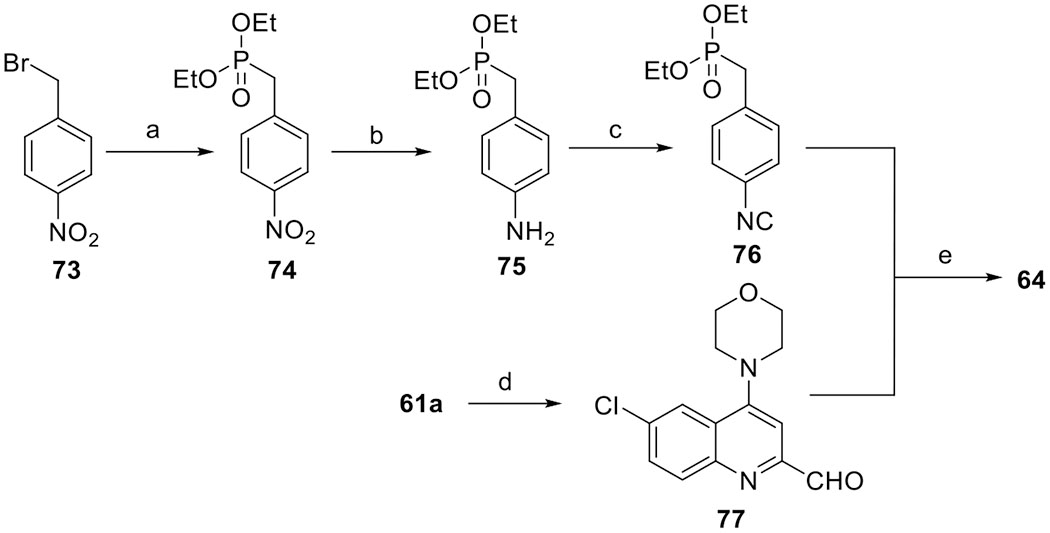
aReagents and conditions: (a) triethylphosphite, reflux, 20 h, 88%; (b) Pd/C, H2, MeOH, overnight, 75%; (c) 50% NaOH, CHCl3, tetrabutylammonium bromide, DCM, rt to 40 °C, 2 h, 80%; (d) SeO2, 1,4-dioxane, 80 °C, 6 h, 32%; and (e) potassium tert-butoxide, anhydrous DMF, rt, 1 h, 58%.
Diversification of the amino group was achieved through nucleophilic substitutions. For example, aminoquinolines 80a–c were prepared by mixing chloroquinoline 4c with the amino alcohol followed by mesylation and substitution. With aminoquinolines 80a–c in hand, 2-arylvinylquinolines 81–87 were obtained by reacting with appropriate aldehydes (Scheme 6).
Scheme 6. Synthesis of 4-Aminoarylvinylquinolines 81–87a.
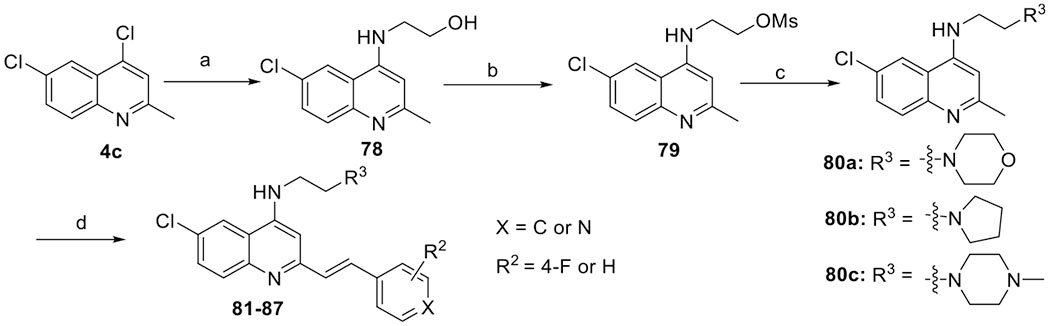
aReagents and conditions: (a) 2-aminoethanol, EtOH, 130 °C, 48 h, 97%; (b) MsCl, Et3N, THF, 0 °C, 1 h, 72%; (c) K2CO3, amines, anhydrous CH3CN, reflux, overnight, 80–94%; and (d) aldehydes 7a, 7h, or 38c, p-TsNH2 xylene, 130 °C, 12 h, 79–91%.
The designed compounds 90–96, containing propylamine and butylamine moieties, were synthesized in two steps (Scheme 7). First, treatment of quinoline 4c with appropriate amines 88a–c furnished aminoquinolines 89a–c, and second, aminoquinolines 89a–c were converted to 90–96 by the corresponding olefination reaction.
Scheme 7. Synthesis of 4-Aminoarylvinylquinolines 90–96a.
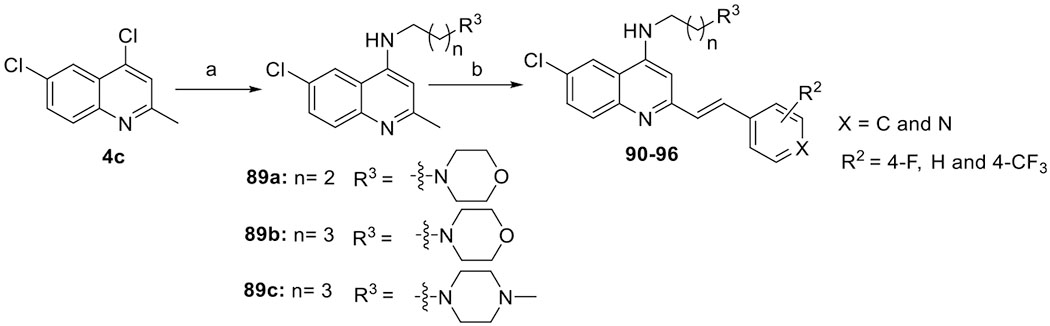
aReagents and conditions: (a) aliphatic amines 88a–c, 130 °C, 24 h, 86–93% and (b) aldehydes 7a, 7h, 7j, or 38c, p-TsNH2, xylene, 130 °C, 12 h, 67–88%.
Next, we turned our attention to the analogues containing various fluorinated substituents and conjugated double bonds. The synthesis of arylvinylquinolines 98–114 was commenced from methylquinoline 89b and the appropriate aldehydes 7i or 97a–p (Scheme 8 and Table 5), using the identical procedures described for compounds 90–96.
Scheme 8. Synthesis of 4-Morpholinobutylaminoarylvinylquinolines 98–114a.
aReagents and conditions: (a) aldehydes 7i and 97a–p, p-TsNH2, xylene, 130 °C, 12 h, 52–92%.
Table 5.
Antiplasmodial Activity of SAR5 Targets against the Dd2 Strain
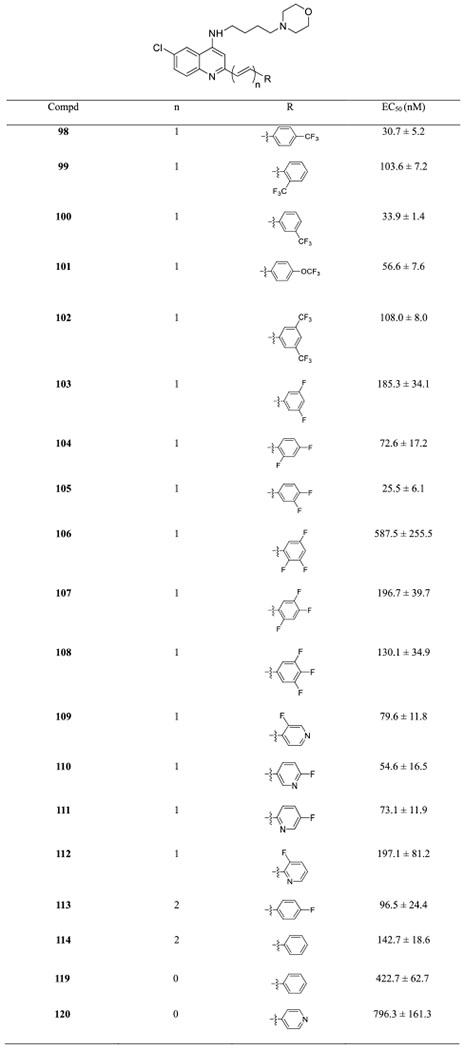 |
To evaluate the importance of the double bond, arylquinolines 119 and 120 were synthesized from 4-chloroaniline 1c (Scheme 9 and Table 5). Briefly, aniline 1c and ethyl benzoylacetate 115 or ethyl isonicotinoyl acetate 116 were condensed in the presence of PPA to give hydroxyquinolines 117a or 117b, which were transformed into chloroarylquinolines 118a or 118b, respectively. Subsequent nucleophilic substitution with 4-morpholinobutanamine furnished 4-aminoarylvinylquinolines 119 and 120, respectively.
Scheme 9. Synthesis of 4-Morpholinobutylaminoarylquinolines 119–120a.
aReagents and conditions: (a) ethyl benzoylacetate 115 or ethyl isonicotinoyl acetate 116, 150 °C, PPA, 6 h; (b) phosphorus oxychloride, 105 °C, 2 h, 20–45% for two steps; and (c) 4-morpholinobutanamine, 130 °C, 24 h, 43–49%.
Biology.
In Vitro Antiplasmodial Activity and Cytotoxicity.
The SAR studies were focused on improving the in vitro activity of 2-arylvinylquinolines against the CQ-resistant Pf Dd2 strain.
SAR1 Targets.
A series of styrylquinoline analogues 8–34 bearing various C6 substituents (R1) and benzenoid substituents (R2) were evaluated for their in vitro activity against the Dd2 strain (Table 1).
In the C6-methoxy series, compound 9 (R2 = 4-NO2, EC50 = 28.6 ± 0.9 nM) exhibited slightly higher inhibitory activity than the unsubstituted compound 8 (R2 = H, EC50 = 41.2 ± 5.3 nM). Moving the nitro group from the para position to ortho or meta position led to decreased activity, as seen with compounds 10 (EC50 = 56.3 ± 8.1 nM) and 11 (EC50 = 49.5 ± 4.0 nM). Replacement of the nitro group with 4-methxoy, 3,4-dimethoxy, and 3,4,5-trimethxoy groups afforded compounds 12–14, respectively, which were less potent than compound 9.
The introduction of a fluorine atom at the C6 position improved antiplasmodial activity over the corresponding methoxylated analogues. For instance, compound 16 (R2 = H, EC50 = 21.0 ± 2.1 nM) and compound 21 (R2 = 3,4,5-trimethoxy, EC50 = 38.6 ± 1.8 nM) showed almost twofold greater activity than the counterparts 8 and 14. Another noteworthy observation was that fluorinated analogue 20 with a 3,4-dimethoxy group exhibited approximately 4.5-fold improved activity as compared to compound 13.
Substitution of the fluorine atom by a chlorine atom led to further enhancement of the antiplasmodial activity. Chlorostyrylquinolines bearing a fluoro or trifluoromethyl group on the benzene ring showed potent activity against the Dd2 strain. Among these analogues, compound 29 (R2 = 4-F) was the most active one with an EC50 value of 4.8 ± 2.0 nM, which was almost 14-fold more potent than UCF501. Compared with compound 29, compounds 24 and 31 demonstrated slightly lower inhibitory activity with EC50 values of 10.9 ± 1.9 and 5.9 ± 1.4 nM, respectively. However, altering the fluorine atom from the para position to the ortho position led to a fivefold decrease in activity as observed in compound 30 (EC50 = 26.0 ± 0.9 nM). For all styrylquinolines (R1 = −OMe, −F, and −Cl), the introduction of electron-rich groups (R2) proved to be detrimental to the antiplasmodial potency, as seen with compounds 12–14, 19–21, and 25–28, which were less active relative to their analogues (R2 = H and 4-NO2).
Removal of the C6 substituent (R1 = H) caused a significant drop in the antiplasmodial activity. For example, compound 32 (R1 = H, EC50 = 80.7 ± 22.4 nM) exhibited a much lower activity than the corresponding analogues 8 (R1 = MeO), 16 (R1 = F), and 22 (R1 = Cl). Therefore, the general trend of the substituents at the C6 position in the order of improved potency is H < OMe < F < Cl.
We also briefly investigated the spacing parameter for the C4 amino side chain of styrylquinolines. As shown in Table 1, significant loss of potency was observed for the compounds containing a dimethylaminobutyl group. It appeared that for styrylquinolines, dimethylaminoethylamine was a superior side chain to dimethylaminobutylamine.
SAR2 Targets.
Having determined the impacts of dimethylaminoalkyl and halogen substituents on the antiplasmodial activity, we next tested if the replacement of the phenyl ring (R) with heterocycles and nonbenzenoid carbocycles would affect the inhibitory activity against the Dd2 strain (Table 2). Substitutions of the benzenoid motif by five-membered aromatic heterocycles including furan (compounds 39, 44, and 47), thiophene (compounds 40, 45, and 48), and thiazole (compound 49) led to reduced antiplasmodial activity. However, replacement of the phenyl group with a 4-pyridyl group (compounds 41, 46, and 50) retained the antiplasmodial potency. Importantly, the position of the nitrogen atom significantly influenced the antiplasmodial activity. For instance, 4-pyridylvinylquinoline 50 was approximately twofold more active than compound 51 bearing the 2-pyridylvinyl group. This manifestation became more evident for methoxylated pyridylvinylquinolines because the activity difference was up to 30-fold, for example, compounds 41 (R = 4-pyridyl, EC50 = 33.6 ± 5.8 nM) and 42 (R = 2-pyridyl, EC50 = 1032.8 ± 232.9 nM).
Moreover, chlorinated arylvinylquinolines were more potent than the corresponding fluorinated and methoxylated analogues in this series. For instance, compound 47 (R1 = Cl, EC50 = 37.0 ± 4.3 nM) showed a twofold higher activity than compounds 39 (R1 = MeO, EC50 = 88.7 ± 2.3 nM) and 44 (R1 = F, EC50 = 82.6 ± 9.4 nM). Accordingly, the work provided further support that the chlorine atom at the C6 position was superior to the fluorine atom and methoxy substituent for antiplasmodial potency.
In addition, we found that replacement of pyridine by other heterocycles such as pyrimidine, indole, and quinoline resulted in a marked loss of potency, as seen with the corresponding arylvinylquinolines 53 (EC50 = 155.0 ± 11.6 nM), 54 (EC50 = 95.9 ± 6.7 nM), and 55 (EC50 = 281.3 ± 40.3 nM). Unfortunately, our search for compounds more active than 22 by incorporating carbocycles, such as naphthalene and saturated cyclohexane, to the vinylquinoline scaffold was unsuccessful.
SAR3 Targets.
Compared with 4-aminoquinolines, for example, CQ, a unique feature of our lead compound is the vinyl group that bridges the quinoline core and the aromatic ring. In this series, we intended to assess the impacts of the double bond on antiplasmodial activity. As illustrated in Table 3, in the absence of the arylvinyl group, aminoquinolines 6a, 6c, and 6e were inactive against the Dd2 strain. Even with an aromatic group, the saturated analogues showed almost an order of magnitude weaker activity than the vinyl analogues, as demonstrated by pyridylethylquinoline 58 (EC50 = 708.7 ± 58.2 nM) versus 41 (EC50 = 38.8 ± 4.7 nM) and pyridylethylquinolines 59 (EC50 = 259.0 ± 15.5 nM) versus 50 (EC50 = 28.8 ± 5.0 nM). Therefore, these data indicated that the absence of the 2-arylvinyl moiety severely reduced antiplasmodial activity, which was in line with recent studies.29,44
SAR4 Targets.
Having identified the optimal substituent at the C6 position and the aromatic motif (phenyl and pyridyl) at C2 position, we shifted our focus to exploring nitrogen-containing groups at C4 position. As a result, the first subset of analogues 62–72 was prepared by incorporating morpholine, pyrrolidine, 1-(2-pyridyl)piperazine, 4-piperidinoaniline, and bipiperidine directly to the arylvinylquinoline scaffold, and they were screened for their antiplasmodial activity (Table 4). These compounds generally showed moderate to low activity against the Dd2 strain, with EC50 values ranging from 428.0 ± 15.0 to 6753.3 ± 1076.0 nM. Unexpectedly, the isonitrile compound 64 did not show significant antiplasmodial activity (EC50 > 1000 nM). Meanwhile, we found that the arylvinylquinoline (R1 = 4-NC) containing the N,N-dimethylaminoethylamino group was less potent than compound 29 (data not shown). Compound 72 bearing a 2-picolylmethylamine moiety, the most active compound in this subset, had an EC50 value of 155.5 ± 34.5 nM.
Inspired by the abovementioned results, the second subset of analogues 81–96 was synthesized by attaching different alkylamines. These analogues displayed promising activities with EC50 values below 100 nM, and the majority of them were more potent than the positive control UCF501. The nitrogen atom spacing was also screened in this subset of analogues. For the morpholinylalkylamine series, arylvinylquinolines with a tetramethylene linker showed a much higher activity than that of di- or trimethylene linkers as demonstrated by the most potent compounds 92 (EC50 = 2.4 ± 1.1 nM) and 93 (EC50 = 9.9 ± 1.3 nM), displaying almost a 2-fold and 13-fold improved potency as compared with the counterparts 81 and 82, respectively. However, this linker length preference was inconclusive for the N-methylpiperazinylalkylamine series. For example, compound 96, containing a 4-carbon linker, was slightly more active compared to the corresponding analogue 87. However, compound 86 bearing a 2-carbon linker was nearly 2.5-fold more potent than analogue 95.
SAR5 Targets.
After C4-substitution optimization, we used arylvinylquinolines containing a 4-morpholinobutanamine motif (92 and 93) as benchmark molecules for further investigation. In this series, we focused on the C2 modification by fluorine-containing aromatics. Given the wide use of fluorine substitution in drug discovery to improve biological activity, permeability, and pharmacokinetic issues45–47 and in light of morpholine as a privileged structure with advantageous physicochemical, biological, and metabolic properties,48 it was expected to produce synergistic effects when these two motifs were combined. For this reason, we prepared arylvinylquinolines 98–114 with the optimal 4-morpholinobutanamine at the C4 position to evaluate their capability to suppress the growth of the Dd2 strain. The results are summarized in Table 5. Compound 100 with a 3-trifluoromethyl group showed comparable activity to its para-positional isomer 98, which was threefold more potent than the ortho isomer 99 (EC50 = 103.6 ± 7.2 nM). This result provided additional evidence that introducing orthosubstituents at the phenyl ring may compromise the antiplasmodial potency. Replacement of the 4-trifluoromethyl group by the 4-trifluoromethoxy substituent afforded compound 101 with a slightly decreased activity. A noteworthy observation was that introduction of the disubstituted fluorinated phenyl ring caused a significant loss of antiplasmodial potency as observed in analogues 102–105. We also observed that compounds 105 and 104 were nearly 7- and 2.5-fold more active than 103, respectively, confirming that the fluorine substituent at the para position of the phenyl ring was favorable to improve antiplasmodial activity. Further fluorination on the phenyl ring dramatically reduced activity, rendering compounds 106–108 less active than the benchmark molecule 92. Collectively, the SAR study demonstrated that monofluorination at the para position is the best choice within the current scope of screening and any positional deviation or excessive fluorination is disadvantageous for antiplasmodial potency. Additionally, these results suggested that there is limited space available for target interactions, with substituent ortho position and steric effects being particularly sensitive.
Deviating from our expectations, the introduction of a fluorine atom at the pyridine ring did not increase antiplasmodial activity. For instance, compound 109 with a 3-fluoro substituent (EC50 = 79.6 ± 11.8 nM) was almost 7.5-fold less active than its counterpart 93 (Table 4, EC50 = 9.9 ± 1.3 nM). Interestingly, both pyridylvinylquinolines 109 and 111 demonstrated much weaker inhibitory activity than compound 110 (EC50 = 54.6 ± 16.5 nM). Additionally, among this series, compound 112 bearing a 3-fluoropyridine moiety displayed the weakest inhibitory effect on the Dd2 strain with an EC50 value of 197.1 ± 16.5 nM. Again, these results implied that the ortho substitution at the aromatic ring could intervene the target interactions that were quite sensitive to the steric effect and the orientation of the aromatic ring.
Analogues with a different number of double bonds (113–114 and 119–120) were also assessed for their in vitro antiplasmodial activity. Compounds 113 and 114 with two double bonds exhibited a much lower activity than the benchmark compounds, implying that a single double bond is a better choice for improving the antiplasmodial potency. Additional support for the crucial role of the vinyl group came from the evaluation of analogues without any double bond. As shown in Table 5, a loss of activity was observed in arylquinolines 119 and 120 (EC50 > 400 nM). Thus, we concluded that a single double bond between the quinoline core and the aromatic ring is required for the antiplasmodial activity, highlighting the uniqueness of our chemical scaffold.
Upon completion of the SAR study on the Dd2 strain, several bioactive arylvinylquinolines were selected for further antiplasmodial activity assay using the CQ-sensitive 3D7 strain (Tables S1 and 6). Methoxylated styrylquinolines (8 and 13–14) bearing the dimethylaminoethylamine moiety exhibited much stronger inhibitory activity against the CQ-sensitive 3D7 strain than the CQ-resistant Dd2 strain (resistance index (RI) ≥ 1.5), whereas all fluorinated and chlorinated analogues were more potent against the Dd2 strain than the 3D7 (RI < 1), except for compound 28 (RI = 1.3), indicative of no cross-resistance induced by these vinylquinolines between the CQ-resistant and CQ-sensitive parasites. In contrast, the RI of CQis almost 10. In this series, the SAR trends observed for the 3D7 strain were similar to those observed for the Dd2 strain. For instance, compound 30 with a 2-fluoro group (EC50 = 55.9 ± 9.5 nM) showed remarkably diminished potency as compared to the corresponding 4-fluoro (compound 29, EC50 = 8.7 ± 0.5 nM) or 3-fluoro analogues (compound 31, EC50 = 23.0 ± 2.8 nM). Besides, removal of the methoxy group or fluorine atom at C6 position of the styrylquinoline scaffold resulted in decreased activity, as seen with compounds 32 versus 8, and 32 versus 16. In addition, 2-pyridylvinylquinoline 51 was less potent than its corresponding isomers 50 and 52 against the 3D7 strain. All pyridylvinylquinolines demonstrated highly potent activity against the Dd2 strain as compared to the 3D7 strain (RI < 1), as seen with compounds 41, 46, and 50–52. Interestingly, substitution of pyridine in compound 50 by other nitrogen-containing heterocycles (e.g., pyrimidine, indole, and quinoline) and carbocycles (e.g., naphthalene and cyclohexane) led to respective compounds 53–57, which displayed a significantly stronger growth-inhibitory effect on the 3D7 strain than the Dd2 strain (RI ≥ 1.3) (Table S1). These results indicated that for the R group, phenyl and pyridyl are favorable to overcome cross-resistance.
Table 6.
Selectivity and Resistance Indices of Synthesized Analogues
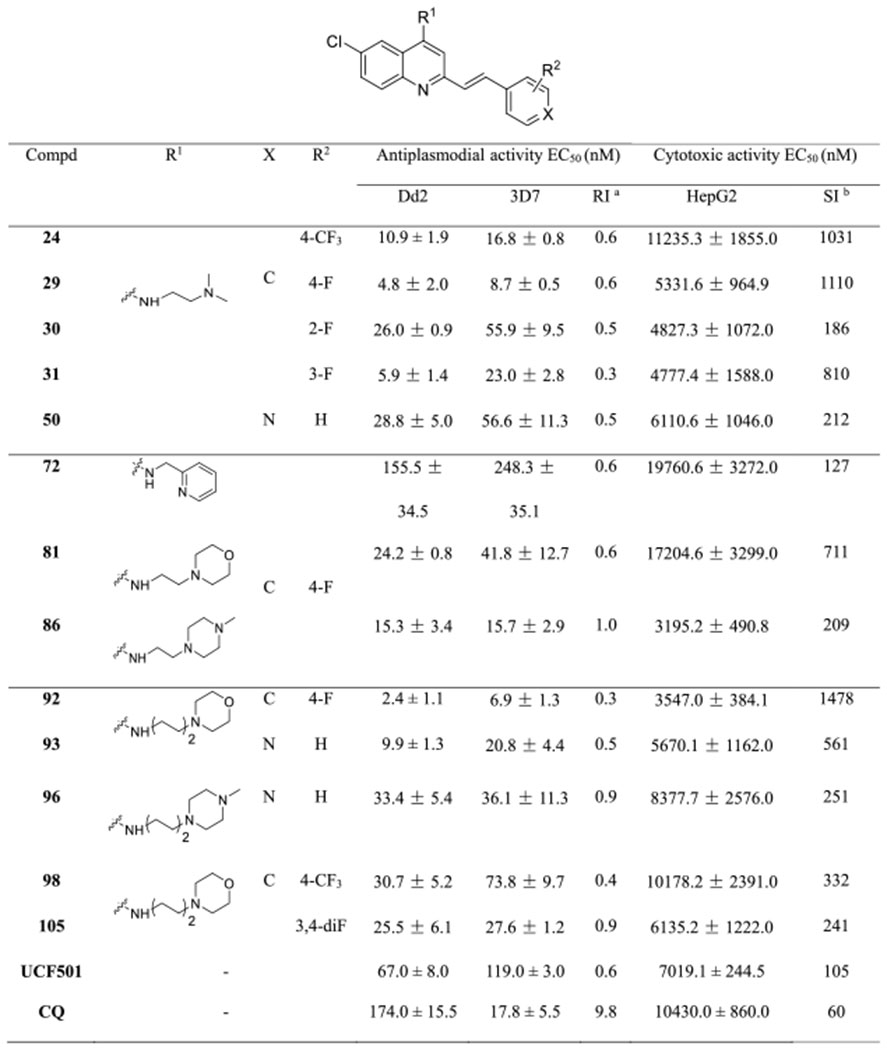 |
RI = Dd2 EC50/3D7 EC50.
SI = HepG2 EC50/Dd2 EC50.
A similar observation was that arylvinylquinolines (64 and 67–72) devoid of the methylene linker exhibited moderate to low activity against the 3D7 strain with EC50 values in the range of 155.0 ± 6.0 to 2643.1 ± 214.8 nM (Table S1). By comparison, incorporation of the morpholinoalkylamino or N-methylpiperazinylalkylamino group into the arylvinylquinoline scaffold markedly enhanced the antiplasmodial activity against the 3D7 strain. These results further confirmed that the introduction of the flexible alkylamine moiety on the arylvinylquinoline scaffold was beneficial to the antiplasmodial activity. It was worth noting that among this subset, arylvinylquinolines (106–107, 110, and 112) containing the 4-morpholinobutanamino motif showed significantly improved inhibitory activity against the 3D7 strain relative to the Dd2 strain. In addition, compound 110 was almost fivefold more potent than compound 112 against the 3D7 strain, further corroborating the result that the positional effect of the nitrogen atom and fluorine atom on the pyridyl ring dramatically influenced the antiplasmodial activity.
Selected compounds were also evaluated for their cytotoxicity against HepG2 cells (Table 6). In most cases, the cytotoxicity profiles of these compounds correlated well with their antiplasmodial activity, as demonstrated by compounds 24 versus 29, 29 versus 72 or 81, 72 versus 81 or 86, 81 versus 92, 86 versus 96, 93 versus 96, and 98 versus 105. For instance, compound 92 containing a tetramethylene linker exhibited a nearly 6-fold increase in inhibitory activity against the 3D7 strain and a 4.8-fold increase in cytotoxicity relative to compound 81. Nevertheless, the cytotoxic activity of compound 24 was comparable to compound 98, although compound 24 demonstrated more potent antiplasmodial activity. Additionally, we observed that the position of the fluorine atom at the phenyl ring has a marginal effect on cytotoxicity, as seen with compounds 29–31. Notably, all compounds tested showed high selectivity profiles (selectivity index (SI) > 120), especially for compounds 24, 29, and 92 (SI > 1000), indicating good safety windows.
Metabolic Stability and Preliminary Metabolite Identification.
To assess the metabolic stability, selected compounds listed in Table 7 were subjected to an in vitro microsomal turnover assay with mouse liver microsomal preparations. This assay determines the percentage of the parent compound residues after 60 min of incubation (Table 7). We identified that compounds bearing the 4-trifluoromethyl group on the phenyl ring exhibited a better metabolic stability than that with the 4-fluoro group, as demonstrated by compounds 24 (t1/2 = 104.2 min) versus 29 (t1/2 = 55.2 min), and 92 (t1/2 = 22.2 min) versus 98 (t1/2 = 64.9 min). The intrinsic stability of the N,N-dimethylaminoethylamino moiety was superior to the 2-pyridinemethanamino or 2-(4-methylpiperaziyl)ethanamino groups, for example, compounds 29 versus 72 (t1/2 = 65.3 min) and 86 (t1/2 = 60.3 min). The morpholine motif appeared to be the most labile group upon enzymatic degradation as seen with compound 92 which had the shortest half-life time (t1/2 = 22.2 min) in this series. Additionally, we observed that replacing the phenyl ring with pyridine in arylvinylquinoline rendered the compound more susceptible to hepatic metabolism, such as in the case of compounds 50 (t1/2 = 22.3 min) and 93 (t1/2 = 14.4 min). This suggested that the pyridyl ring had no advantages over benzenoids in microsomal stability. The assay indicated that the C4 amino side chain and the arylvinyl group significantly influenced compound metabolic stability.
