

Abstract
The residue lysine 28 (K28) is known to form an important salt bridge that stabilizes the Aβ amyloid structure, and acetylation of lysine 28 (K28Ac) slows the Aβ42 fibrillization rate but does not affect fibril morphology. On the other hand, acetylation of lysine 16 (K16Ac) residue greatly diminishes the fibrillization property of Aβ42 peptide and also affects its toxicity. This is due to the fact that lysine 16 acetylated amyloid beta peptide forms amorphous aggregates instead of amyloid fibrils. This is likely a result of increased hydrophobicity of the K16-A21 region due to K16 acetylation, as confirmed by molecular dynamic simulation studies. The calculated results show that the hydrophobic patches of aggregates from acetylated peptides were different when compared to wild-type (WT) peptide. K16Ac and double acetylated (KKAc) peptide aggregates show significantly higher cytotoxicity compared to the WT or K28Ac peptide aggregates alone. However, the heterogeneous mixture of WT and acetylated Aβ42 peptide aggregates exhibited higher free radical formation as well as cytotoxicity, suggesting dynamic interactions between different species could be a critical contributor to Aβ pathology.
Keywords: amyloid β peptide, acetylation, amyloid fibril, molecular dynamics, aggregation, toxicity, post-translational modifications
Graphical Abstract
INTRODUCTION
The extracellular amyloid plaques formed from amyloid β (Aβ) peptides are the major hallmarks of Alzheimer’s disease (AD), and variability in toxicity has been reported for different structures of Aβ aggregates.1–6 However, the relationship between different aggregated structures of Aβ peptides and their associated toxicity is not well understood.7 This is further underscored by the failure of AD treatment based on Aβ aggregates, as these treatments targeted mature amyloid fibril forms but failed to target highly transient and dynamic Aβ species that are naturally formed in the brain.3,8,9
The in vivo “pool” of Aβ contains not only the Aβ peptides of different lengths, but also the post-translationally modified forms,8 which have been determined to play an important role in protein folding and aggregation. Post-translational modifications (PTMs) such as phosphorylation,10–12 truncation, 13–15 isomerization,16,17 and pyroglutamate formation15,18 were found to impact Aβ peptide structure and aggregation propensity. In a recent study, protein PTM levels were found to increase roughly twofold in AD samples compared to normal controls, especially in Aβ and tau enriched areas.19 As one of the most common PTMs in proteins, acetylation is generating renewed interest as it plays a vital role in health issues such as DNA repair, protein structure, signaling, and protein self-assembly and in diseases such as cancer and neurodegenerative diseases.20–33 Interestingly, among all the identified PTM types in AD brains, acetylation affects only ∼10% of the total modified peptides but showed the highest increase in level of Aβ- and tau-containing aggregates.19 In the case of AD, acetylation of tau protein was already identified and proposed as a new therapeutic target.33–35 The Aβ peptide has two potential acetylation sites, lysine 16 (K16) and lysine 28 (K28) (Figure 1A); the effect of K28 acetylation has been investigated, though K16 residue as an acetylation site was often ignored. Lysine 28 was recognized as a key player during Aβfibril formation as it forms a salt bridge with residue A42 in Aβ42 fibrils36 or with residue D23 in Aβ40 fibrils37 to stabilize the β-sheet structure.
Figure 1.

(A) Sequence of Aβ42 peptide (PDB entry: 1Z0Q40). Atomic structures of (B) WT, (C) K16Ac, (D) K28Ac, and (E) KKAc monomer. The two acetylated positions, K16 and K28, are highlighted in ball and stick presentation.
Acetylation of lysine residues in tau has been linked to pathological accumulation of tau and Aβ peptides in Alzheimer’s disease and synaptic dysfunction.29–33 Several studies show that tau protein can be preferentially acetylated at specific residues, thereby affecting tau’s intrinsic property to aggregate and hence its role in the disease process.31,33,38 Although, the role of tau acetylation has been extensively studied in Alzheimer’s disease, the role of Aβ acetylation has not been explored. Therefore, we carried out a systematic study investigating the impact of acetylation of Aβ on protein aggregation and its associated toxicity. This is important because many common drugs and metabolites were recently found to be able to facilitate protein acetylation.20,39 In this study, synthesized Aβ42 peptides were modified with single acetylation on K16 (K16Ac) or K28 (K28Ac) or were double acetylated on both lysine residues (KKAc) (Figure 1B–E). The fibril formation was simulated via assembly of nine strands of Aβ42 peptides. Surprisingly, we found that side chain acetylation of K16 can increase hydrophobicity of Aβ42 peptide, profoundly impair the fibrillization property, and as a consequence, modulate Aβ42 pathogenicity.
RESULTS
Physicochemical Characterization of Aβ Peptide Aggregation.
Fibrillization/aggregation of the four Aβ42 peptides (wild-type (WT), K16Ac, K28Ac, and KKAc) were monitored by ThT fluorescence (Figure 2A–D). K28Ac showed sigmoidal fluorescence trend similar to WT Aβ42 (Figure 2 A, E) and was able to form fibril-like structures in both the homogeneous K28Ac sample (Figure 2A, G) and in the equimolar mixture with WT (Figure 2C, K). In sharp contrast, the two peptides that have acetylation on K16 (K16Ac and KKAc) both showed nonsigmoidal ThT fluorescence trend (Figure 2A) and formed amorphous aggregates after 7 days (Figure 2F, H) that stayed amorphous even after 14 days (Supplementary Figure 1) of incubation. The initial rates of aggregation for K16Ac and KKAc peptides were rapid compared to those of K28Ac and WT peptides, resulting in a sharp early increase in slope that slowed considerably around day 2 of incubation (Figure 2A). In contrast, WT and K28Ac peptides showed a sigmoidal curve for aggregation with slow initial change in the first 2 days (lag phase) (Figure 2A) and then rapid increase in fluorescence from day 2 to 4 (log phase) before slowing again (stationary phase). In the presence of WT Aβ42, K16Ac and KKAc still maintained their aggregation kinetics and properties, and only amorphous aggregates were observed (Figure 2B, D, J, L). These results show that acetylation at K16 can significantly impact aggregation of Aβ42 peptide, resulting in amorphous structure instead of a fibrillar structure. Interestingly, Aβ42 peptide that was single acetylated (K16Ac) or double acetylated (KKAc) formed amorphous aggregates that stayed amorphous even when mixed with WT Aβ42 peptides (1:1 ratio) with distinct morphology (Figure 2F, H, J, L) even upon longer incubation time. By using the structure sensitive probe bis-ANS, we determined that the aggregates of K16Ac peptide have the most flexible structure with a clear bead-like structure among the four amorphous aggregates (Figure 2I, F) studied. Aggregates formed from KKAc peptide are slightly less flexible and are denser compared to K16Ac peptides and have a ThT fluorescence signal comparable to that of WT Aβ42 peptides (Figure 2I, H, A). Furthermore, the mixtures of single and double acetylated peptides with WT Aβ42 peptide (1:1 ratio) formed amorphous aggregates with a fluorescence signal comparable to that of WT Aβ42 aggregates which are more rigid than those formed by pure K16Ac peptides (Figure 2I).
Figure 2.

Fibrillization/aggregation of WT, acetylated, and mixtured Aβ42 peptides were monitored using ThT fluorescence, bis-ANS fluorescence, and SEM. The mixture represents the 1:1 ratio of the acetylated peptides with WT Aβ42 peptides. For ThT fluorescence analysis (A–D), 10 μM peptide samples were incubated with 10 μM ThT. Peak intensities at 487 nm were plotted as a function of time. After 7 day of incubation, the structures of fibrils or aggregates were visualized using SEM (E–H and J–L). Scale bars are 1 μm for all SEM images. Structure flexibility of the fibrils or aggregates was measured by bis-ANS fluorescence (I) by incubating 10 μM peptide samples with 1 μM bis-ANS and measuring peak intensities at 484 nm. Error bars = ±SD (For A-D n = 3; for I n = 3). * represents significant differences (p < 0.05) among 7 d Aβ42 aggregated peptides.
Molecular Aggregation Dynamics and Surface Properties of Aβ Peptides.
