

Abstract
Catalytic enantioselection usually depends on differences in steric interactions between prochiral substrates and a chiral catalyst. We have discovered a carbene Si–H insertion in which the enantioselectivity depends primarily on the electronic characteristics of the carbene substrate, and the log(er) values are linearly related to Hammett parameters. A new class of chiral tetraphosphate dirhodium catalysts was developed; it shows excellent activity and enantioselectivity for the insertion of diarylcarbenes into the Si–H bond of silanes. Computational and mechanistic studies show how the electronic differences between the two aryls of the carbene lead to differences in energies of the diastereomeric transition states. This study provides a new strategy for asymmetric catalysis exploiting the electronic properties of the substrates.
Graphical Abstract
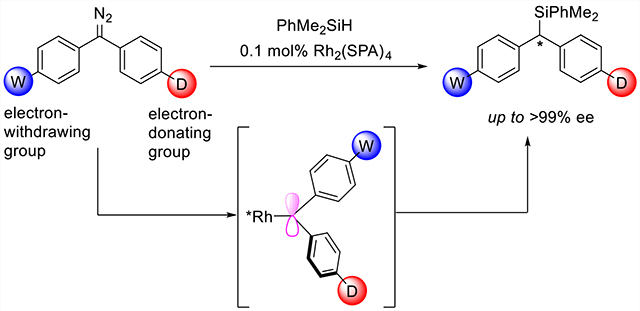
INTRODUCTION
Enantiomers of chiral molecules often exhibit biological activities distinct from one another because of the homochirality of biological molecules (e.g., L-amino acids and D-saccharides). As a result, industrial compounds such as pharmaceuticals, pesticides, flavors, and fragrances often must be a single enantiomer.1 In the past several decades, asymmetric catalysis has become a reliable method for the synthesis of enantiomerically enriched chiral compounds.2 Most current methods of asymmetric catalysis rely on the spatial interactions between the catalyst and substrate for enantiocontrol. Many sterically crowded chiral catalysts have been developed that utilize this principle to achieve high enantioselectivity.3 For unsaturated substrates, the chiral catalyst is often able to achieve enantiocontrol by discriminating between the two prochiral faces of the substrate.4 It is difficult to identify prochiral faces when the reactive center is attached to two sterically similar substituents (Figure 1A). For this reason, only limited successes have been achieved in the catalytic enantioselective reactions, such as hydrogenations of diaryl5 or dialkyl ketones6 or of diaryl or dialkyl ethylenes7 and cycloadditions of diaryl ethylenes.8
Figure 1.

Chiral differentiations of prochiral faces and enantioselective diarylcarbene insertion into Si–H bonds.
Many active intermediates of organic reactions, such as carbocations, carbon radicals, carbenes, and conjugated carbanions, have planar structures. Asymmetric reactions that proceed via these active intermediates often rely on the steric differences between the substituents connected to the prochiral center.9 Catalytic enantiocontrol by discriminating substituent electronics is very rare.10 Recently, Furstner and co-workers11 studied the structures of rhodium-diphenylcarbenes by X-ray single-crystal diffraction: the electron-rich phenyl ring (4-Me2NC6H4) adopts a coplanar orientation with the carbene plane (Θ = 0.9°) to maximize the overlap of the π cloud of the phenyl ring with the (empty) carbene p orbital. The electron-deficient phenyl ring (4-CF3C6H4) lies orthogonal to the carbene plane (Θ = −94.7°) to stabilize the lone pair of the carbene that also donates to the Rh dz2 orbital. In this way, the electronic properties of the substituents determine the degree of conjugation between the aryl rings and the p or lone-pair orbitals of the carbene center. On the basis of this property, we speculated that enantioselective transformation could be achieved by a chiral catalyst that distinguishes the conformations of a prochiral carbene intermediate. This strategy provides a new method for enantiocontrol for chiral transformations of substrates, which have sterically similar substituents. Although stereoselectivity has been achieved in carbene reactions catalyzed by Cu, Fe, and Rh catalysts,12 the carbenes generally have one ester group and one aryl or alkenyl group, or in the work by Shaw, very different substitution patterns on the two aryl groups of diaryl carbenes that cause large steric differences.13 We have now found that dirhodium catalysts modified with chiral spiro phosphate ligands (Figure 1B, Rh2(SPA)4) can differentiate the conformations of diarylcarbenes to achieve highly enantioselective transformations even though the substituents are in positions where they have no different direct interactions with the catalyst.