Table 7.
In Vitro Metabolism in Mouse Liver Microsomes
| compd | % remaining after 60 min | projected t1/2 (min) | (μL/min/mg) |
|---|---|---|---|
| 24 | 67.1 | 104.2 | 13.3 |
| 29 | 47.1 | 55.2 | 25.1 |
| 31 | 38.2 | 43.2 | 32.1 |
| 50 | 15.5 | 22.3 | 62.2 |
| 72 | 52.9 | 65.3 | 21.2 |
| 86 | 50.2 | 60.3 | 23.0 |
| 92 | 15.4 | 22.2 | 62.4 |
| 93 | 5.6 | 14.4 | 96.2 |
| 98 | 52.7 | 64.9 | 21.4 |
| 105 | 19.9 | 25.8 | 53.7 |
| UCF501 | 36.7 | 41.5 | 33.4 |
| 47.8a | 56.2a |
Values reported from the initial study.33
In the context of the present study, the metabolite mixture was analyzed by liquid chromatography–mass spectrometry (LC–MS) after 15, 30, and 60 min of incubation. N-dealkylation from the tertiary terminal amine and oxidation of the arylvinylquinoline scaffold appeared to be the major pathways for metabolic decomposition, which is consistent with the previous studies on 4-aminoquinolines metabolism,4,8,49 and P450 mediated oxidation in liver microsomes.8,50 For example, the microsomal metabolites of UCF501 were primarily derived from N-deethylation of the tertiary amine (monodeethyl UCF501 and UCF501-M1), O-demethylation (UCF501-M2), oxidation of styrylquinoline (UCF501-M3), and the reduction of the nitro group (three minor metabolites, Figure S1). The tentatively assigned metabolites of compound 29 included monodemethylated (29-M1), bi-demethylated (29-M2), and oxidized products (29-M3) (Figure S3). The same decomposition pathway was not observed for compound 24 containing an identical C4 side chain (Figure S2), which could explain the different metabolic stability profiles of 24, 29, and UCF501. Although N-demethylation was the primary metabolic route for compound 24, its half-life time was acceptable as compared to related compounds in other literature reports.8,27,51,52
Demethylated (86-M1) and oxidized products (86-M2) were identified as the possible metabolites of compound 86 (Figure S4). The metabolic instability of arylvinylquinolines 92, 93, 98, and 105 was largely attributed to the breakage of the morpholine ring (alkanolamine metabolite M1 and primary amine metabolite M2). Among this series, compound 93 was the most susceptible to hepatic metabolism, and oxidative metabolites 93-M3 and 93-M4 were observed in addition to ring-opening metabolites M1 and M2 (Figure S5).
2-Arylvinylquinolines Block the Trophozoite Stage in the Pf Asexual Life Cycle.
To understand the antiplasmodial activity of 2-arylvinylquinolines, we decided to establish the developmental stage specific action of the most promising compounds 24, 29, and 86 by microscopic and flow cytometric analysis.33,53 Tightly synchronized cultures were exposed to 5 × EC50 concentration of compound 24 at 6, 18, 30, and 42 hours post invasion (HPI) of the merozoites. Dihydroartemisinin (DHA, 50 nM), atovaquone (6.6 nM), and a dimethyl sulfoxide (DMSO) vehicle were included as controls. Microscopic analysis of Giemsa-stained thin smears and flow cytometric evaluation were performed at 12 h intervals. As seen in Figure 2A, the untreated control cultures underwent normal cell cycle progress through the trophozoite (18 HPI), early schizont (30 HPI), late schizont/segmenter (42 HPI), and reinvaded ring (54 HPI) with an increased peak height (Figure 2A,B). In contrast to untreated cultures, the cycle progression of compound 24-treated cultures was blocked when the compound was added at 6, 18, and 30 HPI (Figure 2A,B). However, the main effect seems to be in the transition between late trophozoite and schizont stages. In contrast, the addition of compound 24 at a later stage (42 HPI) did not affect the schizont maturation or the reinvasion of new merozoites (Figure 2A,B). Similarly, compounds 29 and 86 demonstrated stage-specific action at the late trophozoite phase in Dd2 cultures (Figures S6 and S7). The stage-specific inhibition in the DHA and atovaquone-treated cultures replicated the expected pattern, inhibition of all stages in the case of DHA and no inhibition in late stages with atovaquone (Figures S8 and S9). Thus, these results indicated that 2-arylvinylquinolines inhibit blood stage parasites by acting in the late trophozoite phase in the Pf asexual life cycle.
Figure 2.
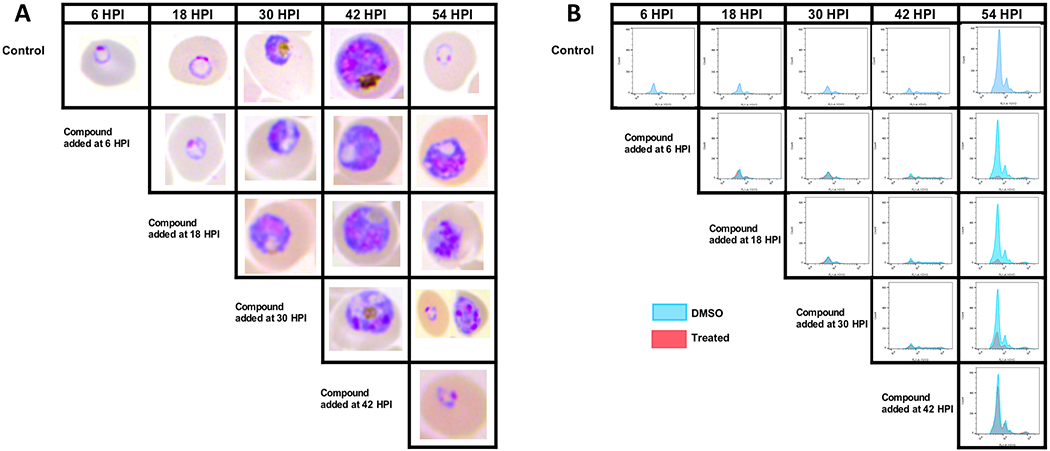
Stage-specific inhibition of Pf growth by compound 24. Tightly synchronized Dd2 parasites were treated at 6, 18, 30, and 42 HPI with compound 24 at 5 × EC50 concentration. Samples for Giemsa staining and flow cytometry were collected every 12 h following compound addition. (A) Microscopic images of Giemsa-stained thin smears. (B) Histogram plot of YOYO-1-labeled cells from flow cytometric analysis. Results shown are representative of three independent biological replicates.
Ultrastructural Effects of 2-Arylvinylquinolines.
A notable morphological change in parasites upon exposure to compound 24 is the appearance of large vacuoles. To explore these morphological changes, parasites were exposed to compound 24 at 5 × EC50 for 1.5, 3, 6, or 12 h and processed for transmission electron microscopy (TEM). Micrographs revealed the appearance of membrane-bound structures within the parasite DV (Figure 3). These structures were observed as early as 1.5 h following compound exposure in the late trophozoite–early schizont stages and persisted until 12 h post exposure (Figure 3). The enlarged food vacuoles (FVs) observed in the Giemsa-stained parasites maybe due to the accumulation of undegraded hemoglobin, as has been reported previously in parasites treated with inhibitors targeting proteases in the parasite FV.54 Similar morphological alterations observed in the DV in TEM images have been reported in the parasite treated with piperaquine.55 Although the mechanism of action of the piperaquine has not been elucidated, genetic changes in plasmepsin II and III have been associated with clinical resistance to this drug.56 Plasmepsin II is a key enzyme in the degradation of the host hemoglobin.57 The presence of this phenotype in parasites treated with compound 24 and the fact that the compound acts on the trophozoite when most hemoglobin degradation occurs suggest that hemoglobin digestion could be a potential cellular target of 24.
Figure 3.
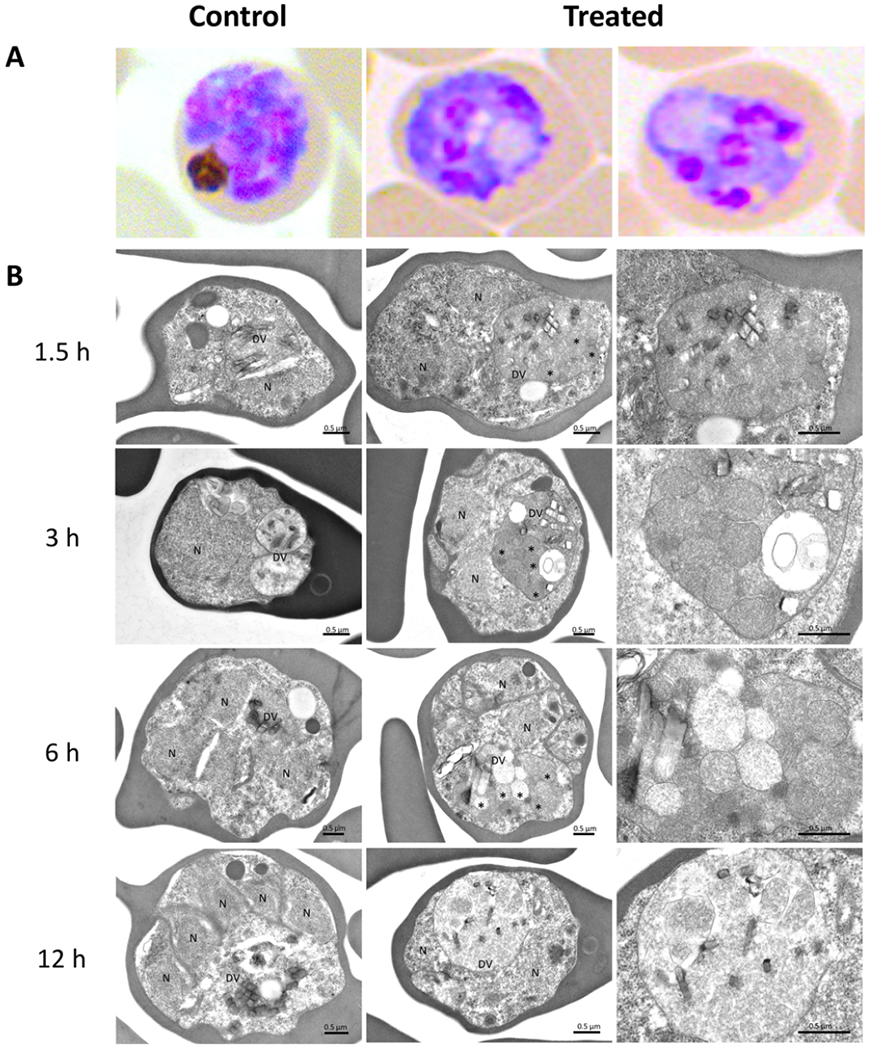
Transmission electron micrograph of Pf Dd2 exposed to 5 × EC50 compound 24. (A) Giemsa-stained image of representative parasites processed for TEM. A large vacuole is observed in the cytosol of treated parasites. (B) Micrographs of the parasite after treatment with compound 24 for increasing periods of time. Parasites treated with 0.15% DMSO used as a control. Micrographs of the treated parasite show membrane-bound structures within DV (*), suggesting undigested hemoglobin vesicles. Details of DV from the treated parasite with undigested vesicles (*) and hemozoin crystals are shown in the far-right panel. Scale bar is 0.5 μm.
2-Arylvinylquinolines are Fast-Acting Parasitocidal Agents.
To study whether 2-arylvinylquinolines exerted their antiplasmodial activity through a parasitocidal or parasitostatic mechanism, time-kill kinetic experiments using the Dd2 strain were performed.33 Asynchronously growing Dd2 cultures were treated with 5 × EC50 concentrations of 2-arylvinylquinolines 24, 29, 86, DHA (50 nM), and atovaquone (6.6 nM) for different lengths of time (6, 12, 24, or 48 h). After removal of the inhibitors after the specified incubation times, culture growth was assessed for 96 h. DHA and atovaquone were used as references for fast- and slow-acting compounds, respectively. A notable reduction in parasitemias was observed following all treatment durations with compounds 24, 29, and 86, which was similar to the kill kinetic profile of DHA (Figure 4A–D).58 In contrast, as can be seen from Figure 4A–D, parasites exposed to atovaquone required a longer exposure (48 h) to induce a reduction in parasitemias, confirming its slow-acting antiplasmodial activity. Overall, these results suggested that 2-arylvinylquinolines produced rapid parasite clearance through a parasitocidal mechanism, making them ideal drug candidates.
Figure 4.
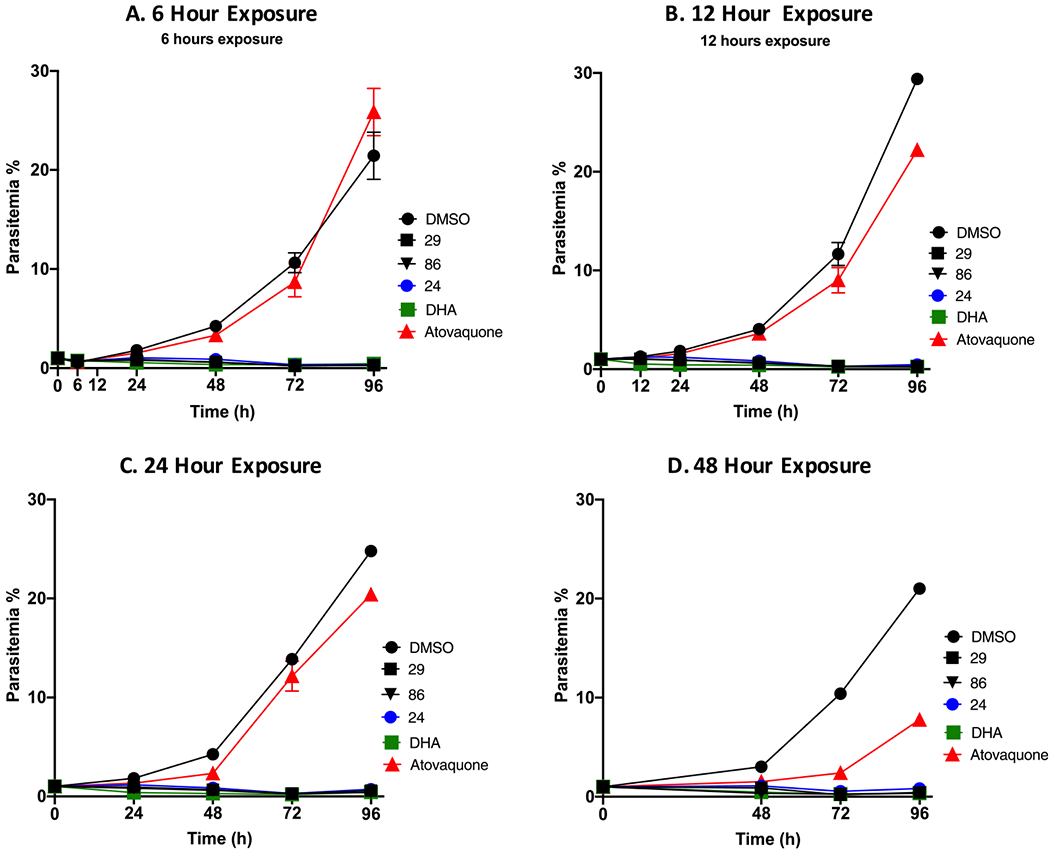
Parasitocidal activity of 2-arylvinylquinolines. Asynchronous Dd2 parasite culture was exposed to 5 × EC50 concentrations of test compounds for (A) 6, (B) 12, (C) 24, and (D) 48 h followed by compound removal. Parasite growth was then monitored daily for 96 h. DHA and atovaquone (50 and 6.6 nM) were included as fast- and slow-acting controls, respectively. Parasitemia was determined by microscopy of Giemsa-stained smear. Results shown are representative of three independent biological replicates. Mean ± SEM of three independent readings.
In Vitro Gametocytocidal Activity of 2-Arylvinylquinolines.
The in vitro gametocytocidal activity of 2-arylvinylquinolines 24, 29, and 86 is summarized in Table 8. All three compounds demonstrated potent inhibitory activity toward early-stage (II–III) gametocytes with sub-micromolar EC50 values. Late-stage (IV–V) gametocytes are more refractory to antimalarial drugs than early-stage gametocytes and blood stage parasites, and thus, few compounds were effective against late-stage Pf gametocytes.59 Encouragingly, all compounds tested displayed strong inhibitory activity toward late-stage gametocytes, among which, compound 24 was the most active molecule with an EC50 value of 393.6 ± 99.4 nM (Table 8 and Figure 5). Therefore, 2-arylvinylquinolines represent promising leads as new antimalarials with promising dual-stage (blood and gametocyte) activity.
Table 8.
Inhibition of Early- and Late-Stage Gametocytes
| compd | early-stage EC50 (nM)a | late-stage EC50 (nM)a |
|---|---|---|
| 24 | 471.5 ± 18.4 | 393.6 ± 99.4 |
| 29 | 590.7 ± 118.7 | 2049.3 ± 113.6 |
| 86 | 909.9 ± 314.2 | 2495.7 ± 423.8 |
| methylene blue | 74.7 ± 21.9 | 107.2 ± 46.3 |
| DHA | 17.4 ± 3.7 | 37.4 ± 8.9 |
EC50 data represent the means and SEMs of three separate experiments.
Figure 5.
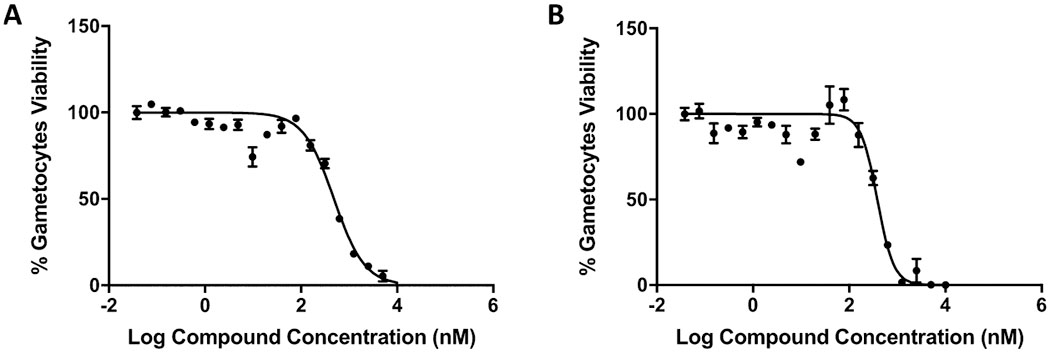
Activity of compound 24 on 3D7 P. falciparum gametocyte stages. The viability of gametocytes after the exposure of compound 24 was evaluated on early (A) or late (B) gametocyte stages of the 3D7-expressing luciferase parasite. EC50 data represent the means and SEMs of three experiments.
β-Hematin Inhibition Activity of 2-Arylvinylquinolines.
CQ and other aminoquinolines are known to act on the malaria parasites by blocking hematin biocrystallization (hemozoin formation) through π–π stacking. As a result, the accumulated toxic heme causes the death of parasites by inducing oxidative membrane damage.9,60 We performed β-hematin inhibition experiments to determine the possible mode of actions of arylvinylquinolines. As shown in Figure 6A,B, CQ strongly inhibited β-hematin formation. Within the three tested compounds, 29 and 86 also showed inhibitory activity toward β-hematin formation in a concentration-dependent manner, although their activity was much weaker than that of CQ. In contrast, compound 24 demonstrated very low activity against β-hematin crystal formation even at the highest concentration tested (200 μM). These results suggested that the potent antimalarial activity of 2-arylvinylquinolines could associate with mechanisms other than the inhibition of heme detoxification.
Figure 6.
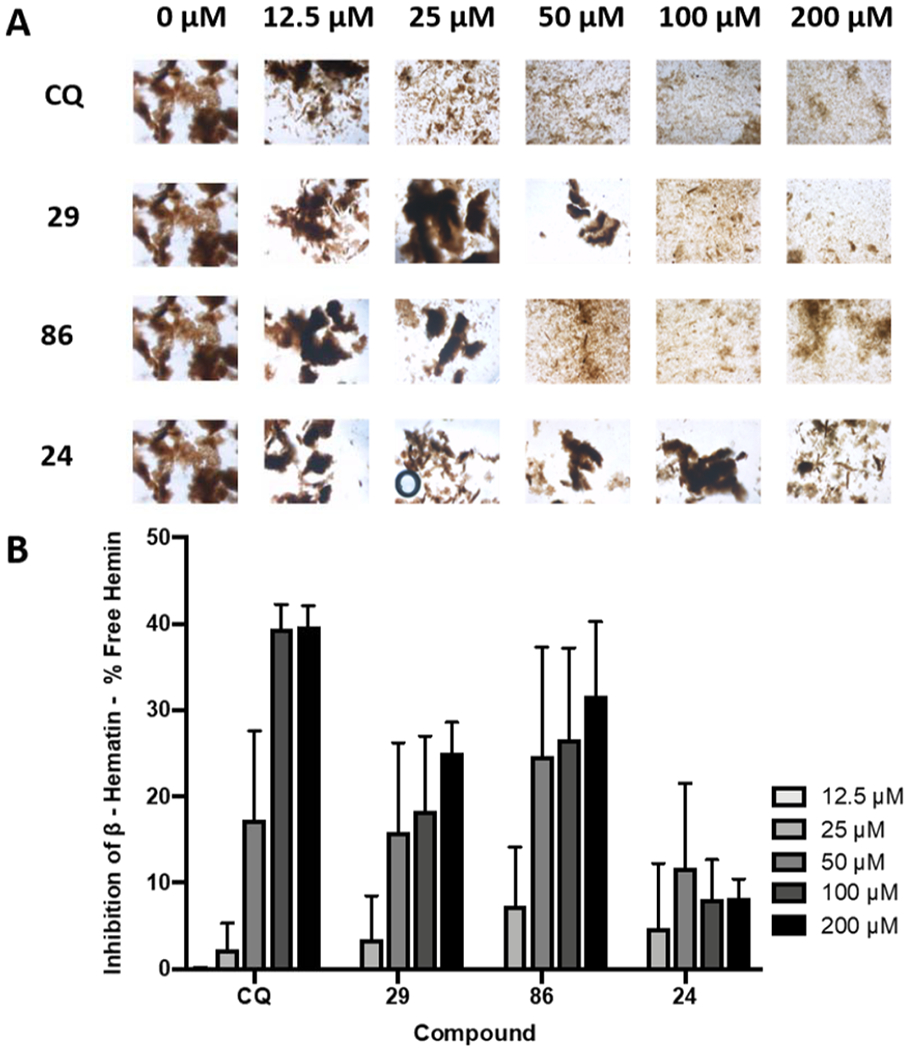
Effect of the 2-arylvinylaminequinoline derivatives on the β-hematin crystal formation. (A) Images of β-hematin crystals following incubation of 100 μM hemin, propionate buffer, phosphatidylcholine, and varying concentrations of the compound for 16 h at 37 °C. Images were taken using a Nikon Eclipse TE200. (B) Free hemin, indicative of inhibition of β-hematin crystal formation, was determined using a linear calibration curve. Data represent mean ± SEM of three independent experiments.
2-Arylvinylquinolines are Well Tolerated and Active in an in Vivo Model.
Given the robust antiplasmodial potency of 2-arylvinylquinolines against the Dd2 and 3D7 strains, and the good microsomal stability profiles of 24, 29, and 86, we assessed the in vivo efficacy of these compounds using the rodent malaria model. The monophosphate salts of 24, 29, and 86 were used in female Swiss Webster mice infected with Plasmodium berghei ANKA strain. As observed from Figure 7A,B, all of these compounds completely cured malaria infection in mice when exposed to 100 mg/kg daily by p.o. administration in a standard Peters’ four-day test, and no parasite bioluminescence was detected in any of the tested mice (Figure 7A). Although a lower dose of the monophosphate salt of the compound 86 (86s) was ineffective for malaria-infected mice, monophosphate salts of 29 (29s)and 24 (24s) effectively cleared the parasites at 25 mg/kg. Although a marginal luminescence signal was detected in compound 29s-treated mice, this signal could arise from a small amount of circulating or dying parasites. Significantly, we found that 24s provided full protection and cure at 25 mg/kg with no bioluminescence signals detected after treatment.
Figure 7.
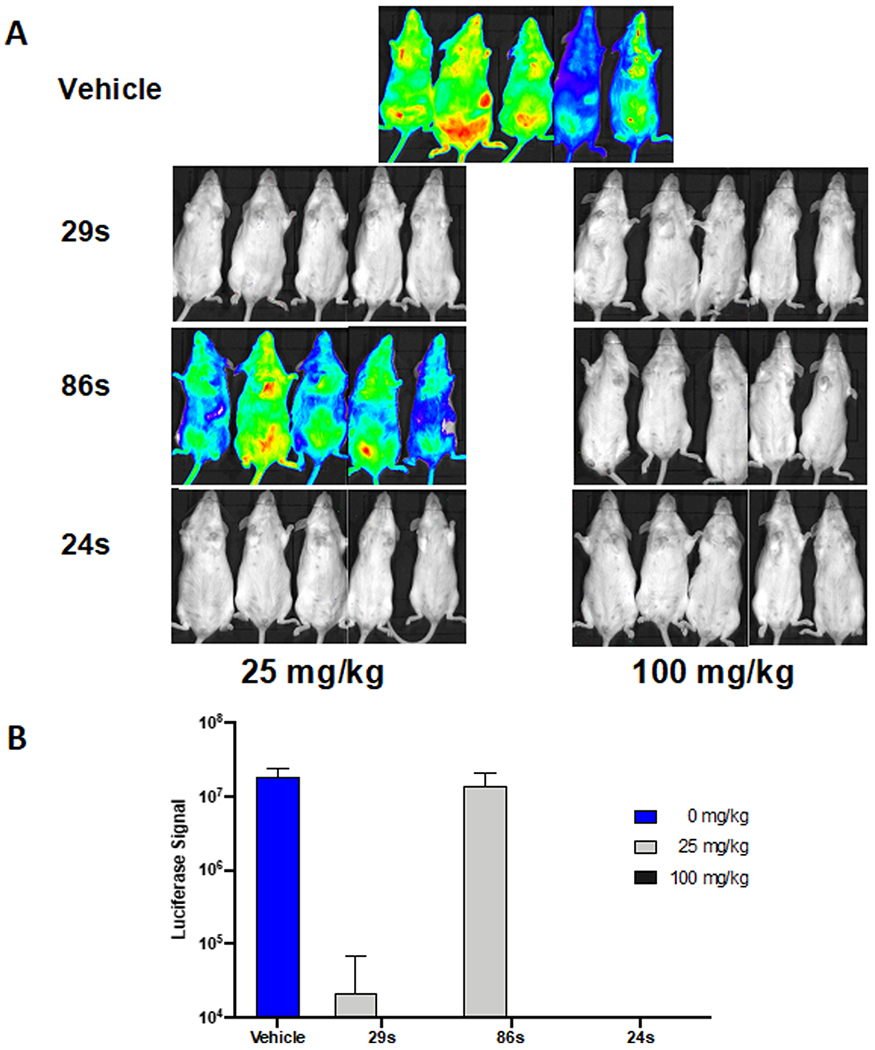
Curative property of 2-arylvinylquinoline derivatives. (A) Swiss Webster female mice were infected with P. berghei ANKA luciferase-expressing strain and treated with 25 and 100 mg/kg p.o. once daily 48 h post infection. A total of 7 days after infection, bioluminescence was detected (A) and quantified (B) using an in vivo imaging system (IVIS).