To gain atomistic insights into the conformation changes, aggregation dynamics, and map the folding pathways of Aβ42 peptides,41–47 we performed molecular dynamics (MD) simulations of WT and acetylated Aβ42 monomers along with nine strand aggregates of all four peptides (WT, K16Ac, K28Ac, and KKAc) in aqueous solution using NAMD program48 and CHARMM27 force field.49 Note that aggregation of nine strands of WT and acetylated peptides were considered as replica models for simulating the formation of fibril-like structures. Supplementary Figure 3A–D displays the structural changes in WT and acetylated Aβ42 monomer in aqueous solution at time = 0 and 100 ns. WT Aβ42 is mostly α-helical at residues 11–23 and 29–37, which agrees well with previously reported results based on the discrete MD simulation method.50 In our case of atomistic MD study, the flexible regions are the first 10 amino acids of N-terminus, residues 38–42 of C-terminus that adopt a random coil structure, and the β-turn region between residues 24–28 (Supplementary Figure 4A). Snapshots of WT and acetylated peptide aggregates at time = 0 and 100 ns elucidate that the nature of fibrillization is distinctive of the modifications: K16Ac and KKAc assemble into an amorphous structure, while K28Ac retains a fibril-like structure similar to that of WT peptides (Figure 3A–D). MD simulation snapshots (Supplementary Figure 7) of K16Ac at 100 ns affirm the formation of amorphous aggregates, as seen in SEM micrographs (Figure 2F). A comparison of the orientation of K16 and K28 residues in acetylated aggregates suggests that in the former structure, K16Ac residues are somewhat embedded within the aggregate, stabilized by interchain hydrophobic interactions, while in the latter, K28Ac residues project outward with reduced interchain interactions (Supplementary Figure 5). Both MD simulation and experimental results thus find that K16Ac presents a flexible structure with loss of α-helicity, and acetylation significantly impacts aggregation dynamics and impedes fibril formation. Root-mean-square deviation (RMSD) analysis was performed for the simulations of each system (Supplementary Figure 10). RMSD of WT and acetylated peptides increases in the first 20 ns and then reaches a plateau, suggesting an overall stability of the system. A slightly higher RMSD of K16Ac correlates to the molecular flexibility and observed conformational transition to an amorphous structure.
Figure 3.

Simulation snapshots depicting the structural changes in aggregation of nine strands of WT and acetylated (K16Ac, K28Ac, and KKAc) Aβ42 peptides in aqueous solution. For each structure, the predicted hydrophobic patches (yellow) were mapped on the molecular surface (violet) using SPDB software. (A) WT, (B) K16Ac, (C) K28Ac, and (D) KKAc aggregates. The water molecules are not shown for clarity.
Hydrophobic patches on a protein surface identify accessible regions of neighboring apolar atoms that may play a crucial role in protein folding, structural dynamics, and protein–ligand interactions.51 Surface hydrophobicity of WT and acetylated peptide aggregates were analyzed for 100 ns structures (Figure 3). Note that acetylation of the positively charged K16 and K28 lysine residues into a hydrophobic moiety destabilizes fibril formation with an overall increase in the surface area (Supplementary Table 1). Interestingly, the surface area of KKAc is intermediate to K16Ac and K28Ac with relatively scattered hydrophobic patches on the molecular surface. K16Ac has the highest surface area and consequently exhibits a greater flexibility to adopt an amorphous structure. WT Aβ42 peptides have 1 patch above 300 Å2 and 9 patches between 100–300 Å2 (Supplementary Table 1). Acetylation leads to loss of the dominant patch above 300 Å2 followed by decreases in size of the hydrophobic patches but increases the number of patches on the molecular surface. For acetylated peptides, the hydrophobic patches range between 100–300 Å2. These findings are consistent with bis-ANS fluorescence data (Figure 2I) that show increased fluorescence for 7 d aggregates of K16Ac peptides compared to 7 d WT Aβ42 peptides.
Surface Hydrophobicity of Aβ Peptides.
Surface hydrophobicity of the peptides and aggregates was also measured by ANS fluorescence as well as hydrophobic bead binding assay (Figure 4). All three fresh acetylated peptides showed ANS fluorescence similar to that of WT Aβ42 peptides. Interestingly, after 3 d of incubation, ANS fluorescence of WT Aβ42 peptides increased significantly compared to acetylated peptides and then decreased upon longer incubation of 7 d to levels comparable to fresh WT Aβ42 peptides (Figure 4A). Differences in hydrophobicity among the four peptides were amplified after 7 days of incubation and were extremely significant for K16Ac peptides that almost doubled compared to 7 d WT Aβ42 peptides (Figure 4A). Seven-day incubated K28Ac Aβ42 peptides showed increased fluorescence signal, and KKAc aggregates showed signals that were much higher compared to 7 d WT Aβ42 peptides (Figure 4A). In addition, for 7 d samples incubated as a mixture, the aggregates showed slightly less hydrophobicity compared to pure K16Ac and KKAc peptides, but signals were significantly higher compared to pure 7 d WT Aβ42 aggregates (Figure 4A). The surface hydrophobicity of 7 d Aβ42 peptides was further confirmed by hydrophobic bead binding assay52 (Figure 4B and C). The 7 d Aβ42 peptides were incubated with hydrophobic beads, washed, eluted in sample buffer, and then analyzed by SDS-PAGE (Figure 4B, Supplementary Figure 11). Interestingly, the peptides bound to hydrophobic beads run as monomers in SDS-PAGE and showed results that were consistent with hydrophobicity measured by ANS fluorescence for 7 d incubated peptides (Figure 4A and C). Aβ42 K16Ac peptides and its mixture with WT Aβ42 showed the highest percentage of peptides bound to hydrophobic beads (Figure 4C and Supplementary Figures 11 and 12). These results are also consistent with bis-ANS fluorescence data (Figure 2I) and molecular dynamics simulation data (Figure 3).
Figure 4.

Hydrophobicity of Aβ42 peptides measured by ANS fluorescence and hydrophobic bead binding assay. (A) Hydrophobicity of fresh Aβ42 peptides and their aggregates measured by ANS fluorescence by incubating 10 μM peptide samples with 10 μM ANS and measuring peak intensities at 485 nm. Mixture represents the 1:1 ratio of the acetylated peptides with WT Aβ42 peptides. Error bars = ±SD (n = 3). *, **, and *** represent significant differences (* = p < 0.05), (** = p < 0.005), and (*** = p < 0.0005) among 7 d Aβ42 aggregated peptides. (B) Seven-day incubated Aβ42 peptide samples (3 μg) were further incubated with phenyl-sepharose beads overnight with gentle agitation at 25 °C. The beads were washed thrice with binding buffer, and then Aβ42 peptides bound to beads were eluted by boiling with denaturing buffer for 3 min. Eluted Aβ42 peptide samples and loading controls (WTC 1–4) were run on SDS-PAGE and visualized by staining with Coomassie blue. (C) The plot shows percentages of Aβ42 peptides bound to hydrophobic beads which were calculated with respect to WT Aβ42 peptides controls (WTC 1–4) loaded on gel.
Cellular Toxicity of Aβ Peptides.
Effect of 5 μM Aβ42 peptides on SH-SY5Y neuroblastoma cells health and 2 μM Aβ42 peptides on primary neuronal cells from rat were measured by incubating cells with fresh peptides and aggregates at 72 h and 7 d time points (Figures 5, 6). We chose these three time points based on the fibrillization/aggregation kinetics data of the peptides as monitored by ThT fluorescence (Figure 2A–D). We monitored the effect of Aβ42 fibrils and aggregates on cell viability of SH-SY5Y neuroblastoma cells by MTS assay (Figure 5A). The data show that compared to pure WT Aβ42 peptides, 7 d aggregates of KKAc peptides or 1:1 mixture of WT:K16Ac and WT:K28Ac inhibit proliferation of SH-SY5Y cells significantly (Figure 5A). Among the group, the 1:1 mixture of WT:KKAc 7 d peptides shows the highest inhibition of SH-SY5Y cells, resulting in significant decrease in cell viability (Figure 5A).
Figure 5.

The effect of fresh Aβ42 peptides and their aggregates on SH-SY5Y neuroblastoma cell health was measured by (A) MTS assay, (B) DCFH-DA assay, and (C) LDH assay. The fresh peptides, 72 h, or 7 d aggregates were added to the cells to a final concentration of 5 μM. Cells were incubated for 48 h in the presence of peptides for MTS and LDH assay and for 24 h for DCFH-DA assay. For the t-BHP control for the DCFH-DA assay, cells were incubated with 50 μM t-BHP for 4 h at 37 °C. The mixtures represent samples containing equimolar (1:1) WT and acetylated peptides. Error bars = ±SD (for A n = 6; for B n = 5; and for C n = 5). *, **, and *** represent significant differences (* = p < 0.05), (** = p < 0.005), and (*** = p < 0.0005) among 7 d Aβ42 aggregated peptides.
Figure 6.

The effect of fresh Aβ42 peptides and their aggregates on primary neuronal cell health was measured by (A) DCFH-DA assay and (B) LDH assay. The fresh peptides, 72 h, or 7 d aggregates were added to the cells to a final concentration of 2 μM. Cells were incubated for 24 h in the presence of peptides for both assays. For the t-BHP control for the DCFH-DA assay, cells were incubated with 50 μM t-BHP for 4 h at 37 °C. The mixtures represent samples containing equimolar (1:1) WT and acetylated peptides. Error bars = ±SD (for A n = 5; and for B n = 4). *, **, and *** represent significant differences (* = p < 0.05), (** = p < 0.005), and (*** = p < 0.0005) among 7 d Aβ42 aggregated peptides.
