

Abstract
Rhesus macaque is an Old World monkey that shared a common ancestor with human ∼25 Myr ago and is an important animal model for human disease studies. A deep understanding of its genetics is therefore required for both biomedical and evolutionary studies. Among structural variants, inversions represent a driving force in speciation and play an important role in disease predisposition. Here we generated a genome-wide map of inversions between human and macaque, combining single-cell strand sequencing with cytogenetics. We identified 375 total inversions between 859 bp and 92 Mbp, increasing by eightfold the number of previously reported inversions. Among these, 19 inversions flanked by segmental duplications overlap with recurrent copy number variants associated with neurocognitive disorders. Evolutionary analyses show that in 17 out of 19 cases, the Hominidae orientation of these disease-associated regions is always derived. This suggests that duplicated sequences likely played a fundamental role in generating inversions in humans and great apes, creating architectures that nowadays predispose these regions to disease-associated genetic instability. Finally, we identified 861 genes mapping at 156 inversions breakpoints, with some showing evidence of differential expression in human and macaque cell lines, thus highlighting candidates that might have contributed to the evolution of species-specific features. This study depicts the most accurate fine-scale map of inversions between human and macaque using a two-pronged integrative approach, such as single-cell strand sequencing and cytogenetics, and represents a valuable resource toward understanding of the biology and evolution of primate species.
Structural variants (SVs) are genomic alterations that involve segments of DNA that are >50 bp (Iafrate et al. 2004; Sebat et al. 2004; Tuzun et al. 2005; Kidd et al. 2008; Mills et al. 2011; Eichler 2019). SVs can include “balanced” rearrangements, such as inversions and translocations, or genomic imbalances (duplications and deletions), commonly referred to as copy number variants (CNVs). Inversions represent an intriguing class of SVs, first identified by Sturtevant in 1917 (Sturtevant 1917), that play a key dual role in primate evolution and predisposition to disease. Chromosome inversions are the most common rearrangements differentiating humans and the great ape species at the karyotypic level (Yunis et al. 1980; Yunis and Prakash 1982; Nickerson and Nelson 1998; Locke et al. 2003). A key evolutionary effect of inversions is that they suppress recombination as heterozygotes. As a consequence, inversions can act as an initial step toward genomic divergence by protecting chromosomal regions from gene flow (Rieseberg 2001). Inversions are also the source of the majority of genetic structure within populations and affect polymorphisms chromosome-wide (Corbett-Detig and Hartl 2012). Despite the importance of inversions as a major mechanism of genome reorganization, we still struggle to understand how and why they evolve almost a century after Sturtevant's initial discovery owing to technical challenges in their discovery.
Recently, new advances in sequencing technologies, optical mapping, and novel assembly algorithms have deepened our understanding of SVs and their role in genome function, evolution, and disease. However, inversions still remain one of the most poorly studied types of genetic variation, mainly because of our insufficient ability to accurately detect them. Their balanced nature and the presence of segmental duplications (SDs) at inversion boundaries pose major challenges for inversion detection. A number of studies have identified and characterized large inversions (>2 Mbp) using laborious target-based cytogenetic studies (Ventura et al. 2001, 2003, 2004, 2007, 2011; Carbone et al. 2002, 2006, 2007, 2008; Kehrer-Sawatzki et al. 2005a,b, Kehrer-Sawatzki and Cooper 2008; Stanyon et al. 2008; Capozzi et al. 2012). With the advent of sequencing, inversions have been inferred from next-generation sequence data by abnormal paired-end mapping and split-read alignment signatures (Tuzun et al. 2005; Kidd et al. 2008). Because the human genome is highly enriched in SDs (Bailey and Eichler 2006), these approaches often lead to false positives or fail to detect inversions flanked by highly identical sequences.
Recently, strand-specific sequencing technologies have been developed and successfully applied to detect inversions in human genomes (Sanders et al. 2016; Chaisson et al. 2019). Single-cell template strand sequencing (Strand-seq) is an amplification-free sequencing technique that selectively sequences the template strands used for DNA replication during a single mitotic cell division. Although Strand-seq resolution is lower in repetitive regions, it is able to detect inversions with a size limit up to 1 kbp (Sanders et al. 2016; Chaisson et al. 2019; Porubsky et al. 2020b). The power of Strand-seq lies in its ability to track the directionality of DNA template strands in every single cell. Inversions are detected solely by identifying DNA sequence strand switches internal to the inverted sequence, readily identifying inversions flanked by large SDs that can be neither assembled nor traversed using standard DNA sequencing technologies. These features make Strand-seq the leading genomic method for nontargeted inversion detection, especially suited for large repeat-embedded variants (Sanders et al. 2016; Chaisson et al. 2019).
In this study, we took advantage of this newly developed method to identify inversions in the rhesus macaque genome. Macaque is an Old World monkey sharing a common ancestor with humans ∼25 Myr ago. This primate showed some similarities with humans in physiology, neurobiology, and susceptibility to infectious diseases, making it one of the most important primate models for studies on human diseases (Rhesus Macaque Genome Sequencing and Analysis Consortium 2007); a deeper understanding of its genetic features can help us better understand these processes. By applying Strand-seq in conjunction with molecular cytogenetics, we generated a complete map of inversions between human and macaque and identified variants affecting key genes that may be essential in understanding the evolution of specific human traits. Thus, this study represents a critical resource for genomic research that fills a major gap in the nonhuman primate research field.
Results
Detection of inversions by Strand-seq
To detect inversions in the macaque genome, we generated 61 high-quality Strand-seq cell libraries for one macaque individual (MMU1). To overcome the low sequence coverage obtained for each single-cell Strand-seq library, selected libraries were merged into a high-coverage and directional composite file for each chromosome (Sanders et al. 2016). Inversions arising between macaque and human were discovered by aligning the macaque data to the human reference assembly genome (GRCh38/hg38) and performing breakpoint (BP) detection on the composite file using breakpointR (R version: 3.3.3, 2017-03-06) (R Core Team 2014; Porubsky et al. 2020a). This allowed us to predict the location and genotype of inversions based on segmental changes in read directionality arising within inverted loci. The BED-formatted composite file was additionally uploaded as custom track onto the UCSC Genome Browser (GRCh38/hg38 release) to facilitate manual curation and analysis of all predicted inversions. Because previous comparative studies were focused on autosomes, we excluded the X and Y Chromosomes from our analysis. By using this approach, we initially identified 373 inversions in the Strand-seq data (Supplemental Table S1) that after validation and literature interrogation were extended to 375 (see “Validation of inversions in macaque” section) (Fig. 1; Supplemental Table S2).
Figure 1.

Genome-wide distribution of 375 inversions detected by Strand-seq between human and macaque genomes. Human chromosomes are shown on the left; orthologous macaque chromosomes, on the right. Orange lines between human and macaque ideograms show inversions detected by a simple strand switch. Green lines represent inversions within inversions, which are apparently direct by Strand-seq.
Inversions ranged in size from 859 bp to 92 Mbp and were distributed along all chromosomes with the highest density (number of inversions every 10 Mbp) on Chromosome 22 and the lowest on Chromosome 1 (Supplemental Fig. S1A). The vast majority of detected inversions (359 out of 375) appeared in homozygous state (i.e., both homologs being inverted and thus showing a “complete” switching of the read directionality within the locus). Conversely, the remaining 16 inversions were found in a heterozygous state (i.e., only one homolog was inverted and thus showed a “mixed” switch in read directionality); these likely represent polymorphic inversions among macaque individuals (Supplemental Fig. S1B). Moreover, 87 out of 375 inversions were nested within larger inversions, in a “matryoshka” configuration, and are apparently direct by Strand-seq (Supplemental Fig. S2A,B). One inversion (Chr7_inv12) flipped twice during evolution and appeared inverted by Strand-seq (Supplemental Fig. S2C).
Comparison of human and macaque assemblies and published literature
We first compared the detected inversions with rearrangements previously reported for macaque (Ventura et al. 2007; Antonacci et al. 2009; Catacchio et al. 2018; Maggiolini et al. 2019) and confirmed the orientation of 48 events, which correspond mainly to larger inversions (>130 kbp) (Supplemental Table S2). Conversely, 327 regions (87%), ranging in size from 859 bp to 9 Mbp and amounting to 55.6 Mbp of inverted DNA, were novel and described here for the first time as human/macaque inversions. However, 58 of these 327 regions were previously found to be inverted in Hominidae in a study in which Strand-seq was applied to discover inversions between humans and great apes (Porubsky et al. 2020b), and 11 out 327 have been described to be polymorphic inversions in human (Antonacci et al. 2009; Chaisson et al. 2019; Giner-Delgado et al. 2019; Puig et al. 2020).
All previously published macaque inversions (Ventura et al. 2007; Catacchio et al. 2018) have been detected by our Strand-seq analysis, except for three regions (Chr1_inv5, Chr16_inv4, and Chr16_inv5) for which Strand-seq shows a direct orientation. However, this is not a Strand-seq error because these inversions have been reported to be potential misassemblies (Chr1_inv5) and minor alleles (Chr16_inv4 and Chr16_inv5) of the human reference genome (GRCh38/hg38) (Antonacci et al. 2010; Sanders et al. 2016; Catacchio et al. 2018), and therefore, the orientation of these three regions is the opposite of what appears by Strand-seq (Supplemental Table S3; Supplemental Fig. S2D).
We also identified four regions (Chr11_inv4, Chr12_inv2, Chr12_inv10, and Chr21_inv6) that appeared inverted by Strand-seq but are reported as assembly errors in the human reference genome (GRCh38/hg38) (Supplemental Table S3; Sanders et al. 2016; Vicente-Salvador et al. 2017; Audano et al. 2019; Chaisson et al. 2019). Also, here the orientation of the region in macaque should be the opposite of what is shown by Strand-seq, and thus, these are not real inversions between human and macaque genomes but are an artifact because the reads are mapped against the human reference genome (Supplemental Table S3; Supplemental Fig. S2E).
Next, we investigated 39 regions previously reported to be errors in the macaque BCM Mmul_8.0.1/rheMac8 release by Catacchio and colleagues (Catacchio et al. 2018) and confirmed all previously reported errors and that all the regions were corrected in the latest Mmul_10/rheMac10 release (Supplemental Table S4).
Validation of inversions in macaque
To validate our novel 327 inversions, we tested 16 by fluorescence in situ hybridization (FISH) in the same Macaca mulatta individual (MMU1) for which Strand-seq data were generated. To also define if an inversion was polymorphic, 14 inversions were tested on two additional macaque individuals (M. mulatta, MMU2; Macaca fascicularis, MFA63). Owing to technical limitations, we were able to test only regions >500 kbp and for which the SD content was not an impediment for the FISH probe selection. In particular, 15 out of 16 inversions were tested by three-color FISH, whereas one, >2 Mbp, was tested by metaphase two-color FISH. Two of these were performed to refine the inversion BPs; for 10 regions, FISH experiments confirmed the inverted orientation in homozygous state in all macaque individuals, whereas for Chr10_inv9, one out of three individuals was found to carry the inversion in the heterozygous state. Moreover, for three inversions (Chr15_inv1, Chr15_inv3, and Chr16_inv3), the orientation could not be determined in all tested individuals (Supplemental Table S5). Testing multiple cell lines should allow investigation of the polymorphic nature of the inversions. However, we only tested three macaque individuals (six chromosomes), and therefore, we were unable to define if an inversion was polymorphic for allele frequencies <16.6%.
To further validate inversions not amenable to FISH, we performed BAC-end sequence (BES) paired mapping of a M. mulatta BAC library (CHORI-250) against the human reference genome. We expect BACs spanning inversion BPs discovered in macaque to be “discordant” when mapped to the human reference genome sequence with their ends mapping farther apart than expected in an incorrect orientation (Ventura et al. 2007; Antonacci et al. 2009; Catacchio et al. 2018; Maggiolini et al. 2019). Seventy-six inversions detected as homozygous by Strand-seq had support from macaque discordant BAC clones spanning at least one BP (Supplemental Table S6). Ten inversions identified as heterozygous by Strand-seq had just concordant (four inversions) or discordant (six inversions) clones spanning the inversion BPs, whereas one homozygous inversion had both concordant and discordant clones, suggesting that the macaque for which BAC ends are available might be heterozygous. Finally, just once BES paired mapping was inconsistent with Strand-seq data, because only one concordant clone was identified at the BPs of a homozygous inversion by Strand-seq. As a more direct means of validation, we selected 13 of these BAC clones for complete sequencing with Illumina as previously described (Tuzun et al. 2005). BAC-insert sequencing was 100% concordant with BES mapping and Strand-seq results (Supplemental Table S6).
Next, we selected 14 regions, without SDs at the BPs and ranging in size from 2 to 54 kbp, for polymerase chain reaction (PCR) (Supplemental Table S7). In all tested regions, both orientations were detected in different species, and macaque was inverted as suggested by Strand-seq.
