

Abstract
Cancer immunotherapy is revolutionizing oncology and has emerged as a promising strategy for the treatment of multiple cancers. Indoleamine 2,3-dioxygenase 1 (IDO1), an immune checkpoint, plays an important role in tumor immune escape through the regulation of multiple immune cells and has been regarded as an attractive target for cancer immunotherapy. Proteolysis Targeting Chimeras (PROTAC) technology has emerged as a new model for drug research and development for its advantageous mechanism. Herein, we reported the application of PROTAC technology in targeted degradation of IDO1, leading to the discovery of the first IDO1 PROTAC degrader 2c, which induced significant and persistent degradation of IDO1 with maximum degradation (dmax) of 93% in HeLa cells. Western-blot based mechanistic studies indicated that IDO1 was degraded by 2c through the ubiquitin proteasome system (UPS). Label-free real-time cell analysis (RTCA) indicated that 2c moderately improved tumor-killing activity of chimeric antigen receptor-modified T (CAR-T) cells. Collectively, these data provide a new insight for the application of PROTAC technology in tumor immune-related proteins and a promising tool to study the function of IDO1.
Key words: IDO1, PROTAC, Protein degradation, Ubiquitin proteasome system, Tumor immune escape
Graphical abstract
The first IDO1 PROTAC degrader 2c was designed and synthesized through conjugation of known IDO1 inhibitor epacadostat and CRBN ligand pomalidomide. 2c induced significant and persistent degradation of IDO1 with maximum degradation (Dmax) of 93% in HeLa cells, and moderately improved the activity of HER2 CAR-T cells.

1. Introduction
Indoleamine 2,3-dioxygenase 1 (IDO1), the key rate-limiting enzyme in l-tryptophan (l-Trp) metabolism through kynurenine pathway, which catalyzes the oxidation of l-Trp to N-formyl kynurenine (NFK)1. Initially, IDO1 was considered as a key immunomodulatory factor in embryonic immune tolerance to protect fetal from the maternal immune system2,3. Recently, a large number of reports have confirmed that IDO1 is overexpressed in a variety of cancers and plays an important role in cancer immune escape4,5. The immune evasion of cancer mediated by IDO1 is the result of comprehensive regulation of multiple immune cells through multiple mechanisms of action, the most important being the regulation of T cells6. Overexpressed IDO1 leads to depletion of l-Trp, which in turn activates the general control nonderepressible 2 kinase (GCN2) to inhibit proliferation and function of T cells. The metabolites of kynurenine pathway could also bind to and activate the aryl hydrocarbon receptor (AHR), leading to a range of biological effects including the proliferation of regulatory T cells, the conversion of dendritic cells (DCs) and macrophages to immunosuppressive phenotype. In addition, the metabolites, such as kynurenine, kynurenic acid and 3-hydroxykynurenine, could also directly inhibit the function of T cells7,8. Therefore, IDO1 has become an important target for cancer immunotherapy. Indeed, several highly potent and selective small molecule IDO1 inhibitors developed in pharmaceutical industry have advanced into the stage of clinical development for the treatment of human cancers (Fig. 1). Epacadostat (Epa) and BMS-986250 have shown significant antitumor efficacy in phase 2 clinical trials in combination with PD1/PD-L1 antibodies9.
Figure 1.

IDO1 inhibitors in clinical research and their characteristics9.