Oxidative stress has been implicated in several neurodegenerative diseases, including Alzheimer’s disease.53–59 Therefore, we wanted to measure the effect of 5 μM Aβ42 peptides (fresh as well as aggregates) on generation of reactive oxygen species (ROS) in cells. We used 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as a fluorescent probe to measure levels of ROS in cells (Figure 5B).60 Compared to untreated SH-SY5Y cells, the cells treated with 7 d WT Aβ42 peptides showed significant increase in ROS production. The 7 d aggregated acetylated peptides K16Ac, K28Ac, and KKAc peptides showed much higher production of ROS in SH-SY5Y cells compared to 7 d WT Aβ42 peptide aggregates (Figure 5B). Interestingly, the 1:1 7 d mixture of WT and all acetylated Aβ42 peptides also showed much higher production of ROS compared to the pure 7 d WT Aβ42 peptide aggregates, with WT:KKAc 7 d peptide aggregates showing the highest ROS production (Figure 5B). To better understand the implication of Aβ42 peptide aggregates on ROS production and hence cell health, we also measured toxicity of these peptides by lactate dehydrogenase (LDH) cytotoxicity assay (Figure 5C).61 The aggregates from pure WT Aβ42, K16Ac, and K28Ac peptides and equimolar mixture with WT (1:1) all show highly significant increase in toxicity for SH-SY5Y cells for 7 d aggregates, consistent with increased ROS production (Figure 5B, C). Interestingly, among the group, double acetylated peptide (KKAc) aggregates showed the highest toxicity (Figure 5C). Equimolar mixtures (1:1) of WT and KKAc 7 d Aβ42 peptide aggregates showed much higher increase in ROS production compared to pure KKAc (Figure 5B). However, the cytotoxicity observed for the mixtures was comparable to that of pure KKAc 7 d aggregates (Figure 5C). This may be because both 7 d aggregates (pure KKAc and 1:1 mixture of WT:KKAc) are extremely toxic, and values are close to the max LDH value, which is the positive control (Figure 5C).
To further validate results on SH-SY5Y neuroblastoma cells, we carried out toxicity and ROS studies on primary neuronal cells from rat (Figure 6). Because 5 μM peptides were extremely toxic to SH-SY5Y neuroblastoma cells, we used a lower concentration of peptides (2 μM) to evaluate cytotoxicity and ROS production for primary neuronal cells. We incubated neuronal cells with 2 μM WT and acetylated Aβ42 peptides for 24 h before the assay. Compared to the untreated neuronal cells, cells treated with 7 d aggregates of WT Aβ42 peptides and K28Ac peptides show a highly significant increase in ROS production (Figure 6A). Cells treated with 7 d aggregates from K16Ac, KKAc, and equimolar mixtures (1:1) of WT and acetylated Aβ42 peptides showed much higher increase in ROS production compared to aggregates from pure 7 d WT Aβ42 peptides (Figure 6A). Consistent with increased ROS production, all neuronal cells treated with 2 μM WT and acetylated Aβ42 peptides and their (1:1 ratio) mixtures showed an extremely significant increase in cytotoxicity, as measured by LDH assay for 7 d aggregated peptides (Figure 6B). Among the group, 7 d aggregates of double acetylated Aβ42 peptides (KKAc) showed the highest increase in cytotoxicity as well as ROS production (Figure 6).
Immunostaining of Primary Neuronal Cells Treated with WT and Acetylated Aβ42 Peptides.
To visualize the effect of WT and acetylated Aβ42 peptides on primary neuronal cells, we carried out immunostaining of cells after treating them with 2 μM 7 d Aβ42 peptide aggregates for 24 h (Figure 7). We chose 2 μM peptide concentration of 7 d aggregates based on our ROS and LDH cytotoxicity assay for neuronal cells (Figure 6). We compared the morphology of neuronal cells treated with 7 d Aβ42 peptide aggregates for 24 h to control cells (untreated cells) that were similarly incubated by immunofluorescence microscopy (Figure 7). We used anti-tubulin antibody (green) to image tubulin protein and anti-tau antibody (red) to image tau protein in cells. The control cells (untreated primary neuronal cells) show a normal morphology with tau signal that is diffused and evenly distributed in neurons (both soma and neurites) (Figure 7). The neuronal cells treated with 2 μM WT, K16Ac, and KKAc Aβ42 peptides for 24 h showed a significantly higher immunofluorescence signal for tau in soma of neurons compared to neurites (Figure 7, Supplementary Figure 13). Cell bodies of primary neurons showed the highest tau signal for cells treated with KKAc peptides, high tau signal for cells treated with WT and K16Ac peptides, and a much lower tau signal for cells treated with K28Ac peptides (Figure 7). These findings are consistent with the ROS production and cytotoxicity data reported for these cells (Figure 6).
Figure 7.

Immunostaining of primary neuronal cells from rat brain. Primary neuronal cells were incubated with 2 μM 7 d WT and acetylated Aβ42 peptides for 24 h at 37 °C. Control indicates cells that are untreated and incubated under conditions identical to cells treated with Aβ42 peptides for comparison. The cells were fixed and then stained for immunofluorescence with antibodies for tubulin (green) and tau (red). The images were acquired using Leica DMIL LED at 20× magnification. Scale bar = 50 μm.
DISCUSSION
Scientific literature suggests that formation of highly ordered amyloid fibrils or less ordered oligomer structures could be an outcome of the competition between hydrogen bonding and hydrophobic interactions, as the higher ordered structures are usually stabilized by a high density of direct interchain hydrogen bonds and steric zipper interactions.62–64 Furthermore, amyloid fibril dynamics could be affected by the chemical nature of the Aβ peptide through side chain interactions and/or post-translation modifications.65 In theory, introducing an acetyl moiety to a lysine residue removes a charge and hence can increase surface hydrophobicity of the peptide. Interestingly, in this study, we showed that acetylation of lysine 16 and 28 on Aβ42 does not affect protein aggregation in the same way; K16 acetylation affects the aggregation morphology as well as kinetics very severely.
We found that the K16Ac peptide is much more hydrophobic with a structure flexibility higher than that of the WT and the other two acetylated peptides (Figuress 2I, 3, and 4). The structural changes on the Aβ42 monomer in aqueous solution (Supplementary Figure 3) agree well with previously reported results based on the discrete MD simulation method.50 Acetylation of Aβ42 monomer at K16 leads to one extra β-turn between residues 14–17 (Supplementary Figure 4B), but acetylation at K28 retains a structure very similar to that of the WT (Supplementary Figure 4A and C). In addition, acetylation of Aβ42 at K16 and K28 residues on KKAc introduces a pleated region between residues 31–37 with loss in the α-helix (Supplementary Figure 4D). In agreement with our MD results, a rapid aggregation kinetics with no lag time was reported in a simulation study of the Aβ16–22 fragment, where the Aβ16–22 oligomers were disordered molten globular structures due to strong hydrophobic interactions62 because K16 is located in the central hydrophobic core, L17-F20 region, and the 16–20 residues (KLVFF) act as a docking motif of Aβfibrillation.40 This fact is also supported by our MD simulations of 9 strand K16Ac aggregates, where the K16Ac residues are embedded inside the aggregate structure, stabilized by the interchain hydrophobic intercalations (Figures 3B, Supplementary Figure 5). The Aβ fibril grows in two distinct steps: a rapid and reversible “dock” phase, followed by a slow but irreversible “lock” phase.64,66 In the case of K16Ac peptide aggregation, as observed in the current study, it is possible that the interchain hydrophobic interactions of the 16–20 region are further enhanced after removal of the positive charge on K16.67 This may result in an initial rapid docking process that becomes irreversible and as a consequence kinetically inhibits the conformational reorganization in the locking phase. Even if the formation of amyloid fibrils is thermodynamically favored, the transition from monomers to amyloid conformations is kinetically limited.64,68 Although this study investigated the effect of acetylation of the Aβ 42 peptide, it is quite likely that acetylation of other Aβ peptides (Aβ 40 and Aβ 39) at K16 may also impact aggregation kinetics and morphology in a similar manner. However, this needs to be determined experimentally as an independent study.
