

Abstract
Congenital generalized lipodystrophy (CGL) is a heterogeneous autosomal recessive disorder characterized by a near complete lack of adipose tissue from birth and, later in life, the development of metabolic complications, such as diabetes mellitus, hypertriglyceridaemia and hepatic steatosis. Four distinct subtypes of CGL exist: type 1 is associated with AGPAT2 mutations; type 2 is associated with BSCL2 mutations; type 3 is associated with CAV1 mutations; and type 4 is associated with PTRF mutations. The products of these genes have crucial roles in phospholipid and triglyceride synthesis, as well as in the formation of lipid droplets and caveolae within adipocytes. The predominant cause of metabolic complications in CGL is excess triglyceride accumulation in the liver and skeletal muscle owing to the inability to store triglycerides in adipose tissue. Profound hypoleptinaemia further exacerbates metabolic derangements by inducing a voracious appetite. Patients require psychological support, a low-fat diet, increased physical activity and cosmetic surgery. Aside from conventional therapy for hyperlipidaemia and diabetes mellitus, metreleptin replacement therapy can dramatically improve metabolic complications in patients with CGL. In this Review, we discuss the molecular genetic basis of CGL, the pathogenesis of the disease’s metabolic complications and therapeutic options for patients with CGL.
Introduction
Lipodystrophies are heterogeneous inherited or acquired disorders that are characterized by selective loss of adipose tissue and a predisposition to developing insulin resistance and its associated complications, such as diabetes mellitus, hypertriglyceridaemia, hepatic steatosis, polycystic ovary syndrome, acanthosis nigricans and hypertension.1 Inherited lipodystrophies are rare disorders with some manifesting at birth, such as congenital generalized lipodystrophy (CGL) and neonatal progeroid syndrome,2–5 whereas fat loss manifests later in life in other lipodystrophies, such as familial partial lipodystrophies and mandibuloacral dysplasia.6,7 The loss of fat can be general (involving nearly all the body fat depots), partial (affecting mainly the limbs) or localized (that is, from discrete areas of the body such as the abdomen or thigh). The severity of the metabolic complications is generally proportional to the extent of body fat loss with the generalized and partial lipodystrophies presenting with severe metabolic derangements, whereas the localized lipodystrophies usually do not predispose patients to metabolic complications.1
Genetic lipodystrophies can be classified into autosomal recessive or autosomal dominant on the basis of the pattern of inheritance (Box 1); however, a few syndromes (such as Hutchinson-Gilford progeria syndrome, atypical progeroid syndrome and mandibular hypoplasia, deafness and progeroid features syndrome) develop as a result of de novo heterozygous variants.8–11 The two most prevalent subtypes of genetic lipodystrophies are CGL and familial partial lipodystrophy (FPLD), each of which has been reported in ~300–500 patients worldwide so far.12–15 By contrast, the other types of lipodystrophy—mandibuloacral dysplasia, autoinflammatory lipodystrophy, progeroid syndromes associated lipodystrophy, SHORT syndrome associated lipodystrophy and Keppen-Lubinsky syndrome associated lipodystrophy—are extremely rare and in total each subtype has been reported in no more than 30 patients (Box 1).10,11,16–30 However, FPLD has autosomal dominant inheritance and some patients with subtle loss of fat from the extremities might not receive an accurate diagnosis; therefore, the prevalence of this lipodystrophy might be higher than is reported. CGL has the most extreme phenotype with loss of nearly all the body fat at birth (Figure 1) and early development of metabolic complications in childhood.13,15 Consequently, in this Review, we discuss the different subtypes of CGL and the implications of understanding the pathophysiological basis of metabolic complications in CGL for patients with generalized and regional obesity.
Box 1 |. Classification of genetic lipodystrophies.
Autosomal recessive
Congenital generalized lipodystrophy (Berardinelli-Seip syndrome; AGPAT2, BSCL2, CAV1, PTRF)
Mandibuloacral dysplasia (LMNA, ZMPSTE24)
Familial partial lipodystrophy (CIDEC, LIPE, WRN, PCYT1A)
Autoinflammatory lipodystrophy (JMP/CANDLE syndrome; PSMB8)
CGL-like phenotypes (PPARG, FOS)
Autosomal dominant
Familial partial lipodystrophy (LMNA, PPARG, AKT2, PLIN1)
Hutchinson-Gilford progeria syndrome (LMNA)
Atypical progeroid syndrome (LMNA)
Neonatal progeroid syndrome (FBN1, CAV1 and others)
Mandibular hypoplasia, deafness, progeroid features (MDP) syndrome (POLD1)
SHORT syndrome associated with lipodystrophy (PIK3R1)
Keppen-Lubinsky syndrome associated with lipodystrophy (KCNJ6)
Abbreviations: CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; CGL, congenital generalized lipodystrophy; JMP, joint contractures, muscle atrophy, microcytic anaemia, and panniculitis-induced lipodystrophy; SHORT, short stature, hyperextensibility, hernia, ocular depression, Rieger anomaly, and teething delay.
Figure 1 |.
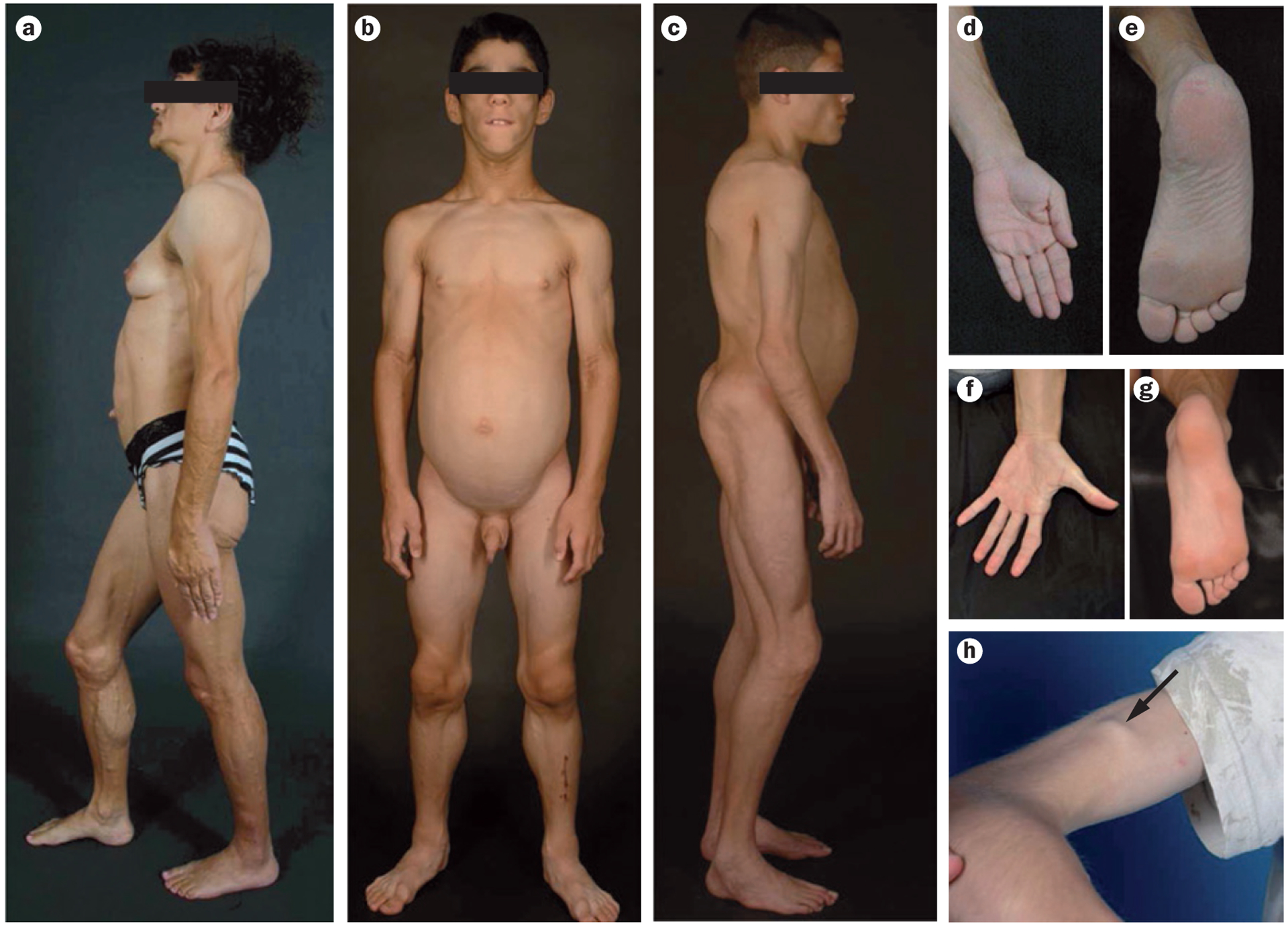
Clinical features of patients with CGL. a | Lateral view of a 34-year-old female with type 1 CGL owing to a homozygous mutation c.589–2A>G (p.Val197Glufs*32) in AGPAT2. This patient has a generalized lack of body fat, extreme muscularity, umbilical prominence and acanthosis nigricans in the axillae and neck. b | Anterior view of an 8-year-old male with type 2 CGL owing to compound heterozygous mutations c.193delCinsGGA (p.Pro65Glyfs*28) and c.325_326insA (p.Thr109Asnfs*5) in BSCL2. This patient has a generalized lack of fat and extreme muscularity. c | Lateral view of an 11-year-old male with type 4 CGL owing to homozygous mutation c.135delG (p.Lys45Asnfs*5) in PTRF This patient has generalized lipodystrophy, prominent muscularity, acromegalic features such as large hands and feet and protuberant abdomen. d | The left palm of a patient with type 1 CGL (the patient in panel a) showing normal subcutaneous fat. e | The sole of the right foot of the patient in panel a with type 1 CGL showing normal subcutaneous fat. f | The left palm of the patient with type 2 CGL in panel b showing loss of subcutaneous fat. g | The right sole of the patient in panel b showing loss of subcutaneous fat. h | Arrow points to percussion-induced muscle mounding on the biceps of the patient shown in panel c. Images are not to the same scale. Abbreviation: CGL, congenital generalized lipodystrophy. Written consent for publication of panel a was obtained from the patient. Written consent for publication of panels b and c was obtained from the patient’s responsible relative. Panel h reproduced with permission from Wiley © Shastry, S. et al. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am. J. Med. Genet. A 152A, 2245–2253 (2010).74
Congenital generalized lipodystrophy
CGL is an autosomal recessive disorder usually recognized at birth or shortly thereafter and was first reported in 1954 in two brothers from Brazil who were aged 2 years and 6 years and presented with marked hepatosplenomegaly, acromegaloid gigantism, fatty liver and hyperlipidaemia.2 Three further patients were subsequently identified in Norway who had generalized lack of body fat from birth and the disorder was termed congenital generalized lipodystrophy.3 Since these initial reports in the 1950s, ~300–500 patients with CGL have been reported in the literature.1,31–33 The precise population prevalence of CGL is unknown, but is estimated by us to be ~1 in 10 million.1 Many cases are reported in consanguineous families from Brazil, Lebanon and Scandinavia, as well as in families of African ancestry; the prevalence of some founder mutations and increased occurrence of consanguinity and endogamy probably accounts for the increased frequency of CGL in these regions and ethnic groups.33–35
Essential diagnostic criteria for CGL have been proposed that are based on the published literature and careful phenotyping using both conventional anthro-pometry including measurements of skinfold thickness by calipers and by whole body MRI.31 These criteria include a generalized lack of body fat with extreme muscularity from birth, confirmed with characteristic body fat distribution on MRI.31 Our knowledge of CGL has expanded considerably since these criteria were published in 2000. Furthermore, four molecularly distinct subtypes have been described and two other closely related phenotypes due to additional loci have been reported.35–40 In view of these developments, we recommend updated diagnostic criteria for CGL (outlined in Box 2). In addition, patients with CGL might have prominent superficial subcutaneous veins due to lack of fat. Children with CGL have accelerated growth owing to a voracious appetite.1,41,42 Prominence of umbilicus or umbilical hernia is often noticed at birth, which is probably due to underlying hepatomegaly and/or splenomegaly.1,41,43 Acanthosis nigricans develops during early childhood or after puberty, and can affect the neck, axillae and groin regions, as well as other locations such as the trunk, hands, knees, elbows and ankles.13,44,45 Patients can have liver enlargement due to hepatic steatosis in infancy.15 Some patients develop steatohepatitis, which can progress to fibrosis, cirrhosis or end-stage liver disease that requires liver transplantation.46,47 Furthermore, hypertrophic cardiomyopathy13,15 and focal segmental glomerulosclerosis have also been reported (Box 3).48
Box 2 |. Diagnostic criteria for CGL.
