

Abstract
The rapid scale-up of research on coronavirus disease 2019 (COVID-19) has spawned a large number of potential vaccines and immunotherapies, accompanied by a commensurately large number of in vitro assays and in vivo models to measure their effectiveness. These assays broadly have the same end-goal — to predict the clinical efficacy of prophylactic and therapeutic interventions in humans. However, the apparent potency of different interventions can vary considerably between assays and animal models, leading to very different predictions of clinical efficacy. Complete harmonization of experimental methods may be intractable at the current pace of research. However, here we analyse a selection of existing assays for measuring antibody-mediated virus neutralization and animal models of infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and provide a framework for comparing results between studies and reconciling observed differences in the effects of interventions. Finally, we propose how we might optimize these assays for better comparison of results from in vitro and animal studies to accelerate progress.
Subject terms: Infectious diseases, Viral infection
Are you new to virus research and trying to interpret the ever-expanding literature on immunity to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)? Here, the authors compare the different assays and animal models used to measure immunity to SARS-CoV-2 infection and reconcile differences in apparent potency of antibodies assessed in different assays.
Introduction
The spread of the coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to rapid progress in the development of potential therapeutics and assays to assess them. However, the nature of this progress means that numerous assays and animal models for measuring antiviral immunity have been independently developed by different groups. Many of these are based on similar approaches and aimed at measuring identical outcomes. However, differences in cell lines and viral isolates (or laboratory adaptation of isolates), as well as in animal species and conditions, across different laboratories may lead to different predictions of the efficacy of interventions. For example, mutations in SARS-CoV-2 spike protein may affect the ability of antibodies to directly bind to this viral protein1,2, alter virus transmission dynamics3 or modulate viral binding to its entry receptor angiotensin-converting enzyme 2 (ACE2)4. Even for seemingly similar in vitro assays that use identical cells and viral isolates, minor details of assay design such as the inoculum size, length of incubation and method used to measure the infection level can have major impacts on interpreting the efficacy of different interventions in reducing infection.
Advancing studies into different animal models adds further complexity as factors such as the initial inoculum size, route of administration and infected cell type may all vary between species and laboratories. An agreement on a set of standardized assays for measuring SARS-CoV-2 immunity would advance the field substantially. However, given the pace of development and diversity of approaches, this may be challenging to achieve in the short term. In the interim, a better understanding of the characteristics and limitations of different in vitro assays and animal models should provide a rational basis for comparison.
In this Review, we provide an overview of different assays and animal models for SARS-CoV-2 infection and provide a theoretical framework for analysis and assessment of these studies. We show that many of the differences between alternative approaches can be understood through a consideration of infection dynamics in vitro and in vivo. This perspective of the dynamics of infection in different assays and animal models not only provides a foundation to understand variation in results between studies but also allows us to extrapolate to likely clinical effects. Finally, we outline key considerations for harmonizing and improving the use of current models to investigate SARS-CoV-2 immunity.
Measuring antiviral activity in vitro
A primary assessment of SARS-CoV-2 immunity involves measuring the neutralization capacity of serum or monoclonal antibodies in vitro5–7. This can be studied by measuring the ability of antibodies to inhibit the binding of the viral receptor binding domain (RBD) to the human protein ACE2 in vitro8,9. However, there may not be a direct relationship between binding inhibition and the level of inhibition of cellular infection. Therefore, numerous assays have been developed to measure neutralization of the infection of cells with either the native SARS-CoV-2 or a pseudotyped reporter virus carrying SARS-CoV-2 spike protein. Infection is measured after a period of co-incubation of virus and serum or antibody, quantifying either the number of infected cells, the production of viral RNA or infectious virus, or the viral cytopathic effect.
Antiviral activity is measured by comparing infection levels in antibody-treated and untreated cultures, and efficacy is often reported as an IC50 (the concentration of antibody required to reduce infection to 50% of that seen in untreated control cultures). The IC50 in these assays is usually interpreted as the concentration of antibody required to neutralize 50% of virions. However, as we show below, depending on factors such as the initial inoculum size, length of incubation and method of measuring infection, we would expect neutralization of anywhere between 10% and 99% of virions to be required to produce an apparent IC50 in different assays. Here, we highlight that different IC50 measurements between assays may arise from predictable differences in what is being measured under the specific assay conditions. We analyse several common assays and provide a framework for comparing assays and for interpreting assay results in a clinical context.
Single-cycle virus neutralization assays
Pseudotyped virus (or pseudovirus) assays involve incorporation of SARS-CoV-2 spike protein onto other viruses such as vesicular stomatitis virus (VSV)1,10,11 or lentiviruses12,13 (Table 1). These chimeric viruses also encode luciferase or other fluorescent reporters, providing a direct read-out of the level of infection in vitro when they are used to infect (transduce) ACE2-expressing cells. Pseudotyped virus assays using SARS-CoV-2 spike protein are only suitable for studying viral entry and the effects of antibodies targeting spike protein, because they do not include other components of the SARS-CoV-2 viral replication machinery.
Table 1.
In vitro models of SARS-CoV-2 infection
| Format | Description | Duration (days) | Primary outcome | Quantification methods | Target outcome | Refs |
|---|---|---|---|---|---|---|
| ACE2–RBD inhibition assays | SARS-CoV-2 spike RBD incubated with soluble hACE2 and Ab at various concentrations | 1 | RBD binding to hACE2 | Binding can be measured by ELISA, FRET, SPR or BLI | Ab concentration required to inhibit RBD and ACE2 binding by 50% | 8,9 |
| Pseudovirus (single cycle) |
VSV lacking G protein is pseudotyped with SARS-CoV-2 spike (usually lacking 18–21 amino-terminal residues) HIV-1 or MLV lacking envelope is pseudotyped with SARS-CoV-2 spike and cultured in susceptible cell lines after the addition of Ab at various concentrations |
1 | Viral entry | Viral entry can be quantified using fluorescent or luciferase reporters | Ab required to reduce viral entry after a single cycle by 50% | 1,10,12,55–57 |
| Chimeric virus (replicating) | VSV with the G gene replaced by SARS-CoV-2 spike (usually lacking 18–21 amino-terminal residues) and cultured in susceptible cell lines after the addition of Ab at various concentrations | 1–2 | Viral entry, viral replication | Infection read-out using GFP or FRNT | Ab required to reduce the viral entry or viral load at the assay end point by 50% | 1,15,58 |
| Live SARS-CoV-2 — quantitative | Live SARS-CoV-2 is cultured in susceptible cell lines after incubation with Ab at various concentrations | 1–4 | Viral replication | Viral antigens (generally N protein) or cell death (measured using LDH) detected by ELISA or microscopy | Ab required to reduce the viral load at the assay end point by 50% | 1,21 |
| Live SARS-CoV-2 — limiting dilution | SARS-CoV-2 (often 100 TCID50) and various concentrations of Ab are co-incubated before addition to susceptible cell lines | 3–5 | Fraction of replicate wells positive for virus | Cytopathic effect or viral antigens (generally N protein) detected by ELISA or microscopy, or fluorescence or luminescence with labelled SARS-CoV-2 | Ab required to inhibit ~99% of inoculum | 22,59–61 |
| PRNT or FRNT | SARS-CoV-2 and Ab (at various concentrations) are co-incubated before addition to a cell monolayer | 3 | Reduction in plaque number | Plaques can be quantified by visual examination aided by colorimetric or fluorescent reporters | Ab required to reduce the number of plaques by 50% (or 90% for PRNT90) | 62 |
Ab, antibody; BLI, biolayer interferometry; ELISA, enzyme-linked immunosorbent assay; FRET, fluorescence resonance energy transfer; FRNT, focus reduction neutralization titre; GFP, green fluorescent protein; hACE2, human angiotensin-converting enzyme 2; LDH, lactate dehydrogenase; MLV, murine leukaemia virus; PRNT, plaque reduction neutralization titre; RBD, receptor binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SPR, surface plasmon resonance; TCID50, 50% tissue culture infectious dose; VSV, vesicular stomatitis virus.
