

Tau pathology and vascular dysfunction are important contributors to Alzheimer's disease (AD), but vascular–tau associations and their effects on cognition are poorly understood.
Keywords: amyloid, cerebral blood flow, cognition, pericyte, PET imaging, tau
Abstract
Tau pathology and vascular dysfunction are important contributors to Alzheimer's disease (AD), but vascular–tau associations and their effects on cognition are poorly understood. We investigated these associations in male and female humans by conducting voxelwise comparisons between cerebral blood flow (CBF) and tau positron emission tomography (PET) images in independent discovery [cognitively normal (CN), 19; mild cognitive impairment (MCI) risk, 43; MCI, 6] and replication (CN,73; MCI, 45; AD, 20) cohorts. In a subgroup, we assessed relationships between tau and soluble platelet-derived growth factor β (sPDGFRβ), a CSF marker of pericyte injury. We tested whether CBF/sPDGFRβ–tau relationships differed based on Montreal Cognitive Assessment (MoCA) global cognition performance, or based on amyloid burden. Mediation analyses assessed relationships among CBF/sPDGFRβ, tau, and cognition. Negative CBF–tau correlations were observed predominantly in temporal-parietal regions. In the replication cohort, early negative CBF–tau correlations increased in spatial extent and in strength of correlation with increased disease severity. Stronger CBF–tau and sPDGFRβ–tau correlations were observed in participants with greater amyloid burden and lower MoCA scores. Importantly, when stratifying by amyloid status, stronger CBF–tau relationships in individuals with lower MoCA scores were driven by amyloid+ participants. Tau PET was a significant mediator CBF/sPDGFRβ–MoCA relationships in numerous regions. Our results demonstrate vascular–tau associations across the AD spectrum and suggest that early vascular–tau associations are exacerbated in the presence of amyloid, consistent with a two-hit model of AD on cognition. Combination treatments targeting vascular health, as well as amyloid-β and tau levels, may preserve cognitive function more effectively than single-target therapies.
SIGNIFICANCE STATEMENT Emerging evidence demonstrates a role for vascular dysfunction as a significant contributor to Alzheimer's pathophysiology. However, associations between vascular dysfunction and tau pathology, and their effects on cognition remain poorly understood. Multimodal neuroimaging data from two independent cohorts were analyzed to provide novel in vivo evidence of associations between cerebral blood flow (CBF), an MRI measure of vascular health, and tau pathology using PET. CBF–tau associations were related to cognition and driven in part by amyloid burden. Soluble platelet-derived growth factor β, an independent CSF vascular biomarker, confirmed vascular–tau associations in a subgroup analysis. These results suggest that combination treatments targeting vascular health, amyloid-β, and tau levels may more effectively preserve cognitive function than single-target therapies.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder with several biological hallmarks, including amyloid-β plaques and hyperphosphorylated tau tangles. Vascular dysfunction has gained traction as another important contributor to AD etiology (Rabin et al., 2018; Bos et al., 2019; Govindpani et al., 2019; Sweeney et al., 2019b). One measure of vascular health is cerebral blood flow (CBF), which decreases consistently throughout the progression of AD, and can be measured noninvasively with MRI (Binnewijzend et al., 2016; Benedictus et al., 2017; Sweeney et al., 2018). Conversely, amyloid-β and tau levels, measured in vivo by positron emission tomography (PET), increase throughout the course of AD, typically with earlier detection of increased amyloid-β deposition followed by elevated levels of tau. Despite seemingly anticorrelated trajectories of CBF and amyloid-β and tau PET measures, associations between these variables and their effects on clinical outcome, are not well understood.
A large body of work describes relationships between CBF and amyloid-β, including recent neuroimaging investigations in humans suggesting that decreased CBF is generally paralleled by increased amyloid-β deposition (Mattsson et al., 2014; McDade et al., 2014; Yan et al., 2018), as well as preclinical studies that indicate bidirectional interactions between amyloid-β and vascular dysfunction (for review, see Govindpani et al., 2019). However, studies focused on associations between tau and the vascular system are less common. Some recent preclinical research suggests that the tau protein also accumulates in cerebral vasculature in AD patients and animal AD models (Castillo-Carranza et al., 2017) and that tau overexpression in animals leads to abnormal angiogenesis (Bennett et al., 2018). Even less is known about the association between tau and vascular health in humans. One recent study indicated that heightened vascular risk factors may influence subsequent elevations in tau (Rabin et al., 2019), but no studies to date have directly investigated the regional association between tau levels and cerebral blood flow or CSF biomarkers of vascular dysfunction.
Despite the wealth of knowledge documenting the involvement of amyloid-β in AD, the causative role for amyloid-β is still open for debate. Other pathologies, such as tau, are consistently shown to be more strongly associated with AD-related outcomes like neurodegeneration and cognitive decline compared with amyloid-β (Hedden et al., 2013; Ossenkoppele et al., 2019). Therefore, the main focus of the present study was understanding the interactions between tau and vascular function on cognition, and also the influence of amyloid burden on these associations. In a subgroup analysis, the relationship between tau uptake and CSF soluble platelet-derived growth factor β (sPDGFRβ), a molecule released from injured pericytes, which are integral to vascular function (Montagne et al., 2015, 2020; Nation et al., 2019), was evaluated. We hypothesized that vascular dysfunction (decreased MRI CBF and increased CSF sPDGFRβ) would be associated with increased tau PET signal, which would predict global cognition, and that these associations would be stronger in individuals with significant amyloid accumulation.
Materials and Methods
Study design
To elucidate interactions between vascular dysfunction and pathologic tau accumulation, we conducted a human study comparing two independent markers of vascular health and tau levels in males and females using a discovery and replication design to ensure that the study findings were replicable and robust.
All data in the discovery cohort (n = 68), which included pooled data from two ongoing research initiatives, were collected at the University of Southern California (USC). Each participant received PET scans with [18F]flortaucipir (FTP) to measure tau and Neuraceq [florbetaben F 18 (FBB)] injection to measure amyloid-β, and a 3D pseudocontinuous arterial spin labeling (pCASL) MRI scan to measure CBF. All scans were conducted on the same PET and MRI scanners, respectively. A subset of the USC discovery cohort underwent lumbar punctures to measure CSF sPDGFRβ (n = 19), a molecule released from damaged pericytes that was recently shown to predict early cognitive decline, even before traditional AD markers (Nation et al., 2019).
Replication data were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) phase 3. The replication dataset consisted of ADNI participant demographics, MRI and PET imaging, and cognitive data (n = 138). Imaging data were selected to match the discovery cohort data as closely as possible with regard to the FTP PET tracer and ASL MRI sequences. Only ADNI participants with both a 3D pCASL and an FTP PET scan collected within a 12 month window (mean, 43.5 ± 53 d apart) were included in the replication cohort. All ADNI participants were also required to have a PET scan with either FBB (n = 60) or [18F]florbetapir (FBP; n = 78), for measuring amyloid-β collected within 12 months of the pCASL scans (mean, 35.1 ± 45 d apart). From the total of 154 participants meeting these criteria, 16 were excluded because of poor data quality, giving a total sample of n = 138.
Participants
USC discovery cohort
The study and procedures were approved by the Institutional Review Board (IRB) at USC, and informed consent was obtained from all participants before enrollment. Participants in the USC cohort were recruited from the greater Los Angeles area, and data were collected between October 2016 and November 2019. All participants were between the ages of 46 and 80 years and had no current or prior history of any major psychiatric illness, organ failure, epilepsy, and hydrocephalus. All participants received a battery of neuropsychological tests assessing attention/executive function and episodic memory and were categorized as cognitively normal (CN) if they scored within 1 SD of published normative values. If participants scored >1 SD outside the normal range on at least one test, they were categorized as being at risk for mild cognitive impairment (MCI-risk), as the 1 SD threshold has been suggested to have high predictive power to determine progression to dementia (Busse et al., 2006). A subset of participants (n = 31) received additional screening for MCI in accordance with National Alzheimer's Coordinating Center (NACC) guidelines (https://www.alz.washington.edu/). Briefly, a clinician or formal consensus panel determines an MCI diagnosis based on each annual visit. MCI criteria include abnormal cognition [global Clinical Dementia Rating (CDR) >0 and/or neuropsychological testing >1.5SD outside of normative values] and no diagnosis of clinical dementia. The NACC MCI diagnostic assessment also incorporates the Petersen criteria, similar to ADNI assessments. No participants had contraindications for MRI scanning.
ADNI replication cohort
ADNI protocols were approved by the IRB of all participating institutions, and written informed consent was obtained from all participants at each site. Data collection occurred between January 2017 and December 2019. Details of clinical diagnoses have been previously described (Petersen et al., 2010; for general methods, see http://adni.loni.usc.edu/adni-3/). Briefly, CN participants had a Mini-Mental State Examination (MMSE) score of ≥24 and a CDR of 0. MCI participants had an MMSE score of ≤26 and a CDR of 0.5. Participants with probable AD had an MMSE score between 20 and 24, and a CDR of 0.5 or 1.0. Additionally, MCI and AD participants were required to express both subjective memory concerns and objective memory loss based on scoring below adjusted cutoffs on the Logical Memory II subscale of the Wechsler Memory Scale–Revised.
Cognitive assessment
Global cognition was measured with the Montreal Cognitive Assessment (MoCA; Nasreddine et al., 2005), as this test was available for all participants across both cohorts.
Quantitative Western blotting of sPDGFRβ in human CSF
The quantitative Western blot analysis was used to detect sPDGFRβ in human CSF (in ng/ml), as previously reported (Montagne et al., 2015; Nation et al., 2019). Briefly, CSF samples and recombinant human PDGFRβ carrier-free protein (catalog #385-PR-100/CF, R&D Systems) were subjected to 4–12% Bis-Tris SDS/PAGE (Thermo Fisher Scientific) for 2 h at 150 V and subsequently were transferred to a nitrocellulose membrane. The membrane was blocked for 1 h with a superblock blocking buffer (catalog #37537, Thermo Fisher Scientific). The membrane was incubated with a primary antibody for PDGFRβ (catalog #AF1042, R&D Systems) overnight, then incubated with donkey anti-goat IgG secondary antibody (1:5000 dilution; catalog #A15999, Thermo Fisher Scientific) for 1 h at room temperature, treated with Western ECL detection solution (catalog #34075, Thermo Fisher Scientific), exposed to CL-Xposure film (catalog #34091, Thermo Fisher Scientific), and developed in an X-Omat 3000 RA film processor (Kodak). Images were acquired, and densitometry analysis was performed using National Institutes of Health ImageJ software.
