

Abstract
Gene expression is determined by a balance between RNA synthesis and RNA degradation. To elucidate the underlying regulatory mechanisms and principles of this, simultaneous measurements of RNA synthesis and degradation are required. Here, we report the development of “Dyrec-seq,” which uses 4-thiouridine and 5-bromouridine to simultaneously quantify RNA synthesis and degradation rates. Dyrec-seq enabled the quantification of RNA synthesis and degradation rates of 4702 genes in HeLa cells. Functional enrichment analysis showed that the RNA synthesis and degradation rates of genes are actually determined by the genes’ biological functions. A comparison of theoretical and experimental analyses revealed that the amount of RNA is determined by the ratio of RNA synthesis to degradation rates, whereas the rapidity of responses to external stimuli is determined only by the degradation rate. This study emphasizes that not only RNA synthesis but also RNA degradation is important in shaping gene expression patterns.
Gene expression is one of the most fundamental regulatory processes determining cellular conditions through regulating the level of proteins. The expression level of mature RNA is one of the major determinants of gene expression, which reflects the balance between the rate of RNA synthesis as the sum of transcription and maturation kinetics and that of RNA degradation (Rabani et al. 2011, 2014; Pérez-Ortín et al. 2013; De Pretis et al. 2015; Maekawa et al. 2015; McManus et al. 2015; Eser et al. 2016; Baptista and Dölken 2018; Kiefer et al. 2018; Duffy et al. 2019; Schmid et al. 2019). To obtain a comprehensive overview of the expression landscape of mature RNAs, the simultaneous quantification of RNA synthesis and degradation is required.
To date, several methods have been developed to quantify the transcription, splicing, and degradation of RNAs at the genome-wide level (Tani et al. 2012; Imamachi et al. 2014; Schwalb et al. 2016; Herzog et al. 2017; Baptista and Dölken 2018; Kiefer et al. 2018; Matsushima et al. 2018; Schofield et al. 2018; Duffy et al. 2019). For example, SLAM seq enables the quantification of RNA synthesis by in situ RNA labeling with 4-thiouridine (4sU) (Herzog et al. 2017). 4sU is alkylated in vitro after the isolation of total RNA from 4sU-labeled cells, which is followed by massively parallel sequencing analysis. Because a guanine (G) rather than an adenine (A) base pairs with alkylated 4sU during the reverse transcription reaction in preparation of the library for massive sequencing, the uridine (thymine [T] in DNA) replaced by 4sU in sequencing data is converted to a cytosine (C; T > C conversion). Bioinformatic detection of this T > C conversion enables 4sU-labeled RNAs to be distinguished from intracellular RNAs. Herzog et al. combined this base conversion with a polyadenylation-dependent 3′-end RNA sequence (QuantSeq) (Moll et al. 2014), which allows the rapid and quantitative access of mature RNA expression profiles, to quantify the comprehensive RNA synthesis rate. By measuring the T > C conversion in mature RNAs at sequential time points after 4sU labeling, RNA synthesis rates can be determined. BRIC-seq enables measurement of the degradation rates of the RNAs using in situ labeling of RNA with 5′-bromouridine (BrU) (Tani et al. 2012; Imamachi et al. 2014). The BrU-labeled RNAs chronologically isolated from cells prelabeled with BrU are immunoprecipitated, and then the immunoprecipitated RNAs are chased by massive sequencing to estimate the degradation rates of the RNAs. However, methods enabling simultaneous measurement of RNA synthesis and degradation by using multiple ribonucleoside analogs are not currently available.
To approach this issue, in this study, we developed a system to simultaneously measure actual RNA synthesis rates, defined as the number of polyadenylated RNA appearances per unit of time, and RNA degradation rates, defined as the ratio of RNA disappearance per unit time, at the genome-wide level by combining SLAM seq and BRIC-seq. We named this simultaneous measurement of RNA synthesis and degradation rates as “sequencing for RNA dynamics recording” (Dyrec-seq). Moreover, we determined the RNA synthesis and degradation rates in the human cervical cancer HeLa cell line by using Dyrec-seq.
Results
Sequential labeling of endogenous RNAs with BrU and 4sU
To quantify RNA synthesis and degradation rates simultaneously, we labeled HeLa cells with BrU followed by 4sU (Fig. 1A). The HeLa cells were precultured in BrU-containing medium for 12 h, and then RNAs were isolated at 0, 15, 30, 45, 60, 120, 240, 480, and 720 min after the BrU-containing medium had been changed to 4sU-containing medium without BrU (Total-RNAs). After 4sU was added and BrU was removed from the medium by changing the medium (time: 0 min), the abundances of BrU-labeled and 4sU-labeled RNAs decreased and increased, respectively, over time. The cell proliferation assay revealed that BrU and 4sU labeling at the concentrations used in this study did not affect the cell growth of HeLa cells (Supplemental Fig. S1A). The isolated RNAs were divided into two samples. One was immunoprecipitated with an anti-BrdU antibody to isolate BrU-labeled RNAs (IP-RNAs), and the other was treated with iodoacetamide (IAA) to alkylate 4sU residues in newly synthesized RNAs (Alkyl-RNAs) in vitro (Fig. 1A,B). Because the alkylated 4sU paired with guanines instead of adenines during the reverse transcription, the 4sU incorporated in the RNAs is detected as specific mutation (T > C conversion). The 3′ ends of RNAs within the Alkyl-RNAs and IP-RNAs were sequenced using QuantSeq, which enabled quantification of the mRNA expression by sequencing the sequences close to the 3′ end of polyadenylated RNA (Supplemental Fig. S1B).
Figure 1.

Simultaneous labeling of intracellular RNAs with BrU and 4sU. (A) Schematic illustration of the labeling procedure. HeLa cells were precultured in medium containing 150 μM BrU, and then the medium was changed to one containing 200 μM 4sU. Black, blue, and red lines indicate kinetics of expression of total RNAs, BrU-labeled RNAs, and 4sU-labeled RNAs, respectively. (B) Preparation and quantification of BrU-labeled and 4sU-labeled RNAs. Total RNAs were isolated and purified from labeled cells in time series and then divided into two samples. One was immunoprecipitated using an anti-BrdU antibody, and the cDNA was reverse-transcribed to be provided to QuantSeq (IP-RNAs). The other was alkylated using IAA, and the cDNA was reverse-transcribed to be provided to QuantSeq (Alkyl-RNAs). Because alkylated 4sU pairs with guanine (G) instead of adenine (A) during reverse transcription, the 4sU-labeled RNAs were identified as those including T > C conversions.