By combining different validation methods, we tested a total of 104 out of 327 novel inversions (Supplemental Fig. S3). Among these, 35 correspond to inversions within larger inversions, which appeared as direct by Strand-seq (Supplemental Figs. S2A, S4A). After validation, the number of polymorphic inversions in macaque increased from 16 to 19 (Supplemental Table S2). All validations, except for one, supported the inversion state identified by Strand-seq. Moreover, by intersecting data from previously published inversions (n = 5) and experimental analyses (n = 26), we confirmed the inverted status of 31 out of 87 nested inversions (Supplemental Fig. S2A,B), which appeared to be in direct orientation by Strand-seq but were mapping within a larger region in an inverted orientation. Eighty-three of these represent cases of simple nested inversions without BP reuse (Supplemental Fig. S2A). With the aforementioned analyses, the total number of inversions changed from 373 to 375. Through these efforts, we compiled a highly curated call-set of inversions that distinguishes macaque and humans, which we used for further analysis.
FISH analyses of complex inversions with BP reuse
Several FISH experiments were performed to better resolve the organization of complex regions. An example is shown in Supplemental Figure S4B, where FISH analysis of a 2.7-Mbp inversion (BP2-BP3 inversion) on Chromosome 10 allowed us to refine the BPs of a previously known 36-Mbp inversion (BP4-BP5 inversion). BP2-BP3 and BP4-BP5 inversions were detected by Strand-seq as two inversions separated by a 940-kbp region in direct orientation between BP3 and BP4. Initial analyses of a BP2-BP3 inversion were performed using a reference probe mapping outside of the inversion and within the direct distal BP3-BP4 region (Supplemental Fig. S4B, experiment 1). FISH experiments showed that this probe (blue) in macaque maps several megabases apart than expected, suggesting that the region detected in direct orientation by Strand-seq (BP3-BP4 region) is internal to the large 36-Mbp inversion (BP4-BP5 inversion). Consequently, the BP3-BP4 inversion appears to be direct by Strand-seq because it is nested within a larger inversion (Supplemental Fig. S4B, experiment 2). Further experimental validations allowed us to define that the proximal BP of the 36-Mbp inversion is not BP4 but is BP3 (∼900 kbp upstream than previously reported) (Supplemental Fig. S4B; Supplemental Table S5; Catacchio et al. 2018).
Notably, the BP2-BP3 inversion is also flanked proximally by a ∼900-kbp region between BP1 and BP2, which was previously reported to be inverted between human and macaque and is still polymorphic in human (Ventura et al. 2007). The BP1-BP2 inversion, however, appeared to be in direct orientation by Strand-seq, leading us to hypothesize that this could be another example of an inversion within an inversion. Further investigation of this region confirmed that the BP1-BP2 inversion is nested within a larger one (BP1-BP3) (Supplemental Fig. S4B, experiments 3 and 4).
In total, we identified and validated four cases (Chr2_inv14, Chr9_inv14, Chr10_inv8, and Chr10_inv9) (Supplemental Figs. S4B, S5) of nested inversions with BP reuse. In all four cases, SDs are flanking recurrently inverted regions. Moreover, we identified three inversions (Chr7_inv11, Chr7_inv13, and Chr10_inv7) for which the reused BPs are shared with adjacent inversions and not with the larger inversions that include them.
Nested inversions analyses
To assess the statistical significance of the observed nested inversions (n = 87), we conducted 100,000 simulations of the 375 observed inversions. First, we shuffled the observed inversion coordinates (n = 375) across the entire GRCh38/hg38 at random, using BEDTools (v2.28.0) (Quinlan and Hall 2010), excluding assembly gaps and centromeres. Second, we limited our shuffling to occur only in the space between inter-chromosomal SD pairs of ≥98% sequence identity, accounting for the biological bias of inversion occurrence between high-identity SD pairs. We observed a trend toward significance for the enrichment in nested inversions (Supplemental Fig. S6A,B). When the shuffling of the 375 inversion coordinates is restricted to the space between inter-chromosomal SD pairs, the enrichment is no longer significant (Supplemental Fig. S6C), suggesting that nested inversions are likely driven by highly identical SD pairs. As expected, we observe that the number of nested inversions depends on the size of the inversion they are nested in Supplemental Figure S7A. We further noticed that Chromosomes 11 and 7 have a comparably high number of nested inversions, considering that there are two large inversions within these chromosomes (Supplemental Fig. S7B,C).
Evolutionary analyses
To resolve the evolutionary history of the inversions detected by Strand-seq, we first took advantage of published data from previous studies (Catacchio et al. 2018) to establish the lineage specificity of 41 inversions (Supplemental Table S2). For 33 inversions, experimental analyses were performed; the remaining four regions were validated using a combination of both experimental and literature (Supplemental Table S2). Specifically, we tested nine regions >500 kbp by FISH in multiple primate cell lines, including two chimpanzees (Pan troglodytes), two gorillas (Gorilla gorilla), two orangutans (Pongo pygmaeus), and three macaques (two M. mulatta and one M. fascicularis); we used marmoset (Callithrix jacchus) as outgroup when necessary (Fig. 2A; Supplemental Table S5).
Figure 2.
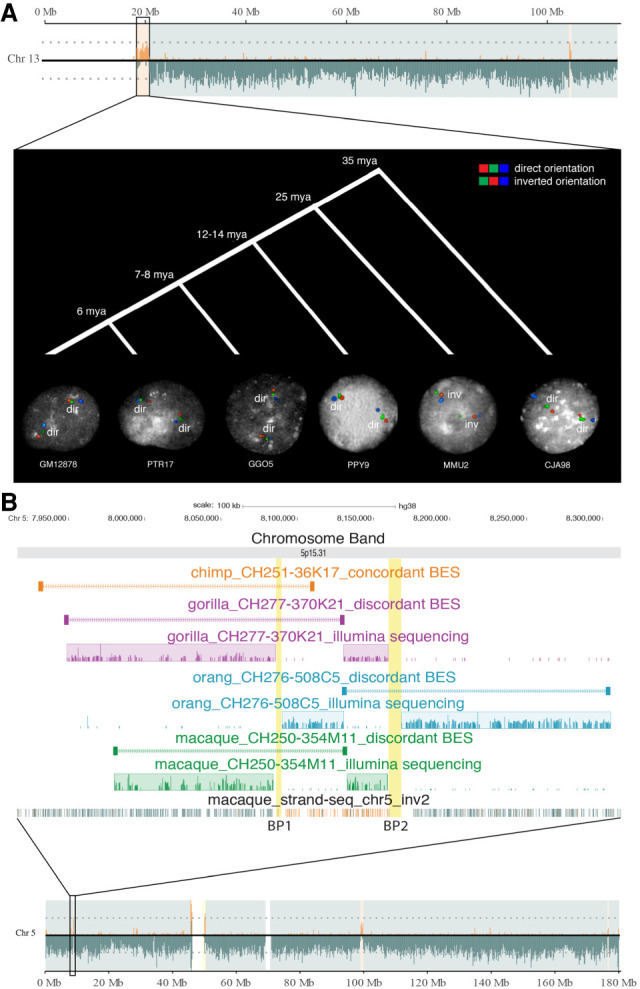
Evolutionary history of two inversions. (A) Strand-seq view of Chromosome 13 shows the switch in orientation of a 2-Mbp region, suggesting the presence of an inversion (Chr13_inv1). The region was tested using FISH in interphase nuclei in multiple primate species and was inverted just in macaque, whereas all the other primates are in direct orientation similar to human. (HSA) Homo sapiens; (PTR) Pan troglodytes; (GGO) Gorilla gorilla; (PPY) Pongo pygmaeus; (MMU) Macaca mulatta; (CJA) Callithrix jacchus. (B) Strand-seq view of a 89-kbp inversion (Chr5_inv2) between BP1 and BP2 is shown. BES mapping and Illumina sequencing of primate clones show that the region is inverted in gorilla, orangutan, and macaque and is direct in chimpanzee.
We also tested 14 regions by PCR in the same species: Four are human specific; one occurred in the human and chimpanzee ancestor and another three in the human, chimpanzee, and gorilla ancestor; and one is macaque specific. Finally, for all the inversions detected by Strand-seq, we checked the BES paired mapping profiling from primate BAC and fosmid clones (CHORI-251, CHORI-277, CHORI-276, CHORI-250, CHORI-259, and CHORI-1277) as previously described (Antonacci et al. 2009; Sanders et al. 2016; Catacchio et al. 2018; Kronenberg et al. 2018; Giner-Delgado et al. 2019; Maggiolini et al. 2019). We selected a total of 405 clones (257 concordant and 148 discordant) spanning the BPs of 176 putative inversions (Supplemental Table S6), and among these, 26 clones were fully sequenced with Illumina (Fig. 2B; Supplemental Table S6).
In total, we reconstructed the evolutionary history of 78 out of 375 regions. Twelve (15.4%) of these are human specific; 10 (12.8%) occurred in the human–chimpanzee common ancestor; 23 (29.5%) occurred in the African great apes ancestor, although three of these show the direct orientation in chimpanzee (which means that either the region in chimpanzee flipped back to the direct orientation or it represents a case of incomplete lineage sorting); eight (10.3%) occurred in the great ape ancestor; and 25 (32.1%) in the macaque lineage (Fig. 3A; Supplemental Table S2).
Figure 3.
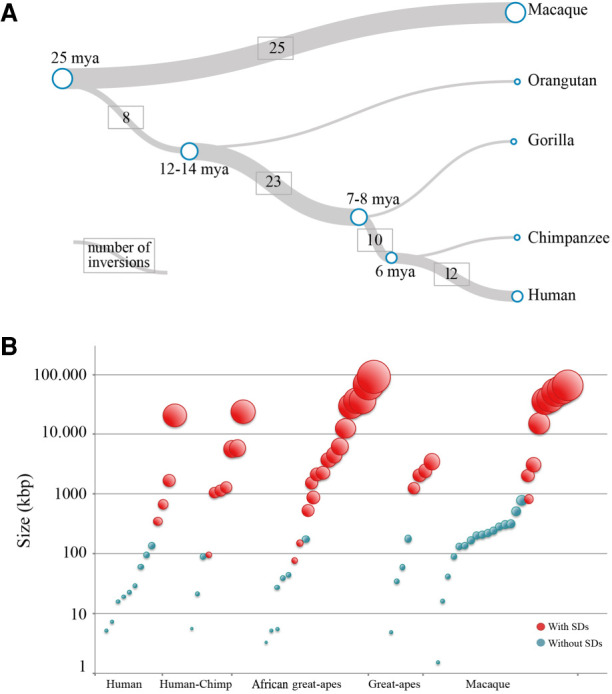
Evolutionary history and segmental duplication (SD) architecture of inverted region. (A) All inversions for which the evolutionary history has been determined are mapped on a phylogenetic tree in which the branch thickness is proportional to the number of inversions. (B) Inversions for which the lineage specificity has been determined are shown. The figure highlights the correlation between the presence of SDs at the inversion BPs and the size of the inversions.
To gather more information regarding the lineage specificity of the inversions, we used inversion calls generated for great apes, also from Strand-seq data (Porubsky et al. 2020b), and net alignments for the most recent releases of genome assemblies of two New World monkey outgroup species (C. jacchus, calJac3; Saimiri boliviensis, saiBol1). This revealed that 49.1% (184/375) of the inversions occurred in the Old World monkeys, whereas 43.5% (163/375) are specific to Hominidae. We were not able to define the lineage specificity in only 28 cases (7.5%) as it was not possible to test the inversions in other species because the region structure makes validation difficult (Supplemental Table S2).
We compared the inversions identified between human and macaque genomes that are flanked by SDs, with the inversion list recently reported for other great ape genomes (Porubsky et al. 2020b). We identified 51 (65%) inversions that are inverted in at least one of the great ape genomes (Supplemental Fig. S8), which we identify as candidate nonallelic homologous recombination (NAHR)–mediated inversions that may undergo recurrent rearrangements in primate genomes.
Analysis of genomic features
Because inversions can directly act on genes via direct breaking of structure and separation of promoters from cis-acting regulatory elements, we searched for human RefSeq genes mapping at the inversion BPs. We found that, of the 375 inversions, 156 have human genes spanning at least one BP. In particular, by removing duplicates, we detected 861 genes from the RefSeq curated subset (Supplemental Table S8) overlapping with our inversion BPs. By considering these genes, we performed a Gene Ontology analysis applying the ToppFun default parameters on the ToppGene portal and found matches for 855 out of 861 genes. Gene Ontology analysis showed a high percentage of defensins, genes involved in the response to bacteria, and an enrichment of the golgin family members (Supplemental Table S9). We also asked whether inversions were less likely to fall on annotated genes and found that protein-coding, but not all, genes were significantly depleted at inversion BPs (P = 0.001, permutation analysis). Within 100 kbp of BPs, no such depletion was observed (Supplemental Fig. S9A–D).