MRI acquisition
USC discovery cohort
All MR images were acquired on a Siemens 3 T Prisma scanner. Structural T1-weighted (T1w) MPRAGE images were acquired with the following parameters: repetition time (TR)/echo time (TE), 2400/2.2 ms; field of view (FOV), 176 × 240 × 256 mm; resolution, 1.0 mm3 isotropic. 3D pCASL images were acquired with background suppression and the following parameters: TR/TE/post-label delay (PLD), 4300/36.7/2000 ms; FOV, 240 × 240 × 120 mm; resolution 2.5 mm3 isotropic; label duration, 1500 ms. Seven label–control image pairs were acquired, as well as an initial M0 image.
ADNI replication cohort
All MR images were acquired on a GE Discovery MR750 3 T scanner, with the exception of eight participants, whose data were acquired on a GE Signa Premier 3 T scanner. All images were acquired according to ADNI3 sequence parameter guidelines (http://adni.loni.usc.edu/adni-3). Briefly, structural T1w images were acquired with the following parameters: TR/TE, 2300 ms/min full echo; FOV, 208 × 240 × 256 mm; resolution, 1 mm3 isotropic. 3D pCASL images were acquired with the following parameters: TR/TE/PLD, 4885/10.5/2000 ms; FOV, 240 × 240 × 160 mm; resolution, 1.9 × 1.9 × 4 mm3; and label duration, 1800 ms.
PET image acquisition
All participants received an IV injection of the tracer outside of the scanner room. For FTP scans, tracer was injected (USC, 10.6 ± 1.2 mCi; ADNI, 10.2 ± 0.7 mCi), and 6 × 5 min frames were acquired beginning 75 min postinjection. For FBB and FBP scans, tracer was injected (USC FBB: −8.11 6 0.6 mCi; ADNI FBB, 7.96 6 0.6 mCi; ADNI FBP: −10.1 6 0.8 mCi) and 4 × 5 min frames were acquired beginning at 90 and 50 min postinjection for FBB and FBP scans, respectively.
Data processing
All data processing was performed using an automated in-house processing pipeline, as detailed below.
T1 MRI
Separate T1w MR templates were created for each cohort from T1w scans using the Advanced Normalization Tools (ANTs) package template building tool (Avants et al., 2010, 2011). Each participant's T1w image was processed with FreeSurfer version 6.0.0 (https://surfer.nmr.mgh.harvard.edu) to generate subject-specific regions of interest (ROIs).
3D PCASL
For USC data, motion correction was performed separately for label and control images by rigid body registration of individual frames to the first respective image. Perfusion-weighted images were obtained by simple subtraction of label–control pairs, followed by principal component analysis-based denoising. CBF time series images were computed based on the kinetic model recommended by Alsop et al. (2015) and averaged to create a mean CBF image. For ADNI data, CBF quantification was performed with the static perfusion-weighted and M0 images available through ADNI, using the same formula as above. For each participant, the M0 image was coregistered to the T1w image, moved into template space, and smoothed with a 5 mm Gaussian kernel. To obtain a global gray matter CBF measure, T1w images were segmented with SPM12 to produce probabilistic tissue maps. The gray matter probabilistic map was thresholded at 60% to reduce partial volume effects (PVEs) and resampled to the resolution of the CBF image, and the average gray matter CBF was extracted in native CBF space. Discovery CBF data were corrected for PVE assuming 2.5 times greater perfusion in gray matter compared with white matter (Johnson et al., 2005). To control for intersubject variability in CBF related to potential confounding factors (e.g., caffeine intake, anxiety, movement), parametric CBF images were normalized by dividing each CBF voxel by the average global gray matter CBF (Aslan and Lu, 2010; Lacalle-Aurioles et al., 2013).
PET
Dynamic FTP, FBB, and FBP images were motion corrected by aligning each frame to an average image. Motion-corrected PET frames were then averaged, coregistered to the T1w image, moved into template space, and smoothed with an 8 mm Gaussian kernel. For FTP data, inferior cerebellar gray matter was used as a reference region (Baker et al., 2017). Cerebellar segmentation was performed with SUIT (spatially unbiased atlas template of the cerebellum and brainstem; http://www.diedrichsenlab.org/imaging/suit.htm), and dorsal regions were removed from the cerebellar ROI. For FBB and FBP data, the whole cerebellum was used as a reference region. Bilateral cerebellar gray and white matter labels from FreeSurfer were combined and eroded by 1 voxel to reduce PVE, then moved into PET space. The average PET signal was extracted from reference ROIs in native PET space. Parametric Parametric FTP standardized uptake value ratio (SUVR) images were created by dividing the PET signal in each voxel by the average signal in the respective reference regions. FTP PET data from the discovery cohort were corrected for PVE with the region-based voxelwise technique, based on the geometric transfer matrix method (Rousset et al., 1998) incorporated in FreeSurfer version 6.0.0 (Greve et al., 2016). To characterize general levels of PET binding, composite ROIs were created. For FTP, two composite ROIs were created to reproduce Braak stage 1-2 and Braak stage 3-4 areas consistent with neuropathological observations. Regions were created according to FreeSurfer labels as defined in Baker et al. (2017). For FBB and FBP, the composite ROI consisted of frontal, parietal, lateral temporal, and cingulate cortices [Landau S, Jagust W, unpublished observations (https://adnibitbucketio/reference/docs/UCBERKELEYAV45/ADNI_AV45_Methods_JagustLab_062515pdf)]. A modified level 3 calibration was used to convert cortical FBB and FBP uptake to Centiloids (CLs) using the formulas given by ADNI (see below). An amyloid CL value of 19 was used as the cutoff for amyloid positivity, as the rate of change in amyloid PET has been shown to reliably increase at this level (Jack et al., 2017).
Amyloid PET Centiloid calibration (level 3)
We performed a modified level 3 CL calibration to verify agreement between a previously validated CL processing method (ADNI, which uses SPM8 for preprocessing of the data) and our processing pipeline (which uses ANTs).
FBP PET and structural MR images for 76 participants, and FBB PET and MR images for 49 participants were downloaded from the ADNI Image Database Archive (IDA). We also downloaded the final SUVR data processed through the ADNI pipeline, which is also available on the IDA. We created cortical SUVR values using the same reference and cortical regions described by ADNI (Landau S, Jagust W, unpublished observations) our nonstandard preprocessing pipeline using the steps described in the Data processing subsection of this article. We subsequently compared our FBB and FBP cortical SUVR values to those obtained from the ADNI processing, using a linear model as suggested in the study by Klunk et al. (2015). We showed that our pipeline did not introduce errors into the data, and the comparisons between SUVR values derived from our pipeline and those derived from the ADNI pipeline are shown Extended Data Fig. 1-1. These equations for each tracer were then used to convert our SUVR values to ADNI-equivalent standard SUVR values, which were then converted to CL values using the equations determined for ADNI data (Kolibash SA, Minhas D Lopresti BJ, unpublished observations, FBB SUVR to CL: CLFBB = 159.08 × SUVRFBB – 151.65; FBP SUVR to CL: CLFBP = 196.9 × SUVRFBP – 196.03). We then plotted all CL values obtained from our processing pipeline against the values obtained through ADNI (Extended Data Fig. 1-2). The fit between the standard CL values and our values was well within the ranges given by Klunk et al. (2015; i.e., slope = 0.997 and r2 = 0.986). Therefore, the CL values reported in the current article meet the specifications delineated by the Centiloid Project.
Experimental design and statistical analyses
Differences in participant characteristics between diagnostic groups within cohorts were assessed with Kruskal–Wallis tests for continuous variables, followed by Bonferroni-corrected post hoc Dunn's tests to decompose statistically significant results. Differences between cohorts were assessed with Quade tests (correcting for diagnosis) for continuous variables, and with χ2 tests for categorical variables.
In the discovery analysis, we performed voxelwise partial correlations between normalized CBF images and FTP SUVR images using the Biological Parametric Mapping toolbox (Yang et al., 2011). Analyses were spatially restricted with a gray matter mask, which was obtained by segmentation of the MR template. For the discovery cohort, correlations between CBF and FTP PET images were compared with or without incorporating PVE correction. Because the results were largely overlapping, all subsequent analyses were conducted with non-PVE-corrected data. For the replication analyses, a binarized mask of significant clusters from the discovery analysis was used to restrict the search volume of the voxelwise replication analysis. Discovery and replication analyses were corrected for age, sex, amyloid burden, and diagnosis.
To probe CBF–tau associations that extended outside of the search volume restricted by the binarized mask used in the replication analyses, we conducted secondary analyses in the ADNI cohorts using a gray matter mask. Secondary analyses were conducted across all participants within a cohort, as well as stratified by diagnosis to determine whether certain patterns of CBF–tau associations were more specific to a given diagnostic group. Because of the relatively small sample size of AD patients, AD and MCI groups were combined. All analyses were corrected for age, sex, amyloid burden, and diagnosis if the analysis contained more than one diagnostic group. A voxelwise threshold of p < 0.001 and a false discovery rate (FDR)-corrected cluster-level threshold of p < 0.05 were used to determine significance for all voxelwise analyses.
Average FTP SUVR and CBF values were extracted from significant clusters in all voxelwise partial correlations for further analysis. To reduce the number of ROIs for multiple comparisons, bilateral clusters and clusters within the same anatomic region [e.g., left and right inferior parietal lobe (IPL)] were combined into one ROI. To correct for potential confounding effects of atrophy, the average gray matter volume was extracted from each ROI by moving the ROI from template space into subject space and multiplying with the gray matter probability from SPM segmentation.
