

Abstract
Extensive manipulations involved in the preparation of DNA samples for sequencing have hitherto made it impossible to determine the precise structure of double-stranded DNA fragments being sequenced, such as the presence of blunt ends, single-stranded overhangs, or single-strand breaks. We here describe MatchSeq, a method that combines single-stranded DNA library preparation from diluted DNA samples with computational sequence matching, allowing the reconstruction of double-stranded DNA fragments on a single-molecule level. The application of MatchSeq to Neanderthal DNA, a particularly complex source of degraded DNA, reveals that 1- or 2-nt overhangs and blunt ends dominate the ends of ancient DNA molecules and that short gaps exist, which are predominantly caused by the loss of individual purines. We further show that deamination of cytosine to uracil occurs in both single- and double-stranded contexts close to the ends of molecules, and that single-stranded parts of DNA fragments are enriched in pyrimidines. MatchSeq provides unprecedented resolution for interrogating the structures of fragmented double-stranded DNA and can be applied to fragmented double-stranded DNA isolated from any biological source. The method relies on well-established laboratory techniques and can easily be integrated into routine data generation. This possibility is shown by the successful reconstruction of double-stranded DNA fragments from previously published single-stranded sequence data, allowing a more comprehensive characterization of the biochemical properties not only of ancient DNA but also of cell-free DNA from human blood plasma, a clinically relevant marker for the diagnosis and monitoring of disease.
Because of advances in DNA sequencing technology and associated sample preparation techniques, great progress has been made in the retrieval of DNA sequences from highly degraded biological material over the past two decades. This is perhaps most impressively illustrated by the recovery of genome-wide sequence data, and even whole-genome sequences, from ancient biological samples. The analysis of sequences that are tens, or sometimes even hundreds of thousands of years old (Orlando et al. 2013; Meyer et al. 2014, 2016), has made important contributions to our understanding of the evolutionary history of humans and other species. In biomedical research, another type of degraded DNA, cell-free DNA (cfDNA), has become established as an important marker in recent years for pathologies such as cancer (Sozzi et al. 2003; Snyder et al. 2016) and is used in prenatal diagnostics to detect aneuploidies (Chiu et al. 2008). Changes in cfDNA quantity in the blood have been linked to organ transplant rejection (De Vlaminck et al. 2014) or increased apoptosis in the trophoblast surrounding the fetus (Tjoa et al. 2006).
Beyond the identification of genetic variants, the sequencing of billions of DNA fragments has allowed inferences about the structures of molecules and characteristics of DNA damage that have proven critical for the development of new analytical techniques. For example, it has been shown that changes from cytosine to thymine (and guanine to adenine if libraries are prepared using double-stranded methods) represent by far the most frequent class of substitutions in ancient DNA sequences. These substitutions are caused by the deamination of cytosine to uracil, or 5′ methylcytosine to thymine, and accumulate predominantly at the ends of DNA molecules (Briggs et al. 2007), hinting at the existence of single-stranded DNA overhangs in which deamination occurs much faster than in double-stranded DNA (Lindahl and Nyberg 1974). In addition to being recognized as a source of error, deamination has been used to enrich genuine ancient DNA fragments in silico or during library preparation (Krause et al. 2010; Meyer et al. 2014; Skoglund et al. 2014) and to reconstruct methylation maps (Gokhman et al. 2014). Similarly, fragmentation patterns in cfDNA sequences are used to reconstruct nucleosome or transcription factor footprints, which are informative about the tissue of origin (Snyder et al. 2016).
Continued analytical and methodological advances in the analysis of degraded DNA are hampered by the extensive modifications of DNA fragments during library preparation, which obscure the true structures of DNA molecules. Double-stranded library preparation methods involve a blunt-end repair step, in which a DNA polymerase with 3′-5′ exonuclease activity is used to remove 3′ and fill in 5′ overhangs before double-stranded adapters are ligated to the molecule ends (Briggs et al. 2007). Blunt-end repair thus preserves only the 5′ ends of the DNA fragments, whereas the 3′ ends are trimmed or extended to match the 5′ end of the complementary strand. When working with ancient DNA, polymerase activity leads to an incorporation of adenines across uracils in 5′ overhangs, which manifest as G-to-A substitutions near the 3′ ends of ancient DNA sequences (and less frequently in the interior of sequences if molecules contain single-strand breaks and a strand-displacing polymerase is used in library preparation) (Briggs et al. 2007). Because single-stranded overhangs are removed during blunt-end repair, their length can only be estimated using deamination-induced C-to-T and G-to-A substitutions as proxies for the presence of single-stranded regions in molecules (Briggs et al. 2007; Jónsson et al. 2013). An alternative method for library preparation, which is particularly efficient for retrieving sequences from extremely short molecules such as those present in ancient biological material, relies on heat denaturation of double-stranded DNA fragments and the independent conversion of the single DNA strands into library molecules (Meyer et al. 2012; Gansauge and Meyer 2013; Gansauge et al. 2017). Whereas single-stranded library preparation does not involve the manipulation of molecule ends, it does not preserve the double-stranded state of DNA molecules.
For any given strand of a double-stranded molecule recovered in a single-stranded library, the probability of also recovering the complementary strand should roughly correspond to the efficiency of library preparation, which has been estimated to be between 10% and 50% based on the conversion rate of synthetic DNA oligonucleotides (Gansauge and Meyer 2013). We therefore reasoned that it may be possible to computationally reconstruct double-stranded DNA fragments from the sequences of individual DNA strands (which may be two or more if a fragment carries single-stranded breaks). However, such an approach does not only pose computational challenges, it also necessitates the adjustment of experimental parameters to increase the chance of identifying sequence pairs that originate from genuine double-stranded ancient DNA fragments: First, single-stranded libraries have to be sequenced very deeply so that each unique DNA strand is observed, on average, multiple times. This is necessary in order to minimize losses of molecules owing to the stochasticity of the sequencing process, as well as biases in library amplification. Second, libraries have to be prepared from small quantities of DNA to minimize the number of sequences that align in close proximity on the reference genome by chance.