We tested for enrichment of protein-coding genes in all inversions identified between human and macaque and did not see any significant enrichment (P-value = 0.198, Z-score −0.82). However, we hypothesized that inversions predicted to be formed by NAHR and potentially undergoing recurrent rearrangement could show an effect on gene content owing to selective pressures acting at these inversions throughout evolution. To address this, we performed an enrichment analysis by focusing on the 51 inversions we identified as potential NAHR candidates, and tested for enrichment of protein-coding genes annotated in the human reference assembly (GRCh38/hg38). This revealed that NAHR-candidate inversions show an enrichment of protein-coding genes (P-value 0.031, Z-score 1.758) in support of the hypothesis (Supplemental Fig. S10).
At least five of the inversions validated by PCR overlap genes: Two inversions overlap protein-coding genes, and the other three overlap lncRNA genes. A detailed analysis of the human and macaque genome sequences (GRCh38/hg38 and rheMac10) shows that Chr17_inv12 and Chr21_inv5 inversions disrupt alternative transcripts of the protein-coding genes CCDC40 and ERG, respectively, which cannot be generated from the macaque genome, although the remaining transcripts would be unaffected by the inversion. Also, four lncRNA genes are truncated by three inversions. Inversion Chr17_inv13 exchanges in macaque the last exons of two lncRNAs, leaving the first exons outside, so if these transcripts exist in macaque, they would be chimeric. In another macaque inversion (Chr4_inv14), all transcripts of lncRNA LINC01094, expressed in brain and placenta in humans, are disrupted by removing the first exon. Finally, lncRNA LINC00605, expressed in the human testis, is disrupted by inversion Chr14_inv16 in macaque and marmoset. In this last case, the generation of the inverted allele would have created the lncRNA, which would not exist in the ancestral inverted allele in its current human form.
We also performed a pairwise dN/dS analysis between macaque (Mmul_10/rheMac10) and human (GRCh38/hg38) in order to investigate what kind of evolutionary forces shaped the inversions. We created two orthologous gene sets, one including pairwise orthologs between macaque and human in inverted regions and the other including pairwise orthologs between macaque and human in noninverted regions. We used PAML (Yang 1997) to calculate the dN/dS value of all orthologs and found that the dN/dS distribution of genes in the inverted regions is not significantly different from the dN/dS distribution of genes in noninverted regions (Wilcoxon rank-sum test, P-value = 0.5201) (Supplemental Fig. S11).
Because duplications play a crucial role in the origin of inversions, we analyzed the SD content at inversion BPs. We found that 77 out of 375 inversions (20.5%) have SDs mapping at their BPs, whereas if we consider just inversions >300 kbp, 91.7% (55/60) have SDs at their BPs. We also investigated the link between SD regions and the lineage specificity of inversions and found that 29.4% of Hominidae-specific inversions have SDs at the BPs, whereas only 8.7% of the macaque-specific inversions have SDs. Of note, when filtering for inversions >300 kbp, the percentage of regions flanked by SDs increased to 100% for Hominidae-specific inversions and to 69% for Old World monkey inversions. When considering regions >1 Mbp, the percentage of Old World monkey–specific inversions flanked by SDs goes up to 89% (Fig. 3B; Supplemental Table S2).
We compared our inversions with the UCSC ClinVar (Newman et al. 2005; Catacchio et al. 2018), development delay (Landrum et al. 2016), and ClinGen CNVs (Cooper et al. 2011; Coe et al. 2014) tracks and found 19 pathogenic CNVs overlapping inverted regions between human and macaque (Table 1).
Table 1.
19 inversions associated with human disease

Previous studies identified nine out of these 19 inversions as polymorphic in human, including the Chromosome 4p16.2-4p16.1 and 8p23 inversions, both of which predispose to further rearrangements leading to complex neurological disorders (Miller et al. 2010; Kaminsky et al. 2011); a 2.5-Mbp inversion involving the 7q11 locus predisposing to the deletion associated with Williams–Beuren syndrome (Giglio et al. 2001, 2002; Antonacci et al. 2009); a 2-Mbp inversion predisposing to RCAD syndrome (Osborne et al. 2001; Schubert 2009); a 1.5-Mbp inversion involving the 10q11 locus (Mefford et al. 2007); two inversions at the 16p12.1 locus associated with deletion and duplication of the same loci (Catacchio et al. 2018); and the inversion of the 15q25 locus predisposing to a deletion associated with developmental delay (Table 1; Miller et al. 2010; Cooper et al. 2011; Kaminsky et al. 2011; Coe et al. 2014; Landrum et al. 2016). For 17 out of 19 regions previously shown to be associated to pathogenic CNVs, we were able to define the lineage specificity of the inversions and show that the Hominidae orientation is always derived (Supplemental Table S2).
Because ancestral duplications, termed core duplicons, have been shown to be hotspots of genomic rearrangements, including large-scale inversion polymorphisms and recurrent CNVs associated with disease (Zody et al. 2008; Giannuzzi et al. 2013; Antonacci et al. 2014; Dennis and Eichler 2016; Nuttle et al. 2016; Maggiolini et al. 2019), we compared genes present at the inversion BPs with gene families mapping at core duplicons reported by Jiang and colleagues (2007). Almost half (nine out of 19) of these regions have one of these genes mapping at the inversion BPs (Table 1; Supplemental Table S2). This is also evident in our Gene Ontology analysis, which highlighted golgin genes, a core duplicon gene family previously implicated in other complex genomic rearrangements on human Chromosome 15 (Jiang et al. 2007; Antonacci et al. 2014; Maggiolini et al. 2019), as being enriched at BPs.
Recombination and heterozygosity
Moreover, because inversions can influence recombination, we analyzed the suppression of recombination over the inverted regions of the genome, relative to the background recombination rates (Wilcoxon rank sum test with continuity correction, P-value < 2.2−16). We observed a significant (<10−15) suppression of recombination in the inverted regions. Also, the recombination suppression effect was particularly pronounced in the case of polymorphic inversions (0.827 × background RC), followed by the fixed inverted regions (0.952 × background RC) (Supplemental Fig. S12).
In addition, we investigated whether there is a difference of heterozygosity on inversions’ flanking regions. We compared the heterozygosity distributions of four types of inversions and found that the polymorphic inversions’ flank regions have higher heterozygosity than random 5-kbp regions’ (P-value 0.02617, random vs. polymorphic inversions with SDs; P-value 1.68 × 10−8, random vs. polymorphic) and fixed inversions (P-value 0.01655, fixed inversions with SDs vs. polymorphic with SDs; P-value 5.40 × 10−7, fixed vs. polymorphic) (Supplemental Fig. S13). However, we did not observe heterozygosity difference between fixed inversions and random regions (P-value 0.944, random vs. fixed with SDs; P-value 0.07333, random vs. fixed).
Effect of inversions on gene regulation
Because inversions have the potential to reorganize genes and regulatory elements, we sought to determine whether macaque inversions impact the expression of nearby genes. By using existing RNA-seq data from human and macaque lymphoblastoid cell lines (LCLs) and primary tissues (heart, kidney, liver, and lung) (Khan et al. 2013; Blake et al. 2020), we identified interspecific differentially expressed genes (DEGs) at a 5% FDR (on average approximately 4800 DEGs per tissue) (Supplemental Tables S10–S14). We tested inversions, inversion BPs, and inversion BPs ±100 kbp for enrichment of DEGs in LCLs and the four tissues, either including or excluding genes overlapping SDs. However, after multiple testing (Benjamini–Hochberg) correction, none of the scenarios showed significant enrichment. A few tests displayed nominally significant enrichment with SDs excluded (uncorrected P ≤ 0.05): LCL DEGs (within inversions and at BPs) and kidney DEGs (BPs ± 100 kbp). When including SDs, LCL DEGs were also associated with inversions. Thus, we conclude that these results are compatible with SV alteration of gene expression reported in other species, but any true signal may be difficult to discern owing to the high overall proportion of DEGs between humans and macaques.
We next searched for changes to chromatin topology to which the observed differential expression may be attributable. By using a set of topologically associated domains (TADs) from the human LCL GM12878, we defined putatively disrupted TADs as those partially overlapping inversions (i.e., excluding those entirely within or containing inversions). We counted 48 inversions intersecting 69 putatively disrupted TADs, a number significantly lower than expected by chance (permutation test; empirical P = 0.001) (Supplemental Fig. S9; Supplemental Table S8). This depletion is also observed when SDs and inversions with BPs overlapping SDs are excluded from the analysis (P = 0.001) (Supplemental Fig. S9F). This is consistent with TAD-altering inversions being subject to negative selection. One such inversion (Chr18_inv4) is depicted in Figure 4, A and B, along with chromatin domains predicted from a parallel analysis of paired human and rhesus Hi-C data generated for this study from LCLs (Methods), as well as previously published data from fibroblasts (Rao et al. 2014; Darrow et al. 2016).
Figure 4.

Comparison of chromatin structure and gene expression at a selected inversion (Chr18_inv4). Coordinates depicted are Chr 18: 9,140,001–13,490,000 (GRCh38). (A) Hi-C heatmap of human (top) and macaque (bottom) LCLs with predicted chromatin domains outlined in yellow, visualized in Juicebox. SDs are shown as colored blocks in the top track (taken from the UCSC Genome Browser). Genes are colored by differential expression: Red genes are up-regulated in macaque relative to human, blue genes are down-regulated, black genes are not differentially expressed, and gray genes were not tested. (B) The same locus is depicted with fibroblast Hi-C data. No differential expression analysis was conducted in fibroblasts.
Macaque-specific chromatin interactions are visible across the inversion from >1 Mbp away in the human reference, and the domain structure appears to be altered at the inversion BPs and associated SDs. In LCLs, in which the gene-expression analysis was performed, most of the genes tested within and adjacent to the inversion are DEGs (Fig. 4B). Because of the lower sequencing depth of our Hi-Ci data, an alternative domain caller was used, which produced continuous annotations of domains, in contrast to the GM12878 TADs, which mostly contain gaps in between. We observed that BPs of large inversions (>100 kbp) often fell on domain boundaries (Supplemental Fig. S14) and confirmed significant enrichment for macaque domain boundaries (permutation test; empirical P < 0.03 for LCL and fibroblast) and human fibroblast domain boundaries (P = 0.002 for fibroblast). As with the depletion of putatively disrupted TADs, this is suggestive of conservation of chromatin structure. Finally, as mentioned previously, many large inversions are flanked by SDs, which cannot be uniquely aligned to or may be missing from macaque. As such, identifying altered domain structure at BPs was not possible owing to missing Hi-C data (Supplemental Fig. S15).
Discussion
Combining single-cell strand sequencing with cytogenetics, we created the most accurate fine-scale map of inversions between human and macaque to date. This approach was efficient in terms of time, cost, and resolution compared with high-throughput sequencing methods used to date. In total, we identified 375 inversions ranging in size from 859 bp to 92 Mbp, distributed along all the autosomes with the highest number on Chromosome 2 and the lowest on Chromosome 21. Despite this, considering the correlation between the size of each chromosome and the overall size of detected inversions for each of them, Chromosome 7 and Chromosome 3 show the highest percentage of inverted sequence (65% and 57%, respectively); indeed, these two chromosomes are two of the most rearranged among great ape and macaque genomes.
Of the 375 inversions, 48 were previously known, whereas the remaining 327 events (87.2%) are described here for the first time, increasing by eightfold the number of reported inversions between human and macaque. The vast majority (89.9%) of the 327 novel inversions are <100 kbp (Supplemental Fig. S16); this highlights the efficiency of Strand-seq in locating inversions, detecting even small events intractable by other methods.
To verify the reliability of Strand-seq, we validated a selection of 104 of the 327 novel inversions. All our results support Strand-seq data except for one case in which BES mapping validation and Strand-seq seemed to be discordant. Yet, this does not exclude that the inversion might be polymorphic in the population, and therefore, the individual for whom the BES data are available may be in direct orientation. However, the size of the inversion and the SD content did not allow us to validate it in additional individuals with other methods.
Although our analysis showed the efficiency of Strand-seq in detecting inversions, it also highlights that Strand-seq analysis must be aware of cytogenetic rearrangements. Indeed, 23% of our inversions appeared direct by Strand-seq because they are nested inversions. Among these, the vast majority are “simple” cases of nested inversions without BP reuse and thus can be easily identified if large-scale cytogenetic inversions are already known. Although in four cases, the regions were much more complex (Supplemental Figs. S4B, S5), and several FISH experiments were necessary to resolve their genomic organization. These were cases of inversions in which BPs have been reused multiple times during evolution, contributing to their complexity. We observed that nested inversions are more likely to occur than expected by chance and that they are likely driven by highly identical SDs (Supplemental Fig. S6).