To assess the curative property of compound 24s, we monitored the survival of P. berghei ANKA-infected mice for 30 days following oral administration of 25 and 100 mg/kg. The monophosphate salt of 24 was well tolerated and did not display apparent adverse symptoms such as hunched posture, hypotrichosis, or reduced mobility. In addition, the administration of 24s by these dosing regiments caused no significant weight loss, whereas the body weight of control groups decreased sharply (Figure S10). The mean survivability in the control group was 8 days (Figure 8A). In contrast, administration of 24s at 100 mg/kg prolonged the lives of the mice by 22 days (the end of experiment, 30 days), and no parasites were detected in Giemsa-stained thin blood smears. Importantly, with p.o. administration of 24s at a lower dose (25 mg/kg), 3/4 mice survived without detection of any malaria parasites in thin blood smears, while all mice succumbed in the control group. Similar results have been reported with CQ administered at 30 mg/kg (MSD: 24 days).61 The curative dose for piperaquine and ferroquine are better (16 and 30 mg/kg, respectively, with a single oral dose) compared to 24s and CQ (Sergio Wittlin, Swiss Tropical Institute, personal communication). In comparison, UCF501 cured malaria infection in 4/5 mice when they were exposed to a high dose of 100 mg/kg twice daily p.o. administration in our previous study,33 suggesting good in vivo efficacy improvement with 24s. This improvement can be explained, to some extent, by the enhanced metabolic stability of 24.
Figure 8.
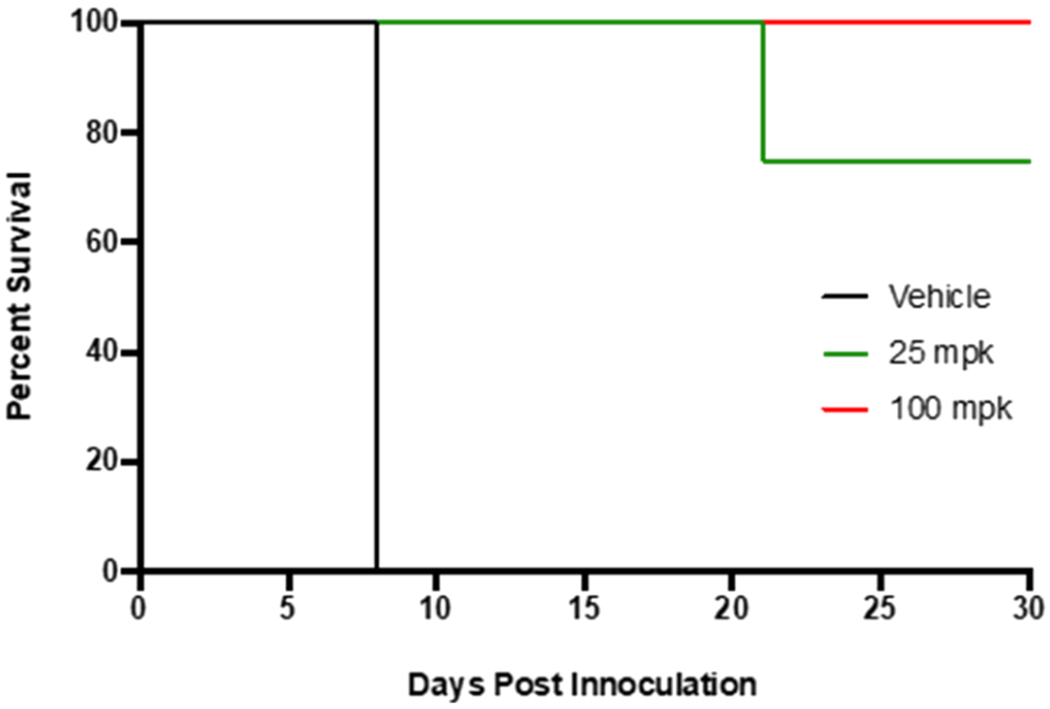
Effect of compound 24s on the survivability of P. berghei ANKA-infected mice. BALB/c females were infected with a P. berghei ANKA luciferase expressing strain and treated 4 h postinfection with 25 or 100 mg/kg given orally once daily for 4 days. There was no statistically significant difference between the 25 and 100 mg/kg treatment group log-rank (Mantel–Cox) test (p = 0.2636).
CONCLUSIONS
In this work, in-depth SAR studies were performed around a quinoline scaffold, leading to the generation of 6-chloroarylvinylquinolines. The SAR trends are summarized in Figure 9. Many promising arylvinylaminoquinolines exhibited more potent antimalarial activity than the positive controls (CQ and UCF501), the most active of which being compounds 24, 29, 31, 86, 92, and 93 with an EC50 ≤ 15 nM. The inhibitory activity of most compounds tested against the CQ-resistant strain was higher than inhibition of the CQ-susceptible strain (RI < 1), suggesting no cross-resistance induced by this chemotype.
Figure 9.
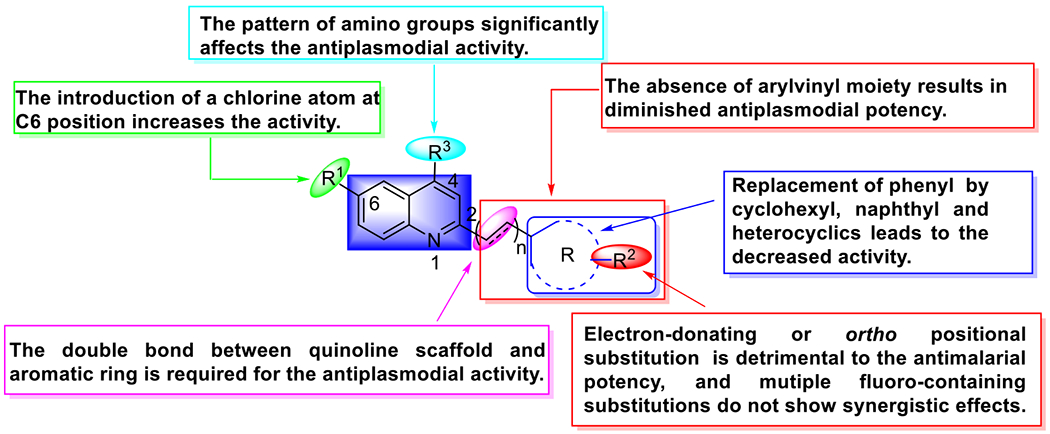
Summary of SAR trends.
The most promising compound 24 (EC50 = 10.9 ± 1.9 nM against the Dd2 strain; t1/2 = 104.2 min) is a fast-acting parasitocidal agent with robust blood and gametocyte stage activity, demonstrating stage-specific action at the trophozoite phase in the Pf asexual life cycle. Importantly, 24s displayed remarkable efficacy in the rodent malaria model, resulting in 100% reduction of parasitemia in 5/5 mice at 100 mg/kg, p.o., and 3/4 mice at 25 mg/kg, p.o., with no apparent signs of toxicity. Future studies will focus on assessment of pharmacokinetic and toxicological liabilities, if any, of 24s. Additionally, compound 24 showed a weaker inhibitory activity toward β-hematin formation as compared with CQ, indicating that the potent antimalarial activity of 24 may associate with different modes of actions. In particular, the accumulations of membrane-bound structures inside the FV in treated parasites suggest that compound 24 interferes with the hemoglobin digestion process. Collectively, 6-chloro-arylvinylquinolines 24 can be considered as a promising lead for the development of new antimalarials.
EXPERIMENTAL SECTION
Chemistry.
Synthesis of Compounds.
All reagents were commercially available and used without further purification. Nuclear magnetic resonance spectra were recorded in CDCl3, MeOD, and DMSO solutions. 1H NMR, 13C NMR, and 19F NMR were recorded on Bruker spectrometers operating at 400 and 500 MHz. Data are reported as follows: chemical shift (δ), multiplicity, integrated intensity, and coupling constant (J) in hertz. Melting points were measured on a Stuart SMP20 melting point apparatus and were uncorrected. High-resolution mass spectroscopy (HRMS) images were obtained on an Agilent 6230 FOF/LC/MS. High-performance LC (HPLC) analysis was performed on an Agilent 1260 system using a ZORBAX C18 column (150 × 4.6 mm, 3.5 μm) at room temperature with a gradient elution using the mobile-phase (A) nanopure water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid. HPLC condition A: 10–70% of B at 0–7 min, 70–95% of B at 7–7.3 min, 95% of B at 7.3–9.3 min, 95–10% of B at 9.3–10 min, and 10% of B at 10–13 min and a flow rate of 0.8 mL/min; HPLC condition B: 45–95% of B at 0–7 min, 95% of B at 7–9.3 min, 95–45% of B at 9.3–10 min, and 45% of B at 10–13 min and a flow rate of 0.5 mL/min. All compounds used for biological evaluation have a purity of ≥95%.
General Procedure 1: Synthesis of Hydroxyquinolines (3a–d).
Method 1A.
A mixture of 4-methoxyaniline (24.4 mmol), ethyl acetoacetate (3.49 g, 26.8 mmol), anhydrous magnesium sulfate (3.51 g, 29.2 mmol), and acetic acid (0.2 mL) in ethanol (30 mL) was heated to 90 °C for 6 h. Once the reaction was completed, as monitored by thin layer chromatography, the resultant mixture was filtered, and the resultant solvent was concentrated under reduced pressure to give a pale brown liquid. This crude product was purified by silica gel column chromatography to afford imine (3.2 g, 56% yield). Then, a solution of imine (3.1 g, 13.2 mmol) in DOWTHERM (5 mL) was heated to 270 °C for 20 min. After cooling, the reaction mixture was diluted in ethyl acetate. The solid was collected by filtration, washed with water, and dried in a vacuum oven to give the hydroxyquinoline 3a (1.9 g, 76% yield) as an off-white solid.
Method 1B.
PPA (80.6 g) was added to a solution of anilines (0.1 mol) and ethyl acetoacetate (12.6 mL, 0.1 mol). The reaction mixture was stirred at 150 °C for 2 h. Upon completion, the reaction mixture was poured into ice water with vigorous stirring. The precipitated solid was collected by filtration and dried in a vacuum oven to get hydroxyquinolines 3a–d. The crude products were then used for the next reaction without further purification.
General Procedure 2 for Synthesis of Chlorinated Quinolines (Chlorination of Hydroxyquinolines) (4a–d).
The hydroxyquinolines (0.10 mol) in phosphorous oxychloride (60 mL) were heated to 105 °C for 2 h. Upon completion, the excess of phosphorous oxychloride was removed under reduced pressure. The residue was quenched into crushed ice and neutralized using a saturated solution of sodium bicarbonate. The solid obtained was then collected by filtration and dried in vacuum to afford chlorinated quinolines 4a–d. The crude products were used for the next step without further purification.
General Procedure 3 for Synthesis of Aminoquinolines (Nucleophilic Substitution of Chlorinated Quinolines) (6a–e, 61a–f, and 89a–c).
Method 3A.
Chlorinated quinolines (2.4 mmol) and amine (3.0 mL) in a pressure tube were heated at 140 °C for 24 h. Upon completion, water was added to the resultant content. The precipitated solid was collected by filtration and dried in vacuum to afford aminoquinolines.
Method 3B.
Chlorinated quinolines (3.0 mmol), secondary amine (27 mmol), and EtOH (2.0 mL) were heated in a pressure tube at 130 °C for 36 h. Upon completion, the resultant solvent was removed to dryness. The residue was dissolved in dichloromethane (DCM) and washed with water and brine. The organic layer was separated, and the aqueous layer was extracted with DCM. The combined organic extracts were then dried over Na2SO4, filtered, and evaporated under reduced pressure to give the desired aminoquinolines.
N1-(6-Methoxy-2-methylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (6a).
The title compound was synthesized from 4a and N1,N1-dimethylethane-1,2-diamine (5a) according to the method described for procedure 3A. Isolated yield: 92%; off-white solid; 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 9.2 Hz, 1H), 7.31 (dd, J = 9.2, 2.8 Hz, 1H), 7.13 (d, J = 2.8 Hz, 1H), 6.25 (s, 1H), 3.95 (s, 3H), 3.39–7.35 (m, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.69 (s, 3H), 2.34 (s, 6H).
Nl-(6-Fluoro-2-methylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (6b).
The title compound was synthesized from 4b and N1,N1-dimethylethane-1,2-diamine (5a) according to the method described for procedure 3A. Isolated yield: 89%; off-white solid; 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 9.2 Hz, 1H), 7.44–7.37 (m, 2H), 6.31 (s, 1H), 3.32 (t, J = 5.8 Hz, 2H), 2.71 (t, J = 6.0 Hz, 2H), 2.66 (s, 3H), 2.33 (s, 6H).
N1-(6-Chloro-2-methylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (6c).
The title compound was synthesized from 4c and N1,N1-dimethylethane-1,2-diamine (5a) according to the method described for procedure 3A. Isolated yield: 93%; off-white solid; 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 9.0 Hz, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 9.0, 2.4 Hz, 1H), 6.31 (s, 1H), 3.38 (q, J = 6.0 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.60 (s, 3H), 2.32 (s, 6H).
N1,N1-Dimethyl-N2-(2-methylquinolin-4-yl)ethane-1,2-diamine (6d).
The title compound was synthesized from 4d and N1,N1-dimethylethane-1,2-diamine (5a) according to the method described for procedure 3B. Isolated yield: 92%; yellow oil; 1H NMR (500 MHz, CDCl3): δ 7.84 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.52 (t, J = 8.4 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 6.22 (s, 1H), 3.25–3.21 (m, 2H), 2.63–2.60 (m, 2H), 2.54 (s, 3H), 2.24 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 159.5, 150.0, 148.0, 129.1, 128.7, 123.8, 119.7, 117.5, 99.1, 57.2, 45.1, 40.0, 25.5.
N1-(6-Chloro-2-methylquinolin-4-yl)-N4,N4-dimethylbutane-1,4-diamine (6e).
The title compound was synthesized from 4c and N1,N1-dimethylbutane-1,4-diamine (5b) according to the method described for procedure 3A. Isolated yield: 87%; off-white solid; 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 9.0, 2.2 Hz, 1H), 6.27 (s, 1H), 3.26–3.21 (m, 2H), 2.60 (s, 3H), 2.40 (t, J = 6.4 Hz, 2H), 2.31 (s, 6H), 1.94–1.89 (m, 2H), 1.75–1.69 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 160.0, 150.4, 146.6, 130.5, 130.0, 129.4, 120.1, 118.8, 99.4, 59.9, 45.9, 43.9, 27.2, 26.3, 25.8.
4-(6-Chloro-2-methylquinolin-4-yl)morpholine (61a).
The title compound was synthesized from 4c and 60a according to the method described for procedure 3A. Isolated yield: 90%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.90–7.86 (m, 2H), 7.53 (dd, J = 9.0, 2.4 Hz, 1H), 6.74 (s, 1H), 3.96 (t, J = 5.6 Hz, 4H), 3.15 (t, J = 5.6 Hz, 4H), 2.65 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.2, 156.3, 148.0, 131.2, 130.9, 130.3, 122.9, 122.7, 110.6, 67.2, 52.9, 25.9.
6-Chloro-2-methyl-4-(pyrrolidin-1-yl)quinoline (61b).
The title compound was synthesized from 4c and 60b according to the method described for procedure 3A. Isolated yield: 92%; brown oil; 1H NMR (400 MHz, CDCl3): δ 8.14 (d, J = 2.4 Hz, 1H), 7.82 (d, J = 9.0 Hz, 1H), 7.48 (dd, J = 9.0, 2.4 Hz, 1H), 6.37 (s, 1H), 3.67–3.63 (m, 4H), 2.58 (s, 3H), 2.06–2.03 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 159.1, 152.4, 148.7, 130.8, 129.5, 128.0, 124.4, 120.8, 104.0, 52.4, 26.3, 25.8.
6-Chloro-2-methyl-N-(4-(piperidin-1-yl)phenyl)quinolin-4-amine (61c).
The title compound was synthesized from 4c and 60c according to the method described for procedure 3B. Isolated yield: 73%; green solid; 1H NMR (400 MHz, CDCl3): δ 10.48 (s, 1H), 9.08 (s, 1H), 8.25 (d, J = 9.0 Hz, 1H), 7.47 (dd, J = 9.0, 1.8 Hz, 1H), 7.23 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 6.38 (s, 1H), 3.16–3.12 (m, 4H), 2.60 (s, 3H), 1.72–1.67 (m, 4H), 1.62–1.57 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 154.7, 154.0, 151.4, 137.8, 133.6, 132.5, 127.6, 126.7, 123.7, 122.3, 117.7, 116.8, 100.3, 50.3, 26.0, 24.5, 20.7.
6-Chloro-2-methyl-4-(4-(pyridin-2-yl)piperazin-1-yl)quinoline (61d).
The title compound was synthesized from 4c and 60d according to the method described for procedure 3B. Isolated yield: 86%; brown solid; 1H NMR (400 MHz, CDCl3): δ 8.24 (dd, J = 4.9, 2.8 Hz, 1H), 7.98 (d, J = 2.4 Hz, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.58–7.55 (m, 1H), 7.55–7.52 (m, 1H), 6.80 (s, 1H), 6.74 (d, J = 8.6 Hz, 1H), 6.71–6.68 (m, 1H), 3.83–3.79 (m, 4H), 3.32–3.29 (m, 4H), 2.67 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.2, 159.8, 156.4, 148.4, 148.0, 138.0, 131.2, 130.9, 130.3, 123.1, 122.8, 114.3, 110.8, 107.7, 52.4, 45.8, 25.9.
6-Chloro-2-methyl-N-(pyridin-2-ylmethyl)quinolin-4-amine (61f).
The title compound was synthesized from 4c and 60f according to the method described for procedure 3B. Isolated yield: 80%; brown solid; 1H NMR (400 MHz, CDCl3): δ 8.67 (d, J = 5.4 Hz, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.72–7.68 (m, 1H), 7.53–7.51 (m, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.26 (t, J = 6.8 Hz, 1H), 6.34 (s, 1H), 4.56 (d, J = 4.4 Hz, 2H), 2.59 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.3, 155.9, 150.0, 148.9, 147.0, 137.2, 130.9, 130.1, 129.9, 123.0, 122.1, 119.6, 118.8, 100.5, 47.8, 26.0.
6-Chloro-2-methyl-N-(3-morpholinopropyl)quinolin-4-amine (89a).
The title compound was synthesized from 4c and 3-morpholinopropan-1-amine (88a) according to the method described for procedure 3B. Isolated yield: 93%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 9.0 Hz, 1H), 7.52 (dd, J = 9.0, 2.2 Hz, 1H), 6.26 (s, 1H), 3.95–3.93 (m, 4H), 3.40–3.37 (m, 2H), 2.66–2.60 (m, 6H), 2.59 (s, 3H), 1.98–1.94 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 160.4, 150.1, 147.0, 131.0, 129.9, 129.6, 119.9, 118.8, 99.3, 67.2, 59.8, 54.5, 44.8, 26.1, 23.5.
6-Chloro-2-methyl-N-(4-morpholinobutyl)quinolin-4-amine (89b).
The title compound was synthesized from 4c and 4-morpholinobutan-1-amine (88b) according to the method described for procedure 3B. Isolated yield: 88%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 2.2 Hz, 1H), 7.52 (dd, J = 9.0, 2.2 Hz, 1H), 6.33 (s, 1H), 3.78–3.75 (m, 4H), 3.32 (q, J = 6.7 Hz, 2H), 2.61 (s, 3H), 2.48–2.42 (m, 6H), 1.87–1.81 (m, 2H), 1.73–1.67 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 160.2, 149.4, 146.9, 131.0, 130.1, 129.8, 119.1, 100.0, 67.3, 58.5, 54.1, 43.6, 26.8, 24.7.
6-Chloro-2-methyl-N-(4-(4-methylpiperazin-1-yl)butyl)quinolin-4-amine (89c).
The title compound was synthesized from 4c and 4-(4-methylpiperazin-1-yl)butan-1-amine (88c) according to the method described for procedure 3B. Isolated yield: 86%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 9.0, 2.2 Hz, 1H), 6.27 (s, 1H), 3.26–3.21 (m, 2H), 2.60 (s, 3H), 2.57–2.47 (m, 6H), 2.43 (t, J = 6.4 Hz, 4H), 2.31 (s, 3H), 1.94–1.89 (m, 2H), 1.75–1.69 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 160.0, 150.4, 146.6, 130.5, 130.0, 129.4, 120.1, 118.8, 99.4, 59.9, 45.9, 43.9, 27.2, 26.3, 25.8.
General Procedure 4 for Synthesis of Arylvinylquinolines (Olefination Reaction) (8–37, 39–57, 62–63, 65–72, 81–87, 90–96, and 98–114).
A mixture of 2-methylquinolines (0.20 mmol), p-TsNH2 (34 mg, 0.20 mmol), and the appropriate aldehyde (1.0 mmol) in m-xylene (2.0 mL) was stirred at 140 °C for 12 h. Upon completion, the cooled mixture was directly loaded and purified on a silica gel column chromatography to afford arylvinylquinolines.
(E)-N1-(6-Methoxy-2-styrylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diaminei (8).
The title compound was synthesized from 6a and 7a. Isolated yield: 85%; yellow solid; mp 153–156 °C; 1H NMR (400 MHz, CDCl3): δ 8.19 (d, J = 9.2 Hz, 1H), 7.77 (d, J = 16.4 Hz, 1H), 7.55 (dd, J = 6.8, 3.2 Hz, 3H), 7.35 (d, J = 16.4 Hz, 1H), 7.31–7.27 (m, 3H), 7.20 (dd, J = 9.2, 2.6 Hz, 1H), 6.56 (s, 1H), 3.91 (s, 3H), 3.65 (t, J = 6.2 Hz, 2H), 2.98 (t, J = 6.0 Hz, 2H), 2.48 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 158.0, 150.6, 143.6, 142.6, 140.9, 133.1, 129.4, 127.8, 126.6, 126.3, 122.6, 121.8, 118.9, 101.0, 96.7, 57.1, 56.3, 45.3, 40.6; HRMS (ESI) m/z: calcd for C22H26N3O, 348.2076 [M + H]+; found, 348.2049; retention time 5.478 min (HPLC condition A).
(E)-N1-(6-Methoxy-2-(4-nitrostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (9).
The title compound was synthesized from 6a and 7b. Isolated yield: 82%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.20 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 9.2 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 16.2 Hz, 1H), 7.41 (d, J = 16.2 Hz, 1H), 7.33 (dd, J = 9.2, 2.8 Hz, 1H), 7.05 (d, J = 2.8 Hz, 1H), 6.65 (s, 1H), 3.95 (s, 3H), 3.41 (q, J = 6.0, 5.8 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.4, 153.2, 149.9, 147.4, 144.2, 143.9, 134.6, 131.5, 130.1, 127.7, 124.5, 120.9, 119.6, 100.3, 98.0, 57.5, 56.0, 45.5, 40.6; HRMS (ESI) m/z: calcd for C22H25N4O3, 393.1927 [M + H]+; found, 393.1936; retention time 2.664 min (HPLC condition B).
(E)-N1-(6-Methoxy-2-(2-nitrostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (10).
The title compound was synthesized from 6a and 7c. Isolated yield: 80%; yellow solid; mp 168–170 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.96 (d, J = 16.4 Hz, 1H), 7.89 (d, J = 9.0 Hz, 1H), 7.63 (t, J = 8.2 Hz, 1H), 7.48–7.42 (m, 2H), 7.33 (dd, J = 9.2, 2.6 Hz, 1H), 7.16 (d, J = 2.6 Hz, 1H), 6.72 (s, 1H), 3.96 (s, 3H), 3.47 (t, J = 6.2 Hz, 2H), 2.81 (t, J = 6.2 Hz, 2H), 2.39 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.8, 152.5, 150.7, 148.5, 141.4, 133.8, 133.1, 132.5, 129.2, 129.0, 125.2, 121.7, 119.3, 100.8, 95.9, 57.3, 56.2, 45.4, 40.5; HRMS (ESI) m/z: calcd for C22H25N4O3, 393.1927 [M + H]+; found, 393.1901; retention time 5.795 min (HPLC condition A).
(E)-N1-(6-Methoxy-2-(3-nitrostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (11).
The title compound was synthesized from 6a and 7d. Isolated yield: 83%; yellow solid; mp 183–185 °C; 1H NMR (400 MHz, CDCl3): δ 8.46 (t, J = 2.2 Hz, 1H), 8.13 (dd, J = 8.2, 2.2 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 16.2 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 16.2 Hz, 1H), 7.34 (dd, J = 9.2, 2.6 Hz, 1H), 7.07 (d, J = 2.6 Hz, 1H), 6.65 (s, 1H), 3.96 (s, 3H), 3.43–3.39 (m, 2H), 2.76 (t, J = 6.2 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.3, 153.4, 149.9, 149.1, 144.2, 139.2, 133.1, 131.5, 130.1, 130.0, 122.8, 121.8, 120.8, 119.5, 100.3, 98.0, 57.5, 56.0, 45.5, 40.6; HRMS (ESI) m/z: calcd for C22H25N4O3, 393.1927 [M + H]+; found, 393.1901; retention time 5.771 min (HPLC condition A).
(E)-N1-(6-Methoxy-2-(4-methoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (12).
The title compound was synthesized from 6a and 7e. Isolated yield: 60%; yellow solid; mp 171–173 °C; 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 9.2 Hz, 1H), 7.57 (d, J = 16.4 Hz, 1H), 7.51 (J = 8.6 Hz, 2H), 7.23 (dd, J = 9.2, 2.8 Hz, 1H), 7.15 (d, J = 16.4 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H), 6.55 (s, 1H), 3.90 (s, 3H), 3.81 (s, 3H), 3.43 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.33 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.5, 159.5, 157.3, 153.1, 150.8, 134.6, 131.0, 129.3, 129.1, 121.5, 118.8, 114.5, 113.9, 101.0, 96.2, 57.4, 56.2, 55.7, 45.5, 40.8; HRMS (ESI) m/z: calcd for C23H28N3O2, 378.2182 [M + H]+; found, 378.2155; retention time 5.649 min (HPLC condition A).
(E)-N1-(2-(3,4-Dimethoxystyryl)-6-methoxyquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (13).
The title compound was synthesized from 6a and 7f. Isolated yield: 64%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 9.2 Hz, 1H), 7.61 (d, J = 16.2 Hz, 1H), 7.25–7.21 (m, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 10.0 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.55 (s, 1H), 5.29 (s, 3H), 3.92 (s, 3H), 3.91 (s, 3H), 3.50 (t, J = 5.8 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.37 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.5, 151.3, 150.5, 149.5, 149.1, 129.8, 129.4, 122.7, 121.9, 121.1, 112.5, 111.4, 109.6, 101.2, 100.7, 95.7, 57.2, 56.3, 53.8, 45.5, 45.4, 40.8; HRMS (ESI) m/z: calcd for C24H30N3O3, 408.2287 [M + H]+; found, 408.2273; retention time 2.609 min (HPLC condition B).
(E)-N1-(6-Methoxy-2-(3,4,5-trimethoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (14).
The title compound was synthesized from 6a and 7g. Isolated yield: 75%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 9.2 Hz, 1H), 7.56 (d, J = 16.4 Hz, 1H), 7.25–7.18 (m, 3H), 6.83 (s, 2H), 6.56 (s, 1H), 3.90 (s, 3H), 3.89 (s, 6H), 3.87 (s, 3H), 3.48 (t, J = 6.0 Hz, 2H), 2.78 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.5, 153.7, 153.2, 152.1, 151.2, 139.3, 135.5, 132.0, 127.5, 125.3, 121.9, 118.7, 104.9, 101.1, 95.9, 61.3, 57.2, 56.5, 56.3, 45.4, 40.8; HRMS (ESI) m/z: calcd for C25H32N3O4, 438.2393 [M + H]+; found, 438.2405; retention time 2.527 min (HPLC condition B).
(E)-N1-(2-(4-Fluorostyryl)-6-methoxyquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (15).
The title compound was synthesized from 6a and 7h. Isolated yield: 71%; yellow solid; mp 161–164 °C; 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 9.2 Hz, 1H), 7.60 (d, J = 16.4 Hz, 1H), 7.58–7.54 (m, 2H), 7.29 (dd, J = 9.2, 2.8 Hz, 1H), 7.21 (d, J = 16.4 Hz, 1H), 7.08 (d, J = 2.6 Hz, 1H), 7.07–7.02 (m, 2H), 6.59 (s, 1H), 3.93 (s, 3H), 3.42 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 157.3, 153.3, 150.5, 142.0, 133.1 (d, J = 3.0 Hz), 132.9, 129.6, 129.2 (d, J = 8.0 Hz), 127.8, 121.2, 119.0, 116.1 (d, J = 21.0 Hz), 100.7, 96.8, 57.4, 56.1, 45.5, 40.7; HRMS (ESI) m/z: calcd for C22H25FN3O, 366.1982 [M + H]+; found, 366.1957; retention time 5.684 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-styrylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (16).