Essential criterion
Near total, generalized lack of body fat and extreme muscularity present at birth or soon thereafter
Characteristic body fat distribution patterns on physical examination and whole-body MRI consistent with the different subtypes of CGL
Confirmatory
Molecular diagnosis based on genotyping
Abbreviation: CGL, congenital generalized lipodystrophy.
Box 3 |. Clinical features of CGL.
Muscular appearance
Prominent veins
Accelerated growth
Voracious appetite
Prominence of umbilicus or umbilical hernias
Hepatomegaly and/or splenomegaly
Acanthosis nigricans
Mild hirsutism and clitoromegaly in female patients
Irregular menstrual periods with polycystic ovaries
Advanced bone age
Abbreviation: CGL, congenital generalized lipodystrophy.
Mild hirsutism, clitoromegaly and irregular menstruation are common in women with CGL and some present with primary or secondary amenorrhoea and polycystic ovaries.32,49,50 Insulin resistance is suspected to be the main cause of excessive ovarian androgen production in these patients.51 Severe leptin deficiency can lead to a loss of the pulsatile secretion of gonadotropins,50,52 but has not been documented in patients with CGL. Anovulation in women with CGL, therefore, is probably the result of a combined effect of irregular luteinizing hormone stimulation and androgenic effects on the ovaries.53 The development of the mammary glands is normal but an overlying subcutaneous fat layer surrounding the tissue is absent.43 Most women with CGL are unable to conceive; however, successful pregnancies have been reported in two patients.53,54 Pregnancy in these patients is high-risk and might require management of diabetes mellitus as well as severe hypertriglyceridaemia. Men with CGL might have a normal reproductive ability,55,56 but teratozoospermia has been reported in one patient.57
Focal lytic lesions in the long bones such as the humerus, femur, radius, ulna, carpal, tarsal or phalangeal bones after puberty have been reported in some patients, which might be confused with polyostotic fibrous dysplasia.42,47,58–60 These patients have an increased risk of pathological fractures in the affected bones and should be advised to avoid contact sports. The inability to make normal bone marrow fat to replace haematopoietic marrow during childhood and adolescence might underlie the development of focal lytic bone lesions.47 Advanced bone age has also been reported in nine individuals.44,61–63
Patients with CGL can develop insulin resistance and associated metabolic abnormalities soon after birth as well as during infancy and early childhood.13,15 The majority of patients develop hyperinsulinaemia,41 and ~45% develop diabetes mellitus during the pubertal years.13,15 Amyloidosis of pancreatic islets affecting >90% of the islets and β-cell atrophy has been seen on autopsy in a 24-year-old female with CGL.64 Interestingly, diabetes mellitus is ketosis resistant, which is probably due to endogenous hyperinsulinaemia. Some patients require extremely high doses of insulin to achieve glycaemic control (up to 3,000 units a day).65,66
Hypertriglyceridaemia is present in >70% of patients with CGL and develops mostly in late childhood and adolescence and only rarely during infancy.67 Severe hypertriglyceridaemia is often associated with eruptive xanthomas and recurrent pancreatitis, particularly in patients with uncontrolled diabetes mellitus.13,43 HDL cholesterol levels also tend to be low.68 An isolated case report has also described stroke in a 5-month-old male infant with a clinical diagnosis of CGL (although this diagnosis was not confirmed by genetic analysis).69 However, other atherosclerotic vascular complications such as coronary heart disease or peripheral vascular disease have not been reported in the literature. The serum levels of leptin and adiponectin are markedly reduced in patients with CGL secondary to near total loss of fat.70,71 In one study, median serum levels of adiponectin in patients with CGL were 1.5 μg/ml and leptin levels were 0.63 ng/ml.70 Severe hypoleptinaemia might induce a voracious appetite, which can further contribute to metabolic complications.72
Genetic subtypes of CGL
Four distinct genetic subtypes of CGL have been reported to date. Type 1 CGL results from mutations in the gene that encodes 1-acyl-sn-glycerol-3-phosphate acyltransferase β (AGPAT2) and type 2 CGL is due to mutations in the gene that encodes seipin (encoded by Berardinelli-Seip congenital lipodystrophy 2; BSCL2); these are the most common subtypes.35 Type 3 CGL, which seems to be due to a homozygous nonsense mutation in the gene that encodes caveolin 1 (CAV1) has been reported in a single patient37 and type 4 CGL, which is the result of mutations in the gene that encodes polymerase I and transcript release factor (PTRF), has been reported in ~30 patients.38,73–76 In addition, phenotypes that are closely related to CGL have been reported that are associated with biallelic mutations in the gene that encodes peroxisome proliferator activated receptor γ (PPARG)39 and a proto-oncogene c-FOS protein (FOS).40,77
Type 1 CGL
Type 1 CGL, (Online Mendelian Inheritance in Man [OMIM] #608594),78 is caused by mutations in AGPAT2. AGPAT2 is located on chromosome 9q34 and encodes lysophosphatidic acid acyltransferase-β, which is involved in triglyceride (triacylglycerol) biosynthesis.79 AGPAT2 has 278 amino acids and contains two highly conserved domains, NHXXXXD and EGTR, which are required for the enzymatic activity.79 The AGPATs catalyse the conversion of lysophosphatidic acid (1-acylglycerol-3-phosphate) to phosphatidic acid (1,2 diacylglycerol-3-phosphate) by esterifying a fatty acyl group at the sn-2 position of the glycerol backbone.80–83 This is a key intermediate step during the biosynthesis of glycerophospholipids and triglycerides from glycerol-3-phosphate.36,45,84 These phosphatidic acids can be further acylated and processed to form triglycerides or other phospholipids (Figure 2).81 AGPAT has 11 known isoforms, each encoded by a different gene.85 Each isoform has a unique tissue expression and the AGPAT2 isoform is highly expressed in adipose tissue.36 Other isoforms, such as AGPAT1, AGPAT3, AGPAT4, AGPAT6, AGPAT8, AGPAT10 and AGPAT11 are also expressed in adipose tissue, albeit at lower levels than AGPAT2.86 Consequently, AGPAT2 deficiency might impair triglyceride and phospholipid biosynthesis in the adipocytes and result in lipodystrophy.
Figure 2 |.