The other lysine residue, K28, is known to form important salt bridges in Aβ fibrils.36,37 Removing the positive charge on K28 by acetylation results in K28Ac peptide having a lag time longer than that of WT Aβ42 during fibril formation. This is consistent with the stabilizing role of the K28 salt bridge on the amyloid fibrils.69 However, the K28Ac peptides were capable of forming amyloid structures upon longer incubation, either as a pure K28Ac peptide sample or in a heterogeneous equimolar mixture with WT Aβ42 (Figure 2A, C, G, K), suggesting the K28 salt bridge may not play a role in the conformational transition from Aβ42 monomers to mature amyloids. This finding is also supported by a previous study of N-ε-amino acetylation on K28 of the Aβ25–35 fragment, where the gross structure of the Aβ25–35 K28Ac fibril was very similar to that of the WT Aβ25–35 fibrils.70
In this study, we identified two major forms of aggregated structures: amorphous and fibrillar, from WT, acetylated Aβ42 peptides, and their mixtures by SEM imaging. The 7 d aggregates from these 7 different Aβ42 peptides (WT, K16Ac, K28Ac, KKAc, and 1:1 mixtures of WT:K16Ac, WT:K28Ac, and WT:KKAc) can be further categorized based on their unique physicochemical properties, as measured by fluorescent dyes, hydrophobic bead binding assay, ROS production, and their associated cytotoxicity (Table 1). Among these 7 different peptides tested, WT Aβ42 peptide 7 d aggregates show well organized fibrillar structure that has low flexibility, low hydrophobicity, moderate ROS production, and moderate toxicity at 5 μM concentrations (Figures 2E, I, 4, and 5B, C). K28Ac peptide 7 d aggregates form fibrils that are similar to the WT peptides but show higher ROS production with high toxicity (Figures 2G, I, 4, and 5B, C). K16Ac peptide 7 d aggregates are amorphous, highly hydrophobic, and flexible (Figures 2F, I and 4). In addition, these aggregates also show cytotoxicity that is much higher than that of the WT peptide with high ROS production (Figure 5B, C). In the case of neuronal cells, 7 d aggregates of 2 μM WT Aβ42 peptides and K28Ac show moderate ROS production but high cytotoxicity (Figure 6). In comparison, K16Ac peptides show much higher ROS production as well as cytotoxicity (Figure 6). Interestingly, 7 d aggregates from KKAc peptides that are amorphous in nature show significantly higher ROS production as well as cytotoxicity for SH-SY5Y as well as primary neuronal cells (Figures 5B, C and 6). Furthermore, the 7 d aggregates of 1:1 mixtures of WT peptides with K16Ac, K28Ac, and KKAc, although they have variable structural morphology, all show very high ROS production and cytotoxicity (Figures 2H, J–L, 5B, C, and 6), with WT:KKAc mixture showing the highest ROS production as well as cytotoxicity (Figures 5B, C and 6). The increased oxidative stress has been considered very important for pathogenesis in Alzheimer’s disease as it can lead to oxidation of proteins, lipid peroxidation, free radical generation, mitochondrial dysfunction, and synaptic dysfunction.71 And neurons are particular sensitive to ROS, which serves as source of oxidative stresses and results in damages in the brain.59,72,73
Table 1.
Comparison of Unique Physicochemical Properties of WT and Acetylated Aβ42 Peptides and Their Mixtures for 7 d Aggregatesa
| Aβ42 peptides | structure type | hydrophobicity | flexibility | cytotoxicity | ROS production |
|---|---|---|---|---|---|
| WT | fibril | + | + | ++ | ++ |
| K16Ac | amorphous | ++++ | +++ | +++ | +++ |
| K28Ac | fibril | ++ | + | ++ | +++ |
| KKAc | amorphous | +++ | ++ | ++++ | ++++ |
| WT + K16Ac (1:1) | amorphous | ++ | + | +++ | +++ |
| WT + K28Ac (1:1) | fibril | ++ | + | +++ | +++ |
| WT + KKAc (1:1) | amorphous | ++ | + | ++++ | ++++ |
Number of + indicates proportional levels: (+) = low; (++) = moderate; (+++) = high; (++++) = highest among the group.
We treated primary neuronal cells from rat with 2 μM 7 d Aβ42 peptide aggregates for 24 h to see resulting cellular changes (Figure 7). The neuronal cells treated with Aβ42 peptides for 24 h all show a significantly higher immunofluorescence signal for tau in soma of neurons compared to neurites (Figure 7, Supplementary Figure 13). This is consistent with other literature reports where a direct relationship between amyloid β oligomers leading to altered tau protein biochemistry is seen.74–78 This altered biochemistry may result in tau that is fragmented, soluble, with/without requiring hyperphosphorylation that may be toxic.74–79 Oligomeric Aβ species have also been implicated as the toxic species in Alzheimer’s disease that can cause synaptic dysfunction directly or mediate their toxicity through tau.39,74,77,78,80 In addition, increased aberrant hydrophobicity of proteins has been implicated in neuronal toxicity.22,52,80–82 Therefore, we measured surface hydrophobicity of 7 d Aβ42 peptide aggregates by hydrophobic bead binding assay to determine the percentage of peptides showing increased surface hydrophobicity (Figure 4B, C). K16Ac peptides showed the highest percentage of hydrophobic peptides bound to beads which is consistent with other experimental findings that show high hydrophobicity for K16Ac Aβ42 peptides (Figures 2I, 4A). Interestingly, the hydrophobic bead bound peptides (7 d Aβ42 peptide aggregates) run as monomer in SDS-PAGE (Figure 4B, Supplementary Figure 11). This suggests that hydrophobic peptides bound to beads may either be oligomers, formed via off-pathway that break down and run as monomer on SDS-PAGE, or monomers83 (Supplementary Figure 11). These oligomeric or monomeric forms of Aβ42 peptides that are hydrophobic may mediate their cytotoxic effect by aberrant hydrophobic interactions directly, as detailed in Figure 8, or through tau.74,76–78,80,84–92
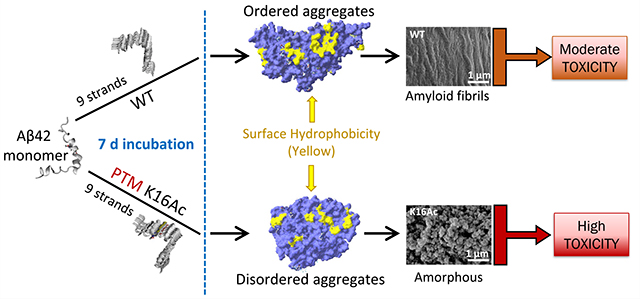
Figure 8.

Suggested schematic model for WT and acetylation modified Aβ42 peptide aggregation and cytotoxicity. WT Aβ42 and K28Ac peptides show a lag and log phase of aggregation kinetics and form ordered aggregates with large surface hydrophobic patches (yellow) that can assemble as amyloid fibrils and have moderate toxicity. Peptides acetylated at K16 (K16Ac or double acetylated KKAc) show rapid aggregation kinetics and form disordered, amorphous, and flexible aggregates that have higher surface hydrophobicity and high toxicity.
Consistent with ROS production and cytotoxicity data of primary neuronal cells (Figure 6), immunostaining data showed significantly higher immunofluorescence signal for tau in soma of neurons compared to neurites with the highest tau signal for cells treated with KKAc peptides (Figure 7, Supplementary Figure 13). This is also consistent with other animal/cell model studies where increased cross talk between Aβ and tau has been reported with Aβ exerting its toxicity via tau that may be fragmented, become oligomeric with or without hyperphosphorylation, and impact synaptotoxicity.77,78,93–96 Our data show that the Aβ42 peptide aggregates lead to increased tau signal in soma of neuronal cells compared to control cells (Figure 7).
Although both K16Ac and KKAc aggregates are amorphous, the K16Ac aggregates are bead-like structures that are slightly more hydrophobic and flexible compared to KKAc (Figures 2F, H, I and 4). Both of these peptide 7 d aggregates show high toxicity, but the difference in ROS production of these two peptides may be related to their subtle structure variations (Table 1).84 This is consistent with recently reported literature findings where the toxicity of misfolded proteins was determined by both size and surface hydrophobicity of the molecule.22 As large assemblies have lower surface hydrophobicity/volume ratios, they have lower diffusional mobility and lower potential to interact with cell membrane and receptors.22 In addition, several studies with Aβ peptide (Aβ40 and/or Aβ42) that form spherical or oligomeric aggregates show that these peptides that are highly hydrophobic can aberrantly interact with the cellular membrane, permeabilize it, and as a consequence, result in cellular toxicity.84,97–103 It is likely in this study that these amorphous Aβ aggregates of K16 and KKAc that are highly hydrophobic may similarly lead to an increase in cellular toxicity by aberrantly interacting with the cellular membrane and disrupting it. An alternative hypothesis suggests that the cytotoxicity may be related to the aggregation process that is dynamic, rather than a single structural species.8,104 In another study, minor shifts in the Aβ40:Aβ42 ratio were demonstrated to be enough to modulate neurotoxicity.105 The nontoxic K16N mutation of Aβ42 exhibits severe toxicity when mixed with WT peptides.106
In summary, WT Aβ42 and acetylated peptides K16Ac, K28Ac, and KKAc, although having different structural morphologies (Figure 2), all show increased ROS production and cytotoxicity (Figures 5B, C and 6). However, the 7 d aggregates formed from pure K16Ac, KKAc peptides, and heterogeneous 1:1 mixtures of WT and acetylated peptides all show a much higher hydrophobicity, increased ROS production, and toxicity compared to pure WT Aβ42 peptides (Figures 4, 5B, C, and 6). Furthermore, the increased tau signal in soma of neurons compared to control cells when treated with Aβ42 peptide aggregates suggests a dynamic interaction between tau and Aβ42 peptides (Figure 7). Overall, the data suggest that in addition to the structural characteristics, the dynamic interactions of different Aβ species may contribute significantly to toxicity directly or through cross-talk with other proteins such as tau.