Quantification of newly synthesized and pre-existing RNAs
Subsequently, we estimated the numbers of newly synthesized and pre-existing RNAs at each time point based on the sequencing of the Alkyl-RNAs and IP-RNAs by QuantSeq. Because 4sU is incorporated into newly synthesized RNAs at a constant rate, the numbers of newly synthesized RNAs are in proportion to the numbers of reads containing T > C mutations. Because newly synthesized RNA is synthesized at a constant rate and degraded at a rate dependent on its concentration per unit time, the number of newly synthesized RNAs increases logarithmically. Fitting of this curve of logarithmic increase indicated that ∼35% of the newly synthesized RNAs are labeled with 4sU (see Methods). Therefore, we estimated the number of newly synthesized RNAs by correcting their count per million mapped reads (CPM) using the labeling efficiency (Supplemental Fig. S1C). Furthermore, we estimated the number of pre-existing RNAs using the total number of newly synthesized RNAs at each time point. Because the total number of intracellular RNAs can be considered as constant and independent of time, the sum of the number of newly synthesized and pre-existing RNAs should be constant. Thus, we estimated the number of pre-existing RNAs at each time point by subtracting the number of newly synthesized RNAs at each time point from 1 million (Supplemental Fig. S1D).
Extraction of RNAs in a steady state
In this study, we assumed that the expression of genes is in a steady state during labeling with modified ribonucleosides. However, some genes’ expression was changed after changing medium form BrU-containing medium to 4sU-containing medium. Because the cells were not treated with any specific stimuli, the altered expressed genes should be caused by medium change procedure, such as serum stimuli or mechanical stress. We excluded the altered expressed genes based on the statistical significance of their trends of expression (Supplemental Fig. S1E), comparing their empirical distribution with that of a randomized gene set with preserved expression level (see Methods). A statistical test indicated that, among 19,485 genes, 6830 (35.1%) were not expressed at all time points, 2203 (11.3%) were expressed differentially (FDR < 0.01), and 10,452 (53.6%) were in a steady state (FDR > 0.01) (Supplemental Fig. S1F). We extracted these 10,452 genes in a steady state as subjects of further analysis.
Simultaneous evaluation of RNA synthesis and degradation rates
Next, we estimated the RNA synthesis and degradation rates for individual genes simultaneously. The RNA synthesis rate of each gene is defined as the number of polyadenylated RNAs synthesized per unit time and calculated based on the time series of the number of newly synthesized RNAs estimated from the sequencing of the Alkyl-RNAs. The degradation rate of each RNA is defined as the ratio of the RNAs degraded per unit time calculated based on the time series of the number of pre-existing RNAs estimated from the sequencing of the IP-RNAs. Because the medium change removes BrU and adds 4sU, the numbers of reads derived from newly synthesized and pre-existing RNAs increase and decrease in RNA synthesis rate–dependent and degradation rate–dependent manners, respectively.
That is, the kinetics of the read numbers derived from newly synthesized RNAs (xt) and pre-existing RNAs (yt) at each time point is as follows:
where ks, kd,, α, β1, and β2 are the RNA synthesis rate, degradation rate, time lag, scaling factor, basal value for newly synthesized RNA-derived reads, and basal value for pre-existing RNA-derived reads, respectively (Fig. 2A–C). We estimated the RNA synthesis and degradation rates of individual genes at the genome-wide level by fitting the time series of the read numbers to these curves and extracting well-fitted RNAs based on the significance of correlation coefficients between read numbers and estimated values (see Methods). Because 4sU-labeled RNAs, particularly those with short half-lives, are being degraded as well as synthesized, the amount of 4sU-labeled RNAs does not directly reflect RNA synthesis rates. Our method of estimating RNA synthesis and degradation rates developed in this study enables the correction of the effects of degradation. As a result, we obtained the RNA synthesis and degradation rates for 4702 genes in HeLa cells (Supplemental Table S1). The estimated RNA synthesis rates obeyed a log normal-like unimodal distribution with a median of 1.17 × 10−1 CPM/min (Fig. 2D). The estimated degradation rates obeyed a log normal-like unimodal distribution with a median of 3.38 × 10−3 min−1, equivalent to a half-life of 205.0 min (Fig. 2E).
Figure 2.

Estimation of RNA synthesis and degradation rates of individual genes. (A) Kinetics model of gene expression. In this model, a transcript was synthesized according to the RNA synthesis rate, ks (CPM/min) and degraded according to the degradation rate, kd (min−1). (B) Synthesis rates were estimated by fitting of the time series of the amount of newly synthesized RNAs from individual genes to a logarithmically increasing curve (see Methods). β1 and t0 indicate the basal value of newly synthesized RNAs and the time delay, respectively. (C) Degradation rates were estimated by fitting of the time series of the amount of pre-existing RNAs from individual genes to an exponentially decreasing curve (see Methods). β2 indicates the basal value of pre-existing RNAs. (D,E) Distribution of estimated RNA synthesis rate (D) and degradation rate (E). (F,G) Gene Set Enrichment Analysis (GSEA) for genes with ks values (F) or with kd values (G).
