

Abstract
Objective
To determine predicting factors and frequency of phenoconversion from pure autonomic failure (PAF) into a synucleinopathy with motor or cognitive involvement of multiple system atrophy (MSA), Parkinson disease (PD), or dementia with Lewy bodies (DLB).
Methods
We performed a retrospective review of all patients with PAF from 2001 to 2011 evaluated at Mayo Clinic, Rochester. Clinical follow-up and patient telephone calls were used to assess for development of symptoms and diagnosis of MSA, PD, or DLB. Clinical and laboratory variables were extracted with factors predictive of evolution assessed using group comparison, odds ratio, and logistical regression.
Results
Among 275 patients with PAF at presentation, 67 (24%) phenoconverted to a synucleinopathy with motor or cognitive involvement; 34 met criteria for MSA, while 33 met criteria for PD or DLB. Age at onset was younger in MSA phenoconverters. Clinical features at presentation influenced phenoconversion: severe bladder symptoms were more common in MSA phenoconverters; subtle motor signs were more frequent in MSA and PD/DLB phenoconverters. MSA phenoconverters were more likely to have higher supine norepinephrine levels and preganglionic pattern of anhidrosis. Presentation variables predicting MSA phenoconversion included subtle motor signs, supine norepinephrine levels, severe bladder symptoms, and dream enactment behavior. Presentation variables predictive of PD/DLB phenoconversion included subtle motor signs, dream enactment behavior, and constipation.
Conclusions
Our findings suggest that at least a quarter of patients with PAF phenoconvert to MSA, PD, or DLB. Presentation features determine patients at risk for evolution with specific patterns indicative of phenoconversion to MSA vs PD/DLB.
Classification of evidence:
This study provides Class II evidence that several presentation variables including subtle motor signs, severe bladder symptoms, and dream enactment behavior are associated with an increased risk of developing a synucleinopathy with motor or cognitive involvement.
The synucleinopathies are a collection of neurodegenerative disorders characterized by aggregation of α-synuclein in the central and peripheral nervous system that leads to clinically distinct diseases based on the cellular location of α-synuclein aggregates and affected neuronal populations.1,2 Pure autonomic failure (PAF) is characterized by predominantly peripheral accumulation of α-synuclein as neuronal cytoplasmic inclusions.3,4 The neuropathologic hallmark of multiple system atrophy (MSA) is oligodendroglial cytoplasmic inclusions,5,6 while neuronal α-synuclein inclusions of Lewy bodies and Lewy neurites are the major pathologic feature of Parkinson disease (PD) and dementia with Lewy bodies (DLB).1 The synucleinopathies have variable manifestations and severity of autonomic failure with shared clinical features of REM sleep behavior disorder (RBD).7–9 The role of RBD and autonomic failure as prodromal phases in the synucleinopathies is becoming increasingly well-defined.10
Clinically, PAF is characterized by neurogenic orthostatic hypotension.11 PAF was originally known as Bradbury-Eggleston syndrome based on the seminal description in 1925, which described 3 men with orthostatic hypotension, fixed heart rate, constipation, anhidrosis, and erectile dysfunction.12 In the original report, one patient had subtle motor signs of hyperreflexia and bilateral Babinski signs in addition to autonomic failure. By definition, patients with PAF have no evidence of CNS dysfunction although the presence of RBD is indicative of central involvement.11 Since the 1925 description, it is well-established that a proportion of patients with PAF may later develop motor or cognitive symptoms leading to the diagnosis of a synucleinopathy such as MSA, PD, or DLB.13–16 Therefore, identifying patients with PAF at risk for phenoconverting to MSA or Lewy body disorders represents an opportunity for early intervention with future disease-modifying therapies, and has prognostic implications.
We have previously reported a phenoconversion rate from PAF to a synucleinopathy with motor or cognitive involvement of at least 12%, with different variables predicting which patients are at risk for phenoconversion to MSA vs PD and DLB.14 A prospective natural history study of PAF in the United States reported phenoconversion rates of 34% based on 25 out of 74 patients phenoconverting to MSA or PD/DLB,15 which was similar to an Italian study based on follow-up from 50 patients with PAF.16 We sought to expand follow-up in our original cohort to determine long-term phenoconversion rate and risk factors, and to create a clinical tool to allow clinicians to identify patients with PAF at risk for phenoconversion to MSA or PD/DLB at the time of initial PAF presentation.