METHODS
Preparation of Amyloid β Peptide Samples.
Synthetic wild-type (WT), K16Ac, K28Ac, and KKAc Aβ42 peptides were purchased from Biomatik (Canada). The identity and purity (>95%) of all four peptides were confirmed by mass spectrometry and RP-HPLC. Preparation of aggregate-free monomers followed the method from Roychaudhuri et al.107 In brief, the lyophilized peptide was first dissolved in 10% (v/v) of 60 mM NaOH, followed by 45% (v/v) Milli-Q water. The pH was adjusted by adding 45% (v/v) of 10 mM sodium phosphate buffer (pH 7.4). After 10 min centrifuge at 16 000g at 4 °C, the supernatant fluid was filtered through a 0.22 μm membrane. Concentration of peptide stock was determined using ε214 = 75 887 M−1 cm−1. Samples (50 μM peptide in 50 mM phosphate buffer, 300 mM NaCl, pH 7.4) were incubated at 37 °C with agitation for the indicated time. All of the solutions and Milli-Q water were filtered through a 0.22 μm membrane before use. Peptide stocks were stored at −80 °C until use.
Fluorescence Measurements.
Samples were diluted by phosphate buffer (10 mM, pH 7.4) to a final concentration of 10 μM. Fluorescent probes Thioflavin T (ThT), 8-anilino-1-naphthalene sulfonic acid (ANS), and 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS) were prepared in ethanol and then added to samples to final concentrations of 10 μM (ThT), 10 μM (ANS), and 1 μM (bis-ANS). Emission spectra were collected using a Horiba Jobin Yvon spectrofluorometer (Fluoromax-4) at room temperature. Samples containing ThT were excited at 450 nm, and emission spectra were collected from 460–700 nm. For samples with ANS and bis-ANS, spectra were acquired from 400–700 nm with excitation at 380 and 360 nm, respectively.
Field Emission Scanning Electron Microscopy (FESEM).
Fibrils and aggregates of peptides were analyzed using a cold field emission high-resolution scanning electron microscope, Hitachi S-4700 FESEM. After incubation, fibrils or aggregates were pelleted by centrifuging at 9000g for 45 min at 25 °C. To wash off salts, the pellets were suspended in Milli-Q water (0.22 μm membrane filtered) and centrifuged at 9000g for 45 min at 25 °C. Washed fibrils or aggregates were applied on SEM stubs and air-dried at room temperature. The SEM samples were then coated with 10 nm platinum. For FESEM imaging, 10 kV of acceleration voltage and 5 μA of emission current were used.
Cell Viability (MTS) Assay.
SH-SY5Y human neuroblastoma cells (from ATCC) were cultured in Medium 199 and DMEM/F-12 medium, respectively, with 10% FBS and 100 U/ml penicillin–streptomycin at 37 °C in 5% CO2 humidified environment and used within the first 10 passages. Cells were plated at 2 × 104 cells/well (SH-SY5Y) on 96-well plates and allowed to grow overnight. The next day, cells were washed with 1× PBS buffer (pH 7.4) twice. Then, 100 μL of fresh media containing peptide samples were added. Six replicates were prepared per sample. Media without any peptide were used as controls. After 48 h of incubation, 20 μL of CellTiter 96 AQueous One Solution Cell Proliferation (MTS) Assay kit (Promega) was added and incubated for 4 h. Absorption at 490 nm was collected using an ELISA plate reader (BioTek Instruments, Inc.). Blanks containing media and peptide samples but no cells were similarly prepared and used for background subtraction.
Cytotoxicity (LDH) Assay.
SH-SY5Y human neuroblastoma cells (from ATCC) were cultured as described above and plated at 2 × 104 cells/well on 96-well plates and allowed to grow overnight. The next day, cells were washed with 1× PBS buffer (pH 7.4) twice. Then, 100 μL of fresh media containing peptide samples were added. Six replicates were prepared per sample. After 48 h incubation, 10 μL of lysis solution (CytoTox 96 Non-Radioactive Cytotoxicity Assay (LDH) Assay kit, Promega) was added to max LDH release control wells and incubated for 45 min at 37 °C in 5% CO2. Fifty microliters of supernatant from each well was transferred into a new plate, and to each of those wells was added 50 μL of LDH reagent. The plate was further incubated in the dark for 30 min. After incubation, 50 μL of stop solution was added to each well. Absorption at 490 nm was collected using a plate reader (BioTek Instruments, Inc.). Appropriate blanks containing media and peptide samples (without cells) were similarly prepared and used for background subtraction. Max LDH control was represented as 100% cytotoxic, and all samples were normalized to max LDH control.61 Primary neuronal cells were prepared as described in the immunostaining protocol (below). The LDH assay was carried out for neuronal cells as described for SH-SY5Y cells above with the following variations. Neuronal cells were plated at 1× 104 cells/well in 96-well plate, and the cells were incubated with 2 μM peptides in 100 μL of media for 24 h.
2′,7′-Dichlorofluorescin Diacetate (DCFH-DA) Fluorescence Assay.
SH-SY5Y human neuroblastoma cells (from ATCC) were cultured and plated at 2 × 104 cells/well on 96-well plates and allowed to grow overnight. The next day, cells were washed with 1× PBS buffer (pH 7.4) twice. Then, cells were incubated with 20 μM DCFH-DA for 45 min at 37 °C in the dark. After incubation, the cells were washed twice with 1× PBS (pH 7.4), and then 100 μL of fresh media containing peptide samples was added to DCFH-DA (Sigma) treated cells in 5 replicates and incubated for 24 h at 37 °C in 5% CO2 humidified environment. Cells without any treatment and blanks were also incubated under identical conditions. For positive control, cells after DCFH-DA treatment were incubated with 50 μM t-BHP for 4 h at 37 °C. Finally, the fluorescence intensity was measured using a fluorescence plater reader (Fluoroskan Ascent, Thermo Scientific) at excitation wavelength of 485 nm and emission wavelength of 538 nm. All data were normalized to t-BHP, wherein t-BHP treated cells were represented as 100% free radical generating.60 DCFH-DA assay for primary neuronal cells was the same as that detailed for SH-SY5Y cells above with the following variations. Neuronal cells were plated at 1× 104 cells/well in a 96-well plate, and the cells were incubated with 2 μM peptides in 100 μL of media for 24 h.
Immunostaining of Primary Neuronal Cells.
Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, United States) and used in our breeding colony to generate 1-day-old pups which were euthanized via overdose of pentobarbital. Hippocampus and cortex from 1-day old rats were dissected, combined, and neuron cells dissociated, as detailed previously.108 All animal work and experiments were carried out under the guidelines of the National Institutes of Health guide for the care and use of laboratory animals with approval of IACUC, Michigan Technological University, Houghton, MI. Cells were plated in poly-L-lysine coated 24-well plates at the seeding density of 5 × 104 cells/well and incubated at 37 °C incubator in neurobasal medium containing B27 supplement (Thermo Fisher Scientific). Culture medium was changed every 3 days, and the cells were allowed to grow for 10–14 days prior to their use in experiment. During the experiment, 500 μL of fresh media containing 2 μM 7 d Aβ42 peptide aggregates (WT and acetylated) was added to neuronal cells and incubated for 24 h at 37 °C. Control cells did not have any peptides added to them but were incubated similarly. After 24 h, the cells were washed twice with PBS. Then, cells were fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were then washed with PBS three times for 5 min each. Blocking solution (5% horse serum and 0.3% Triton X-100 in PBS) was added to each well and incubated for 1 h. After 1 h, cells were further incubated overnight in a cocktail consisting of mouse anti-tau antibody (Invitrogen, catalog # AHB0042; 1:200 dilution) and rabbit anti-β-tubulin antibody (Cell-Signaling, catalog # 15115S; 1:100 dilution) in PBS buffer (containing 1% BSA and 0.3% Triton X-100) on a shaker at 4 °C. The cells were washed thrice for 5 min each with PBS and then further incubated overnight at 4 °C in a mixture of secondary antibodies (Alexa Fluor 594 donkey antimouse IgG and Alexa Fluor 488 donkey antirabbit IgG, both in 1:300 dilution). The sections were mounted in Vectashield (Vector Laboratories, Burlingame, CA), and images were acquired on a Leica fluorescence microscope (LEICA DM IL LED) at 20× and were analyzed using ImageJ software.109,110
Hydrophobic Bead Binding Assay and SDS-PAGE.