Based on these considerations, we developed MatchSeq, a method for assessing the true structure of double-stranded DNA fragments on a single-molecule level. The method relies on deep sequencing of single-stranded libraries produced from diluted DNA samples containing double-stranded DNA fragments, followed by the computational matching of sequences that map in close proximity on the reference genome in the same or reverse complementary strand orientation (Fig. 1A). By applying this method to Neanderthal DNA, we infer new aspects of ancient DNA degradation that are dependent on DNA structure and time.
Figure 1.

Identification of sequences from DNA strands originating from the same double-stranded DNA molecules. (A) Schematic overview of MatchSeq: DNA fragments isolated from ancient or recent biological material are diluted to a relatively small number, denatured by heat, converted into a single-stranded library, amplified, and deeply sequenced. Double-stranded fragments are then reconstructed in silico by identifying overlapping sequences mapping to the reference genome in reverse complementary orientation or in close proximity on the same strand. Sequences <35 bp cannot be confidently mapped to the reference and are discarded. The same analysis is performed for an equal number of control sequences obtained from shallow sequencing of a vast amount of artificially fragmented modern human DNA, in which sequences are expected to be distributed randomly across the reference genome. (B) Distribution of distances between the 5′ end of each sequence to the nearest 3′ end on the opposing strand obtained from Neanderthal DNA (Vindija 33.19; gray bars) and the control (red line). (C) Distribution of distances between sequence alignments on the same strand. Molecular structures putatively underlying the observed signals are indicated by schematic drawings. Filled DNA strands indicate sequences that were recovered; unfilled strands are hypothesized to be present but were not sequenced.
Results
Reconstructing double-stranded DNA fragments on a single-molecule level
To determine whether it is possible to reconstruct double-stranded DNA fragments from single-stranded libraries, we extracted DNA from a small quantity of bone powder (3 mg) of a well-preserved Neanderthal bone fragment from Vindija Cave, Croatia (Vindija 33.19) from which a high-coverage genome sequence was previously obtained (Prüfer et al. 2017). DNA extraction was performed using a buffer exchange (BE) method that avoids denaturation or loss of short DNA fragments (Glocke and Meyer 2017). An aliquot of the extract was then converted into a single-stranded library (Gansauge and Meyer 2013), which was amplified by PCR and deeply sequenced. Of the 235 million full-length molecule sequences generated from this library, 6.5% mapped to the human reference genome (hg19/GRCh37) when requiring a minimum length of 35 bp and a map quality score of 25 or greater (Supplemental Table S1). The mapped sequences were represented by 2.6 sequence duplicates on average, indicating that the library had been exhaustively sequenced. After duplicate removal, sequences from 2.7 million single-stranded DNA molecules were available for further analysis. By using sites in which all currently known Neanderthal genomes differ from 90% of the genomes of present-day humans, we estimated the contribution of modern human contamination to these sequences to <1.35% (95% confidence interval [C.I.] 0.90%–1.93%) (Meyer et al. 2016).
To determine whether there are more overlaps between sequences aligning to the reference genome in forward and reverse orientation than expected by chance, we plotted the distribution of distances between the 5′ end of each sequence and the 3′ end of the nearest sequence on the opposing strand (Fig. 1B). For comparison, we created a control data set of sequences randomly distributed across the reference genome by down-sampling previously published sequences (de Filippo et al. 2018) that were generated from artificially fragmented modern human DNA using single-stranded library preparation to an equal number of unique sequences. Unlike the Neanderthal library, which contains only minute amounts of hominin DNA, this control library had been produced from ∼10 ng of human DNA (corresponding to approximately 3000 copies of the haploid genome) and was not sequenced to exhaustion. It can therefore be assumed that nearly all sequenced DNA strands in the control data originate from independent double-stranded DNA fragments. In congruence with this expectation, we did not detect an overrepresentation of overlapping sequence alignments in the control data. In contrast, the Vindija 33.19 data show a strong accumulation of sequence alignments that start and end in close proximity on opposing strands, a signal that is most pronounced at distances between −2 and 2, corresponding to 3′ overhangs of 1 or 2 nucleotides (nt), blunt ends, and 5′ overhangs of up to 2 nt (Fig. 1B). Taken together, these structures are at least 12.8 times more frequent than in the control data at the same sequence depth (Supplemental Table S2), indicating that more than ∼92% of the inferred double-stranded molecules with short overhangs and blunt ends represent genuine double-stranded DNA molecules (for examples of reconstructed double-stranded molecules, see Fig. 2). Taken together, 89,555 (3.3%) of the 5′ ends of all mapped sequences were assigned to one of the above categories. We note that the distribution of overhangs is not fully symmetrical as slightly more molecules with single-nucleotide 5′ overhangs were identified than with 3′ overhangs.
Figure 2.

Examples of double-stranded Neanderthal DNA fragments reconstructed from deep sequencing of a single-stranded DNA library. The presence of uracil was inferred by a C-to-T substitution in one of the sequenced strands, whereas the G on the other strand matched the human reference genome.