Transition-metal-catalyzed carbene insertion into Si–H bonds is a powerful method for the synthesis of optically active silanes. However, high enantioselectivity has been achieved only with the use of carbenes with markedly different substituents (e.g., one substituent is an alkyl, alkenyl, or aryl group and the other substituent is an electron-withdrawing group).14 Enantioselective Si–H bond insertion of diarylcarbenes remains undeveloped. The challenge in the enantioselective Si–H bond insertion of diarylcarbene stems from the difficulty of precisely distinguishing the Re and Si faces of the carbene. We sought to overcome this limitation using a chiral catalyst capable of distinguishing the electronic-induced conformations of the prochiral carbene intermediate (Figure 1B).
RESULTS AND DISCUSSION
The insertion of 4-nitrophenyl phenyl diazomethane (D1) into dimethylphenylsilane (S1) was performed using several chiral dirhodium catalysts commonly used in asymmetric carbene transformations15 (see the Supporting Information for more details). The best performing catalyst for this reaction, Rh2(S-PTTL)4, gave high yield but only modest enantioselectivity, prompting further investigation. A new type of dirhodium catalyst that contains spiro phosphate ligands C1–C5 (see Supporting Information for details), was developed.16 Of the spiro phosphate dirhodium catalysts, C2 afforded both good yield and the highest enantioselectivity (Scheme 1). Even 0.01 mol % catalyst is sufficient for excellent results.
Scheme 1. Enantioselective Si–H Bond Insertion of 4-Nitrophenylphenyl Diazomethane Catalyzed by Chiral Dirhodium Catalystsa.

aReaction conditions: Catal/D1/S1 = 0.0002:0.2:0.24 (mmol), 1 mL solution of D1 was dropped into a 2 mL solution of S1 and catalyst at 0 °C, <3 min, isolated yield.
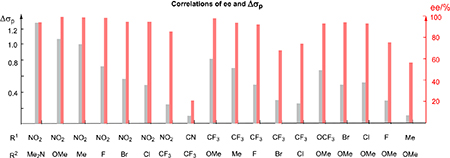
A variety of diphenyl diazomethylenes D1–D12 bearing electronically different para substituents were evaluated in the reaction with silane S1 catalyzed by C2 (Table 1). The substrates with a strong electron-withdrawing group or a strong electron-donating group at the para position of one phenyl ring exhibited high enantioselectivity (entries 1–4 and 12), whereas the substrates having a moderate or weak electronic effect exhibited lower enantioselectivity (entries 5–11). Moreover, the substrates with electron-withdrawing groups (D3, R = CF3) or electron-donating groups (D11, R = OMe) afforded Si–H bond insertion products with opposite absolute configurations (entry 3 and entry 11). When the Hammett substituent constant (σp)17 differences of the para substituents of two phenyl rings are over 0.5, the ee values of the corresponding Si–H bond insertion products are over 90%. Moreover, a plot of log(er) values against Hammett’s σp values is shown in Figure S1 (slope 2.86, R2 = 0.96). These results clearly indicate that the enantioselectivity of this reaction is directly related to the electronic properties of carbene intermediates.
Table 1.
Rhodium-Catalyzed Enantioselective Si–H Bond Insertions of Diphenyl Diazomethylenes D1–D12
 | |||||
|---|---|---|---|---|---|
| entrya | R | Δσp (R-H)b | product | yield (%) | ee (%) |
| 1 | NO2 | 0.78 | P1 | 92 | >99 |
| 2 | CN | 0.66 | P2 | 96 | 98 |
| 3c | CF3 | 0.54 | P3 | 91 | 95 (S)d |
| 4c | SCF3 | 0.50 | P4 | 68 | 97 |
| 5c | OCF3 | 0.35 | P5 | 66 | 76 |
| 6 | Cl | 0.28 | P6 | 95 | 66 |
| 7 | Br | 0.23 | P7 | 79 | 77 |
| 8 | I | 0.18 | P8 | 62 | 82 |
| 9c | F | 0.06 | P9 | 88 | 25 |
| 10 | Me | −0.17 | P10 | 51 | 18 |
| 11 | OMe | −0.27 | P11 | 47 | 64 (R)d |
| 12 | NMe2 | −0.60 | P12 | 45 | 91 |
Reaction conditions: Condition A (for entries 1–3, 6, 7, 10, and 11): diazo compound (0.2 mmol), PhMe2SiH (1.2 equiv), C2 (0.1 mol %), DCM, 0 °C. Condition B (for entries 4, 5, 8, 9, and 12): hydrazone (0.2 mmol), MnO2 (3.5 equiv), MgSO4, DCM, rt, 5–6 h; then PhMe2SiH (1.2 equiv), C2 (0.1 mol %), DCM, 0 °C. Isolated yields were given. The ee values were determined by chiral HPLC.