Are these rates of RNA synthesis and degradation related to gene function? To approach this issue, we performed Gene Set Enrichment Analysis (GSEA) (Mootha et al. 2003; Subramanian et al. 2005) for the estimated RNA synthesis and degradation rates (Supplemental Table S2, S3). GSEA statistically tests the homogeneity of the ks values (synthesis rates) and kd values (degradation rates) of genes related to specific biological terms: A uniform distribution of the ks or kd values of genes related to a term makes the P-value larger, whereas a biased distribution of these values makes it smaller. For the RNA synthesis rates, GSEA detected that the genes showing only large ks values (fast RNA synthesis) are particularly related to several signaling pathways, such as MTOR complex 1 (MTORC1) signaling, TNFA signaling, TGFB signaling, and PI3K/AKT/MTOR signaling (Fig. 2F). For the degradation rates, GSEA detected that the genes showing only small kd values (slow degradation) are particularly related to several metabolic pathways, such as adipogenesis, xenobiotic metabolism, glycolysis, and fatty acid metabolism (Fig. 2G). TNFA signaling and MTORC1 signaling were related only to genes showing large kd values (fast degradation) and those with small kd values (slow degradation), respectively (Fig. 2G). An integrated interpretation of these functional analyses indicates the following: The RNAs involved in the inflammatory response (TNFA signaling via NF-kB, inflammatory response) are synthesized faster, and their transcripts are degraded faster; the genes involved in cell growth and survival (E2F targets, MYC targets [v1], MTORC1 signaling) are synthesized faster, but their transcripts are degraded slower (Fig. 2F,G). These results indicate that the rates of RNA synthesis and degradation are closely related to the biological function of the gene, and especially, signaling factors tend to be synthesized faster and metabolic enzymes tend to be degraded slower.
Comparison of RNA synthesis and degradation rates
To examine how the combination of RNA synthesis and degradation rates is related to the function and behavior of individual genes, we classified the 4702 RNAs into four classes based on their RNA synthesis and degradation rates; fast RNA synthesis and fast degradation (Class I, 277 genes), fast RNA synthesis and slow degradation (Class II, 372 genes), slow RNA synthesis and fast degradation (Class III, 291 genes), and slow RNA synthesis and slow degradation (Class IV, 229 genes) (Supplemental Table S4). To avoid the effects of estimation error, first and third quartiles of RNA synthesis rates (0.280 and 0.048 CPM/min, respectively) and those of degradation rates (0.006 and 0.002 min−1, respectively) were adopted as thresholds of the classification. To examine the relationship of the combination of RNA synthesis and degradation rates with the biological function of the genes, we performed functional enrichment analysis of the genes included in each class. This analysis of the genes using the DAVID tool (Huang et al. 2009a, 2009b) provided several functional terms significantly enriched in the individual classes (Fig. 3A,B; Supplemental Table S5). In Class I (fast RNA synthesis and fast degradation), the terms related to signaling pathways such as “serine/threonine-protein kinase” and those related to DNA repair were significantly enriched. In Class II (fast RNA synthesis and slow degradation), the terms were related to posttranscriptional regulation such as “mRNA splicing,” “mRNA processing,” and “RNA-binding.” The terms related to some signaling pathways such as NF-kB signaling and Wnt signaling were also significantly enriched in Class II. The terms “alternative splicing” and “phosphoprotein” (indicating proteins to be phosphorylated) were significantly enriched in both Class I and Class II (classes of genes with fast RNA synthesis). The terms of transcriptional regulation including “zinc-finger” were significantly enriched in both Class I and Class III (classes of genes with fast degradation). The terms “acetylation” and “mitochondrion” were significantly enriched in both Class II and Class IV (classes of genes degraded slowly).
Figure 3.

Joint distribution of RNA synthesis and degradation rates. (A) Joint distribution of RNA synthesis and degradation rates. Histograms of individual rates and a scatter plot are shown. The x- and y-axes indicate RNA synthesis rate (ks) and degradation rate (kd), respectively. The first and third quartiles of RNA synthesis and degradation rates indicated with black dashed lines were adopted as thresholds of the classification. (B) GO enrichment analysis of the genes classified using the RNA synthesis and degradation rates. Only representative terms significantly enriched for each class are shown.
In summary, functional enrichment analysis suggested the following. First, the RNAs related to signaling are generally synthesized rapidly, which is consistent with the results of GSEA, but the rapidity of degradation varies depending on the signaling pathways. Second, the genes related to posttranscriptional regulation such as splicing are synthesized rapidly and degraded slowly. Third, the genes related to transcriptional regulation are degraded rapidly, whereas those encoding proteins to be acetylated are degraded slowly. These results show that genes have optimized regulation of transcription and degradation according to their functions and physiological roles.
Combination of RNA synthesis and degradation rates determines expression level
Theoretically, the expression level of a gene in a steady state is determined by the ratio of its synthesis and degradation rates (Hargrove and Schmidt 1989). Thus, we examined how the RNA synthesis and degradation rates of individual genes affect the expression level using these estimated rates.
Generally, the expression rate of genes whose regulation is expressed as per the model in Figure 2A is described as follows:
where x, t, ks, and kd represent the expression level, time, RNA synthesis rate, and degradation rate of an RNA, respectively. Because the expression level does not change during a steady state,
Thus, the expression level of the RNA in a steady state is described as
To verify the appropriateness of the theoretical prediction, we compared the experimentally estimated ks and kd values with the expression levels of individual genes estimated based on the sequencing of the Alkyl-RNAs. As expected, we observed strong positive correlation between the ratios of ks and kd values (ks/kd) and the expression levels (Pearson's correlation of R2 = 0.85, P-value < 0.01) (Fig. 4A), suggesting that the combination of RNA synthesis and degradation rates determines the expression levels of individual genes. This indicates that not only regulation of RNA synthesis but also the regulation of degradation is an important factor determining the level of gene expression.
Figure 4.

Effect of RNA synthesis and degradation rates on behavior of gene expression. (A) Comparison of ratios of RNA synthesis and degradation rates, with expression levels. The x- and y-axes indicate the ratio of RNA synthesis and degradation rates (ks/kd) and the expression level calculated based on the sequencing of Alkyl-RNAs (total RNA-seq reads with/without T > C conversion), respectively. Black solid line is the regression line based on log-transformed values. (B) Simulations of expression time series of genes imitating the characteristics of Class I (fast RNA synthesis and fast degradation), Class II (fast RNA synthesis and slow degradation), Class III (slow RNA synthesis and fast degradation), and Class IV (slow RNA synthesis and slow degradation). In each simulation, the RNA synthesis rates doubled at 0 min (red dotted line). (C) Time constants of expression of genes showing various RNA synthesis and degradation rates. The x- and y-axes indicate RNA synthesis rate (ks) and degradation rate (kd). Colors indicate the time constants (τ). (D,E) GSEA of degradation rate (D) and RNA synthesis rate (E) showing enrichment of “estrogen response (early)” and “estrogen response (late).” (NES) Normalized enrichment score. (F) Comparison of degradation rates (kd) and time constants (τ). The x- and y-axes indicate the degradation rate and time constants, respectively. Red dashed line is a regression line based on log-transformed values. (G) Comparison of RNA synthesis rates (ks) and time constants (τ). The x- and y-axes indicate the RNA synthesis rate and time constants, respectively. Red dashed line is a regression line based on log-transformed values.