To reconstruct the lineage specificity of inversions, we tested several primate species by combining different methods and determined that 49% of the inversions occurred in Old World monkeys, whereas 44% are specific to Hominidae (Supplemental Table S2). Our analysis of the duplications at the inversion BPs suggests that NAHR mediated by SDs promoted most (89%) inversions >1 Mbp in Old World monkeys and all inversions >300 kbp in Hominidae (Supplemental Table S2). This observation is concordant with the expansion of SDs after the divergence of Hominidae from Old World monkeys and strongly suggests a link between SD expansion and the emergence of inversions. The use of Strand-seq is mandatory to resolve these structural variations as a genomic technique not hampered by SDs.
Although our knowledge on the impact of inversions on human health is limited, the strong correlation between some inversions and neurocognitive disorders (Khan et al. 2013) is well documented. Thus, we searched for disease regions that are recurrently rearranged in humans and found 19 overlapping with our inversions, with nine being still polymorphic in humans (Table 1). For 17 of these regions, we were able to determine the lineage specificity. In 100% of the cases, the inversions are specific of the Hominidae, reinforcing the hypothesis that SDs played a fundamental role in generating inversions in humans and great apes that today, through their peculiar genomic structure, predispose to disease-causing rearrangements in humans.
Because SDs are frequently organized around core duplicons, we searched for their presence at the inversion BPs and found a total of 13 regions associated with cores. Among them, nine map at the BPs of inversions that overlap the aforementioned disease-associated regions (Table 1). Core duplicons have been previously described to be associated with the burst of SDs in the human–great ape ancestral lineage (Rao et al. 2014). This SD expansion likely set the stage for large-scale inversions to occur, ultimately leading to recurrent rearrangements associated to disease in humans.
Another interesting aspect about inversions is that they suppress recombination in heterokaryotype individuals. This results in independent genome evolution of direct and inverted arrangements and opportunities for divergence and speciation (Kirkpatrick and Barton 2006; Kirkpatrick 2010). Our results show a significant difference (<10−15) in recombination suppression between inverted versus noninverted regions in a size-independent way. Notably, by comparing homozygous and heterozygous inversions, we quantified how much the recombination was suppressed in fixed (∼5% lower than background) versus polymorphic (∼18% lower than background) inversions (Supplemental Fig. S12). This supports the role of inversions as a direct driving force in speciation because they suppress recombination when in heterozygous state. In addition, we observed a higher heterozygosity in polymorphic inversion flanking regions rather than in fixed inversions and random regions, supporting the idea that balancing selection has an important role in the maintenance of inversion polymorphisms, as previously reported (Wellenreuther and Bernatchez 2018; Mérot et al. 2020).
Because of the impact that inversions could have on the structure of genes, we searched for genes that can be altered by the presence of inversions. We identified 861 human genes overlapping with 156 inversion BPs (Supplemental Table S8); these include genes belonging to several groups, including members involved in the response to bacteria, genes with chemokine receptor binding activity, and golgin family members (Supplemental Table S9). At least five of the inversions that were validated by PCR overlap genes, with two located close to protein-coding genes and the other three in lncRNA genes. Future studies may evaluate the functional consequences of inversions on these genes in contributing to phenotypic differences among humans, great apes, and macaques.
Our assessment of the impact of inversions on gene regulation largely agrees with previous works that find structural variation alters gene expression in humans and nonhuman primates (Marques-Bonet et al. 2009; Lazar et al. 2018). Although expression analysis was limited to a single cell type, we report an enrichment of DEGs within and nearby (<100 kbp) inversion BPs that suggests that inversions between human and macaque may have the same functional impact reported in other species. In parallel, we also report that macaque inversions tend to avoid disrupting chromatin domain structure, as is true for deletions and rearrangements in other primates (Giglio et al. 2001; Osborne et al. 2001; Zody et al. 2008; Stankiewicz and Lupski 2010; Lazar et al. 2018; Maggiolini et al. 2019). Chromatin domains are thought to play a role in orchestrating promoter–enhancer interactions, and their disruption is associated with pathological phenotypes in humans (Marques-Bonet and Eichler 2009). Together, these findings support a view in which inversions impacting critical genes or altering regulation are likely to be deleterious. At the same time, inversions between human and macaque are associated with differential expression of nearby genes. This study provides a list of 48 inversions that are candidates for driving rhesus-specific expression patterns (Supplemental Table S10), although this is by no means exhaustive given that TAD annotations vary by algorithm and that TAD alterations per se are not required to alter transcription.
In conclusion, our approach based on the combination of Strand-seq and cytogenetic data offered us the opportunity to create a complete and detailed map of genomic inversions between human and macaque. We identified many hotspots of genomic instability that pinpoint regions with complex rearrangement activity, likely implicated in evolutionary innovations, as well as medical conditions.
Methods
Strand-seq detection of inversions
High-quality Strand-seq single-cell libraries (Iskow et al. 2012; Kronenberg et al. 2018) were obtained from an LCL derived from one macaque (M. mulatta, MMU1). The cells were maintained using standard culture conditions, and 40 µM of BrdU was added to the media for 23 h before sorting. Single cells were deposited in 96-well plate using the BD FACSMelody cell sorter, and Strand-seq library construction was pursued for single cells following the protocol previously described (Fudenberg and Pollard 2019; Huynh and Hormozdiari 2019). Libraries were sequenced on a NextSeq 500 (MID-mode, 75-bp paired-end protocol) and demultiplexed, and data were aligned to GRCh38/hg38 (BWA 0.7.15). Low-quality libraries, such as those with high background reads, were excluded from analysis, and 61 high-quality cells were obtained for inversion analysis. For each selected cell, only chromosomes inherited in the WW (plus-plus) or CC (minus-minus) state were considered and compiled into a directional composite file as previously described (Lupiáñez et al. 2015; Franke et al. 2016). The composite files were processed using breakpointR (v.1.2.0) (Falconer et al. 2012) to locate putative inversion BPs. To curate BPs and inversion calls, composite files were BED-formatted, uploaded to the UCSC Genome Browser, and manually inspected.
FISH analysis
Metaphases and interphase nuclei were obtained from two humans, two chimpanzees (P. troglodytes), two gorillas (G. gorilla), two orangutans (P. pygmaeus), three macaques (two M. mulatta and one M. fascicularis), and one marmoset (C. jacchus). Two-color and three-color FISH experiments were performed using human fosmid (n = 18) or BAC (n = 39) clones (Supplemental Table S5) directly labeled by nick-translation with Cy3-dUTP (PerkinElmer), Cy5-dUTP (PerkinElmer), and fluorescein-dUTP (Enzo) as previously described (Sanders et al. 2017), with minor modifications. Briefly, 300 ng of labeled probe was used for the FISH experiments; hybridization was performed at 37°C in 2× SSC, 50% (v/v) formamide, 10% (w/v) dextran sulphate, and 3 mg sonicated salmon sperm DNA in a volume of 10 mL. Posthybridization washing was at 60°C in 0.1 × SSC (three times, high stringency, for hybridizations on human, chimpanzee, gorilla, and orangutan) or at 37°C in 2 × SSC and 42°C in 2 × SSC, 50% formamide (three times each, low stringency, for hybridizations on macaque and marmoset). Nuclei were simultaneously DAPI stained. Digital images were obtained using a Leica DMRXA2 epifluorescence microscope equipped with a cooled CCD camera (Princeton Instruments). DAPI, Cy3, Cy5, and fluorescein fluorescence signals, detected with specific filters, were recorded separately as grayscale images. Pseudocoloring and merging of images were performed using Adobe Photoshop software. For interphase three-color FISH, each region >500 kbp was interrogated using two probes within the predicted inversion and a reference probe outside. A change in the order of the probes mapping within the inversion was indicative of the presence of the inversion. For inversions >2 Mbp, two-color FISH on metaphase chromosomes was performed using two probes within the inverted region (Supplemental Table S5).
BES and fosmid-end sequence paired mapping
BESs of chimpanzee, gorilla, orangutan, and macaque BAC libraries (CHORI-251, CHORI-277, CHORI-276, CHORI-250, and CHORI-259) and fosmid-end sequences of gorilla fosmid library (CHORI-1277) were obtained from the NIH trace repository and mapped against the human reference (GRCh38/hg38) following a protocol optimized by Sanders and colleagues (Sanders et al. 2016) for fosmids and adapted to BAC insert sizes as previously described (Porubsky et al. 2020a). BAC clones spanning regions in the same orientation as in human are concordant in size and orientation of the ends, whereas clones spanning inversion BPs are discordant because they have end pairs that are incorrectly oriented and map abnormally far apart when mapped to the human reference genome sequence. BES and fosmid-end sequence profiling of 372 BAC and 33 fosmid clones was used to study the orientation of 176 regions in different species.
Illumina sequencing of BAC clones
DNA from three CH251, five CH277, seven CH276, 13 CH250, and two CH259 (Supplemental Table S6) BAC clones was isolated, prepped into sequencing libraries, and sequenced (PE250) on an Illumina MiSeq using a Nextera protocol (Lichter et al. 1990). DNA from one clone was barcoded before library preparation, whereas DNA from 25 clones mapping to different chromosomes and free of SDs was pooled two at a time before library preparation and then barcoded and sequenced. Sequencing data were mapped with mrsFAST (Tuzun et al. 2005) to the human reference genome, and singly unique nucleotide (SUN) identifiers were used to discriminate between highly identical SDs (Catacchio et al. 2018). Illumina sequencing data of the BAC clones were accessed from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA429373.
Polymerase chain reaction
PCR was used to test 14 inversions <54 kbp (Supplemental Table S7) with simple BPs without large repeats. To validate inversions between different species, the first step was to identify the exact location of the inversion BPs through the NCBI “Blast2seq” tool to find the exact position range of the BPs for each species. Alignments were performed for human and chimpanzee, human and gorilla, human and orangutan, and human and macaque. After that, we designed four different primers (A, B, C, and D) that amplify two regions for each haplotype and include the BPs so that in the direct haplotype the BP1 is inside the AB amplicon and the BP2 inside CD. The inverted haplotype instead is revealed by amplification of primers A and C and of primers B and D. In some cases, additional primers were required to detect one of the orientations owing to the presence of indels associated to the inversion. Primers were designed with “Primer 3 Plus” (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) in order to amplify regions of 500–1000 bp. PCR amplification across inversion BPs was performed with genomic DNA from two humans (NA12878 and NA20528), two chimpanzees (PTR12 and N457/03), two gorillas (GGO2 and Z02/03), two orangutans (PPG9 and PPG10), two rhesus macaques (MMU1 and MMU2), one crab-eating macaque (MFA63), and one marmoset (CJA98), if needed. DNA N457/03 and Z02/03 were isolated from frontal cortex tissue samples of the Banc de Teixits Animals de Catalunya. PCR conditions were 30 sec at 94°C, 30 sec at 60°C–64°C, and 0.5–2 min at 72°C in 25-μL reactions with 100 ng of genomic DNA, 200 μM dNTPs, 10 pmol of each primer, and 1 U of Taq polymerase (Roche).
Simulations of nested inversions
In both simulation scenarios, we counted the number of times a nested inversion occurred, which was defined as an inversion that is 100% contained within another larger inversion. The null distributions from each scenario were constructed using the counts of the simulated nested inversions across 100,000 simulations. The empirical P-value was calculated after Z-score transformation using a one-tailed test, and the enrichment factor was estimated using 87/µ, where µ is the observed mean nested inversion count.
Gene Ontology analysis
Genes at the inversion BPs were extracted from the curated subset of the RefSeq track from the UCSC Genome Browser. The obtained gene list has been analyzed using the ToppGene portal (Chen et al. 2009; https://toppgene.cchmc.org/), which is a one-stop portal for gene list enrichment analysis and candidate gene prioritization based on functional annotations and protein interaction networks. In particular, the ToppFun function has been used to detect functional enrichment of genes based on transcriptome, proteome, regulome (TFBS and miRNA), ontologies (GO, Pathway), phenotype (human disease and mouse phenotype), pharmacome (Drug-Gene associations), literature cocitation, and other features.
Recombination analysis
The 375 inversions were annotated as fixed (n = 356) and polymorphic (n = 19) following conversion of genomic coordinates from GRCh38/hg38 to MGSC Merged 1.0/rheMac2 using liftOver; because of large structural differences between these two genome assemblies, some of the coordinates failed to convert, resulting in 11 fixed and 214 polymorphic inversions successfully mapped onto rheMac2 space (60% liftOver success rate). All recombination data were obtained from the latest recombination estimates of the macaque genome (Xue et al. 2016).
Heterozygosity analysis
We downloaded the macaque whole-genome sequencing (WGS) population data and selected 94 Indian macaque individuals for which sequence coverage was greater than 10× (Xue et al. 2016). We used PLINK (v1.9) (Purcell et al. 2007) to calculate fixed/fixed + SD/polymorphic/polymorphic + SD 5-kbp flank regions heterozygosity. Moreover, we used BEDTools (v2.29.0) (Quinlan and Hall 2010) to randomly choose 200 5-kbp regions excluding inversions flanking regions, and we used PLINK to calculate their heterozygosity. We used a t-test to perform statistical analysis in R.