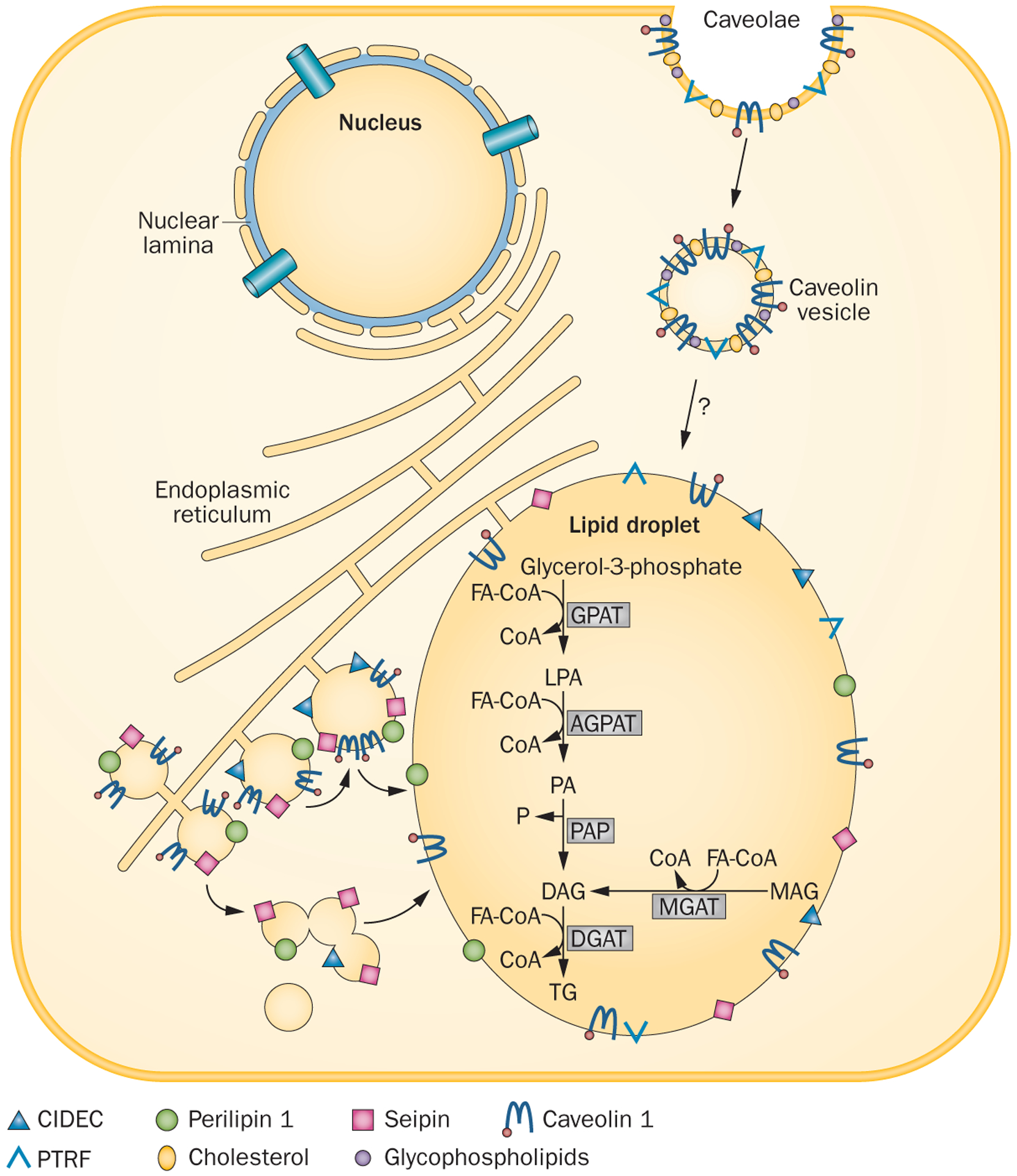
Lipid droplet formation in adipocytes. Lipid droplets are organelles that store triglycerides within the cell. They form as budding vesicles at the endoplasmic reticulum that fuse in adipocytes to form a single large lipid droplet. Many proteins, such as CIDEC, seipin and perilipin 1 are present on the lipid droplet membrane. CIDEC and seipin might be involved in the fusion of lipid droplets to form a larger droplet, whereas perilipin 1 is essential for lipid storage and hormone-mediated lipolysis. Caveolae are formed from lipid rafts on the cell surface, which include cholesterol, glycosphingolipids and caveolin 1. Endocytosis of caveolae forms caveolin vesicles that might directly merge with lipid droplets and translocate fatty acids to the lipid droplets. In the adipose tissue, triglyceride synthesis requires glycerol-3-phosphate as the initial substrate (classic pathway), whereas in the small intestine, triglyceride synthesis can occur via an alternative pathway using monoacylglycerol as the initial substrate. Acylation of glycerol-3-phosphate using FA-CoA at the sn-1 position is catalysed by GPAT, which results in the formation of 1-acylglycerol-3-phosphate or LPA. LPA is then acylated at the sn-2 position by AGPATs to yield phosphatidic acid. Removal of a phosphate group from phosphatidic acid by PAP produces DAG. Further acylation of DAG at the sn-3 position by DGAT finally produces triglyceride. In the alternative pathway, MAG is acylated to DAG by MGAT, which is then further converted to triglyceride. Abbreviations: AGPAT, 1-acyl-sn-glycerol-3-phosphate acyltransferase; CIDEC, cell death activator CIDE-3; CoA, coenzyme A; DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; FA-CoA, fatty acyl coenzyme A; GPAT, glycerol-3-phosphate acyltransferase; LPA, lysophosphatidic acid; MGAT, monoacylglycerol acyltransferase; P, phosphate; PA, phosphatidic acid; PAP, phosphatidic acid phosphatase; TG, triglyceride. Reproduced with permission from Endocrine Society © Garg, A. Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 96, 3313–3325 (2011).32
More than 90% of patients with type 1 CGL have null mutations,13,36,55,87,88 with no enzymatic activity detectable in vitro.89 However, 4% of patients with type 1 CGL are compound heterozygotes with a null and a missense mutation (with some residual enzymatic activity) and only 2% of patients have been reported to have homozygous missense mutations.13,36,55,87,88 However, the type of mutation does not seem to determine the phenotype or the severity of fat loss. Almost all patients of African origin have the founder mutation in intron 4, c.589–2A>G (p.Val197Glufs*32, also designated as p.Gln196fsX228), of one or both alleles. This mutation might be a founder variant in populations of African origin.13
Patients with type 1 CGL have a characteristic body fat distribution and might present with acromegaloid features with enlarged mandible, hands and feet (Figure 1a).13 These patients lack metabolically active adipose tissue in most of the subcutaneous areas, intra-abdominal regions, intra-thoracic regions and bone marrow, whereas adipose tissue in the palms, soles, under the scalp, orbital, peri-articular regions, perineum, vulva and pericalyceal regions of the kidneys is present (Figure 1d,e).43,90 In our opinion, the preservation of mechanical adipose tissue in patients with AGPAT2 mutations might be due to the increased expression of other AGPAT isoforms or expression of other genes in these adipose-tissue depots. These patients also have increased predisposition to develop lytic lesions in the appendicular skeleton (Table 1).15
Table 1 |.
Unique clinical features in CGL subtypes
| Complication | Type 1 CGL | Type 2 CGL | Type 3 CGL | Type 4 CGL |
|---|---|---|---|---|
| Adipose tissue | Absent metabolically active adipose tissue Preserved mechanical adipose tissue |
Absent metabolically active and mechanical adipose tissue | Absent metabolically active adipose tissue Preserved mechanical and bone marrow adipose tissue |
Absent metabolically active adipose tissue Preserved mechanical and bone marrow adipose tissue |
| Cardiovascular complications | N/A | Cardiomyopathy | N/A | Cardiomyopathy Ventricular arrhythmia, extended QT interval and sudden death |
| Bones, joints and movement | Focal lytic lesions in the long bones after puberty | Spastic gait (rare) | Short stature | Osteopenia, distal metaphyseal deformation with joint stiffness, atlanto-axial instability |
| Gastrointestinal complications | N/A | N/A | Functional megaoesophagus | Congenital pyloric stenosis |
| Skeletal muscle | N/A | N/A | N/A | Congenital myopathy |
| Other features | Acromegaloid features with enlarged mandible, hands and feet | Teratozoospermia | Hypocalcaemia due to vitamin D resistance | Late onset of lipodystrophy in infancy |
Abbreviations: CGL, congenital generalized lipodystrophy; N/A, not applicable.
Agpat2−/− mice develop severe lipodystrophy that affects the white and brown adipose tissue, these animals also have extreme insulin resistance, diabetes mellitus and hepatic steatosis.80 Owing to the total lack of adipose tissue, Agpat2−/− mice cannot be used to understand the mechanisms related to the development of lipodystrophy, instead this model enables the study of severe hepatic insulin resistance and steatosis in CGL. The expression of lipogenic genes and rates of de novo fatty acid biosynthesis were increased up to fourfold in Agpat2−/− mouse livers.80 mRNA and protein levels of monoacylglycerol acyltransferase isoform 1 (Mogat1) were increased by 25-fold to 48-fold and fivefold to sevenfold, respectively in the livers of these mice, which suggests that an alternative monoacylglycerol pathway for triglyceride biosynthesis is activated.80 Feeding a fat-free diet to Agpat2−/− mice reduced the level of triglycerides in the liver by ~50%, which suggests that both dietary and endogenous hepatic triglycerides might contribute to hepatic steatosis.80 Agpat2−/− mice also develop severe whole-body insulin resistance with reduced hepatic expression of insulin receptor (Insr), insulin receptor substrate 1 (Irs1) and insulin receptor substrate 2 (Irs2); these mice also have marked islet hypertrophy (which is probably a compensatory phenomenon).80
In vitro studies reveal that AGPAT2 mRNA expression is increased by 30-fold during adipocyte differentiation and that AGPAT2 enzymatic activity is required for triacylglycerol accumulation in mature adipocytes.91 The investigators of this study suggested that an AGPAT2-mediated metabolic pathway might be important for adipocyte differentiation.91 In another study comparing adipogenesis in muscle-derived multipotent cells from patients with type 1 CGL and healthy individuals, AGPAT2 was shown to have a role in regulating early stages of adipogenesis by modulating the lipome, as it altered the normal activation of the phosphatidylinositol 3-kinase/Akt and PPAR³ pathways.92 In type 1 CGL, phosphatidylinositol synthesis might also be reduced. Phosphatidylinositol has an important role in the metabolic actions of insulin such as glucose transport, especially in the adipose tissue (more so than in the liver or skeletal muscle).81
Type 2 CGL
Type 2 CGL, (OMIM #269700),93 is caused by mutations in BSCL2.The BSCL2 gene, located on chromosome 11q13, encodes a 398 amino acid transmembrane protein called seipin.35,94 Seipin has a CAAX motif at the C-terminus and a glycosylation site, NVS, at amino acid positions 88–90.94 Seipin function is complex and different mechanisms have been suggested regarding its role in lipid homeostasis. Seipin has been postulated to have an important role in lipid droplet assembly and in adipocyte differentiation.95 Furthermore, seipin is a transmembrane protein in the endoplasmic reticulum that concentrates at the junction with nascent lipid droplets and might, therefore, function in the fusion of lipid droplets (Figure 2).32,95 Seipin might be a docking protein to facilitate trafficking of lipids and/or proteins between the endoplasmic reticulum and the lipid droplet,95,96 and might also help to regulate lipid droplet biogenesis and adipogenesis.97,98 Seipin binds phosphatidic acid phosphatase LPIN1 (also known as lipin-1) and can also interact with AGPAT2.99,100 Consequently, this transmembrane protein might also be involved in phospholipids and triglyceride synthesis.101 Indeed, mutated forms of seipin that are truncated are unable to bind lipin-1, whereas missense mutants were able to interact with this protein.100
Nearly three-quarters of the mutations in BSCL2 reported in patients with type 2 CGL are null.35 However, approximately one-quarter have been missense mutations. So far, no phenotypic differences have been reported between those with null and missense mutations. All reported patients with CGL who are of Lebanese origin have a c.315_319delGTATC (p.Tyr106Cysfs*6) homozygous BSCL2 mutation.13 Type 2 CGL has also been reported in patients various ethnicities of including individuals originating from Europe, Mediterranean and Middle Eastern Arabs and Japanese.102
Patients with type 2 CGL have an almost total lack of body fat with both metabolically active and mechanical adipose tissue being absent (Figure 1b,g,h, Table 1).90 These patients have an increased prevalence of cardiomyopathy and mild mental retardation,13,15 and have lower median serum levels of leptin (0.01 ng/ml) and adiponectin (3.3 μg/ml) than healthy individuals (who had median serum levels of 4.6 ng/ml and 7.8 μg/ml for leptin and adiponectin, respectively).71 Teratozoospermia has been reported in a single patient with type 2 CGL, with sperm defects including abnormal head morphology and bundled sperm with two or more sperm connected to each other with large ectopic lipid droplets.57 Spastic gait due to upper motor neuron lesion has also been reported in three patients of a family from Pakistan with BSCL2 mutations.103 Four patients have also been reported with an early onset, fatal neurodegenerative syndrome with a CGL phenotype, with null mutations in BSCL2.104 Interestingly, gain-of-function heterozygous BSCL2 mutations located in the N-glycosylation motif can lead to distal hereditary motor neuropathy.105