Proteolysis targeting chimeras (PROTAC) technology utilizes hetero-bifunctional chimeric molecule to mimic the endogenous protein homeostasis machinery, the ubiquitin proteasome system (UPS), to achieve post-translational selective degradation of target proteins in eukaryote10. This concept was first proposed by the pioneers Crews and coworkers11 in 2001, and has been developed as a new model for drug research and development. PROTAC molecule comprises a target protein binding ligand, an E3 ligase-binding ligand, and a linker to join them together. The two ends of PROTAC molecule could simultaneously bind to the target protein and E3 ligase, respectively, and pull them closer to form a relatively stable ternary complex, thereby inducing polyubiquitination of target protein and its subsequent recognition and degradation by proteasome through UPS12, 13, 14. Initially, due to the lack of small molecule E3 ligase ligands, early PROTAC molecules utilized E3 ligase affinity polypeptides as binding ligands for E3 ligase11,15. However, the low affinity, poor membrane permeability and stability of peptides extremely limited the application and development of PROTAC technology. Until 2008, PROTAC technology entered a rapid development period owe to the discovery of highly potential small molecule ligands for E3 ligase, such as MDM2, CRBN and VHL16, 17, 18, 19. To date, PROTAC technology has been widely used for the selective degradation of proteins, such as kinases20,21, transcription factors22,23, and epigenetic readers24, and has shown great therapeutic potential for related diseases25,26, especially cancers27, 28, 29, 30. However, PROTAC technology has not been applied in immune related proteins. There are several potential theoretical advantages for PROTAC molecule over traditional small molecule inhibitor, such as sub-stoichiometric catalytic activity, restoration of function of target protein demanding resynthesis and an additional layer of specificity, providing favorable pharmacology for PROTAC molecule31, 32, 33. Considering the important role of IDO1 in cancer immune escape and the unique mechanism of PROTAC technology, we sought to apply PROTAC technology for targeted degradation of IDO1.
2. Results and discussion
2.1. Design of IDO1 PROTAC degraders
As a hot target in the field of cancer immunotherapy, both academic and pharmaceutical institutions have great enthusiasm for IDO1, and several highly potent and selective small molecule IDO1 inhibitors have entered clinical trials (Fig. 1). In the design of IDO1 PROTAC, it is critical to identify suitable ligands for IDO1 and E3 ligase as well as the site for tethering. To obtain effective IDO1 PROTAC, we chose Epa, which is currently in clinical phase 3 as IDO1 ligand because of its highly activity and selectivity (Fig. 1)34,35. The co-crystal structure of IDO1 and Epa shows that the sulfamide of Epa is projected into solvent-exposed region. It was confirmed that a large number of substituents on this group were well tolerated, making it a potentially suitable site for tethering in the design of IDO1 PROTAC, which is exactly what other IDO1 inhibitors lack34,36. The active site of IDO1 is located in a deep cavity, the distance from amino at tethering site to outer interface is > 10 Å, which suggests that longer linker is required to avoid steric hindrance between IDO1 and the E3 ligase (Fig. 2A). Therefore, amphiphilic polyethylene glycol (PEG) was chosen as the linker for its good ductility in both hydrophobic and hydrophilic environments. On average, the CRBN E3 ligase based PROTACs with thalidomide or its derivatives as E3 ligase ligand come closer to “drug-like” space with their average H-bond donor and clogP values falling within the Lipinski's “rule-of-5” boundaries due to the distinct starting properties of E3 ligase warhead37. Therefore, we designed and synthesized 7 potential IDO1 PROTAC degraders with the highly potent and selective IDO1 inhibitor Epa as IDO1 ligand, pomalidomide (Pom) and lenalidomide that have been widely used in PROTAC design as E3 ligase ligand, and different length of PEG (2–9 PEG units) as linkers (Fig. 2B and Table 1). The synthetic route of IDO1 PROTACs was shown in Scheme 1.
Figure 2.

Design of IDO1 PROTAC degraders. (A) The co-crystal structure of IDO1 (green, PDB: 5WN8) and CRBN (pink, PDB: 5FQD) with their ligand Epa and lenalidomide respectively. Linker was attached via the solvent exposed site. (B) Structure of IDO1 PROTACs based on Epa and CRBN ligands.
Table 1.