Most pseudotyped virus assays involve a replication-defective virus (because SARS-CoV-2 spike protein is included in trans), and thus they measure the number of cells infected during a single infection cycle11,12,14 (replication-competent pseudoviruses are discussed below). This has the major benefit of requiring a lower level of laboratory containment. To test antibody-mediated inhibition, the virus and the antibody are pre-incubated for a period before being applied to cells (typically using methods such as spinoculation or polybrene treatment to improve infection efficiency), and inhibition is measured as the relative reduction in reporter signal, usually 24 h later. Fitting of the relationship between antibody concentration and reporter signal is then used to estimate the IC50 of an antibody. This assay can provide a direct read-out of the decrease in successful viral entry during a single round of infection as a result of treatment (Fig. 1a). However, the use of a pseudovirus also raises numerous challenges. Factors such as the folding, cleavage, density and geometry of spike proteins on the virion can affect both the mechanics of cell entry and the ability of antibodies to bind to (pseudo)virions and neutralize infectivity15,16 and may differ from those of the native virus17–20.
Fig. 1. In vitro assays for measuring viral inhibition.

a | Single-cycle pseudotyped virus assays involve co-incubation of virus and cells and measurement of the number of infected cells by a fluorescent reporter construct. They can provide a direct measure of the proportion of virus entry neutralized by serum or antibodies. b | Multi-cycle assays use either replication-competent pseudoviruses or native severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and measure the spread of infection over multiple cycles of infection in vitro. The level of infection can be measured using detection of a fluorescent reporter construct, viral antigen in infected cells or free virus in the supernatant. Some assays reach saturation before the end of the incubation and are thus insensitive to small changes in initial inoculum or viral growth rate. Once saturation is overcome, the fraction reduction in initial infectious viral levels is reflected as an equivalent fold-change in final viral levels (left hand panels). By contrast, small changes in viral growth rate are amplified over multiple rounds of infection, leading to large changes in final viral levels (right hand panels). c | Plaque reduction neutralization assays involve co-incubation of virus and antibody followed by plating out of virus onto an immobilized cell monolayer and incubation. The number of infectious virions remaining in the inoculum is enumerated by counting plaques of infected cells. d | An alternative limiting dilution approach involves co-incubation of antibody and virus followed by splitting into multiple wells to observe the proportion of wells infected. Cytopathic effect is commonly used as a read-out. The apparent IC50 (the concentration of antibody required to reduce infection to 50% of that seen in untreated control cultures) of the assay is highly dependent on the initial inoculum size. Inhibition of the cytopathic effect is only observed when the initial viral titres are reduced to <1 TCID50 (50% tissue culture infectious dose) in some wells. For this reason, limiting dilution-based assays can estimate a very different IC50 compared with single-cycle pseudotyped viral assays. Note that in the cytopathic effect assay, for a given input level of V0 infectious units, the IC50 occurs when the fraction of virions neutralized is 0.5(1/V0).
Multi-cycle virus neutralization assays
Numerous assays involve measuring the ability of antibodies to inhibit the replication of virus over several days. Both replicating VSV/SARS-CoV-2 chimeric viruses1,15 and native SARS-CoV-2 (refs1,21) have been used to infect susceptible cell lines, with subsequent measurement of the level of infection after several days of incubation by quantifying reporter protein expression, viral antigen in infected cells or free virus in the supernatant (see Table 1). These assays can then be used to measure antibody neutralization by pre-incubation of different concentrations of antibody with the viral inoculum and measuring the relationship between antibody concentration and inhibition of infection.
Depending on the construct, a replicating chimeric virus may require lower-level containment than native SARS-CoV-2 but suffers from the same issues as single-cycle pseudovirus regarding the quality of spike protein. In addition, it is important to recognize that all aspects of viral replication, except receptor binding, are mediated by the parental (VSV) viral proteins. Therefore, the assay may have very different replication kinetics to native SARS-CoV-2. A major advantage of the use of native SARS-CoV-2 is the ability to measure the effects of agents acting at different parts of the viral life cycle, typically over multiple life cycles in vitro.
The use of a multi-cycle assay introduces several potential confounders compared with the single-cycle assays. First, for 50% neutralization of the inoculum to translate into 50% reduction in final infection levels, viral growth must not ‘saturate’ before the end of the assay. Saturation can frequently occur if a large proportion of cells become infected and, thus, the lack of uninfected cells limits viral expansion. If viral levels are saturated at the end of the assay (as is likely to be the case for most live virus neutralization assays), then reducing the initial inoculum will not reduce the final level of virus, instead simply making the maximal viral level occur later (see Fig. 1b). Such assays will be insensitive to low levels of neutralization and not directly comparable with single-cycle assays. Thus, care must be taken that growth remains exponential throughout the assay.
A second important consideration is whether the antibody acts only on the initial inoculum or remains throughout the assay. If the antibody remains present during the assay, then it can act not only to neutralize a proportion of the inoculum but also to inhibit the subsequent spread of virus in the culture. If the final read-out is the ‘level of total infection relative to control’, this will be very sensitive to small changes (per cycle) in the viral growth rate during culture. For example, a 10% reduction in viral growth over six cycles of infection will lead to a 50% reduction in the final infection. Thus, the apparent IC50 (in the assay) may occur when antibody neutralizes only 10% of virions (on each cycle), leading to very different estimates of antibody efficacy between assays (see Fig. 1b, lower panels). The situation can become even more complex because the level of inhibition may not be constant over time, as the stoichiometry of antibody to virus varies over the course of incubation. This can potentially be avoided by removing the antibody during incubation. However, a more definitive approach may simply be to focus on measuring the outcome of interest and choose assays accordingly. For example, if one wishes to measure neutralization (of an inoculum), use of a single-cycle assay measures neutralization of a single round of infection. At present, single-cycle assays typically depend on non-replicating pseudovirus, which has inherent differences to native virus. However, single-cycle live SARS-CoV-2 assays are also possible if viral infection is limited to a single cycle either by short incubation or by addition of antibodies early after initial infection to prevent further viral spread.