For all voxelwise analyses, we conducted follow-up tests to determine whether the slope of CBF–tau associations differed as a function of cognitive performance. To this end, we conducted univariate general linear model (GLM) analyses with MoCA and regional CBF as independent variables, with a MoCA * CBF interaction term. Regional FTP SUVR identified from the discovery/replication or secondary gray matter masked analyses was the dependent variable, which were run in parallel models. Age, sex, education, diagnosis, amyloid burden, and gray matter volume were included as covariates. GLM analyses were also used to investigate the effect of amyloid burden on CBF–tau associations. Amyloid CL and regional CBF were entered as independent variables, with an amyloid * CBF interaction term. Regional FTP SUVR identified from the discovery/replication or secondary gray matter masked analyses was entered as the dependent variable. Age, sex, diagnosis, amyloid burden, and gray matter volume were included as covariates. In the subgroup of USC participants with CSF sPDGFRβ data, we used ROIs from the discovery analysis to extract FTP SUVR to conduct sPDGFRβ–tau partial correlations and GLM analyses analogous to those above. The GLMs included FTP SUVR as the dependent variable, CSF sPDGFRβ as the independent variable, as well as MoCA * sPDGFRβ and amyloid-β * sPDGFRβ interaction effects in parallel models. Significance for interaction effects in all analyses was set at p < 0.05, FDR corrected.
To further examine the effects of amyloid, MoCA * CBF interactions were assessed independently in amyloid+ and amyloid– groups. Stratified analyses for amyloid positivity were not conducted for MoCA * sPDGFRβ interactions because of the limited number of participants with sPDGFRβ data.
To better understand the order of events between vascular markers (CBF/sPDGFRβ), tau, and cognition, we conducted causal mediation analyses. We analyzed the following two hypothesized pathologic models leading to impaired cognition: (1) CBF/sPDGFRβ → tau PET → MoCA (tau mediates vascular dysfunction); and (2) tau PET → CBF/sPDGFRβ → MoCA (vascular dysfunction mediates tau). Mediation models were compared using the entire group sample, as well as between amyloid+ and amyloid– groups. Age, sex, diagnosis, amyloid burden, gray matter volume, and education were included as covariates. Significance for mediation effects in all analyses was set at p < 0.05, FDR corrected. Tests assessing differences between and within groups and the GLM analyses following voxelwise comparison were conducted with SPSS. Causal mediation analyses were performed using the “mediate” function of the “mediation” package in R (https://cran.rstudio.com/web/packages/mediation/mediation.pdf).
Finally, as an exploratory analysis and to compare our results to previously published studies investigating CBF–amyloid relationships (Mattsson et al., 2014; McDade et al., 2014; Yan et al., 2018), we performed voxelwise partial correlations between normalized CBF images and FBB SUVR images. Analyses were spatially restricted with a gray matter mask, and age, sex, Braak stage 1-2 FTP SUVR, and diagnosis were included as covariates. Analyses were conducted in the USC cohort (n = 68), and in a subgroup of the ADNI dataset for which FBB PET scans were available (n = 60).
Data availability
The data that support the findings of this study will be made available on request to the corresponding author in accordance with the existing data-sharing agreement with the National Institute on Aging and the ADNI committee.
Results
Participant characteristics
All participant characteristics are shown in Table 1. In the USC discovery cohort, there were 19 CN (average ± SD, 62.7 ± 9.1 years of age), 43 MCI-risk (66.6 ± 6.9), and 6 MCI (68.7 ± 5.5) participants. In the ADNI replication cohort, there were 73 CN (72.7 ± 6.6), 45 MCI (74.3 ± 7.5), and 20 AD (76.6 ± 7.3) participants.
Table 1.
Participant characteristics
| USC discovery cohort (n = 68) |
ADNI replication cohort (n = 138) |
|||||
|---|---|---|---|---|---|---|
| CN (n = 19) | MCI-risk (n = 43) | MCI (n = 6) | CN (n = 73) | MCI (n = 45) | AD (n = 20) | |
| Age (years)c | 62.7 ± 9.1 | 66.6 ± 6.9 | 68.7 ± 5.5 | 72.7 ± 6.6 | 74.3 ± 7.5 | 76.6 ± 7.3 |
| Sexc | 16 F (84%) | 30 (70%) | 4 F (67%) | 39 F (53%) | 18 (40%) | 6 (29%) |
| Global GM CBFc | 40.4 ± 6.4 | 41.0 ± 8.6 | 40.1 ± 14 | 48.3 ± 11 | 43.5 ± 11 | 40.7 ± 16a |
| Education (years) | 16.9 ± 1.6 | 16.9 ± 2.5 | 15.5 ± 2.5 | 16.8 ± 2.4 | 16.6 ± 2.7 | 16.0 ± 2.6 |
| APOE4 carrier* | 11 (61%) | 11 (26%)a | 4 (67%)b | 24 (34%) | 11 (27%) | 8 (44%) |
| MoCA | 28.3 ± 1.5 | 26.5 ± 2.5a | 20.3 ± 4.2a,b | 25.8 ± 2.6 | 23.2 ± 3.2a | 17.8 ± 4.4a,b |
| Amyloid (CLs) | 13.1 ± 15 | 17.1 ± 26 | 59.6 ± 44 | 20.9 ± 30 | 31.9 ± 44 | 89.7 ± 35a,b |
| Braak stage 1/2 FTP SUVR | 1.15 ± 0.1 | 1.19 ± 0.1 | 1.54 ± 0.3a,b | 1.19 ± 0.1 | 1.22 ± 0.2 | 1.40 ± 0.2 |
| Braak stage 3/4 FTP SUVR | 1.13 ± 0.1 | 1.13 ± 0.1 | 1.52 ± 0.4a,b | 1.13 ± 0.1 | 1.19 ± 0.2 | 1.50 ± 0.3a,b |
| Injected dose – FTP (mCi)c | 10.5 ± 0.4 | 10.6 ± 1.5 | 10.4 ± 0.6 | 10.1 ± 0.8 | 10.2 ± 0.5 | 10.4 ± 0.3a,b |
F, Female.
aSignificantly different from CN group within cohort, p < 0.05.
bSignificantly different from MCI-risk (USC) or MCI (ADNI) group within cohort, p < 0.05.
cSignificant difference between USC and ADNI cohort, p < 0.05.
*Data unavailable for 10 participants.
Comparisons between cohorts
Compared with the USC discovery cohort, the ADNI replication cohort had a higher frequency of males (χ2(1) = 15.0, p < 0.001), higher global CBF (F(1,205) = 4.04, p = 0.046), older age (F(1,205) = 10.2, p = 0.002), and higher injected FTP dose (F(1,205) = 5.97, p = 0.015).
Comparisons between diagnoses within cohort
Within the USC discovery cohort, MoCA score (χ2(2) = 17.6, p < 0.001), Braak stage 1-2 FTP SUVR (χ2(2) = 11.0, p = 0.04), Braak stage 3-4 FTP SUVR (χ2(2) = 8.77, p = 0.012), and frequency of APOE4 carriers (χ2(2) = 8.90, p = 0.012) were significantly different across groups. Post hoc tests demonstrated lower MoCA scores and higher Braak stage 1-2/3-4 FTP SUVR in MCI patients compared with the CN groups (p < 0.001, p = 0.003, p = 0.012) and MCI-risk groups (p = 0.019, p = 0.014, p = 0.017), lower MoCA scores in MCI-risk compared with CN participants (p = 0.029), and lower frequency of APOE4 carriers in MCI-risk patients (p = 0.003). Within the ADNI replication cohort, global gray matter CBF (χ2(2) = 12.7, p = 0.002), MoCA (χ2(2) = 48.7, p < 0.001), FTP injected dose (χ2(2) = 6.36, p = 0.042), amyloid burden (χ2(2) = 33.2, p < 0.001), Braak stage 1-2 FTP SUVR (χ2(2) = 17.1, p < 0.001), and Braak stage 3-4 FTP SUVR (χ2(2) = 29.9, p < 0.001) were significantly different across groups. Post hoc tests revealed that global CBF was significantly higher in CN compared with AD patients (p = 0.003); MoCA scores were higher in CN compared with both MCI patients (p < 0.001) and AD patients (p < 0.001), and were higher in MCI compared with AD patients (p = 0.002); and amyloid burden and Braak stage 1-2/3-4 FTP SUVRs were higher in AD compared with MCI groups (p values < 0.003) and CN groups (p values < 0.001), respectively.
CBF and sPDGFRβ are associated with tau
Discovery analysis
In the discovery cohort, we observed several significant negative correlations between CBF and FTP SUVR in cortical regions, including inferior temporal gyrus (ITG), middle temporal gyrus (MTG), superior temporal gyrus (STG), and superior frontal gyrus (SFG; Fig. 1A, Extended Data Fig. 1-3A).
Figure 1.

CBF and tau are negatively correlated in discovery and replication analyses. A, Clusters shown in blue color scale depict significant negative correlations between CBF (normalized by global gray matter CBF) and FTP SUVR in the discovery cohort (n = 68). B, Blue clusters show significant CBF–tau correlations in the ADNI replication cohort (n = 138), masked with significant results from the discovery analysis (A). All analyses were covaried for age, sex, diagnosis, and amyloid CL. CL calibration is shown in Extended Data Figures 1-1 and 1-2. For visualization purposes, average values from selected significant clusters were extracted, adjusted for covariates, and plotted below each analysis. Coordinates and statistics for all significant clusters are listed in Extended Data Figure 1-3. Note: because the voxelwise statistical threshold was set at p < 0.001, the p value for each of the plots is <0.001.
A, B, Scatterplots of ADNI standard SUVR values for FBP (A) and FBB (B) scans. Download Figure 1-1, TIF file (1.7MB, tif) .
Scatterplot of ADNI standard CL values against CL values obtained without processing pipeline. Download Figure 1-2, TIF file (2.9MB, tif) .
A, B, Coordinates of clusters showing significant negative relationships between [18F]flortaucipir and CBF in the USC discovery (A; n = 68) and ADNI replication cohorts (B; n = 138). Download Figure 1-3, DOCX file (15.1KB, docx) .