Hammett substituent constant for para substituents.
The ee value was determined by the corresponding alcohol obtained through the oxidation of the Si–H insertion product.
The absolute configuration was assigned by analogy with the reported data (see Supporting Information for details).
Next, various diphenyl diazomethanes bearing different para substituents were studied (Table 2). These results show that the enantioselectivity of the reaction is directly correlated with the differences in substrate electronics. Again, when the difference of the Hammett substituent constant of the two para substituents is equal or greater than 0.5, the ee of the corresponding Si–H bond insertion product is equal or greater than 90% (entries 1–6, 9–11, and 14–16). Moreover, sterically similar substituents (e.g., p-NO2 vs p-NMe2, entry 1; p-CF3 vs p-CH3, entry 10; p-OCF3 vs p-OCH3, entry 14) were precisely differentiated, indicating that the observed enantioselectivity is not a result of steric effects.
Table 2.
Rhodium-Catalyzed Enantioselective Si–H Bond Insertion of Diphenyl Diazomethanes D13–D30
 | ||||||
|---|---|---|---|---|---|---|
| entrya | R1 | R2 | Δσp (R1-R2)b | product | yield(%) | ee(%) |
| 1 | NO2 | NMe2 | 1.38 | P13 | 66 | 96 |
| 2 | NO2 | OMe | 1.05 | P14 | 78 | 99 |
| 3 | NO2 | Me | 0.95 | P15 | 87 | 99 |
| 4 | NO2 | F | 0.72 | P16 | 86 | 98 |
| 5 | NO2 | Br | 0.55 | P17 | 80 | 95 |
| 6 | NO2 | CI | 0.5 | P18 | 88 | 96 |
| 7 | NO2 | CF3 | 0.24 | P19 | 58 | 86 |
| 8 | CN | CF3 | 0.12 | P20 | 42 | 36 |
| 9 | CF3 | OMe | 0.81 | P21 | 78 | 98 |
| 10c | CF3 | Me | 0.71 | P22 | 85 | 94(S) |
| 11c | CF3 | F | 0.48 | P23 | 63 | 92 |
| 12c | CF3 | Br | 0.31 | P24 | 71 | 67 |
| 13c | CF3 | CI | 0.26 | P25 | 73 | 76(R) |
| 14 | OCF3 | OMe | 0.62 | P26 | 58 | 93 |
| 15 | Br | OMe | 0.50 | P27 | 58 | 94 |
| 16 | CI | OMe | 0.55 | P28 | 59 | 93 |
| 17c | F | OMe | 0.33 | P29 | 43 | 77(R) |
| 18 | Me | OMe | 0.10 | P30 | 40 | 56 |
 | ||||||
Reaction conditions: hydrazone (0.2 mmol), MnO2 (3.5 equiv), MgSO4, DCM, rt, 5–6 h; then PhMe2SiH (1.2 equiv), C2 (0.1 mol %), DCM, 0 °C. Isolated yields were given. The ee values were determined by chiral HPLC analysis.
The Hammett substituent constant difference for para substituents.
The ee value was determined by the corresponding alcohol obtained through the oxidation of Si–H insertion product. A plot of log(er) vs Δσp (R1-R2) is shown in Figure S2.