Degradation rate is a key factor determining the rapidity of response
Finally, to examine how the RNA synthesis and degradation rates affect the behavior of individual RNAs (Koh et al. 2019), we simulated RNA expression dynamics in response to a change of RNA synthesis using the mathematical model shown in Figure 2A and numerically analyzed the behavior of RNA expression. The dynamics of gene expression was simulated independently for four classes whose ks and kd values imitate the representative values of those in Classes I to IV. In this simulation, the RNA synthesis rate doubled at 0 min. The rapidity of expression was evaluated using a time constant (τ) defined as the time when RNA expression reaches 1 − e−1 (≈ 63.2%) of the final value (Supplemental Fig. S1G). Theoretically, the time constant (τ) is represented as follows:
depending on not the RNA synthesis rate but only the degradation rate, indicating that the time constants are larger for the genes in the classes whose degradation is fast, such as Classes I and III, and they are smaller for the RNAs in the classes whose degradation is slow, such as Classes II and IV. In accordance with the theoretical prediction, the time constants simulated in Classes I and III (fast degradation) are smaller than those simulated in Classes II and IV (slow degradation) (Fig. 4B). Moreover, the time constants were determined depending on kd, not on ks (Fig. 4C). These results suggest that the degradation rate of each gene is a key factor determining not only their expression level but also their rapidity of differential expression caused by changes in the extracellular environment.
To confirm the above idea experimentally, we compared the estimated degradation rates with the time constants calculated based on previously published RNA-seq data. The GSEA for the degradation rates indicated that, although “estrogen response (early)” was significantly enriched for the large kd values (fast degradation), “estrogen response (late)” was significantly enriched for the small kd values (slow degradation) (FDR < 0.01) (Fig. 4D). In contrast, the GSEA for the RNA synthesis rates showed a uniform distribution of those terms (Fig. 4E). This indicates that the products of most genes related to late periods in estrogen response are degraded slowly, suggesting that the rapidity of expression of genes in response to estrogen is determined by their degradation rates. Note that because estrogen-responsive genes include those with both fast and slow degradation, as described above, Gene Ontology (GO) terms related to estrogen response were not significantly enriched in any class (Fig. 3B). Because estrogen receptor is a nuclear receptor directly regulating transcriptional activity and its target genes might not be affected by the rapidity of signal transduction, the rapidity of expression of genes in response to estrogen is likely to reflect the degradation rates of the products of individual genes. Therefore, we calculated time constants from the time series of RNA-seq of human breast cancer–derived MCF-7 cells stimulated with estradiol (E2), a type of estrogen (Baran-Gale et al. 2016), and compared those with the estimated degradation rates. To avoid the effect of outliers of estimated expression levels, we redefined the time constant (τ) as the time when RNA expression reaches half of the final value.
As expected, we observed a significant negative correlation between the degradation rates and time constants (Pearson's correlation of r = −0.326, P-value = 1.65 × 10−17) (Fig. 4F), whereas the RNA synthesis rates were not significantly correlated (Pearson's correlation of r = 0.033, P-value = 0.40) (Fig. 4G). This suggests that the rapidity of the response of gene expression to estrogen stimulation is at least partially determined in a manner dependent on the degradation rate. However, some genes showed extremely fast or slow time constants independent of their degradation rates, suggesting that the rapidity of expression of such genes affects secondary signal transduction and/or other signaling pathways.
Discussion
Recent developments in procedures to unravel the kinetics of gene expression at the genome-wide level provide several insights into the regulation of gene expression. For instance, BRIC-seq (Tani et al. 2012; Imamachi et al. 2014) enables comprehensive clarification of the regulation of degradation through chase experiments of BrU-labeled RNAs. Moreover, SLAM seq (Herzog et al. 2017) enables comprehensive clarification of RNA synthetic regulation through the quantification of 4sU-labeled RNAs by identifying IAA-induced T > C mutations. In this study, we developed “Dyrec-seq,” a system to simultaneously measure RNA synthesis and degradation rates of the comprehensive set of RNAs in HeLa cells, by chasing BrU- and 4sU-labeled RNAs. It provided simultaneous measurement of the RNA synthesis and degradation rates of 4702 genes in HeLa cells. Moreover, functional enrichment analysis of the genes classified by the synthesis and degradation rates showed that these rates of genes related to various cellular functions are regulated in common, suggesting that the synthesis and degradation rates of individual genes are closely associated with their biological functions. To examine how such regulation is associated with the biological roles of the genes, we constructed a mathematical model. This theoretical approach indicated that (1) the ratios of synthesis rates to degradation rates of individual genes determine their expression levels, and (2) the degradation rate is a key factor determining the rapidity of expression of each gene. Taking these results together, the following insights can be obtained (Fig. 5A,B): (1) Posttranscriptional factors, including mainly RNA-binding proteins in Class II, are constitutively expressed constitutively at extremely high levels but respond slowly to extracellular stimuli; (2) transcription factors in Classes I and III show various expression levels but respond rapidly to extracellular stimuli; (3) genes encoding phosphorylated proteins, mainly including signaling factors in Classes I and II, are generally constitutively expressed at extremely high levels; and (4) genes encoding proteins under posttranslational regulation, such as acetylation in Class II and IV, that are degraded slowly and do not respond rapidly to extracellular stimuli in terms of gene expression levels, likely show posttranslational regulation. Terms related to signaling pathways, such as NF-kB signaling and Wnt signaling, and serine/threonine protein kinases were not significantly enriched in genes with average transcription and degradation factors. This indicates that the synthesis and degradation rates of genes involved in the signaling pathways are strictly regulated to ensure high expression, enabling rapid responses to posttranslational regulation such as phosphorylation. These insights explain at least in part how the differences of expression levels are regulated and why the responses to extracellular stimuli differ according to the function of the genes. Simultaneous measurement of RNA synthesis and degradation rates using Dyrec-seq provides an integrated understanding of the functions of genes and their behavior of expression.