The cytotoxicity and IDO1 inhibitory activity of IDO1 PROTAC degraders. Data are expressed as mean ± SD (n = 3). ND, not detected.
| Compd. | X | Linker | Cell-based potency IDO1 IC50 (μmol/L) | Cytotoxicity IC50 (μmol/L) | IDO1 degradation |
|---|---|---|---|---|---|
| 1a | CH2 | 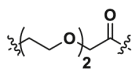 |
9.36 | >40 | ND |
| 1b | CH2 | 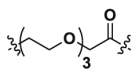 |
8.16 | >40 | ND |
| 1c | CH2 | 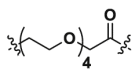 |
5.91 | >40 | ND |
| 2a | CO | 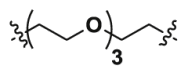 |
8.21 | 34.27 | ND |
| 2b | CO | 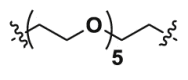 |
4.48 | 35.38 | ND |
| 2c | CO | 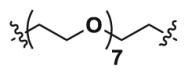 |
1.07 | 37.43 | YES |
| 2d | CO | 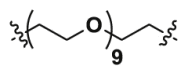 |
7.56 | >40 | ND |
| 2c-CH3 | CO | 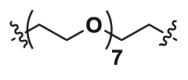 |
7.23 | >40 | ND |
| Pom-8PEG | CO | 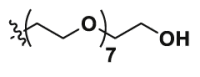 |
ND | >40 | ND |
| Epa-8PEG | 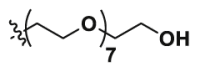 |
3.82 | >40 | ND | |
| Epa | 0.036 | >40 | ND |
Scheme 1.

Synthetic route of IDO1 PROTACs.
2.2. The cytotoxicity and IDO1 inhibitory activity of IDO1 PROTAC degraders
The cytotoxicity was detected before evaluating the degradation ability of IDO1 PROTAC degraders. MTT assay revealed that the cytotoxicity of newly synthesized 7 IDO1 PROTAC degraders was fairly weak with IC50 > 30 μmol/L (Table 1 and Supporting Information Fig. S1), indicating that the expression or activity of IDO1 would not be interfered by these compounds. It was reported that a phthalimide bearded “MDM2 PROTAC degrader” worked as a molecular glue, inducing degradation of GSPT1 to achieve its potent anticancer activity38. Therefore, 7 IDO1 PROTAC degraders were not suspected GSTP1 degraders as the degradation of GSTP1 usually leads to strong anti-proliferative phenotype in a variety of tumor cell lines39. Then, HeLa cells-based IDO1/kynurenine analysis illustrated that the inhibitory activity of 7 IDO1 PROTAC degraders significantly decreased compared to the parent compound (Table 1 and Supporting Information Fig. S2). Considering the depth of the binding site, the initial length of linker was set to 2 PEG units, and then gradually increased to 9 PEG units. In general, there is an optimal linker for a PROTAC degrader, which usually exerts higher affinity for target protein and capacity of degradation. Interestingly, with the extension of the linker from 2 to 9 PEG units, the IDO1 inhibitory activity increased first and then decreased. The most potent IDO1 PROTAC degrader 2c contains 7 PEG units. While the potency of its analogs with the same linker as 2c, Epa-8PEG (Epa + linker) and 2c-CH3 (inactive form for CRBN of 2c) decreased significantly. These data indicated that 7 PEG units might be preferred for IDO1 PROTAC and 2c might be a potent IDO1 degrader. However, the molecular enlargement tends to inevitably lead to a significant decrease in affinity for IDO1, which is a great concern in design of PROTAC, especially for those proteins with deep binding pocket.