Methods
The primary research questions were as follows: What is the long-term phenoconversion rate of PAF? Which factors increase the risk for phenoconversion to MSA or PD/DLB at the time of PAF presentation? The classification of evidence is Class II for each of those questions.
Patients
We performed a retrospective review of all patients evaluated by an autonomic specialist at Mayo Clinic Rochester between July 1, 2001, and July 1, 2011, who fulfilled the following criteria.
Inclusion criteria included the presence of orthostatic hypotension (OH), defined as a systolic blood pressure (BP) drop of ≥30 mm Hg or diastolic BP drop ≥15 mm Hg during a 5-minute head-up tilt and patient age 18–80 years at time of onset of symptoms.
Exclusion criteria included (1) autonomic testing not consistent with neurogenic OH (normal BP recovery time on Valsalva maneuver); (2) clinical evidence of central neurodegeneration (parkinsonism, cerebellar ataxia, or dementia) at initial presentation for PAF; (3) evidence of clinically significant peripheral neuropathy, Adie pupils, or prominent upper gastrointestinal symptoms; (4) medications or medical conditions that could interfere with the results of autonomic testing, including significant heart disease, neck radiation, and chemotherapy; and (5) expert diagnosis not consistent with PAF.
Standard protocol approvals, registrations, and patient consents
This study was reviewed and approved by the Mayo Clinic institutional review board.
Diagnostic outcomes
Patients who fulfilled inclusion and exclusion criteria were considered to have PAF. At subsequent evaluations, diagnostic clinical criteria were used to make the diagnosis of MSA,17 PD,18 and DLB.19 In order to achieve long-term follow-up in those who had not phenoconverted at the time of last in-person follow-up, patients were contacted by telephone and verbally given a survey assessing development of motor, cognitive, and additional autonomic symptoms, as well as diagnosis of other neurologic disorders including MSA, PD, and DLB, and the date of diagnosis. When patients were deceased, next of kin were asked questions regarding symptoms prior to death, diagnosis at the time of death, and cause of death. Phone assessments were conducted between September 2016 and January 2018, so that a minimum follow-up duration of 5 years was achieved for living patients. Disease duration was calculated based on time from onset of symptoms until death or last follow-up.
Survival
Survival data were obtained from the clinical record or phone call to next of kin. When data on survival were not available, the social security death index was queried to obtain the date of death.
Clinical variables
The presence or absence of clinical features from the time of presentation was extracted from the medical record including symptom type and onset date. Patients completed questionnaires at the time of evaluation, which assessed the presence or absence of orthostatic intolerance, urinary symptoms, bowel symptoms, and falls. Bladder symptoms of frequency, urgency, and nocturia were recorded as mild while incontinence, retention, and use of catheterization were recorded as severe. Subtle motor findings were based on clinical examination and defined as bradykinesia or incoordination that was too mild to definitely discern from a normal variant or normal aging. Subtle motor findings included isolated unilateral reduction in arm swing with walking, mild slowing, reduction or incoordination of rapid alternating movements, isolated increase in muscle tone, and isolated hyperreflexia or extensor plantar response. The presence and timing of dream enactment behavior was recorded from the patient or patient bed partner's report. While assessment of autonomic symptoms was completed for all patients with clinical documentation and patient-completed questionnaires at the time of evaluation, physician documentation of the presence or absence of dream enactment behavior was recorded in 131/275 (48%) patients during the study period.
Autonomic function testing
All patients underwent standardized autonomic function testing with autonomic reflex screen (ARS), which tests postganglionic sudomotor, cardiovagal, and cardiovascular adrenergic function.20 Postganglionic sudomotor axon reflex testing was assessed using quantitative sudomotor axon reflex testing (QSART), which involves iontophoresis of an acetylcholine solution at 4 sites.21 Cardiovagal testing included heart rate response to deep breathing and heart rate response to Valsalva maneuver. Cardiovascular adrenergic function was evaluated by assessing blood pressure responses to Valsalva maneuver and passive head up tilt.22 Patients are routinely instructed to discontinue medications that may interfere with autonomic function for at least 48 hours prior to autonomic function testing and abstain from alcohol and caffeine use.