Bscl2−/− mice have complete loss of white adipose tissue and display most metabolic complications of human type 2 CGL, including hyperinsulinaemia, insulin resistance and hepatic steatosis;106–108 however, these mice have lower plasma levels glucose and triglyceride upon fasting than wild-type mice, but have postprandial hypertriglyceridaemia.106 The liver of Bscl2−/− mice has impaired acute insulin signalling after a short term (4 h) fast, but this effect disappeared after 16 h of fasting.109 Interestingly, mice with an adipose-tissue-specific Bscl2 knock out have adipocyte hypertrophy with enlarged lipid droplets, reduced lipolysis, adipose tissue inflammation, progressive loss of white and brown adipose tissue, insulin resistance and hepatic steatosis.110 Conversely, rats with a homozygous null p.Leu20* mutation in Bscl2 have hypertriglyceridaemia alongside other metabolic abnormalities.111 The lack of hypertriglyceridaemia in Bscl2−/− mice was in contrast to the severe hypertriglyceridaemia seen in patients with type 2 CGL. However, data from the rat model is concordant with human data.111 Whether development of hypertriglyceridaemia is species dependent or related to the manner in which seipin-deficient animals were generated remains unclear.111
In in vitro experiments with Bscl2−/− murine embryonic fibroblasts (MEFs) and stromal vascular cells revealed failure of cells to terminally differentiate due to unbridled cyclic AMP (cAMP)-dependent protein kinase A (PKA)-activated lipolysis.107,112 This effect led to loss of lipid droplets and silencing of the expression of adipose-tissue-specific transcription factors. Such defects in differentiation could be rescued by inhibitors of lipolysis but not by a PPARγ agonist.107,112 However, thiazolidinediones can rescue the adipogenesis but not the alteration in lipolysis in Bscl2−/− MEFs.108 An increase in lipid droplet number, but a decrease in size, has been reported in in lymphoblastoid cell lines from 12 patients with type 2 CGL who had null BSCL2 mutations compared with control cell lines.113 These investigators also observed an increased proportion of saturated fatty acids in hepatic triglycerides and phosphatidylethanolamine at the expense of the corresponding monounsaturated fatty acids, which suggests a defect in acyl-CoA desaturase activity.
Type 3 CGL
Type 3 CGL, (OMIM #612526),114 is associated with mutations in CAV1, which is located on chromosome 7q31 and encodes caveolin 1.115 Caveolin 1 is a major component of caveolae, which are specialized plasma membrane microdomains appearing as 50–100 nm vesicular invaginations.116,117 CAV1 is ubiquitously expressed, but is particularly highly expressed in adipocytes, endothelial cells and fibroblasts (Figure 2).116 Caveolae maintain the integrity and function of the lipid droplets, are responsible for binding, transport and/or storage of fatty acids and cholesterol, augment insulin signalling and inhibit PKA signalling.118 The pool of caveolin 1 protein also affects phospholipid and surface protein composition of lipid droplets and enables the expansion of lipid droplet size.119 In adipose cell lines and mice, expression of Cav1 is crucial to increase the density of caveolae, to increase the ability of adipocytes to accommodate large lipid droplets and to promote cell expansion by increased glucose utilization.120 Caveolinproteins bind cholesterol, and the protein’s ability to move between cellular compartments helps control intracellular cholesterol fluxes.121
A homozygous nonsense mutation in CAV1 has been reported in a 20-year-old woman from Brazil with generalized lipodystrophy, short stature, functional megaoesophagus and hypocalcaemia, which was presumed to be secondary to vitamin D resistance.37 She had acanthosis nigricans, severe hypertriglyceridaemia, primary amenorrhoea, chronic diarrhoea, hepatic steatosis and splenomegaly, she had also developed diabetes mellitus at age 13 years. Metabolic adipose tissue was absent but mechanical adipose tissue and fat in the bone marrow were not affected by this particular lipodystrophy (Table 1).
Cav1−/− mice are hyperphagic with overt resistance to diet-induced obesity.118 With increasing age, a systemic decompensation in lipid accumulation occurs that results in dramatically smaller fat pads, reduced adipocyte cell diameter and a poorly differentiated and/or hypercellular white adipose parenchyma.118 Although serum levels of insulin, glucose and cholesterol are normal, Cav1−/− mice have severely elevated triglyceride and free fatty acid levels, especially in the postprandial state.118 Loss of fat in caveolin 1 deficiency might be due to autophagy, probably as the result of defective insulin and lipolytic response in adipocytes.119 Cav1−/− mice also have decreased levels of circulating total and high molecular weight adiponectin, and a reduced ability to change substrate use in response to feeding and fasting conditions.122 These mice also exhibit cardiovascular diseases, diabetes mellitus, breast cancer,123 atherosclerosis, pulmonary hypertension and pulmonary fibrosis.124,125 Indeed, heterozygous CAV1 mutations have been reported to be associated with pulmonary hypertension,126 as well as with rare neonatal onset lipodystrophy and progeroid syndrome in humans.22
Type 4 CGL
Type 4 CGL (OMIM #613327)127 is caused by mutations in PTRF, which is located at 17q21.2 and encodes PTRF (also known as cavin-1), an essential factor in the biogenesis of caveolae. Cavin-1 co-localizes with caveolin 1 in adipocytes (Figure 2).128 The presence of cavin-1 on the inside surface of caveolae stabilizes these structures, probably via interaction with the cell’s cytoskeleton.38 Cavin-1 also regulates adipocyte differentiation and is a determinant of adipose tissue expandability.129
About 30 patients with type 4 CGL owing to PTRF mutations have been identified.38,73,74 These patients might not have severe lipodystrophy at birth and might lose body fat progressively during infancy. In addition to having generalized lipodystrophy (Figure 1c), patients have congenital myopathy with high serum levels of creatine kinase.38,74,130 Other clinical features include osteopenia and distal metaphyseal deformation with joint stiffness, pyloric stenosis, atlanto-axial instability, percussion induced, local protracted muscle contractions (known as ‘mounding’; Figure 1h).73,130,131 Individuals with type 4 CGL also have a predisposition to serious arrhythmias such as catecholaminergic polymorphic ventricular tachycardia, long QT interval and sudden death.73,74 Nephrosis and transient immunoglobulin A deficiency has also been reported in a single patient.38 Mechanical and bone marrow fat has been reported to be well preserved in three patients (Table 1).130,131
Mice lacking Ptrf do not have morphologically detectable caveolae, in addition to a markedly diminished protein expression of all three caveolin isoforms.132 Ptrf−/− mice are viable and of normal weight but have high circulating levels of triglyceride and considerably reduced adipose tissue mass compared with wild-type controls, but retain muscle mass, have glucose intolerance and hyperinsulinaemia—characteristics that constitute a lipodystrophic phenotype.38,128 Ptrf−/− mice are also resistant to diet-induced obesity and have abnormal lipid metabolism in white and brown adipose tissue and liver.133 These lipolytic defects in white adipocytes were the result of impaired perilipin phosphorylation, and the reduced triglyceride accumulation was caused by decreased fatty acid uptake and incorporation as well as the virtual absence of insulin-stimulated glucose transport.133
Differential diagnosis of CGL
PPARG mutations
A 30-year-old female patient with generalized and infan-tile onset lipodystrophy and compound heterozygous mutations, c.413_416delAATG (p.Glu138Valfs*168) and c.490C>T (p.Arg164Trp), in PPARG has been identified.39 However, in contrast to a CGL phenotype, the patient had some facial adipose tissue. This individual developed hypertriglyceridaemia, eruptive xanthomata, acanthosis nigricans and marked hepatosplenomegaly at age 12 years, she had multiple episodes of pancreatitis during her teenage years and later developed severe insulin resistant diabetes mellitus, end-stage renal disease and irregular menstruation. Whether other individuals with homozygous or compound heterozygous PPARG mutations will also reveal similar phenotypes remains to be seen.
c-fos mutation
A single female patient with CGL and a homozygous point mutation in the promoter of the c-fos gene has been described;40 c-fos is an essential transcription factor required to initiate adipocyte differentiation. However, as both parents of the patient did not carry this variant, the likelihood of de novo homozygous variant in the patient seems highly improbable. The patient did not thrive, and had lipodystrophy and acanthosis at ~1 year of age. Other clinical features included hypertrichosis and hepatosplenomegaly. Interestingly, she developed clinically significant hypercholesterolaemia but with only mild hypertriglyceridaemia and impaired glucose tolerance at 5 years of age. She died at age 8 years during a hyperacute varicella infection.
Other lipodystrophies
CGL should be differentiated from acquired generalized lipodystrophy (AGL), atypical progeroid syndrome, Rabson-Mendenhall syndrome, Donohue syndrome and neonatal progeroid syndrome (Box 4). Correct diagnosis of CGL is important for predicting the future course of the disease, the complications and choosing therapeutic options.
Box 4 |. Differential diagnosis of CGL.