By contrast, if the outcome of interest is a reduction in viral growth, then a multi-cycle assay can be used to measure growth directly. This can be done by measuring virus levels at different time points during the assay and estimating growth over time. Although time consuming, this is the only direct way to allow a comparison of the concentration of antibody or serum required to achieve a given level of viral growth inhibition in vitro. Importantly, it is likely that the relationship between neutralization and growth inhibition will vary between culture systems owing to changes in factors such as cellular infectivity and viral burst size, and whether these reflect the dynamics of viral replication and inhibition in human infection is unclear.
Plaque neutralization and limiting dilution assays
The multi-cycle assays described above aim to measure the level of infection at the end of the assay. Other approaches aim to directly quantify the degree of neutralization of the virus in the initial SARS-CoV-2 inoculum. In this case, viral replication in culture is only important inasmuch as it allows visualization of infection arising from a single initial virion. The plaque reduction neutralization test, for example, involves incubating virus and antibody and quantifying the number of infectious virions by counting ‘plaques’ of infected cells after immobilization in a gel and incubation. The IC50 is determined as the concentration of antibody that reduces the number of plaques by 50% (Fig. 1c). This approach can be technically challenging as it involves forming a monolayer of cells in gel, plaque counting (which may include an element of operator subjectivity) and may also be affected by antibody persistence during incubation. However, it aims to give a direct read-out of the proportion of the inoculum neutralized by antibodies.
A similar assay involves a limiting dilution approach to measure the amount of infectious virus remaining in the initial inoculum. This requires incubating the virus and antibody and then splitting the virus–antibody mixture into several wells and using the viral cytopathic effect as a read-out of infectivity22. In this case, individual wells are scored at the end of the assay as having a binary outcome of either ‘infection’ or ‘no infection’ (Fig. 1d). The IC50 is then calculated using the Reed–Muench method to determine the antibody concentration where growth is inhibited in 50% of wells10,11. However, the degree of viral neutralization that is required to see ‘no growth’ in a culture well is highly dependent on the initial inoculum in culture. For a typical inoculum of 100 TCID50 (50% tissue culture infectious dose), ‘no infection’ will only be observed when all infectious doses are neutralized. Therefore, infection in 50% of wells (that is, IC50) will only be observed when ∼99% of the inoculum is neutralized (more generally, the observed IC50 is seen when the proportion of virus neutralized is equal to 0.5(1/inoculum)).
A major advantage of both of these assays is that they aim to measure the number of infectious units neutralized at the start of incubation (even if they require viral growth for the final read-out of infection). That is, the assays rely on the ability of a single infectious unit at the start of the assay either to form a visible plaque or to mediate a widespread cytopathic effect by the end of the assay, both of which involve extensive viral replication. Thus, if the antibody remains present during the assay and is able to inhibit viral replication, it will have the same appearance as complete neutralization of the inoculum. For example, if the initial inoculum is 100 TCID50 and each infected cell produces 10 infectious virions over subsequent rounds of infection, it would be possible to prevent viral growth and cytopathic effects with an antibody that neutralizes just over 9 out of every 10 virions.
Comparing neutralization between in vitro assays
The choice of cell line and virus (or pseudovirus) can clearly play a major role in the apparent efficacy of antibodies, as they can affect factors such as viral entry route and burst size during infection. This can be further compounded by batch variation of viral stocks. However, it is clear from the discussion above that even when the cell line and virus are standardized, the apparent IC50 of an antibody in an assay can require between 10% and 99% of virions to be neutralized depending on the assay design. Importantly, this relationship cannot be simply scaled between assays. That is, the antibody with the highest IC50 in one assay does not necessarily rank as having the highest IC50 in a different assay. This is because antibodies can vary greatly in the shape of the dose–response relationship between antibody concentration and ‘proportion of virions inhibited’ (Box 1). Therefore, some degree of harmonization or standardization is urgently required to allow better comparison between both serological levels of immunity and antibody products in development.
Choosing an optimal in vitro assay may depend on the proposed use of the intervention — prophylactic or therapeutic. Single-cycle assays are most suitable for predicting prophylactic efficacy, as they measure ‘protective efficacy’ of antibodies against small inocula such as might be encountered in community transmission (Fig. 2a). By contrast, multi-cycle assays that measure viral growth inhibition can be used to predict efficacy as post-exposure prophylaxis or treatment of established infection. In either case, single-cycle pseudovirus assays are suitable for high-throughput screens owing to their lower level of biocontainment required. However, the relationship between pseudovirus and live SARS-CoV-2 virus infection assays should be established and the results of pseudovirus assays should be confirmed in live SARS-CoV-2 virus infection assays23, where viral characteristics are more physiological.
Fig. 2. In vivo control of SARS-CoV-2 infection.

a | Goals and challenges of intervention at different stages of infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the potential differences between animal models and human infection. b | Relationship between the level of neutralization or inhibition of the viral inoculum and observed protective efficacy following challenge with different-sized inocula. c | Schematic of how the time from treatment to peak viral load limits the observed effect of treatment on peak viral load. High inocula in animal models shorten the time to peak and limit the impact of therapies that reduce viral growth rate. d | The rate of decline in viral titres after peak is significantly faster in animal models than in human infection (P = 0.0007), suggesting differences in the rate of infected cell death or the degree of ongoing infection after peak. For details of published data used for viral decay analysis, see Supplementary information.
Box 1 Scaling between assays.
Single-cycle or plaque reduction neutralization titre (PRNT) assays tend to provide a continuous read-out of the proportion of virions neutralized in a single round of infection. By contrast, limiting dilution assays using native severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) involve an initial inoculum of 100 TCID50 (50% tissue culture infectious dose) and assess the ability of serum or antibodies to neutralize this and prevent a viral cytopathic effect. As a result, a higher apparent IC50 (the concentration of antibody required to reduce infection to 50% of that seen in untreated control cultures) is usually estimated in limiting dilution assays than in single-cycle assays6 because the assays are measuring different outcomes.