Based on deamination signals in the interior of ancient DNA sequences, it has been hypothesized that single-stranded breaks exist in ancient DNA (Glocke and Meyer 2017), namely, double-stranded DNA fragments consisting of three (or more) single-stranded molecules. If such fragments exist, it is highly unlikely that all three molecules would be recovered during library preparation and sequencing and that they would all be long enough to be confidently mapped to the reference genome. Nonetheless, even if only two out of three molecules are recovered, strand breaks are expected to leave distinct patterns in our data. First, if the two molecules originate from opposing strands of the DNA fragment, the missing third molecule would render the reconstructed double-stranded DNA fragment into one with a long overhang. In agreement with this prediction, the distribution of inferred overhang lengths is not solely characterized by a sharp peak around short overhangs as described above but appears to contain a second, underlying distribution, which is wider and extends to overhang lengths of up to ∼50 nt (Fig. 1B). Second, if two molecules are recovered from the same strand of a DNA fragment that carries a strand break, we expect an accumulation of sequences that align in close proximity on the reference genome. To investigate this second prediction, we plotted the distribution of distances between the 3′ terminus of each sequence to the 5′ terminus of the nearest sequence aligning downstream in the reference genome (Fig. 1C). Although the control data set yielded a flat distribution as expected, the Vindija 33.19 data show an excess of alignments that are separated by a gap of 1 bp, and a small excess of alignments separated by slightly longer gaps. This analysis thus provides direct evidence for the presence of gaps, but not nicks, in ancient DNA fragments.
Dependence of cytosine deamination rates on structural context
Having established that double-stranded DNA fragments can successfully be reconstructed from the Vindija 33.19 sequence data, we next examined the frequency of substitutions and the base composition of sequences separately for each structural context (short overhangs, blunt ends, and single-strand breaks). We also performed a background correction in all subsequent analyses in order to determine the robustness of the results to small amounts of noise in the data that are contributed by sequences that by chance align in close proximity on the same or opposing strands. For this purpose, we inferred the signal-to-noise ratio for each structural context separately by dividing the number of sequences assigned to the respective context in the Vindija 33.19 library by the number of sequences in the control library (Supplemental Table S2). We then used the average base composition and substitution frequencies determined from the complete set of aligned sequences without stratification for structural context as a proxy for the noise and subtracted its contribution according to the signal-to-noise ratio. We note that the background subtraction leads to only minor changes in numbers and hence present the background corrected results in the Supplemental Material only (see Supplemental Figs. S1–S3).
In congruence with the hypothesis that deamination proceeds faster in single-stranded than in double-stranded DNA, C-to-T substitutions are greatly elevated in inferred single-stranded 5′ and 3′ overhangs (Fig. 3; Supplemental Fig. S1). For example, 71% of the 5′ terminal positions that match a C in the reference genome and are inferred to be located in 1-nt single-stranded overhangs carry a T, compared with 46% without stratification for structural context. However, elevated signals of deamination are also present at termini located in blunt and recessed ends, that is, in a double-stranded context. This signal is strongest at 5′ termini, where C-to-T substitution rates range from 19.2% to 20.7%, compared with the 3′ termini, where the rates are between 9.1% and 12.8% in these structural contexts. We also find that C-to-T substitution frequencies are higher at the second position of sequences located in 1-nt 5′ overhangs (13.8%; 95% C.I. 12.6%–15.1%) than at the third position of sequences located in 2-nt 5′ overhangs (6.9%; 95% C.I. 5.8%–8.1%) (see Fig. 3; Supplemental Tables S3, S4), each corresponding to the first position in a double-stranded context. This observation suggests that cytosine deamination occurs less frequently in double-stranded context if the distance to the 5′ terminus is >2 nt. This pattern is less pronounced at 3′ termini, where deamination in a double-stranded context is less frequent in general.
Figure 3.

C-to-T substitution frequencies and reference base composition for the termini of Neanderthal sequences located in various structural contexts. The composition of the reference genome around alignment start and end points is shown in sequence logo plots, where the relative size of the letters is proportional to the frequency of each base. The total height of the letters (in bits) indicates how much the base composition deviates from randomness. The structure of the termini is indicated by schematic drawings. The strand that was used for calculating the reference base composition and substitution frequencies is marked in black; the complementary strand, in gray. The first (upper left) plot shows C-to-T substitution frequencies and reference base composition of all aligned sequences without stratification for structural context.
The frequency of nucleotide substitutions can also be investigated in the proximity of single-strand breaks. We first focused on sequences that directly flank 1-nt gaps (Fig. 4) and observed lower C-to-T substitution frequencies (5′ ends: 22.7% [95% C.I. 19.3%–26.3%], 3′ ends: 13.1% [95% C.I. 10.0%–16.8%]) than averaged across the whole data set (5′ ends: 45.7% [95% C.I. 45.6%–45.8%], 3′ ends: 33.9% [95% C.I. 33.7%–34.0%]), an observation that is in line with lower deamination rates in a double-stranded context. Next, we investigated the frequency of nucleotide substitutions on the strand opposing the gap. Assuming that the vast majority of long overhangs represent molecules with single-strand breaks (see Fig. 1B), we isolated and analyzed molecules inferred to carry 20-nt overhangs as representative examples for such molecules. In both 20-nt 5′ and 3′ overhangs, elevated C-to-T substitution frequencies are present in the three positions preceding the start of the opposing strand, consistent with that they derived from DNA fragments containing a short single-stranded gap (Fig. 5; Supplemental Figs. S3, S4). The presence of C-to-T substitutions opposite the gap corroborates that deamination proceeds faster in single-stranded than in double-stranded DNA also in the interior of molecules. The frequency of C-to-T substitutions at the first position downstream from the gap is low (5′ overhangs: 3.9% [95% C.I. 2.3%–6.0%], 3′ overhangs: 3.9% [95% C.I. 2.0%–6.7%]), again indicating that deamination occurs in a double-stranded context only close to the ends of DNA strands. We also attempted to quantify the effect of methylation on the rate of cytosine deamination, but CpG dinucleotides were too rare in our relatively small data set (Supplemental Fig. S5).
Figure 4.