Acquired generalized lipodystrophy
Patients have normal adipose tissue distribution at birth but lose fat during early childhood. This loss mainly affects subcutaneous adipose tissue and visceral adipose tissue is preserved. Three subtypes of acquired generalized lipodystrophy exist: panniculitis-associated, autoimmune and idiopathic.32
Atypical progeroid syndrome
Patients have progeroid manifestations such as short stature, beaked nose, premature greying, partial alopecia, high-pitched voice and skin atrophy over the hands and feet. Some patients develop generalized lipodystrophy with variable loss of adipose tissue, metabolic complications and skeletal anomalies such as mandibular hypoplasia and mild acro-osteolysis. Atypical progeroid syndrome is the result of heterozygous de novo missense mutations in LMNA.10
Rabson-Mendenhall syndrome and Donohue syndrome
Patients have extreme insulin resistance, acanthosis nigricans and growth retardation during childhood. Rabson-Mendenhall syndrome and Donohue syndrome are caused by homozygous or compound heterozygous mutations in INSR.164
Neonatal progeroid syndrome
This syndrome is heterogeneous and manifests with generalized lipodystrophy that usually spares the buttocks, hands and feet; failure to thrive; hypotrichosis of the scalp hair, eyebrows and eyelashes; triangular face; micrognathia and variable clinical features like ear dysplasia, laryngomalacia and hyperpigmentation of the skin.165,166 Metabolic complications do not usually occur. Two subtypes have been reported to be due to de novo heterozygous mutations in FBN1 and CAV1.21,22
Abbreviation: CGL, congenital generalized lipodystrophy.
Genetic testing and counselling
A number of clinical and research laboratories offer genetic testing for CGL.134 The penetrance of CGL in homozygotes or compound heterozygotes is 100%; however, the severity of metabolic complications increases with age and is worse in female patients compared with male patients.13,15,135 Some patients have a CGL with no known genetic cause so far, which suggests further genetic heterogeneity and additional loci for this disease might exist. Next-generation sequencing might reveal these additional loci for such unexplained CGL in patients.13
As additional loci for CGL might exist, negative genetic tests should not rule out the diagnosis. In fact, patients with a CGL and their family members should be referred to research laboratories for further investigations and search for additional loci. Because CGL has a striking phenotype, screening family members who do not display the phenotype is unnecessary. Genetic counselling of patients and their families must also consider the fact that our understanding of the natural history of CGL and the complications of the disease is incomplete. Prenatal screening should also take into account parents’ ethical beliefs and interplay of environmental factors. Development of metabolic complications might be hastened in patients living in developed countries compared with those in underdeveloped or developing countries.
Metabolic complications in CGL
The major mechanism for developing metabolic complications in CGL is related to the near total lack of adipose tissue for efficient storage of circulating triglycerides (both from diet and endogenous synthesis in the liver).136 This lack of storage results in spillover of excess triglycerides to other organs such as the liver and skeletal muscles, which results in steatosis (Figure 3). This contention is supported by observations that hepatic steatosis is not improved in Agpat2−/− mice upon overexpression of Agpat2 in the liver.137 Furthermore, in mice with a liver-specific inactivation of Bscl2, no hepatic steatosis is seen, even under high-fat diet conditions.109 However, how hepatic steatosis and increased intramyocellular lipids induce insulin resistance has not been elucidated. In our own observations in Agpat2−/− mice, elevated levels of some types of phosphatidic acid (such as, C16:0/18:1 and C18:1/20:4) can increase hepatic glucose production.138
Figure 3 |.
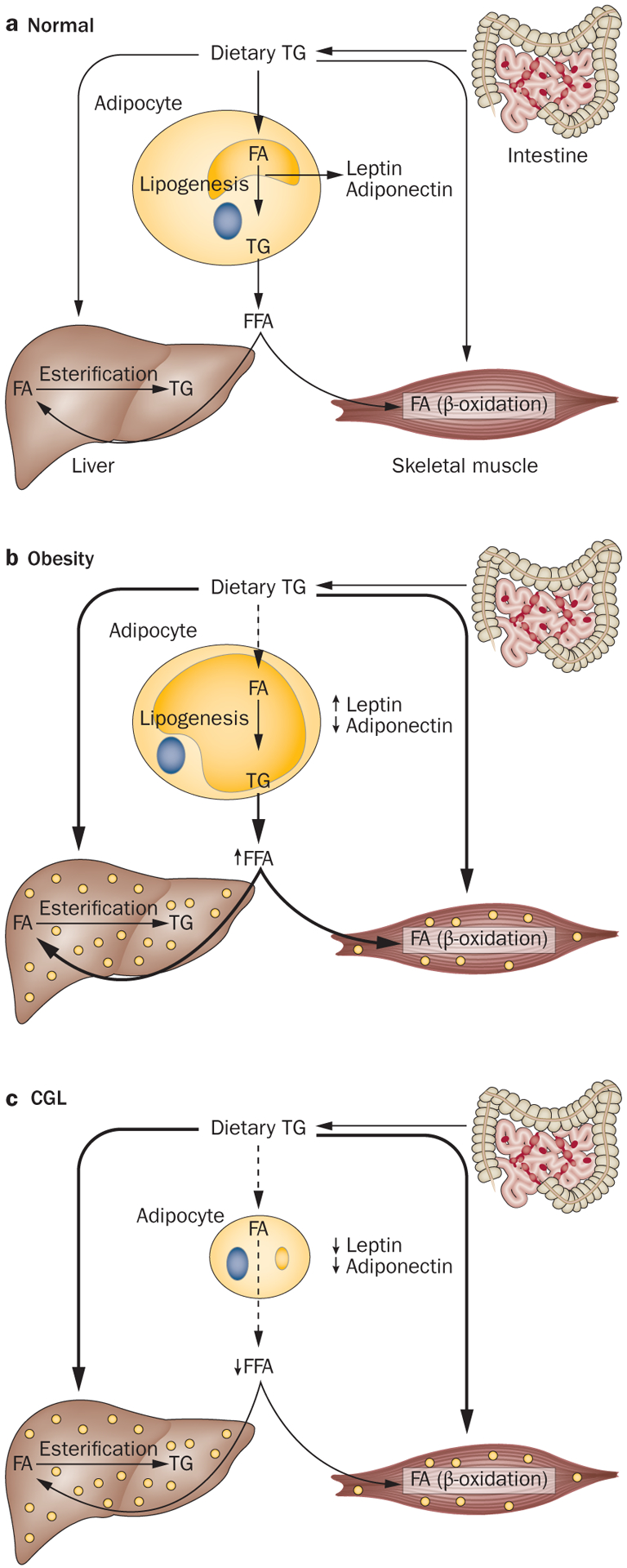
Mechanism of developing metabolic complications in obesity and CGL. a | Under normal conditions, dietary TGs are carried in chylomicrons and provide a source of FFA to the adipose tissue, liver and muscle for further storage and metabolism. Normally, adipocytes have plenty of capacity to store excess dietary TG and thus fewer TGs are directed to the liver and skeletal muscles. Skeletal muscles utilize FA for energy production (β-oxidation). During energy deprivation, the stored TGs are released from the adipocytes to deliver FA to the liver, skeletal muscle and other organs. b | In individuals with generalized or regional adiposity, adipocyte size enlarges and owing to the limited capacity to store more TG, dietary TG might be stored in sites such as the liver and skeletal muscles. Furthermore, lipolysis of excess TG stored in the adipocytes can also contribute to increased FA flux, which can contribute to TG storage in ectopic sites. Uptake of FFA and glucose depends on insulin action, thus in insulin resistance, uptake of FFA and glucose in these tissues is reduced, which might induce hypertriglyceridaemia, hyperglycaemia and hepatic steatosis. c | Patients with CGL lack adipocytes that can store TGs, which limits the disposal of excess dietary TG in the remaining adipocytes and consequently TGs are stored in ectopic sites such as the liver and skeletal muscles. This ectopic storage leads to severe insulin resistance and its complications. A lack of leptin induces hyperphagia, which further exacerbates ectopic storage of TG. Abbreviations: CGL, congenital generalized lipodystrophy; FA, fatty acid; FFA, free fatty acid; TG, triglyceride.
Similar mechanisms might be involved in patients with generalized or regional (for example, truncal) obesity, in whom circulating triglycerides are diverted to ectopic sites, owing to adipocytes already being overfilled with triglycerides. However, some unique metabolic derangements are observed in patients with CGL. No data have been published relating to circulating levels of free fatty acids (FFA) exclusively in patients with CGL. Some investigators have reported slightly high circulating levels of FFA in patients with CGL, acquired generalized lipodystrophy, familial partial lipodystrophy and acquired partial lipodystrophy.139,140 These patients had uncontrolled diabetes mellitus and severe hypertriglyceridaemia secondary to lipodystrophy, which can further affect circulating levels of FFA. However, both mouse models of Agpat2 and Bscl2 deficiency have low levels of FFA.80,109 By contrast, patients with obesity and metabolic derangements have high circulating levels of FFA.141 Profound hypoleptinaemia in patients with CGL further exacerbates metabolic derangements by inducing hyperphagia.
Management
The treatment of CGL is directed towards the specific symptoms that are apparent in each individual. Management requires the coordinated efforts of a team of specialists such as paediatricians, surgeons, cardiologists, endocrinologists and nutritionists. Diagnosis can cause anxiety and extreme psychological distress, and psychological support is recommended for the patients and their families. Special education might be necessary for individuals who also have an intellectual disability. Cosmetic surgery might be beneficial in improving appearance as the characteristic loss of adipose tissue in individuals with CGL cannot be reversed. Patients with CGL can also undergo reconstructive facial surgery. Bilateral gluteus maximus muscle flap advancement can be used for deformity of the buttocks.142
No evidence from clinical trials has been presented that supports specific nutritional recommendations or drug therapy (except the use of metreleptin, a recombinant analogue of human leptin) for the management of metabolic complications in patients with CGL.143 Dietary management is of extreme importance and individuals with CGL are encouraged to follow a high carbohydrate, low-fat diet.144,145 Such a diet can improve chylomicronaemia but might increase VLDL triglyceride levels. Importantly, children should still consume sufficient energy for proper growth and development and engage in regular exercise. Patients with type 4 CGL should avoid strenuous exercise and can be treated with β-adrenergic blockers and other anti-arrhythmic medications to prevent catecholaminergic polymorphic ventricular tachycardia. Some patients with type 4 CGL might even benefit from an implantable pacemaker or defibrillator.146 Whether patients with type 2 CGL and cardiomyopathy should avoid exercise is unclear.