It is possible to provide a crude scaling between the two assays based on an understanding of their methods. The native limiting dilution assay result is roughly equivalent to IC99 of the single-cycle or PRNT assay. However, the relationship between IC50 and IC99 of an assay is strongly affected by the shape of the sigmoid neutralization curve (that is, the Hill coefficient). The figure, part a, shows a theoretical example of the neutralization of infection by three antibodies with different IC50 and Hill coefficients, measured in a single-cycle assay. In this example, the measured inhibition of infection on the y axis corresponds to the proportion of virus neutralized. The figure, part b, predicts the estimated neutralization curves of the same antibodies measured in a limiting dilution assay measuring neutralization of the viral cytopathic effect (vCPE) (assuming a standard inoculum of 100 TCID50). In this case, the y axis indicates the proportion of culture wells with no viral growth (that is, where all of the inoculum has been neutralized). Both the estimated IC50 and the ranking of the antibodies (in terms of potency) is reversed between the two assays. The figure, part c, predicts the general scaling between IC50 estimated by single-cycle and limiting dilution cytopathic effect assays (assuming an inoculum of 100 TCID50), with the scaling of the three theoretical antibodies indicated. Estimates of IC50 with the two assays will be more similar for antibodies with a steep increase in inhibition with increasing concentration (that is, a large Hill coefficient). However, antibodies with less steep relationships will have very different IC50 estimates under the two approaches. Note that by assuming an inoculation size and Hill coefficient , the scaling factor can be estimated as .
Animal models of COVID-19
The next stage of assessing antibody efficacy typically involves animal testing, where it is hoped that key elements of infection such as viral replication, pathogenesis and immunity may mimic those observed in human infection. Various species have been used as models of SARS-CoV-2 infection24 (Table 2). A major challenge with attempts to recapitulate human infection dynamics is the benign course of SARS-CoV-2 infection in most human subjects and the large variability of outcomes in older individuals, as such variability of outcomes in animal models would require intractably large study sizes to observe statistically significant treatment effects. As a result, animal models tend to be designed either towards eliciting pathological outcomes (aimed to be prevented by treatment) or as studies of virological rather than clinical end points to assess the effects of treatment. Similar to in vitro models, a major question in choosing an animal model is the therapeutic intent of an intervention: does the study aim to measure prophylactic, post-exposure or therapeutic efficacy (Fig. 2)? Depending on the therapeutic goal, numerous factors need to be considered.
Table 2.
Animal challenge models for SARS-Cov-2 infection
| Species | Challenge dose and route | Pathology | Viral dynamics | Lethal vs non-lethal | Ref. |
|---|---|---|---|---|---|
| Mouse hACE2 (mouse Ace2 promoter) |
IN: 105 TCID50 BetaCoV/Wuhan/IVDC-HB-01/2020 |
Lung pathology peaked 5 dpi | vRNA and infectious titre peaked in the lungs 1–3 and 3 dpi, respectively (102.44 TCID50 in 100 μl) | Non-lethal | 52 |
| Mouse hACE2 (Hfh4 promoter) |
IN: 3 × 104 TCID50 IN: 7 × 105 TCID50 (re-exposure) SARS-CoV-2 isolate USA-WA1/2020 |
Lethal infection with respiratory distress and neurological symptoms Pulmonary lesions peaked 3–5 dpi |
vRNA and infectious titre peaked in the lungs 5 dpi vRNA also detected in the eyes, heart and brain |
Lethal | 45 |
| Mouse hACE2 (K18 promoter) |
IN and IV: 2.5 × 104 PFU 2019n-CoV/USA_WA1/2020 |
Lung pathology peaked 7 dpi Histopathology revealed abnormalities in the brain, heart, liver and kidney of infected mice |
Infectious titre and vRNA load peaked in the lungs 2–4 and 2–7 dpi, respectively | Non-lethal | 63 |
| Mouse hACE2 (adenoviral vector) |
IN: 105 FFU IN or IV: 105 FFU 2019n-CoV/USA_WA1/2020 |
Lung pathology peaked 8 dpi Histopathology consistent with severe viral pneumonia found in patients with COVID-19 |
vRNA load peaked in the lungs 1–4 dpi and was detected in the heart, brain, liver, spleen, kidney, serum and gastrointestinal tract Infectious virus was detected in the lungs 4 dpi (∼106 PFU/g) |
Non-lethal | 64 |
| Mouse hACE2 (CRISPR–Cas9 knock-in) |
IN: 4 × 105 PFU BetaCoV/Wuhan/AMMS01/2020 |
Young and aged mice developed interstitial pneumonia 6 dpi |
No longitudinal viral load data available vRNA detected in the lungs, trachea and brain 6 dpi |
Non-lethal | 65 |
| Golden Syrian hamster |
IN: 8 × 104 TCID50 BetaCoV/Hong Kong/VM20001061/2020 |
Lung consolidation peaked 7 dpi Transmission from infected animals to naive animals via direct and indirect contact |
vRNA and infectious titre peaked in the lungs 2–5 and 2 dpi, respectively | Non-lethal | 28 |
| Syrian hamster |
IN: 105 PFU Hong Kong isolate |
Lung pathology and consolidation peaked 7 dpi Transmission from infected animals to naive animals in direct contact |
vRNA and infectious titre peaked in the lungs and nasal turbinate 2–4 dpi | Non-lethal | 29 |
| Syrian hamster |
IN or ocular: 103–105.3PFU SARS-CoV-2/UT-NCGM02/Human/2020/Tokyo |
Lung pathology peaked 6–8 dpi for low-dose and high-dose infected animals High-dose infected animals had higher severity scores (by CT scan) |
Infectious virus titre peaked in the lungs, nasal turbinate and brain 3 dpi | Non-lethal | 66 |
| Ferret |
IN: 105.5 TCID50 NMC-nCoV02 |
No longitudinal lung pathology data available Acute bronchiolitis 4 dpi Transmission from infected animals to naive animals in direct and indirect contact |
vRNA and infectious titre peaked in nasal turbinate and lungs 4 dpi | Non-lethal | 27 |
| Ferret |
IN or IT: 105 PFU SARS-CoV-2/CTan/human/2020/ Wuhan SARS-CoV2/F13/environment/ 2020/Wuhan |
No longitudinal lung pathology data available Severe lymphoplasmacytic perivasculitis and vasculitis in the lungs 13 dpi |
vRNA and infectious titre peaked in nasal wash 6 and 4 dpi, respectively vRNA and infectious virus were not detected in the lower respiratory tract |
Non-lethal | 67 |
| Rhesus macaque |
IT: 1 × 106 TCID50 SARS-CoV-2/WH-09/human/2020/CHN |
Mild-to-moderate interstitial infiltration in animals with pneumonia | vRNA load peaked in nasal swabs 3 dpi (106.5 RNA copies/ml) | Non-lethal | 68 |
| Rhesus macaque |
IN and IT: 1.1 × 104–1.1 × 106 PFU SARS-CoV-2 isolate USA-WA1/2020 |
Peak lung inflammation and pneumonia 2 dpi, diminished by 4 dpi | vRNA load peaked in nasal swabs and BAL 2 dpi | Non-lethal | 26 |