The chiral spiro phosphate dirhodium catalyst C2 is also efficient for the enantioselective Si–H bond insertion of other diazo compounds containing two different aryl groups (Scheme 2). In every case, the electronic property of the substituents on the aryl rings significantly affected the enantioselectivity of the reaction. For instance, the naphthyl phenyl diazomethane afforded the Si–H bond insertion product P31 with 53% ee; however, introducing a p-NO2 at the phenyl ring dramatically increased the ee value of product P32 to 96%. Similarly, 4-nitrophenyl 2′-methoxylphenyl diazomethane provided much higher enantioselectivity (P34, >99% ee) than 2-methoxylphenyl phenyl diazomethane (P33, 32% ee). Moreover, if one aryl ring of the substrates has a strong electron-withdrawing para substituent (e.g., NO2 or CF3) and the other aryl ring is a heteroaryl ring (P36–P38), excellent enantioselectivity can be obtained. Excellent enantioselectivity can also be achieved when the diaryl diazomethane substrates have an ortho substituent (P39 and P40). Presumably because of the small radius of the fluorine atom, the 2-fluoro-substituted diazo compound afforded relatively lower ee (83% ee, P41). However, the enantioselectivity can be increased by introducing an electron-donating group (4-OMe) at the other aryl ring of the substrate (91% ee, P42). In addition to dimethylphenylsilane (Si), other silanes can also be used in the Si–H bond insertion reaction with diaryl diazomethane D1, affording Si–H bond insertion products with excellent enantioselectivtiy (P43–P49). It is worth mentioning that the alkynyl silane underwent Si–H bond insertion reaction, giving the corresponding product P47 with high yield (93%) and excellent enantioselectivity (>99% ee). The absolute configuration of (S)-P47 was determined by single-crystal X-ray diffraction analysis.
Scheme 2. Substrate Scope of Diarylcarbene Insertion into Si–H Bondsa.

aReaction conditions: hydrazone (0.2 mmol), MnO2 (3.5 equiv), MgSO4, DCM, rt, 5–6 h; then PhMe2SiH (1.2 equiv), C2 (0.1 mol %), DCM, 0 °C. Isolated yields were given. The ee values were determined by chiral HPLC analysis. bDimethylchlorosilane was used and the hydrolysis product was isolated after workup.
MECHANISTIC STUDIES
A kinetic isotopic study was carried out; in a competition experiment using dimethylphenylsilane and deuterated dimethylphenylsilane, the kinetic isotope effect (KIE) was found to be 1.5 (see Supporting Information for details). This result is similar to the values reported in the Si–H insertions of aryl diazoesters catalyzed by transition metals such as Ir,14c Rh,14d and Cu;18 these reactions proceed through concerted three- center transition states. To gain insights about the origin of enantioselectivity from the catalyst and the electronic effects of substrates, we also conducted a computational investigation. C1 was used as the model catalyst.19 X-ray structure analysis of the catalyst C1 reveals that the presence of four identical chiral ligands around the dirhodium core results in a rigid catalyst with higher symmetry (D4 symmetry) than the ligands themselves (C2 symmetry).16 This symmetry causes both sides of the dirhodium catalyst to be identical, limiting the number of possible conformations for the transition state of Si–H insertion.
Initially, a dirhodium-tetraformate catalyst (Rh2(O2CH)4) was computed to observe transition-state geometries in the absence of any steric effects caused by the ligands (see Supporting Information for details). Figure 2A shows that the Si–H insertion proceeds via a concerted three-center transition state. Calculated transition-state geometries for diphenylcarbene insertion into the Si–H bond of dimethylphenylsilane show that one aryl ring rotates to a near-coplanar orientation with the carbene empty p orbital, while the other phenyl ring rotates to a near orthogonal orientation. For substituted diphenylcarbenes, two transition-state geometries are possible, depending on whether the substituted aryl ring is conjugated (TS-1) or orthogonal (TS-2) to the empty p orbital. These two transition states are not equal in energy and the favored transition state always results when the electron-rich aryl ring is nearly coplanar with the carbene plane. Plotting the energy difference (ΔΔG‡) vs the corresponding Hammett substituent constant σp shows a linear correlation (Figure 2A), indicating that this energy difference is greater when the electronic difference between aryl rings is more pronounced.
Figure 2.

Computational studies. (A) Optimized transition-state structures for carbene insertion into the Si–H bond with a tetraformate dirhodium (Rh2(O2CH)4) as a model catalyst. (B) Structures of optimized transition states for the Si–H bond insertion of asymmetrically substituted diphenylcarbene formed from C1. ONIOM partitioning of the transition-state geometry, with the atoms shown opaque modeled with density functional theory (DFT), and the atoms shown transparent modeled with the universal force field (UFF). The relative Gibbs free energies with single-point corrections are given in kcal mol−1. (C) Favorable enantio-determining transition- state structure for the Si–H bond insertion of asymmetrically substituted diphenylcarbene (R1 = NO2, R2 = OMe) formed from C1.