2.3. The degradation of IDO1 by PROTAC degraders
As is known to all, IFN-γ is an efficient IDO1 inducing factor and can significantly upregulate the expression of cellular IDO1 via IFN-γ-STAT1/3 pathway5. Therefore, the levels of mRNA and protein of IDO1 in HeLa cells treated with different concentration of IFN-γ with 2c were measured by qPCR and Western blot assay to explore appropriate IFN-γ concentration and quantify the degradative capacity of IDO1 degraders in mRNA and protein level. After treatment with IFN-γ for 24 h, the IDO1 mRNA markedly increased as the concentration of IFN-γ increased, and tended to be saturated at 10 ng/mL of IFN-γ (Fig. 3B). IDO1 protein also significantly increased after stimulation by IFN-γ (Fig. 3A). Compound 2c (10 μmol/L) notably decreased IDO1 level induced by IFN-γ (5 ng/mL), and the corresponding IDO1 mRNA was 105 times than that of unstimulated HeLa cells and even higher than that of the internal reference GAPDH. However, the amount of IDO1 induced by higher concentrations (10 and 20 ng/mL) of IFN-γ exceeded the capability of 2c mediated UPS-dependent degradation. These results suggested that there might be saturation in the degradation of proteins induced by PROTAC strategy via UPS, which is worthy of attention in PROTAC related research, especially for those proteins with extremely high expression level.
Figure 3.

The degradation of IDO1 by PROTAC degraders. (A) Degradation of IDO1 by 2c under different concentration of IFN-γ. HeLa cells were treated with 2c and IFN-γ for 24 h. (B) IFN-γ upregulate the expression of cellular IDO1 mRNA. Data are expressed as the mean ± SD (n = 3), error was shown as shaded area. (C) Capacity of degradation of degraders. 10 μmol/L of degraders and 5 ng/mL of IFN-γ were incubated with HeLa cells for 24 h. As determined by Western blot, almost complete degradation of IDO1 was observed in 2c. (D) Determination of degradative ability of 2c. Compound 2c and IFN-γ (5 ng/mL) were incubated with HeLa cells for 24 h, and a significant dose-dependent degradation was observed. (E) The determination of degradative ability of 2c by immunoblot band intensity. Data are expressed as the mean ± SD (n = 3).
Then, the degradation activity of 7 IDO1 PROTAC degraders was determined, as expected, only 2c induced significant degradation of IDO1 (Fig. 3C), indicating that 7 PEG units was preferred linker for IDO1 PROTAC in aspect of length. The degradative potency of 2c was measured, and the DC50 of 2c was found to be 2.84 μmol/L and a maximum degradation (Dmax) of 93% was achieved based on the immunoblot band intensity (Fig. 3D and E).
2.4. Compound 2c induced persistent degradation of IDO1 through UPS
As an IDO1 degrader based on PROTAC technology, depletion of IDO1 protein by 2c should require its binding to IDO1 and CRBN E3 ligase simultaneously through its Epa and Pom segments to introduce IDO1 into UPS. Accordingly, we explored the effect of 2c related control compounds 2c-CH3, Epa-8PEG and Pom-8PEG on the degradation of IDO1 (Fig. 4A), only 2c induced degradation of IDO1 (Fig. 4B). It was expected that excessive Epa or Pom, proteasome inhibitor MG132 and neddylation inhibitor MLN4924, could abolish the degradation of IDO1 induced by 2c. Western blot analysis showed that the degradation of IDO1 was significantly blocked after pretreatment with excessive Epa, lenalidomide, MG132 or MLN4924 (Fig. 4C). In aspect of kinetics, the expression of IDO1 was evident after treatment with IFN-γ (5 ng/mL) for 4 h and continuous increased until 48 h, while 2c induced sustained and almost complete IDO1 degradation until 48 h (Fig. 4D and E). The above results demonstrated that 2c is an effective IDO1 degrader that hijacks IDO1 to CRBN E3 ligase to introduce IDO1 into UPS and eventually achieve ubiquitination and degradation.
Figure 4.