| Rhesus macaque |
IN, IT, ocular and oral: 2.6 × 106 TCID50 SARS-CoV-2 isolate USA-WA1/2020 |
Radiograph scores peaked 3–5 dpi |
vRNA load peaked in nasal swabs 1–3 dpi and BAL 1 dpi Infectious virus isolated in nasal swabs, BAL and throat swabs 1–3 dpi |
Non-lethal | 46 |
| Cynomolgus macaque |
IN and IT: 1 × 106 TCID50 BetaCoV/Munich/BavPat1/2020 |
Histopathological changes characteristic of acute and advanced diffuse alveolar damage 4 dpi | vRNA load peaked 2 dpi in young animals and 4 dpi in old animals | Non-lethal | 69 |
| Cynomolgus macaque |
IB: 3.65 × 106 PFU 2019-nCoV USA-WA1-A12/2020 |
Percent change in lung hyperdensity peaked 2–8 dpi CT lung scores peaked 2–6 dpi |
vRNA load peaked in nasal, rectal and oral swabs 2 dpi | Non-lethal | 70 |
| African green monkey |
Aerosol: 2–2.5 × 103 PFU IT, IN, oral and CJ: 3.61 × 106 PFU SARS-CoV-2 isolate USA-WA1/2020 |
2 of 4 animals (16 years) developed pneumonia, ARDS and a cytokine storm Animals euthanized 8 and 22 dpi owing to rapidly declining clinical condition |
vRNA load peaked in nasal and pharyngeal swabs 3–7 dpi | Lethal | 71 |
| African green monkey |
IT and IN: 5 × 105 PFU SARS-CoV-2/INMI1-Isolate/2020/Italy |
Thoracic radiographs inconclusive 2–5 dpi Necropsy of 3 animals 5 dpi showed varying degrees of pulmonary consolidation with hyperaemia and multifocal lesions with evidence of diffuse alveolar damage |
vRNA load peaked in nasal swabs 2–4 dpi and in BAL 3–5 dpi Infectious titre peaked in nasal swabs 2 dpi and in BAL 3–5 dpi vRNA and infectious titre peaked in oral swabs 3 dpi |
Non-lethal | 72 |
| African green monkey |
Aerosol: 103–104 PFU IN, IT, oral and ocular: 106 PFU SARS-CoV-2/München-1.1/2020/929 |
Peak lung inflammation and pathology (in PET or CT scan) 4 dpi, lesions resolved by 11 dpi |
vRNA load peaked in oral, nasal and conjunctival swabs 2–7 dpi Infectious virus isolated from oral, nasal, rectal and ocular swabs 2–4 dpi |
Non-lethal | 47 |
| Baboon |
IN, IT and ocular: 1.05 × 106 PFU SARS-CoV-2 isolate USA-WA1/2020 |
Bronchitis observed 14–17 dpi No longitudinal lung pathology data available |
vRNA load peaked in nasopharyngeal swabs and BAL 3 dpi | Non-lethal | 73 |
| Marmoset |
IN, IT and ocular: 1.05 × 106 PFU SARS-CoV-2 isolate USA-WA1/2020 |
Very mild pathology No longitudinal lung pathology data available |
vRNA load peaked in nasal wash samples 3 dpi | Non-lethal | 73 |
ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; CJ, conjunctival; COVID-19, coronavirus disease 19; CT, computed tomography; dpi, days post inoculation; FFU, focus forming units; hACE2, human angiotensin-converting enzyme 2; IB, intrabronchial; IN, intranasal; IT, intratracheal; IV, intravenous; PET, positron emission tomography; PFU, plaque forming units; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TCID50, 50% tissue culture infectious dose; vRNA, viral RNA.
Assessing prophylactic interventions in animal models
Preventing the establishment of infection in animal models is a key goal of many vaccine or prophylactic treatment studies. In most current models, animals are infected with ~104–106 TCID50 via different routes into the respiratory tract (Table 2) (although in vivo infectivity may be lower than in vitro infectivity owing to in vitro sequence artefacts25). It is not clear how these doses relate to the minimal infectious dose for a particular species and challenge route. However, current challenge doses may be many orders of magnitude higher than either the minimal dose for infection or the dose received in natural transmission. To completely prevent the establishment of infection requires neutralizing enough of the inoculum to leave less than one infectious dose remaining (Fig. 2b). This may lead to a significant overestimation of the concentration of antibody required to neutralize natural transmission (Supplementary Box 1). In addition, use of high-dose inocula may lead to difficulties differentiating between residual virus from the inoculum and new viral replication, requiring assays to detect sub-genomic mRNA26.
The absence of detectable viral growth in an animal does not necessarily indicate complete neutralization of the inoculum. Apparent sterilizing immunity will also be observed if treatment can block the spread of infection from cell to cell (even if the initial inoculum had not been fully neutralized). Thus, if the per-cell production of infectious virus from infected cells in vivo is less than from the initial inoculum, neutralizing cell to cell spread may be an easier mechanism to produce an apparent sterilizing treatment. The degree of viral inhibition required for apparent sterilizing immunity may vary between species because of differences in viral production and spread. As a result, prophylactic treatments may look more effective in models with lower viral replication.
When assessing protective efficacy in animal models, we suggest it may be beneficial to use a low-dose challenge model with sequence-verified virus stocks, in which animals are infected with something approximating the minimal animal infectious dose. Similarly, more physiological transmission could be modelled using nebulized virus or co-housing of infected and uninfected animals to allow direct animal to animal transmission27–29, rather than direct installation of virus in liquid suspension into the airways. A major impediment to this approach is that truly low-dose infection may lead to only a proportion of control animals being uninfected, which greatly reduces the statistical power in treatment studies. However, modifications such as serial or parallel low-dose challenge have the potential to provide a greater sensitivity to detect the protective efficacy of an intervention (Box 2). Low-dose challenge models for SARS-CoV-2 are yet to be reported, although these have become common in animal models of HIV and tuberculosis30–32. Challenge with more physiological inocula, in principle, should be more reflective of the level of immunity needed in vaccination or prophylactic treatment in humans (Supplementary Box 1). Interestingly, human challenge models of SARS-CoV-2 infection have also been proposed33,34. The need to calibrate the dose would be even more important in human studies, as there may be a dose-dependent effect on infection outcome. However, in human studies there may be less temptation towards the use of high challenge doses and the human infectious dose can be determined by dose escalation studies35. This further highlights the need to develop comparable low-dose challenge studies in animal models.
Box 2 Measuring protection against low-dose challenge.