No significant positive correlations were observed. Using regions showing significant CBF–tau correlations as ROIs, we found significant positive associations between CSF sPDGFRβ and FTP SUVR in ITG/MTG (r = 0.642, p = 0.040), MTG/STG (r = 0.592, p = 0.040), and MTG (r = 0.578, p = 0.040) regions.
Replication analysis
The majority of significant negative CBF–tau correlations observed in lateral temporal areas in the discovery analysis were replicated in the ADNI cohort (Fig. 1B, Extended Data Fig. 1-3A). There were no significant positive CBF–tau correlations.
Secondary analyses of ADNI cohort
Using a gray matter mask to restrict the search volume, we observed significant negative CBF–tau associations in numerous cortical regions, including temporal, parietal, and frontal areas (Fig. 2A, Extended Data Fig. 2-1A). Within the AD-MCI subgroup (n = 65), significant negative correlations between CBF and FTP SUVR were observed in regions similar to those in the whole-group analysis (Fig. 2B, Extended Data Fig. 2-1B). Within the CN subgroup (n = 73), significant negative CBF–tau associations were observed in a smaller subset of regions observed in the whole-group analysis, including dorsolateral prefrontal cortex (dlPFC), temporal-parietal cortex, and lateral parietal areas (Fig. 2C, Extended Data Fig. 2-1C). No positive correlations were detected for any of the secondary CBF–tau analyses.
Figure 2.
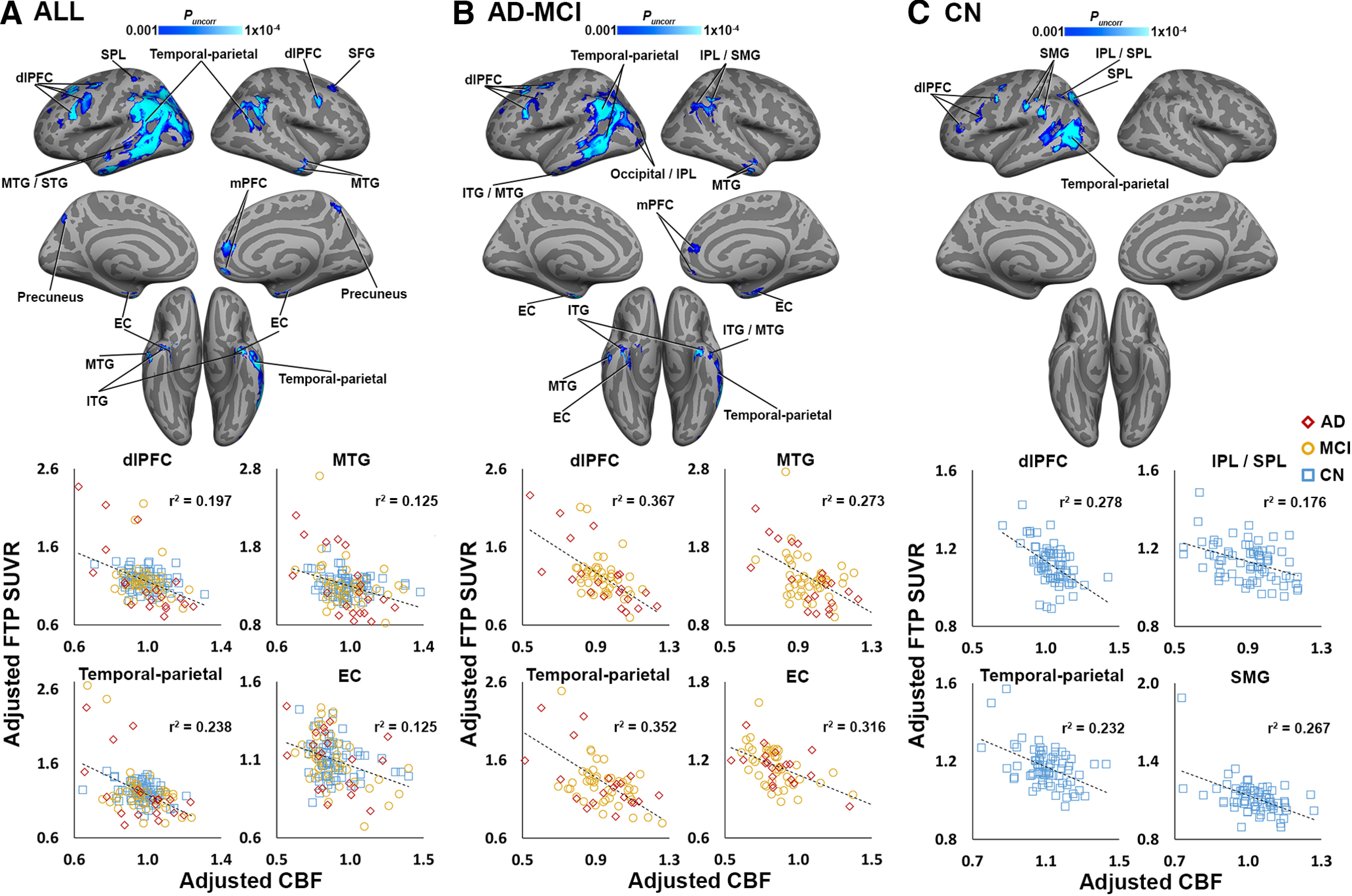
CBF and tau are negatively correlated in secondary gray matter-masked ADNI analyses. A, Clusters shown in blue color scale depict significant negative correlations between CBF (normalized by global gray matter CBF) and FTP SUVR across the whole group (n = 138). B, Blue clusters show significant CBF–tau correlations in the ADNI AD-MCI subgroup (n = 65). C, Significant negative CBF–tau correlations in the ADNI CN subgroup (n = 73). All analyses were covaried for age, sex, and amyloid CL (and diagnosis in A and B). For visualization purposes, average CBF and FTP SUVRs from selected significant clusters in the voxelwise analysis were extracted, adjusted for covariates, and plotted below voxelwise results. Coordinates and statistics for all significant clusters are listed in Extended Data Figure 2-1. Note: because the voxelwise statistical threshold was set at p < 0.001, the p value for each of the plots is <0.001. EC – entorhinal cortex; IPL/SPL – inferior/superior parietal lobe; SMG – supramarginal gyrus; mPFC – medial prefrontal cortex.
Coordinates of clusters showing significant negative relationships between [18F]flortaucipir and CBF in the secondary, gray matter-masked analyses of the ADNI FTP cohort (n = 138). Download Figure 2-1, DOCX file (18.9KB, docx) .
CBF- and sPDGFRβ–tau correlations differ depending on cognitive performance
CBF–tau correlations
From the USC discovery analysis, significant MoCA * CBF interaction effects on FTP SUVR were observed in all five lateral temporal and frontal ROIs (Fig. 3A, Extended Data Fig. 3-1A), indicating that individuals with poorer global cognition have a steeper negative relationship between CBF and tau levels. In the replication analysis, both MTG/STG and ITG ROIs exhibited a significant MoCA * CBF interaction (Fig. 3B, Extended Data Fig. 3-1B). In the secondary analysis of the ADNI cohort, significant MoCA * CBF interaction effects on FTP SUVR were detected in all temporal, parietal, and frontal regions showing significant negative CBF–tau associations (Fig. 2A), with the exception of entorhinal cortex (EC; Fig. 3C, Extended Data Fig. 3-1C) across all participants. In the AD-MCI ADNI subgroup analysis, significant interactions between MoCA performance and CBF were observed in many of the same regions observed in the whole group (Extended Data Figs. 3-1C, 3-2A). In the CN ADNI subgroup, a significant MoCA * CBF interaction was observed only in the temporal-parietal cortex (Extended Data Figs. 3-1C, 3-2B).
Figure 3.

Participants with low global cognition show stronger relationships between CBF and sPDGFRβ. A–D, MoCA * CBF interaction effects on FTP SUVR in discovery (A), replication (B), and secondary GM-masked ADNI (C) analyses. MoCA * sPDGFRβ interaction effects on FTP SUVR from discovery analysis ROIs (D). Average CBF and FTP from significant clusters in the voxelwise analysis, or CSF sPDGFRβ, were entered into GLMs. The predicted FTP SUVRs from the models are plotted against regional CBF (normalized by global gray matter CBF) or sPDGFRβ (D). All analyses were covaried for age, sex, diagnosis, gray matter volume, and amyloid CL. MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA score group (shown in purple) and a high-MoCA score group (shown in green). Detailed statistics for all MoCA * CBF/sPDGFRβ interaction terms are listed in Extended Data Figure 3-1. Plots for AD-MCI and CN subgroups of the ADNI replication cohort are displayed in Extended Data Figure 3-2. Note: the p values shown on the scatterplots is for display purposes only and is not a statistical result from the primary GLM analyses including MoCA as a continuous variable.
MoCA * CBF/sPDGFRβ interactions. S1, Primary somatosensory cortex; SPL, superior parietal lobe. Download Figure 3-1, DOCX file (19KB, docx) .
Participants with low global cognition show stronger relationships between CBF and sPDGFRβ in ADNI subgroup analyses. A, B, MoCA * CBF interaction effects on FTP SUVR in secondary GM-masked ADNI AD-MCI (A) and CN (B) subgroups. Average CBF and FTP from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, gray matter volume, amyloid CL, and diagnosis (in A). MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA group (shown in purple) and a high-MoCA group (shown in green). Download Figure 3-2, TIF file (4.5MB, tif) .
sPDGFRβ–tau correlations
Significant MoCA * sPDGFRβ interactions were detected in ITG, MTG, and STG regions (Fig. 3D, Extended Data Fig. 3-1D), indicating that individuals with poorer cognitive performance exhibit a steeper positive relationship between tau and pericyte deficiency.