Composite autonomic severity score (CASS) was derived from ARS. CASS is a validated instrument that quantifies severity and distribution of autonomic failure and is composed of 3 subdomains: sudomotor (score range 0–3), cardiovagal (0–3), and adrenergic (0–4). The total CASS therefore may range from 0 to 10, with 10 indicating severe autonomic failure.20 When patients underwent multiple ARS, the ARS from the time of presentation for PAF was used for analysis and CASS.
Thermoregulatory sweat test (TST) assesses the integrity of the central and peripheral thermoregulatory system and is often used in conjunction with ARS. TST is a standardized clinical procedure that involves warming the body to a core temperature of 38°C while the patient is lying supine with exposed body surface covered with an indicator powder that changes when exposed to sweat. Digital photography is used to quantify the percentage anhidrosis of the anterior body service.23
The results of QSART and TST were used to determine whether the anhidrosis pattern was preganglionic (central), postganglionic (peripheral), or mixed. A region of reduced or absent sweating on TST in the context of normal QSART response was determined preganglionic (central). An area of reduced or absent sweating on TST with absent QSART values was determined postganglionic (peripheral). The lesion site was deemed mixed when there was anhidrosis on TST with diminished, but not absent, corresponding QSART values or when a patient had both postganglionic and preganglionic patterns at different QSART sites. TST results were available in 243/275 (88%) patients.
Laboratory variables
Laboratory variables were extracted from the clinical record from the time of presentation for PAF and included supine and orthostatic norepinephrine levels (pg/mL) performed as part of plasma catecholamine testing. Catecholamine data were available in 135/275 (49%) patients.
Statistics
Summary statistics were used to describe demographic and clinical variables including mean (SD), median (interquartile range [IQR]), and frequency (percentage). Categorical variables were analyzed using χ2 test or Fisher exact test, as appropriate. Continuous variables were compared using Student t test or Wilcoxon rank-sum test, as appropriate. We assessed the odds of conversion (odds ratio [OR]) for those variables with significant differences between groups (stable PAF vs MSA and stable PAF vs PD/DLB) to identify the strength of predictors. We used univariable and multivariable logistic regression analysis with conversion as the outcome of interest where stable PAF was used as the reference group.
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
Of 275 patients who initially presented with PAF, 170 (62%) were men, with a median age at onset of 65 years (range, 56–72). All patients had symptoms of orthostatic intolerance. The mean systolic BP drop at 5 minutes of tilt was 61.1 mm Hg (±31.3) with a diastolic drop of 31.4 mm Hg (±46.1). The mean heart rate change at 5 minutes of tilt was 11.5 beats per minute (±13.8). Telephone contact, consent, and phone survey were completed for 73 patients with PAF or their next of kin (27%).
Of those 275 patients who initially were diagnosed with PAF, 67 (24%) phenoconverted: 34 to MSA and 33 to a Lewy body disorder of PD or DLB (figure and table 1). Median time to conversion to MSA was 5.88 years (range, 3.86–9.26), while the median time to phenoconversion to PD/DLB was 8.00 years (5.23–14.16; p = 0.0128). Patients who phenoconverted were more likely to be deceased and overall had a shorter disease duration than those with stable PAF (table 1). Mean follow-up time for patients with stable PAF was 5.5 years (95% confidence interval, 4.7–6.3 years), while mean follow-up time for phenoconverters was 6.9 years (range, 5.6–8.3 years). MSA phenoconverters had mean follow-up time of 5.2 years (range, 3.6–6.9 years), while PD/DLB phenoconverters had mean follow-up time of 8.7 years (range, 7.2–10.2 years).
Figure. Study flow chart.

DLB = dementia with Lewy bodies; MSA = multiple system atrophy; OH = orthostatic hypotension; PAF = pure autonomic failure; PD = Parkinson disease.
Table 1.
Features of patients with stable pure autonomic failure (PAF) and those who convert to a different synucleinopathy

Clinical features at presentation varied between those with stable PAF and those who phenoconverted (table 1). Phenoconverters were more likely to present with features of severe bladder symptoms and constipation, exhibit subtle motor signs on examination, and report dream enactment behavior. Laboratory features of supine norepinephrine level and norepinephrine rise upon standing differed between patients with stable PAF and phenoconverters. Autonomic function testing showed severe autonomic failure in both patients with stable PAF and phenoconverters (table 1).