Compound 2c is an effective and persistent IDO1 degrader. (A) Structures of 2c, 2c-CH3, Epa-8PEG and Pom-8PEG. (B) Effect of control compounds on IDO1 degradation. HeLa cells were treated with 5 μmol/L of 2c or control compounds and IFN-γ (5 ng/mL) for 24 h. (C) Confirmation of UPS based mechanism in driving IDO1 degradation upon 2c treatment. HeLa cells were pre-treated with 1 μmol/L of Epa, Pom, MG132 or MLN4924 for 2 h, then 2c (5 μmol/L) and IFN-γ (5 ng/mL) were incubated with HeLa cells for 24 h. (D) and (E) HeLa cells were treated with or without 2c (5 μmol/L) in the presence of IFN-γ (5 ng/mL) for 48 h. Compound 2c leads to efficient and sustained degradation of IDO1.
2.5. Compound 2c is an effective IDO1 PROTAC degrader
Additionally, immunofluorescence and flow cytometry analysis were conducted to visualize and further confirm the degradation of IDO1. After stimulation with IFN-γ, the fluorescence signal of IDO1 was significantly enhanced, which indicating that the expression of IDO1 in HeLa cells significantly increased (Fig. 5A). While after treatment with 2c, the intracellular IDO1 fluorescence signal dramatically decreased to the level similar to that of control. The results of flow cytometry also showed that IFN-γ induced a significant increase of IDO1, and 2c decreased IDO1 to a level comparable to the control (Fig. 5B). These data further confirmed that 2c is an effective IDO1 degrader.
Figure 5.

Compound 2c induced significant degradation of IDO1. (A) HeLa cells were pre-treated with 2c (5 μmol/L) and INF-γ (5 ng/mL) for 24 h before immunostaining. Scale bar, 20 μm. (B) The results of flow cytometry analysis that IFN-γ induced a significant increase of IDO1, and 2c reduced IDO1 to a level comparable to the control. Scale bar: 20 μm.
2.6. Compound 2c moderately improved the activity of HER2 chimeric antigen receptor-modified T (CAR-T)
CAR-T cell therapy has shown remarkable antitumor efficacy for the treatment of tumors, especially for hematological malignancies. Whereas, tumor cells, solid tumors especially, utilize IDO1 to inhibit the proliferation and function of T cells, cause immune tolerance through multiple ways. It has been proved that the therapeutic strategy to inhibit IDO1 can delay tumor growth. Compared with the mechanism of traditional small molecule inhibitors, PROTAC degraders modulate the biological function of target protein by degrading the protein through UPS. This strategy has obvious kinetic advantages because cancer cells need to undergo more processes to restore biological function by re-synthesizing proteins. Herein, we employed label-free real-time cell analysis (RTCA) system to explore the effects of different IDO1 inhibition strategies (including IDO1 degrader 2c and traditional inhibitor Epa to act on HeLa cells, respectively) on the killing activity of HER-2 specific CAR-T cells for HeLa (Supporting Information Fig. S3, HER-2 positive) cells. According to real-time monitoring results, it was found that all the cell indexes of HeLa cells treated with HER-2 CAR-T cells decreased. It's particularly noteworthy that the cells treated with IDO1 degrader 2c, whose decline trend of cell index was more obvious than that of the control group and the Epa group (Fig. 6). These data demonstrated that IDO1 degrader 2c combined with CAR-T cells can improve the tumor-killing activity of HER-2 CAR-T cells, further indicating that PROTAC strategy based IDO1 degrader 2c has a kinetic advantage over IDO1 inhibitor.
Figure 6.

Compound 2c moderately improved the activity of HER2 CAR-T. HeLa cells were treated with IFN-γ (5 ng/mL)+2c (5 μmol/L) or Epa (5 μmol/L) for 24 h, then the medium was replace to wash out IFN-γ and 2c, and HER2 CAR-T cells were add. Data are expressed as the mean ± SD (n = 3).