CBF–tau and sPDGFRβ–tau correlations are affected by amyloid levels
CBF–tau correlations
From the USC discovery analysis, significant amyloid * CBF interaction effects on FTP SUVR were observed in three of the five lateral temporal and frontal ROIs (Fig. 4A, Extended Data Fig. 4-1A), which indicates that higher amyloid levels are reflected by stronger negative CBF–tau relationships. In the replication analysis, only MTG/STG exhibited a significant amyloid * CBF interaction (Fig. 4B, Extended Data Fig. 4-1B). From the secondary analysis of the ADNI cohort, significant amyloid * CBF interaction effects on FTP SUVR were detected in all ROIs except primary somatosensory cortex, medial prefrontal cortex (mPFC), ITG, and MTG/STG (Fig. 4C, Extended Data Fig. 4-1C). In the AD-MCI ADNI group analysis, a significant amyloid * CBF interaction was observed only in the IPL/ supramarginal gyrus (SMG) ROI (Extended Data Figs. 4-1C, 4-2A). In the CN ADNI group, significant amyloid * CBF interactions were observed in all ROIs except dlPFC (Extended Data Figs. 4-1C, 4-2B).
Figure 4.

Participants with high amyloid burden show stronger relationships between CBF and sPDGFRβ. A–C, Amyloid * CBF interaction effects on FTP SUVR in discovery (A), replication (B), and secondary GM-masked ADNI (C) analyses. D, Amyloid * sPDGFRβ interaction effects on FTP SUVR from discovery analysis ROIs. Average CBF and FTP SUVR from significant clusters in the voxelwise analysis, or CSF sPDGFRβ, were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF) or sPDGFRβ (D). All analyses were covaried for age, sex, diagnosis, gray matter volume, and amyloid CL. Amyloid CL was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-amyloid group (shown in blue) and a high-amyloid group (shown in red). Detailed statistics for all amyloid * CBF/sPDGFRβ interaction terms are listed in Extended Data Figure 4-1. Plots for AD-MCI and CN subgroups of the ADNI replication cohort are displayed in Extended Data Figure 4-2. Note: the p values shown on the scatterplots is for display purposes only and is not a statistical result from the primary GLM analyses including amyloid CL as a continuous variable.
Amyloid * CBF/sPDGFRβ interactions. *Significant at p < 0.05, uncorrected Download Figure 4-1, DOCX file (18.7KB, docx) .
Participants with high amyloid burden show stronger relationships between CBF and sPDGFRβ in ADNI subgroup analyses. A, B, Amyloid * CBF interaction effects on FTP SUVR in secondary GM-masked ADNI AD-MCI (A) and CN (B) subgroups. Average CBF and FTP SUVR from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, gray matter volume, amyloid CL, and diagnosis (in A). Amyloid CL was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-amyloid group (shown in blue) and a high-amyloid group (shown in red). Download Figure 4-2, TIF file (8.5MB, tif) .
sPDGFRβ–tau correlations
Significant amyloid * sPDGFRβ interactions were detected in ITG, MTG, and STG regions (Fig. 4D, Extended Data Fig. 4-1D), demonstrating that individuals with higher amyloid burden exhibit a steeper positive relationship between tau levels and pericyte deficiency.
Interactions between cognitive performance and CBF–tau correlations are driven by amyloid positivity
Based on our results suggesting that both cognitive performance and amyloid burden affect the relationship between vascular markers and tau levels (Figs. 3, 4), we wanted to further determine whether the MoCA * CBF interactions differed as a function of amyloid positivity. When groups were stratified by amyloid positivity (>19 CLs), we observed significant MoCA * CBF interactions only in amyloid+ groups. These were present in the MTG/STG ROI from the replication analysis; MTG/STG, SFG, and temporal-parietal ROIs from the secondary analysis in the ADNI group (Fig. 5, Extended Data Fig. 5-1B,C); and the ITG/MTG ROI from the ADNI AD-MCI subgroup (Extended Data Figs. 5-1B, 5-2). Several additional regions in the amyloid+ group showed significant MoCA * CBF interactions but did not survive multiple-comparisons correction (Extended Data Fig. 5-1).
Figure 5.

Interactions between MoCA and CBF are stronger in amyloid+ individuals. Amyloid * CBF interaction effects on FTP SUVR in replication (A), and secondary GM-masked ADNI (B) analyses, split by amyloid status (amyloid+ on the left, amyloid– on the right). Average CBF and FTP SUVR from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). All analyses were covaried for age, sex, diagnosis, gray matter volume, and amyloid CL. MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA group (shown in orange) and a high-MoCA group (shown in green). Detailed statistics for all MoCA * CBF/sPDGFRβ interaction terms for amyloid+ and amyloid– groups are listed in Extended Data Figure 5-1. Plots for the AD-MCI subgroup of the ADNI replication cohort are displayed in Extended Data Figure 5-2. Note: the p values shown on the scatterplots is for display purposes only and is not a statistical result from the primary GLM analyses including MoCA as a continuous variable.
MoCA * CBF/sPDGFRβ interactions split by amyloid status. *Significant at p < 0.05, uncorrected. Download Figure 5-1, DOCX file (20.3KB, docx) .
Interactions between MoCA and CBF are stronger in amyloid+ individuals in ADNI subgroup analyses. Amyloid * CBF interaction effects on FTP SUVR in the secondary GM-masked ADNI AD-MCI subgroup, split by amyloid status (amyloid+ on the left, amyloid– on the right). Average CBF and FTP SUVRs from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, diagnosis, gray matter volume, and amyloid CL. MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA group (shown in orange) and a high-MoCA group (shown in green). Download Figure 5-2, TIF file (2.3MB, tif) .
Tau mediates the association between CBF/sPDGFRβ and cognition
To investigate the order of events related to tau accumulation and vascular dysfunction on cognition, we conducted parallel causal mediation models following two potential pathways leading to impaired cognition. Tau was identified as a significant mediator in the pathway from CBF/sPDGFRβ to MoCA performance for a majority of the ROIs examined, with significant mediating effects present in all subgroups except for the ADNI CN group (Fig. 6A, Extended Data Fig. 6-1). CBF/sPDGFRβ was a significant mediator between tau SUVR and MoCA only for the mPFC in the ADNI AD-MCI group (Extended Data Fig. 6-2). When assessing the mediating effects of tau on the CBF–MoCA pathway separately in amyloid+ and amyloid– groups, we discovered significant effects only for the amyloid+ group, in almost of all the same regions showing significance in the analysis combining all participants (Fig. 6B, Extended Data Fig. 6-3), which showed that the effects were largely driven by the amyloid+ participants.
Figure 6.

In amyloid+ individuals, tau SUVR mediates the relationship between CBF/sPDGFRβ and MoCA. A, B, Mediation models assessing the relationship among CSF/sPDGFRβ, FTP SUVR, and MoCA in the whole USC (n = 68) or ADNI (n = 138) cohorts (A), and within the cohorts split by amyloid status (B). The a, b, and c′ path values are shown for one representative region, the temporal-parietal cortex, identified by the white stars on the surface projections below. Each region exhibiting a significant mediating effect is shown in red-yellow color scale on the surface projections. Mediation effects for the pathway displayed here (CBF/sPDGFRβ → tau PET → MoCA) were substantially stronger than for the alternative pathway (tau PET → CBF/sPDGFRβ → MoCA). Detailed results from the mediation analyses for model 1 and model 2, and model 1 split by amyloid status are listed in Extended Data Figures 6-1, 6-2, and 6-3.
Mediation analyses from model 1 (CBF/sPDGFRβ → tau → MoCA). *Significant at p < 0.05, uncorrected. Download Figure 6-1, DOCX file (19.2KB, docx) .
Mediation analyses from model 1 (tau → CBF/sPDGFRβ → MoCA). Significant at p < 0.05, uncorrected. Download Figure 6-2, DOCX file (19.2KB, docx) .
Mediation analyses from model 1 (CBF/sPDGFRβ → Tau → MoCA) split by amyloid status. *Significant at p < 0.05, uncorrected. Download Figure 6-3, DOCX file (20.7KB, docx) .
CBF is associated with amyloid-β
In confirmatory analyses of previous studies, we observed significant negative correlations between FBB SUVR and CBF in several cortical regions (Fig. 7). Similar to associations between CBF and tau, we observed several negative correlations in lateral temporal and frontal areas in both USC and ADNI groups, although the spatial extent of the associations was largely reduced. In general, associations were stronger and more widespread in less impaired groups (Fig. 7A,D) compared with more impaired groups (Fig. 7C). In the ADNI cohort, a few significant positive CBF–FBB associations were observed in posterior cingulate cortex, orbitofrontal cortex, and fusiform gyrus that were likely driven by the AD-MCI subgroup (data not shown).
Figure 7.