The title compound was synthesized from 6b and 7a. Isolated yield: 75%; yellow solid; mp 114–116 °C; 1H NMR (400 MHz, CDCl3): δ 8.05 (dd, J = 9.2, 5.6 Hz, 1H), 7.68–7.60 (m, 3H), 7.42 (dd, J = 10.2, 2.4 Hz, 1H), 7.40–7.35 (m, 3H), 7.33–7.27 (m, 2H), 6.65 (s, 1H), 3.41 (t, J = 5.8 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 159.9 (d, J = 242.0 Hz), 155.8, 150.4 (d, J = 4.0 Hz), 145.0, 136.9, 134.2, 131.6 (d, J = 9.0 Hz), 129.1, 128.8, 127.6, 119.5 (d, J = 25.0 Hz), 119.1 (d, J = 9.0 Hz), 104.6 (d, J = 23.0 Hz), 97.4, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C21H23FN3, 336.1876 [M + H]+; found, 336.1848; retention time 4.999 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(4-nitrostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (17).
The title compound was synthesized from 6b and 7b. Isolated yield: 79%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.22 (d, J = 9.0 Hz, 2H), 7.98 (dd, J = 9.6, 5.6 Hz, 1H), 7.74–7.66 (m, 3H), 7.45–7.36 (m, 3H), 6.62 (s, 1H), 3.36 (q, J = 5.8 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.1 (d, J = 245.0 Hz), 154.9, 150.3 (d, J = 5.0 Hz), 147.5, 145.8, 143.6, 134.4, 132.4 (d, J = 8.0 Hz), 130.9, 127.9, 124.5, 119.6 (d, J = 25.0 Hz), 119.4 (d, J = 9.0 Hz), 104.5 (d, J = 23.0 Hz), 98.3, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C21H22FN4O2, 381.1727 [M + H]+; found, 381.1709; retention time 2.666 min (HPLC condition B).
(E)-N1-(6-Fluoro-2-(4-fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (18).
The title compound was synthesized from 6b and 7h. Isolated yield: 88%; yellow solid; mp 120–122 °C; 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 10.0, 5.4 Hz, 1H), 7.70 (d, J = 16.2 Hz, 1H), 7.60–7.56 (m, 2H), 7.52 (dd, J = 9.8, 2.4 Hz, 1H), 7.41–7.36 (m, 1H), 7.23 (d, J = 16.4 Hz, 1H), 7.05 (t, J = 8.6 Hz, 2H), 6.64 (s, 1H), 3.52 (t, J = 5.8 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 2.45 (s, 6H); 13C NMR (100MHz, CDCl3): δ 163.1 (d, J = 247.0 Hz), 160.0 (d, J = 244.0 Hz), 155.7, 150.3 (d, J = 5.0 Hz), 145.2, 133.2 (d, J = 3.0 Hz), 132.8, 129.1 (d, J = 8.0 Hz), 119.6 (d, J = 25.0 Hz), 119.1, 116.2, 116.0, 115.4, 115.2, 104.7 (d, J = 23.0 Hz), 97.3, 57.1, 45.1, 40.2;; HRMS (ESI) m/z: calcd for C21H22F2N3, 354.1782 [M + H]+; found, 354.1754; retention time 5.223 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(4-methoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (19).
The title compound was synthesized from 6b and 7e. Isolated yield: 86%; yellow solid; mp 141–144 °C; 1H NMR (400 MHz, CDCl3): δ 8.00 (dd, J = 10.0, 5.4 Hz, 1H), 7.60 (d, J = 16.2 Hz, 1H), 7.58–7.54 (m, 2H), 7.41–7.37 (m, 2H), 7.16 (d, J = 16.2 Hz, 1H), 6.92 (d, J = 8.8 Hz, 2H), 6.63 (s, 1H), 3.85 (s, 3H), 3.38 (t, J = 5.8 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.3, 159.8 (d, J = 244.0 Hz), 159.5, 150.2 (d, J = 4.0 Hz), 149.2, 133.6, 132.0 (d, J = 9.0 Hz), 131.1, 129.8, 128.9, 119.4 (d, J = 25.0 Hz), 119.2, 114.6, 113.8, 104.5 (d, J = 22.0 Hz), 97.3, 57.4, 55.7, 45.4, 40.4; HRMS (ESI) m/z: calcd for C22H25FN3O, 366.1982 [M + H]+; found, 366.1954; retention time 5.034 min (HPLC condition A).
(E)-N1-(2-(3,4-Dimethoxystyryl)-6-fluoroquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (20).
The title compound was synthesized from 6b and 7f. Isolated yield: 83%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.05 (dd, J = 9.0, 5.6 Hz, 1H), 7.39 (dd, J = 9.0, 2.0 Hz, 2H), 6.97 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 6.87 (d, J = 12.6 Hz, 1H), 6.78 (s, 1H), 6.76 (d, J = 5.0 Hz, 1H), 6.41 (s, 1H), 3.87 (s, 3H), 3.62 (s, 3H), 2.99 (t, J = 5.8 Hz, 2H), 2.57 (t, J = 6.0 Hz, 2H), 2.28 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.2 (d, J = 245.0 Hz), 156.5, 149.6 (d, J = 6.0 Hz), 149.2, 148.7, 134.5, 131.3, 129.8, 129.3, 122.8, 119.6 (d, J = 26.0 Hz), 118.7 (d, J = 9.0 Hz), 112.6, 111.1, 104.6 (d, J = 23.0 Hz), 51.1, 56.2, 56.0, 45.3, 40.0; HRMS (ESI) m/z: calcd for C23H27FN3O2, 396.2087 [M + H]+; found, 396.2048; retention time 2.552 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(3,4,5-trimethoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (21).
The title compound was synthesized from 6b and 7g. Isolated yield: 80%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.05 (dd, J = 9.0, 5.6 Hz, 1H), 7.57 (d, J = 16.2 Hz, 1H), 7.44–7.37 (m, 2H), 7.22 (d, J = 16.2 Hz, 1H), 6.86 (s, 2H), 6.67 (s, 1H), 3.91 (s, 6H), 3.88 (s, 3H), 3.41 (t, J = 5.8 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.0 (d, J = 245.0 Hz), 155.5, 153.7, 153.2, 150.5 (d, J = 4.0 Hz), 139.1, 134.3 (d, J = 7.0 Hz), 132.5, 131.1, 128.2, 119.7 (d, J = 25.0 Hz), 119.0 (d, J = 8.0 Hz), 106.8, 104.7 (d, J = 23.0 Hz), 96.8, 61.3, 57.2, 56.5, 45.4, 40.4; HRMS (ESI) m/z: calcd for C24H29FN3O3, 426.2193 [M + H]+; found, 426.2173; retention time 5.251 min (HPLC condition A).
(E)-N1-(6-Chloro-2-styrylquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (22).
The title compound was synthesized from 6c and 7a. Isolated yield: 82%; yellow solid; mp 135–136 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.72 (d, J = 2.2 Hz, 1H), 7.66 (d, J = 13.6 Hz, 1H), 7.62 (d, J = 6.8 Hz, 2H), 7.56 (dd, J = 9.0, 2.4 Hz, 1H), 7.42–7.37 (m, 2H), 7.32 (d, J = 7.4 Hz, 1H), 7.29 (d, J = 16.0 Hz, 1H), 6.65 (s, 1H), 3.40–3.35 (m, 2H), 2.75–2.71 (m, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.9, 149.8, 147.2, 137.0, 134.1, 131.4, 130.4, 130.2, 129.8, 129.1, 128.8, 127.6, 119.6, 98.0, 57.4, 45.5, 40.4; HRMS (ESI) m/z: calcd for C21H23ClN3, 352.1581 [M + H]+; found, 352.1580; retention time 5.818 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-nitrostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (23).
The title compound was synthesized from 6c and 7b. Isolated yield: 85%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.25 (d, J = 9.0 Hz, 2H), 7.93 (d, J = 9.0 Hz, 1H), 7.76–7.71 (m, 4H), 7.59 (dd, J = 9.0, 2.4 Hz, 1H), 7.38 (d, J = 16.2 Hz, 1H), 6.63 (s, 1H), 3.40–3.35 (m, 2H), 2.77–2.72 (m, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 155.6, 150.0, 147.6, 143.6, 134.2, 131.7, 131.3, 130.8, 130.7, 127.92, 124.5, 119.6, 98.7, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H22ClN4O2, 397.1431 [M + H]+; found, 397.1403; retention time 6.526 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-(trifluoromethyl)styryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (24).
The title compound was synthesized from 6c and 7i. Isolated yield: 83%; yellow solid; mp 171–172 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.72 (d, J = 6.4 Hz, 1H), 7.70–7.63 (m, 4H), 7.57 (dd, J = 9.0, 2.2 Hz, 1H), 7.33 (d, J = 16.2 Hz, 1H), 6.64 (s, 1H), 3.39 (t, J = 5.6 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.1, 149.9, 147.2, 140.5, 132.3, 132.1, 131.5, 130.6, 130.5, 127.6, 126.0 (q, J = 4.0 Hz), 123.1, 119.6, 119.5, 98.3, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C22H22ClF3N3, 420.1454 [M + H]+; found, 420.1463; retention time 2.399 min (HPLC condition B).
(E)-N1-(6-Chloro-2-(4-methoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (25).
The title compound was synthesized from 6c and 7e. Isolated yield: 79%; yellow solid; mp 166–168 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.61 (d, J = 16.2 Hz, 1H), 7.58–7.53 (m, 3H), 7.14 (d, J = 16.2 Hz, 1H), 6.92 (d, J = 8.8 Hz, 2H), 6.63 (s, 1H), 3.85 (s, 3H), 3.37 (q, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.3, 159.5, 156.3, 150.2, 133.6, 131.1, 130.3, 129.8, 128.9, 119.5, 114.6, 113.8, 104.6, 100.6, 97.3, 57.4, 55.7, 45.4, 40.4; HRMS (ESI) m/z: calcd for C22H25ClN3O, 382.1686 [M + H]+; found, 382.1656; retention time 5.988 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(3,4-dimethoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-l,2-diamine (26).
The title compound was synthesized from 6c and 7f. Isolated yield: 81%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.57 (d, J = 16.2 Hz, 1H), 7.56 (dd, J = 9.0, 2.4 Hz, 1H), 7.22 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 16.2 Hz, 1H), 7.14 (dd, J = 4.0, 2.2 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.66 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.40–3.35 (m, 2H), 2.75–2.71 (m, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 158.1, 150.0, 149.5, 148.9, 133.9, 131.4, 130.2, 122.9, 121.5, 119.6, 112.6, 111.5, 111.0, 109.3, 100.9, 57.3, 56.2, 56.0, 45.3, 40.1; HRMS (ESI) m/z: calcd for C23H27ClN3O2, 412.1792 [M + H]+; found, 412.1775; retention time 5.910 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(3,4,5-trimethoxystyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (27).
The title compound was synthesized from 6c and 7g. Isolated yield: 75%; yellow solid; mp 187–189 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.72 (d, J = 2.2 Hz, 1H), 7.56 (dd, J = 9.0, 2.4 Hz, 1H), 7.54 (d, J = 16.2 Hz, 1H), 7.19 (d, J = 16.2 Hz, 1H), 6.87 (s, 2H), 6.67 (s, 1H), 3.92 (s, 6H), 3.89 (s, 3H), 3.41–3.36 (m, 2H), 2.75–2.71 (m, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.9, 153.8, 149.7, 147.3, 139.0, 133.9, 132.7, 131.4, 130.4, 130.2, 129.4, 119.6, 119.5, 104.6, 97.5, 61.3, 57.4, 56.5, 45.5, 40.4; HRMS (ESI) m/z: calcd for C24H29ClN3O3, 442.1897 [M + H]+; found, 442.1868; retention time 6.390 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-(dimethylamino)styryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (28).
The title compound was synthesized from 6c and 7j. Isolated yield: 84%; red solid; mp 159–162 °C; 1H NMR (400 MHz, CD3OD): δ 8.17 (d, J = 2.2 Hz, 1H), 7.69–7.65 (m, 3H), 7.40 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 16.2 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.61 (d, J = 8.2 Hz, 1H), 6.60 (s, 1H), 3.65 (t, J = 5.6 Hz, 2H), 2.97 (s, 6H), 2.81 (t, J = 5.6 Hz, 2H), 2.43 (s, 6H); 13C NMR (100 MHz, CD3OD): δ 154.0, 152.9, 152.1, 140.1, 132.4, 131.0, 130.7, 129.5, 123.5, 123.2, 121.6, 118.2, 115.9, 111.9, 95.1, 56.7, 44.3, 40.7, 39.2; HRMS (ESI) m/z: calcd for C23H28ClN4, 395.2002 [M + H]+; found, 395.1984; retention time 7.245 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (29).
The title compound was synthesized from 6c and 7h. Isolated yield: 88%; yellow solid; mp 177–179 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.63 (d, J = 16.2 Hz, 1H), 7.60–7.55 (m, 3H), 7.18 (d, J = 16.2 Hz, 1H), 7.10–7.05 (m, 2H), 6.62 (s, 1H), 3.38 (q, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 156.7, 149.8, 147.2, 133.2, 132.8, 131.4, 130.5, 129.5, 129.1 (d, J = 8.0 Hz), 119.6, 119.5, 116.1 (d, J = 22.0 Hz), 98.0, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H22ClFN3, 370.1486 [M + H]+; found, 370.1496; retention time 2.341 min (HPLC condition B).
(E)-N1-(6-Chloro-2-(2-fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (30).
The title compound was synthesized from 6c and 7k. Isolated yield: 82%; yellow solid; mp 116–118 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 16.4 Hz, 1H), 7.74 (d, J = 2.2 Hz, 1H), 7.70 (dd, J = 7.6, 1.8 Hz, 1H), 7.55 (dd, J = 7.6, 2.2 Hz, 1H), 7.35 (d, J = 16.4 Hz, 1H), 7.31–7.27 (m, 1H), 7.17 (dd, J = 7.6, 1.0 Hz, 1H), 7.14–7.08 (m, 1H), 6.67 (s, 1H), 3.38 (t, J = 5.6 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 161.1 (d, J = 249.0 Hz), 156.7, 149.9, 147.0, 132.0, 131.2, 130.5, 130.4, 130.0 (d, J = 8.0 Hz), 128.2 (d, J = 3.0 Hz), 126.4 (d, J = 3.0 Hz), 124.9 (d, J = 12.0 Hz), 124.7 (d, J = 4.0 Hz), 119.6, 119.5, 116.2 (d, J = 22.0 Hz), 97.8, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H22ClFN3, 370.1486 [M + H]+; found, 370.1463; retention time 5.710 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(3-fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (31).
The title compound was synthesized from 6c and 7l. Isolated yield: 82%; yellow solid; mp 154–157 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.62 (d, J = 16.2 Hz, 1H), 7.56 (dd, J = 9.0, 2.2 Hz, 1H), 7.39–7.29 (m, 4H), 7.24 (d, J = 16.2 Hz, 1H), 6.61 (s, 1H), 3.37 (t, J = 5.6 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 163.5 (d, J = 244.0 Hz), 156.3, 149.9, 147.1, 139.4 (d, J = 7.0 Hz), 132.8 (d, J = 3.0 Hz), 131.4, 130.9, 130.5 (d, J = 2.0 Hz), 130.4 (d, J = 8.0 Hz), 123.5 (d, J = 3.0 Hz), 119.6, 119.5, 115.5 (d, J = 22.0 Hz), 113.8 (d, J = 22.0 Hz), 98.2, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H22ClFN3, 370.1486 [M + H]+; found, 370.1459; retention time 5.803 min (HPLC condition A).
(E)-N1,N1-Dimethyl-N2-(2-styrylquinolin-4-yl)ethane-1,2-diamine (32).
The title compound was synthesized from 6d and 7a. Isolated yield: 89%; yellow solid; mp 158–161 °C; 1H NMR (400 MHz, CDCl3): δ 8.26 (d, J = 8.6 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 16.2 Hz, 1H), 7.62 (d, J = 4.0 Hz, 1H), 7.59 (dd, J = 8.4, 2.8 Hz, 1H), 7.41–7.36 (m, 2H), 7.33–7.28 (m, 3H), 6.64 (s, 1H), 3.53 (t, J = 6.0 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 153.8, 152.4, 143.6, 137.3, 136.0, 131.4, 129.6, 129.1, 128.0, 12.5.7, 120.8, 117.6, 96.1, 57.0, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H24N3, 318.1970 [M + H]+; found, 318.1955; retention time 6.292 min (HPLC condition A).
(E)-N1,N1-Dimethyl-N2-(2-(4-nitrostyryl)quinolin-4-yl)ethane-1,2-diamine (33).
The title compound was synthesized from 6d and 7b. Isolated yield: 87%; yellow solid; mp 152–154 °C; 1H NMR (400 MHz, CDCl3): δ 8.22 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.76–7.72 (m, 3H), 7.69–7.64 (m, 1H), 7.46–7.41 (m, 2H), 6.65 (s, 1H), 3.40 (t, J = 5.8 Hz, 2H), 2.75 (t, J = 5.8 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 155.1, 150.9, 147.5, 143.6, 134.2, 131.3, 130.1, 129.7, 127.9, 125.3, 124.4, 120.1, 118.8, 97.9, 57.4, 45.4, 40.3; HRMS (ESI) m/z: calcd for C21H23N4O2, 363.1821 [M + H]+; found, 363.1798; retention time 6.133 min (HPLC condition A).
(E)-N1-(2-(4-Fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (34).
The title compound was synthesized from 6d and 7h. Isolated yield: 84%; yellow solid; mp 194–198 °C; 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 16.2 Hz, 1H), 7.64–7.55 (m, 3H), 7.39 (t, J = 7.4 Hz, 1H), 7.26 (d, J = 16.2 Hz, 1H), 7.00 (t, J = 8.6 Hz, 2H), 6.64 (s, 1H), 3.46 (t, J = 5.8 Hz, 2H), 2.75 (t, J = 5.8 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 163.3 (d, J = 247.0 Hz), 155.6, 151.2, 133.6, 133.3 (d, J = 3.0 Hz), 130.3, 129.3 (d, J = 8.0 Hz), 128.4, 128.1, 125.0, 120.2, 118.3, 116.1 (d, J = 21.0 Hz), 96.9, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C21H23FN3, 336.1876 [M + H]+; found, 336.1853; retention time 6.245 min (HPLC condition A).
(E)-N1-(6-Chloro-2-styrylquinolin-4-yl)-N4,N4-dimethylbutane-1,4-diamine (35).
The title compound was synthesized from 6e and 7a. Isolated yield: 85%; yellow solid; mp 102–105 °C; 1H NMR (400 MHz, CD3OD): δ 8.08 (d, J = 2.2 Hz, 1H), 7.71 (d, J = 9.0 Hz, 1H), 7.60–7.57 (m, 2H), 7.56 (d, J = 5.8 Hz, 1H), 7.51 (dd, J = 9.0, 2.2 Hz, 1H), 7.35 (d, J = 5.8 Hz, 1H), 7.31 (d, J = 16.4 Hz, 1H), 7.15 (d, J = 16.4 Hz, 1H), 6.71 (s, 1H), 3.36 (t, J = 6.8 Hz, 2H), 2.64 (t, J = 6.8 Hz, 2H), 2.44 (s, 6H), 1.77–1.68 (m, 4H); 13C NMR (100 MHz, CD3OD): δ 156.2, 151.3, 145.3, 136.6, 135.1, 130.5, 130.1, 128.9, 128.8, 128.4, 127.4, 127.3, 120.8, 119.2, 96.3, 58.7, 46.7, 43.7, 25.9, 24.0; HRMS (ESI) m/z: calcd for C23H27ClN3, 380.1894 [M + H]+; found, 380.1876; retention time 6.078 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-nitrostyryl)quinolin-4-yl)-N4,N4-dimethylbutane-1,4-diamine (36).
The title compound was synthesized from 6e and 7b. Isolated yield: 76%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CD3OD): δ 8.17 (d, J = 8.8 Hz, 2H), 8.15 (d, J = 2.2 Hz, 1H), 7.78 (dd, J = 8.8, 3.6 Hz, 3H), 7.72 (d, J = 16.4 Hz, 1H), 7.59 (dd, J = 9.0, 2.2 Hz, 1H), 7.36 (d, J = 16.4 Hz, 1H), 6.86 (s, 1H), 3.50 (t, J = 6.4 Hz, 2H), 3.03 (t, J = 6.4 Hz, 2H), 2.75 (s, 6H), 1.87–1.82 (m, 2H); 13C NMR (100 MHz, CD3OD): δ 155.2, 151.5, 147.8, 145.3, 143.1, 132.6, 131.7, 130.8, 130.6, 128.6, 127.9, 123.9, 120.9, 97.1, 58.0, 42.8, 42.4, 25.5, 22.9; HRMS (ESI) m/z: calcd for C23H26ClN4O2, 425.1744 [M + H]+; found, 425.1724; retention time 6.177 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(4-fluorostyryl)quinolin-4-yl)-N4,N4-dimethylbutane-1,4-diamine (37).
The title compound was synthesized from 6e and 7h. Isolated yield: 81%; yellow solid; mp 145–147 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 2.2 Hz, 1H), 7.65 (d, J = 16.4 Hz, 1H), 7.59 (d, J = 5.8 Hz, 1H), 7.58 (d, J = 5.8 Hz, 1H), 7.53 (dd, J = 9.0, 2.0 Hz, 1H), 7.18 (d, J = 16.4Hz, 1H), 7.06 (t, J = 8.6 Hz, 2H), 6.55 (s, 1H), 3.32 (t, J = 6.8 Hz, 2H), 2.45 (t, J = 6.8 Hz, 2H), 2.35 (s, 6H), 1.97–1.92 (m, 2H), 1.78–1.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 156.4, 150.7, 146.7, 133.2 (d, J = 3.0 Hz), 133.0, 130.7, 130.4, 130.0, 129.2 (d, J = 8.0 Hz), 120.3, 119.6, 116.1 (d, J = 21.0 Hz), 97.3, 59.8, 45.8, 43.9, 27.1, 26.2; HRMS (ESI) m/z: calcd for C23H26ClFN3, 398.1799 [M + H]+; found, 398.1779; retention time 6.641 min (HPLC condition A).
(E)-N1-(2-(2-(Furan-2-yl)vinyl)-6-methoxyquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (39).
The title compound was synthesized from 6a and 38a. Isolated yield: 62%; yellow solid; mp 114–116 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 9.2 Hz, 1H), 7.52 (d, J = 16.2 Hz, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.20 (dd, J = 9.2, 2.8 Hz, 1H), 7.16 (d, J = 2.6 Hz, 1H), 7.07 (d, J = 16.2 Hz, 1H), 6.51 (d, J = 3.4 Hz, 1H), 6.44–6.41 (m, 2H), 3.89 (s, 3H), 3.40 (t, J = 6.2 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.32 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.3, 152.9, 152.3, 151.0, 143.7, 129.2, 128.4, 126.4, 122.3, 121.7, 118.8, 112.4, 101.0, 100.9, 97.1, 57.3, 56.2, 45.5, 40.8; HRMS (ESI) m/z: calcd for C20H24N3O2, 338.1869 [M + H]+; found, 338.1867; retention time 2.497 min (HPLC condition B).
(E)-N1-(6-Methoxy-2-(2-(thiophen-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (40).
The title compound was synthesized from 6a and 38b. Isolated yield: 64%; yellow solid; mp 123–126 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 9.2 Hz, 1H), 7.74 (d, J = 16.2 Hz, 1H), 7.25–7.19 (m, 3H), 7.14 (d, J = 2.8 Hz, 1H), 7.04–6.99 (m, 2H), 6.45 (s, 1H), 3.89 (d, J = 1.2 Hz, 3H), 3.39 (t, J = 6.0 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.33 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.2, 152.8, 150.6, 142.4, 141.8, 129.1, 128.4, 128.2, 127.3, 127.1, 126.3, 121.4, 119.0, 100.8, 96.8, 57.4, 56.1, 45.5, 40.8; HRMS (ESI) m/z: calcd for C20H24N3OS, 354.1640 [M + H]+; found, 354.1617; retention time 5.373 min (HPLC condition A).
(E)-N1-(6-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (41).
The title compound was synthesized from 6a and 38c. Isolated yield: 80%; yellow solid; mp 168–170 °C; 1H NMR (400 MHz, CDCl3): δ 8.54 (d, J = 5.2 Hz, 2H), 7.96 (d, J = 8.8 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 16.4 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.23 (s, 1H), 6.65 (s, 1H), 3.89 (s, 3H), 3.59 (t, J = 6.0 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 2.44 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.9, 150.6, 143.6, 142.6, 140.9, 129.3, 126.6, 126.3, 122.6, 121.8, 118.9, 101.0, 96.7, 57.1, 56.3, 45.3, 40.6; HRMS (ESI) m/z: calcd for C21H25N4O, 349.2028 [M + H]+; found, 349.2056; retention time 2.355 min (HPLC condition B).
(E)-N1-(6-Methoxy-2-(2-(pyridin-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (42).
The title compound was synthesized from 6a and 38d. Isolated yield: 83%; brown solid; mp 147–150 °C; 1H NMR (400 MHz, CD3OD): δ 8.63 (dd, J = 4.8, 2.4 Hz, 1H), 7.96 (d, J = 16.4 Hz, 1H), 7.90–7.85 (m, 1H), 7.83 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.71–7.67 (m, 2H), 7.51 (dd, J = 9.2, 2.6 Hz, 1H), 7.19 (s, 1H), 3.99 (s, 3H), 3.29–3.28 (m, 4H), 2.77 (s, 6H); 13C NMR (100 MHz, CD3OD): δ 159.0, 154.5, 153.5, 150.0, 149.1, 137.8, 125.0, 124.7, 124.6, 122.6, 118.6, 102.1, 95.9, 56.0, 43.5, 39.5; HRMS (ESI) m/z: calcd for C21H25N4O, 349.2028 [M + H]+; found, 349.2009; retention time 2.744 min (HPLC condition A).
(E)-N1-(6-Methoxy-2-(2-(pyridin-3-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (43).
The title compound was synthesized from 6a and 7e. Isolated yield: 76%; brown solid; mp 127–129 °C; 1H NMR (400 MHz, CDCl3): δ 8.79 (d, J = 2.2 Hz, 1H), 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.95–7.92 (m, 1H), 7.62 (d, J = 16.4 Hz, 1H), 7.37 (d, J = 16.4 Hz, 1H), 7.32 (dd, J = 9.2, 2.6 Hz, 2H), 7.12 (d, J = 2.6 Hz, 1H), 6.65 (s, 1H), 3.95 (s, 3H), 3.48–3.44 (m, 2H), 2.81 (t, J = 5.6 Hz, 2H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.5, 150.8, 150.5, 149.6, 149.4, 148.7, 136.7, 133.6, 132.7, 130.2, 124.0, 123.2, 121.4, 119.2, 100.7, 97.1, 57.4, 56.2, 45.4, 40.6; HRMS (ESI) m/z: calcd for C21H25N4O, 349.2028 [M + H]+; found, 349.2001; retention time 2.918 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(2-(furan-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (44).
The title compound was synthesized from 6b and 38a. Isolated yield: 65%; yellow solid; mp 107–110 °C; 1H NMR (400 MHz, CDCl3): δ 8.03 (dd, J = 9.0, 5.6 Hz, 1H), 7.56 (d, J = 16.0 Hz, 1H), 7.45 (d, J = 1.8 Hz, 1H), 7.42–7.35 (m, 2H), 7.14 (d, J = 16.0 Hz, 1H), 6.54 (s, 1H), 6.52 (d, J = 3.6 Hz, 1H), 6.46 (dd, J = 3.6, 1.8 Hz, 1H), 3.37 (t, J = 5.8 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 159.9 (d, J = 244.0 Hz), 155.2, 153.2, 150.4 (d, J = 4.0 Hz), 143.4, 131.4 (d, J = 6.0 Hz), 126.8, 121.9, 119.6 (d, J = 25.0 Hz), 119.0 (d, J = 9.0 Hz), 112.3, 111.4, 104.6 (d, J = 23.0 Hz), 98.1, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C19H21FN3O, 326.1669 [M + H]+; found, 326.1649; retention time 4.859 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(2-(thiophen-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (45).
The title compound was synthesized from 6b and 38b. Isolated yield: 69%; yellow solid; mp 121–123 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (dd, J = 9.0, 5.6 Hz, 1H), 7.83 (d, J = 16.0 Hz, 1H), 7.43–7.36 (m, 2H), 7.27 (d, J = 5.6 Hz, 1H), 7.22 (d, J = 3.6 Hz, 1H), 7.07 (d, J = 16.0 Hz, 1H), 7.03 (dd, J = 5.2, 3.6 Hz, 1H), 6.56 (s, 1H), 3.38 (t, J = 5.8 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 159.9 (d, J = 244.0 Hz), 155.3, 150.4 (d, J = 4.0 Hz), 145.0, 142.5, 131.4 (d, J = 9.0 Hz), 128.3, 128.1, 127.3, 126.2, 119.6 (d, J = 25.0 Hz), 119.0 (d, J = 9.0 Hz), 104.6 (d, J = 23.0 Hz), 97.6, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C19H21FN3S, 342.1440 [M + H]+; found, 342.1421; retention time 4.899 min (HPLC condition A).