In the chiral environment of C1, the transition state for the Si–H bond approaching the asymmetrical diphenylcarbene (R1 = NO2, R2 = OMe) from its Re face (TS-3) is the lowest in energy (Figure 2B,C). This transition state is favored by 5.6 kcal mol−1 over TS-5 because of the different steric effects of the two aryl substituents on carbene; this steric difference originates from the electronic effects of substituents that orient electron-rich and electron-deficient aryl rings differently. Moreover, TS-3 is favored by 2.2 kcal mol−1 over TS-4 (which would lead to the opposite enantiomer) because of the steric effects that the chiral catalyst has on the transition-state conformations. In these structures, the electron-rich aryl ring (R2 = OMe) is able to adopt a more coplanar orientation in TS-3 (Θ = 19.5°) than in TS-4 (Θ = 31.6°). The last possible transition state, TS-6, is 7.4 kcal mol−1 higher in energy than TS-3 because of both unfavorable electronics and steric clashes with the catalyst. Moreover, when two aryls of the carbene have similar electronic properties, as in the case for R1 = CN and R2 = CF3, the energy difference between TS′-3 and TS′-5 is smaller (2.4 kcal/mol), which is consistent with the low enantioselectivity observed in the experiment (see Supporting Information for details).20
These calculations show that the chiral environment of the catalyst C1 can fix the conformations of the transition state in the Si–H bond insertion reaction. On this basis, the energy difference of the transition states, which relates to the enantioselectivity of the reaction, is primarily due to the electronic difference between two aryls of carbene.
CONCLUSION
In summary, we have achieved a method for highly enantioselective carbene insertion into Si–H bonds that relies primarily on the electronic properties of the substrates. It represents the first highly enantioselective diarylcarbene insertion into the heteroatom–hydrogen bonds. A new class of D4 symmetric dirhodium catalysts bearing chiral spiro phosphate ligands was developed, and computational studies demonstrate that the chiral environment of these catalysts can differentiate the various possible transition-state conformations. The chiral induction observed in this study not only enables the unprecedented enantioselective transformations of carbenes having spatially similar groups but also inspires the development of new strategies for chiral transformations of charged active intermediates, such as carbocations, carbanions, and carbon radicals.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Natural Science Foundation of China (21625204, 21790332, 21532003, and 21971119), the “111” project (B06005) of the Ministry of Education of China, and the National Program for Special Support of Eminent Professionals, the National Science Foundation of the USA (CHE-176328 to K.N.H.) and the National Institute of Health (T32GM008496 to D.E.) for financial support.
Footnotes
The authors declare no competing financial interest.
Geometrical parameters for the structure of reagents C3 and P47 are available from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference numbers CCDC 1888695 and CCDC 1888689, respectively.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04725.
Materials and methods, supplementary text, spectral data, calculations data, and additional references (PDF)
X-ray crystallographic data (CIF)
X-ray crystallographic data (CIF)
Contributor Information
Liang-Liang Yang, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
Declan Evans, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095-1569, United States.
Bin Xu, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
Wen-Tao Li, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
Mao-Lin Li, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
Shou-Fei Zhu, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095-1569, United States.
Qi-Lin Zhou, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China.
REFERENCES
- (1).Nicolaou KC; Montagnon T Molecules that Changed the World; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- (2).Jacobsen EN; Pfaltz A; Yamamoto H Comprehensive Asymmetric Catalysis I—III; Springer Verlag: Berlin, 1999. [Google Scholar]
- (3).For reviews, see:Desimoni G; Faita G; Jørgensen KA C2-Symmetric Chiral Bis(oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2011, 111, PR284–PR437.Parmar D; Sugiono E; Raja S; Rueping M Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2014, 114, 9047–9153.Shaw S; White JD Asymmetric Catalysis Using Chiral Salen—Metal Complexes: Recent Advances. Chem. Rev. 2019, 119, 9381–9426.