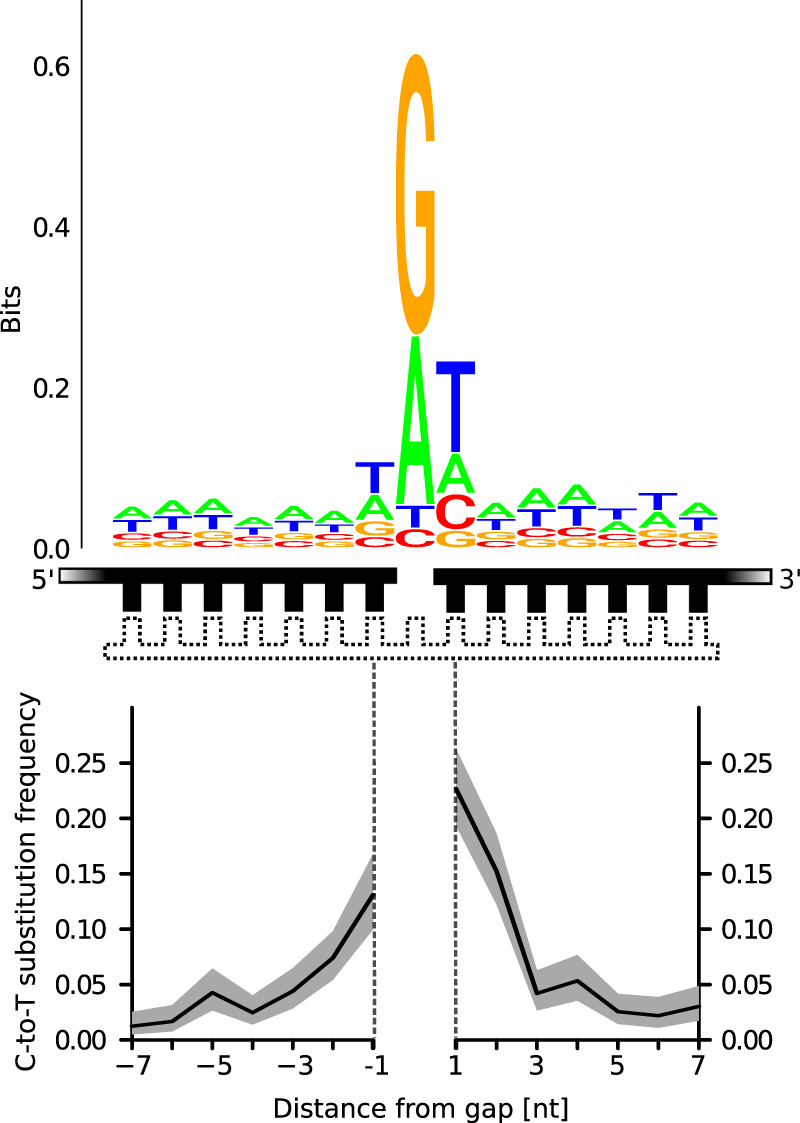
C-to-T substitution frequencies and sequence logo plot of the reference base composition around putative 1-nt gaps in Neanderthal DNA fragments. The strands used for calculating reference base compositions and substitution frequencies are marked black in the schematic drawing; the complementary strand that was putatively present, in white. The gray area surrounding the C-to-T substitution frequencies denotes the 95% binomial confidence interval.
Figure 5.

C-to-T substitution frequencies and sequence logo plot of the reference base composition around Neanderthal molecules with 20-nt 5′ overhangs. A schematic representation of the alignments shows the strand used for calculating reference base composition and substitution frequencies (marked in black), the identified complementary strand (marked in gray), and a second complementary strand of unknown length that was putatively present (white fill). Note that the size of the gap is unknown. The gray area surrounding the C-to-T substitution frequencies denotes the 95% binomial confidence interval.
Although cytosine deamination-induced C-to-T substitutions are a common feature of authentic ancient DNA, other base substitutions have been observed at such low frequencies that it is difficult to determine whether they are the result of rare base damage, sequence errors, or evolutionary sequence differences between the sample and the reference genome. If other miscoding base damage existed in ancient DNA and if it occurred predominantly in single-stranded regions of DNA fragments, stratification of sequences by the structural context in which they appear may provide additional power to detect non-C-to-T substitutions. Indeed, whereas the frequency of such substitutions does not exceed 0.5% when combining all mapped sequences in the analysis, we observe small but statistically significant increases in the frequency of non-C-to-T substitutions in 5′ overhangs (up to 2.4%) (see Supplemental Table S3) and, to a lesser extent, also in 3′ overhangs (up to 1.5%) (see Supplemental Table S4). These substitutions are from G and A to T in both 5′ and 3′ overhangs, as well as from G to C in 5′ overhangs (Supplemental Fig. S6). Although the chemical modifications causing these substitutions cannot be determined directly, it is known that most polymerases, including the Klenow fragment of Escherichia coli DNA polymerase I that was used here, preferentially incorporate adenine across abasic sites (Strauss et al. 1982; Shibutani et al. 1997). Substitutions from G and A to T may thus point to elevated rates of DNA depurination in single-stranded overhangs.
DNA fragmentation and signatures of DNA decay in various ancient DNA data sets
It has been shown that sequence alignments of ancient DNA molecules preferentially start and end next to purines in the reference genome (Briggs et al. 2007; Sawyer et al. 2012), suggesting that depurination and subsequent breakage of the sugar-phosphate backbone is at least partially responsible for postmortem DNA fragmentation. This pattern is also present in the Vindija 33.19 data analyzed here (Fig. 3; Supplemental Fig. S1), in which guanine and, to a lesser extent, adenine are overrepresented in the flanking bases of the reference genome, irrespective of the inferred structure of the double-stranded DNA fragment. The overrepresentation of guanine and adenine is particularly pronounced in 1-nt gaps (Fig. 4), suggesting that depurination is the main mechanism initiating the creation of gaps in ancient DNA molecules. Single-stranded overhangs at the end of molecules are enriched in pyrimidines (T and C) (Fig. 3), which may indicate a higher stability of those bases in a single-stranded context. However, to elucidate how the base composition of DNA fragments changes as degradation proceeds, it would be necessary to compare the Vindija 33.19 sequences to similar data from other specimens that show different states of preservation.