Differential gene expression
Gene expression was quantified in using RNA-seq data from LCLs (macaque N = 5 individuals; human N = 6) (Khan et al. 2013) and primary tissues (N = 4 each) (Blake et al. 2020). TrimGalore (v0.6.0; http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used to trim FASTQ files using the flags “-q 20 ‐‐phred33 ‐‐length 20.” Transcriptome indices were built (Salmon v1.1.0) (Sudmant et al. 2010) from species-specific sequences of all orthologous transcripts previously published (Chen et al. 2009; Zhu et al. 2014), and the most recent reference genomes (GRCh38/hg38 and Mmul_10/rheMac10) were included as decoy sequences. Transcripts per million (TPM) values were estimated using Salmon using “‐‐validateMappings.” To compute counts at the gene level for a total of 28,372 coding and noncoding genes, tximport (Khan et al. 2013) was used with the setting ‘“countsFromAbundance = ‘lengthScaledTPM.’” A total of 15,920 genes were tested for differential expression after excluding those with less than one count per million in all samples. Length-normalized counts were passed to limma-voom (Patro et al. 2017), and each gene was fitted with a linear model accounting for species and sex. DEGs were called at a 5% FDR with no fold-change filter.
Chromatin conformation analyses
TADs were defined as a set of predictions generated from high-depth Hi-C of the human LCL GM12878 (approximately 4.9 billion Illumina reads) (Zhu et al. 2014). Coordinates of 9262/9274 TADs were converted to GRCh38 using the liftOver utility from the UCSC Genome Browser. The 5-kbp windows (resolution of the TAD-calling analysis) centered on the start and end coordinates of each TAD were considered to be TAD boundaries.
For an interspecies comparison of chromatin domain structure, we produced Hi-C libraries for LCLs of both species using a DNase-based method (Soneson et al. 2015). Three human (GM12878, GM20818, GM20543, analyzed together) and one rhesus macaque (MM290-96) individual were included. Valid Hi-C contacts on the human reference (GRCh38/hg38) were produced with the Juicer pipeline (Law et al. 2014; Ritchie et al. 2015). Human alignments were down-sampled to about 200 million reads to match the number of macaque Hi-C contacts passing the MAPQ filter of 30 (BWA) (Li and Durbin 2009). Hi-C interaction matrices were generated using Juicer tools (MAPQ > 30, Knight–Ruiz normalization) (Durand et al. 2016) at a resolution of 50 kbp. TopDom (Rao et al. 2014) was used to identify chromatin domains with the default window size of five. The measure of concordance (MoC) as implemented by Zufferey et al. (2018) was used to quantify similarity between domain sets on a scale of zero (no concordance) to one (identical) using Chromosome 1 matrices. Hi-C contact maps with coverage normalization and domain calls were visualized together in Juicebox (v1.11.08). Domain boundaries were defined as 50-kbp windows centered on the domain start and end coordinates and were considered to be shared between species if they intersected or were adjacent to a boundary in the other. This analysis was repeated in fibroblast cell lines using human IMR-90 (Durand et al. 2016) and macaque (Shin et al. 2016) data (about 230 million reads). Human Hi-C data from LCLs are available under NCBI BioProject accession number PRJEB36949.
Enrichment and depletion analyses
Permutation tests were conducted to identify over- and underrepresentation of genomic features (genes and boundaries) at and within 100 kbp of inversion BPs. Inversions were shuffled (BEDTools v2.25.0) (Quinlan and Hall 2010) 1000 times in GRCh38/hg38, preserving the sizes and distances of BPs, and the number of features intersecting BPs was counted in each set. Empirical P-values were calculated as P = (M + 1)/(N + 1), where M is the number of iterations yielding an equal or more extreme count than observed (greater for enrichment or fewer for depletion), and N is the number of permutations. To test BP regions for enrichment of DEGs, a hypergeometric test was implemented to compare the ratio of DEGs at or near BPs to the overall ratio of DEGs.
Data access
The Strand-seq library sequence data generated from this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA625922. Illumina sequencing data of the BAC clones generated in this study have been submitted to BioProject database under accession numbers PRJNA627588. Rhesus Hi-C data from LCLs have been uploaded to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser) under accession number PRJEB37908.
Competing interest statement
E.E.E. was on the scientific advisory board (SAB) of DNAnexus, Inc.
Supplementary Material
Acknowledgments
We thank T. Brown for critical review of the manuscript, Alejandra Delprat for help with the PCR validations, and Mira Mastoras and Pietro D'Addabbo for bioinformatics support. This work was supported by “Fondi di Ateneo, University of Bari” grant (grant number CUP H92F17000190005) to F.A., by the Agencia Estatal de Investigación (AEI, Spain) and the European Regional Development Fund (FEDER, EU; grant number BFU2016-77244-R) grant to M.C., by European Research Council Consolidator Grant MOSAIC grant (grant number 773026) to J.O.K., by the National Institutes of Health, National Institute of Neurological Disorders and Stroke (R00NS083627) grant to M.Y.D., and by U.S. National Institutes of Health (NIH) Clinical Center grants (grant numbers HG002385 and HG010169) to E.E.E. E.E.E. is an investigator of the Howard Hughes Medical Institute, and M.Y.D. is a Sloan fellow (FG-2016-6814).
Author contributions: F.A. and F.A.M.M. designed the study. A.D.S., F.A.M.M., and C.R.C. performed Strand-seq single-cell libraries construction. A.D.S. performed Illumina sequencing and data analysis. F.A.M.M., M.D., D.Pa., and L.M. performed FISH experiments. F.A.M.M. and C.R.C. performed Illumina sequencing of BAC clones. M.P. and M.B. performed PCR experiments. C.J.S. and M.Y.D. performed differential gene expression and chromatin conformation analyses. D.P. and E.E.E. contributed to evolutionary analysis. A.S. and D.P. performed simulations of nested inversions. Y.M. performed heterozygosity analysis. F.A., F.A.M.M., A.D.S., M.V., C.J.S., M.Y.D., M.P., M.C., J.O.K., and E.E.E. contributed to data interpretation. F.A. and F.A.M.M. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.265322.120.