Demographic, clinical, laboratory, and autonomic function testing features of patients with stable PAF compared to those who phenoconvert are separated based on whether phenoconversion was to MSA or PD/DLB (table 2). Compared to patients with stable PAF, age at onset was younger in those who phenoconverted to MSA, whereas those who phenoconverted to PD/DLB were older. Severe bladder symptoms at presentation were seen in phenoconversion to MSA, but not PD/DLB. Subtle motor signs at presentation were also more common in MSA phenoconversion, but still frequently seen in phenoconversion to PD/DLB. Laboratory features were distinct between phenoconverters with higher supine norepinephrine levels in MSA compared to stable PAF and lower in PD/DLB phenoconverters. Autonomic function testing results showed notable differences on TST; MSA phenoconverters demonstrated a preganglionic pattern of sweat loss and slightly higher heart rate rise at 1 minute of tilt whereas PD/DLB phenoconverters were similar to stable PAF (table 2).
Table 2.
Demographic, clinical, autonomic function, and laboratory features of patients with stable pure autonomic failure (PAF) compared to those who convert to multiple system atrophy (MSA) and Parkinson disease (PD)/dementia with Lewy bodies (DLB)

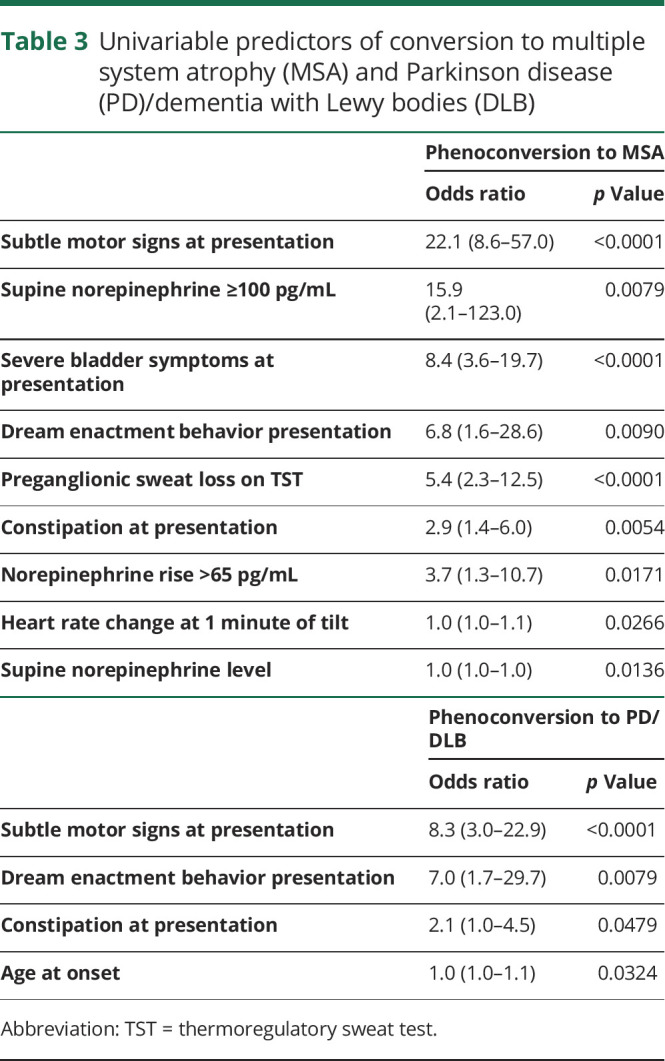
Based on the differences among patients with stable PAF, those with phenoconversion to MSA, and those with phenoconversion to PD/DLB, independent variables were identified as predictors of phenoconversion (table 3). Variables predictive of phenoconversion to MSA were subtle motor signs at presentation, severe bladder symptoms at presentation, and variables that suggest a central lesion, including supine norepinephrine levels, norepinephrine rise, heart rate change at 1 minute, and preganglionic pattern on TST. Subtle motor signs at presentation were also the strongest predictor of phenoconversion to PD/DLB, followed by presentation features of dream enactment behavior and constipation, and age at onset.
Table 3.