Although the majority of previously published single-stranded ancient DNA sequences was generated from libraries with a relatively low content of genomic DNA, these libraries were much less exhaustively sequenced than the Vindija 33.19 library produced in this study. To determine the sequence depth that is required to reconstruct double-stranded DNA fragments from single-stranded molecule sequences, we down-sampled the Vindija 33.19 data set to lower average numbers of duplicates per unique sequence. This test revealed that double-stranded DNA fragments can be reconstructed even at duplication rates as low as 1.14 (Supplemental Fig. S7). We therefore compiled a data set of published sequences from 36 single-stranded libraries from eight Neanderthal individuals (Goyet Q56-1 [Hajdinjak et al. 2018], Hohlenstein-Stadel [HST] [Peyrégne et al. 2019], Les Cottés Z4-1514 [Hajdinjak et al. 2018], Mezmaiskaya 1 and 2 [Prüfer et al. 2014; Hajdinjak et al. 2018], Scladina I-4A [Peyrégne et al. 2019], Spy 94a [Hajdinjak et al. 2018], and Sima de los Huesos [Meyer et al. 2016]) (see Supplemental Table S2) and evaluated their suitability for fragment reconstruction via MatchSeq by computing the distance between the 5′ end of each alignment and the closest 3′ end of an alignment on the opposing strand. To ensure sufficient confidence in the reconstruction of double-stranded DNA fragments, we required the number of inferred single-nucleotide 5′ overhangs to be at least 1000, and two times higher than in a set of control sequences down-sampled to the same number of unique sequences. This left 22 libraries from five specimens for further analysis (HST, Les Cottés Z4-1514, Scladina I-4A, Spy 94a, and Sima de los Huesos) (see Supplemental Table S2). These specimens are from Europe and Siberia and cover a time period from ∼39 to ∼430 thousand years ago.
Previous studies have shown that age alone is not a good predictor for the level of deamination and DNA fragmentation observed in ancient biological material (Sawyer et al. 2012; Kistler et al. 2017) as preservation conditions (temperature, pH, etc.) vary greatly across archeological sites. To test whether deamination rates in specific structural contexts produce a better correlation with sample age than those obtained from all molecules combined, we determined the frequency of C-to-T substitutions in 1-nt 5′ overhangs and blunt ends and plotted them against the presumed ages of the samples (Supplemental Figs. S8, S9). In addition, we plotted the base composition in 1-nt 5′ overhangs against age (Supplemental Fig. S10). Both plots did not produce significant correlations between the biochemical properties of the DNA and sample age. Assuming that deamination is a better proxy for the degree of DNA degradation than age, we next plotted the reference base composition in 1-nt 5′ overhangs against the C-to-T substitution frequency (Fig. 6). This analysis indeed yielded a significant correlation between the frequency of T nucleotides in single-stranded overhangs and deamination rate (P-value 0.01). In contrast to this, the frequency of C's decreases (P-value 0.04), although just barely significantly so. The frequency of purines is not significantly correlated with C-to-T substitution frequency (P-value G: 0.40, P-value A: 0.55).
Figure 6.
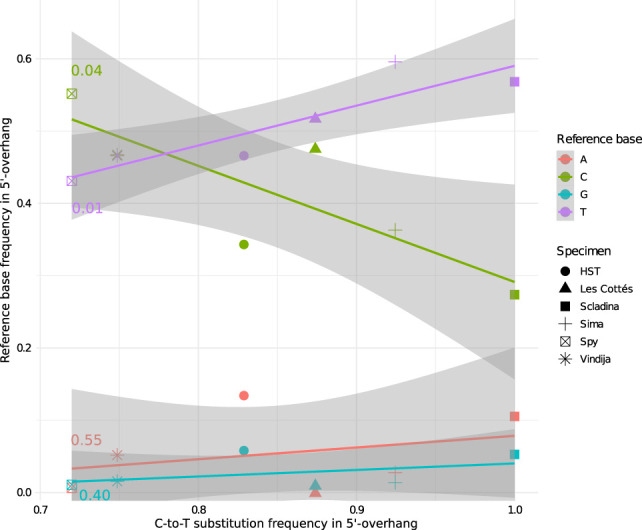
Reference base composition versus C-to-T substitution frequencies in 1-nt 5′ overhangs in libraries from various Neanderthal specimens (N = 6). As some libraries show substantial levels of human DNA contamination, the analysis was restricted to putatively endogenous, deaminated fragments by requiring a C-to-T substitution at the 3′ end of sequences. Background noise was subtracted using the signal-to-noise ratio determined via comparison to a modern control sample (see Supplemental Table S2). For specimens for which more than one library was available, the library producing the largest number of DNA fragments with an inferred 1-nt 5′ overhang was chosen. Moderately significant positive or negative correlations were found for T (purple; one-tailed F-statistic linear regression on 1° and 4° of freedom: 17.81, P-value 0.01 [R2:0.8166]) and C (green; one-tailed F-statistic linear regression on 1° and 4° of freedom: 8.80, P-value 0.04 [R2: 0.6874]), but not for A or G (F: 0.44, P-value 0.55 [R2: 0.09875] and F: 0.89, P-value 0.40 [R2: 0.1825]).
By leveraging the fact that many more double-stranded DNA fragments could be reconstructed from the previously published Neanderthal data than from the Vinidja 33.19 sequences generated in the present study, we used the combined data to investigate the frequency of C-to-T substitutions in CpG and non-CpG contexts (representing deamination of 5-methylcytosine to thymine and of cytosine to uracil, respectively) in various structural contexts. Although the deamination rate in CpG context is slightly higher than outside CpG's at 5′ ends as expected (Seguin-Orlando et al. 2015), we observed no striking differences between the frequencies of the two types of deamination depending on the structure in which they occur (Supplemental Fig. S11). At 3′ ends, C-to-T substitution frequencies in a CpG context are slightly lower than outside CpGs, probably owing to a bias against the ligation of 3′ uracils in library preparation (Gansauge et al. 2017).