(E)-N1-(6-Fluoro-2-(2-(pyridin-4-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (46).
The title compound was synthesized from 6b and 38c. Isolated yield: 84%; yellow solid; mp 167–169 °C; 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 5.8 Hz, 2H), 8.03 (dd, J = 9.6, 5.6 Hz, 1H), 7.60 (d, J = 16.2 Hz, 1H), 7.47 (d, J = 2.2 Hz, 2H), 7.46–7.39 (m, 3H), 6.65 (s, 1H), 3.41 (t, J = 5.8 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.2 (d, J = 245.0 Hz), 154.9, 150.6, 150.3 (d, J = 5.0 Hz), 145.7, 144.4, 134.2, 132.3 (d, J = 9.0 Hz), 130.8, 121.7, 119.7 (d, J = 24.0 Hz), 119.4 (d, J = 9.0 Hz), 104.6 (d, J = 23.0 Hz), 98.1, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C20H22FN4, 337.1828 [M + H]+; found, 337.1812; retention time 2.430 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(furan-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (47).
The title compound was synthesized from 6c and 38a. Isolated yield: 69%; yellow solid; mp 118–120 °C; 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.56 (d, J = 2.2 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 2.2 Hz, 1H), 7.14 (d, J = 16.2 Hz, 1H), 6.53 (s, 1H), 6.52 (d, J = 3.8 Hz, 1H), 6.46 (dd, J = 3.6, 1.8 Hz, 1H), 3.34 (q, J = 6.0 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.5, 153.3, 149.8, 147.3, 143.3, 131.4, 130.4, 130.0, 127.6, 121.6, 119.5, 119.4, 112.2, 111.2, 98.7, 57.4, 45.4, 40.3; HRMS (ESI) m/z: calcd for C19H21ClN3O, 342.1373 [M + H]+; found, 342.1350; retention time 5.965 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(thiophen-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (48).
The title compound was synthesized from 6c and 38b. Isolated yield: 65%; yellow solid; mp 131–132 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.82 (d, J = 16.2 Hz, 1H), 7.72 (d, J = 2.2 Hz, 1H), 7.55 (dd, J = 9.0, 2.4 Hz, 1H), 7.28 (s, 1H), 7.22 (d, J = 3.6 Hz, 1H), 7.07 (d, J = 11.6 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.56 (s, 1H), 3.36 (q, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.4, 149.8, 147.2, 142.6, 131.3, 130.5, 130.1, 128.9, 128.2, 128.1, 127.1, 126.1, 119.6, 98.1, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C19H21ClN3S, 358.1145 [M + H]+; found, 358.1113; retention time 6.280 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(thiazol-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-l,2-diamine (49).
The title compound was synthesized from 6c and 38f. Isolated yield: 62%; yellow solid; mp 108–110 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.87 (s, 1H), 7.84 (d, J = 16.2 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.57 (dd, J = 4.0, 2.2 Hz, 1H), 7.54 (d, J = 16.2 Hz, 1H), 7.33 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 3.35 (t, J = 5.6 Hz, 2H), 2.74 (t, J = 5.6 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 166.9, 155.4, 149.9, 147.2, 144.2, 135.2, 131.6, 130.8, 130.6, 126.4, 119.7, 119.6, 98.3, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C18H20ClN4S, 359.1097 [M + H]+; found, 359.1073; retention time 3.304 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(pyridin-4-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (50).
The title compound was synthesized from 6c and 38c. Isolated yield: 85%; yellow solid; mp 174–175 °C; 1H NMR (400 MHz, CDCl3): δ 8.62 (d, J = 6.2 Hz, 2H), 7.92 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.59 (d, J = 16.2 Hz, 1H), 7.14 (dd, J = 4.0, 2.2 Hz, 1H), 7.46 (d, J = 6.4 Hz, 2H), 7.42 (d, J = 16.2 Hz, 1H), 6.63 (s, 1H), 3.39–3.34 (m, 2H), 2.76–2.71 (m, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 155.7, 150.7, 150.0, 147.3, 144.4, 134.2, 131.7, 131.1, 130.7, 130.6, 121.7, 119.6, 119.6, 98.5, 57.3, 45.4, 40.4; HRMS (ESI) m/z: calcd for C20H22ClN4, 353.1533 [M + H]+; found, 353.1530; retention time 2.784 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(pyridin-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (51).
The title compound was synthesized from 6c and 38d. Isolated yield: 83%; yellow solid; mp 114–116 °C; 1H NMR (400 MHz, CDCl3): δ 8.64 (d, J = 4.8 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 16.2 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.69 (dd, J = 8.0, 2.4 Hz, 1H), 6.69 (s, 1H), 7.57 (d, J = 2.2 Hz, 1H), 7.55 (m, 1H), 3.34 (t, J = 5.8 Hz, 2H), 2.73 (t, J = 5.8 Hz, 2H), 2.34 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.1, 155.5, 150.1, 150.0, 137.0, 133.6, 133.3, 131.4, 130.5, 123.1, 119.7, 119.5, 99.0, 57.3, 45.4, 40.3; HRMS (ESI) m/z: calcd for C20H22ClN4, 353.1533 [M + H]+; found, 353.1530; retention time 2.844 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(pyridin-3-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (52).
The title compound was synthesized from 6c and 38e. Isolated yield: 86%; yellow solid; mp 128–132 °C; 1H NMR (400 MHz, CDCl3): δ 8.81 (d, J = 1.8 Hz, 1H), 8.53 (d, J = 4.8 Hz, 1H), 7.93 (d, J = 4.8 Hz, 1H), 7.91 (d, J = 16.2 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.65 (d, J = 16.2 Hz, 1H), 7.56 (dd, J = 9.0, 2.2 Hz, 1H), 7.33–7.29 (m, 2H), 6.64 (s, 1H), 3.41 (t, J = 5.8 Hz, 2H), 2.78 (t, J = 5.8 Hz, 2H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 155.8, 150.1, 149.7, 149.5, 146.7, 133.7, 132.6, 131.3, 131.0, 130.8, 130.7, 130.6, 124.0, 119.8, 119.4, 98.1, 57.2, 45.4, 40.3; HRMS (ESI) m/z: calcd for C20H22ClN4, 353.1533 [M + H]+; found, 353.1520; retention time 2.844 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(pyrimidin-5-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (53).
The title compound was synthesized from 6c and 38g. Isolated yield: 67%; yellow solid; mp 171–173 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.09 (s, 2H), 9.07 (s, 1H), 8.29 (d, J = 2.2 Hz, 1H), 7.81 (d, J = 6.0 Hz, 1H), 7.77 (d, J = 16.2 Hz, 1H), 7.62 (dd, J = 9.0, 2.2 Hz, 2H), 7.52 (d, J = 16.2 Hz, 1H), 6.93 (s, 1H), 3.68 (t, J = 5.6 Hz, 2H), 3.28 (t, J = 5.6 Hz, 2H), 2.76 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ 158.2, 156.0, 155.7, 150.4, 147.3, 133.6, 132.4, 131.6, 131.1, 130.8, 129.6, 127.3, 121.9, 120.0, 98.9, 55.5, 43.7, 40.5; HRMS (ESI) m/z: calcd for C19H21ClN5, 354.1485 [M + H]+; found, 354.1463; retention time 3.162 min (HPLC condition A).
(E)-N1-(2-(2-(1H-Indol-2-yl)vinyl)-6-chloroquinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (54).
The title compound was synthesized from 6c and 38h. Isolated yield: 63%; green solid; mp 148–150 °C; 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 2.2 Hz, 1H), 7.68–7.66 (dd, J = 4.0, 2.2 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.0 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 6.81 (d, J = 13.2 Hz, 1H), 6.71 (s, 1H), 6.45 (s, 1H), 6.39 (d, J = 13.2 Hz, 1H), 3.35 (t, J = 6.0 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.35 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.1, 150.0, 146.0, 137.3, 136.9, 130.9, 130.4, 129.8, 129.0, 126.4, 125.3, 123.3, 121.2, 119.8, 118.8, 112.1, 107.6, 102.5, 57.2, 45.4, 40.2; HRMS (ESI) m/z: calcd for C23H24ClN4, 391.1689 [M + H]+; found, 391.1680; retention time 6.631 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(6-methoxyquinolin-2-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (55).
The title compound was synthesized from 6c and 38i. Isolated yield: 66%; brown solid; mp 151–154 °C; 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.6 Hz, 1H), 7.96 (dd, J = 9.0, 2.2 Hz, 2H), 7.86 (d, J = 16.4 Hz, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 16.4 Hz, 2H), 7.57 (dd, J = 9.2, 2.2 Hz, 1H), 7.38 (dd, J = 9.2, 2.8 Hz, 1H), 7.08 (d, J = 2.8 Hz, 1H), 6.85 (s, 1H), 3.95 (s, 3H), 3.40 (t, J = 6.0 Hz, 2H), 2.78 (t, J = 6.0 Hz, 2H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 158.3, 156.3, 153.5, 150.0, 147.0, 146.6, 144.6, 135.6, 134.8, 133.9, 131.1, 130.7, 129.0, 122.9, 120.1, 119.8, 119.6, 105.6, 97.6, 57.3, 56.0, 45.4, 40.4; HRMS (ESI) m/z: calcd for C25H26ClN4O, 433.1795 [M + H]+; found, 433.1812; retention time 6.204 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-(naphthalen-1-yl)vinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (56).
The title compound was synthesized from 6c and 38j. Isolated yield: 60%; brown solid; mp 113–115 °C; 1H NMR (400 MHz, CDCl3): δ 8.49 (d, J = 16.0 Hz, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.87 (q, J = 9.2, 8.2 Hz, 4H), 7.74 (d, J = 2.2 Hz, 1H), 7.59 (dd, J = 5.8 Hz, 2.2 Hz, 1H), 7.55 (dd, J = 5.8 Hz, 2.2 Hz, 1H), 7.53–7.49 (m, 2H), 7.33 (d, J = 16.0 Hz, 1H), 6.72 (s, 1H), 3.42 (t, J = 7.0 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.36 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 156.8, 150.0, 147.0, 134.5, 134.1, 132.3, 131.9, 131.3, 130.5, 130.3, 129.2, 129.0, 126.6, 126.3, 126.0, 124.5, 124.3, 119.6, 119.4, 98.3, 57.3, 45.5, 40.4; HRMS (ESI) m/z: calcd for C25H25ClN3, 402.1737 [M + H]+; found, 402.1723; retention time 5.609 min (HPLC condition A).
(E)-N1-(6-Chloro-2-(2-cyclohexylvinyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (57).
The title compound was synthesized from 6c and 38k. Isolated yield: 48%; yellow solid; mp 88–91 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (d, J = 9.2 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 9.2, 2.2 Hz, 1H), 6.82 (d, J = 16.2 Hz, 1H), 6.60 (d, J = 16.2 Hz, 1H), 6.51 (s, 1H), 3.39 (t, J = 6.0 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.41–2.38 (m, 1H), 2.35 (s, 6H), 1.88–1.76 (m, 6H), 1.35–1.28 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 150.4, 144.9, 142.3, 131.0, 130.5, 129.1, 127.3, 126.7, 119.8, 119.0, 96.6, 57.1, 45.4, 41.6, 32.8, 30.1, 26.3; HRMS (ESI) m/z: calcd for C21H29ClN3, 358.2050 [M + H]+; found, 358.2024; retention time 6.114 min (HPLC condition A).
(E)-4-(6-Chloro-2-(4-fluorostyryl)quinolin-4-yl)morpholine (62).
The title compound was synthesized from 61a and 7h. Isolated yield: 89%; yellow solid; mp 173–175 °C; 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 9.0 Hz, 1H), 7.93 (d, J = 2.2 Hz, 1H), 7.64 (d, J = 16.2 Hz, 1H), 7.61–7.57 (m, 3H), 7.21 (d, J = 16.2 Hz, 1H), 7.11–7.07 (m, 3H), 4.03–4.00 (m, 4H), 3.26–3.23 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 163.3 (d, J = 247.0 Hz), 156.8, 156.6, 148.4, 133.6, 132.9 (d, J = 4.0 Hz), 131.8, 131.4, 130.7, 129.3, 129.2, 128.9 (d, J = 3.0 Hz), 123.8, 122.8, 116.2 (d, J = 22.0 Hz), 108.3, 67.2, 53.0; HRMS (ESI): calcd for C21H19ClFN2O [M + H]+ m/z, 369.1170; found, 369.1153; retention time 8.342 min (HPLC condition A).
(E)-4-(6-Chloro-2-(2-(pyridin-4-yl)vinyl)quinolin-4-yl)morpholine (63).
The title compound was synthesized from 61a and 38c. Isolated yield: 84%; yellow solid; mp 195–198 °C; 1H NMR (400 MHz, CDCl3): δ 8.63 (d, J = 5.4 Hz, 2H), 7.98 (d, J = 9.0 Hz, 1H), 7.94 (d, J = 2.4 Hz, 1H), 7.62–7.58 (m, 2H), 7.46–7.43 (m, 3H), 7.09 (s, 1H), 4.3–4.00 (m, 4H), 3.26–3.23 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 156.9, 155.7, 150.7, 148.4, 144.0, 133.5, 132.0, 131.9, 130.9, 124.0, 122.8, 121.7, 108.7, 67.2, 53.0; HRMS (ESI): calcd for C2oH19ClN3O [M + H]+ m/z, 352.1217; found, 352.1196; retention time 6.309 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-4-(pyrrolidin-1-yl)quinoline (65).
The title compound was synthesized from 61b and 7h. Isolated yield: 85%; yellow solid; mp 161–163 °C; 1H NMR (400 MHz, CDCl3): δ 8.15 (d, J = 2.2 Hz, 1H), 7.89 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 16.2 Hz, 1H), 7.59–7.56 (m, 2H), 7.51 (dd, J = 8.6, 2.2 Hz, 1H), 7.14 (d, J = 16.2 Hz, 1H), 7.07 (t, J = 8.0 Hz, 2H), 6.64 (s, 1H), 3.72–3.68 (m, 4H), 2.08–2.06 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 163.1 (d, J = 247.0 Hz), 155.8, 152.6, 149.1, 133.3 (d, J = 3.0 Hz), 132.4, 131.4, 129.8, 129.5 (d, J = 3.0 Hz), 129.1, 129.0, 128.4, 124.5, 121.6, 116.1 (d, J = 22.0 Hz), 102.3, 52.5, 26.3; HRMS (ESI): calcd for C21H19ClFN2 [M + H]+ m/z, 353.1221; found, 353.1205; retention time 8.792 min (HPLC condition A).
(E)-6-Chloro-2-(2-(pyridin-4-yl)vinyl)-4-(pyrrolidin-1-yl)quinoline (66).
The title compound was synthesized from 61b and 38c. Isolated yield: 81%; yellow solid; mp 175–177 °C; 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 6.0 Hz, 2H), 8.19 (d, J = 2.2 Hz, 1H), 7.92 (d, J = 9.0 Hz, 1H), 7.60 (d, J = 16.2 Hz, 1H), 7.54 (dd, J = 9.0, 2.2 Hz, 1H), 7.46 (d, J = 6.0 Hz, 2H), 7.40 (d, J = 16.2 Hz, 1H), 6.68 (s, 1H), 3.76–3.73 (m, 4H), 2.12–2.08 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 152.8, 150.6, 144.4, 134.0, 131.4, 131.0, 130.1, 129.0, 124.5, 121.7, 102.7, 52.6, 26.4; HRMS (ESI): calcd for C20H19ClN3 [M + H]+ m/z, 336.1268; found, 336.1241; retention time 6.372 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-4-(4-(pyridin-2-yl)piperazin-1-yl)-quinoline (67).
The title compound was synthesized from 61c and 7h. Isolated yield: 74%; yellow solid; mp 154–155 °C; 1H NMR (400 MHz, CDCl3): δ 8.27 (ddd, J = 5.0, 2.0, 0.8 Hz, 1H), 8.00 (d, J = 2.4 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.67 (s, 1H), 7.62–7.53 (m, 5H), 7.22 (d, J = 16.2 Hz, 1H), 7.11 (s, 1H), 7.08 (d, J = 8.6 Hz, 1H), 6.76 (d, J = 8.6 Hz, 1H), 6.72 (ddd, J = 7.2, 5.0, 0.8 Hz, 1H), 3.87–3.83 (m, 4H), 3.38–3.34 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 163.3 (d, J = 248.0 Hz), 159.8, 156.8, 156.7, 148.4, 138.0, 133.5, 133.0 (d, J = 4.0 Hz), 131.8, 131.4, 130.7, 129.3, 129.2, 129.0 (d, J = 2.0 Hz), 124.0, 122.9, 116.2 (d, J = 22.0 Hz), 114.4, 108.5, 107.7, 52.4, 45.9; HRMS (ESI): calcd for C26H23ClFN4 [M + H]+ m/z, 445.1595; found, 445.1588; retention time 7.497 min (HPLC condition A).
(E)-6-Chloro-4-(4-(pyridin-2-yl)piperazin-1-yl)-2-(2-(pyridin-4-yl)-vinyl)quinoline (68).
The title compound was synthesized from 61c and 38c. Isolated yield: 71%; brown solid; mp 163–166 °C; 1H NMR (400 38f Hz, CDCl3): δ 8.65 (d, J = 3.2 Hz, 2H), 8.26 (ddd, J = 5.0, 2.0, 0.8 Hz, 1H), 8.02 (d, J = 2.4Hz, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.60 (d, J = 4.2 Hz, 1H), 7.58–7.54 (m, 1H), 7.49–7.45 (m, 3H), 7.14 (s, 1H), 6.77 (d, J = 8.6 Hz, 1H), 6.72 (ddd, J = 7.2, 5.0, 0.8 Hz, 1H), 3.87–3.84 (m, 4H), 3.40–3.37 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 159.8, 157.0, 150.7, 148.4, 144.0, 138.0, 133.5, 132.0, 131.9, 130.9, 124.2, 123.0. 121.7, 114.4, 108.9, 107.8, 52.4, 45.9; HRMS (ESI): calcd for C21H19ClFN2 [M + H]+ m/z, 428.1642; found, 428.1621; retention time 5.680 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(4-(piperidin-1-yl)phenyl)-quinolin-4-amine (69).
The title compound was synthesized from 61d and 7h. Isolated yield: 48%; yellow solid; mp 154–156 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.59 (dd, J = 9.0, 2.2 Hz, 1H), 7.55 (d, J = 5.4 Hz, 1H), 7.53 (d, J = 2.8 Hz, 1H), 7.51 (d, J = 16.2 Hz, 1H), 7.22 (d, J = 7.0 Hz, 2H), 7.09–7.02 (m, 5H), 6.92 (s, 1H), 3.24–3.21 (m, 4H), 1.80–1.74 (m, 4H), 1.65 1.62 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 156.5, 150.7, 149.0, 133.1, 131.4, 130.8, 130.5, 129.2 (d, J = 8.0 Hz), 129.0, 125.9, 119.6, 119.3, 117.7, 116.1 (d, J = 21.0 Hz), 100.5, 51.0, 26.2, 24.6; HRMS (ESI): calcd for C28H26ClFN3 [M + H]+ m/z, 458.1799; found, 458.1787; retention time 9.254 min (HPLC condition A).
(E)-6-Chloro-N-(4-(piperidin-1-yl)phenyl)-2-(2-(pyridin-4-yl)-vinyl)quinolin-4-amine (70).
The title compound was synthesized from 61d and 38c. Isolated yield: 45%; brown solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.59 (s, br, 2H), 7.96 (d, J = 9.0 Hz, 1H), 7.88 (d, J = 2.2 Hz, 1H), 7.62 (dd, J = 9.0, 2.2 Hz, 1H) 7.48 (d, J = 16.2 Hz, 1H), 7.41 (d, J = 5.6 Hz, 2H), 7.31 (d, J = 16.2 Hz, 1H), 7.22 (d, J = 9.0 Hz, 2H), 7.04 (d, J = 9.0 Hz, 2H), 6.93 (s, 1H), 3.25–3.22 (m, 4H), 1.80–1.74 (m, 4H), 1.66–1.60 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.5, 150.8, 150.6, 149.3, 147.7, 144.3, 133.9, 131.8, 131.3, 131.2, 130.9, 130.3, 126.1, 121.7, 119.4, 117.7, 101.0, 51.0, 26.2, 24.6; HRMS (ESI): calcd for C27H26ClN4 [M + H]+ m/z, 441.1846; found, 441.1805; retention time 6.342 min (HPLC condition A).
(E)-4-([1,4′-Bipiperidin]-1′-yl)-6-chloro-2-(4-fluorostyryl)-quinoline (71).
The title compound was synthesized from 61e and 7h. Isolated yield: 65%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 9.0 Hz, 1H), 7.84 (d, J = 2.2 Hz, 1H), 7.62 (d, J = 16.2 Hz, 1H), 7.60–7.55 (m, 3H), 7.18 (d, J = 16.2 Hz, 1H), 7.10–7.05 (m, 3H), 3.69–3.66 (m, 2H), 2.98–2.86 (m, 7H), 2.31–2.28 (m, 2H), 2.10–2.04 (m, 2H), 1.98–1.91 (m, 4H), 1.64–1.57 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.3 (d, J = 247.0 Hz), 156.7, 156.5, 133.6, 132.9 (d, J = 3.0 Hz), 131.8, 131.4, 130.6, 129.3, 129.2, 128.9 (d, J = 3.0 Hz), 123.9, 122.7, 116.2 (d, J = 19.0 Hz), 108.6, 63.7, 52.1, 50.6, 27.5, 24.8, 23.9; HRMS (ESI): calcd for C27H30ClFN3 [M + H]+ m/z, 450.2112; found, 450.2064; retention time 6.414 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(pyridin-2-ylmethyl)quinolin-4-amine (72).
The title compound was synthesized from 61f and 7h. Isolated yield: 58%; yellow solid; mp 188–190 °C; 1H NMR (400 MHz, CDCl3): δ 8.68 (d, J = 5.0 Hz, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.74–7.70 (m, 1H), 7.60–7.55 (m, 3H), 7.53 (d, J = 3.2 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.29 (dd, J = 4.8, 3.6 Hz, 1H), 7.13 (d, J = 16.2 Hz, 1H), 7.08–7.03 (m, 2H), 6.62 (s, 1H), 4.63 (d, J = 4.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 163.3 (d, J = 247.0 Hz), 156.8, 156.6, 148.4, 133.6, 132.9 (d, J = 4.0 Hz), 131.8, 131.4, 130.7, 129.3, 129.2, 128.9 (d, J = 3.0 Hz), 122.8, 116.2 (d, J = 22.0 Hz), 108.3, 67.2, 53.0; HRMS (ESI): calcd for C23H18ClFN3 [M + H]+ m/z, 390.1173; found, 390.1150; retention time 7.926 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(2-morpholinoethyl)quinolin-4-amine (81).
The title compound was synthesized from 80a and 7h. Isolated yield: 80%; yellow solid; mp 147–149 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.60 (s, 1H), 7.59–7.55 (m, 3H), 7.16 (d, J = 16.2 Hz, 1H), 7.07 (t, J = 8.7 Hz, 2H), 6.61 (s, 1H), 3.79 (t, J = 4.6 Hz, 4H), 3.40 (q, J = 5.4 Hz, 2H), 2.84–2.80 (m, 2H), 2.59–2.55 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 246.0 Hz), 156.7, 149.7, 147.1, 133.1 (d, J = 3.0 Hz), 133.0, 131.4, 130.5, 130.4, 129.3, 129.1 (d, J = 8.0 Hz), 119.4, 119.3, 116.1 (d, J = 22.0 Hz), 98.1, 67.4, 56.4, 53.5, 39.2; HRMS (ESI) m/z: calcd for ĈHĈlFÔ, 412.1592 [M + H]+; found, 412.1565; retention time 6.037 min (HPLC condition A).
(E)-6-Chloro-N-(2-morpholinoethyl)-2-(2-(pyridin-4-yl)vinyl)-quinolin-4-amine (82).
The title compound was synthesized from 80a and 38c. Isolated yield: 83%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 6.0 Hz, 2H), 7.93 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 6.2 Hz, 2H), 7.41 (d, J = 16.2 Hz, 1H), 6.64 (s, 1H), 5.86 (t, J = 4.2 Hz, 1H), 3.81–3.78 (m, 4H), 3.43–3.39 (m, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.59–2.56 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 155.7, 150.7, 149.8, 147.2, 144.3, 134.1, 131.7, 131.3, 130.7, 121.7, 119.4, 98.7, 67.4, 56.4, 53.5, 39.2; HRMS (ESI) m/z: calcd for C22H24ClN4O, 395.1639 [M + H]+; found, 395.1613; retention time 2.839 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(2-(pyrrolidin-1-yl)ethyl)-quinolin-4-amine (83).
The title compound was synthesized from 80b and 7h. Isolated yield: 88%; yellow solid; mp 157–159 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 9.0 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H), 7.63 (d, J = 16.2 Hz, 1H), 7.59 (dd, J = 8.6, 5.4 Hz, 2H), 7.55 (dd, J = 9.0, 2.2 Hz, 1H), 7.17 (d, J = 16.2 Hz, 1H), 7.07 (t, J = 8.6 Hz, 2H), 6.62 (s, 1H), 3.47 (t, J = 6.0 Hz, 2H), 2.98 (t, J = 6.0 Hz, 2H), 2.73–2.68 (m, 4H), 1.91–1.86 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 156.4, 150.0, 146.7, 133.2, 133.1 (d, J = 4.0 Hz), 130.9, 130.6, 130.4, 129.2 (d, J = 8.0 Hz), 128.9, 119.9, 116.1 (d, J = 22.0 Hz), 97.8, 54.2, 41.6, 30.1, 23.9; HRMS (ESI) m/z: calcd for C23H24ClFN3, 396.1643 [M + H]+; found, 396.1618; retention time 6.000 min (HPLC condition A).
(E)-6-Chloro-N-(2-(pyrrolidin-1-yl)ethyl)-2-styrylquinolin-4-amine (84).
The title compound was synthesized from 80b and 7a. Isolated yield: 90%; yellow solid; mp 164–166 °C; 1H NMR (400 MHz, CDCl3): δ 8.51 (d, J = 9.0 Hz, 1H), 8.36 (s, 1H), 8.24 (d, J = 16.2 Hz, 1H), 8.18 (d, J = 7.7 Hz, 2H), 8.12 (dd, J = 9.0, 2.0 Hz, 1H), 7.95 (t, J = 7.5 Hz, 2H), 7.90–7.81 (m, 2H), 6.63 (s, 1H), 4.05 (t, J = 5.8 Hz, 2H), 3.55 (t, J = 5.8 Hz, 2H), 3.30–3.24 (m, 4H), 2.48–2.43 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 156.7, 150.0, 146.9, 136.9, 134.3, 131.1, 130.5, 130.3, 129.4, 129.1, 128.9, 127.6, 119.8, 119.5, 97.8, 54.2, 41.6, 23.9; HRMS (ESI) m/z: calcd for C23H25ClN3, 378.1737 [M + H]+; found, 378.1750, retention time 5.904 min (HPLC condition A).
(E)-6-Chloro-2-(2-(pyridin-4-yl)vinyl)-N-(2-(pyrrolidin-1-yl)ethyl)-quinolin-4-amine (85).
The title compound was synthesized from 80b and 38c. Isolated yield: 91%; yellow solid; mp 197–199 °C; 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 6.0 Hz, 2H), 7.92 (d, J = 9.0 Hz, 1H), 7.82 (d, J = 2.2 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 2.8 Hz, 1H), 7.47–7.45 (m, 2H), 7.42 (d, J = 16.2 Hz, 1H), 6.64 (s, 1H), 3.50 3.46 (m, 2H), 3.01–2.97 (m, 2H), 2.72 (t, J = 6.2 Hz, 4H), 1.92–1.87 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 155.6, 150.6, 150.0, 147.1, 131.2, 131.2, 130.8, 130.7, 121.7, 119.9, 119.6, 98.4, 54.2, 54.1, 41.5, 23.9; HRMS (ESI) m/z: calcd for C22H24ClN4, 379.1689 [M + H]+; found, 379.1664; retention time 2.908 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(2-(4-methylpiperazin-1-yl)-ethyl)quinolin-4-amine (86).