References
- Antonacci F, Kidd JM, Marques-Bonet T, Ventura M, Siswara P, Jiang Z, Eichler EE. 2009. Characterization of six human disease-associated inversion polymorphisms. Hum Mol Genet 18: 2555–2566. 10.1093/hmg/ddp187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, Girirajan S, Alkan C, Campbell CD, Vives L, Malig M, et al. 2010. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet 42: 745–750. 10.1038/ng.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M, et al. 2014. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet 46: 1293–1302. 10.1038/ng.3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. 2019. Characterizing the major structural variant alleles of the human genome. Cell 176: 663–675.e19. 10.1016/j.cell.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Eichler EE. 2006. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 7: 552–564. 10.1038/nrg1895 [DOI] [PubMed] [Google Scholar]
- Blake LE, Roux J, Hernando-Herraez I, Banovich NE, Perez RG, Hsiao CJ, Eres I, Cuevas C, Marques-Bonet T, Gilad Y. 2020. A comparison of gene expression and DNA methylation patterns across tissues and species. Genome Res 30: 250–262. 10.1101/gr.254904.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi O, Carbone L, Stanyon RR, Marra A, Yang F, Whelan CW, de Jong PJ, Rocchi M, Archidiacono N. 2012. A comprehensive molecular cytogenetic analysis of chromosome rearrangements in gibbons. Genome Res 22: 2520–2528. 10.1101/gr.138651.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone L, Ventura M, Tempesta S, Rocchi M, Archidiacono N. 2002. Evolutionary history of chromosome 10 in primates. Chromosoma 111: 267–272. 10.1007/s00412-002-0205-5 [DOI] [PubMed] [Google Scholar]
- Cardone MF, Alonso A, Pazienza M, Ventura M, Montemurro G, Carbone L, de Jong PJ, Stanyon R, D'Addabbo P, Archidiacono N, et al. 2006. Independent centromere formation in a capricious, gene-free domain of chromosome 13q21 in Old world monkeys and pigs. Genome Biol 7: R91 10.1186/gb-2006-7-10-r91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone MF, Lomiento M, Teti MG, Misceo D, Roberto R, Capozzi O, D'Addabbo P, Ventura M, Rocchi M, Archidiacono N. 2007. Evolutionary history of chromosome 11 featuring four distinct centromere repositioning events in catarrhini. Genomics 90: 35–43. 10.1016/j.ygeno.2007.01.007 [DOI] [PubMed] [Google Scholar]
- Cardone MF, Jiang Z, D'Addabbo P, Archidiacono N, Rocchi M, Eichler EE, Ventura M. 2008. Hominoid chromosomal rearrangements on 17q map to complex regions of segmental duplication. Genome Biol 9: R28 10.1186/gb-2008-9-2-r28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catacchio CR, Maggiolini FAM, D'Addabbo P, Bitonto M, Capozzi O, Lepore Signorile M, Miroballo M, Archidiacono N, Eichler EE, Ventura M, et al. 2018. Inversion variants in human and primate genomes. Genome Res 28: 910–920. 10.1101/gr.234831.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 1784 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. 2009. Toppgene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37: W305–W311. 10.1093/nar/gkp427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, et al. 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46: 1063–1071. 10.1038/ng.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, Dougherty JD, Lein ES, Hormozdiari F, Bernier RA, et al. 2019. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 51: 106–116. 10.1038/s41588-018-0288-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. 2011. A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846. 10.1038/ng.909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Hartl DL. 2012. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet 8: e1003056 10.1371/journal.pgen.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M, et al. 2016. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc Natl Acad Sci 113: E4504–E4512. 10.1073/pnas.1609643113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, Eichler EE. 2016. Human adaptation and evolution by segmental duplication. Curr Opin Genet Dev 41: 44–52. 10.1016/j.gde.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, Aiden EL. 2016. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3: 95–98. 10.1016/j.cels.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE. 2019. Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med 381: 64–74. 10.1056/NEJMra1809315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer E, Hills M, Naumann U, Poon SS, Chavez EA, Sanders AD, Zhao Y, Hirst M, Lansdorp PM. 2012. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat Methods 9: 1107–1112. 10.1038/nmeth.2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan WL, et al. 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538: 265–269. 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Pollard KS. 2019. Chromatin features constrain structural variation across evolutionary timescales. Proc Natl Acad Sci 116: 2175–2180. 10.1073/pnas.1808631116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannuzzi G, Siswara P, Malig M, Marques-Bonet T, NISC Comparative Sequencing Program, Mullikin JC, Ventura M, Eichler EE. 2013. Evolutionary dynamism of the primate LRRC37 gene family. Genome Res 23: 46–59. 10.1101/gr.138842.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, et al. 2001. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 68: 874–883. 10.1086/319506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, et al. 2002. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet 71: 276–285. 10.1086/341610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giner-Delgado C, Villatoro S, Lerga-Jaso J, Gayà-Vidal M, Oliva M, Castellano D, Pantano L, Bitarello BD, Izquierdo D, Noguera I, et al. 2019. Evolutionary and functional impact of common polymorphic inversions in the human genome. Nat Commun 10: 4222 10.1038/s41467-019-12173-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh L, Hormozdiari F. 2019. TAD fusion score: discovery and ranking the contribution of deletions to genome structure. Genome Biol 20: 60 10.1186/s13059-019-1666-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. 2004. Detection of large-scale variation in the human genome. Nat Genet 36: 949–951. 10.1038/ng1416 [DOI] [PubMed] [Google Scholar]