Figure 5.

Effect of RNA synthesis and degradation rates on behavior of gene expression. (A) The combination of the rates of RNA synthesis and degradation regulates the temporal expression profiles. The ratio of the RNA synthesis and degradation rates determines the expression level (diagonal arrow), and the degradation rate affects the rapidity of response of gene expression (vertical arrow). (B) Schematic diagram of temporal expression profiles. The temporal expression profiles correspond to the points in A.
To measure the RNA synthesis and degradation rates simultaneously, we labeled the intracellular RNAs using two uridine analogs, BrU and 4sU. Because it is known that uridines are particularly abundant in the 3′ untranslated region (3′ UTR) in RNAs (Shaw and Kamen 1986; Chen and Shyu 1995; Barreau et al. 2005; Vlasova and Bohjanen 2008; Vlasova et al. 2008; Gruber et al. 2011), we used QuantSeq, which specifically detects 3′ UTR using oligo(dT) primer (Moll et al. 2014), to ensure efficient detection. The efficiency of incorporation of nucleotide analogs and the effects of cellular functions might be influenced by the cell type and conditions. Thus, there is a need to verify effects specific to cell types sufficiently to identify labeling conditions that do not influence the gene expression of cells. Moreover, the ability to estimate the RNA synthesis and degradation rates depends on the inherent kinetics of gene expression and library sequencing depth. Therefore, consideration of these parameters is needed to design experiments effectively. Additionally, not only RNA synthesis, but also RNA degradation, is under control of the cell cycle. Because we used unsynchronized cells in this study, further examination is necessary to evaluate RNA synthesis and degradation rates of cell cycle–regulated genes.
Further extension of Dyrec-seq with other kinds of nucleotide analogs would enable the evaluation of additional types of regulation such as splicing and processing (Windhager et al. 2012; Barrass et al. 2015). Moreover, this procedure can be used for understanding the temporal contributions of multiple types of regulation such as RNA synthesis and degradation during transient or continuous changes of gene expression. Recent studies suggested that the regulation of both RNA synthesis and degradation can affect the dynamics of gene expression (Alonso 2012). For example, upon regulation of the gene expression profile in the long term, such as occurs in cell differentiation, the contribution of mechanisms regulating RNA synthesis and degradation might change depending on the stage of differentiation.
In conclusion, we propose the concept of “RNA dynamics recording” to encode the dynamics of RNAs onto the RNAs themselves by using multiple ribonucleoside analogs. This RNA dynamics recording enables clarification of the temporal contributions of multiple forms of regulation independently. Therefore, this approach should provide substantial insights into how gene expression is regulated during differentiation or by extracellular stimuli such as hormones and cytokines.
Methods
Metabolic labeling of endogenous RNAs and RNA isolation
HeLa cells (female, RRID: CVCL_0030) were seeded at a density of 6 × 105 cells per 6-cm dish (Thermo Fisher Scientific) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic at 37°C. In the next day, the medium was replaced with fresh medium containing 150 μM BrU, and then the cells were precultured for 12 h to label pre-existing RNAs. After the preculture, the cells were washed twice with the medium, and then the medium was replaced with one containing 200 μM 4sU to label newly synthesized RNAs until 12 h. After incubating cells in the 4sU-containing medium, the cells were washed twice with PBS, and then total RNA was isolated using RNAiso Plus (TAKARA).
Sample preparation
To quantify 4sU-labeled and BrU-labeled RNAs individually, the isolated total RNAs were divided into two samples, as follows.
RNA samples to quantify 4sU-labeled RNAs
From the isolated total RNAs, 5 μM RNA was alkylated using SLAM seq kinetics kit (Lexogen), in accordance with the manufacturer's instructions. Hereafter, the alkylated RNA samples are referred to as “Alkyl-RNAs.” The quality of Alkyl-RNAs was assessed using Agilent RNA nano 6000 kit (Agilent) on the Agilent Bioanalyzer 2100 (Agilent).
RNA samples to quantify BrU-labeled RNAs
To normalize the quantity of BrU-labeled RNAs among the time points during preanalysis, 1.0 ng of BrU-labeled luciferase RNA was added to 10 μg of extracted total RNA as a spike-in control to serve as an internal standard. The RNA mixtures were diluted to a final volume of 100 μL with TE buffer (a final concentration of 10 mM Tris–HCl at pH 7.0 and 1.0 mM EDTA). BrU-labeled RNAs were immunoprecipitated using anti-BrdU antibody-conjugated beads (MBL, clone 2B1), in accordance with previous reports (Tani et al. 2012; Imamachi et al. 2014). Briefly, the BrU-labeled RNAs were added to the antibody-conjugated protein G agarose suspended with 100 μL of ice-cold PBS containing 0.1% bovine serum albumin (BSA), 1% Triton X-100, 100 U of RNasin plus RNase inhibitor (Promega), and 5 mg/mL heparin. BrU-labeled RNAs were isolated using ISOGEN LS (NIPPON GENE), in accordance with the manufacturer's instructions. Because the amount of purified BrU-labeled RNAs was low, 60 μg of glycogen was added to the mixture during the precipitation of RNA. Hereafter, the immunoprecipitated RNA samples are referred to as “IP-RNAs.” The quality of IP-RNAs was assessed using Agilent RNA nano 6000 kit (Agilent) on the Agilent Bioanalyzer 2100 (Agilent).
Cell proliferation assay
Cell proliferation was determined by measuring intracellular levels of NADH using the cell counting kit-8 (DOJINDO). The absorbance at 450 nm was measured using a GloMax discover microplate reader (Promega).