Serial low-dose challenge involves individual animals being challenged at intervals with a low (commonly 50%) infectious dose of virus. A survival analysis is then used to compare the time to infection between treated (vaccinated) and untreated (unvaccinated) animals and to calculate the relative risk of infection (see the figure, part a). Serial challenges are often spaced by several days to prevent undue stress on animals and to identify which challenge resulted in infection. Although serial challenge implies a potential risk of ‘priming’ an endogenous response through multiple challenges, in practice this has not been observed in the setting of HIV74,75. A similar, parallel low-dose challenge approach involves challenge with multiple strains simultaneously (each at a low (typically 50%) infectious dose). This leads to around half of strains establishing infection in control animals, and inhibition is measured by a reduction in the proportion of strains successfully initiating infection in treated animals76,77 (see the figure, part b). This could be done either with naturally occurring viral strains (and strain-specific PCR to detect the number of strains infecting) or through genetic barcoding78. In this case, protection is measured by the reduction in the number of successfully infecting strains in treated animals. An advantage of the parallel rather than serial approach is that the animals are held for a shorter period.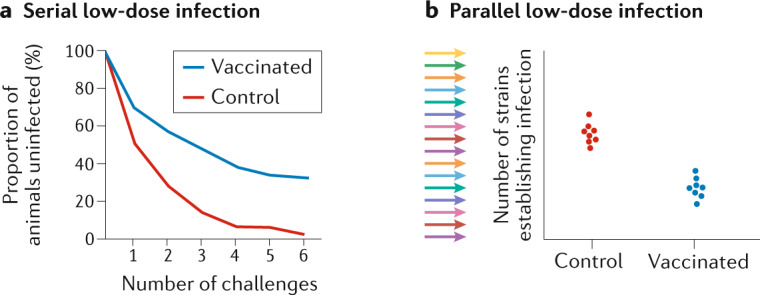
Post-exposure treatment or prophylaxis in animal models
An important use of therapies for COVID-19 is as a post-exposure prophylaxis for close contacts of infected individuals to prevent or control early viral growth, reduce disease and limit forward transmission. This can be modelled in animal studies by treatment after the establishment of infection but before the peak of viraemia. The efficacy of treatment in slowing viral growth depends on the reduction of infectious virus titres in each round of replication, as well as in the number of rounds of replication the virus undergoes (Fig. 2c). For example, if a treatment can reduce viral expansion on each cycle of infection by 50%, over 8 rounds of infection the virus titres will be reduced 256-fold in treated animals. High challenge doses of virus may reduce the time (and number of viral replication cycles) between infection and peak viral loads36 and thus limit the potential impact of ‘growth-reducing’ therapies. The impact of high inoculum size is demonstrated by the earlier peak in viral loads following inoculation in most animal models (2–4 days post infection (Table 2)) compared with animal to animal transmission37 or time to diagnosis in human infection (4–6 days, with the time to severe illness even longer)38,39. If antiviral effects on viral growth are the desired outcome of a study, then clearly the most important measure is a direct comparison of viral growth rates in treated versus untreated infection.
Therapeutic interventions in animal models
The therapeutic use of antiviral agents has the potential to reduce mortality and/or shorten the disease course in infected individuals40. Studies of immunotherapies in animal models might therefore focus on either a reduction in pathology or changes in viral dynamics as a result of treatment. However, several differences in the infection course observed in animal studies make it challenging to directly predict the effects in humans. Studies in patients infected with SARS-CoV-2 show that virus titres decline from the time of symptom onset, suggesting that initial presentation often occurs in the second week of infection at or after the peak in viral replication41,42. However, clinical progression often occurs while viral loads are declining and may be associated with immunopathology43,44. Different animal models show varying degrees of pathology, but in the majority of cases maximal pathology is observed within the first week of infection, up to a few days after the peak in initial viral levels 26,29,45–47 (Table 2). Both the viral peak and peak pathology occur earlier in many animal models than in humans, suggesting potential differences in the underlying pathophysiology. This might occur, in part, because of the altered timing of the viral peak and immune response and the mode of infection. In any event, the differences in pathophysiology raise a question of whether changes in pathological outcomes in animal models will have the same effects for severe COVID-19 in humans.
An alternative approach to measure the efficacy of therapeutic interventions is to directly study their effects on the dynamics of viral clearance. Analysis of patients with COVID-19 suggests that both a higher peak and a slower decay of viral load are seen in more severe infection48–50 and that antiviral treatment can improve outcome40. This suggests that inducing a faster decline in virus titre may improve outcome (although this causality has not been established). The decline in virus from the peak in other viral infections is typically thought to reflect the balance of any ongoing new infection of cells and the underlying death or shutdown of virus-producing cells (rather than the clearance of free virus, which is typically rapid)51. Thus, a faster decline in virus titre can be achieved by mechanisms such as increasing the death rate of infected cells, reducing the rate of production of virus from infected cells (through cytokine inhibition of viral production) or blocking any ongoing infection of cells (through virus neutralization or antiviral effects). An important consideration is whether the underlying mechanisms of decline in virus titre are similar between animal models and human infection. For example, the cell types infected may be quite different in some human ACE2 transgenic mouse models (with ubiquitous expression of human ACE2 driven by a constitutive promoter)52, which may lead to differences in cell susceptibility to viral cytopathic effects and/or differences in immune control of infection in different sites. Indeed, analysis of viral decay rates in animal models suggests these may be significantly faster than in human infection (Fig. 2d; Supplementary Methods). It is unclear whether the slower rates of viral decline in human infection reflect long-lived infected cells continually producing virus or ongoing rounds of infection of new cells. Again, how alterations in viral clearance translate into clinical outcome is uncertain, as immunopathology rather than virus-mediated destruction of infected cells may be a major factor driving severe illness in patients with COVID-19 (ref.43).
Immunity, immunopathology and immune recall in animal models
The early peak of infection in most animal models also affects the relative timing between viral and immune kinetics (Supplementary Box 2). In primary infection in animal models, the peak in viral infection occurs earlier than in human infection and likely reduces the role of acquired immune responses (which typically take 7–10 days to develop) in the early control of viral replication and decay of virus titres. By contrast, the later peak in virus titres in human infection means acquired immunity may play a larger role in driving viral decay. In addition, as the coexistence of high viral loads and high immune responses may contribute to immunopathology, these differences in timing may also limit immunopathology in animal models.
Altered infection kinetics in animal models may also affect the ability of vaccine-induced recall responses to control peak viral levels, as the activation and expansion of recall responses may be delayed for a few days after challenge53,54. Because the earlier the peak viral level occurs, the less time these responses have to act on it, high inocula may inherently limit the ability of vaccination to control peak viral loads (Supplementary Box 2).