The title compound was synthesized from 80c and 7h. Isolated yield: 84%; yellow solid; mp 157–159 °C; 1H NMR (400 MHz, CDCl3): δ 8.13 (t, J = 8.0 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 2.2 Hz, 1H), 7.64 (d, J = 16.2 Hz, 1H), 7.58–7.51 (m, 3H), 7.17 (d, J = 16.2 Hz, 1H), 7.05 (t, J = 7.4 Hz, 2H), 6.58 (s, 1H), 3.45 (m, 2H), 2.78 (t, J = 6.0 Hz, 2H), 2.69–2.61 (m, 8H), 2.42 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 163.4 (d, J = 247.0 Hz), 155.9, 150.3, 134.0, 132.9 (d, J = 3.0 Hz), 132.4, 132.3, 130.9, 130.7, 129.3 (d, J = 8.0 Hz), 127.8, 119.8, 119.2, 116.2, 116.0, 115.2 (d, J = 21.0 Hz), 97.6, 55.7, 55.0, 52.4, 45.7, 39.6; HRMS (ESI) m/z: calcd for C24H27ClFN4, 425.1908 [M + H]+; found, 425.1908; retention time 2.319 min (HPLC condition B).
(E)-6-Chloro-N-(2-(4-methylpiperazin-1-yl)ethyl)-2-(2-(pyridin-4-yl)vinyl)quinolin-4-amine (87).
The title compound was synthesized from 80c and 38c. Isolated yield: 79%; yellow solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.49 (d, J = 6.0 Hz, 2H), 8.08 (d, J = 2.2 Hz, 1H), 7.75 (s, 1H), 7.60 (d, J = 16.2 Hz, 1H), 7.58 (d, J = 1.6 Hz, 2H), 7.56 (dd, J = 6.8 Hz, J = 1.6 Hz, 1H), 7.42 (d, J = 16.2 Hz, 1H), 6.85 (s, 1H), 3.54 (t, J = 6.8 Hz, 2H), 2.78 (t, J = 6.8 Hz, 2H), 2.74–2.65 (m, 8H), 2.42 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 155.3, 151.2, 149.5, 145.7, 145.3, 133.1, 131.6, 130.7, 129.1, 122.0, 120.8, 119.4, 97.2, 55.7, 54.4, 52.0, 44.4, 40.0; HRMS (ESI) m/z: calcd for C23H27ClN5, 408.1955 [M + H]+; found, 408.1958; retention time 3.101 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(3-morpholinopropyl)quinolin-4-amine (90).
The title compound was synthesized from 89a and 7h. Isolated yield: 88%; yellow solid; mp 170–171 °C; 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.65 (d, J = 16.4Hz, 1H), 7.60 (d, J = 5.4Hz, 1H), 7.58–7.55 (m, 2H), 7.18 (d, J = 16.2 Hz, 1H), 7.07 (t, J = 8.6 Hz, 2H), 6.55 (s, 1H), 3.96 (t, J = 4.8 Hz, 4H), 3.48 (q, J = 5.4 Hz, 2H), 2.70–2.67 (m, 2H), 2.65–2.61 (m, 2H), 2.04–1.98 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 163.2 (d, J = 247.0 Hz), 153.4, 150.7, 133.3, 133.2, 133.1 (d, J = 4.0 Hz), 130.9, 130.6, 130.3, 129.2 (d, J = 8.0 Hz), 120.1, 119.4, 116.1 (d, J = 22.0 Hz), 97.1, 67.2, 59.9, 54.6, 45.0, 23.3; HRMS (ESI) m/z: calcd for C24H26ClFN3O, 426.1748 [M + H]+; found, 426.1718; retention time 6.087 min (HPLC condition A).
(E)-6-Chloro-N-(3-morpholinopropyl)-2-(4-(trifluoromethyl)-Styryl)quinolin-4-amine (91).
The title compound was synthesized from 89a and 7i. Isolated yield: 84%; yellow solid; mp 144–146 °C; 1H NMR (400 MHz, CDCl3): δ 7.94–7.91 (m, 2H), 7.71 (d, J = 3.2 Hz, 1H), 7.69–7.62 (m, 4H), 7.58–7.55 (m, 1H), 7.32 (d, J = 16.2 Hz, 1H), 6.57 (s, 1H), 3.96 (t, J = 4.8 Hz, 4H), 3.47 (q, J = 5.8 Hz, 2H), 2.67 (t, J = 5.4 Hz, 2H), 2.63–2.60 (m, 2H), 2.03–1.98 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.2, 150.6, 147.1, 140.5, 132.3, 132.2, 131.5, 130.4 (d, J = 4.0 Hz), 130.1, 127.6, 126.0 (q, J = 4.0 Hz), 123.1, 120.1, 119.6, 97.58, 67.2, 59.9, 54.6, 45.0, 23.3; HRMS (ESI) m/z: calcd for C25H26ClF3N3O, 476.1716 [M + H]+; found, 476.1692; retention time 6.598 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(4-morpholinobutyl)quinolin-4-amine (92).
The title compound was synthesized from 89b and 7h. Isolated yield: 78%; yellow solid; mp 185–187 °C; 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.64 (d, J = 16.2 Hz, 1H), 7.58 (dd, J = 8.8, 5.6 Hz, 2H), 7.53 (dd, J = 9.0, 2.2 Hz, 1H), 7.16 (d, J = 16.2 Hz, 1H), 7.08 (d, J = 8.6 Hz, 2H), 6.58 (s, 1H), 3.78–3.75 (m, 4H), 3.42–3.38 (m, 2H), 2.50–2.44 (m, 6H), 1.90–1.85 (m, 2H), 1.76–1.70 (m, 2H); 13C NMR (100 MHz,CDCl3): δ 163.3 (d, J = 247.0 Hz), 156.1, 150.0, 133.5, 133.0 (d, J = 3.0 Hz), 130.8, 130.7, 130.5, 129.5 (d, J = 8.0 Hz), 128.5, 119.5, 119.1, 116.1 (d, J = 21.0 Hz), 97.7, 67.3, 58.5, 54.1, 43.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C25H28ClFN3O, 440.1905 [M + H]+; found, 440.1878; retention time 6.257 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(2-(pyridin-4-yl)vinyl)-quinolin-4-amine (93).
The title compound was synthesized from 89b and 38c. Isolated yield: 73%; yellow solid; mp 174–176 °C; 1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 6.0 Hz, 2H), 8.02 (d, J = 9.0 Hz, 1H), 7.89 (d, J = 2.0 Hz, 1H), 7.67 (d, J = 16.4 Hz, 1H), 7.54 (dd, J = 9.0, 2.2 Hz, 1H), 7.46–7.42 (m, 3H), 6.62 (s, 1H), 3.79–3.76 (m, 4H), 3.42–3.38 (m, 2H), 2.55–2.48 (m, 6H), 1.92–1.85 (m, 2H), 1.79–1.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 153.3, 150.6, 150.1, 146.8, 144.2, 133.6, 131.5, 131.4, 130.8, 124.1, 121.7, 119.5, 119.4, 98.4, 67.2, 58.5, 54.1, 43.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C24H28ClN4O, 423.1952 [M + H]+; found, 423.1941; retention time 2.812 min (HPLC condition A).
(E)-6-Chloro-N-(4-(4-methylpiperazin-1-yl)butyl)-2-styrylquinolin-4-amine (94).
The title compound was synthesized from 89c and 7a. Isolated yield: 74%; yellow solid; mp 122–123 °C; 1H NMR (400 MHz, CDCl3): δ 8.24 (d, J = 2.2 Hz, 1H), 7.80 (d, J = 5.4 Hz, 1H), 7.76 (d, J = 16.4 Hz, 1H), 7.68–7.65 (m, 1H), 7.63 (dd, J = 7.8, 1.6 Hz, 2H), 7.39–7.34 (m, 3H), 7.20 (d, J = 16.4 Hz, 1H), 6.88 (s, 1H), 3.51 (t, J = 7.0 Hz, 2H), 3.20–3.15 (m, 2H), 2.76–2.69 (m, 6H), 2.57 (t, J = 7.0 Hz, 2H), 2.43 (s, 3H), 1.83–1.78 (m, 2H), 1.73–1.67 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 153.5, 153.1, 141.2, 138.1, 135.9, 132.2, 129.7, 129.0, 127.7, 125.0, 123.3, 121.5, 118.4, 96.3, 57.4, 53.8, 51.7, 44.1, 43.2, 25.9, 23.6; HRMS (ESI) m/z: calcd for C26H32ClN4, 435.2315 [M + H]+; found, 435.2290; retention time 5.789 min (HPLC condition A).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(4-(4-methylpiperazin-1-yl)-butyl)quinolin-4-amine (95).
The title compound was synthesized from 89c and 7h. Isolated yield: 71%; yellow solid; mp 133–135 °C; 1H NMR (400 MHz, CDCl3): δ 8.21 (d, J = 2.2 Hz, 1H), 7.79 (d, J = 9.4 Hz, 1H), 7.75 (d, J = 16.4 Hz, 1H), 7.67 (d, J = 2.2 Hz, 1H), 7.66–7.63 (m, 2H), 7.12 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 9.0 Hz, 2H), 6.86 (s, 1H), 3.51 (t, J = 7.0 Hz, 2H), 3.21–3.15 (m, 2H), 2.79–2.70 (m, 6H), 2.612.57 (m, 2H), 2.45 (s, 3H), 1.83–1.77 (m, 2H), 1.74–1.68 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.8 (d, J = 248.0 Hz), 153.4, 153.1, 141.1, 136.7, 132.3 (d, J = 3.0 Hz), 132.2, 131.2, 129.7 (d, J = 8.0 Hz), 125.0, 123.2, 121.5, 118.4, 115.8 (d, J = 22.0 Hz), 96.3, 57.4, 53.8, 51.7, 44.0, 43.2, 25.9, 23.6; HRMS (ESI) m/z: calcd for C26H31ClN4F, 453.2221 [M + H]+; found, 453.2222; retention time 6.115 min (HPLC condition A).
(E)-6-Chloro-N-(4-(4-methylpiperazin-1-yl)butyl)-2-(2-(pyridin-4-yl)vinyl)quinolin-4-amine (96).
The title compound was synthesized from 89c and38c. Isolated yield: 67%; brown solid; mp >200 °C; 1H NMR (400 MHz, CDCl3): δ 8.50 (d, J = 6.2 Hz, 2H), 8.16 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 9.4 Hz, 1H), 7.62 (d, J = 16.4 Hz, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 5.6, 1.8 Hz, 2H), 7.44 (d, J = 16.4 Hz, 1H), 6.83 (s, 1H), 3.45 (t, J = 6.8 Hz, 2H), 3.20–3.14 (m, 2H), 2.74–2.66 (m, 6H), 2.57 (t, J = 7.6 Hz, 2H), 2.42 (s, 3H), 1.81–1.76 (m, 2H), 1.72–1.67 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 154.7, 151.8, 149.6, 145.2, 145.1, 132.5, 131.9, 130.9, 130.7, 128.5, 122.0, 121.0, 119.3, 97.2, 57.5, 53.9, 51.8, 44.1, 42.8, 26.0, 23.7; HRMS (ESI) m/z: calcd for C25H31ClN, 436.2268 [M + H]+; found, 436.2254; retention time 2.741 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(4-(trifluoromethyl)-Styryl)quinolin-4-amine (98).
The title compound was synthesized from 89b and 7i. Isolated yield: 87%; brown solid; mp 153–155 °C; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 1.3 Hz, 1H), 7.71–7.67 (m, 4H), 7.62 (d, J = 8.2 Hz, 2H), 7.56 (dd, J = 9.0, 2.2 Hz, 1H), 7.31 (d, J = 16.2 Hz, 1H), 6.63 (s, 1H), 3.79–3.76 (m, 4H), 3.40 (d, J = 4.9 Hz, 2H), 2.50–2.44 (m, 6H), 1.91–1.86 (m, 2H), 1.76–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.9, 149.9, 146.9, 140.4, 132.6, 131.8, 131.4, 130.7 (d, J = 3.0 Hz), 130.5, 130.2, 127.6, 126.0 (d, J = 4.0 Hz), 123.1, 119.3 (d, J = 4.0 Hz), 98.3, 67.3, 58.5, 54.1, 43.7, 26.7, 24.8; HRMS (ESI) m/z: calcd for C26H28ClF3N3O, 490.1873 [M + H]+; found, 490.1838; retention time 6.612 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(2-(trifluoromethyl)-Styryl)quinolin-4-amine (99).
The title compound was synthesized from 89b and 97a. Isolated yield: 83%; brown solid; mp 110–112 °C; 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 2.8 Hz, 1H), 7.90 (s, 1H), 7.72 (d, J = 2.2 Hz, 2H), 7.57 (dd, J = 9.0, 2.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.28 (d, J = 16.2 Hz, 2H), 6.70 (s, 1H), 3.78–3.76 (m, 4H), 3.43–3.40 (m, 2H), 2.51–2.48 (m, 4H), 2.46 (d, J = 7.2 Hz, 2H), 1.92–1.86 (m, 2H), 1.78–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.2, 149.9, 146.6, 135.9, 133.5, 132.4, 131.3, 130.8, 130.7, 130.0, 129.1, 128.4, 127.9, 127.2, 126.3 (d, J = 6.0 Hz), 119.3, 97.0, 67.3, 58.5, 54.1, 46.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C26H28ClF3N3O, 490.1873 [M + H]+; found, 490.1837; retention time 6.324 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(3-(trifluoromethyl)-Styryl)quinolin-4-amine (100).
The title compound was synthesized from 89b and 97b. Isolated yield: 81%; brown solid; mp 130–132 °C; 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 9.0 Hz, 1H), 7.87 (s, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.73–7.68 (m, 2H), 7.57 (dd, J = 9.0, 2.2 Hz, 2H), 7.51 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 16.2 Hz, 1H), 6.64 (s, 1H), 3.79–3.77 (m, 4H), 3.44–3.39 (m, 2H), 2.52–2.49 (m, 4H), 2.48 (t, J = 7.8 Hz, 2H), 1.92–1.87 (m, 2H), 1.78–1.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.9, 149.9, 146.9, 137.7, 132.6, 131.7, 131.4, 130.6 (d, J = 3.0 Hz), 129.6, 125.8, 125.2 (d, J = 3.0 Hz), 124.1 (q, J = 4.0 Hz), 123.0, 119.3 (d, J = 3.0 Hz), 98.1, 67.2, 58.5, 54.1, 43.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C26H28ClF3N3O, 490.1873 [M + H]+; found, 490.1868; retention time 6.606 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(4-(trifluoromethoxy)-Styryl)quinolin-4-amine (101).
The title compound was synthesized from 89b and 97c. Isolated yield: 80%; brown solid; mp 140–142 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.67 (d, J = 16.2 Hz, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.55 (dd, J = 9.0, 2.2 Hz, 1H), 7.25 (d, J = 16.2 Hz, 1H), 7.21 (t, J = 7.6 Hz, 2H), 6.60 (s, 1H), 3.79–3.76 (m, 4H), 3.43–3.39 (m, 2H), 2.51–2.45 (m, 6H), 1.91–1.86 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.8, 150.12, 149. 6 (d, J = 2.0 Hz), 146.8, 135.5, 133.1, 130.8, 130.6, 128.9, 122.1, 121.5 (d, J = 2.0 Hz), 119.5 (d, J = 3.0 Hz), 119.1, 97.9, 67.2, 58.5, 54.1, 43.8, 26.7, 25.7. HRMS (ESI) m/z: calcd for C26H28ClF3N3O2, 506.1822 [M + H]+; found, 506.1789; retention time 6.712 min (HPLC condition A).
(E)-2-(3,5-Bis(trifl uoromethyl)styryl)-6-chloro-N-(4-morpholinobutyl)quinolin-4-amine (102).
The title compound was synthesized from 89b and 97d. Isolated yield: 74%; yellow solid; mp 143–144 °C; 1H NMR (400 MHz, CDCl3): δ 8.03 (s, 2H), 7.92 (d, J = 9.0 Hz, 1H), 7.79 (s, 1H), 7.73 (d, J = 16.2 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 7.58 (dd, J = 9.0, 2.2 Hz, 1H), 7.36 (d, J = 16.2 Hz, 1H), 6.63 (s, 1H), 3.79–3.76 (m, 4H), 3.42–3.38 (m, 2H), 2.51–2.45 (m, 6H), 1.90–1.86 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.4, 149.8, 147.3, 139.2, 133.5, 132.6, 132.3, 131.8, 130.8, 130.6 (d, J = 7.0 Hz), 127.1 (q, J = 4.0 Hz), 125.0, 121.8 (q, J = 4.0 Hz), 119.4, 119.2, 98.5, 67.3, 58.5, 54.1, 43.7, 26.8, 24.8; 19F NMR (376 MHz, CDCl3): δ −63.0; HRMS (ESI) m/z: calcd for C27H27ClF6N3O, 558.1747 [M + H]+; found, 558.1719; retention time 7.321 min (HPLC condition A).
(E)-6-Chloro-2-(3,5-difluorostyryl)-N-(4-morpholinobutyl)-quinolin-4-amine (103).
The title compound was synthesized from 89b and 97e. Isolated yield: 86%; yellow solid; mp 120–122 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.60 (d, J = 10.6 Hz, 1H), 7.57 (dd, J = 8.0, 2.2 Hz, 1H), 7.22 (d, J = 16.2 Hz, 1H), 7.13–7.10 (m, 2H), 6.78–6.73 (m, 1H), 6.60 (s, 1H), 3.78 (t, J = 4.8 Hz, 4H), 3.42–3.37 (m, 2H), 2.50–2.48 (m, 4H), 2.47 (s, 2H), 1.91–1.86 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.6 (dd, J = 246.0, 3.0 Hz), 155.8, 149.7, 147.3, 132.2, 131.8, 131.7, 130.6 (d, J = 3.0 Hz), 119.4, 119.2, 110.1 (d, J = 26.0 Hz), 103.9 (t, J = 26.0 Hz), 98.5, 67.3, 58.5, 54.1, 43.7, 26.8, 24.8; HRMS (ESI) m/z: calcd for C25H27ClF2N3O, 458.1811 [M + H]+; found, 458.1775; retention time 6.418 min (HPLC condition A).
(E)-6-Chloro-2-(2,4-difluorostyryl)-N-(4-morpholinobutyl)-quinolin-4-amine (104).
The title compound was synthesized from 89b and 97f. Isolated yield: 82%; yellow solid; mp 105–106 °C; 1H NMR (400 MHz, CDCl3): δ 8.22 (s, 1H), 8.01 (s, 1H), 7.91 (d, J = 9.8 Hz, 1H), 7.75 (d, J = 16.8 Hz, 1H), 7.65–7.59 (m, 1H), 7.42 (dd, J = 8.6, 1.8 Hz, 1H), 7.24 (d, J = 16.8 Hz, 1H), 6.86 (dd, J = 7.8, 3.8 Hz, 1H), 6.29 (s, 1H), 3.83–3.81 (m, 4H), 3.43 (t, J = 6.6 Hz, 2H), 2.732.68 (m, 4H), 2.65 (t, J = 7.6 Hz, 2H), 1.91–1.86 (m, 2H), 1.84–1.79 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 164.4 (dd, J = 251.0, 11.0 Hz), 161.5 (dd, J = 244.0, 12.0 Hz), 153.1, 151.8, 139.6, 133.8, 133.7, 132.2, 131.9, 130.2, 124.6, 122.3, 118.2, 112.5 (d, J = 25.0 Hz), 111.2 (d, J = 24.0 Hz), 104.8 (d, J = 26.0 Hz), 94.5, 66.4, 58.1, 53.4, 43.8, 26.0, 23.6; HRMS (ESI) m/z: calcd for C25H27ClF2N3O, 458.1811 [M + H]+; found, 458.1785; retention time 6.296 min (HPLC condition A).
(E)-6-Chloro-2-(3,4-difluorostyryl)-N-(4-morpholinobutyl)-quinolin-4-amine (105).
The title compound was synthesized from 89b and 97g. Isolated yield: 80%; yellow solid; mp 146–147 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.69 (d, J = 2.2 Hz, 1H), 7.59 (d, J = 11.9 Hz, 1H), 7.57–7.55 (m, 1H), 7.45–7.40 (m, 1H), 7.33–7.30 (m, 1H), 7.20–7.12 (m, 2H), 6.59 (s, 1H), 3.79–3.76 (m, 4H), 3.42–3.38 (m, 2H), 2.51–2.48 (m, 4H), 2.46 (t, J = 7.2 Hz, 2H), 1.91–1.86 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.1, 152.0, 149.8, 147.1, 134.2 (t, J = 5.0 Hz), 131.9, 131.5, 130.6, 130.5, 124.0, 123.9 (d, J = 5.0 Hz), 119.3, 119.2, 117.9 (dd, J = 13.0, 6.0 Hz), 115.7 (dd, J = 15.0, 3.0 Hz), 98.2, 67.3, 58.5, 54.1, 43.7, 26.8, 24.8; HRMS (ESI) m/z: calcd for C25H27ClF2N3O, 458.1811 [M + H]+; found, 458.1787; retention time 6.345 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(2,3,5-trifluorostyryl)-quinolin-4-amine (106).
The title compound was synthesized from 89b and 97h. Isolated yield: 73%; yellow solid; mp 125–126 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 16.4 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.57 (dd, J = 9.0, 2.2 Hz, 1H), 7.31 (d, J = 16.4 Hz, 1H), 7.19–7.14 (m, 1H), 6.90–6.83 (m, 1H), 6.62 (s, 1H), 3.79–3.75 (m, 4H), 3.40 (q, J = 6.6 Hz, 2H), 2.51–2.48 (m, 4H), 2.45 (t, J = 7.2 Hz, 2H), 1.92–1.86 (m, 2H), 1.77–1.71 (m, 2H); 13CNMR (100 MHz, CDCl3): δ 159.2 (m), 156.8 (m), 155.7, 152.3 (m), 149.8, 1747.2, 134.4 (d, J = 5.0 Hz), 131.8, 130.8, 130.6, 124.2 (d, J = 4.0 Hz), 119.4, 119.2, 108.8 (d, J = 24.0 Hz), 105.2 (d, J = 21.0 Hz), 105.1 (d, J = 21.0 Hz), 98.3, 67.3, 58.5, 54.1, 43.7, 26.8, 24.7; 19F NMR (376 MHz, CDCl3): δ −115.4, −133.5, −147.0; HRMS (ESI) m/z: calcd for C23H26ClF3N3O, 476.1716 [M + H]+; found, 476.1689; retention time 6.239 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(2,4,5-trifluorostyryl)-quinolin-4-amine (107).
The title compound was synthesized from 89b and 97h. Isolated yield: 78%; yellow solid; mp 150–152 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.71–7.66 (m, 2H), 7.56 (dd, J = 9.0, 2.2 Hz, 1H), 7.53–7.46 (m, 1H), 7.22 (d, J = 16.4 Hz, 1H), 7.01–6.94 (m, 1H), 6.62 (s, 1H), 3.79–3.76 (m, 4H), 3.42–3.38 (m, 2H), 2.51–2.48 (m, 4H), 2.47 (t, J = 7.2 Hz, 2H), 1.91–1.86 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.0, 149.7, 147.2, 132.8, 131.8, 130.6 (d, J = 6.0 Hz), 124.3, 119.4, 119.2, 115.4 (d, J = 5.0 Hz), 115.2(d, J = 4.0 Hz), 106.4 (d, J = 20.0 Hz), 106.2 (d, J = 20.0 Hz), 98.1, 67.3, 58.5, 54.1, 43.7, 26.8, 24.7; HRMS (ESI) m/z: calcd for C23H26ClF3N3O, 476.1716 [M + H]+; found, 476.1691; retention time 6.304 min (HPLC condition A).
(E)-6-Chloro-N-(4-morpholinobutyl)-2-(3,4,5-trifluorostyryl)-quinolin-4-amine (108).
The title compound was synthesized from 89b and 97j. Isolated yield: 71%; yellow solid; mp 158–159 °C; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.58–7.56 (m, 1H), 7.54 (d, J = 16.0 Hz, 1H), 7.21 (dd, J = 8.8, 6.6 Hz, 2H), 7.13 (d, J = 16.0 Hz, 1H), 6.57 (s, 1H), 3.79–3.76 (m, 4H), 3.41–3.36 (m, 2H), 2.51–2.48 (m, 4H), 2.47 (t, J = 7.2 Hz, 2H), 1.91–1.87 (m, 2H), 1.77–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.6, 153.0 (m), 150.5 (m), 149.8, 147.2, 133.3, 131.8, 131.7, 130.9, 130.6, 130.5, 119.2 (d, J = 6.0 Hz), 119.1 (d, J = 5.0 Hz), 98.5, 67.3, 58.5, 54.1, 43.7, 26.8, 24.8; HRMS (ESI) m/z: calcd for C25H26ClF3N3O, 476.1716 [M + H]+; found, 476.1688; retention time 5.946 min (HPLC condition A).
(E)-6-Chloro-2-(2-(3-fluoropyridin-4-yl)vinyl)-N-(4-morpholinobutyl)quinolin-4-amine (109).
The title compound was synthesized from 89b and 97k. Isolated yield: 79%; yellow solid; mp 140–142 °C; 1H NMR (400 MHz, CDCl3): δ 8.50 (d, J = 2.4 Hz, 1H), 8.42 (d, J = 5.0 Hz, 1H), 7.93 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 16.4 Hz, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 7.4, 1.6 Hz, 1H), 7.55 (d, J = 4.0 Hz, 1H), 7.51 (d, J = 16.4 Hz, 1H), 6.66 (s, 1H), 3.79–3.76 (m, 4H), 3.43–3.39 (m, 2H), 2.51–2.45 (m, 6H), 1.91–1.87 (m, 2H), 1.78–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.4, 154.3, 149.9, 147.2, 146.24 (d, J = 5.0 Hz), 139.2(d, J = 25.0Hz), 136.6(d, J = 6.0 Hz), 132.0, 131.8, 131.0, 130.7, 123.7 (d, J = 3.0 Hz), 121.6, 119.5, 119.3, 98.5, 67.27, 58.5, 54.1, 43.7, 26.7, 24.7; 19F NMR (376 MHz, CDCl3): δ -131.7; HRMS (ESI) m/z: calcd for C24H27ClFN4O, 441.1857 [M + H]+; found, 441.1846; retention time 5.365 min (HPLC condition A).
(E)-6-Chloro-2-(2-(6-fluoropyridin-3-yl)vinyl)-N-(4-morpholinobutyl)quinolin-4-amine (110).
The title compound was synthesized from 89b and 97l. Isolated yield: 84%; yellow solid; mp 129–130 °C; 1H NMR (400 MHz, CDCl3): δ 8.41 (d, J = 2.2 Hz, 1H), 8.07–8.03 (m, 1H), 7.93 (d, J = 9.0 Hz, 1H), 7.72 (d, J = 2.2 Hz, 1H), 7.63 (d, J = 16.2 Hz, 1H), 7.57 (dd, J = 9.0, 2.2 Hz, 1H), 7.23 (d, J = 16.2 Hz, 1H), 6.97 (dd, J = 8.6, 2.8 Hz, 1H), 6.61 (s, 1H), 3.79–3.76 (m, 4H), 3.43–3.39 (m, 2H), 2.51–2.45 (m, 6H), 1.91–1.86 (m, 2H), 1.78–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 163.7 (d, J = 240.0 Hz), 155.6, 150.0, 147.3, 147.1, 138.8 (d, J = 8.0 Hz), 131.3, 131.1, 130.9 (d, J = 4.0 Hz), 130.8, 130.7, 129.1, 119.4, 119.3, 110.4, 110.0, 98.1, 67.2, 58.5, 54.1, 43.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C24H27ClFN4O, 441.1857 [M + H]+; found, 441.1875; retention time 5.741 min (HPLC condition A).
(E)-6-Chloro-2-(2-(5-fluoropyridin-2-yl)vinyl)-N-(4-morpholinobutyl)quinolin-4-amine (111).
The title compound was synthesized from 89b and 97m. Isolated yield: 92%; brown solid; mp 132–134 °C; 1H NMR (400 MHz, CDCl3): δ 8.48 (d, J = 3.0 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 16.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.59 (d, J = 16.4 Hz, 1H), 7.55 (d, J = 5.3 Hz, 1H), 7.53 (dd, J = 7.4, 1.6 Hz, 1H), 7.44–7.39 (m, 1H), 6.65 (s, 1H), 3.77–3.75 (m, 4H), 3.39–3.35 (m, 2H), 2.50–2.47 (m, 4H), 2.44 (t, J = 7.0 Hz, 2H), 1.87–1.82 (m, 2H), 1.73–1.68 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 159.2 (d, J = 256.0 Hz), 155.6, 151.8, 150.1, 146.5, 138.4 (d, J = 24.0 Hz), 132.6 (d, J = 7.0 Hz), 131.0, 130.72, 130.7 (d, J = 5.0 Hz), 124.0 (d, J = 4.0 Hz), 123.8, 123.7, 119.5, 119.2, 98.8, 67.3, 58.5, 54.1, 43.7, 26.7, 24.6; HRMS (ESI) m/z: calcd for C24H27ClFN4O, 441.1857 [M + H]+; found, 441.1835; retention time 5.666 min (HPLC condition A).