3. Conclusions
In summary, we successfully applied PROTAC technology to the targeted degradation of IDO1. We designed, synthesized and screened the first potent IDO1 degrader 2c, with a maximum degradation (Dmax) of 93%. Due to the deep binding pocket in IDO1, longer linker is necessary for IDO1 degrader to avoid steric hindrance, however, it inevitably leads to a significant decrease in affinity. This suggests that flexible and short linker is preferred in PROTAC degrader. Although 2c was an efficient IDO1 degrader (DC50 = 2.84 μmol/L), the amount of IDO1 protein induced by excessive IFN-γ exceeded the capability of 2c mediated UPS-dependent degradation. These results indicated that there might be saturation in the degradation of proteins induced by PROTAC strategy via UPS. Thus, PROTAC strategy-based degrader might be inapplicable for those proteins with extremely high expression level. Following immunofluorescence and flow cytometry, analysis further confirmed that 2c induced sufficient degradation of IDO1. RTCA analysis indicated that 2c moderately improved the tumor-killing activity of H ER2 CAR-T cells. These results provide new insight for the application of PROTAC technology in tumor immune related proteins. In addition, 2c could also serve as a biological probe to study the function of IDO1 in tumor immune escape.
4. Experimental
4.1. Chemistry
All materials were obtained from commercial suppliers and used without further purification. All solvents were dried according to the standard methods prior to use. The melting points were determined with an SGWX-4 melting point apparatus (Shanghai optical instrument factory, Shanghai, China). 1H NMR and 13C NMR spectra were measured on a Bruker AV-400 (Bruker Inc., Karlsruhe, Germany) NMR spectrometer. HRMS spectral data were recorded on a Bruker Daltonics Bio TOF mass spectrometer (Bruker Inc., Karlsruhe, Germany). High-performance liquid chromatography (HPLC) analysis was performed with a Waters Alliance 2695e equipped with PDA 2998 detector (Waters Inc., Milford, MA, USA). The synthesis and characterization of linkers for IDO1 PROTACs, Epa and its derivatives could be found in Supporting Information.
4.1.1. Synthesis and characterization of IDO1 PROTACs
4.1.1.1. Synthesis of compound 16
Lenalidomide (500 mg, 1.93 mmol), compound 5 (4 mmol), and DMAP (488 mg, 4 mmol) were dissolved in N,N-dimethylformamide (DMF, 10 mL) and stirred for 5 h at 60 °C. DMF was removed by evaporation and the residue was purified (DCM/MeOH = 100:1) to give the Cl-PEGs-Lenalidomide. Sodium iodide (628 mg, 9.65 mmol) was added to the acetonitrile solution of Cl-PEGs-Lenalidomide and refluxed for 24 h. The acetonitrile was removed by evaporation, the residue was re-dissolved in ethyl acetate, and was washed with saturated brine. The organic phase was concentrated to afford compound 15 without further purification.
16a: Yield 536.73 mg, 54%. 1H NMR (400 MHz, DMSO-d6), δ 11.00 (s, 1H), 9.67 (s, 1H), 7.75 (dd, J = 7.7, 1.3, 1H), 7.66–7.44 (m, 2H), 5.14 (dd, J = 13.3, 5.1, 1H), 4.48–4.24 (m, 2H), 4.15 (s, 2H), 3.78–3.61 (m, 8H), 3.00–2.84 (m, 1H), 2.60 (dt, J = 17.2, 2.8, 1H), 2.37 (qd, J = 13.2, 4.5, 1H), 2.02 (dtd, J = 12.5, 5.2, 2.2, 1H). MS (ESI), Calcd. for C19H22N3O6I [M+H]+: 516.1, Found: 516.2.
16b: Yield 616.06 mg, 57%.1H NMR (400 MHz, DMSO-d6), δ 11.00 (s, 1H), 9.66 (s, 1H), 7.75 (dd, J = 7.7, 1.3, 1H), 7.60–7.47 (m, 2H), 5.14 (dd, J = 13.3, 5.1, 1H), 4.53–4.26 (m, 2H), 4.14 (s, 2H), 3.76–3.48 (m, 10H), 3.27 (t, J = 6.5, 2H), 2.92 (ddd, J = 17.2, 13.6, 5.4, 1H), 2.65–2.56 (m, 1H), 2.37 (qd, J = 13.2, 4.4, 1H), 2.05–1.97 (m, 1H). MS (ESI), Calcd. for C21H26 N3O7I [M+H]+: 560.1, Found: 560.1.