Individuals with extreme hypertriglyceridaemia can be treated with fibric acid derivatives such as gemfi-brozil, fenofibrate, bezafibrate or long chain n-3 poly-unsaturated fatty acids from fish oils.14,144 In some patients, low-dose statins can be added to reduce non-HDL cholesterol levels. Niacin exacerbates insulin resistance and should be avoided. In women, oral estrogens are contraindicated because they might induce extreme hypertriglyceridaemia and acute pancreatitis.136
Drugs such as metformin and sulphonylureas are the first-line therapy for diabetes mellitus in patients with CGL.14 Preclinical studies suggest a potential efficacy of thiazolidinediones that still needs to be assessed in clinical trials.108,147 High doses of insulin are needed for most patients and some patients might experience difficulty injecting insulin because they lack subcutaneous fat in the abdomen or thighs. Patients with long-standing diabetes mellitus might have diabetic nephropathy and end-stage renal disease requiring haemodialysis and kidney transplantation.54
In multiple prospective studies, treatment with metreleptin, has been shown to be beneficial for improving metabolic complications, such as diabetes mellitus, hypertriglyceridaemia and hepatic steatosis in CGL.65,148–152 A study conducted at the NIH in the USA suggested that metreleptin has effects over 12 months in patients with CGL (n = 32).153 The dose of subcutaneous metreleptin injections varied from 0.06 mg/kg per day to 0.24 mg/kg per day. Mean HbA1c decreased by 2.2% from a baseline value of 9% in 20 patients and median triglyceride levels decreased by 60.7% from a baseline value of 5.11 mmol/l in 22 patients. Mean alanine aminotransferase levels decreased by 52.8 U/l from a baseline of 130.4 U/l and aspartate aminotransferase by 35.7 U/l from a baseline of 94.1 U/l in 31 patients with generalized lipodystrophy including both AGL and CGL. The most common adverse effects of metreleptin therapy were hypoglycaemia, headache, nausea, decreased weight and abdominal pain. Neutralizing antileptin antibodies might develop, which could lead to severe infections or loss of treatment effectiveness, but this possibility is rare.153 T cell lymphoma was reported in three patients with AGL while they were receiving metreleptin therapy.153 In one study, in which six male patients with CGL and one female patient with CGL (aged 2.4–13.6 years) were treated with metreleptin for 4 months, a 63% reduction in circulating levels of triglycerides, 30% increase in insulin sensitivity and 20% reduction in liver volume were seen.149 The only adverse effect reported was transient local inflammation at the site of injection. Recombinant leptin therapy has been studied in five patients, three with type 2 CGL and two patients with CGL but negative for mutations in AGPAT2 or BSCL2 (two male patients and three female patients, aged 15–33 years) in Japan over a period of 2–36 months.152 In this study, fasting glucose levels (mean ± SE, 9.55 ± 1.11 mmol/l to 6.66 ± 0.67 mmol/l) and triglyceride levels (7.91 ± 3.07 mmol/l to 2.94 ± 1.11 mmol/l) were both improved within 1 week. During long-term follow up of variable duration (between 18 months and 36 months), fasting plasma levels of glucose and HbA1c levels were well-controlled and insulin sensitivity improved. Antileptin antibodies were detected in two patients but did not demonstrate neutralizing activity.152 A significant reduction in HbA1c (from 10.4% to 7.1%), plasma triglyceride levels (by 76%) and hepatic enzymes (>65%) with metreleptin therapy has been reported in three male patients with type 2 CGL and four female individuals, two of whom had type 2 CGL, one had type 1 CGL and one an unknown subtype (these patients were aged between 23 months and 22 years), from Spain over a median period of 3 years.148 One patient had transient proximal lower limb myopathy, which was considered unrelated to their use of metreleptin, and no other major adverse effects were reported.148
Metreleptin therapy improves metabolic parameters mainly by its action in the hypothalamus to reduce appetite.154 This contention is supported by the observation that selective deletion of leptin receptors in hepatocytes did not prevent the positive metabolic actions of leptin in Agpat2−/− mice.155 Metreleptin therapy can ameliorate macroalbuminuria, microalbuminuria and hyperfiltration.48,152 An improvement in sex hormone profile with leptin therapy has been observed with metreleptin, which is likely to occur both from increasing insulin sensitivity and from restoring luteinizing hormone pul-satility.50 In female patients with type 1 or type 2 CGL, serum levels of free testosterone were decreased, sex hormone-binding globulin was increased and restoration of normal menstrual cycles seen; while in male patients, serum levels of testosterone and sex hormone-binding globulin increased.50 Interestingly, investigators have reported a young woman with type 1 CGL treated with metreleptin who experienced menarche, conceived normally and delivered a live born male baby.53 BMD is not affected by metreleptin therapy.63,152 Of note, only patients with type 1 CGL or type 2 CGL have been treated with leptin, because type 3 CGL and type 4 CGL are extremely rare. In 2014, the FDA approved the use of metreleptin, in addition to dietary changes, in patients with CGL and AGL.143 In 2013, the regimen was also approved for patients with lipodystrophy in Japan.156
Potential novel therapies
Patients with CGL have extreme hypertriglyceridaemia, which is challenging to treat with conventional therapeutic agents; consequently, novel therapies directed towards lowering triglyceride levels are needed. Loss-of-function variants in apolipoprotein C3 (encoded by APOC3) and angiopoietin-like protein 3 have been associated with markedly low serum levels of triglyceride, making these proteins targets for novel therapies.157–159 Several therapeutic agents are being developed that affect one of these targets. For example, a phase II clinical trial of antisense oligonucleotide that hybridizes with APOC3 mRNA in patients with hypertriglyceridaemia is currently underway;160 and a phase I clinical trial has started to assess the safety and tolerability of angiopoietin-like protein 3 antibody in patients with hypertriglyceridaemia.161 Furthermore, no therapy has been approved by any health-care regulatory body for managing severe hepatic steatosis in patients with CGL. In the FLINT trial,162 obeticholic acid, a selective farnesoid X receptor agonist, improved the histological features of nonalcoholic steatohepatitis in patients who did not have a lipodystrophy.163 Consequently, evaluating the safety and efficacy of obeticholic acid for improving hepatic steatosis in patients with CGL might be of interest. Finally, we hope that in the future, gene therapy might be available to treat and correct the specific molecular defects, such as AGPAT2 enzyme deficiency, in patients with CGL.
Conclusions
CGL is a rare heterogeneous (both genetically and phenotypically) autosomal recessive disorder characterized by near total, generalized lack of body fat and extreme muscularity since birth or soon thereafter. Patients develop metabolic complications, such as diabetes mellitus, hypertriglyceridaemia and hepatic steatosis later in life. Studies of patients and mouse models of CGL has provided insights into the molecular mechanisms of extreme insulin resistance and associated metabolic complications, and has important implications in understanding the pathogenesis of metabolic dysregulation in obesity. Patients with CGL have low FFA and leptin levels compared with patients who have obesity. Excess accumulation of triglyceride in the liver and skeletal muscle is the main mechanism of metabolic complication in CGL, which can be exacerbated by severe hypoleptinaemia. Patients with CGL still have limited treatment options; however, metreleptin replacement therapy, in addition to conventional therapy, has the potential to drastically improve metabolic complications and is now approved for use in the USA and Japan.
Key points.
Congenital generalized lipodystrophy (CGL) is a heterogeneous autosomal recessive disorder characterized by near total absence of body fat and extreme muscularity present at birth or soon thereafter
Patients with CGL are extremely hypoleptinaemic and are predisposed to develop metabolic complications, such as diabetes mellitus, hypertriglyceridaemia and hepatic steatosis
Four distinct subtypes of CGL exist: type 1 is associated with AGPAT2 mutations; type 2 is associated with BSCL2 mutations; type 3 is associated with CAV1 mutation; and type 4 is associated with PTRF mutations
Therapeutic options for patients with CGL include conventional lipid-lowering and antihyperglycaemic drugs, as well as metreleptin replacement therapy
Acknowledgments
A.G. acknowledges grant support from the NIH RO1 DK105448, CTSA Grant UL1 RR024982 and Southwest Medical Foundation. We thank P.-Y. Tseng for help with illustrations and mutational screening.
Footnotes
Competing interests
A.G. co-holds a patent regarding the use of leptin for treating human lipoatrophy and the method of determining predisposition to this treatment but receives no financial compensation. A.G. has received research grants from Aegerion, Astra-Zeneca, Bristol-Myers-Squibb and Pfizer and is a consultant for Amgen, Back Bay Life Sciences, Biomarin Pharmaceuticals, BioMedical Insights, Clearview Healthcare, Eli Lilly, Engage Health, Gerson Lehrman Group, Health Advances, Ipsen Pharmaceuticals, Intellisphere, Medscape and Tekmira. A.G. is also an advisory board member for AstraZeneca. N.P. declares no competing interests.