Univariable predictors of conversion to multiple system atrophy (MSA) and Parkinson disease (PD)/dementia with Lewy bodies (DLB)
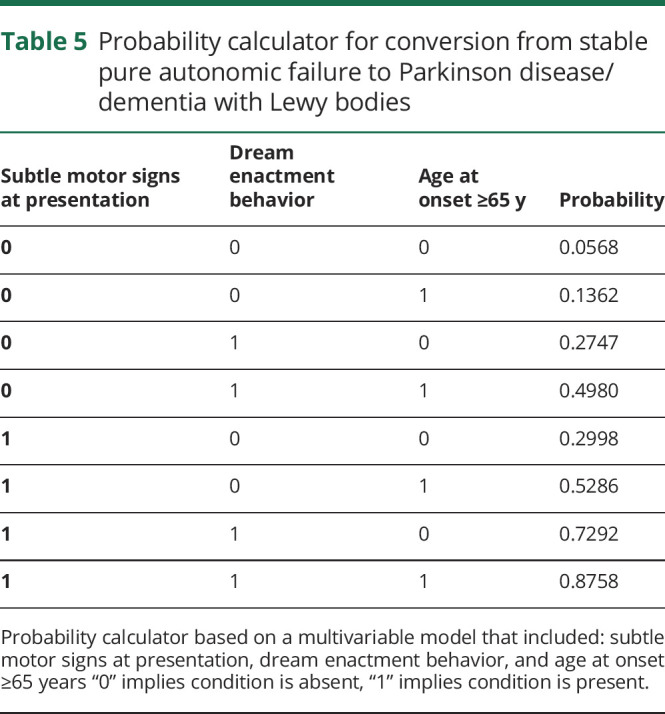
We then created a probability calculator based on a multivariable logistic regression model that included the strongest variables predictive of phenoconversion to MSA (table 4) and phenoconversion to PD/DLB (table 5). This tool can be used to predict probability of future phenoconversion in patients who present with PAF. For instance, in a patient with PAF who presents with subtle motor signs, supine norepinephrine levels ≥100 pg/mL, and severe bladder symptoms, the probability that he or she may later meet diagnostic criteria for MSA is approximately 98%.
Table 4.
Probability calculator for conversion from stable pure autonomic failure to multiple system atrophy
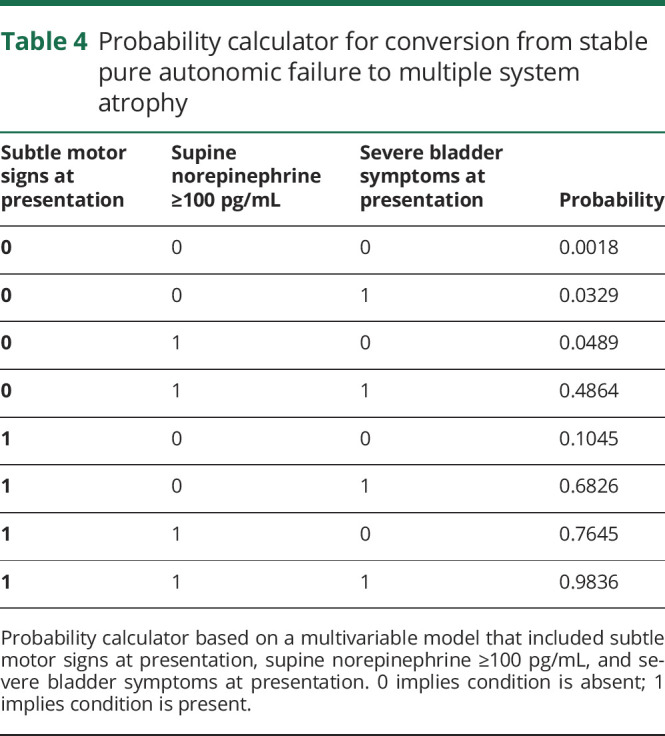
Table 5.
Probability calculator for conversion from stable pure autonomic failure to Parkinson disease/dementia with Lewy bodies
In the 67 patients with PAF who phenoconverted, 6 variables predicted the risk of phenoconverting to MSA vs to PD/DLB in logistic regression analysis: supine norepinephrine ≥100 pg/mL, preganglionic or preganglionic/mixed pattern on TST, age <65 at time of PAF presentation, systolic BP drop at 5 minutes, and heart rate change at 1 minute of tilt. A multivariable model using patients with catecholamine data (which were only available in a subset, 33 out of 67 converters) led to supine norepinephrine ≥100 pg/mL and age below 65 as predictors of phenoconversion to MSA rather than PD/DLB with area under the curve of 0.876. An alternative model that excluded supine norepinephrine ≥100 pg/mL (therefore used 5 out of 6 statistically significant predictors) evaluated data from 62/67 phenoconverters and found variables of age <65 years and preganglionic/mixed pattern on TST predicted phenoconversion to MSA rather than PD/DLB with an area under the curve of 0.753.