- (4).Walsh PJ; Kozlowski MC Fundamentals of Asymmetric Catalysis; University Science Books: Sausalito, CA, 2008. [Google Scholar]
- (5).Touge T; Nara H; Fujiwhara M; Kayaki Y; Ikariya T Efficient Access to Chiral Benzhydrols via Asymmetric Transfer Hydrogenation of Unsymmetrical Benzophenones with Bifunctional Oxo-Tethered Ruthenium Catalysts. J. Am. Chem. Soc. 2016, 138, 10084–10087. [DOI] [PubMed] [Google Scholar]
- (6).(a) Jiang Q; Jiang Y; Xiao D; Cao P; Zhang X Highly Enantioselective Hydrogenation of Simple Ketones Catalyzed by a Rh-PennPhos Complex. Angew. Chem., Int. Ed. 1998, 37, 1100–1103. [DOI] [PubMed] [Google Scholar]; (b) Garbe M; Junge K; Walker S; Wei Z; Jiao H; Spannenberg A; Bachmann S; Scalone M; Beller M Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand. Angew. Chem., Int. Ed. 2017, 56, 11237–11241. [DOI] [PubMed] [Google Scholar]
- (7).(a) Mazuela J; Verendel JJ; Coll M; Schäffner B; Börner A; Andersson PG; Pàmies O; Diéguez M Iridium Phosphite-Oxazoline Catalysts for the Highly Enantioselective Hydrogenation of Terminal Alkenes. J. Am. Chem. Soc. 2009, 131, 12344–12353. [DOI] [PubMed] [Google Scholar]; (b) Mazuela J; Norrby P-O; Andersson PG; Pàmies O; Diéguez M Pyranoside Phosphite-Oxazoline Ligands for the Highly Versatile and Enantioselective Ir-Catalyzed Hydrogenation of Minimally Functionalized Olefins. A Combined Theoretical and Experimental Study. J. Am. Chem. Soc. 2011, 133, 13634–13645. [DOI] [PubMed] [Google Scholar]; (c) Besset T; Gramage-Doria R; Reek JNH Remotely Controlled Iridium-Catalyzed Asymmetric Hydrogenation of Terminal 1,1-Diaryl Alkenes. Angew. Chem., Int. Ed. 2013, 52, 8795–8797. [DOI] [PubMed] [Google Scholar]
- (8).Wiest JM; Conner ML; Brown MK Allenoates in Enantioselective [2 + 2] Cycloadditions: From a Mechanistic Curiosity to a Stereospecific Transformation. J. Am. Chem. Soc. 2018, 140, 15943–15949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For a review, see: Mohr JT; Hong AY; Stoltz BM Enantioselective Protonation. Nat. Chem. 2009, 1, 359–369. For representative examples, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (a) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wendlandt AE; Vangal P; Jacobsen EN Quaternary Stereocentres via an Enantioconver- gent Catalytic Sn1 Reaction. Nature 2018, 556, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Legros J-Y; Fiaud J-C Electronic Control of Enantioselectivity in the Palladium-Catalyzed Asymmetric Allylic Substitution of trans 4-tButyl-1-vinylcyclohexyl Benzoates. Tetrahedron 1994, 50, 465–474. [Google Scholar]; (b) Corey EJ; Helal CJ Novel Electronic Effects of Remote Substituents on the Oxazaborolidine-Catalyzed Enantioselective Reduction of Ketones. Tetrahedron Lett. 1995, 36, 9153–9156. [Google Scholar]; (c) Zhang H; Xue F; Mak TCW; Chan KS Enantioselectivity Increases with Reactivity: Electronically Controlled Asymmetric Addition of Diethylzinc to Aromatic Aldehydes Catalyzed by a Chiral Pyridylphenol. J. Org. Chem. 1996, 61, 8002–8003. [DOI] [PubMed] [Google Scholar]; (d) Wong HL; Tian Y; Chan KS Electronically Controlled Asymmetric Cyclopropanation Catalyzed by a New Type of Chiral 2,2′-Bipyridine. Tetrahedron Lett. 2000, 41, 7723–7726. [Google Scholar]
- (11).Werlé C; Goddard R; Philipps P; Farès C; Fürstner A Structures of Reactive Donor/Acceptor and Donor/Donor Rhodium Carbenes in the Solid State and Their Implications for Catalysis. J. Am. Chem. Soc. 2016, 138, 3797–3805. [DOI] [PubMed] [Google Scholar]
- (12).For reviews, see:Zhu S-F; Zhou Q-L Transition-Metal-Catalyzed Enantioselective Heteroatom-Hydrogen Bond Insertion Reactions. Acc. Chem. Res. 2012, 45, 1365–1377.Keipour H; Carreras V; Ollevier T Recent Progress in the Catalytic Carbene Insertion Reactions into the Silicon–Hydrogen Bond. Org. Biomol. Chem. 2017, 15, 5441–5456.