Seven-day samples of Aβ42 peptides were incubated with phenyl-sepharose 6 Fast Flow high substitution beads for measuring the extent of binding of peptides to hydrophobic beads.52 Three micrograms of each Aβ42 peptide sample in 50 μL of binding buffer (50 mM phosphate buffer, 300 mM NaCl, pH 7.4) was added to 20 μL of beads and then incubated overnight with gentle agitation at 25 °C. The next day, the beads were washed thrice with the binding buffer, and then proteins bound to hydrophobic beads were eluted by boiling with denaturing sample buffer (62 mM Tris at pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, and 0.05% bromophenol blue) for 3 min. The protein samples eluted in sample buffer were run for 3 h at 80 V on a 15% polyacrylamide gel for SDS-PAGE and then stained with Coomassie Blue (0.1% Coomassie Blue R-250 in 50% methanol and 10% glacial acetic acid). Fresh WT Aβ42 peptides were run as controls at varying concentrations (WTC 1 = 0.5, WTC 2 = 1, WTC 3 = 2 and WTC 4 = 4 μg/lane). The gel images were acquired using a scanner and then analyzed by ImageJ software. The percentage of Aβ42 peptides bound to hydrophobic beads were then calculated with respect to WT Aβ42 peptides controls (WTC 1–4) loaded on gel.
Statistical Analysis.
Statistical analysis was performed using one-way ANOVA and posthoc t test. The p values < 0.05 or greater were considered as significant and are represented as *, **, and *** (* = p < 0.05), (** = p < 0.005), and (*** = p < 0.0005). In this study, only 7 d data for Aβ42 peptides were analyzed for statistical significance. Details of statistical analysis of 7 d data are provided in the Supporting Information.
Computational Methods.
To construct the acetylated (K16Ac, K28Ac, and KKAc) peptides, we started with the WT Aβ42 monomer, as shown in Figure 1B (PDB entry: 1Z0Q). The NH3+ group of lysine 16 and lysine 28 residues of the Aβ42 monomer was modified by substituting with an acetyl group in the side chain. Likewise, initial configurations of 9-strand aggregates of Aβ42 fibrils (WT and acetylated) were constructed by aligning the individual Aβ42 monomers parallel to one other, avoiding any close contacts or atomic clashes during the assembly. The Aβ42 monomers and fibril structures were initially placed in an periodic orthorhombic box and solvated with water molecules defined using the TIP3P model.111 The H atoms of water molecules were constrained using the SETTLE algorithm,112 and counterions were added to maintain overall charge neutrality of the system. The details of system size are provided in Supporting Information, Table 2.
Atomistic MD simulations of WT and acetylated Aβ42 peptides were performed in the isothermal–isobaric (NPT) ensemble using the NAMD program and CHARMM27 force field. The simulation temperature (310 K) and pressure (101.3 kPa) were maintained using the Langevin dynamics and Langevin piston Nose–Hoover method.113 The long-range electrostatic interactions were calculated using the particle mesh Ewald method with a cutoff of 12.0 Å.114 For each system, we performed energy minimization for 2000 steps at a time step of 1.0 fs using the conjugate gradient method, followed by 100 ns of production run at a time-step of 1 fs. We performed a total of 800 ns runs for all systems and each simulation run was repeated for reproducibility of the results. Simulation snapshots corresponding to structural changes, and convergence of MD simulation trajectory was analyzed using the backbone RMSD for the peptides using the VMD 1.9.2. program.115 The surface hydrophobic patches for 9 strands of WT and acetylated (K16Ac, K28Ac, and KKAc) Aβ42 peptides in aqueous solution were analyzed using the SPDB software.
Supplementary Material
ACKNOWLEDGMENTS
Helpful discussions with Dr. Siegfried Hoefinger and Dr. S. Gowtham are acknowledged. The MD simulations were performed on the SUPERIOR High Performance Computing Cluster at Michigan Technological University.
Funding
This work was supported by the Research Excellence Fund, Michigan Technological University, Linda J. Horton Laboratory Research Fund, and Protein Misfolding Diseases Research Fund to A.T., NIH R15 HL129213 to Z.S. R.A. gratefully acknowledges finishing fellowship support from the graduate school, Michigan Technological University. The authors would like to thank the Department of Chemistry, Michigan Technological University for support in various forms that allowed the completion of this work.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00069.
Detailed MD simulation method, ANS binding sites on acetylated Aβ42, SEM images of samples after 7–14 days of incubation, zoomed-in images of primary neuronal cells from rats, SDS PAGE of 7 d aggregates of Aβ42 peptides, and statistical analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00069
Contributor Information
Rashmi Adhikari, Department of Chemistry, Michigan Technological University, Houghton, Michigan 49931, United States.
Mu Yang, Department of Chemistry, Michigan Technological University, Houghton, Michigan 49931, United States.
Nabanita Saikia, Department of Physics, Michigan Technological University, Houghton, Michigan 49931, United States.
Colina Dutta, Department of Chemistry, Michigan Technological University, Houghton, Michigan 49931, United States.
Wafa F.A. Alharbi, Department of Biological Sciences, Michigan Technological University, Houghton, Michigan 49931, United States
Zhiying Shan, Department of Kinesiology and Integrative Physiology, Michigan Technological University, Houghton, Michigan 49931, United States.
Ravindra Pandey, Department of Physics, Michigan Technological University, Houghton, Michigan 49931, United States.
Ashutosh Tiwari, Department of Chemistry, Michigan Technological University, Houghton, Michigan 49931, United States.
REFERENCES
- (1).Assoc A (2015) Alzheimer’s Association Report 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 11 (3), 332–384. [DOI] [PubMed] [Google Scholar]
- (2).Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, and Jones E (2011) Alzheimer’s disease. Lancet 377 (9770), 1019–1031. [DOI] [PubMed] [Google Scholar]
- (3).Kodali R, Williams AD, Chemuru S, and Wetzel R (2010) A beta(1–40) Forms Five Distinct Amyloid Structures whose beta-Sheet Contents and Fibril Stabilities Are Correlated. J. Mol. Biol 401 (3), 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Glabe CG (2008) Structural Classification of Toxic Amyloid Oligomers. J. Biol. Chem 283 (44), 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Atwood CS, Obrenovich ME, Liu TB, Chan H, Perry G, Smith MA, and Martins RN (2003) Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res. Rev 43 (1), 1–16. [DOI] [PubMed] [Google Scholar]
- (6).National Plan to Address Alzheimer’s Disease: 2015 Update; U.S. Department of Health and Human Service, 2015. [Google Scholar]
- (7).Orner BP, Liu L, Murphy RM, and Kiessling LL (2006) Phage display affords peptides that modulate beta-amyloid aggregation. J. Am. Chem. Soc 128 (36), 11882–9. [DOI] [PubMed] [Google Scholar]
- (8).Hubin E, van Nuland NAJ, Broersen K, and Pauwels K (2014) Transient dynamics of A beta contribute to toxicity in Alzheimer’s disease. Cell. Mol. Life Sci 71 (18), 3507–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Hoffmann VP, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, and Siemers E (2018) Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med 378 (4), 321–330. [DOI] [PubMed] [Google Scholar]
- (10).Kumar S, Singh S, Hinze D, Josten M, Sahl HG, Siepmann M, and Walter J (2012) Phosphorylation of Amyloid-beta Peptide at Serine 8 Attenuates Its Clearance via Insulin-degrading and Angiotensin-converting Enzymes. J. Biol. Chem 287 (11), 8641–8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wullnner U, Glebov K, Heneka MT, Walsh DM, Zweckstetter M, and Walter J (2011) Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 30 (11), 2255–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kumar S, Wirths O, Theil S, Gerth J, Bayer TA, and Walter J (2013) Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease. Acta Neuropathol. 125 (5), 699–709. [DOI] [PubMed] [Google Scholar]
- (13).Jarrett JT, Berger EP, and Lansbury PT (1993) The Carboxy Terminus of the Beta-Amyloid Protein Is Critical for the Seeding of Amyloid Formation - Implications for the Pathogenesis of Alzheimers-Disease. Biochemistry 32 (18), 4693–4697. [DOI] [PubMed] [Google Scholar]
- (14).Sullivan CP, Berg EA, Elliott-Bryant R, Fishman JB, Mckee AC, Morin PJ, Shia MA, and Fine RE (2011) Pyroglutamate-A beta 3 and 11 colocalize in amyloid plaques in Alzheimer’s disease cerebral cortex with pyroglutamate-A beta 11 forming the central core. Neurosci. Lett 505 (2), 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bouter Y, Dietrich K, Wittnam JL, Rezaei-Ghaleh N, Pillot T, Papot-Couturier S, Lefebvre T, Sprenger F, Wirths O, Zweckstetter M, and Bayer TA (2013) N-truncated amyloid beta (A beta) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol 126 (2), 189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Murakami K, Uno M, Masuda Y, Shimizu T, Shirasawa T, and Irie K (2008) Isomerization and/or racemization at Asp23 of A beta 42 do not increase its aggregative ability, neurotoxicity, and radical productivity in vitro. Biochem. Biophys. Res. Commun 366 (3), 745–751. [DOI] [PubMed] [Google Scholar]
- (17).Shimizu T, Watanabe A, Ogawara M, Mori H, and Shirasawa T (2000) Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch. Biochem. Biophys 381 (2), 225–234. [DOI] [PubMed] [Google Scholar]