16c: Yield 606.17 mg, 52%. 1H NMR (400 MHz, DMSO-d6), δ 11.02 (s, 1H), 9.69 (s, 1H), 7.74 (dd, J = 7.7, 1.3, 1H), 7.62–7.42 (m, 2H), 5.15 (dd, J = 13.3, 5.1, 1H), 4.57–4.26 (m, 2H), 4.14 (s, 2H), 3.75–3.59 (m, 8H), 3.58–3.41 (m, 8H), 2.92 (ddd, J = 17.2, 13.6, 5.5, 1H), 2.67–2.56 (m, 1H), 2.37 (qd, J = 13.3, 4.5, 1H), 2.05–1.95 (m, 1H). MS (ESI), Calcd. for C23H30N3O8I [M+H]+: 604.1, Found: 604.3.
4.1.1.2. Synthesis of compound 1
Compound 16 (0.2 mmol) was dissolved in anhydrous DMF (3 mL), compound 14 (71.6 mg, 0.2 mmol) and triethylamine (20.2 mg, 0.2 mmol) were added. The mixture was stirred at 60 °C overnight. DMF was removed by evaporation and the residue was purified (DCM/MeOH/NH3·H2O = 100:5:0.5) to give the compound 1.
1a: Yield 67.12 mg, 45%, m.p. 156.3–160.1 °C, HPLC >95%.1H NMR (400 MHz, CD3OD), δ 7.75–7.62 (m, 2H), 7.52 (t, J = 7.7, 1H), 7.10 (dd, J = 6.0, 2.7, 1H), 7.02 (t, J = 8.7, 1H), 6.82 (ddd, J = 8.9, 4.1, 2.7, 1H), 5.15 (dd, J = 13.3, 5.2, 1H), 4.50 (s, 2H), 4.21 (d, J = 7.6, 2H), 3.88–3.60 (m, 7H), 3.40 (t, J = 6.2, 2H), 2.98–2.81 (m, 4H), 2.76 (ddd, J = 17.6, 4.7, 2.4, 1H), 2.46 (qd, J = 13.2, 4.7, 1H), 2.16 (dtd, J = 12.8, 5.3, 2.4, 1H). 13C NMR (100 MHz, DMSO-d6), δ 172.24, 169.38, 167.72, 156.10, 155.41, 153.03, 146.92, 146.82, 140.33, 139.71, 138.41, 138.38, 136.69, 132.55, 125.24, 121.92, 121.85, 117.89, 116.48, 111.15, 109.69, 107.59, 70.46, 70.31, 70.25, 70.09, 69.35, 49.61, 47.79, 44.08, 42.19, 42.09, 31.57, 27.04, 21.85, 21.76, 14.53. HR-MS Calcd. for C30H33BrFN9O8 [M+H]+: 746.1619, Found: 746.1719.