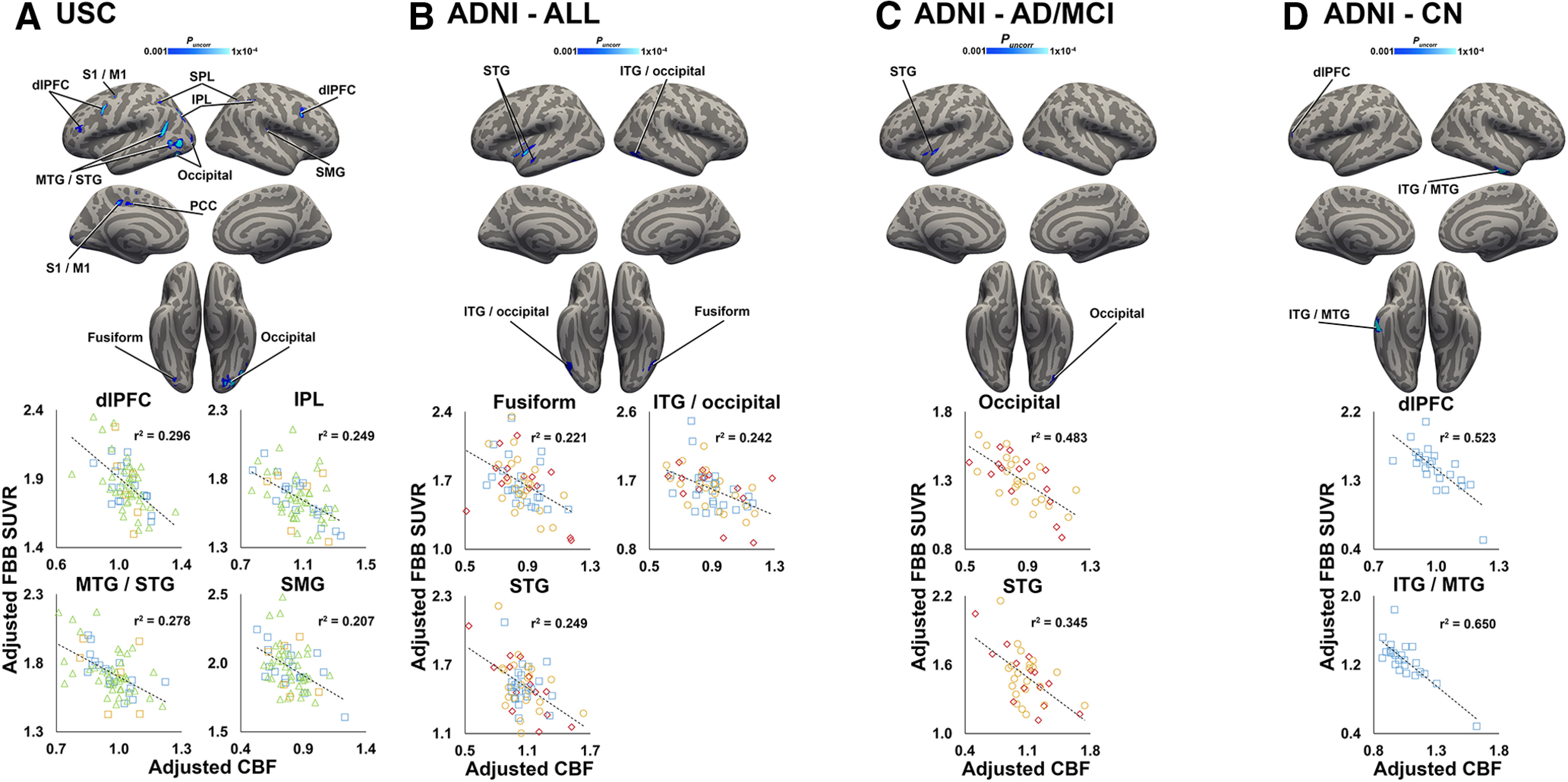
CBF and amyloid-β are negatively correlated. A–D, Clusters shown in blue colorscale depict significant negative correlations between CBF (normalized by global gray matter CBF) and FBB SUVR in the USC cohort (A; n = 68), in the entire ADNI cohort (B; n = 60), and in ADNI AD-MCI (C; n = 36) and CN (D; n = 24) subgroups. All analyses were covaried for age, sex, Braak stage 1-2 FTP SUVR, and diagnosis (A–C only). For visualization purposes, average values from selected significant clusters were extracted, adjusted for covariates, and plotted below each analysis. Note: because the voxelwise statistical threshold was set at p < 0.001, the p value for each of the plots is <0.001. S1/M1, primary somatosensory/motor cortex.
Discussion
In the current study, we showed the following: (1) that two markers of vascular health (MRI CBF and CSF sPDGFRβ) were significantly associated with tau PET levels, and that this effect was present in two independent cohorts; (2) that linear CBF–tau/sPDGFRβ–tau PET relationships had steeper slopes as a function of poorer cognition and higher amyloid burden in independent analyses; (3) that MoCA–CBF/sPDGFRβ interactions on tau PET appeared to be driven by amyloid positivity; (4) that causal mediation analyses suggested that vascular dysfunction may relate to cognitive dysfunction through a tau-mediated pathway, particularly in the presence of amyloid positivity; and (5) that CBF–tau associations are stronger than and spatially distinct from CBF–amyloid associations. Together, these results show important associations among the following three key pathologic factors in AD: tau, amyloid-β, and vascular dysfunction.
A major finding of the current study is that several cortical regions showed negative CBF–tau PET associations (Figs. 1, 2), which were largely localized to temporal-parietal and frontal areas. Although there are no published studies reporting correlations between elevated tau PET signal and decreased ASL-derived CBF to our knowledge, support for these associations comes from PET studies using tau ligands and [18F]fluorodeoxyglucose (FDG), which is a marker of glucose metabolism, a process coupled to CBF. Previous studies documented negative tau–FDG PET relationships within regions similar to those reported in the current study (Saint-Aubert et al., 2016; Hanseeuw et al., 2017; Kang et al., 2017; Whitwell et al., 2018), which may indicate vascular dysfunction [e.g., impaired glucose transport across the blood–brain barrier (BBB)], in addition to neuronal dysfunction (Sweeney et al., 2019a). It is noteworthy that although CBF measured with ASL MRI and FDG PET are generally positively correlated spatially and show similar diagnostic accuracy for AD (Chen et al., 2011; Musiek et al., 2012), several studies reported large variability in regional correlations across modalities (Cha et al., 2013; Yan et al., 2018), emphasizing differences between the two measures. Importantly, the regions showing negative CBF–tau associations in the current study consistently exhibited hypoperfusion, as measured with ASL MRI or [15O]-water PET, throughout the progression of AD, from early CBF decreases in healthy aging individuals (Chen et al., 2011) to MCI and AD dementia (Alexopoulos et al., 2012; Wang et al., 2013; Okonkwo et al., 2014; Binnewijzend et al., 2016). Temporal-parietal and frontal regions also exhibited elevated tau levels in cognitively unimpaired individuals that increased during AD progression (Pontecorvo et al., 2017; Lowe et al., 2018; Harrison et al., 2019) and showed early associations with cognitive performance (Ossenkoppele et al., 2019). The combined evidence from this study and previous studies demonstrating negative CBF–tau associations suggests that increasing tau deposition and decreasing CBF may be linked mechanistically.
Mechanisms underlying CBF–tau associations are poorly understood. Evidence from human patients and animal stroke models suggested that chronic hypoperfusion can induce tau hyperphosphorylation and accumulation (Qiu et al., 2016; Pluta et al., 2018; Shimada et al., 2019). Conversely, data from human postmortem studies and animal models of AD showed that tau oligomers deposit in microvasculature (Castillo-Carranza et al., 2017) and that pathologic tau can induce abnormal remodeling of brain vasculature (Merlini et al., 2016; Bennett et al., 2018). CBF is strongly linked to vascular health, but, as mentioned above, it is also coupled with other physiological processes such as metabolism and synaptic function; therefore, it is difficult to determine which process is most associated with elevated tau. However, a strength of the study is that we also showed significant relationships between lateral temporal tau levels and an additional vascular marker, CSF sPDGFRβ. PDGFRβ in the adult brain is expressed predominantly in pericytes, cells that are integral to BBB maintenance and regulation of CBF (Kisler et al., 2017). Pericyte damage results in the cleavage of soluble PDGFRβ, and increased CSF sPDGFRβ has recently been observed in early stages of cognitive impairment (Nation et al., 2019). Importantly, a pericyte-deficient AD mouse model exhibited tau accumulation (Sagare et al., 2013), further supporting a link between vascular dysfunction and tau accumulation.
Another major finding of the study is that CBF/sPDGFRβ–tau associations differed as a function of amyloid burden (Fig. 4, Extended Data Fig. 4-1). Numerous postmortem and in vivo human studies posit an interactive model where early amyloid accumulation potentially promotes tau tangle formation (Ingelsson et al., 2004; Hanseeuw et al., 2019; Jack et al., 2019). The current study augments this model by uncovering a vascular component, and it appears that not only does tau increase in the presence of amyloid, but CBF and sPDGFRβ also undergo dynamic changes. It is possible that amyloid * CBF interactions are driven by the presence of high tau levels predominantly in individuals with high amyloid. Because few participants exhibited high tau but low amyloid levels, consistent with primary age-related tauopathy, the restricted dynamic tau range in these individuals may make it more difficult to accurately estimate the slope. However, not every region that displayed a significant voxelwise CBF/sPDGFRβ–tau correlation also showed a significant amyloid * CBF/sPDGFRβ interaction, suggesting that some regional CBF/sPDGFRβ–tau relationships arise before substantial amyloid accumulation. This is in line with postmortem evidence of early arterial tau deposition in the absence of amyloid-β (Merlini et al., 2016). Though some initial CBF/sPDGFRβ–tau associations appeared in advance and independent of amyloid-β positivity, the presence of significant amyloid in the system greatly strengthened the relationship. Because amyloid-β itself is known to have vasoactive properties, and in many cases leads to vascular deposits in dementia patients (Govindpani et al., 2019), it is possible that amyloid interacts with the vascular system in tau-independent and tau-dependent pathways. A tau-independent pathway is supported by recent evidence that amyloid deposits act directly on pericytes to reduce CBF through pericyte-mediated capillary constriction (Nortley et al., 2019). Further research is needed to disentangle the cellular interactions among tau, vascular function, and amyloid-β.
Finally, we reported differential associations between CBF/sPDGFRβ and tau PET as a function of global cognition. Across cohorts, participants with lower global cognition, as measured by MoCA performance, displayed steeper CBF/sPDGFRβ–tau relationships (Fig. 3, Extended Data Fig. 3-1). Investigating tau and CBF independently in aging or cognitively impaired cohorts, the tau PET signal in temporal, parietal, and lateral frontal regions was negatively correlated with cognitive performance (Saint-Aubert et al., 2016; Kang et al., 2017; Ossenkoppele et al., 2019), while CBF in similar regions showed positive correlations with cognitive performance (Mak et al., 2012; Wang et al., 2013; Okonkwo et al., 2014; Binnewijzend et al., 2016; Benedictus et al., 2017). However, instead of assessing cognition-imaging correlations separately for CBF and tau PET as has been done previously, the current study investigated CBF and tau together and asked whether the slope of observed CBF–tau relationships differed according to cognitive function. Our observations indicated that, in participants demonstrating relatively lower global cognition, the negative CBF–tau association was stronger than in participants with higher cognitive performance, suggesting that the link between CBF and tau levels is clinically meaningful. Furthermore, we also showed in the majority of regions showing MoCA * CBF interactions that this effect was largely driven by amyloid+ individuals (Fig. 5, Extended Data Fig. 5-1). This is consistent with a two-hit vascular hypothesis of AD (Zlokovic, 2011), as CBF–tau relationships appear to precede amyloid, but are exacerbated with the accumulation of amyloid-β. Subsequently, CBF–tau relationships are strengthened even further in amyloid+ individuals with poorer global cognition, suggesting that cognitive deficits are worse when all three pathologic factors are present. Results from the mediation analysis were consistent with evidence that vascular dysfunction occurs early in the progression of AD, followed by the accumulation of misfolded proteins and clinical decline (Iturria-Medina et al., 2016). However, the cross-sectional nature of the study makes it difficult to dissect the causal order of these events and our interpretation of these mediating effects needs to be validated in longitudinal studies.