Sequencing
The 3′ ends of RNAs within the Alkyl- and IP-RNAs were sequenced using the QuantSeq 3′ mRNA-seq library prep kit (Lexogen), in accordance with the manufacturer's instructions. Briefly, 5.0 ng from each Alkyl- or IP-RNA sample was used for reverse transcription. The 4sU incorporated into the Alkyl-RNAs paired with guanines instead of adenines during the reverse transcription. The RNA was subsequently removed, and second-strand synthesis was initiated by a random primer, containing Illumina-compatible linker sequences and appropriate in-line barcodes, followed by magnetic bead–based purification. The resulting library was amplified using PCR with 12 cycles for Alkyl-RNAs and 18 cycles for IP-RNAs, and then purified using AMPure XP. Library quality and quantity were assessed on a Bioanalyzer using DNA high sensitivity kit reagents (Agilent Technologies). Standard Illumina protocols were used to generate 100-bp end read libraries that were sequenced on the HiSeq 3000 platform (Illumina).
Quantification of 4sU- and BrU-labeled RNAs
The newly synthesized and pre-existing polyadenylated RNAs were quantified based on the sequence data obtained from the Alkyl- and IP-RNAs for each time point by using SlamDunk v0.3.3, a pipeline for SLAM seq data analysis, with the default parameters (Herzog et al. 2017). Because QuantSeq targets mainly the 3′ end of individual RNAs in a poly(A) tail-dependent manner, the sequence data were aligned on genome-wide 3′ UTR sequences generated based on the human genome sequence and annotation data (GRCh38) obtained from the Ensembl database (release 92). Twelve bases from the 5′ end were trimmed as adaptor-clipped reads, and then four or more subsequent adenines from the 3′ end were regarded as the remaining poly(A) tail and removed. Multiply mapped reads were allowed up to 100 of regions. In VarScan 2.4.1 (Koboldt et al. 2012), included in the SlamDunk tool, an SNP was called in the case of a mismatch exceeding a variant fraction of 0.8 and a coverage cutoff of 10-fold. Through these filters, we calculated the total number of reads and the number of those including non-SNP T > C conversions aligned on the 3′ UTR of individual genes. Because 4sU pairs with guanine (G) during reverse transcription, the 4sU-labeled reads were identified as those including T > C conversion. Because one read is generated from one RNA, the total number of reads and the number of those including T > C conversions correspond to the numbers of total RNAs and 4sU-labeled RNAs, respectively. Therefore, the numbers of reads including T > C conversion from Alkyl-RNAs and those of reads from IP-RNAs were counted as the numbers of 4sU-labeled and BrU-labeled RNAs, respectively, and normalized to CPM.
Identification of RNAs in a steady state
Although we did not stimulate the cells in this study and the genes should have been in a steady state, we observed that some of the genes showed differential expression. Because the expression of a gene in a steady state changes dependent on only white noise, the sum of angles formed by the lines connecting each time point is relatively small, whereas the sum of angles in a differentially expressed gene (DEG) with a constant trend is relatively large (Supplemental Fig. S1E). Therefore, we designed a filter to exclude the DEGs with constant trends. First, we calculated the sum of the angles formed by the lines connecting certain time points and neighboring ones, within a time series of total read numbers for each gene calculated from Alkyl-RNAs. Second, we generated an empirical distribution of the sum of angles by rearranging all time points randomly. Finally, we calculated the probability that the sum of angles in the empirical distribution was larger than the actual value (empirical P-value). We calculated FDR from the empirical P-values using Storey's procedure (Storey et al. 2004). Among the genes expressed at all time points, those with FDR values greater than 0.01 were identified as genes in a steady state.
Correction of newly synthesized and pre-existing RNA numbers
Because the 4sU added to the medium labels only part of newly synthesized RNAs owing to the incorporation efficiency, we estimated the incorporation efficiency based on the time series of total 4sU-labeled RNAs. In a steady state, constant copies of 4sU-labeled RNAs are generated, and their level decreases at a constant rate per unit time. Therefore, the time series of 4sU-labeled RNAs in CPM obeys the following:
where x, , and indicate the number of 4sU-labeled RNAs in CPM, the RNA synthesis rate, and the degradation rate of the total 4sU-labeled RNAs, respectively. In addition, this time series asymptotically approaches the ratio of to (). Because the number of newly synthesized RNAs in CPM can approximate 106, the incorporation efficiency (η) can be presented as
Therefore, the numbers of newly synthesized RNAs of total and individual genes are calculated by dividing the number of 4sU-labeled RNAs in CPM by the estimated incorporation efficiency.
Moreover, we estimated the number of pre-existing RNAs at each time point. Because the number of intracellular RNAs in a steady state can be regarded as constant, the total number of pre-existing RNAs in CPM can be determined by subtracting the total number of newly synthesized RNAs in CPM from 1 million. Therefore, the number of pre-existing RNAs of individual genes was calculated by correcting the number of BrU-labeled RNAs of individual genes in CPM by the total number of pre-existing RNAs.
Estimation of RNA synthesis and degradation rates
The numbers of newly synthesized and pre-existing RNAs increase and decrease, respectively, in a time-dependent manner, according to the RNA synthesis and degradation rates. Therefore, we estimated the RNA synthesis and degradation rates simultaneously by fitting the time series of the estimated newly synthesized and pre-existing RNA levels to
and
where ks, kd, α, t0, β1, and β2 indicate the RNA synthesis rate, degradation rate, scaling factor, time delay, basal value for newly synthesized RNAs, and basal value for pre-existing RNAs of individual genes, respectively. The scaling factor is incorporated as the number of pre-existing RNAs at 0 min. The time delay is incorporated as the time when the newly synthesized RNAs begin to be synthesized after 4sU addition. The basal value for newly synthesized RNAs is incorporated as the number of RNAs including the inherent T > C mutant. The basal value for pre-existing RNAs is incorporated as the background of immunoprecipitation of BrU-labeled RNAs.