- Iskow RC, Gokcumen O, Abyzov A, Malukiewicz J, Zhu Q, Sukumar AT, Pai AA, Mills RE, Habegger L, Cusanovich DA, et al. 2012. Regulatory element copy number differences shape primate expression profiles. Proc Natl Acad Sci 109: 12656–12661. 10.1073/pnas.1205199109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. 2007. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet 39: 1361–1368. 10.1038/ng.2007.9 [DOI] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, et al. 2011. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13: 777–784. 10.1097/GIM.0b013e31822c79f9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. 2008. Molecular mechanisms of chromosomal rearrangement during primate evolution. Chromosome Res 16: 41–56. 10.1007/s10577-007-1207-1 [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Sandig C, Chuzhanova N, Goidts V, Szamalek JM, Tänzer S, Müller S, Platzer M, Cooper DN, Hameister H. 2005a. Breakpoint analysis of the pericentric inversion distinguishing human chromosome 4 from the homologous chromosome in the chimpanzee (Pan troglodytes). Hum Mutat 25: 45–55. 10.1002/humu.20116 [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Sandig CA, Goidts V, Hameister H. 2005b. Breakpoint analysis of the pericentric inversion between chimpanzee chromosome 10 and the homologous chromosome 12 in humans. Cytogenet Genome Res 108: 91–97. 10.1159/000080806 [DOI] [PubMed] [Google Scholar]
- Khan Z, Ford MJ, Cusanovich DA, Mitrano A, Pritchard JK, Gilad Y. 2013. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342: 1100–1104. 10.1126/science.1242379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64. 10.1038/nature06862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick M. 2010. How and why chromosome inversions evolve. PLoS Biol 8: e1000501 10.1371/journal.pbio.1000501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick M, Barton N. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434. 10.1534/genetics.105.047985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJP, Dougherty ML, et al. 2018. High-resolution comparative analysis of great ape genomes. Science 360 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. 2016. Clinvar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–D868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar NH, Nevonen KA, O'Connell B, McCann C, O'Neill RJ, Green RE, Meyer TJ, Okhovat M, Carbone L. 2018. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res 28: 983–997. 10.1101/gr.233874.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter P, Tang CJ, Call K, Hermanson G, Evans GA, Housman D, Ward DC. 1990. High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247: 64–69. 10.1126/science.2294592 [DOI] [PubMed] [Google Scholar]
- Locke DP, Archidiacono N, Misceo D, Cardone MF, Deschamps S, Roe B, Rocchi M, Eichler EE. 2003. Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biol 4: R50 10.1186/gb-2003-4-8-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiolini FAM, Cantsilieris S, D'Addabbo P, Manganelli M, Coe BP, Dumont BL, Sanders AD, Pang AWC, Vollger MR, Palumbo O, et al. 2019. Genomic inversions and GOLGA core duplicons underlie disease instability at the 15q25 locus. PLoS Genet 15: e1008075 10.1371/journal.pgen.1008075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Eichler EE. 2009. The evolution of human segmental duplications and the core duplicon hypothesis. Cold Spring Harb Symp Quant Biol 74: 355–362. 10.1101/sqb.2009.74.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Girirajan S, Eichler EE. 2009. The origins and impact of primate segmental duplications. Trends Genet 25: 443–454. 10.1016/j.tig.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, et al. 2007. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81: 1057–1069. 10.1086/522591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérot C, Llaurens V, Normandeau E, Bernatchez L, Wellenreuther M. 2020. Balancing selection via life-history trade-offs maintains an inversion polymorphism in a seaweed fly. Nat Commun 11: 670 10.1038/s41467-020-14479-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. 2010. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86: 749–764. 10.1016/j.ajhg.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65. 10.1038/nature09708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman TL, Tuzun E, Morrison VA, Hayden KE, Ventura M, McGrath SD, Rocchi M, Eichler EE. 2005. A genome-wide survey of structural variation between human and chimpanzee. Genome Res 15: 1344–1356. 10.1101/gr.4338005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson E, Nelson DL. 1998. Molecular definition of pericentric inversion breakpoints occurring during the evolution of humans and chimpanzees. Genomics 50: 368–372. 10.1006/geno.1998.5332 [DOI] [PubMed] [Google Scholar]
- Nuttle X, Giannuzzi G, Duyzend MH, Schraiber JG, Narvaiza I, Sudmant PH, Penn O, Chiatante G, Malig M, Huddleston J, et al. 2016. Emergence of a Homo sapiens–specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 536: 205–209. 10.1038/nature19075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, et al. 2001. A 1.5 million-base pair inversion polymorphism in families with Williams–Beuren syndrome. Nat Genet 29: 321–325. 10.1038/ng753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo O, Palumbo P, Palladino T, Stallone R, Miroballo M, Piemontese MR, Zelante L, Carella M. 2012. An emerging phenotype of interstitial 15q25.2 microdeletions: clinical report and review. Am J Med Genet A 158A: 3182–3189. 10.1002/ajmg.a.35631 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Sanders AD, Taudt A, Colomé-Tatché M, Lansdorp PM, Guryev V. 2020a. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics 36: 1260–1261. 10.1093/bioinformatics/btz681 [DOI] [PubMed] [Google Scholar]