Discussion
This is the largest study on the natural history of PAF to date, which allowed for calculation of phenoconversion rates to synucleinopathies with motor or cognitive involvement of 24%. We have previously reported a rate of at least 12% of phenoconversion based on patients with PAF with in-person follow-up of at least 3 years while a prospective natural history study in North America reported 34% and a well-characterized Italian cohort reported 32% phenoconversion rate.14–16 Longer duration of follow-up likely increases the phenoconversion rate, particularly for phenoconversion to Lewy body disorders.
Whereas calculating the phenoconversion rate from PAF is meaningful, our findings differentiating variables predictive of phenoconversion has implications on understanding disease mechanisms in PAF. For instance, patients who phenoconvert to MSA have a distinct presentation: earlier age at onset, subtle motor features, more severe bladder involvement, and more severe autonomic failure with central autonomic involvement based on laboratory and autonomic function testing. This suggests that patients with PAF presenting with central features may represent a premotor state or prodromal MSA. In contrast, patients with PAF with predominantly postganglionic/peripheral involvement and less severe autonomic features at presentation may retain the PAF presentation for years, with possible later development of motor or cognitive symptoms leading to the diagnosis of PD or DLB. This differentiation has significant clinical implications as well as identification of an opportunity for intervention with future disease-modifying therapy. In order to differentiate and classify these patients at the time of presentation for PAF, we have created a clinical tool to predict probability of conversion to MSA or PD/DLB. This tool allows clinicians to counsel patients at the time of PAF presentation regarding risk for phenoconversion and identifies patients who may benefit from future disease-modifying therapies.
Our results confirm earlier studies with findings that patients at risk for MSA phenoconversion have younger age at onset15 and clinical features of early and severe urinary symptoms13–16 with relatively preserved cardiovagal function.14–16 Patients converting to PD/DLB tend to do so later in the disease course.15 Similar to previous studies, patients with stable PAF were less likely to have dream enactment behavior at presentation15,16 with autonomic function and laboratory evidence of peripheral denervation.14,15 When evaluating only the patients who phenoconvert, younger age and evidence of central autonomic involvement (supine norepinephrine ≥100 pg/mL and central pattern on TST) differentiates patients more likely to phenoconvert to MSA rather than a Lewy body disorder.
Compared with previous studies, our phenoconversion percentage of 24% is lower than the 32% and 34% reported in earlier studies.15,16 We suspect that the 24% phenoconversion rate is in the lower range for phenoconversion. Although we had a good rate of contact for a phone survey study, there was significantly longer follow-up time for phenoconverters compared to those with stable PAF. This suggests that longer follow-up of patients with PAF would likely lead to increased rate of phenoconversion. This phenomenon is appreciated when comparing the rate of phenoconversion with our earlier study, which had a higher percentage of phenoconverters to MSA compared to PD/DLB.14 By extending the follow-up, we captured considerably more PD/DLB cases. This may also relate to underlying disease mechanism with phenoconversion to PD/DLB occurring later than phenoconversion to MSA. Another consideration is that phenoconversion to Lewy body disorders may occur in some patients decades after onset of autonomic failure, which means that some patients may not manifest motor or cognitive symptoms in their natural lifetime.
Limitations to our study include the retrospective collection of presentation data, which could lead to lack in standardization between providers, although we attempted to mitigate this by limiting to patients seen by a neurologist with autonomic subspecialty expertise. The time frame of the study (2001–2011) influenced the awareness and collection of certain variables, such as dream enactment behavior, as this was less likely to be reported in the clinical history prior to widespread knowledge of the importance of RBD in synucleinopathies. As a tertiary referral center, we may see more severe cases of PAF, which may increase the risk for phenoconversion to MSA.
We report a 24% rate of phenoconversion from PAF to a synucleinopathy with motor or cognitive involvement and provide accessible calculators to determine the probability of phenoconverting to MSA or PD/DLB based on readily available data obtained at presentation for PAF. As evolution of synucleinopathies becomes increasingly well-defined, it will be of utmost importance to identify patients who may benefit from disease-modifying therapies to delay progression and development of motor or cognitive symptoms.