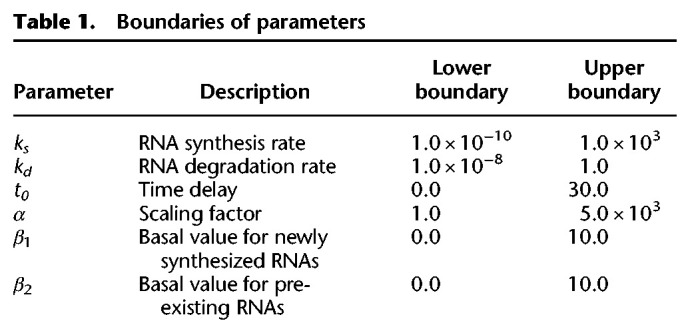
The fittings were performed with the combination of an evolutionary algorithm (genetic algorithm) and hill climbing (L-BFGS-B algorithm) with evaluation by the least squares method in Python 2.7. The genetic algorithm was implemented using the DEAP library with a generation number of 200, population number of 50, crossover probability of 0.5, and mutation probability of 0.2. The boundaries of parameters are shown in Table 1. The L-BFGS-B algorithm was implemented using the minimize module in the SciPy package, in which the parameters estimated by the genetic algorithm are given as initial parameters. The fitness in each gene was evaluated as the correlation of actual newly synthesized and pre-existing RNA levels with estimated values. The probability of the null hypothesis that a population correlation coefficient is equivalent to zero was calculated for each gene using the OLS module in the StatsModels package, and the RNA synthesis and degradation rates of the genes whose FDR as determined by Storey's procedure (Storey et al. 2004) was less than 10−5 were extracted.
Table 1.
Boundaries of parameters
Gene Set Enrichment Analysis
The functions of genes and their associations with the estimated RNA synthesis and degradation rates were independently examined using GSEA 4.0.3. We set one as the control value for individual genes and sorted the genes by the ratio of the ks or kd values to the control values (i.e., the original ks or kd values). The enrichment scores (ESs) for the terms included in GSEA hallmark were calculated using the default parameters, and empirical P-values were calculated by comparing those with the distributions of ES values from 10,000 randomized gene sets. The empirical P-values were corrected as FDR. The terms with FDRs of less than 0.05 were considered as significantly enriched.
Classification of genes according to RNA synthesis and degradation rates
Genes were classified according to associated RNA synthesis and degradation rates. To avoid the effects of estimation error, first and third quartiles of RNA synthesis rates and degradation rates were adopted as thresholds of the classification. The first and third quartiles were calculated for the RNA synthesis rate (ks) and degradation rate (kd) independently: Qs1, first quartile of ks values; Qs3, third quartile of ks values; Qd1, first quartile of kd values; and Qd3, third quartile of kd values. By using these thresholds, we classified the genes into four classes: Class I, fast RNA synthesis (ks > Qs3) and fast degradation (kd > Qd3); Class II, fast RNA synthesis and slow degradation (kd < Qd1); Class III, slow RNA synthesis (ks < Qs1) and fast degradation; and Class IV, slow RNA synthesis and slow degradation.
Functional enrichment analysis
The biological functions of the genes classified by the RNA synthesis and degradation rates were statistically determined using the DAVID tool v6.8 (Huang et al. 2009a,b; https://david.ncifcrf.gov/), by examining the GO categories of biological process (GOTERM_BP_DIRECT), cellular component (GOTERM_CC_DIRECT), and molecular function (GOTERM_MF_DIRECT), as well as UniProt keywords (UP_KEYWORDS). The P-values of enrichment were calculated by modified Fisher's exact test (Fisher 1922; Huang et al. 2009a, 2009b). The whole human genome (Homo sapiens) was used as a background (default). The biological functions whose FDR values were less than 0.01 were identified as those that were significantly enriched.
Estimation of time constants from published database
The time constants (τ) were calculated as a criterion of the rapidity of the gene expression response, defined as the time when the change in expression level reaches a particular magnitude (Supplemental Fig. S1G). In the mathematical model, τ was defined as the time when the change in expression level reaches 1 − e−1 (≈63.2%) of the final value. For experimental validation, a prequantified data set of RNA-seq of human breast cancer–derived MCF-7 cells stimulated with E2 (Baran-Gale et al. 2016) was downloaded from NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE78169. This prequantified RNA-seq data set consists of the expression levels at 0, 1, 2, 3, 4, 5, 6, 8, 12, and 24 h after E2 stimulation with triplicate data for individual time points. First, the significance of the changes of the expression levels at each time point after stimulation relative to those without stimulation (0 h) was tested by Welch's t-test using the ttest_ind function of the stats module in SciPy library (Virtanen et al. 2020). The genes whose expression levels were significantly changed in at least one time point were extracted as DEGs. For each DEG, we calculated the τ, redefined as the time when the expression reaches half of the final expression level to avoid the effect of outliers of estimated expression levels. The time constants whose identifiers were replaced with Ensembl Gene ID from NCBI gene ID based on db2db on the bioDBnet web service (Mudunuri et al. 2009; https://biodbnet-abcc.ncifcrf.gov/) were compared with the estimated RNA synthesis and degradation rates.
Data access
The high-throughput sequencing data of Alkyl- and IP-RNAs generated in this study have been submitted to the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA; https://ddbj.nig.ac.jp/DRASearch/) under accession number DRA008497.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank our laboratory members for critically reading this manuscript and for their technical assistance with the experiments. We also thank Kouta Watanabe, Kiyomi Imamura, Terumi Horiuchi, and Yuta Kuze for performing NGS experiments and analysis. The computational analysis of this work was performed on the National Institute of Genetics (NIG) supercomputer system at Research Organization of Information and Systems (ROIS). This manuscript was edited by Edanz (www.edanzediting.co.jp). This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 17KK0163, 18H02570, 18KT0016, 16H06279, and 20H04838). K.K. received funding from JSPS KAKENHI (grant number 19K16635) and Takeda Science Foundation. T.Y. received the JSPS Research Fellowship for Young Scientists and is supported by a grant-in-aid for JSPS fellows (17J11266). M.S. was supported by JSPS KAKENHI (grant number 16H06279 [PAGS] and 19K16108). Y.S. was supported by JSPS KAKENHI (grant number 16H06279 [PAGS]).
Author contributions: K.K. and N.A. conceived the project. H.W., H.H., and N.A. designed and performed the experiments. K.K., T.Y., and K.T. analyzed the data. M.S. and Y.S. performed the RNA-seq experiments. K.K, T.Y., K.T., and N.A. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.264408.120.