References
- 1.Garg A Acquired and inherited lipodystrophies. N. Engl. J. Med 350, 1220–1234 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Berardinelli W An undiagnosed endocrinometabolic syndrome: report of 2 cases. J. Clin. Endocrinol. Metab 14, 193–204 (1954). [DOI] [PubMed] [Google Scholar]
- 3.Seip M Lipodystrophy and gigantism with associated endocrine manifestations: a new diencephalic syndrome? Acta Paediatrica 48, 555–574 (1959). [PubMed] [Google Scholar]
- 4.Wiedemann HR Newly recognized congenital progeroid disorder. Am. J. Med. Genet 42, 857 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Rautenstrauch T, Snigula F & Wiedemann HR Neonatal progeroid syndrome (Wiedemann-Rautenstrauch). A follow-up study [German]. Klin. Padiatr 206, 440–443 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Dunnigan MG, Cochrane MA, Kelly A & Scott JW Familial lipoatrophic diabetes with dominant transmission. A new syndrome. Q. J. Med 43, 33–48 (1974). [PubMed] [Google Scholar]
- 7.Young LW et al. New syndrome manifested by mandibular hypoplasia, acroosteolysis, stiff joints and cutaneous atrophy (mandibuloacral dysplasia) in two unrelated boys. Birth Defects Orig. Artic. Ser 7, 291–297 (1971). [PubMed] [Google Scholar]
- 8.Eriksson M et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Sandre-Giovannoli A et al. Lamin A truncation in Hutchinson-Gilford progeria. Science 300, 2055 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Garg A et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J. Clin. Endocrinol. Metab 94, 4971–4983 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelosini C et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism 63, 1385–1389 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Capeau J et al. Human lipodystrophies: genetic and acquired diseases of adipose tissue. Endocr. Dev 19, 1–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwal AK et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab 88, 4840–4847 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Chan JL & Oral EA Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr. Pract 16, 310–323 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Van Maldergem L et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med. Genet 39, 722–733 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thauvin-Robinet C et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am. J. Hum. Genet 93, 141–149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyment DA et al. Mutations in PIK3R1 cause SHORT syndrome. Am. J. Hum. Genet 93, 158–166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chudasama KK et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am. J. Hum. Genet 93, 150–157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masotti A et al. Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am. J. Hum. Genet 96, 295–300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shastry S et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J. Clin. Endocrinol. Metab 95, E192–E197 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garg A & Xing C De novo heterozygous FBN1 mutations in the extreme C-terminal region cause progeroid fibrillinopathy. Am. J. Med. Genet. A 164A, 1341–1345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg A et al. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am. J. Med. Genet. A 10.1002/aimg.a.37115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prokocimer M, Barkan R & Gruenbaum Y Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 12, 533–543 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Ahmad Z, Zackai E, Medne L & Garg A Early onset mandibuloacral dysplasia due to compound heterozygous mutations in ZMPSTE24. Am. J. Med. Genet. A 152A, 2703–2710 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simha V, Agarwal AK, Oral EA, Fryns JP & Garg A Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J. Clin. Endocrinol. Metab 88, 2821–2824 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Agarwal AK et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet 87, 866–872 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farhan SM et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can. J. Cardiol 30, 1649–1654 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Saha B et al. A novel LMNA mutation causes altered nuclear morphology and symptoms of familial partial lipodystrophy (Dunnigan variety) with progeroid features. Mol. Syndromol 1, 127–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donadille B et al. Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Orphanet J. Rare Dis 8, 106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Payne F et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc. Natl Acad. Sci. USA 111, 8901–8906 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garg A Lipodystrophies. Am. J. Med 108, 143–152 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Garg A Clinical review: Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab 96, 3313–3325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nolis T Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J. Hum. Genet 59, 16–23 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Pardini VC et al. Leptin levels, β-cell function, and insulin sensitivity in families with congenital and acquired generalized lipoatrophic diabetes. J. Clin. Endocrinol. Metab 83, 503–508 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Magre J et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet 28, 365–370 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Agarwal AK et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet 31, 21–23 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Kim CA et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J. Clin. Endocrinol. Metab 93, 1129–1134 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Hayashi YK et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J. Clin. Invest 119, 2623–2633 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dyment DA et al. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur. J. Med. Genet 57, 524–526 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Knebel B et al. A mutation in the c-fos gene associated with congenital generalized lipodystrophy. Orphanet J. Rare Dis 8, 119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seip M & Trygstad O Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatrica Suppl 413, 2–28 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Westvik J Radiological features in generalized lipodystrophy. Acta Paediatrica Suppl 413, 44–51 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Garg A, Fleckenstein JL, Peshock RM & Grundy SM Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab 75, 358–361 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Seip M & Trygstad O Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr. Suppl 413, 2–28 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Garg A et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J. Clin. Endocrinol. Metab 84, 3390–3394 (1999). [DOI] [PubMed] [Google Scholar]
- 46.[No authors listed]. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 1–1975. N. Engl. J. Med 292, 35–41 (1975). [DOI] [PubMed] [Google Scholar]
- 47.Chandalia M, Garg A, Vuitch F & Nizzi F Postmortem findings in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab 80, 3077–3081 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Javor ED et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. J. Clin. Endocrinol. Metab 89, 3199–3207 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Upreti V, Dhull P, Patnaik SK & Kumar KV An unusual cause of delayed puberty: Berardinelli-Seip syndrome. J. Pediatr. Endocrinol. Metab 25, 1157–1160 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Musso C et al. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism 54, 255–263 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Lungu AO, Zadeh ES, Goodling A, Cochran E & Gorden P Insulin resistance is a sufficient basis for hyperandrogenism in lipodystrophic women with polycystic ovarian syndrome. J. Clin. Endocrinol. Metab 97, 563–567 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abel BS, Muniyappa R, Skarulis MC, Gorden P & Brown RJ Recombinant human leptin (metreleptin) administration augments nocturnal LH secretion in lipodystrophy patients Presented at The Endocrine Society’s 97th Annual Meeting & Expo (2015). [Google Scholar]
- 53.Maguire M, Lungu A, Gorden P, Cochran E & Stratton P Pregnancy in a woman with congenital generalized lipodystrophy: leptin’s vital role in reproduction. Obstet. Gynecol 119, 452–455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNally M et al. Successful renal transplantation in a patient with congenital generalized lipodystrophy: a case report. Am. J. Transplant 4, 447–449 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Miranda DM et al. Novel mutations of the BSCL2 and AGPAT2 genes in 10 families with Berardinelli-Seip congenital generalized lipodystrophy syndrome. Clin. Endocrinol. (Oxf.) 71, 512–517 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Gomes KB et al. Mutations in the Seipin and AGPAT2 genes clustering in consanguineous families with Berardinelli-Seip congenital lipodystrophy from two separate geographical regions of Brazil. J. Clin. Endocrinol. Metab 89, 357–361 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Jiang M et al. Lack of testicular seipin causes teratozoospermia syndrome in men. Proc. Natl Acad. Sci. USA 111, 7054–7059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunzell JD, Shankle SW & Bethune JE Congenital generalized lipodystrophy accompanied by cystic angiomatosis. Ann. Intern. Med 69, 501–516 (1968). [DOI] [PubMed] [Google Scholar]
- 59.Fleckenstein JL, Garg A, Bonte FJ, Vuitch MF & Peshock RM The skeleton in congenital, generalized lipodystrophy: evaluation using whole-body radiographic surveys, magnetic resonance imaging and technetium-99m bone scintigraphy. Skeletal Radiol 21, 381–386 (1992). [DOI] [PubMed] [Google Scholar]
- 60.Guell-Gonzalez JR et al. Bone lesions in congenital generalised lipodystrophy. Lancet 2, 104–105 (1971). [DOI] [PubMed] [Google Scholar]
- 61.Shinya T et al. Computed tomography findings of congenital generalized lipodystrophy: multiple nodular fatty liver and diffuse sclerosis of bones. Radiat. Med 25, 484–487 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Shirwalkar HU et al. Congenital generalized lipodystrophy in an Indian patient with a novel mutation in BSCL2 gene. J. Inherit Metab. Dis 31(Suppl. 2), S317–S322 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Christensen JD et al. Bone mineral content in patients with congenital generalized lipodystrophy is unaffected by metreleptin replacement therapy. J. Clin. Endocrinol. Metab 99 E1493–E1500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garg A, Chandalia M & Vuitch F Severe islet amyloidosis in congenital generalized lipodystrophy. Diabetes Care 19, 28–31 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Oral EA et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med 346, 570–578 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Diker-Cohen T, Cochran E, Gorden P & Brown RJ Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J. Clin. Endocrinol. Metab 100, 1802–1810 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garg A in Dyslipidemias: Pathophysiology, Evaluation and Management (ed. Garg A) 287–302 (Springer, 2015). [Google Scholar]
- 68.Rego AG et al. Cardiometabolic abnormalities in patients with Berardinelli-Seip syndrome [Portuguese]. Arq. Bras. Cardiol 94, 109–118 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Indumathi CK, Lewin S & Ayyar V Berardinelli Seip syndrome with insulin-resistant diabetes mellitus and stroke in an infant. Indian J. Endocrinol. Metab 15, S62–S64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haque WA, Shimomura I, Matsuzawa Y & Garg A Serum adiponectin and leptin levels in patients with lipodystrophies. J. Clin. Endocrinol. Metab 87, 2395–2398 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Antuna-Puente B et al. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-O-acyltransferase-2 deficiency. J. Clin. Endocrinol. Metab 95, 1463–1468 (2010). [DOI] [PubMed] [Google Scholar]
- 72.McDuffie JR et al. Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J. Clin. Endocrinol. Metab 89, 4258–4263 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajab A et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet 6, e1000874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shastry S et al. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am. J. Med. Genet. A 152A, 2245–2253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ardissone A et al. Novel PTRF mutation in a child with mild myopathy and very mild congenital lipodystrophy. BMC Med. Genet 14, 89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dwianingsih EK et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol. Genet. Metab 101, 233–237 (2010). [DOI] [PubMed] [Google Scholar]
- 77.UniProt. FOS - FOS Protein - Homo sapiens (human) [online], http://www.uniprot.org/uniprot/Q6FG41 (2015).
- 78.Online Mendelian Inheritance in Man©. #608594 Lipodystrophy, congenital generalized, type 1; CGL1 [online], http://omim.org/entry/608594 (2014).
- 79.Garg A & Agarwal AK Lipodystrophies: disorders of adipose tissue biology. Biochim. Biophys. Acta 1791, 507–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cortes VA et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab 9, 165–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Agarwal AK & Garg A Congenital generalized lipodystrophy: significance of triglyceride biosynthetic pathways. Trends Endocrinol. Metab 14, 214–221 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Takeuchi K & Reue K Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab 296, E1195–E1209 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leung DW The structure and functions of human lysophosphatidic acid acyltransferases. Front. Biosci 6, D944–D953 (2001). [DOI] [PubMed] [Google Scholar]
- 84.Agarwal AK, Barnes RI & Garg A Genetic basis of congenital generalized lipodystrophy. Int. J. Obes. Relat. Metab. Disord 28, 336–339 (2004). [DOI] [PubMed] [Google Scholar]
- 85.Agarwal AK & Garg A Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: upregulated in breast and cervical cancers. J. Lipid Res 51, 2143–2152 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agarwal AK Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol 23, 290–302 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Fu M et al. Mutations in Gng3lg and AGPAT2 in Berardinelli-Seip congenital lipodystrophy and Brunzell syndrome: phenotype variability suggests important modifier effects. J. Clin. Endocrinol. Metab 89, 2916–2922 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Magre J et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes 52, 1573–1578 (2003). [DOI] [PubMed] [Google Scholar]
- 89.Haque W, Garg A & Agarwal AK Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem. Biophys. Res. Commun 327, 446–453 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Simha V & Garg A Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy due to mutations in the AGPAT2 or Seipin genes. J. Clin. Endocrinol. Metab 88, 5433–5437 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Gale SE et al. A regulatory role for O-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J. Biol. Chem 281, 11082–11089 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Subauste AR et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes 61, 2922–2931 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Online Mendelian Inheritance in Man©. #269700 Lipodystrophy, congenital generalized, type 2; CGL2 [online], http://omim.org/entry/269700 (2014).