Study funding
Supported in part by NIH (P01NS44233, U54NS065736, K23NS075141, R01 FD004789, R01 NS092625, NS 65736 FP00101673), Mayo CCaTS (UL1TR000135), and Cure PSP Foundation. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
Glossary
- ARS
autonomic reflex screen
- BP
blood pressure
- CASS
composite autonomic severity score
- DLB
dementia with Lewy bodies
- IQR
interquartile range
- MSA
multiple system atrophy
- OH
orthostatic hypotension
- OR
odds ratio
- PAF
pure autonomic failure
- PD
Parkinson disease
- QSART
quantitative sudomotor axon reflex testing
- RBD
REM sleep behavior disorder
- TST
thermoregulatory sweat test
Appendix. Authors
Footnotes
Class of Evidence: NPub.org/coe
Podcast: NPub.org/ojrusd
References
- 1.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 2.Campbell BC, McLean CA, Culvenor JG, et al. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J Neurochem 2001;76:87–96. [DOI] [PubMed] [Google Scholar]
- 3.Hague K, Lento P, Morgello S, Caro S, Kaufmann H. The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol 1997;94:192–196. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann H, Hague K, Perl D. Accumulation of alpha-synuclein in autonomic nerves in pure autonomic failure. Neurology 2001;56:980–981. [DOI] [PubMed] [Google Scholar]
- 5.Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 1994;117:235–243. [DOI] [PubMed] [Google Scholar]
- 6.Inoue M, Yagishita S, Ryo M, Hasegawa K, Amano N, Matsushita M. The distribution and dynamic density of oligodendroglial cytoplasmic inclusions (GCIs) in multiple system atrophy: a correlation between the density of GCIs and the degree of involvement of striatonigral and olivopontocerebellar systems. Acta Neuropathol 1997;93:585–591. [DOI] [PubMed] [Google Scholar]
- 7.Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 2013;12:443–453. [DOI] [PubMed] [Google Scholar]
- 8.Boeve BF, Silber MH, Ferman TJ, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 2013;14:754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coon EA, Cutsforth-Gregory JK, Benarroch EE. Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord 2018;33:349–358. [DOI] [PubMed] [Google Scholar]
- 10.Fereshtehnejad SM, Yao C, Pelletier A, Montplaisir JY, Gagnon JF, Postuma RB. Evolution of prodromal Parkinson's disease and dementia with Lewy bodies: a prospective study. Brain 2019;142:2051–2067. [DOI] [PubMed] [Google Scholar]
- 11.The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996;46:1470. [DOI] [PubMed] [Google Scholar]
- 12.Bradbury S, Eggleston C. Postural hypotension: an autopsy upon a case. Am Heart J 1927;3:105–106. [Google Scholar]
- 13.Mabuchi N, Hirayama M, Koike Y, et al. Progression and prognosis in pure autonomic failure (PAF): comparison with multiple system atrophy. J Neurol Neurosurg Psychiatry 2005;76:947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer W, Berini SE, Sandroni P, et al. Pure autonomic failure: predictors of conversion to clinical CNS involvement. Neurology 2017;88:1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufmann H, Norcliffe-Kaufmann L, Palma JA, et al. The natural history of pure autonomic failure: a U.S. prospective cohort. Ann Neurol 2017;81:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannini G, Calandra-Buonaura G, Asioli GM, et al. The natural history of idiopathic autonomic failure: the IAF-BO cohort study. Neurology 2018;91:e1245–e1254. [DOI] [PubMed] [Google Scholar]
- 17.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 19.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc 1993;68:748–752. [DOI] [PubMed] [Google Scholar]
- 21.Low PA, Caskey PE, Tuck RR, Fealey RD, Dyck PJ. Quantitative sudomotor axon reflex test in normal and neuropathic subjects. Ann Neurol 1983;14:573–580. [DOI] [PubMed] [Google Scholar]
- 22.Low PA. Autonomic nervous system function. J Clin Neurophysiol 1993;10:14–27. [DOI] [PubMed] [Google Scholar]
- 23.Fealey RD, Low PA, Thomas JE. Thermoregulatory sweating abnormalities in diabetes mellitus. Mayo Clinic Proc 1989;64:617–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.