- (18).Jawhar S, Wirths O, and Bayer TA (2011) Pyroglutamate Amyloid-beta (A beta): A Hatchet Man in Alzheimer Disease. J. Biol. Chem 286 (45), 38825–38832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ayyadevara S, Balasubramaniam M, Parcon PA, Barger SW, Griffin WST, Alla R, Tackett AJ, Mackintosh SG, Petricoin E, Zhou WD, and Reis RJS (2016) Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer’s hippocampus from normal controls. Aging Cell 15 (5), 924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tokikawa R, Loffredo C, Uemi M, Machini MT, and Bechara EJH (2014) Radical acylation of L-lysine derivatives and L-lysine-containing peptides by peroxynitrite-treated diacetyl and methylglyoxal. Free Radical Res. 48 (3), 357–370. [DOI] [PubMed] [Google Scholar]
- (21).Dueramae I, Yoneyama M, Shinyashiki N, Yagihara S, and Kita R (2017) Self -assembly of acetylated dextran with various acetylation degrees in aqueous solutions: Studied by light scattering (vol 159, pg 171, 2017). Carbohydr. Polym 161, 306–306. [DOI] [PubMed] [Google Scholar]
- (22).Mannini B, Mulvihill E, Sgromo C, Cascella R, Khodarahmi R, Ramazzotti M, Dobson CM, Cecchi C, and Chiti F (2014) Toxicity of Protein Oligomers Is Rationalized by a Function Combining Size and Surface Hydrophobicity. ACS Chem. Biol 9 (10), 2309–2317. [DOI] [PubMed] [Google Scholar]
- (23).Auburger G, Gispert S, and Jendrach M (2014) Mitochondrial Acetylation and Genetic Models of Parkinson’s Disease. Prog. Mol. Biol. Transl. Sci 127, 155–182. [DOI] [PubMed] [Google Scholar]
- (24).Srivastava OPSK, Chaves JM, and Gill AK (2017) Post-translationally modified human lens Crystallin fragments show aggregation in vitro. Biochem. Biophys. Rep 10, 94–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Drazic A, Myklebust LM, Ree R, and Arnesen T (2016) The world of protein acetylation. Biochim. Biophys. Acta, Proteins Proteomics 1864 (10), 1372–1401. [DOI] [PubMed] [Google Scholar]
- (26).Gil J, Ramirez-Torres A, and Encarnacion-Guevara S (2017) Lysine acetylation and cancer: A proteomics perspective. J. Proteomics 150, 297–309. [DOI] [PubMed] [Google Scholar]
- (27).Ali I, Conrad RJ, Verdin E, and Ott M (2018) Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev 118 (3), 340–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Narita T, Weinert BT, and Choudhary C (2019) Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol 20 (3), 156–174. [DOI] [PubMed] [Google Scholar]
- (29).Hettinger JC, and Cirrito JR (2016) Bad Acetylated Tau. Neuron 90 (2), 205–6. [DOI] [PubMed] [Google Scholar]
- (30).Mattson MP (2010) Acetylation unleashes protein demons of dementia. Neuron 67 (6), 900–2. [DOI] [PubMed] [Google Scholar]
- (31).Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, and Lee VM (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, and Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67 (6), 953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cook C, Stankowski JN, Carlomagno Y, Stetler C, and Petrucelli L (2014) Acetylation: a new key to unlock tau’s role in neurodegeneration. Alzheimer’s Res. Ther 6 (3), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, and Mucke L (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci 18 (8), 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gorsky MK, Burnouf S, Dols J, Mandelkow E, and Partridge L (2016) Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci. Rep 6, 22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xiao YL, Ma BY, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, and Ishii Y (2015) A beta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol 22 (6), 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kajava AV, Baxa U, and Steven AC (2010) beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 24 (5), 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, and Trojanowski JQ (2013) Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol 183 (2), 344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ayyadevara S, Balasubramaniam M, Kakraba S, Alla R, Mehta JL, and Reis RJS (2017) Aspirin-Mediated Acetylation Protects Against Multiple Neurodegenerative Pathologies by Impeding Protein Aggregation. Antioxid. Redox Signaling 27 (17), 1383–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, and Nordstedt C (1996) Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem 271 (15), 8545–8548. [DOI] [PubMed] [Google Scholar]
- (41).Daggett V (2002) Molecular dynamics simulations of the protein unfolding/folding reaction. Acc. Chem. Res 35 (6), 422–429. [DOI] [PubMed] [Google Scholar]
- (42).Brown AM, and Bevan DR (2016) Molecular Dynamics Simulations of Amyloid beta-Peptide (1–42): Tetramer Formation and Membrane Interactions. Biophys. J 111 (5), 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, and Shea JE (2006) Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci. 15 (3), 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Matthes D, Gapsys V, Brennecke JT, and de Groot BL (2016) An Atomistic View of Amyloidogenic Self-assembly: Structure and Dynamics of Heterogeneous Conformational States in the Prenucleation Phase. Sci. Rep 6, 33156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, and Teplow DB (2003) Amyloid beta-protein (A beta) assembly: A beta 40 and A beta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U. S. A 100 (1), 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Xu YC, Shen JJ, Luo XM, Zhu WL, Chen KX, Ma JP, and Jiang HL (2005) Conformational transition of amyloid beta-peptide. Proc. Natl. Acad. Sci. U. S. A 102 (15), 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yang MF, and Teplow DB (2008) Amyloid beta-Protein Monomer Folding: Free-Energy Surfaces Reveal Alloform-Specific Differences. J. Mol. Biol 384 (2), 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, and Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102 (18), 3586–3616. [DOI] [PubMed] [Google Scholar]
- (50).Urbanc B, Cruz L, Ding F, Sammond D, Khare S, Buldyrev SV, Stanley HE, and Dokholyan NV (2004) Molecular Dynamics Simulation of Amyloid β Dimer Formation. Biophys. J 87 (4), 2310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lijnzaad P, and Argos P (1997) Hydrophobic patches on protein subunit interfaces: Characteristics and prediction. Proteins: Struct., Funct., Genet 28 (3), 333–343. [PubMed] [Google Scholar]
- (52).Tiwari A, Xu Z, and Hayward LJ (2005) Aberrantly increased hydrophobicity shared by mutants of Cu, Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J. Biol. Chem 280 (33), 29771–9. [DOI] [PubMed] [Google Scholar]
- (53).Tonnies E, and Trushina E (2017) Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis 57 (4), 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, and Tyagi N (2016) Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol 53 (1), 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Angelova PR, and Abramov AY (2017) Alpha-synuclein and beta-amyloid different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun 483 (4), 1110–1115. [DOI] [PubMed] [Google Scholar]
- (56).Liu ZW, Zhou TY, Ziegler AC, Dimitrion P, and Zuo L (2017) Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Medicine and Cellular Longevity 2017, 2525967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Smith MA, Rottkamp CA, Nunomura A, Raina AK, and Perry G (2000) Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta, Mol. Basis Dis 1502 (1), 139–144. [DOI] [PubMed] [Google Scholar]
- (58).Zhao Y, and Zhao BL (2013) Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longevity 2013, 316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chen XP, Guo CY, and Kong JM (2012) Oxidative stress in neurodegenerative diseases. Neural Regener. Res 7 (5), 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Aranda A, Sequedo L, Tolosa L, Quintas G, Burello E, Castell JV, and Gombau L (2013) Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay: A quantitative method for oxidative stress assessment of nanoparticle-treated cells. Toxicol. In Vitro 27 (2), 954–963. [DOI] [PubMed] [Google Scholar]
- (61).Vetten MA, Tlotleng N, Rascher DT, Skepu A, Keter FK, Boodhia K, Koekemoer LA, Andraos C, Tshikhudo R, and Gulumian M (2013) Label-free in vitro toxicity and uptake assessment of citrate stabilised gold nanoparticles in three cell lines. Part. Fibre Toxicol 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Cheon M, Chang I, Mohanty S, Luheshi LM, Dobson CM, Vendruscolo M, and Favrin G (2007) Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLoS Comput. Biol 3 (9), 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hartl FU, and Hayer-Hartl M (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol 16 (6), 574–581. [DOI] [PubMed] [Google Scholar]
- (64).Brender JR, Ghosh A, Kotler SA, Krishnamoorthy J, Bera S, Morris V, Sil TB, Garai K, Reif B, Bhunia A, and Ramamoorthy A (2019) Probing transient non-native states in amyloid beta fiber elongation by NMR. Chem. Commun. (Cambridge, U. K.) 55 (31), 4483–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kellermayer MSZ, Grama L, Karsai A, Nagy A, Kahn A, Datki ZL, and Penke B (2005) Reversible mechanical unzipping of amyloid beta-fibrils. J. Biol. Chem 280 (9), 8464–8470. [DOI] [PubMed] [Google Scholar]
- (66).Nguyen PH, Li MS, Stock G, Straub JE, and Thirumalai D (2007) Monomer adds to preformed structured oligomers of A beta-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. U. S. A 104 (1), 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Vivekanandan S, Brender JR, Lee SY, and Ramamoorthy A (2011) A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun 411 (2), 312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, and Maggio JE (2000) Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39 (21), 6288–6295. [DOI] [PubMed] [Google Scholar]