Application to cfDNA
Although the main focus of this study is on ancient DNA, MatchSeq should in principle be applicable to any type of degraded double-stranded DNA. To show this, we reanalyzed published sequence data from cfDNA from the plasma of healthy donors that had been converted into single-stranded libraries using the SRSLY method (Troll et al. 2019). As expected, the experimental design chosen by Troll and colleagues was not ideal for our purpose, as the sample DNA had not been diluted before library preparation, and libraries had not been exhaustively sequenced. Nonetheless, an analysis of the library that yielded the highest number of sequence duplicates (“SR-05”; 1.13 duplicates on average for sequences amounting to 3.4-fold coverage of the genome) revealed that several structural contexts are overrepresented relative to the control (Supplemental Fig. S12A), producing a highly irregular pattern of single-stranded overhangs, albeit at signal-to-noise ratios of 1.34 or less (Supplemental Table S5). In addition, we found that nicks are far more prominent than short gaps in cfDNA fragments (Supplemental Fig. S12B), a signal that is in stark contrast to the patterns observed with ancient DNA, and that the base composition around nicks appears to be highly nonrandom (Supplemental Fig. S13). More optimal data (produced by deeper sequencing of libraries prepared from lower DNA input amounts) would be required to elucidate the patterns of degradation in cfDNA at higher resolution and to determine whether these patterns vary among samples from different patients and tissues.
Discussion
The analysis of MatchSeq data from several Neanderthal specimens provides new insights about the state in which DNA survives in ancient biological material. We show that the ends of ancient DNA molecules are predominantly composed of blunt ends and small single-stranded overhangs and that small gaps of 1 to 3 nt exist in the interior of ancient DNA molecules, whereas evidence for nicks in the sugar phosphate backbone is elusive. In positions where a gap was detected, the most abundant bases in the reference genome were guanine and adenine, providing further evidence for depurination as the main mechanism driving fragmentation in ancient DNA. Furthermore, an analysis of the base composition in single-stranded overhangs suggests that purines, and in particular guanines, are unstable in overhangs and that cytosine is less stable than thymine as DNA degradation proceeds. In addition to revealing the structure of ancient DNA fragments, the reconstruction of double-stranded molecules allowed us to elucidate at high resolution the presence of miscoding damage in ancient DNA. Most noticeably perhaps, our analyses showed that deamination is not restricted to single-stranded regions of molecules but also occurs in a double-stranded context at the end of DNA strands if the distance to the terminus is small. This had previously been speculated (Briggs et al. 2007; Jónsson et al. 2013) but was never directly shown. Although miscoding damage that cannot be explained by cytosine deamination is generally rare, an increased rate of adenine misincorporation opposite of guanine and adenine points to the existence of abasic sites or other base damage in single-stranded overhangs.
The reconstruction of double-stranded DNA molecules from published Neanderthal and cfDNA sequence data shows that fragment reconstruction via MatchSeq is possible under a broad range of experimental parameters and as a side product of data generation for other purposes if a few requirements are met: First, libraries must be prepared using a single-stranded method that preserves the native 5′ and 3′ ends of DNA strands. Specifically, we showed that MatchSeq is compatible with the ssDNA 2.0 library preparation method, used here for the analysis of ancient DNA (Gansauge et al. 2020), and the SRSLY method (commercialized by Claret Bioscience) (Troll et al. 2019), which was used to generate the cfDNA data analyzed here. Second, the total genomic coverage in the library should be relatively low so that random mapping of sequences in close proximity is rare. Although MatchSeq can in principle be performed using libraries containing multifold coverage (up to approximately threefold, and possibly more) of the genome of interest, very high signal-to-noise ratios (greater than 10) were obtained only for libraries containing less than approximately 0.1-fold coverage of the genome (compare Supplemental Table S2). Third, the library should be sequenced deeply enough so that more than one sequence is generated from at least 20% (preferably more) of the unique molecules in the library. To satisfy these conditions, it may be necessary to perform library preparation twice, in which the first experiment is used to determine library complexity by qPCR and shallow shotgun sequencing (Gansauge et al. 2017, 2020), allowing for the optimal input volume of DNA extract to be determined for a second experiment. This titration would only need to be performed once on a single sample or on a few samples when working with sets of samples with similar DNA concentrations and degradation states.
Apart from allowing investigations into the biochemical properties of ancient DNA and the mechanism leading to its decay, the reconstruction of ancient DNA fragments from sequence data opens the door for a number of future methodological advances: First, MatchSeq provides an ideal tool for monitoring the success of enzymatic reactions that could be used to seal gaps in ancient DNA molecules. Ancient DNA repair has been attempted (Pusch et al. 1998) but has not yet led to major improvements in sequence recovery when coupled with high-throughput sequencing (Mouttham et al. 2015). If successful, it could potentially extend the temporal range of ancient DNA studies. Second, sequences from the two complementary strands of a DNA fragment could be used to determine a small number of high-quality haploid genotypes from specimens with insufficient DNA preservation for high-coverage genome sequencing. Confident genotype calling critically depends on the presence of at least two independently sequenced DNA strands in order to allow identification of errors resulting from base damage and library amplification, in addition to reducing sequencing error by overlap merging of paired-end reads and consensus calling from duplicates reads. For example, of the approximately 127 million positions of the Vindija 33.19 genome covered by sequence data generated in this study, ∼2.7 Mbp is represented by overlapping complementary strands putatively originating from DNA fragments of which both strands were recovered (i.e., strands with alignment start and end coordinates mapping in a distance of two or less on at least one end), corresponding to an approximately 0.001-fold coverage of the genome for which high-quality genotypes might be obtained. Although this number is small, newly sequenced archaic human genomes can be expected to carry a few hundred thousand unique substitutions that distinguish them from previously sequenced genomes (Meyer et al. 2016). Thus, even in a data set as small as the one generated here, it may be possible to identify with confidence a few hundred sites that are highly informative for resolving the phylogenetic relationship among archaic humans.