- (13).Soldi C; Lamb KN; Squitieri RA; González-López M; Di Maso MJ; Shaw JT Enantioselective Intramolecular C–H Insertion Reactions of Donor–Donor Metal Carbenoids. J. Am. Chem. Soc. 2014, 136, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For representative examples, see:Davies HML; Hansen T; Rutberg J; Bruzinski PR Rhodium(II) (S)-N-(Arylsulfonyl)-prolinate Catalyzed Asymmetric Insertion of Vinyl- and Phenylcarbenoids into the Si–H Bond. Tetrahedron Lett. 1997, 38, 1741 — 1744.Zhang Y-Z; Zhu S-F; Wang L-X; Zhou Q-L Copper-Catalyzed Highly Enantioselective Carbenoid Insertion into Si–H Bonds. Angew. Chem., Int. Ed. 2008, 47, 8496–8498.Yasutomi Y; Suematsu H; Katsuki T Iridium(III)-Catalyzed Enantioselective Si–H Bond Insertion and Formation of an Enantioenriched Silicon Center. J. Am. Chem. Soc. 2010, 132, 4510–4511.Chen D; Zhu D-X; Xu M-H Rhodium(I)-Catalyzed Highly Enantioselective Insertion of Carbenoid into Si–H: Efficient Access to Functional Chiral Silanes. J.Am. Chem. Soc. 2016, 138, 1498–1501.Hyde S; Veliks J; Liégault B; Grassi D; Taillefer M; Gouverneur V Copper-Catalyzed Insertion into Heteroatom-Hydrogen Bonds with Trifluorodiazoalkanes. Angew. Chem. Int. Ed. 2016, 55, 3785–3789.
- (15).Doyle MP; McKervey MA; Ye T Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; Wiley: New York, 1998.For representative reviews, see:Ford A; Miel H; Ring A; Slattery CN; Maguire AR; McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981 – 10080.Davies HML; Liao K Dirhodium Tetracarboxylates as Catalysts for Selective Intermolecular C–H Functionalization. Nat. Rev. Chem. 2019, 3, 347–360.
- (16).Xu B; Zhu S-F; Zhang Z-C; Yu Z-X; Ma Y; Zhou Q-L Highly Enantioselective S–H Bond Insertion Cooperatively Catalyzed by Dirhodium Complexes and Chiral Spiro Phosphoric Acids. Chem. Sci 2014, 5, 1442–1448. [Google Scholar]
- (17).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- (18).Dakin LA; Ong PC; Panek JS; Staples RJ; Stavropoulos P Speciation and Mechanistic Studies of Chiral Copper(I) Schiff Base Precursors Mediating Asymmetric Carbenoid Insertion Reactions of Diazoacetates into the Si–H Bond of Silanes. Organometallics 2000, 19, 2896–2908. [Google Scholar]
- (19).Both the dirhodium catalyst C1 and C2 exhibit good enantioselectivity for this insertion reaction. And to avoid the conformation complexity from the relative rotation of the outermost phenyl of C2.
- (20).During submission of this manuscript, Davies and co-workers reported a rhodium-catalyzed asymmetric cyclopropanation of diarylcarbenes with styrene. In that elegant work a correlation of the distortion angle of the two aryl rings of diarylcarbenes with the diastereoselectivity of the reaction was also observed.Lee M; Ren Z; Musaev DG; Davies HML Rhodium-Stabilized Diarylcarbenes Behaving as Donor/Acceptor Carbenes. ACS Catal 2020, 10, 6240–6247.For steric effect of in-plane and out-of-plane phenyl groups, see:Charton M Steric Effects. I. Esterification and Acid-Catalyzed Hydrolysis of Esters. J. Am. Chem. Soc. 1975, 97, 1552–1556.Charton M Steric Effects. II. Base-Catalyzed Ester Hydrolysis. J. Am. Chem. Soc. 1975, 97, 3691–3693.Charton M Steric Effects. 7. Additional v Constants. J. Org. Chem. 1976, 41, 2217–2220.Sigman MS; Miller JJ Examination of the Role of Taft-Type Steric Parameters in Asymmetric Catalysis. J. Org. Chem 2009, 74, 7633–7643.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.