(E)-6-Chloro-2-(2-(3-fluoropyridin-2-yl)vinyl)-N-(4-morpholinobutyl)quinolin-4-amine (112).
The title compound was synthesized from 89b and 97n. Isolated yield: 90%; brown solid; mp 113–115 °C; 1H NMR (400 MHz, CDCl3): δ 8.45 (d, J = 4.6 Hz, 1H), 7.01 (d, J = 16.2 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.88 (d, J = 16.2 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.55–7.52 (m, 1H), 7.45–7.40 (m, 1H), 7.26–7.22 (m, 1H), 6.65 (s, 1H), 3.78–3.76 (m, 4H), 3.41–3.37 (m, 2H), 2.52–2.49 (m, 4H), 2.47 (t, t, J = 7.6 Hz, 2H), 1.89–1.85 (m, 2H), 1.76–1.69 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 158.0 (d, J = 261.0 Hz), 155.5, 150.1, 145.8 (d, J = 6.0 Hz), 143.9, 143.8, 134.5, 131.2, 130.7, 125.9, 124.3 (d, J = 4.0 Hz), 123.9, 123.7, 119.5, 119.3, 99.5, 67.2, 58.5, 54.1, 43.7, 26.7, 24.6; HRMS (ESI) m/z: calcd for C24H27ClFN4O, 441.1857 [M + H]+; found, 441.1852; retention time 5.642 min (HPLC condition A).
6-Chloro-2-((1E,3E)-4-(4-fluorophenyl)buta-1,3-dien-1-yl)-N-(4 morpholinobutyl)-quinolin-4-amine (113).
The title compound was synthesized from 89b and 97o. Isolated yield: 58%; brown solid; mp 141–144 °C; 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.6 Hz, 1H), 7.76–7.71 (m, 1H), 7.54–7.48 (m, 1H), 7.44 (dd, J = 8.6, 5.4 Hz, 2H), 7.6 (t, J = 8.6 Hz, 2H), 6.92 (d, J = 15.4 Hz, 1H), 6.86 (d, J = 15.4 Hz, 1H), 6.78 (d, J = 15.4 Hz, 1H), 6.49 (s, 1H), 3.78–3.76 (m, 4H), 3.40 (t, J = 7.8 Hz, 2H), 2.51–2.48 (m, 4H), 2.47 (s, 2H), 1.90–1.85 (m, 2H), 1.75–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 162.9 (d, J = 246.0 Hz), 157.3, 149.6, 136.3, 135.4, 131.4, 130.4, 128.9, 128.8 (d, J = 8.0 Hz), 126.8, 119.4, 118.5, 116.0 (d, J = 22.0 Hz), 101.2, 67.2, 58.5, 54.1, 43.7, 26.7, 24.6; HRMS (ESI) m/z: calcd for C27H30ClFN3O, 466.2061 [M + H]+; found, 466.2039; retention time 6.383 min (HPLC condition A).
6-Chloro-N-(4-morpholinobutyl)-2-((1E,3E)-4-phenylbuta-1,3-dien-1-yl)quinolin-4-amine (114).
The title compound was synthesized from 89b and 97p. Isolated yield: 52%; brown solid; mp 135–137 °C; 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 8.6 Hz, 1H), 7.57–7.47 (m, 3H), 7.44 (d, J = 8.6 Hz, 2H), 7.35 (t, J = 8.6 Hz, 2H), 7.29 (d, J = 7.2 Hz, 1H), 7.01 (d, J = 15.4 Hz, 1H), 6.89 (d, J = 15.4 Hz, 1H), 6.76 (d, J = 15.4 Hz, 1H), 6.36 (s, 1H), 3.76 (t, J = 5.6 Hz, 4H), 3.41 (t, J = 7.2 Hz, 2H), 2.52–2.46 (m, 4H), 1.89–1.84 (m, 2H), 1.75–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 156.8, 150.0, 145.7, 138.0, 137.6, 135.9, 130.7, 129.1, 129.0, 128.5, 128.4, 127.4, 127.3, 126.5, 119.7, 118.4, 100.9, 67.1, 58.4, 53.9, 43.6, 26.6, 24.4; HRMS (ESI) m/z: calcd for C27H31ClN3O, 448.2156 [M + H]+; found, 448.2133; retention time 6.203 min (HPLC condition A).
General Procedure (Hydrazine Hydrate Reduction Reaction) for Synthesis of Compounds 58 and 59.
A solution of pyridylvinylquinolines (0.2 mmol) and hydrazine hydrate (1.50 mL) in EtOH (6.0 mL) was heated at 80 °C for 36 h. After completion of the reaction, the solvents were removed to dryness. The residue was dissolved in DCM and washed with water and brine. The organic layer was separated, and the aqueous layer was extracted with DCM. The combined organic layers were then dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography.
N1-(6-Methoxy-2-(2-(pyridin-4-yl)ethyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (58).
The title compound was synthesized from pyridylvinylquinoline 41. Isolated yield: 81%; white solid; mp 104–106 °C; 1H NMR (400 MHz, CDCl3): δ 8.45 (d, J = 5.2 Hz, 2H), 8.29 (d, J = 9.2 Hz, 1H), 7.44 (d, J = 2.6 Hz, 1H), 7.33 (dd, J = 9.2, 2.6 Hz, 1H), 7.22 (d, J = 6.0 Hz, 2H), 6.19 (s, 1H), 3.97 (s, 3H), 3.49 (t, J = 6.0 Hz, 2H), 3.34–3.30 (m, 2H), 3.22–3.18 (m, 2H), 2.89 (t, J = 6.0 Hz, 2H), 2.47 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 157.9, 156.2, 155.0, 150.2, 149.9, 126.1, 124.4, 122.8, 117.9, 101.3, 98.4, 56.8, 56.4, 45.2, 40.4, 36.9, 35.4; HRMS (ESI): calcd for C21H27N4O [M + H]+ m/z, 351.2185; found, 351.2162; retention time 2.078 min (HPLC condition A).
N1-(6-Chloro-2-(2-(pyridin-4-yl)ethyl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (59).
The title compound was synthesized from pyridylvinylquinoline 50. Isolated yield: 83%; white solid; mp 115–117 °C; 1H NMR (400 MHz, CDCl3): δ 8.47 (d, J = 5.6 Hz, 2H), 8.13 (d, J = 9.0 Hz, 1H), 7.94 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 9.0, 2.2 Hz, 1H), 7.20 (d, J = 6.0 Hz, 2H), 6.21 (s, 1H), 3.35 (t, J = 6.0 Hz, 2H), 3.27–3.23 (m, 2H), 3.20–3.16 (m, 2H), 2.78 (t, J = 6.8 Hz, 2H), 2.47 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 160.1, 150.7, 150.4, 150.1, 145.4, 130.9, 130.6, 129.9, 124.4, 119.8, 118.6, 99.5, 57.0, 46.1, 45.3, 40.2, 35.3; HRMS (ESI): calcd for C20H24ClN4 [M + H]+ m/z, 355.1689; found, 355.1667; retention time 2.203 min (HPLC condition A).
Diethyl (4-Nitrobenzyl)phosphonate (74).
The mixture of 1-(bromomethyl)-4-nitrobenzene (73) (2.44 g, 11.3 mmol) and triethylphosphite (5.0 mL, 28.79 mmol) was refluxed under a nitrogen atmosphere for 20 h. After cooling, the reaction mixture was evaporated in vacuo to give the product 74 (2.72 g, 88% yield) as a brown oil. 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.8 Hz, 2H), 7.47 (dd, J = 8.8, 2.6 Hz, 2H), 4.09–4.01 (m, 4H), 3.24 (d, J = 22.4 Hz, 2H), 1.26 (t, J = 7.0 Hz, 6H).
Diethyl (4-Aminobenzyl)phosphonate (75).
Diethyl (4-nitrobenzyl)phosphonate (74) (273 mg, 1.0 mmol) was dissolved in MeOH (20 mL) under an inert atmosphere. Then, 10% Pd/C (108 mg, 10% mol) was added carefully. After being stirred at room temperature overnight, the reaction mixture was filtered off on a celite pad and washed with MeOH. The resultant solvents were evaporated under vacuum to give compound 75 (190 mg, 76% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 6.96 (dd, J = 8.4, 2.6 Hz, 2H), 6.52 (d, J = 8.4 Hz, 2H), 3.95–3.85 (m, 4H), 2.94 (d, J = 22.0 Hz, 2H), 1.14 (t, J = 7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 145.9 (d, J = 3.0 Hz), 130.8 (d, J = 7.0 Hz), 120.8 (d, J = 9.0 Hz), 115.5 (d, J = 3.0 Hz), 62.3 (d, J = 6.0 Hz), 33.0 (d, J = 138.0 Hz), 16.7 (d, J = 6.0 Hz).
Diethyl (4-Isocyanobenzyl)phosphonate (76).
To a stirred solution of aniline 75 (150 mg, 0.62 mmol), chloroform (50 μL, 0.62 mmol), and benzyltriethylammonium chloride (2.0 mg) in DCM (3.0 mL) was added 50% aqueous NaOH solution (0.71 mmol) dropwise. Then, the reaction mixture was refluxed for 2 h. Upon completion, the resultant content was diluted with water (5 mL) and extracted with DCM. The combined organic layers were washed with water and brine and dried over Na2SO4. The resultant solvents were evaporated to give compound 76 (125 mg, 80% yield) as a yellow oil. 1H NMR (400 MHz, CDQ3): δ 7.29 (s, br, 4H), 4.02–3.97 (m, 4H), 3.12 (d, J = 22.0 Hz, 2H), 1.22 (t, J = 7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 134.0 (d, J = 9.0 Hz), 131.1 (d, J = 7.0 Hz), 130.9 (d, J = 7.0 Hz), 126.8 (d, J = 3.0 Hz), 115.6 (d, J = 3.0 Hz), 62.6 (d, J = 7.0 Hz), 33.9 (d, J = 137.0 Hz), 16.7 (d, J = 5.0 Hz).
6-Chloro-4-morpholinoquinoline-2-carbaldehyde (77).
To a solution of 4-(6-chloro-2-methylquinolin-4-yl)morpholine (61a) (150 mg, 0.55 mmol) in dioxane (5 mL) was added SeO2 (443 mg, 3.99 mmol) slowly. The resultant mixture was heated at 80 °C for 6 h under a nitrogen atmosphere. After cooling, the resultant solvents were removed in vacuo. The residue was purified by column chromatography to furnish compound 77 (50 mg, 32% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 10.13 (s, 1H), 8.15 (d, J = 9.4 Hz, 1H), 8.02 (d, J = 2.4 Hz, 1H), 7.70 (dd, J = 9.0, 2.4 Hz, 1H), 7.51 (s, 1H), 4.01 (t, J = 5.6 Hz, 4H), 3.28 (t, J = 5.6 Hz, 4H); 13C NMR (100 MHz, CDCl3): δ 194.1, 157.5, 153.7, 148.1, 134.5, 133.0, 131.4, 126.1, 123.3, 105.9, 67.1, 52.9.
(E)-4-(6-Chloro-2-(4-isocyanostyryl)quinolin-4-yl)morpholine (64).
A solution of quinoline-2-carbaldehyde 77 (43 mg, 0.16 mmol) and diethyl (4-isocyanobenzyl)phosphonate (76) (18 mg, 0.16 mmol) in anhydrous dimethylformamide (DMF) (1.5 mL) was added dropwise to a solution of potassium tert-butoxide (46 mg, 0.28 mmol) in anhydrous DMF (1.5 mL). The resultant mixture was stirred at room temperature for 1 h and then poured onto crushed ice. The precipitated solid was collected by filtration and dried. The crude product obtained was purified by silica gel column chromatography to give compound 64 (35 mg, 58% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 2.2 Hz, 1H), 7.66 (d, J = 16.2Hz, 1H), 7.63 (dd, J = 4.0, 2.4 Hz, 2H), 7.61 (dd, J = 9.0, 2.4 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H), 7.31 (d, J = 16.2 Hz, 1H), 7.09 (s, 1H), 4.02 (t, J = 5.6 Hz, 4H), 3.26 (t, J = 5.6 Hz, 4H); 13C NMR (100 MHz, CDCl3): δ 165.4, 156.8, 156.1, 148.4, 138.0, 132.7, 131.9, 131.8, 131.4, 130.8, 128.4, 127.2, 123.9, 122.8, 108.6, 67.2, 53.0; HRMS (ESI): calcd for C22H19ClN3O [M + H]+ m/z, 376.1217; found, 376.1186; retention time 5.905 min (HPLC condition B).
2-((6-Chloro-2-methylquinolin-4-yl)amino)ethyl Methanesulfonate (79).
A mixture of 4,6-dichloroquinoline (840 mg, 4.0 mol), EtOH (1.0 mL), and 2-aminoethanol (6.0 mL, 99.4 mmol) was heated with stirring at 130 °C for 48 h. After cooling, the reaction mixture was poured into ice water. The resulting precipitate was collected by filtration and dried to give 78 (920 mg, 97% yield) as an off-white solid.
To a suspension of 78 (700 mg, 2.96 mmol) in anhydrous tetrahydrofuran (THF, 12 mL) under a nitrogen atmosphere was added triethylamine (820 μL, 5.92 mmol). The mixture was cooled to below 0 °C. Methanesulfonyl chloride (365 μL, 4.74 mmol) was added slowly keeping the temperature below 5 °C, and the reaction mixture was stirred in an ice bath for 1 h. Then, the reaction contents were quenched by the addition of saturated solution of sodium bicarbonate and brine. The reaction contents were then transferred to a separatory funnel, diluting with ethyl acetate. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were then dried over Na2SO4, filtered, and evaporated to give 79 (172 mg, 72% yield) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.55 (dd, J = 9.0, 2.2 Hz, 1H), 6.35 (s, 1H), 4.59–4.56 (m, 2H), 3.70 (q, J = 5.0 Hz, 2H), 3.09 (s, 3H), 2.62 (s, 3H).
General Procedure for Synthesis of 2-Methyl-4-aminoquinolines (80a–c).
A mixture of 2-((6-chloro-2-methylquinolin-4-yl)amino)-ethyl methanesulfonate (79) (172 mg, 0.5 mmol), appropriate amines (2.5 mmol), and potassium carbonate (415 mg, 3.0 mmol) in anhydrous CH3CN (6 mL) was stirred at 82 °C overnight. Upon completion, the resultant mixture was cooled down to room temperature. The resultant solvents were removed. The residue was dissolved in DCM and washed with water and brine. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were then dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel.
6-Chloro-2-methyl-N-(2-morpholinoethyl)quinolin-4-amine (80a).
The title compound was synthesized from precursor 79 and morpholine. Isolated yield: 94%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 2.2 Hz, 1H), 7.52 (dd, J = 9.0, 2.2 Hz, 1H), 6.30 (s, 1H), 3.76 (t, J = 6.4 Hz, 4H), 3.30 (d, J = 6.2 Hz, 2H), 2.76 (t, J = 6.8 Hz, 2H), 2.59 (s, 3H), 2.55–2.51 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 160.2, 149.3, 146.9, 131.0, 130.1, 129.8, 119.1, 118.7, 100.3, 67.4, 56.5, 53.5, 39.1, 26.0.
6-Chloro-2-methyl-N-(2-(pyrrolidin-1-yl)ethyl)quinolin-4-amine (80b).
The title compound was synthesized from precursor 79 and pyrrolidine. Isolated yield: 92%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.52 (dd, J = 8.9, 2.2 Hz, 1H), 6.31 (s, 1H), 3.34–3.30 (m, 2h), 2.88–2.85 (m, 2H), 2.60 (t, J = 6.4 Hz, 7H), 1.85–1.82 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 160.2, 149.8, 147.0, 130.9, 130.0, 129.6, 119.5, 118.7, 100.1, 54.1, 54.2, 41.7, 26.0, 23.9.
6-Chloro-2-methyl-N-(2-(4-methylpiperazin-1-yl)ethyl)quinolin-4-amine (80c).
The title compound was synthesized from precursor 79 and N-methylpiperidine. Isolated yield: 80%; brown solid; 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 8.8 Hz, 1H), 7.63 (s, 1H), 7.44 (d, J = 8.0 Hz, 1H), 6.22 (s, 1H), 5.83 (s, 1H), 3.25–3.19 (m, 2H), 2.69 (t, J = 6.4 Hz, 2H), 2.52 (s, 3H), 2.51–2.27 (m, 8H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.1, 149.5, 146.7, 130.7, 130.0, 129.7, 119.4, 118.7, 100.1, 55.8, 55.5, 52.9, 46.4, 39.4, 25.8.
General Procedure for Synthesis of 4,6-Dichloro-2-arylquinolines (118a-b).
A stirred mixture of 4-chloroaniline (2.6 g, 20.4 mmol) and appropriate ethyl acetates (2.60 mmol) in PPA (14 g) was heated at 150 °C for 6 h. After cooling, the reaction mixture was poured into ice water with vigorous stirring. The resulting precipitate was collected by filtration, washed with water, and dried in a vacuum oven to give 2-arylhydroxyquinolines 117a or 117b.
2-Arylhydroxyquinolines (2.0 mol) in phosphorous oxycholoride (2.0 mL) was heated to 105 °C for 2 h. The excess of phosphorous oxychloride was removed under reduced pressure. The residue was quenched into crushed ice, neutralized using saturated solution of sodium bicarbonate, and extracted with DCM. The combined organic extracts were washed with brine, dried over Na2SO4, filtrated, and evaporated. The residue was purified by column chromatography on silica gel.
4,6-Dichloro-2-phenylquinoline (118a).
Isolated yield: 20% (for 2 steps); brown solid; 1H NMR (400 MHz, CDCl3): δ 8.22 (d, J = 2.4 Hz, 1H), 8.15 (dd, J = 6.6, 1.8 Hz, 2H), 8.12 (d, J = 9.0 Hz, 1H), 7.99 (s, 1H), 7.73–7.70 (m, 1H), 7.55 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 157.8, 147.8, 142.4, 138.5, 133.6, 132.1, 131.9, 130.4, 129.4, 127.8, 126.4, 123.3, 120.1.
4,6-Dichloro-2-(pyridin-4-yl)quinoline (118b).
Isolated yield: 45% (for 2 steps); brown solid; 1H NMR (400 MHz, CDCl3): δ 8.81 (d, J = 6.0 Hz, 2H), 8.25 (d, J = 2.4 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 1.6 Hz, 1H), 8.03 (s, 2H), 7.76 (dd, J = 9.0, 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 155.0, 151.0, 147.8, 145.5, 143.1, 134.8, 132.3, 130.3, 127.0, 123.4, 121.7, 119.7.
6-Chloro-N-(4-morpholinobutyl)-2-phenylquinolin-4-amine (119).
The title compound was synthesized from 118a according to the method described for procedure 3B. Isolated yield: 43%; white solid; mp 141–143 °C; 1H NMR (400 MHz, CDCl3): δ 8.05 (dd, J = 8.2, 1.4 Hz, 2H), 8.02 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.56 (dd, J = 9.0, 2.4 Hz, 1H), 7.52–7.48 (m, 2H), 7.47–7.43 (m, 1H), 6.84 (s, 1H), 3.78–3.75 (m, 4H), 3.45–3.40 (m, 2H), 2.51–2.45 (m, 6H), 1.90 1.86 (m, 2H), 1.77–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 158.6, 150.2, 146.7, 140.3, 131.5, 130.5, 129.7, 129.1, 127.9, 119.3, 97.6, 67.2, 58.5, 54.1, 43.7, 26.7, 24.6; HRMS (ESI) m/z: calcd for C23H27ClN3O, 396.1843 [M + H]+; found, 396.1830; retention time 5.243 min (HPLC condition A).
6-Chloro-N-(4-morpholinobutyl)-2-(pyridin-4-yl)quinolin-4-amine (120).
The title compound was synthesized from 118b according to the method described for procedure 3B. Isolated yield: 49%; white solid; mp 185–186 °C; 1H NMR (400 MHz, CDCl3): δ 8.74 (d, J = 6.4 Hz, 2H), 7.97 (t, J = 7.2 Hz, 3H), 7.76 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 9.0, 2.2 Hz, 1H), 6.84 (s, 1H), 3.77–3.74 (m, 4H), 3.43–3.38 (m, 2H), 2.49–2.43 (m, 6H), 1.90–1.84 (m, 2H), 1.76–1.71 (m, 2H); 13C NMR (100 MHz, CDCI3): δ 156.0, 150.6, 150.3, 148.1, 147.4, 132.4, 131.2, 130.6, 122.1, 119.4, 119.3, 97.0, 67.2, 58.5, 54.1, 43.7, 26.7, 24.7; HRMS (ESI) m/z: calcd for C22H26ClN4O, 397.1795 [M + H]+; found, 397.1771; retention time 2.651 min (HPLC condition A).
General Procedure for Preparation of the Monophosphate of 2-Arylvinylaminoquinolines (24s, 29s, and 86s).
To a 50 mL signal flask were charged 2-arylethenylaminoquinolines (0.81 mmol), acetonitrile (13.0 mL), and 0.1 Mphosphoricacid (10 mL, 1.0 mmol). The reaction mixture was stirred at room temperature for 24 h. Then, the resultant solution was filtered, and the solvents were dried with a lyophilizer under vacuum (0.135 mbar) with a temperature of −46 °C for 48 h to afford phosphate salts 24s, 29s, and 86s.
(E)-N1-(6-Chloro-2-(4-(trifluoromethyl)styryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine Monophosphate Salt (24s).
The title compound was synthesized from compound 24. Yield: 90%; light-yellow solid; mp >200 °C; 1HNMR (500 MHz, DMSO-d6): δ 8.27 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.77 (d, J = 16.2 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.55 (dd, J = 9.0, 2.4 Hz, 1H), 7.41 (d, J = 16.2 Hz, 1H), 7.28 (s, 1H), 6.83 (s, 1H), 3.50 (t, J = 6.0 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.39 (s, 6H); 13C NMR (125 MHz, DMSO-d6): δ 155.9, 150.2, 147.2, 141.1, 132.8, 131.7, 131.3, 130.2, 128.9, 128.1, 126.1 (q, J = 4.0 Hz), 123.7, 121.5, 119.6, 98.6, 56.5, 44.8, 40.3; HRMS (ESI) m/z: calcd for C22H22ClF3N3, 420.1454 [M + H]+; found, 420.1463; retention time 2.399 min (HPLC condition B).
(E)-N1-(6-Chloro-2-(4-fluorostyryl)quinolin-4-yl)-N2,N2-dimethylethane-1,2-diamine Monophosphate Salt (29s).
The title compound was synthesized from compound 29. Yield: 83%; light-yellow solid; mp >200 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.24 (d, J = 2.2 Hz, 1H), 7.70 (d, J = 6.6 Hz, 1H), 7.71–7.64 (m, 4H), 7.52 (dd, J = 9.0, 2.2 Hz, 1H), 7.22–7.16 (m, 3H), 7.01 (t, J = 5.2 Hz, 1H), 6.71 (s, 1H), 3.383.34 (m, 2H), 2.53 (t, J = 6.8 Hz, 2H), 2.18 (s, 6H); 13C NMR (125 MHz, DMSO-d6): δ 162.3 (d, J = 245.0 Hz), 156.4, 150.2, 147.4, 133.6, 132.0, 131.4, 130.1, 129.9, 129.5 (d, J = 8.0 Hz), 128.4, 121.4, 119.5, 116.2 (d, J = 23.0 Hz), 98.1, 57.5, 45.8, 41.1; HRMS (ESI) m/z: calcd for C21H22ClFN3, 370.1486 [M + H]+; found, 370.1502; retention time 2.70 min (HPLC condition B).
(E)-6-Chloro-2-(4-fluorostyryl)-N-(2-(4-methylpiperazin-1-yl)-ethyl)quinolin-4-amine Monophosphate Salt (86s).
The title compound was synthesized from compound 86. Yield: 80%; light-yellow solid; mp >200 °C; 1H NMR (500MHz, DMSO-d6): δ 8.23 (d, J = 2.2 Hz, 1H), 7.71–7.65 (m, 4H), 7.52 (dd, J = 9.0, 2.2 Hz, 1H), 7.21–7.16 (m, 3H), 7.04 (t, J = 5.4Hz, 1H), 6.73 (s, 1H), 3.40–3.36 (m, 2H), 3.30–3.25 (m, 2H), 2.60 (t, J = 6.8 Hz, 2H), 2.51–2.45 (m, 2H), 2.40–2.28 (m, 4H), 2.14 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 163.6 (d, J = 245.0 Hz), 161.2, 156.4, 147.3, 133.6, 132.0, 131.3, 130.0, 129.8 (d, J = 9.0 Hz), 129.5, 128.5, 121.3, 119.5, 116.2 (d, J = 21.0 Hz), 98.2, 56.3, 55.0, 53.0, 45.9, 40.7; HRMS (ESI) m/z: calcd for C24H27ClFN4, 425.1908 [M + H]+; found, 425.1910; retention time 2.334 min (HPLC condition B).
Biology.
In Vitro Antiplasmodial Activity.
Antiplasmodial activity was assessed by SYBR green I fluorescence, as described previously.33 Twofold serial dilutions of compounds were incubated with Plasmodium falciparum cultures at 1% parasitemia and 1% hematocrit in 96-well plates (Santa Cruz Biotechnology, Dallas, TX) at 37 °C under a 5% CO2 humidified atmosphere. The maximum DMSO concentration in assays was 0.1%. DMSO and CQ were used as growth and inhibition controls, respectively. After 72 h of incubation, plates were frozen at −80 °C, thawed, and incubated with 100 μL of lysis buffer (20 mM Tris-HCl, 0.008% saponin, 5 mM EDTA, 0.08% Triton X-100, and 0.01% SYBR Green I). Following incubation in the dark at room temperature for 1 h, the fluorescence signal was measured at 485 nM excitation and 530 nM emission using a Synergy Neo2 multimode reader (BioTek, Winooski, VT, USA). GraphPad Prism 8.0 software was used to generate nonlinear dose–response curves and calculate the EC50 values for each compound.
In Vitro Cytotoxicity Assay.
Test compounds were screened for cytotoxic activity in human hepatocyte cells (HepG2) seeded in 384-well clear bottom plates (Santa Cruz Biotechnology) at 2250 cells per well for 24 h at 37 °C in a 5% CO2 humidified atmosphere. Serial dilutions of test compounds were added starting at 25 μM, and plates were incubated for an additional 48 h. Subsequently, 10 μL of [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophen-yl)-2H-tetrazolium)] (MTS, CellTiter 96 Aqueous One, Promega, Madison, WI) was added to each well, and cell viabilitywas determined after 3 h of incubation at 37 °C by measuring the absorbance at 490 nm using a Synergy Neo2 multimode reader (BioTek, Winooski, VT, USA). GraphPad Prism 8.0 software was used to generate nonlinear dose–response curves and calculate the EC50 values for each compound.
In Vitro Gametocytocidal Activity.
Highly synchronous gametocyte cultures of the P. falciparum 3D7 line luc7 were obtained using an established method of gametocyte induction.62,63 Briefly, gametocytes induced by nutrient starvation were continually cultured in a heparin-treated medium to eliminate asexual-stage parasites. Gametocytes at the early stage (II-III) and late stage (IV-V) were purified via Percoll gradient centrifugation. Culture (100 μL) containing 50,000 gametocytes was added into each well in black 96-well plate and exposed for 72 h to a 19-point dose–response gradient of test compounds (concentration range of 0.038 nM to 10 μM). Luciferase expression was determined using the ONE-Glo luciferase assay system (Promega, Madison, WI, USA) according to the manufacturer instructions. The reagent (100 μL) was added to the culture, incubated for 5 min, and mixed well before luciferase intensity detection using a plate reader (PerkinElmer). EC50 values were determined in GraphPad Prism 8.0 software using a nonlinear dose–response curve fitting model.
In Vitro Metabolic Stability.
The metabolic stability of compounds was evaluated in mouse liver microsomes (Sigma-Aldrich) with an NADPH-regenerating system.64 Microsomes were suspended in a 0.1 M phosphate buffer (pH = 7.4) at a final protein concentration of 0.5 mg/mL and incubated with test compounds (1 μM) at 37 °C. An NADPH-regenerating system (1.3 mM NADP, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, and 0.4 U/mL glucose 6-phosphate dehydrogenase in 0.1 M potassium phosphate buffer, pH = 7.4) was added to initiate the metabolic reactions. The mixture was incubated on a shaking platform at 37 °C, and aliquots were withdrawn and then quenched with the addition of an equal volume of cold acetonitrile at different time periods. Additionally, samples were incubated in the absence of the NADPH-regenerating system to monitor for noncytochrome P450-mediated metabolism in the microsomal matrix. To remove debris, samples were centrifuged at 14,000 g for 10 min at 20 °C, and the amount of the parent compound remaining in the supernatant was monitored via LC–MS. The compound stability is calculated as the percent remaining of the unchanged parent compound at each time point (T = 60 min) relative to the peak area at T = 0 min.