In vivo veritas? Optimizing animal models
It is clear that there are several significant differences between the pathogenesis and kinetics of human infection and animal models, and there is currently no single, simple and optimal animal model for SARS-CoV-2 infection. In addition, it is also not clear which is the best outcome metric to study — for example, should an intervention aim to reduce the viral titre, pathology or lethality? The most suitable animal model and outcome measure for a particular application depends on the therapeutic intention, as well as the cost, timing and availability. Control of viral levels in the lower airways is clearly a metric that can be used across different animal models, even if they lack a pathological phenotype. Designing studies aimed at reducing pathology can be difficult, as the pathological outcomes are often quite variable between individuals (requiring potentially large group sizes). Syrian golden hamsters currently provide a more consistent lung disease phenotype among animal models described to date (Table 2). These animals, however, suffer from limited genetic diversity and a limited repertoire of available reagents compared with more widely used animal models. Non-human primates are most physiologically similar to humans but disease in these animals is typically mild, although sporadic fatal lung disease has been reported in aged African green monkeys. One question is whether the relatively benign course of infection seen in otherwise healthy non-human primates accurately models human infection, where disease severity is increased in older people with co-morbidities. Developing similar non-human primate models of COVID-19 in obese, diabetic and/or aged animals will likely be near impossible in the foreseeable future, so alternative methods of simulating disease that causes hospitalization in people are urgently needed.
Concluding remarks
Rapid progress is being made in understanding immunity to SARS-CoV-2, as well as in the development of novel prophylactic and therapeutic interventions. Assays to measure naturally acquired immunity and test the efficacy of immune interventions are key to this progress. The goal of the present work is not to criticize or dismiss particular assays or animal models. Instead, it is to state the importance of identifying what we want to measure and matching these goals to our experimental design. If we want to measure neutralization and prophylaxis, we need to choose assays and models that are optimized to quantify this. On the other hand, if we want to understand the effects of an intervention on viral growth, we need to measure growth directly. In most cases, this can be achieved by modifications to existing methods. For example, measuring the effects of interventions on viral growth rates and viral decay rates, rather than simply the peak viral load or time to viral clearance, should provide a clearer metric for comparison between different models and provide a more direct guide to predict therapeutic efficacy in human infection.
It is important to bear in mind that no matter how precisely we can measure the effects of interventions in vitro and in vivo, these assays and animal models remain imperfect mimics of human infection. Regardless of how sophisticated or internally valid an experimental system may be, it may still mislead us in prioritizing interventions in humans. Until correlates of protection can be established in clinical cohorts, our current approach must rely on assumptions and predictions from examples of other infections. However, a thoughtful approach to the use and interpretation of current systems should, ultimately, greatly enhance our ability to understand and predict the impact of immune interventions on SARS-CoV-2 infection.
Supplementary information
Acknowledgements
The authors are supported by National Health and Medical Research Council (NHMRC) Fellowship and Investigator grants 1173027 (to M.P.D.), 1136322 (to S.J.K.), 1141921 (to D.S.K.), 1173528 (to D.C.), 1177174 (to K.S.) and 1173433 (to A.K.W.). The Melbourne World Health Organization (WHO) Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health. SARS-CoV-2 work by D.H.O’C. is supported by US National Institutes of Health (NIH) awards P51OD011106 and R24 OD017850. Additional funding was provided by the Wisconsin National Primate Research Center, the Wisconsin Partnership Program and the Wisconsin Alumni Research Foundation COVID-19 Challenge.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Immunology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41577-020-00471-1.
References
- 1.Schmidt F, et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020;217:e20201181. doi: 10.1084/jem.20201181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Preprint at bioRxiv10.1101/2020.09.10.292078 (2020). [DOI] [PMC free article] [PubMed]
- 3.Korber B, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starr TN, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu L, et al. Potent neutralizing antibodies directed to multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- 6.Zost SJ, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584:443–449. doi: 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amanat F, et al. An in vitro microneutralization assay for SARS-CoV-2 serology and drug screening. Curr. Protoc. Microbiol. 2020;58:e108. doi: 10.1002/cpmc.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan CW, et al. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2-spike protein–protein interaction. Nat. Biotechnol. 2020;38:1073–1078. doi: 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- 9.Abe KT, et al. A simple protein-based surrogate neutralization assay for SARS-CoV-2. JCI Insight. 2020;5:e142362. doi: 10.1172/jci.insight.142362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong HL, et al. Robust neutralization assay based on SARS-CoV-2 S-protein-bearing vesicular stomatitis virus (VSV) pseudovirus and ACE2-overexpressing BHK21 cells. Emerg. Microbes Infect. 2020;9:2105–2113. doi: 10.1080/22221751.2020.1815589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford KHD, et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses. 2020;12:513. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinlan, B. D. et al. The SARS-CoV-2 receptor-binding domain elicits a potent neutralizing response without antibody-dependent enhancement. Preprint at bioRxiv10.1101/2020.04.10.036418 (2020).
- 14.Li Q, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020;182:1284–1294. doi: 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieterle ME, et al. A replication-competent vesicular stomatitis virus for studies of SARS-CoV-2 spike-mediated cell entry and its inhibition. Cell Host Microbe. 2020;28:486–496. doi: 10.1016/j.chom.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case JB, et al. Replication-competent vesicular stomatitis virus vaccine vector protects against SARS-CoV-2-mediated pathogenesis in mice. Cell Host Microbe. 2020;28:465–474. doi: 10.1016/j.chom.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuste E, Johnson W, Pavlakis GN, Desrosiers RC. Virion envelope content, infectivity, and neutralization sensitivity of simian immunodeficiency virus. J. Virol. 2005;79:12455–12463. doi: 10.1128/JVI.79.19.12455-12463.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeSantis MC, Kim JH, Song H, Klasse PJ, Cheng W. Quantitative correlation between infectivity and Gp120 density on HIV-1 virions revealed by optical trapping virometry. J. Biol. Chem. 2016;291:13088–13097. doi: 10.1074/jbc.M116.729210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stano A, et al. Dense array of spikes on HIV-1 virion particles. J. Virol. 2017;91:e00415–e00417. doi: 10.1128/JVI.00415-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu P, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 21.Conzelmann C, et al. An enzyme-based immunodetection assay to quantify SARS-CoV-2 infection. Antivir. Res. 2020;181:104882. doi: 10.1016/j.antiviral.2020.104882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juno JA, et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 2020;26:1428–1434. doi: 10.1038/s41591-020-0995-0. [DOI] [PubMed] [Google Scholar]