- 94.Agarwal AK & Garg A Seipin: a mysterious protein. Trends Mol. Med 10, 440–444 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Szymanski KM et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl Acad. Sci. USA 104, 20890–20895 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fei W et al. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol 180, 473–482 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang W et al. BSCL2/seipin regulates adipogenesis through actin cytoskeleton remodelling. Hum. Mol. Genet 23, 502–513 (2014). [DOI] [PubMed] [Google Scholar]
- 98.Cartwright BR et al. Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol. Biol. Cell 26, 726–739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Talukder MM, Sim MF, O’Rahilly S, Edwardson JM & Rochford JJ Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol. Metab 4, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sim MF et al. Analysis of naturally occurring mutations in the human lipodystrophy protein seipin reveals multiple potential pathogenic mechanisms. Diabetologia 56, 2498–2506 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sim MF et al. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol. Metab 2, 38–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wee K, Yang W, Sugii S & Han W Towards a mechanistic understanding of lipodystrophy and seipin functions. Biosci. Rep 3, e00141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schuster J et al. Exome sequencing circumvents missing clinical data and identifies a BSCL2 mutation in congenital lipodystrophy. BMC Med. Genet 15, 71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guillen-Navarro E et al. A new seipin-associated neurodegenerative syndrome. J. Med. Genet 50, 401–409 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Windpassinger C et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet 36, 271–276 (2004). [DOI] [PubMed] [Google Scholar]
- 106.Cui X et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet 20, 3022–3030 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Chen W et al. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol. Cell Biol 32, 1099–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Prieur X et al. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin-deficient mice. Diabetologia 56, 1813–1825 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Chen W, Zhou H, Saha P, Li L & Chan L Molecular mechanisms underlying fasting modulated liver insulin sensitivity and metabolism in male lipodystrophic Bscl2/Seipin-deficient mice. Endocrinology 155, 4215–4225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu L et al. Adipose-specific knockout of Seipin/Bscl2 results in progressive lipodystrophy. Diabetes 63, 2320–2331 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Ebihara C et al. Seipin is necessary for normal brain development and spermatogenesis in addition to adipogenesis. Hum. Mol. Genet 24, 4238–4249 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Chen W et al. Altered lipid metabolism in residual white adipose tissues of Bscl2 deficient mice. PLoS ONE 8, e82526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boutet E et al. Seipin deficiency alters fatty acid Δ9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie 91, 796–803 (2009). [DOI] [PubMed] [Google Scholar]
- 114.Online Mendelian Inheritance in Man©. #612526 Lipodystrophy, congenital generalized, type 3; CGL3 [online], http://omim.org/entry/612526 (2014).
- 115.Engelman JA, Zhang XL, Galbiati F & Lisanti MP Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, −2, and −3). Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett 429, 330–336 (1998). [DOI] [PubMed] [Google Scholar]
- 116.Stan RV Structure of caveolae. Biochim. Biophys. Acta 1746, 334–348 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Parton RG & Simons K The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol 8, 185–194 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Razani B & Lisanti MP Caveolin-deficient mice: insights into caveolar function human disease. J. Clin. Invest 108, 1553–1561 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blouin CM et al. Lipid droplet analysis in caveolin-deficient adipocytes: alterations in surface phospholipid composition and maturation defects. J. Lipid Res 51, 945–956 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Briand N et al. Caveolin-1 expression and cavin stability regulate caveolae dynamics in adipocyte lipid store fluctuation. Diabetes 63, 4032–4044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bosch M et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol 21, 681–686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Asterholm IW, Mundy DI, Weng J, Anderson RG & Scherer PE Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab 15, 171–185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Williams TM & Lisanti MP Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Cell Physiol 288, C494–C506 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Le Lay S & Kurzchalia TV Getting rid of caveolins: phenotypes of caveolin-deficient animals. Biochim. Biophys. Acta 1746, 322–333 (2005). [DOI] [PubMed] [Google Scholar]
- 125.Drab M et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452 (2001). [DOI] [PubMed] [Google Scholar]
- 126.Austin ED et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet 5, 336–343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Online Mendelian Inheritance in Man©. #613327 Lipodystrophy, congenital generalized, type 4; CGL4 [online], http://omim.org/entry/613327 (2013).
- 128.Liu L & Pilch PF A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J. Biol. Chem 283, 4314–4322 (2008). [DOI] [PubMed] [Google Scholar]
- 129.Perez-Diaz S et al. Polymerase I and transcript release factor (PTRF) regulates adipocyte differentiation and determines adipose tissue expandability. FASEB J 28, 3769–3779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rajab A, Heathcoate K, Joshi S, Jeffery S & Patton M Heterogeneity for congenital generalized lipodystrophy in seventeen patients from Oman. Am. J. Hum. Genet 110, 219–225 (2002). [DOI] [PubMed] [Google Scholar]
- 131.Simha V, Agarwal AK, Aronin PA, Iannaccone ST & Garg A Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am. J. Med. Genet. A 146A, 2318–2326 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu L et al. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab 8, 310–317 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ding SY et al. Pleiotropic effects of cavin-1 deficiency on lipid metabolism. J. Biol. Chem 289, 8473–8483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.GeneTests [online], http://www.genetests.org (2015).
- 135.Cortes VA et al. Divergent metabolic phenotype between two sisters with congenital generalized lipodystrophy due to double AGPAT2 homozygous mutations. A clinical, genetic and in silico study. PLoS ONE 9, e87173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Agarwal AK & Garg A Genetic basis of lipodystrophies and management of metabolic complications. Annu. Rev. Med 57, 297–311 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Agarwal AK et al. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2−/− gene lipodystrophic mice. J. Biol. Chem 286, 37676–37691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sankella S, Garg A, Horton JD & Agarwal AK Hepatic gluconeogenesis is enhanced by phosphatidic acid which remains uninhibited by insulin in lipodystrophic Agpat2−/− mice. J. Biol. Chem 289, 4762–4777 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Petersen KF et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Invest 109, 1345–1350 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Muniyappa R et al. Effects of leptin replacement therapy on pancreatic β-cell function in patients with lipodystrophy. Diabetes Care 37, 1101–1107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Boden G Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes 18, 139–143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Okada E, Iwahira Y & Maruyama Y Buttock deformity repair for congenital generalized lipodystrophy. Plast. Reconstr. Surg 95, 744–746 (1995). [DOI] [PubMed] [Google Scholar]
- 143.[No authors listed]. Metreleptin (Myalept): a leptin analog for generalized lipodystrophy. Med. Lett. Drugs Ther 57, 13–14 (2015). [PubMed] [Google Scholar]
- 144.Simha V & Garg A Inherited lipodystrophies and hypertriglyceridemia. Curr. Opin. Lipidol 20, 300–308 (2009). [DOI] [PubMed] [Google Scholar]
- 145.Handelsman Y et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr. Pract 19, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Napolitano C & Priori SG Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 4, 675–678 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Prieur X, Le May C, Magre J & Cariou B Congenital lipodystrophies and dyslipidemias. Curr. Atheroscler. Rep 16, 437 (2014). [DOI] [PubMed] [Google Scholar]
- 148.Araujo-Vilar D et al. Recombinant human leptin treatment in genetic lipodystrophic syndromes: the long-term Spanish experience. Endocrine 49, 139–147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Beltrand J et al. Metabolic correction induced by leptin replacement treatment in young children with Berardinelli-Seip congenital lipoatrophy. Pediatrics 120, e291–e296 (2007). [DOI] [PubMed] [Google Scholar]
- 150.Chan JL et al. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr. Pract 17, 922–932 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Safar Zadeh E et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J. Hepatol 59, 131–137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ebihara K et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J. Clin. Endocrinol. Metab 92, 532–541 (2007). [DOI] [PubMed] [Google Scholar]
- 153.FDA. Endocrinologic and metabolic drugs advisory committee briefing document: METRELEPTIN (BLA STN125390) [online], http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm377929.pdf. (2013). [Google Scholar]
- 154.Oral EA & Chan JL Rationale for leptin-replacement therapy for severe lipodystrophy. Endocr. Pract 16, 324–333 (2010). [DOI] [PubMed] [Google Scholar]
- 155.Cortes VA et al. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2−/− lipodystrophic mice independent of hepatocyte leptin receptors. J. Lipid Res 55, 276–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chou K & Perry CM Metreleptin: first global approval. Drugs 73, 989–997 (2013). [DOI] [PubMed] [Google Scholar]
- 157.Barter PJ et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med 357, 2109–2122 (2007). [DOI] [PubMed] [Google Scholar]
- 158.Okamoto H et al. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature 406, 203–207 (2000). [DOI] [PubMed] [Google Scholar]
- 159.Robciuc MR et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler. Thromb. Vasc. Biol 33, 1706–1713 (2013). [DOI] [PubMed] [Google Scholar]
- 160.US National Library of Medicine. ClinicalTrials.gov [online], http://clinicaltrials.gov/ct2/show/NCT01529424 (2014). [DOI] [PubMed]
- 161.US National Library of Medicine. ClinicalTrials.gov [online], http://clinicaltrials.gov/ct2/show/NCT01749878 (2015). [DOI] [PubMed]
- 162.US National Library of Medicine. ClinicalTrials.gov [online], http://clinicaltrials.gov/ct2/show/NCT01265498 (2014). [DOI] [PubMed]
- 163.Neuschwander-Tetri BA et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Musso C et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore) 83, 209–222 (2004). [DOI] [PubMed] [Google Scholar]
- 165.Pivnick EK et al. Neonatal progeroid (Wiedemann-Rautenstrauch) syndrome: report of five new cases and review. Am. J. Med. Genet 90, 131–140 (2000). [PubMed] [Google Scholar]
- 166.Hou JW Natural course of neonatal progeroid syndrome. Pediatr. Neonatol 50, 102–109 (2009). [DOI] [PubMed] [Google Scholar]