Although ancient DNA is particularly well suited to show the types of inferences that can be made using MatchSeq, the method may also be highly relevant for biomedical research. By using publicly available data, we were able to show that it is possible to determine the structure of molecule ends in cfDNA. In addition to the already established analysis of nucleosome and transcription factor footprints, as well as simple fragmentation patterns (Snyder et al. 2016), MatchSeq may enable inferences about the apoptotic and necrotic pathways underlying the release of DNA (e.g., Didenko et al. 2003), adding a layer of information that might further advance the use of cfDNA in clinical diagnostics.
Methods
DNA extraction and library preparation
Work was performed in a dedicated ancient DNA laboratory at the Max Planck Institute for Evolutionary Anthropology in Leipzig. Three milligrams of bone powder was removed from a Neanderthal femur fragment (Vindija 33.19) using a sterile dentistry drill. A lysate was prepared by adding 500 µL 0.5 M EDTA and incubating at room temperature. DNA was extracted using the BE method without silica purification (Glocke and Meyer 2017), yielding 100 µL DNA extract. One microliter of the extract was converted into a single-stranded library as previously described (Gansauge et al. 2020), using automated liquid handling. As determined by quantitative PCR (qPCR), the yield of library molecules was only three times higher than that obtained from the library preparation negative control that was included in the experiment, indicating that the complexity of the sample library was low (Supplemental Table S1). Following amplification of the library with a pair of primers carrying sample-specific 7-bp indices (Gansauge et al. 2020), the indexed library was size-selected by gel excision before sequencing to remove short artifacts from library preparation as well as library molecules with inserts <35 bp (Gansauge et al. 2020). The size-selected library was sequenced on an Illumina HiSeq 2500 with a 2 × 76-bp configuration with two 7-bp index reads.
Data processing and analysis
Sequence reads showing perfect matches to the expected index combination were adapter trimmed and overlap-merged into full-length molecule sequences using leeHom (Renaud et al. 2014); sequences not successfully merged were discarded. After mapping to the human reference genome (hg19/GRCh37) with the Burrows–Wheeler aligner (BWA) (Li and Durbin 2009) using parameters optimized for ancient DNA (-n 0.01 –o 2 –l 16500) (Meyer et al. 2012), sequences <35 bp were discarded and PCR duplicates removed using bam-rmdup (https://bitbucket.org/ustenzel/biohazard-tools). The older version of the human reference genome was chosen over hg38/GRCh38 because it is the version commonly used for reconstructing Neanderthal genomes (Prüfer et al. 2014, 2017; Hajdinjak et al. 2018; Mafessoni et al. 2020), and there are few differences between those reference genomes in regions where short reads can be confidently mapped. Sequences not mapping to one of the autosomes or the X Chromosome with a minimum mapping quality of 25 were discarded. Modern human contamination was computed as described elsewhere (Meyer et al. 2016). Distances between sequence alignments, the reference base composition, and substitution frequencies were computed using custom Perl scripts (https://github.com/mpieva/matchseq). After sorting DNA sequences according to their inferred structural context based on alignment distances, the proportion of sequences aligning in that distance by chance (background) was estimated using the modern human control sequences down-sampled to the same number of aligned sequences. Base and substitution frequencies of the background were assumed to equal those of all sequences of that specimen, irrespective of structural context. The background contribution was then subtracted, and substitution and reference base frequencies were calculated from the corrected counts. Sequence logo plots were generated using the ggseqlogo package (https://github.com/omarwagih/ggseqlogo) in R (R Core Team 2018).
Data access
All raw and processed sequence data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena) under accession number PRJEB36197. Perl scripts used to calculate substitution and reference base frequencies, as well as minimal alignment distances on the same and opposing strands of the reference genome, are available at GitHub (https://github.com/mpieva/matchseq) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank P. Rudan, Ž. Kućan, and I. Gušić for contributing material; Sarah Nagel for help with library preparation; and Antje Weihmann and Barbara Schellbach for help with DNA sequencing. We thank C. de Filippo, J. Kelso, S. Pääbo, B. Vernot, and E. Zavala for helpful discussions and comments on the manuscript. This study was funded by the Max Planck Society.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.263863.120.