- Porubsky D, Sanders AD, Höps W, Hsieh P, Sulovari A, Li R, Mercuri L, Sorensen M, Murali SC, Gordon D, et al. 2020b. Recurrent inversion toggling and great ape genome evolution. Nat Genet 52: 849–858. 10.1038/s41588-020-0646-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Lerga-Jaso J, Giner-Delgado C, Pacheco S, Izquierdo D, Delprat A, Gayà-Vidal M, Regan JF, Karlin-Neumann G, Cáceres M. 2020. Determining the impact of uncharacterized inversions in the human genome by droplet digital PCR. Genome Res 30: 724–735. 10.1101/gr.255273.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: 222–234. 10.1126/science.1139247 [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol Evol 16: 351–358. 10.1016/S0169-5347(01)02187-5 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AD, Hills M, Porubský D, Guryev V, Falconer E, Lansdorp PM. 2016. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res 26: 1575–1587. 10.1101/gr.201160.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AD, Falconer E, Hills M, Spierings DCJ, Lansdorp PM. 2017. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat Protoc 12: 1151–1176. 10.1038/nprot.2017.029 [DOI] [PubMed] [Google Scholar]
- Schubert C. 2009. The genomic basis of the Williams–Beuren syndrome. Cell Mol Life Sci 66: 1178–1197. 10.1007/s00018-008-8401-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. 2004. Large-scale copy number polymorphism in the human genome. Science 305: 525–528. 10.1126/science.1098918 [DOI] [PubMed] [Google Scholar]
- Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. 2016. Topdom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res 44: e70 10.1093/nar/gkv1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD. 2015. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4: 1521 10.12688/f1000research.7563.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. 2010. Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437–455. 10.1146/annurev-med-100708-204735 [DOI] [PubMed] [Google Scholar]
- Stanyon R, Rocchi M, Capozzi O, Roberto R, Misceo D, Ventura M, Cardone MF, Bigoni F, Archidiacono N. 2008. Primate chromosome evolution: ancestral karyotypes, marker order and neocentromeres. Chromosome Res 16: 17–39. 10.1007/s10577-007-1209-z [DOI] [PubMed] [Google Scholar]
- Sturtevant AH. 1917. Genetic factors affecting the strength of linkage in Drosophila. Proc Natl Acad Sci 3: 555–558. 10.1073/pnas.3.9.555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, 1000 Genomes Project, et al. 2010. Diversity of human copy number variation and multicopy genes. Science 330: 641–646. 10.1126/science.1197005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732. 10.1038/ng1562 [DOI] [PubMed] [Google Scholar]
- Ventura M, Archidiacono N, Rocchi M. 2001. Centromere emergence in evolution. Genome Res 11: 595–599. 10.1101/gr.152101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Mudge JM, Palumbo V, Burn S, Blennow E, Pierluigi M, Giorda R, Zuffardi O, Archidiacono N, Jackson MS, et al. 2003. Neocentromeres in 15q24-26 map to duplicons which flanked an ancestral centromere in 15q25. Genome Res 13: 2059–2068. 10.1101/gr.1155103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Weigl S, Carbone L, Cardone MF, Misceo D, Teti M, D'Addabbo P, Wandall A, Bjorck E, de Jong PJ, et al. 2004. Recurrent sites for new centromere seeding. Genome Res 14: 1696–1703. 10.1101/gr.2608804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Antonacci F, Cardone MF, Stanyon R, D'Addabbo P, Cellamare A, Sprague LJ, Eichler EE, Archidiacono N, Rocchi M. 2007. Evolutionary formation of new centromeres in macaque. Science 316: 243–246. 10.1126/science.1140615 [DOI] [PubMed] [Google Scholar]
- Ventura M, Catacchio CR, Alkan C, Marques-Bonet T, Sajjadian S, Graves TA, Hormozdiari F, Navarro A, Malig M, Baker C, et al. 2011. Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Res 21: 1640–1649. 10.1101/gr.124461.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Salvador D, Puig M, Gaya-Vidal M, Pacheco S, Giner-Delgado C, Noguera I, Izquierdo D, Martinez-Fundichely A, Ruiz-Herrera A, Estivill X, et al. 2017. Detailed analysis of inversions predicted between two human genomes: errors, real polymorphisms, and their origin and population distribution. Hum Mol Genet 26: 567–581. 10.1093/hmg/ddw415 [DOI] [PubMed] [Google Scholar]
- Wellenreuther M, Bernatchez L. 2018. Eco-evolutionary genomics of chromosomal inversions. Trends Ecol Evol 33: 427–440. 10.1016/j.tree.2018.04.002 [DOI] [PubMed] [Google Scholar]
- Xue C, Raveendran M, Harris RA, Fawcett GL, Liu X, White S, Dahdouli M, Rio Deiros D, Below JE, Salerno W, et al. 2016. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res 26: 1651–1662. 10.1101/gr.204255.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13: 555–556. 10.1093/bioinformatics/13.5.555 [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Prakash O. 1982. The origin of man: a chromosomal pictorial legacy. Science 215: 1525–1530. 10.1126/science.7063861 [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Sawyer JR, Dunham K. 1980. The striking resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science 208: 1145–1148. 10.1126/science.7375922 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Li M, Sousa AM, Šestan N. 2014. XSAnno: a framework for building ortholog models in cross-species transcriptome comparisons. BMC Genomics 15: 343 10.1186/1471-2164-15-343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. 2008. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 40: 1076–1083. 10.1038/ng.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey M, Tavernari D, Oricchio E, Ciriello G. 2018. Comparison of computational methods for the identification of topologically associating domains. Genome Biol 19: 217 10.1186/s13059-018-1596-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
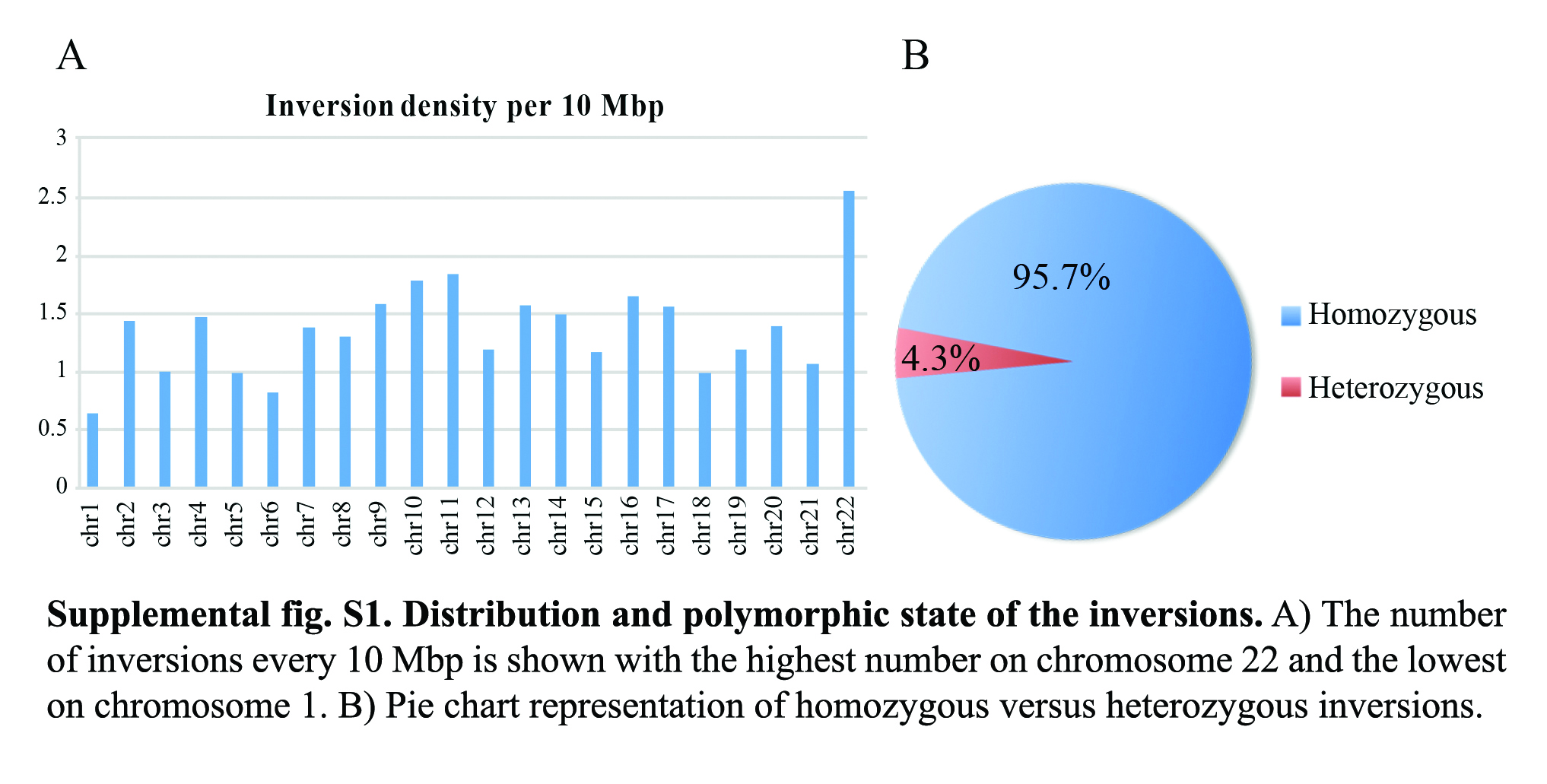{kind=link}
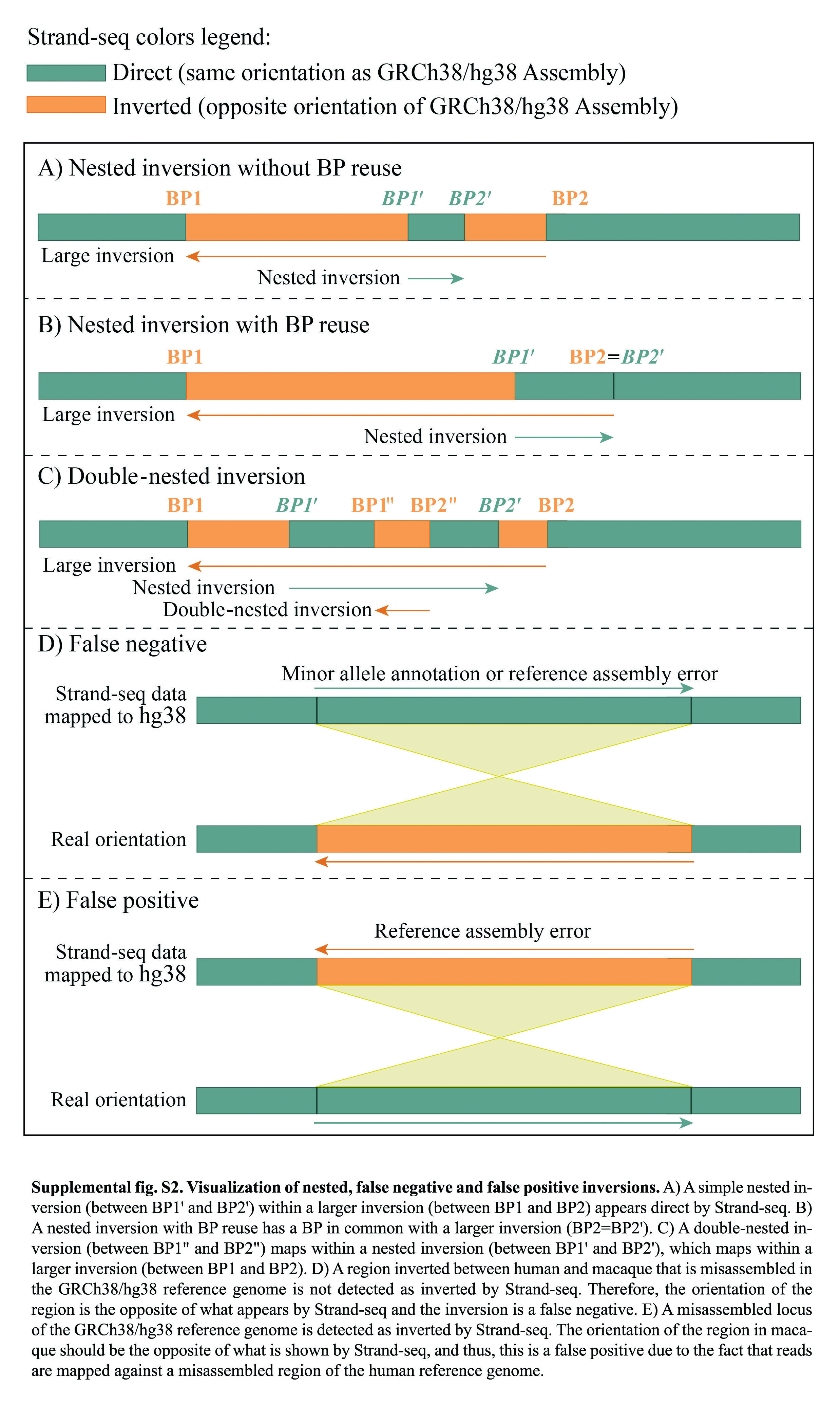{kind=link}
{kind=link}
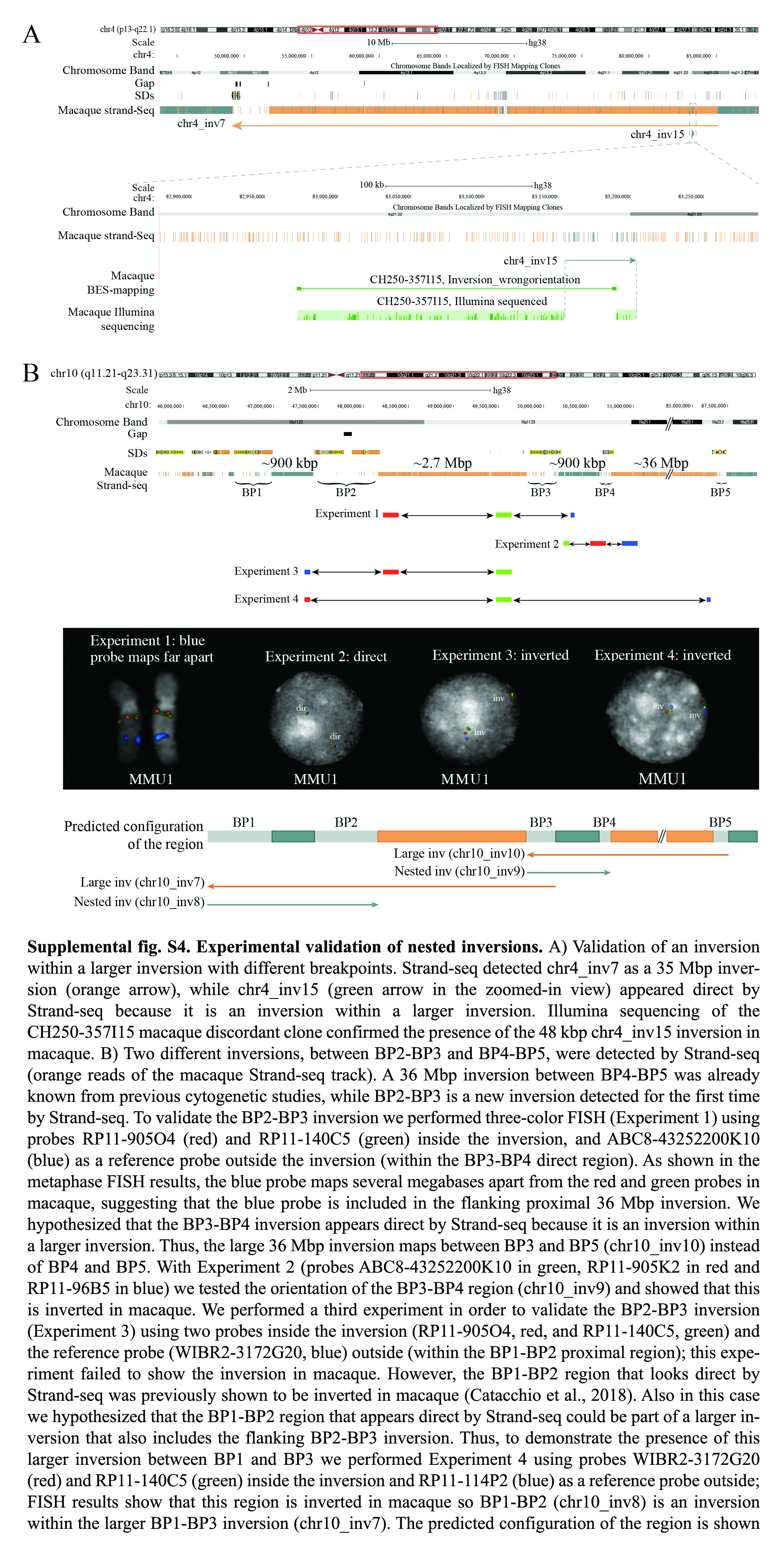{kind=link}
{kind=link}
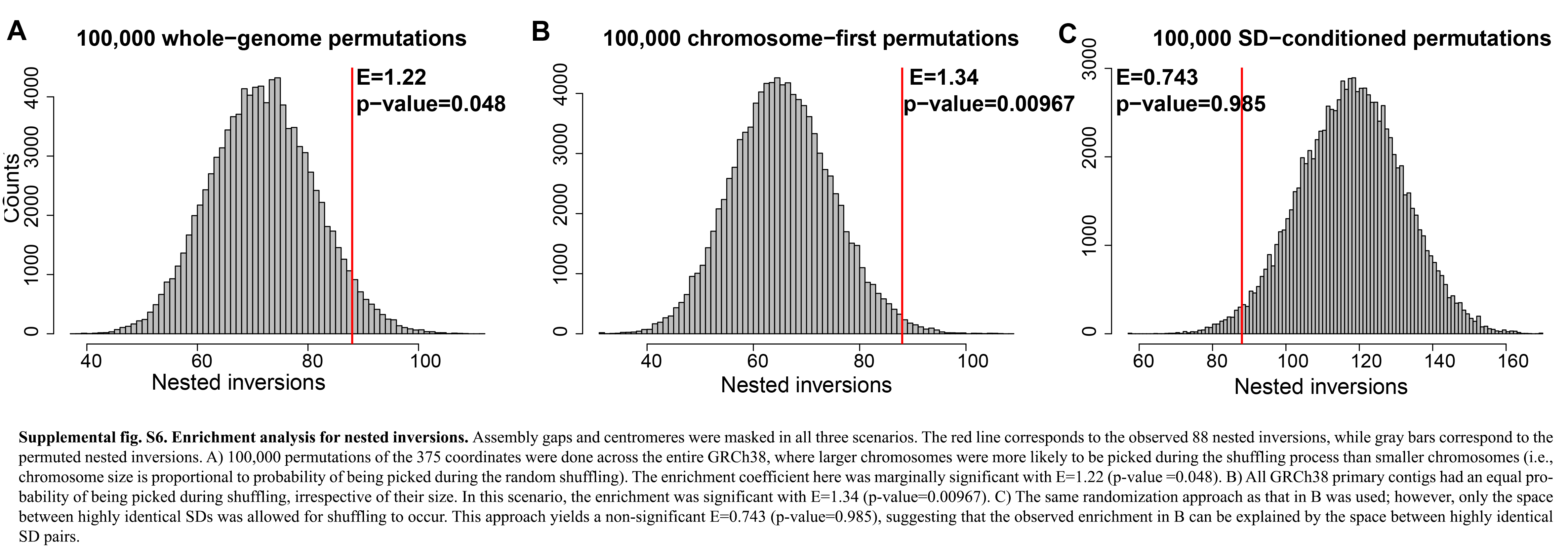{kind=link}
{kind=link}
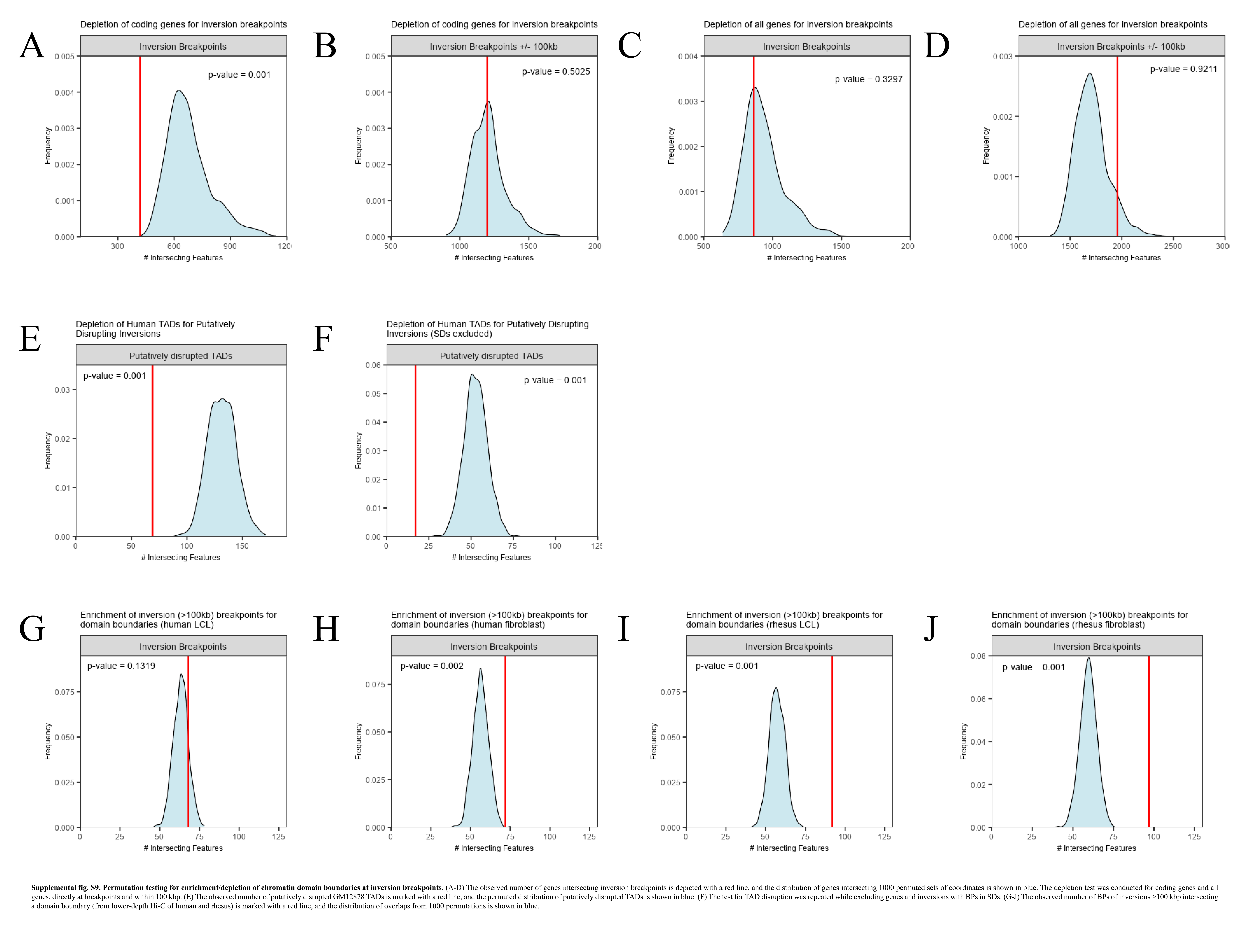{kind=link}
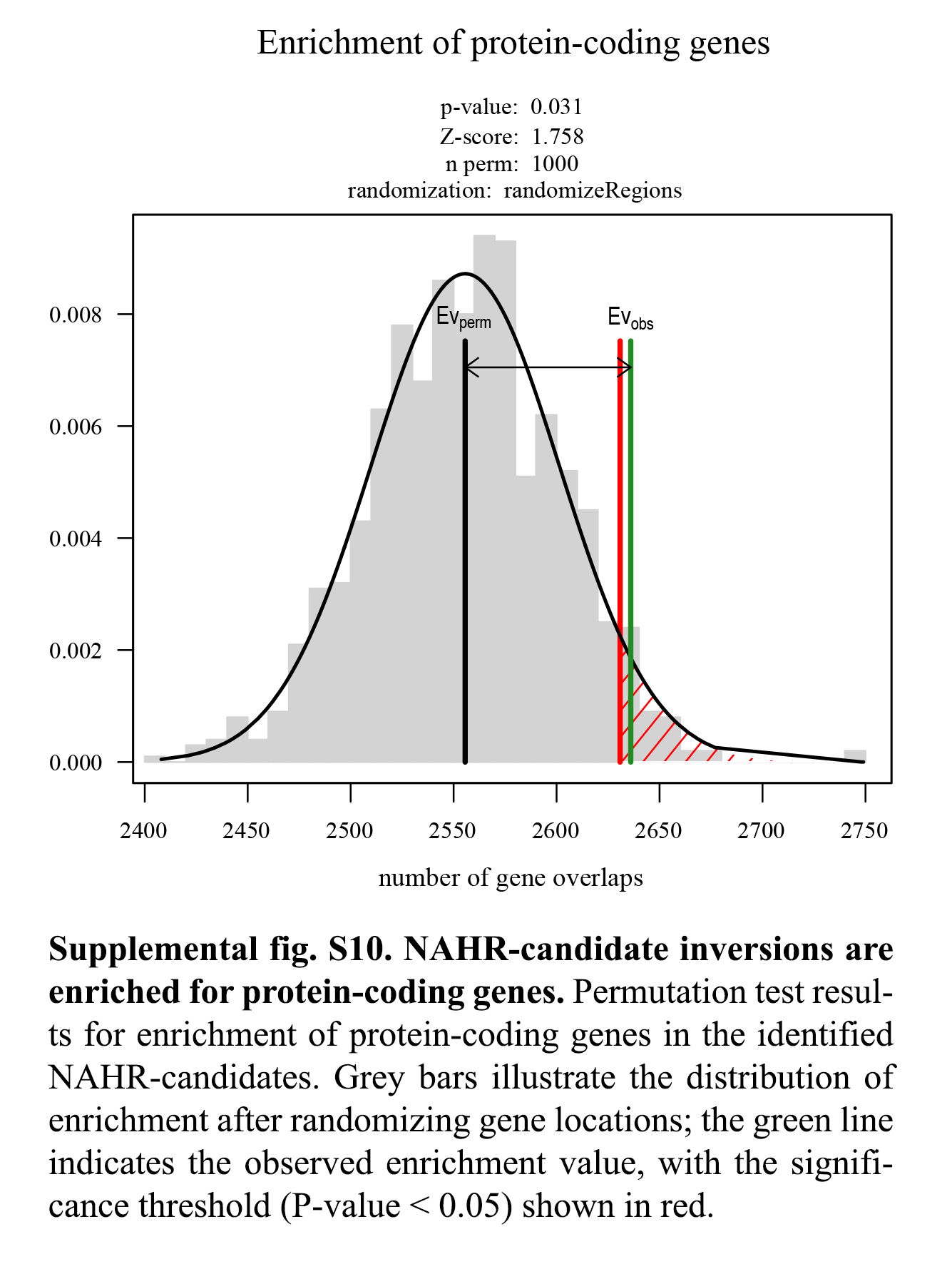{kind=link}
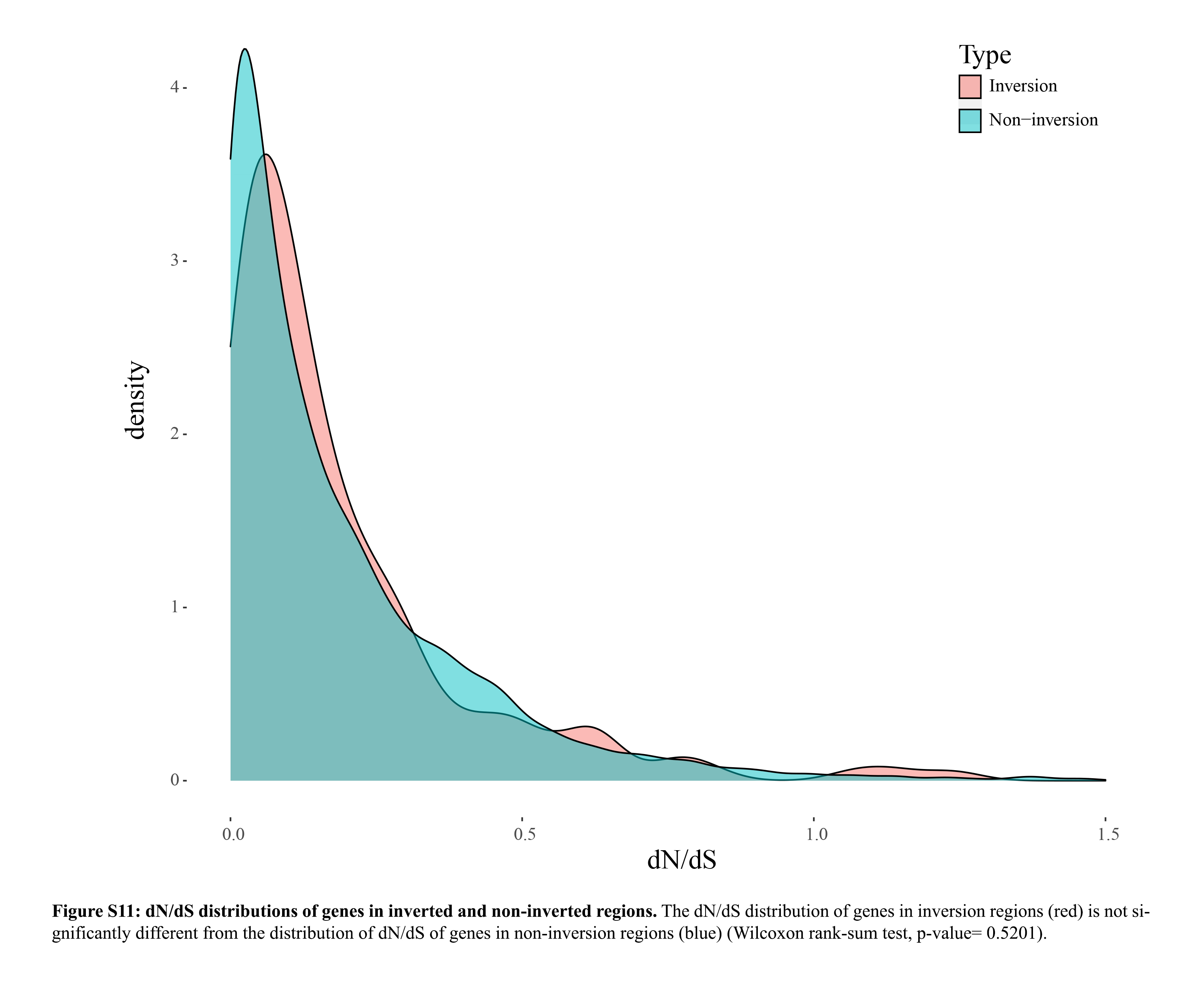{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
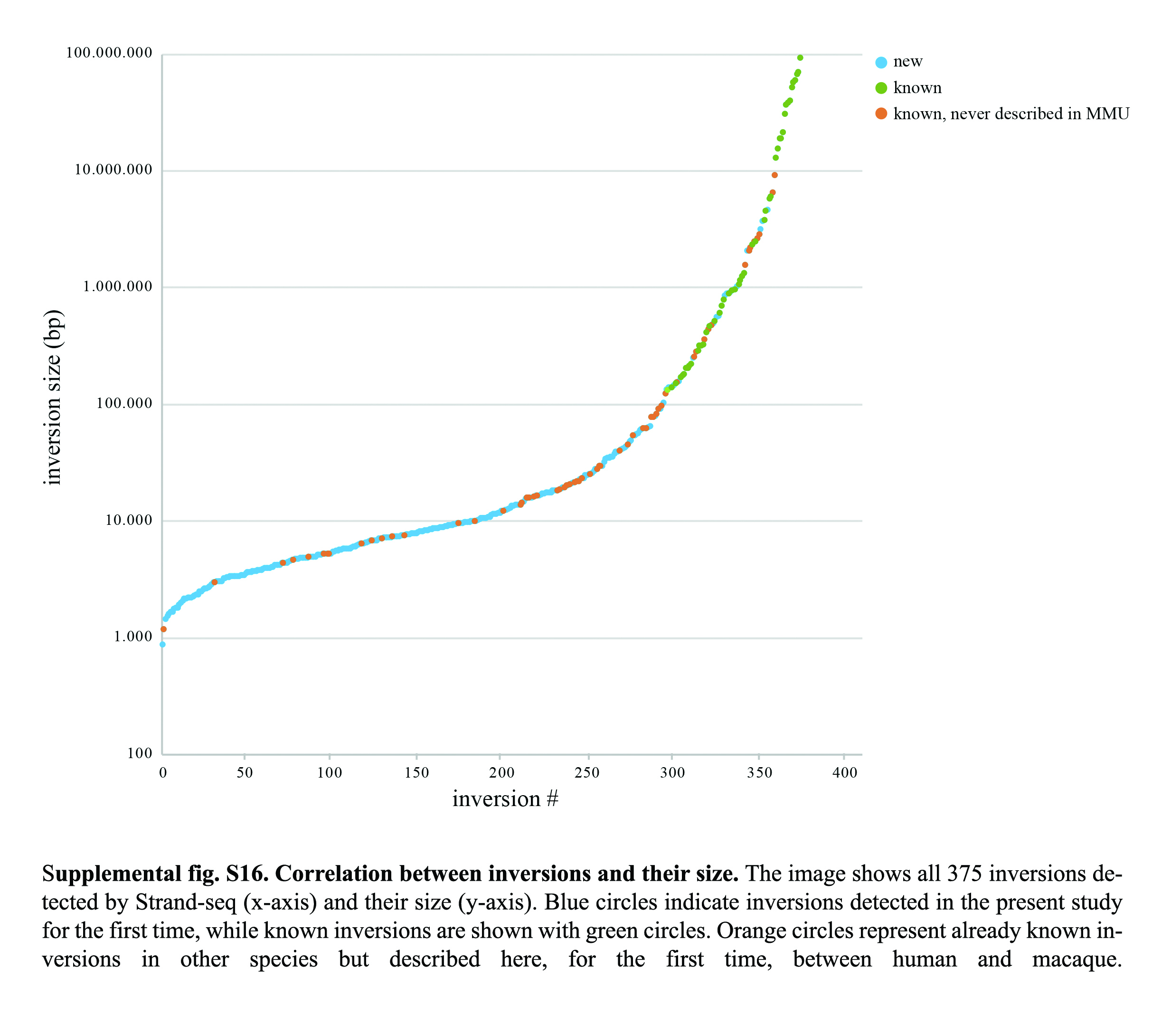{kind=link}