References
- Alonso CR. 2012. A complex “mRNA degradation code” controls gene expression during animal development. Trends Genet 28: 78–88. 10.1016/j.tig.2011.10.005 [DOI] [PubMed] [Google Scholar]
- Baptista MAP, Dölken L. 2018. RNA dynamics revealed by metabolic RNA labeling and biochemical nucleoside conversions. Nat Methods 15: 171–172. 10.1038/nmeth.4608 [DOI] [PubMed] [Google Scholar]
- Baran-Gale J, Purvis JE, Sethupathy P. 2016. An integrative transcriptomics approach identifies miR-503 as a candidate master regulator of the estrogen response in MCF-7 breast cancer cells. RNA 22: 1592–1603. 10.1261/rna.056895.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrass JD, Reid JEA, Huang Y, Hector RD, Sanguinetti G, Beggs JD, Granneman S. 2015. Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biol 16: 282 10.1186/s13059-015-0848-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res 33: 7138–7150. 10.1093/nar/gki1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CYA, Shyu A-B. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465–470. 10.1016/S0968-0004(00)89102-1 [DOI] [PubMed] [Google Scholar]
- De Pretis S, Kress T, Morelli MJ, Melloni GEM, Riva L, Amati B, Pelizzola M. 2015. INSPEcT: a computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA- and 4sU-seq time course experiments. Bioinformatics 31: 2829–2835. 10.1093/bioinformatics/btv288 [DOI] [PubMed] [Google Scholar]
- Duffy EE, Schofield JA, Simon MD. 2019. Gaining insight into transcriptome-wide RNA population dynamics through the chemistry of 4-thiouridine. Wiley Interdiscip Rev RNA 10: e1513 10.1002/wrna.1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser P, Wachutka L, Maier KC, Demel C, Boroni M, Iyer S, Cramer P, Gagneur J. 2016. Determinants of RNA metabolism in the Schizosaccharomyces pombe genome. Mol Syst Biol 12: 857 10.15252/msb.20156526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1922. On the Interpretation of χ2 from contingency tables, and the calculation of P. J R Stat Soc 85: 87 10.2307/2340521 [DOI] [Google Scholar]
- Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. 2011. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res 39: D66–D69. 10.1093/nar/gkq990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove JL, Schmidt FH. 1989. The role of mRNA and protein stability in gene expression. FASEB J 3: 2360–2370. 10.1096/fasebj.3.12.2676679 [DOI] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, Ameres SL. 2017. Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods 14: 1198–1204. 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Imamachi N, Tani H, Mizutani R, Imamura K, Irie T, Suzuki Y, Akimitsu N. 2014. BRIC-seq: a genome-wide approach for determining RNA stability in mammalian cells. Methods 67: 55–63. 10.1016/j.ymeth.2013.07.014 [DOI] [PubMed] [Google Scholar]
- Kiefer L, Schofield JA, Simon MD. 2018. Expanding the nucleoside recoding toolkit: revealing RNA population dynamics with 6-thioguanosine. J Am Chem Soc 140: 14567–14570. 10.1021/jacs.8b08554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh WS, Porter JR, Batchelor E. 2019. Tuning of mRNA stability through altering 3′-UTR sequences generates distinct output expression in a synthetic circuit driven by p53 oscillations. Sci Rep 9: 5976 10.1038/s41598-019-42509-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S, Imamachi N, Irie T, Tani H, Matsumoto K, Mizutani R, Imamura K, Kakeda M, Yada T, Sugano S, et al. 2015. Analysis of RNA decay factor mediated RNA stability contributions on RNA abundance. BMC Genomics 16: 154 10.1186/s12864-015-1358-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima W, Herzog VA, Neumann T, Gapp K, Zuber J, Ameres SL, Miska EA. 2018. SLAM-ITseq: sequencing cell type-specific transcriptomes without cell sorting. Development 145: dev164640 10.1242/dev.164640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus J, Cheng Z, Vogel C. 2015. Next-generation analysis of gene expression regulation-comparing the roles of synthesis and degradation. Mol Biosyst 11: 2680–2689. 10.1039/C5MB00310E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll P, Ante M, Seitz A, Reda T. 2014. QuantSeq 3′ mRNA sequencing for RNA quantification. Nat Methods 11: i–iii. 10.1038/nmeth.f.376 [DOI] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273. 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Mudunuri U, Che A, Yi M, Stephens RM. 2009. bioDBnet: the biological database network. Bioinformatics 25: 555–556. 10.1093/bioinformatics/btn654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Ortín JE, Medina DA, Chávez S, Moreno J. 2013. What do you mean by transcription rate?: The conceptual difference between nascent transcription rate and mRNA synthesis rate is essential for the proper understanding of transcriptomic analyses. Bioessays 35: 1056–1062. 10.1002/bies.201300057. [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. 2011. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29: 436–442. 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, Hacohen N, Schier AF, Blackshear PJ, Friedman N, et al. 2014. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159: 1698–1710. 10.1016/j.cell.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Tudek A, Jensen T. 2019. Preparation of RNA 3′ end sequencing libraries of total and 4-thiouracil labeled RNA for simultaneous measurement of transcription, RNA synthesis and decay in S. cerevisiae. Bio Protoc 9: e3189. 10.21769/BioProtoc.3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield JA, Duffy EE, Kiefer L, Sullivan MC, Simon MD. 2018. TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat Methods 15: 221–225. 10.1038/nmeth.4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, Cramer P. 2016. TT-seq maps the human transient transcriptome. Science 352: 1225–1228. 10.1126/science.aad9841 [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R. 1986. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46: 659–667. 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Storey JD, Taylor JE, Siegmund D. 2004. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J R Stat Soc Ser B (Statistical Methodol) 66: 187–205. 10.1111/j.1467-9868.2004.00439.x [DOI] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, Wakamatsu A, Isogai T, Suzuki Y, Akimitsu N. 2012. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res 22: 947–956. 10.1101/gr.130559.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. 2020. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17: 261–272. 10.1038/s41592-019-0686-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova IA, Bohjanen PR. 2008. Posttranscriptional regulation of gene networks by GU-rich elements and CELF proteins. RNA Biol 5: 201–207. 10.4161/rna.7056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, et al. 2008. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell 29: 263–270. 10.1016/j.molcel.2007.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhager L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kaufmann S, Malterer G, L'Hernault A, Schilhabel M, Schreiber S, et al. 2012. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res 22: 2031–2042. 10.1101/gr.131847.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.