The current study has several limitations. Because of difficulties associated with a multisite study, study sites used different MRI sequences and MRI/PET scanners, which is likely responsible for some of the differences among cohorts, such as global gray matter CBF. However, identifying a relatively large sample of participants with 3D pCASL sequences and the same PET tracers is a strength of this multimodal imaging study with a CSF subcomponent, and the fact that many of the discovery findings were replicable is evidence of the robustness of the findings. Another limitation is that only data uncorrected for PVE were included in the main results. We chose to proceed with non-PVE-corrected data, because associations between PVE-corrected CBF and FTP data in the discovery cohort were not different from those using non-PVE-corrected data, indicating that these associations were likely PVE independent. The inclusion of gray matter volume as a covariate in the ROI analyses also limits the impact of atrophy-induced PVE. An additional limitation is that CSF levels of sPDGFRβ were quantified with Western blot analysis, which can result in less accurate quantification compared with techniques like Meso Scale Discovery (MSD). However, we have previously shown strong positive associations between CSF sPDGFRβ levels quantified by MSD and Western blot (Sweeney et al., 2020), indicating that our Western blot analysis provides accurate quantification.
In conclusion, results from the current study provide the first evidence of associations between elevated tau PET signal and vascular dysfunction, reflected by decreased CBF and increased CSF sPDGFRβ, two independent measures of impaired vascular health. Furthermore, CBF/sPDGFRβ–tau relationships were steeper in amyloid+ individuals and also in individuals with poorer cognitive performance, who were largely dependent on amyloid status. Together, combination treatments targeting vascular health, as well as amyloid-β and tau levels, may be more effective in preserving cognitive function than single-target therapies.
Footnotes
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. (A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.) Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company, enabled the use of the flortaucipir F 18 tracer by providing the tracer, but did not provide direct funding and was not involved in data analysis or interpretation. Life Molecular Imaging enabled the use of the florbetaben F 18 injection (Neuraceq) by providing the tracer, but did not provide direct funding and was not involved in data analysis or interpretation. The authors report no other competing financial interests.
This work was supported by National Institutes of Health Grants R01-AG-046928, R01-AG-054617, P01-AG-052350, P50-AG-005142; Alzheimer's Association Strategic 509279 Grant; and the Foundation Leducq Translational Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference 16 CVD 05. Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01-AG-024904) and Department of Defense Award W81XWH-12-2-0012. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica; Biogen; Bristol-Myers Squibb Company; CereSpir; Cogstate; Eisai; Elan Pharmaceuticals; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche and its affiliated company Genentech; Fujirebio; GE Healthcare; IXICO; Janssen Alzheimer Immunotherapy Research & Development; Johnson & Johnson Pharmaceutical Research & Development; Lumosity; Lundbeck; Merck & Company; Meso Scale Diagnostics; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer; Life Molecular Imaging (FNA Piramal Imaging); Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
References
- Alexopoulos P, Sorg C, Förschler A, Grimmer T, Skokou M, Wohlschläger A, Perneczky R, Zimmer C, Kurz A, Preibisch C (2012) Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer's disease measured by pulsed arterial spin labeling MRI. Eur Arch Psychiatry Clin Neurosci 262:69–77. 10.1007/s00406-011-0226-2 [DOI] [PubMed] [Google Scholar]
- Alsop DC, Detre JA, Golay X, Günther M, Hendrikse J, Hernandez-Garcia L, Lu H, MacIntosh BJ, Parkes LM, Smits M, van Osch MJ, Wang DJ, Wong EC, Zaharchuk G (2015) Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: a consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn Reson Med 73:102–116. 10.1002/mrm.25197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan S, Lu H (2010) On the sensitivity of ASL MRI in detecting regional differences in cerebral blood flow. Magn Reson Imaging 28:928–935. 10.1016/j.mri.2010.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Yushkevich P, Pluta J, Minkoff D, Korczykowski M, Detre J, Gee JC (2010) The optimal template effect in hippocampus studies of diseased populations. Neuroimage 49:2457–2466. 10.1016/j.neuroimage.2009.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC (2011) A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54:2033–2044. 10.1016/j.neuroimage.2010.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ (2017) Considerations and code for partial volume correcting. Data Brief 15:648–657. 10.1016/j.dib.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedictus MR, Leeuwis AE, Binnewijzend MA, Kuijer JP, Scheltens P, Barkhof F, van der Flier WM, Prins ND (2017) Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer's disease. Eur Radiol 27:1169–1175. 10.1007/s00330-016-4450-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT (2018) Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci U S A 115:E1289–E1298. 10.1073/pnas.1710329115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewijzend MA, Benedictus MR, Kuijer JP, van der Flier WM, Teunissen CE, Prins ND, Wattjes MP, van Berckel BN, Scheltens P, Barkhof F (2016) Cerebral perfusion in the predementia stages of Alzheimer's disease. Eur Radiol 26:506–514. 10.1007/s00330-015-3834-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos I, Vos SJB, Schindler SE, Hassenstab J, Xiong C, Grant E, Verhey F Morris JC Visser PJ Fagan AM (2019) Vascular risk factors are associated with longitudinal changes in cerebrospinal fluid tau markers and cognition in preclinical Alzheimer's disease. Alzheimers Dement 15:1149–1159. 10.1016/j.jalz.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse A, Hensel A, Gühne U, Angermeyer MC, Riedel-Heller SG (2006) Mild cognitive impairment: long-term course of four clinical subtypes. Neurology 67:2176–2185. 10.1212/01.wnl.0000249117.23318.e1 [DOI] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, Gerson JE, Sengupta U, Abisambra J, Nelson P, Troncoso J, Ungvari Z, Galvan V, Kayed R (2017) Cerebral microvascular accumulation of tau oligomers in Alzheimer's disease and related tauopathies. Aging Dis 8:257–266. 10.14336/AD.2017.0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha YH, Jog MA, Kim YC, Chakrapani S, Kraman SM, Wang DJ (2013) Regional correlation between resting state FDG PET and pCASL perfusion MRI. J Cereb Blood Flow Metab 33:1909–1914. 10.1038/jcbfm.2013.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Rosas HD, Salat DH (2011) Age-associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 55:468–478. 10.1016/j.neuroimage.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindpani K, McNamara LG, Smith NR, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A (2019) Vascular dysfunction in Alzheimer's disease: a prelude to the pathological process or a consequence of it? J Clin Med 8:651 10.3390/jcm8050651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve DN, Salat DH, Bowen SL, Izquierdo-Garcia D, Schultz AP, Catana C, Becker JA, Svarer C, Knudsen GM, Sperling RA, Johnson KA (2016) Different partial volume correction methods lead to different conclusions: an (18)F-FDG-PET study of aging. Neuroimage 132:334–343. 10.1016/j.neuroimage.2016.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Schultz AP, Papp KV, Mormino EC, Sepulcre J, Bark JS, Cosio DM, LaPoint M, Chhatwal JP, Rentz DM, Sperling RA, Johnson KA (2017) Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol 81:583–596. 10.1002/ana.24910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, Buckley RF, Papp KV, Amariglio RA, Dewachter I, Ivanoiu A, Huijbers W, Hedden T, Marshall GA, Chhatwal JP, Rentz DM, Sperling RA, Johnson K (2019) Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol 76:915 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison TM, La Joie R, Maass A, Baker SL, Swinnerton K, Fenton L, Mellinger TJ, Edwards L, Pham J, Miller BL, Rabinovici GD, Jagust WJ (2019) Longitudinal tau accumulation and atrophy in aging and Alzheimer disease. Ann Neurol 85:229–240. 10.1002/ana.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Oh H, Younger AP, Patel TA (2013) Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80:1341–1348. 10.1212/WNL.0b013e31828ab35d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC (2004) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62:925–931. 10.1212/01.wnl.0000115115.98960.37 [DOI] [PubMed] [Google Scholar]
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC (2016) Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun 7:11934. 10.1038/ncomms11934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC (2017) Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 16:435–444. 10.1016/S1474-4422(17)30077-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, Graff-Radford J, Jones DT, Ferman TJ, Boeve BF, Kantarci K, Lowe VJ, Vemuri P, Mielke MM, Fields JA, Machulda MM, Schwarz CG, Senjem ML, Gunter JL, Petersen RC (2019) The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 142:3230–3242. 10.1093/brain/awz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, Gorno-Tempini ML, Schuff N (2005) Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234:851–859. 10.1148/radiol.2343040197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JM, Lee S-Y, Seo S, Jeong HJ, Woo S-H, Lee H, Lee Y-B, Yeon BK, Shin DH, Park KH, Kang H, Okamura N, Furumoto S, Yanai K, Villemagne VL, Seong J-K, Na DL, Ido T, Cho J, Lee K-M, et al. (2017) Tau positron emission tomography using. Neurobiol Aging 59:210–219. 10.1016/j.neurobiolaging.2017.08.008 [DOI] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18:419–434. 10.1038/nrn.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, Rowe CC, Skovronsky DM, Mintun MA (2015) The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 11:15.e1-4. 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacalle-Aurioles M, Alemán-Gómez Y, Guzmán-De-Villoria JA, Cruz-Orduña I, Olazarán J, Mateos-Pérez JM, Martino ME, Desco M (2013) Is the cerebellum the optimal reference region for intensity normalization of perfusion MR studies in early Alzheimer's disease? PLoS One 8:e81548. 10.1371/journal.pone.0081548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC, et al. (2018) Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 141:271–287. 10.1093/brain/awx320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak HK, Chan Q, Zhang Z, Petersen ET, Qiu D, Zhang L, Yau KK, Chu LW, Golay X (2012) Quantitative assessment of cerebral hemodynamic parameters by QUASAR arterial spin labeling in Alzheimer's disease and cognitively normal elderly adults at 3-tesla. J Alzheimers Dis 31:33–44. 10.3233/JAD-2012-111877 [DOI] [PubMed] [Google Scholar]
- Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR, Beckett LA, Donohue M, Jagust W, Schuff N, Weiner MW (2014) Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain 137:1550–1561. 10.1093/brain/awu043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade E, Kim A, James J, Sheu LK, Kuan DC, Minhas D, Gianaros PJ, Ikonomovic S, Lopez O, Snitz B, Price J, Becker J, Mathis C, Klunk W (2014) Cerebral perfusion alterations and cerebral amyloid in autosomal dominant Alzheimer disease. Neurology 83:710–717. 10.1212/WNL.0000000000000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Wanner D, Nitsch RM (2016) Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer's disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131:737–752. 10.1007/s00401-016-1560-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302. 10.1016/j.neuron.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, Pachicano M, Joe E, Nelson AR, D'Orazio LM, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Reiman EM, Caselli RJ, Chui HC, et al. (2020) APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581:71–76. 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Chen Y, Korczykowski M, Saboury B, Martinez PM, Reddin JS, Alavi A, Kimberg DY, Wolk DA, Julin P, Newberg AB, Arnold SE, Detre JA (2012) Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer's disease. Alzheimers Dement 8:51–59. 10.1016/j.jalz.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53:695–699. 10.1111/j.1532-5415.2005.53221.x [DOI] [PubMed] [Google Scholar]
- Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV (2019) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 25:270–276. 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D (2019) Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 365:eaav9518 10.1126/science.aav9518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, Birdsill AC, Palotti M, Wharton W, Hermann BP, LaRue A, Bendlin BB, Rowley HA, Asthana S, Sager MA, Johnson SC (2014) Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer's disease. Cereb Cortex 24:978–988. 10.1093/cercor/bhs381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Smith R, Ohlsson T, Strandberg O, Mattsson N, Insel PS, Palmqvist S, Hansson O (2019) Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology 92:e601–e612. 10.1212/WNL.0000000000006875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW (2010) Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74:201–209. 10.1212/WNL.0b013e3181cb3e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ (2018) Tau protein dysfunction after brain ischemia. J Alzheimers Dis 66:429–437. 10.3233/JAD-180772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo MJ, Devous MD, Navitsky M, Lu M, Salloway S, Schaerf FW, Jennings D, Arora AK, McGeehan A, Lim NC, Xiong H, Joshi AD, Siderowf A, Mintun MA (2017) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140:748–763. 10.1093/brain/aww334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L, Ng G, Tan EK, Liao P, Kandiah N, Zeng L (2016) Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer's disease mice. Sci Rep 6:23964. 10.1038/srep23964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin JS, Schultz AP, Hedden T, Viswanathan A, Marshall GA, Kilpatrick E, Klein H, Buckley RF, Yang HS, Properzi M, Rao V, Kirn DR, Papp KV, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP (2018) Interactive associations of vascular risk and β-amyloid burden with cognitive decline in clinically normal elderly individuals: findings from the Harvard Aging Brain Study. JAMA Neurol 75:1124–1131. 10.1001/jamaneurol.2018.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin JS, Yang HS, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, Gatchel JR, Marshall GA, Kilpatrick E, Klein H, Rao V, Buckley RF, Yau WW, Kirn DR, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP (2019) Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol 85:272–279. 10.1002/ana.25399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC (1998) Correction for partial volume effects in PET: principle and validation. J Nucl Med 39:904–911. [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013) Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4:2932. 10.1038/ncomms3932 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saint-Aubert L, Almkvist O, Chiotis K, Almeida R, Wall A, Nordberg A (2016) Regional tau deposition measured by. Alzheimers Res Ther 8:38. 10.1186/s13195-016-0204-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Shindo A, Matsuyama H, Yata K, Niwa A, Sasaki R, Ayaki T, Maki T, Wakita H, Tomimoto H (2019) Chronic cerebral hypoperfusion upregulates leptin receptor expression in astrocytes and tau phosphorylation in tau transgenic mice. Neurosci Lett 704:133–140. 10.1016/j.neulet.2019.04.009 [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV (2018) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21:1318–1331. 10.1038/s41593-018-0234-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV (2019a) Blood–brain barrier: from physiology to disease and back. Physiol Rev 99:21–78. 10.1152/physrev.00050.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, et al. (2019b) Vascular dysfunction-The disregarded partner of Alzheimer's disease. Alzheimers Dement 15:158–167. 10.1016/j.jalz.2018.07.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, Pachicano M, Harrington MG, Joe E, Chui HC, Schneider LS, Montagne A, Ringman JM, Fagan AM, Morris JC, Pa J, Nation DA, Toga AW, Zlokovic BV (2020) A novel sensitive assay for detection of a biomarker of pericyte injury in cerebrospinal fluid. Alzheimers Dement 16:821–830. 10.1002/alz.12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Das SR, Xie SX, Arnold SE, Detre JA, Wolk DA (2013) Arterial spin labeled MRI in prodromal Alzheimer's disease: a multi-site study. Neuroimage Clin 2:630–636. 10.1016/j.nicl.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Graff-Radford J, Tosakulwong N, Weigand SD, Machulda MM, Senjem ML, Spychalla AJ, Vemuri P, Jones DT, Drubach DA, Knopman DS, Boeve BF, Ertekin-Taner N, Petersen RC, Lowe VJ, Jack CR, Josephs KA (2018) Imaging correlations of tau, amyloid, metabolism, and atrophy in typical and atypical Alzheimer's disease. Alzheimers Dement 14:1005–1014. 10.1016/j.jalz.2018.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Liu CY, Wong KP, Huang SC, Mack WJ, Jann K, Coppola G, Ringman JM, Wang DJJ (2018) Regional association of pCASL-MRI with FDG-PET and PiB-PET in people at risk for autosomal dominant Alzheimer's disease. Neuroimage Clin 17:751–760. 10.1016/j.nicl.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Beason-Held L, Resnick SM, Landman BA (2011) Biological parametric mapping with robust and non-parametric statistics. Neuroimage 57:423–430. 10.1016/j.neuroimage.2011.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12:723–738. 10.1038/nrn3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, B, Scatterplots of ADNI standard SUVR values for FBP (A) and FBB (B) scans. Download Figure 1-1, TIF file (1.7MB, tif) .
Scatterplot of ADNI standard CL values against CL values obtained without processing pipeline. Download Figure 1-2, TIF file (2.9MB, tif) .
A, B, Coordinates of clusters showing significant negative relationships between [18F]flortaucipir and CBF in the USC discovery (A; n = 68) and ADNI replication cohorts (B; n = 138). Download Figure 1-3, DOCX file (15.1KB, docx) .
Coordinates of clusters showing significant negative relationships between [18F]flortaucipir and CBF in the secondary, gray matter-masked analyses of the ADNI FTP cohort (n = 138). Download Figure 2-1, DOCX file (18.9KB, docx) .
MoCA * CBF/sPDGFRβ interactions. S1, Primary somatosensory cortex; SPL, superior parietal lobe. Download Figure 3-1, DOCX file (19KB, docx) .
Participants with low global cognition show stronger relationships between CBF and sPDGFRβ in ADNI subgroup analyses. A, B, MoCA * CBF interaction effects on FTP SUVR in secondary GM-masked ADNI AD-MCI (A) and CN (B) subgroups. Average CBF and FTP from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, gray matter volume, amyloid CL, and diagnosis (in A). MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA group (shown in purple) and a high-MoCA group (shown in green). Download Figure 3-2, TIF file (4.5MB, tif) .
Amyloid * CBF/sPDGFRβ interactions. *Significant at p < 0.05, uncorrected Download Figure 4-1, DOCX file (18.7KB, docx) .
Participants with high amyloid burden show stronger relationships between CBF and sPDGFRβ in ADNI subgroup analyses. A, B, Amyloid * CBF interaction effects on FTP SUVR in secondary GM-masked ADNI AD-MCI (A) and CN (B) subgroups. Average CBF and FTP SUVR from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, gray matter volume, amyloid CL, and diagnosis (in A). Amyloid CL was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-amyloid group (shown in blue) and a high-amyloid group (shown in red). Download Figure 4-2, TIF file (8.5MB, tif) .
MoCA * CBF/sPDGFRβ interactions split by amyloid status. *Significant at p < 0.05, uncorrected. Download Figure 5-1, DOCX file (20.3KB, docx) .
Interactions between MoCA and CBF are stronger in amyloid+ individuals in ADNI subgroup analyses. Amyloid * CBF interaction effects on FTP SUVR in the secondary GM-masked ADNI AD-MCI subgroup, split by amyloid status (amyloid+ on the left, amyloid– on the right). Average CBF and FTP SUVRs from significant clusters in the voxelwise analysis were entered into GLMs. Predicted FTP SUVR from the models is plotted against regional CBF (normalized by global gray matter CBF). Analyses were covaried for age, sex, diagnosis, gray matter volume, and amyloid CL. MoCA was entered as a continuous variable in the model, but for visualization purposes a median split was used to group participants into a low-MoCA group (shown in orange) and a high-MoCA group (shown in green). Download Figure 5-2, TIF file (2.3MB, tif) .
Mediation analyses from model 1 (CBF/sPDGFRβ → tau → MoCA). *Significant at p < 0.05, uncorrected. Download Figure 6-1, DOCX file (19.2KB, docx) .
Mediation analyses from model 1 (tau → CBF/sPDGFRβ → MoCA). Significant at p < 0.05, uncorrected. Download Figure 6-2, DOCX file (19.2KB, docx) .
Mediation analyses from model 1 (CBF/sPDGFRβ → Tau → MoCA) split by amyloid status. *Significant at p < 0.05, uncorrected. Download Figure 6-3, DOCX file (20.7KB, docx) .
Data Availability Statement
The data that support the findings of this study will be made available on request to the corresponding author in accordance with the existing data-sharing agreement with the National Institute on Aging and the ADNI committee.