- 23.Martin JE, et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a phase I clinical trial. Vaccine. 2008;26:6338–6343. doi: 10.1016/j.vaccine.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munoz-Fontela C, et al. Animal models for COVID-19. Nature. 2020;586:509–515. doi: 10.1038/s41586-020-2787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson AD, et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12:68. doi: 10.1186/s13073-020-00763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandrashekar A, et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. 2020;369:812–817. doi: 10.1126/science.abc4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y, et al. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe. 2020;27:704–709. doi: 10.1016/j.chom.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sia S, et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature. 2020;583:834–838. doi: 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan JF, et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hessell AJ, et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 2009;15:951–954. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe S, et al. Ultra low dose aerosol challenge with Mycobacterium tuberculosis leads to divergent outcomes in rhesus and cynomolgus macaques. Tuberculosis. 2016;96:1–12. doi: 10.1016/j.tube.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Regoes RR, Longini IM, Feinberg MB, Staprans SI. Preclinical assessment of HIV vaccines and microbicides by repeated low-dose virus challenges. PLoS Med. 2005;2:e249. doi: 10.1371/journal.pmed.0020249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deming ME, Michael NL, Robb M, Cohen MS, Neuzil KM. Accelerating development of SARS-CoV-2 vaccines — the role for controlled human infection models. N. Engl. J. Med. 2020;383:e63. doi: 10.1056/NEJMp2020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen LC, et al. Evaluating use cases for human challenge trials in accelerating SARS-CoV-2 vaccine development. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine MM, et al. Viewpoint of a WHO advisory group tasked to consider establishing a closely-monitored challenge model of COVID-19 in healthy volunteers. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamner L, et al. High SARS-CoV-2 attack rate following exposure at a choir practice — Skagit County, Washington, March 2020. MMWR. 2020;69:606–610. doi: 10.15585/mmwr.mm6919e6. [DOI] [PubMed] [Google Scholar]
- 37.Schlottau K, et al. SARS-CoV-2 in fruit bats, ferrets, pigs, and chickens: an experimental transmission study. Lancet Microbe. 2020;5:e218–e225. doi: 10.1016/S2666-5247(20)30089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan WJ, et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou F, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beigel JH, et al. Remdesivir for the treatment of COVID-19 — preliminary report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2007764. [DOI] [PubMed] [Google Scholar]
- 41.Zou L, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.To KK-W, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect. Dis. 2020;20:565–574. doi: 10.1016/S1473-3099(20)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Domingo P, et al. The four horsemen of a viral apocalypse: the pathogenesis of SARS-CoV-2 infection (COVID-19) EBioMedicine. 2020;58:102887. doi: 10.1016/j.ebiom.2020.102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang R, et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell. 2020;182:50–58. doi: 10.1016/j.cell.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munster V, et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature. 2020;585:268–272. doi: 10.1038/s41586-020-2324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartman AL, et al. SARS-CoV-2 infection of African green monkeys results in mild respiratory disease discernible by PET/CT imaging and shedding of infectious virus from both respiratory and gastrointestinal tracts. PLOS Pathog. 2020;16:e1008903. doi: 10.1371/journal.ppat.1008903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magleby R, et al. Impact of SARS-CoV-2 viral load on risk of intubation and mortality among hospitalized patients with coronavirus disease 2019. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He X, et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020;26:672–675. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
- 50.Lescure F-X, et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect. Dis. 2020;20:697–706. doi: 10.1016/S1473-3099(20)30200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramratnam B, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 52.Bao L, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020;583:830–833. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- 53.Davenport MP, Ribeiro RM, Perelson AS. Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J. Virol. 2004;78:10096–10103. doi: 10.1128/JVI.78.18.10096-10103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reynolds MR, et al. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: too late and too little. J. Virol. 2005;79:9228–9235. doi: 10.1128/JVI.79.14.9228-9235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nie J, et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 2020;9:680–686. doi: 10.1080/22221751.2020.1743767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oguntuyo KY, et al. Quantifying absolute neutralization titers against SARS-CoV-2 by a standardized virus neutralization assay allows for cross-cohort comparisons of COVID-19 sera. medRxiv. 2020 doi: 10.1101/2020.08.13.20157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinto D, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 58.Case JB, et al. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host Microbe. 2020;28:475–485.e5. doi: 10.1016/j.chom.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manenti A, et al. Evaluation of SARS-CoV-2 neutralizing antibodies using a CPE-based colorimetric live virus micro-neutralization assay in human serum samples. J. Med. Virol. 2020 doi: 10.1002/jmv.25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corbett KS, et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med. 2020;383:1544–1555. doi: 10.1056/NEJMoa2024671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu J, et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science. 2020;369:806–811. doi: 10.1126/science.abc6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okba NMA, et al. Severe acute respiratory syndrome coronavirus 2-specific antibody responses in coronavirus disease patients. Emerg. Infect. Dis. 2020;26:1478–1488. doi: 10.3201/eid2607.200841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winkler E, et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020;21:1327–1335. doi: 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hassan A, et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell. 2020;182:744–753. doi: 10.1016/j.cell.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun S, et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe. 2020;28:124–133. doi: 10.1016/j.chom.2020.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imai M, et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl Acad. Sci. USA. 2020;117:16587–16595. doi: 10.1073/pnas.2009799117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi J, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science. 2020;368:1016–1020. doi: 10.1126/science.abb7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng W, et al. Primary exposure to SARS-CoV-2 protects against reinfection in rhesus macaques. Science. 2020;369:818–823. doi: 10.1126/science.abc5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rockx B, et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science. 2020;368:1012–1015. doi: 10.1126/science.abb7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finch C, et al. Characteristic and quantifiable COVID-19-like abnormalities in CT- and PET/CT-imaged lungs of SARS-CoV-2-infected crab-eating macaques (Macaca fascicularis) bioRxiv. 2020 doi: 10.1101/2020.05.14.096727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blair RV, et al. Acute respiratory distress and cytokine storm in aged, SARS-CoV-2 infected african green monkeys, but not in rhesus macaques. bioRxiv. 2020 doi: 10.1101/2020.06.18.157933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woolsey, C. et al. Establishment of an African green monkey model for COVID-19. Preprint at bioRxiv10.1101/2020.05.17.100289 (2020).
- 73.Singh DK, et al. SARS-CoV-2 infection leads to acute infection with dynamic cellular and inflammatory flux in the lung that varies across nonhuman primate species. bioRxiv. 2020 doi: 10.1101/2020.06.05.136481. [DOI] [Google Scholar]
- 74.Regoes RR. The role of exposure history on HIV acquisition: insights from repeated low-dose challenge studies. PLoS Comput. Biol. 2012;8:e1002767. doi: 10.1371/journal.pcbi.1002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henning TR, et al. Short communication: viremic control is independent of repeated low-dose SHIVSF162p3 exposures. AIDS Res. Hum. Retroviruses. 2014;30:1125–1129. doi: 10.1089/aid.2014.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parsons MS, et al. Partial efficacy of a broadly neutralizing antibody against cell-associated SHIV infection. Sci. Transl. Med. 2017;9:eaaf1483. doi: 10.1126/scitranslmed.aaf1483. [DOI] [PubMed] [Google Scholar]
- 77.Khanal S, et al. In vivo validation of the viral barcoding of SIVmac239 and the development of new barcoded SIV and subtype B and C SHIVs. J. Virol. 2019;94:e01420-19. doi: 10.1128/JVI.01420-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deleage C, et al. Defining early SIV replication and dissemination dynamics following vaginal transmission. Sci. Adv. 2019;5:eaav7116. doi: 10.1126/sciadv.aav7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.