References
- Briggs AW, Stenzel U, Johnson PL, Green RE, Kelso J, Prufer K, Meyer M, Krause J, Ronan MT, Lachmann M, et al. 2007. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci 104: 14616–14621. 10.1073/pnas.0704665104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu RWK, Chan KCA, Gao Y, Lau VYM, Zheng W, Leung TY, Foo CHF, Xie B, Tsui NBY, Lun FMF, et al. 2008. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci 105: 20458–20463. 10.1073/pnas.0810641105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Filippo C, Meyer M, Prüfer K. 2018. Quantifying and reducing spurious alignments for the analysis of ultra-short ancient DNA sequences. BMC Biol 16: 121. 10.1186/s12915-018-0581-9 [DOI] [PMC free article] [PubMed]
- De Vlaminck I, Valantine HA, Snyder TM, Strehl C, Cohen G, Luikart H, Neff NF, Okamoto J, Bernstein D, Weisshaar D, et al. 2014. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med 6: 241ra77 10.1126/scitranslmed.3007803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didenko VV, Ngo H, Baskin DS. 2003. Early necrotic DNA degradation: presence of blunt-ended DNA breaks, 3′ and 5′ overhangs in apoptosis, but only 5′ overhangs in early necrosis. Am J Pathol 162: 1571–1578. 10.1016/S0002-9440(10)64291-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansauge MT, Meyer M. 2013. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat Protoc 8: 737–748. 10.1038/nprot.2013.038 [DOI] [PubMed] [Google Scholar]
- Gansauge MT, Gerber T, Glocke I, Korlevic P, Lippik L, Nagel S, Riehl LM, Schmidt A, Meyer M. 2017. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res 45: e79 10.1093/nar/gkx033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansauge M-T, Aximu-Petri A, Nagel S, Meyer M. 2020. Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA. Nat Protoc 15: 2279–2300. 10.1038/s41596-020-0338-0 [DOI] [PubMed] [Google Scholar]
- Glocke I, Meyer M. 2017. Extending the spectrum of DNA sequences retrieved from ancient bones and teeth. Genome Res 27: 1230–1237. 10.1101/gr.219675.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhman D, Lavi E, Prüfer K, Fraga MF, Riancho JA, Kelso J, Pääbo S, Meshorer E, Carmel L. 2014. Reconstructing the DNA methylation maps of the Neandertal and the Denisovan. Science 344: 523–527. 10.1126/science.1250368 [DOI] [PubMed] [Google Scholar]
- Hajdinjak M, Fu Q, Hübner A, Petr M, Mafessoni F, Grote S, Skoglund P, Narasimham V, Rougier H, Crevecoeur I, et al. 2018. Reconstructing the genetic history of late Neanderthals. Nature 555: 652–656. 10.1038/nature26151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L. 2013. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29: 1682–1684. 10.1093/bioinformatics/btt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler L, Ware R, Smith O, Collins M, Allaby RG. 2017. A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res 45: 6310–6320. 10.1093/nar/gkx361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko A, Pääbo S. 2010. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol 20: 231–236. 10.1016/j.cub.2009.11.068 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Nyberg B. 1974. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13: 3405–3410. 10.1021/bi00713a035 [DOI] [PubMed] [Google Scholar]
- Mafessoni F, Grote S, de Filippo C, Slon V, Kolobova KA, Viola B, Markin SV, Chintalapati M, Peyrégne S, Skov L, et al. 2020. A high-coverage Neandertal genome from Chagyrskaya Cave. Proc Natl Acad Sci 117: 15132–15136. 10.1073/pnas.2004944117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prufer K, de Filippo C, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338: 222–226. 10.1126/science.1224344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Fu Q, Aximu-Petri A, Glocke I, Nickel B, Arsuaga JL, Martínez I, Gracia A, de Castro JM, Carbonell E, et al. 2014. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505: 403–406. 10.1038/nature12788 [DOI] [PubMed] [Google Scholar]
- Meyer M, Arsuaga JL, de Filippo C, Nagel S, Aximu-Petri A, Nickel B, Martínez I, Gracia A, Bermudez de Castro JM, Carbonell E, et al. 2016. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531: 504–507. 10.1038/nature17405 [DOI] [PubMed] [Google Scholar]
- Mouttham N, Klunk J, Kuch M, Fourney R, Poinar H. 2015. Surveying the repair of ancient DNA from bones via high-throughput sequencing. BioTechniques 59: 19–25. 10.2144/000114307 [DOI] [PubMed] [Google Scholar]
- Orlando L, Ginolhac A, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, et al. 2013. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499: 74–78. 10.1038/nature12323 [DOI] [PubMed] [Google Scholar]
- Peyrégne S, Slon V, Mafessoni F, de Filippo C, Hajdinjak M, Nagel S, Nickel B, Essel E, Le Cabec A, Wehrberger K, et al. 2019. Nuclear DNA from two early Neandertals reveals 80,000 years of genetic continuity in Europe. Sci Adv 5: eaaw5873 10.1126/sciadv.aaw5873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505: 43–49. 10.1038/nature12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, de Filippo C, Grote S, Mafessoni F, Korlević P, Hajdinjak M, Vernot B, Skov L, Hsieh P, Peyrégne S, et al. 2017. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358: 655–658. 10.1126/science.aao1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch CM, Giddings I, Scholz M. 1998. Repair of degraded duplex DNA from prehistoric samples using Escherichia coli DNA polymerase I and T4 DNA ligase. Nucleic Acids Res 26: 857–859. 10.1093/nar/26.3.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Renaud G, Stenzel U, Kelso J. 2014. leeHom: adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res 42: e141 10.1093/nar/gku699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. 2012. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One 7: e34131 10.1371/journal.pone.0034131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin-Orlando A, Gamba C, Sarkissian CD, Ermini L, Louvel G, Boulygina E, Sokolov A, Nedoluzhko A, Lorenzen ED, Lopez P, et al. 2015. Pros and cons of methylation-based enrichment methods for ancient DNA. Sci Rep 5: 11826 10.1038/srep11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. 1997. Translesional synthesis on DNA templates containing a single abasic site: a mechanistic study of the “A rule”. J Biol Chem 272: 13916–13922. 10.1074/jbc.272.21.13916 [DOI] [PubMed] [Google Scholar]
- Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Pääbo S, Krause J, Jakobsson M. 2014. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc Natl Acad Sci 111: 2229–2234. 10.1073/pnas.1318934111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill Andrew J, Daza Riza M, Shendure J. 2016. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164: 57–68. 10.1016/j.cell.2015.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, Roz E, Cirenei N, Bellomi M, Pelosi G, et al. 2003. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol 21: 3902–3908. 10.1200/JCO.2003.02.006 [DOI] [PubMed] [Google Scholar]
- Strauss B, Rabkin S, Sagher D, Moore P. 1982. The role of DNA polymerase in base substitution mutagenesis on non-instructional templates. Biochimie 64: 829–838. 10.1016/S0300-9084(82)80138-7 [DOI] [PubMed] [Google Scholar]
- Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. 2006. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol 169: 400–404. 10.2353/ajpath.2006.060161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troll CJ, Kapp J, Rao V, Harkins KM, Cole C, Naughton C, Morgan JM, Shapiro B, Green RE. 2019. A ligation-based single-stranded library preparation method to analyze cell-free DNA and synthetic oligos. BMC Genomics 20: 1023 10.1186/s12864-019-6355-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.