Stage-Specific Action.
P. falciparum Dd2 cultures tightly synchronized by magnetic separation and sorbitol treatment were exposed to test compounds at 5 × EC50 at 6, 18, 30, or 42 h postinvasion.33 DHA (Sigma-Aldrich, 50 nM), atovaquone (Sigma-Aldrich, 6.6 nM), and DMSO (Sigma-Aldrich, 0.1%) were included as controls. Samples were taken after treatment at 12 h intervals to prepare Giemsa-stained thin smears for microscopic assessment of intraerythrocytic development. Simultaneously, samples were collected and processed for flow cytometric cell cycle analysis using a method previously described.65 Briefly, samples were fixed with 0.0075% glutaraldehyde (Sigma-Aldrich) and 4% paraformaldehyde (Sigma-Aldrich) in phosphate-buffered saline (PBS), permeabilized with 0.25% Triton X-100, treated with RNAse (Sigma-Aldrich) (50 μg/mL), and stained with YOYO-1 DNA binding dye (Invitrogen, Waltham, MA, USA) at a final concentration of 500 nM. Flow data were acquired using the CytoFLEX flow cytometer with 500,000 events per sample (Beckman Coulter, Indianapolis, IN, USA) and analyzed with FlowJo software 10.0 (FlowJo LLC, Ashland, OR, USA).
In Vitro Killing Profile
P. falciparum Dd2 asynchronous cultures at 1% parasitemia and 2% hematocrit were exposed to 5 × EC50 of test compounds for 6, 12, 24, or 48 h. Parasites treated with 0.15% DMSO, DHA (50 nM), and atovaquone (6.6 nM) were included as negative, fast-acting, and slow-acting controls, respectively. After exposure, compounds were removed through three washes with RPMI. Parasitemia was followed for 4 days after the addition of compounds via Giemsa-stained thin smears.
β-Hematin Formation.
Interference of the test compounds in the β-hematin crystal formation was evaluated under physiological conditions in the presence of the lipid catalyst using a 96-well plate assay, as previously described.66 A mixture of 10 μL of 2 mM hemin (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.1 M NaOH and 180 μL of propionate buffer at pH 5.2, and 10 μL of a previously sonicated phosphatidylcholine (Sigma-Aldrich, St. Louis, MO) suspension (10 mg/mL) was incubated with different concentrations of the test compound at 37 °C with gentle shaking for 16 h. DMSO and CQ were used as controls. After incubation, the reaction was stopped by the addition of 100 μL of a solution of sodium dodecyl sulfate (SDS), 7.5% (w/v) dissolved in 0.1 M bicarbonate buffer (pH 9.1). Plates were gently mixed and incubated at room temperature for 10 min. An aliquot of 50 μL from each well was transferred to a second plate preloaded with 200 μL/well of SDS solution (2.5% w/v in 0.1 M bicarbonate buffer), and the absorbance at 405 nm was read using a Synergy Neo2 multimode reader (BioTek). Free hemin in solution (as indicative of inhibition of β-hematin formation) was estimated using a linear calibration curve determined for each assay. Hemin stocks with concentrations ranging from 0 to 16 μM were freshly prepared in 2.5% SDS-0.1 M bicarbonate buffer (pH 9.1). Images of crystals were taken before quenching the reaction with SDS using a Nikon Eclipse TE200 inverted microscope.
In Vivo Antimalarial Activity. Effect on the Elimination of Existing Infection.
This evaluation was conducted at the Anti-Infectives Screening Core at New York Langone Medical Center, New York University (NYU). Briefly, Swiss-Webster female mice were injected with 103 red blood cells infected with P. berghei ANKA strain expressing luciferase. 48 h postinfection, mice were separated in groups of five mice and treated with compounds 24s, 29s, or 86s. Compounds were administrated via oral gavage in 200 μL aliquots of a vehicle containing 2% methylcellulose (Sigma-Aldrich, St. Louis, MO) and 0.5% Tween 80 (Sigma-Aldrich, St. Louis, MO) once daily for 5 days. Compounds were tested at 25 and 100 mg/kg. Compound vehicle was administered to the control mice group. On day 7 postinfection, mice were injected with 150 mg/kg of the D-luciferin potassium salt in PBS (Goldbio, St. Louis, MO), and luminescence was detected using an IVIS Lumina II imager. Results were expressed as the luciferin signal which is proportional to the load of parasites. The protocol used was approved by the NYU institutional animal care and use committee (IACUC).
Assessment of the Curative Property.
BALB/c female mice (10 week old, ~20 g) were inoculated intraperitoneally with 106 parasitized red blood cells with P. berghei ANKA 676m1cl1 (MRA-868, BEI Resources, Manassas, VA). Three experimental groups of five animals each were included: vehicle control, compound 24s at 25, and 100 mg/ kg dose. Treatment was administrated once daily via oral gavage in the same vehicle previously mentioned for 4 days starting at 4 h postinfection. Infection was monitored from day 4 postinoculation by Giemsa-stained thin smear of blood collected from the tail vein. Animals were sacrificed when parasitemia reached 40% or when symptoms of severe malaria were present. Animals with a negative blood smear 30 days post infection were considered cured. Survival data was analyzed using the Kaplan–Meier statistical method with the log-rank significance test. The protocol used in the study was approved by UCF IACUC.
Transmission Electron Microscopy.
P. falciparum Dd2 cultures were exposed to compound 24 at 5 × EC50 for 1.5, 3, 6, or 12 h. Culture treated with 0.15% DMSO was included as a control. For ultrastructural analysis, samples at each time point were centrifuged at low speed and washed once with PBS (pH = 7.4) and then resuspended in 1 mL of 2% paraformaldehyde/2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at a pH of 7.4 (Electron Microscopy Sciences, Hatfield, PA) and incubated at room temperature for 2 h with gently shaking. Following incubation, cells were pelleted and gently resuspended in 0.2 M sodium cacodylate buffer (pH = 7.4) (Electron Microscopy Sciences, Hatfield, PA). Subsequently, buffer was replaced for 1% osmium tetroxide (Polysciences Inc.) and incubated for 1 h. Samples were then rinsed extensively in dH2O prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., Redding, CA) for 1 h. Following several rinses in dH2O, samples were dehydrated in a graded series of ethanol solutions and embedded in Eponate 12 resin (Ted Pella Inc.). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc., Peabody, MA) equipped with an AMT 8 megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques, Woburn, MA).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from NIH/NIAID AI131398. We thank Bryan Kirk at UCF for his technical support during the in vivo study and Monica Bohmer, Sabrina Yu-Alfonzo, and Andy Nguyen for their help in the project. We also thank Ana Rodriguez and Julian Sherman at New York University, New York, NY for the IVIS imaging experiment; Wandy Beatty at Washington University Department of Microbiology, St. Louis, MO for the TEM imaging; and Sergio Wittlin, Swiss Tropical and Public Health Institute, Basel, Switzerland for sharing unpublished results.
ABBREVIATIONS
- Pf
Plasmodium falciparum
- CQ
chloroquine
- ACTs
artemisinin-based combination therapies
- DV
digestive vacuole
- Pf CRT
Plasmodium falciparum chloroquine resistance transporter
- RI
resistance index
- SI
selectivity index
- FV
food vacuole
- TEM
transmission electron microscopy
- SAR
structure–activity relationship
- DHA
dihydroartemisinin
- PPA
polyphosphoric acid
- p-TsNH2
p-toluenesulfonamide
- p.o.
per os
- MSD
mean survival time (in days)
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00858.
Antiplasmodial activity of synthesized analogues; metabolic stability profiles of selected compounds UCF501, 24, 29, 86, and 93; stage specific action of compounds 29, 86, DHA, and atovaquone; mice body weight data for compound 24s; and 1H NMR and 13C NMR spectra and LC–MS data of selected compounds (PDF)
Molecular formula strings and associated biological data (CSV)
Complete contact information is available at: https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00858
The authors declare no competing financial interest.
Contributor Information
Guang Huang, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
Claribel Murillo Solano, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Joel Melendez, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Justin Shaw, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Jennifer Collins, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Robert Banks, Research Program Services, University of Central Florida, Orlando, Florida 32816, United States.
Arash Keshavarzi Arshadi, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Rachasak Boonhok, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, Florida 33612, United States; Department of Medical Technology, School of Allied Health Science, Walailak University, Nakhon Si Thammarat 80160, Thailand.
Hui Min, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, Florida 33612, United States.
Jun Miao, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, Florida 33612, United States.
Debopam Chakrabarti, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Yu Yuan, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
REFERENCES
- (1).WHO. World Malaria Report, 2019.
- (2).Mishra M; Mishra VK; Kashaw V; Iyer AK; Kashaw SK Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2017, 125, 1300–1320. [DOI] [PubMed] [Google Scholar]
- (3).Tilley L; Rosenthal PJ Malaria parasites fine-tune mutations to resist drugs. Nature 2019, 576, 217–219. [DOI] [PubMed] [Google Scholar]
- (4).Stocks PA; Raynes KJ; Bray PG; Park BK; O’Neill PM; Ward SA Novel short chain chloroquine analogues retain activity against chloroquine resistant K1 Plasmodium falciparum. J. Med. Chem. 2002, 45, 4975–4983. [DOI] [PubMed] [Google Scholar]
- (5).Madrid PB; Wilson NT; DeRisi JL; Guy RK Parallel synthesis and antimalarial screening of a 4-aminoquinoline library. J. Comb. Chem. 2004, 6, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kondaparla S; Soni A; Manhas A; Srivastava K; Puri SK; Katti SB Antimalarial activity of novel 4-aminoquinolines active against drug resistant strains. Bioorg. Chem. 2017, 70, 74–85. [DOI] [PubMed] [Google Scholar]
- (7).Burgess SJ; Selzer A; Kelly JX; Smilkstein MJ; Riscoe MK; Peyton DH A Chloroquine-like Molecule Designed to Reverse Resistance in Plasmodium falciparum. J. Med. Chem. 2006, 49, 5623–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Joshi MC; Okombo J; Nsumiwa S; Ndove J; Taylor D; Wiesner L; Hunter R; Chibale K; Egan TJ 4-Aminoquinoline antimalarials containing a benzylmethylpyridylmethylamine group are active against drug resistant Plasmodium falciparum and exhibit oral activity in mice. J. Med. Chem. 2017, 60, 10245–10256. [DOI] [PubMed] [Google Scholar]
- (9).Hu Y-Q; Gao C; Zhang S; Xu L; Xu Z; Feng L-S; Wu X; Zhao F Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2017, 139, 22–47. [DOI] [PubMed] [Google Scholar]
- (10).Lombard MC; N’Da DD; Breytenbach JC; Smith PJ; Lategan CA Artemisinin-quinoline hybrid-dimers: synthesis and in vitro antiplasmodial activity. Bioorg. Med. Chem. Lett. 2010, 20, 6975–6977. [DOI] [PubMed] [Google Scholar]
- (11).Çapcı A; Lorion MM; Wang H; Simon N; Leidenberger M; Borges Silva MC; Moreira DRM; Zhu Y; Meng Y; Chen JY; Lee YM; Friedrich O; Kappes B; Wang J; Ackermann L; Tsogoeva SB Artemisinin-(iso)quinoline hybrids by C-H activation and click chemistry: combating multidrug-resistant malaria. Angew. Chem., Int. Ed. 2019, 58, 13066–13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lombard MC; N’Da DD; Breytenbach JC; Smith PJ; Lategan CA Synthesis, in vitro antimalarial and cytotoxicity of artemisinin-aminoquinoline hybrids. Bioorg. Med. Chem. Lett. 2011, 21, 1683–1686. [DOI] [PubMed] [Google Scholar]
- (13).Ruiz J; Mallet-Ladeira S; Maynadier M; Vial H; André-Barrès C Design, synthesis and evaluation of new tricyclic endoperoxides as potential antiplasmodial agents. Org. Biomol. Chem. 2014, 12, 5212–5221. [DOI] [PubMed] [Google Scholar]
- (14).Basco LK; Dechy-Cabaret O; Ndounga M; Meche FS; Robert A; Meunier B In vitro activities of DU-1102, a new trioxaquine derivative, against Plasmodium falciparum isolates. Antimicrob. Agents Chemother. 2001, 45, 1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Opsenica I; Opsenica D; Lanteri CA; Anova L; Milhous WK; Smith KS; Šolaja BA New chimeric antimalarials with 4-aminoquinoline moiety linked to a tetraoxane skeleton. J. Med. Chem. 2008, 51, 6216–6219. [DOI] [PubMed] [Google Scholar]
- (16).Biot C; Daher W; Ndiaye CM; Melnyk P; Pradines B; Chavain N; Pellet A; Fraisse L; Pelinski L; Jarry C; Brocard J; Khalife J; Forfar-Bares I; Dive D Probing the role of the covalent linkage of ferrocene into a chloroquine template. J. Med. Chem. 2006, 49, 4707–4714. [DOI] [PubMed] [Google Scholar]
- (17).Xiao J; Sun Z; Kong F; Gao F Current scenario of ferrocene-containing hybrids for antimalarial activity. Eur. J. Med. Chem. 2020, 185, 111791. [DOI] [PubMed] [Google Scholar]
- (18).Guantai EM; Ncokazi K; Egan TJ; Gut J; Rosenthal PJ; Bhampidipati R; Kopinathan A; Smith PJ; Chibale K Enone- and chalcone-chloroquinoline hybrid analogues: in silico guided design, synthesis, antiplasmodial activity, in vitro metabolism, and mechanistic studies. J. Med. Chem. 2011, 54, 3637–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raj R; Saini A; Gut J; Rosenthal PJ; Kumar V Synthesis and in vitro antiplasmodial evaluation of 7-chloroquinoline-chalcone and 7-chloroquinoline-ferrocenylchalcone conjugates. Eur. J. Med. Chem. 2015, 95, 230–239. [DOI] [PubMed] [Google Scholar]
- (20).Chu X-M; Wang C; Wang W-L; Liang L-L; Liu W; Gong K-K; Sun K-L Triazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019, 166, 206–223. [DOI] [PubMed] [Google Scholar]
- (21).Cornut D; Lemoine H; Kanishchev O; Okada E; Albrieux F ; Beavogui AH; Bienvenu A-L; Picot S; Bouillon J-P; Médebielle M Incorporation of a 3-(2,2,2-trifluoroethyl)-gamma-hydroxy-gamma-lactam motif in the side chain of 4-aminoquinolines. syntheses and antimalarial activities. J. Med. Chem. 2013, 56, 73–83. [DOI] [PubMed] [Google Scholar]
- (22).Kalaria PN; Karad SC; Raval DK A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. [DOI] [PubMed] [Google Scholar]
- (23).Davis TME; Hung T-Y; Sim I-K; Karunajeewa HA; Ilett KF Piperaquine - a resurgent antimalarial drug. Drugs 2005, 65, 75–87. [DOI] [PubMed] [Google Scholar]
- (24).McCarthy JS; Ruckle T; Djeriou E; Cantalloube C; Ter-Minassian D; Baker M; O’Rourke P; Griffin P; Marquart L; Hooft van Huijsduijnen R; Mohrle JJ APhase IIpilot trial to evaluate safety and efficacy of ferroquine against early Plasmodium falciparum in an induced blood-stage malaria infection study. Malar. J. 2016, 15, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Held J; Supan C; Salazar CLO; Tinto H; Bonkian LN; Nahum A; Moulero B; Sié A; Coulibaly B; Sirima SB; Siribie M; Otsyula N; Otieno L; Abdallah AM; Kimutai R; Bouyou-Akotet M; Kombila M; Koiwai K; Cantalloube C; Din-Bell C; Djeriou E; Waitumbi J; Mordmuller B; Ter-Minassian D; Lell B; Kremsner PG Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: a phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. Lancet Infect. Dis. 2015, 15, 1409–1419. [DOI] [PubMed] [Google Scholar]
- (26).Mushtaque M; Shahjahan. Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: A review. Eur. J. Med. Chem. 2015, 90, 280–295. [DOI] [PubMed] [Google Scholar]
- (27).Aguiar ACC; Panciera M; Simão dos Santos EF; Singh MK; Garcia ML; de Souza GE; Nakabashi M; Costa JL; Garcia CRS; Oliva G; Correia CRD; Guido RVC Discovery of marinoquinolines as potent and fast-acting Plasmodium falciparum inhibitors with in vivo activity. J. Med. Chem. 2018, 61, 5547–5568. [DOI] [PubMed] [Google Scholar]
- (28).Baragaña B; Hallyburton I; Lee MCS; Norcross NR; Grimaldi R; Otto TD; Proto WR; Blagborough AM; Meister S; Wirjanata G; Ruecker A; Upton LM; Abraham TS; Almeida MJ; Pradhan A; Porzelle A; Martínez MS; Bolscher JM; Woodland A; Luksch T; Norval S; Zuccotto F; Thomas J; Simeons F; Stojanovski L; Osuna-Cabello M; Brock PM; Churcher TS; Sala KA; Zakutansky SE; Jiménez-Díaz MB; Sanz LM; Riley J; Basak R; Campbell M; Avery VM; Sauerwein RW; Dechering KJ; Noviyanti R; Campo B; Frearson JA; Angulo-Barturen I; Ferrer-Bazaga S; Gamo FJ; Wyatt PG; Leroy D; Siegl P; Delves MJ; Kyle DE; Wittlin S; Marfurt J; Price RN; Sinden RE; Winzeler EA; Charman SA; Bebrevska L; Gray DW; Campbell S; Fairlamb AH; Willis PA; Rayner JC; Fidock DA; Read KD; Gilbert IH A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015, 522, 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Staderini M; Piquero M; Abengózar MÅ; Nachér-Vázquez M; Romanelli G; López-Alvarado P; Rivas L; Bolognesi ML; Menéndez JC Structure-activityrelationships and mechanistic studies of novel mitochondria-targeted, leishmanicidal derivatives of the 4-aminostyrylquinoline scaffold. Eur. J. Med. Chem. 2019, 171, 38–53. [DOI] [PubMed] [Google Scholar]
- (30).Mrozek-Wilczkiewicz A; Kuczak M; Malarz K; Cieślik W; Spaczyńska E; Musiol R The synthesis and anticancer activity of 2-styrylquinoline derivatives. A p53 independent mechanism of action. Eur. J. Med. Chem. 2019, 177, 338–349. [DOI] [PubMed] [Google Scholar]
- (31).Polanski J; Zouhiri F; Jeanson L; Desmaële D; d’Angelo J; Mouscadet J-F; Gieleciak R; Gasteiger J; Le Bret M Use of the kohonen neural network for rapid screening of ex vivo anti-HIV activity of styrylquinolines. J. Med. Chem. 2002, 45, 4647–4654. [DOI] [PubMed] [Google Scholar]
- (32).Xia C-L; Wang N; Guo Q-L; Liu Z-Q; Wu J-Q; Huang S-L; Ou T-M; Tan J-H; Wang H-G; Li D; Huang Z-S Design, synthesis and evaluation of 2-arylethenyl-N-methylquinolinium derivatives as effective multifunctional agents for Alzheimer’s disease treatment. Eur. J. Med. Chem. 2017, 130, 139–153. [DOI] [PubMed] [Google Scholar]
- (33).Roberts BF; Zheng Y; Cleaveleand J; Lee S; Lee E; Ayong L; Yuan Y; Chakrabarti D 4-Nitro styrylquinoline is an antimalarial inhibiting multiple stages of Plasmodium falciparum asexual life cycle. Int. J. Parasitol.: Drugs Drug Resist. 2017, 7, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Thomas KD; Adhikari AV; Shetty NS Design, synthesis and antimicrobial activities of some new quinoline derivatives carrying 1,2,3-triazole moiety. Eur. J. Med. Chem. 2010, 45, 3803–3810. [DOI] [PubMed] [Google Scholar]
- (35).Nayak N; Ramprasad J; Dalimba U Synthesis and antitubercular and antibacterial activity of some active fluorine containing quinoline-pyrazole hybrid derivatives. J. Fluorine Chem. 2016, 183, 59–68. [Google Scholar]
- (36).Barilli A; Atzeri C; Bassanetti I; Ingoglia F; Dall’Asta V; Bussolati O; Maffini M; Mucchino C; Marchiò L Oxidative stress induced by copper and iron complexes with 8-hydroxyquinoline derivatives causes paraptotic death of HeLa cancer cells. Mol. Pharm. 2014, 11, 1151–1163. [DOI] [PubMed] [Google Scholar]
- (37).Wright AD; Wang H; Gurrath M; König GM; Kocak G; Neumann G; Loria P; Foley M; Tilley L Inhibition of heme detoxification processes underlies the antimalarial activity of terpene isonitrile compounds from marine sponges. J. Med. Chem. 2001, 44, 873–885. [DOI] [PubMed] [Google Scholar]
- (38).Singh C; Srivastav NC; Puri SK In vivo active antimalarial isonitriles. Bioorg. Med. Chem. Lett. 2002, 12, 2277–2279. [DOI] [PubMed] [Google Scholar]
- (39).Young RM; Adendorff MR; Wright AD; Davies-Coleman MT Antiplasmodial activity: The first proof of inhibition of heme crystallization by marine isonitriles. Eur. J. Med. Chem. 2015, 93, 373–380. [DOI] [PubMed] [Google Scholar]
- (40).Daub ME; Prudhomme J; Ben Mamoun C; Le Roch KG; Vanderwal CD Antimalarial properties of simplified kalihinol analogues. ACS Med. Chem. Lett. 2017, 8, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Al-Riyami L; Pineda MA; Rzepecka J; Huggan JK; Khalaf AI; Suckling CJ; Scott FJ; Rodgers DT; Harnett MM; Harnett W Designing anti-inflammatory drugs from parasitic worms: a synthetic small molecule analogue of the acanthocheilonema viteae product ES-62 prevents development of collagen-induced arthritis. J. Med. Chem. 2013, 56, 9982–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gopinath VS; Pinjari J; Dere RT; Verma A; Vishwakarma P; Shivahare R; Moger M; Kumar Goud PS; Ramanathan V; Bose P; Rao MVS; Gupta S; Puri SK; Launay D; Martin D Design, synthesis and biological evaluation of 2-substituted quinolines as potential antileishmanial agents. Eur. J. Med. Chem. 2013, 69, 527–536. [DOI] [PubMed] [Google Scholar]
- (43).Gerold J; Holzenkamp U; Meier H Bis-, tris-, and tetrakis(squaraines) linked by stilbenoid scaffolds. Eur. J. Org. Chem. 2001, 2001, 2757–2763. [Google Scholar]
- (44).Huang G; Solano CM; Su Y; Ezzat N; Matsui S; Huang L; Chakrabarti D; Yuan Y Microwave-assisted, rapid synthesis of 2-vinylquinolines and evaluation of their antimalarial activity. Tetrahedron Lett. 2019, 60, 1736–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (46).Dalvit C; Vulpetti A Ligand-based fluorine NMR screening: principles and applications in drug discovery projects. J. Med. Chem. 2019, 62, 2218–2244. [DOI] [PubMed] [Google Scholar]
- (47).O’Neill PM; Harrison AC; Storr RC; Hawley SR; Ward SA; Park BK The effect of fluorine substitution on the metabolism and antimalarial activity of amodiaquine. J. Med. Chem. 1994, 37, 1362–1370. [DOI] [PubMed] [Google Scholar]
- (48).Kourounakis AP; Xanthopoulos D; Tzara A Morpholine as a privileged structure: a review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 2020, 40, 709–752. [DOI] [PubMed] [Google Scholar]
- (49).Ridley RG; Hofheinz W; Matile H; Jaquet C; Dorn A; Masciadri R; Jolidon S; Richter WF; Guenzi A; Girometta MA; Urwyler H; Huber W; Thaithong S; Peters W 4-Aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob. Agents Chemother. 1996, 40, 1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Coulup SK; Huang DS; Wong HL; Georg GI Identification of the metabolic profile of the alpha-tubulin-binding natural product (−)-Pironetin. J. Med. Chem. 2019, 62, 1684–1689. [DOI] [PubMed] [Google Scholar]
- (51).Delves MJ; Miguel-Blanco C; Matthews H; Molina I; Ruecker A; Yahiya S; Straschil U; Abraham M; Leon ML; Fischer OJ; Rueda-Zubiaurre A; Brandt JR; Cortes A; Barnard A; Fuchter MJ; Calderon F; Winzeler EA; Sinden RE; Herreros E; Gamo FJ; Baum J A high throughput screen for next-generation leads targeting malaria parasite transmission. Nat. Commun. 2018, 9, 3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhan W; Visone J; Ouellette T; Harris JC; Wang R; Zhang H; Singh PK; Ginn J; Sukenick G; Wong T-T; Okoro JI; Scales RM; Tumwebaze PK; Rosenthal PJ; Kafsack BFC; Cooper RA; Meinke PT; Kirkman LA; Lin G Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J. Med. Chem. 2019, 62, 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Roberts BF; Iyamu ID; Lee S; Lee E; Ayong L; Kyle DE; Yuan Y; Manetsch R; Chakrabarti D Spirocyclic chromanes exhibit antiplasmodial activities and inhibit all intraerythrocytic life cycle stages. Int. J. Parasitol.: Drugs Drug Resist. 2016, 6, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rosenthal PJ Plasmodium falciparum: effects of proteinase inhibitors on globin hydrolysis by cultured malaria parasites. Exp. Parasitol. 1995, 80, 272–281. [DOI] [PubMed] [Google Scholar]
- (55).Sachanonta N; Chotivanich K; Chaisri U; Turner GDH; Ferguson DJP; Day NPJ; Pongponratn E Ultrastructural and real-time microscopic changes in P. falciparum-infected red blood cells following treatment with antimalarial drugs. Ultrastruct. Pathol. 2011, 35, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ross LS; Fidock DA Elucidating mechanisms of drug-resistant Plasmodium falciparum. Cell Host Microbe 2019, 26, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Silva AM; Lee AY; Gulnik SV; Maier P; Collins J; Bhat TN; Collins PJ; Cachau RE; Luker KE; Gluzman IY; Francis SE; Oksman A; Goldberg DE; Erickson JW Structure and inhibition of plasmepsin II, a hemoglobin-degrading enzyme from Plasmodium falciparum. Proc. Natl. Acad. Sci U.S.A. 1996, 93, 10034–10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Alin MH; Bjorkman A Concentration and time dependency of artemisinin efficacy against Plasmodium falcium in vitro. Am. J. Trop. Med. Hyg. 1994, 50, 771–776. [DOI] [PubMed] [Google Scholar]
- (59).Peatey CL; Leroy D; Gardiner DL; Trenholme KR Antimalarial drugs: how effective are they against Plasmodium falciparum gametocytes? Malar. J. 2012, H, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Weissbuch I; Leiserowitz L Interplay between malaria, crystalline hemozoin formation, and antimalarial drug action and design. Chem. Rev. 2008, 108, 4899–4914. [DOI] [PubMed] [Google Scholar]
- (61).Le Manach C; Gonzàlez Cabrera D; Douelle F; Nchinda AT; Younis Y; Taylor D; Wiesner L; White KL; Ryan E; March C; Duffy S; Avery VM; Waterson D; Witty MJ; Wittlin S; Charman SA; Street LJ; Chibale K Medicinal chemistry optimization of antiplasmodial imidazopyridazine hits from high throughput screening of a Soft Focus kinase library: part 1. J. Med. Chem. 2014, 57, 2789–2798. [DOI] [PubMed] [Google Scholar]
- (62).Cui L; Miao J; Wang J; Li Q; Cui L Plasmodium falciparum: development of a transgenic line for screening antimalarials using firefly luciferase as the reporter. Exp. Parasitol. 2008, 120, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Miao J; Wang Z; Liu M; Parker D; Li X; Chen X; Cui L Plasmodium falciparum: generation of pure gametocyte culture by heparin treatment. Exp. Parasitol. 2013, 135, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Di L; Kerns EH; Hong Y; Kleintop TA; Mc Connell OJ; Huryn DM Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates. J. Biomol Screening 2003, 8, 453–462. [DOI] [PubMed] [Google Scholar]
- (65).Bouillon A; Gorgette O; Mercereau-Puijalon O; Barale JC Screening and evaluation of inhibitors of Plasmodium falciparum merozoite egress and invasion using cytometry. Methods Mol. Biol. 2013, 923, 523–534. [DOI] [PubMed] [Google Scholar]
- (66).Gorka AP; Alumasa JN; Sherlach KS; Jacobs LM; Nickley KB; Brower JP; de Dios AC; Roepe PD Cytostatic versus cytocidal activities of chloroquine analogues and inhibition of hemozoin crystal growth. Antimicrob. Agents Chemother. 2013, 57, 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.