- (69).Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, and Bowers MT (2009) Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem 1 (4), 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Karsai A, Nagy A, Kengyel A, Martonfalvi Z, Grama L, Penke B, and Kellermayer MSZ (2005) Effect of lysine-28 side-chain acetylation on the nanomechanical behavior of Alzheimer amyloid beta 25–35 fibrils. J. Chem. Inf. Model 45 (6), 1641–1646. [DOI] [PubMed] [Google Scholar]
- (71).Ahmad W, Ijaz B, Shabbiri K, Ahmed F, and Rehman S (2017) Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/RNS generation. J. Biomed. Sci 24, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Uttara B, Singh AV, Zamboni P, and Mahajan RT (2009) Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol 7 (1), 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Xie HR, Hu LS, and Li GY (2010) SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J 123 (8), 1086–1092. [PubMed] [Google Scholar]
- (74).Rapoport M, Dawson HN, Binder LI, Vitek MP, and Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U. S. A 99 (9), 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, Baker-Nigh A, Benzinger TLS, Miller TM, Karch CM, and Bateman RJ (2018) Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 97 (6), 1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Reifert J, Hartung-Cranston D, and Feinstein SC (2011) Amyloid beta-Mediated Cell Death of Cultured Hippocampal Neurons Reveals Extensive Tau Fragmentation without Increased Full-length Tau Phosphorylation. J. Biol. Chem 286 (23), 20797–20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Bloom GS (2014) Amyloid-beta and Tau The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 71 (4), 505–508. [DOI] [PubMed] [Google Scholar]
- (78).Nisbet RM, Polanco JC, Ittner LM, and Gotz J (2015) Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 129 (2), 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, and Bloom GS (2013) Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci 126 (5), 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Benilova I, Karran E, and De Strooper B (2012) The toxic A beta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci 15 (3), 349–357. [DOI] [PubMed] [Google Scholar]
- (81).Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, and Chiti F (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol 6 (2), 140–7. [DOI] [PubMed] [Google Scholar]
- (82).Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TP, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM, and Cremades N (2015) Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. U. S. A 112 (16), E1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Breydo L, and Uversky VN (2015) Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 589 (19 Pt A), 2640–8. [DOI] [PubMed] [Google Scholar]
- (84).Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, and Sato K (2003) Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3 beta. Proc. Natl. Acad. Sci. U. S. A 100 (11), 6370–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Yu Y, Jans DC, Winblad B, Tjernberg LO, and Schedin-Weiss S (2018) Neuronal A beta 42 is enriched in small vesicles at the presynaptic side of synapses. Life Sci. Alliance 1 (3), No. e201800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Funke S (2011) Detection of Soluble Amyloid-β Oligomers and Insoluble High-Molecular-Weight Particles in CSF: Development of Methods with Potential for Diagnosis and Therapy Monitoring of Alzheimer’s Disease. Int. J. Alzheimer’s Dis 2011, 151645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Pryor NE, Moss MA, and Hestekin CN (2012) Unraveling the Early Events of Amyloid-beta Protein (A beta) Aggregation: Techniques for the Determination of A beta Aggregate Size. Int. J. Mol. Sci 13 (3), 3038–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Jan A, Gokce O, Luthi-Carter R, and Lashuel HA (2008) The ratio of monomeric to aggregated forms of A beta 40 and A beta 42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J. Biol. Chem 283 (42), 28176–28189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Guglielmotto M, Monteleone D, Piras A, Valsecchi V, Tropiano M, Ariano S, Fornaro M, Vercelli A, Puyal J, Arancio O, Tabaton M, and Tamagno E (2014) Abeta1–42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy 10 (10), 1827–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Puzzo D, and Arancio O (2012) Amyloid-beta peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimer’s Dis 33 (Suppl 1), S111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Manassero G, Guglielmotto M, Zamfir R, Borghi R, Colombo L, Salmona M, Perry G, Odetti P, Arancio O, Tamagno E, and Tabaton M (2016) Beta-amyloid 1–42 monomers, but not oligomers, produce PHF-like conformation of Tau protein. Aging Cell 15 (5), 914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Upadhaya AR, Lungrin I, Yamaguchi H, Fandrich M, and Thal DR (2012) High-molecular weight Abeta oligomers and protofibrils are the predominant Abeta species in the native soluble protein fraction of the AD brain. J. Cell. Mol. Med 16 (2), 287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Small SA, and Duff K (2008) Linking A beta and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 60 (4), 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Nieweg K, Andreyeva A, van Stegen B, Tanriover G, and Gottmann K (2015) Alzheimer’s disease-related amyloid-beta induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 6, No. e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Rajmohan R, and Reddy PH (2017) Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis 57 (4), 975–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Guerrero-Munoz MJ, Gerson J, and Castillo-Carranza DL (2015) Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front. Cell. Neurosci 9, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Thundimadathil J, Roeske RW, Jiang HY, and Guo L (2005) Aggregation and porin-like channel activity of a beta sheet peptide. Biochemistry 44 (30), 10259–70. [DOI] [PubMed] [Google Scholar]
- (98).Ono K, Condron MM, and Teplow DB (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. U. S. A 106 (35), 14745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Butterfield SM, and Lashuel HA (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem., Int. Ed 49 (33), 5628–54. [DOI] [PubMed] [Google Scholar]
- (100).Kotler SA, Walsh P, Brender JR, and Ramamoorthy A (2014) Differences between amyloid-beta aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev 43 (19), 6692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairi M, Maglia G, and Carulla N (2016) Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. U. S. A 113 (39), 10866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, and Ramamoorthy A (2017) Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-beta. J. Biol. Chem 292 (11), 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Bode DC, Freeley M, Nield J, Palma M, and Viles JH (2019) Amyloid-beta oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J. Biol. Chem 294 (19), 7566–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Ross CA, and Poirier MA (2005) What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol 6 (11), 891–898. [DOI] [PubMed] [Google Scholar]
- (105).Pauwels K, Williams TL, Morris KL, Jonckheere W, Vandersteen A, Kelly G, Schymkowitz J, Rousseau F, Pastore A, Serpell LC, and Broersen K (2012) Structural Basis for Increased Toxicity of Pathological A beta(42):A beta(40) Ratios in Alzheimer Disease. J. Biol. Chem 287 (8), 5650–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Kaden D, Harmeier A, Weise C, Munter LM, Althoff V, Rost BR, Hildebrand PW, Schmitz D, Schaefer M, Lurz R, Skodda S, Yamamoto R, Arlt S, Finckh U, and Multhaup G (2012) Novel APP/A ss mutation K16N produces highly toxic heteromeric A ss oligomers. EMBO Mol. Med 4 (7), 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Roychaudhuri R, Yang MF, Deshpande A, Cole GM, Frautschy S, Lomakin A, Benedek GB, and Teplow DB (2013) C-Terminal Turn Stability Determines Assembly Differences between A beta 40 and A beta 42. J. Mol. Biol 425 (2), 292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Shan ZY, Shi P, Cuadra AE, Dong Y, Lamont GJ, Li QH, Seth DM, Navar LG, Katovich MJ, Sumners C, and Raizada MK (2010) Involvement of the Brain (Pro)renin Receptor in Cardiovascular Homeostasis. Circ. Res 107 (7), 934–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9 (7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Huber MJ, Fan YY, Jiang ES, Zhu FL, Larson RA, Yan JQ, Li NJ, Chen QH, and Shan ZY (2017) Increased activity of the orexin system in the paraventricular nucleus contributes to salt-sensitive hypertension. Am. J. Physiol.: Heart Circ. Physiol 313 (6), H1075–H1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 79 (2), 926–935. [Google Scholar]
- (112).Miyamoto S, and Kollman PA (1992) Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem 13 (8), 952–962. [Google Scholar]
- (113).Feller SE, Zhang YH, Pastor RW, and Brooks BR (1995) Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys 103 (11), 4613–4621. [Google Scholar]
- (114).Darden T, York D, and Pedersen L (1993) Particle Mesh Ewald - an N. Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98 (12), 10089–10092.ss [Google Scholar]
- (115).Humphrey W, Dalke A, and Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.