1b: Yield 66.36 mg, 42%, m.p. 148.6–151.9 °C, HPLC >95%. 1H NMR (400 MHz, CD3OD), δ 7.70 (ddd, J = 10.1, 7.7, 1.0, 2H), 7.54 (t, J = 7.7, 1H), 7.19–6.97 (m, 2H), 6.84 (ddd, J = 8.8, 4.1, 2.7, 1H), 5.17 (dd, J = 13.3, 5.2, 1H), 4.51 (s, 2H), 4.24 (s, 2H), 3.83–3.47 (m, 12H), 3.15 (t, J = 6.0, 2H), 3.03 (dd, J = 6.0, 4.3, 2H), 2.94–2.83 (m, 1H), 2.77 (ddd, J = 17.6, 4.7, 2.4, 1H), 2.52–2.39 (m, 1H), 2.24–2.12 (m, 1H). 13C NMR (100 MHz, CD3OD), δ 173.27, 172.28, 170.14, 169.77, 169.28, 168.43, 167.90, 156.41, 155.74, 154.01, 146.82, 146.76, 140.57, 139.48, 137.67, 135.83, 135.70, 132.59, 132.49, 126.37, 122.50, 122.43, 116.90, 116.71, 115.52, 115.29, 110.66, 110.55, 110.13, 109.91, 107.50, 107.28, 70.27, 70.23, 70.20, 70.16, 70.14, 70.12, 70.10, 69.89, 69.24, 69.09, 43.15, 41.90, 30.82, 30.34, 29.34, 23.74, 23.56, 22.34. HR-MS Calcd. for C32H37BrFN9O9 [M+H]+: 790.1822, Found: 790.1791.
1c: Yield 66.72 mg, 40%, m.p. 142.8–143.9 °C, HPLC >95%. 1H NMR (400 MHz, CD3OD), δ 7.78–7.64 (m, 2H), 7.54 (t, J = 7.7, 1H), 7.11 (dd, J = 6.0, 2.7, 1H), 7.03 (t, J = 8.7, 1H), 6.83 (ddd, J = 8.8, 4.1, 2.7, 1H), 5.16 (dd, J = 13.3, 5.1, 1H), 4.50 (s, 2H), 4.19 (s, 2H), 3.81–3.44 (m, 17H), 2.95–2.76 (m, 5H), 2.51–2.39 (m, 1H), 2.17 (dtd, J = 12.9, 5.3, 2.4, 1H). 13C NMR (100 MHz, CD3OD), δ 173.20, 170.19, 169.80, 169.29, 168.44, 167.90, 155.73, 154.04, 146.78, 146.73, 140.60, 139.47, 137.62, 135.83, 135.70, 132.46, 126.44, 122.56, 122.49, 116.88, 116.70, 115.51, 115.28, 110.69, 110.59, 110.12, 109.89, 107.51, 107.29, 70.22, 70.16, 70.09, 70.06, 69.85, 69.23, 68.46, 42.71, 41.87, 31.34, 30.81, 30.34, 29.38, 23.66, 22.33, 13.03. HR-MS Calcd. for C34H41BrFN9O10 [M+H]+: 834.2144, Found: 834.2029.
4.1.1.3. Synthesis of compound 19
Compounds 17a and 17b were synthesized according to the reported method40. Compound 17 (2 mmol) was dissolved in DMF (10 mL), compound 11 (3 mmol) and triethylamine (202 mg, 2 mmol) were added. The mixture was stirred at 90 °C overnight. DMF was removed by evaporation, the residue was re-dissolved in ethyl acetate and washed with saturated brine. The organic layer was concentrated and purified on silica gel column (DCM/MeOH = 100:1) to give compound 18. Then, compound 18 was treated with excess toluene sulfonyl chloride in the presence of DIPEA in CH2Cl2. After stirring overnight at room temperature, the mixture was washed with 1 mol/L HCl, saturated sodium bicarbonate and saturated brine. The solvent was evaporated to give compound 19 without further purification.
Pom-8PEG: Yield 725.32 mg, 58%, m.p. 113.1–116.9 °C, HPLC >95%. 1H NMR (400 MHz, DMSO-d6), δ 11.08 (s, 1H), 7.58 (dd, J = 8.6, 7.1, 1H), 7.15 (d, J = 8.6, 1H), 7.04 (d, J = 7.0, 1H), 6.60 (t, J = 5.8, 1H), 5.06 (dd, J = 12.9, 5.4, 1H), 4.55 (t, J = 5.4, 1H), 3.70–3.41 (m, 32H), 2.89 (ddd, J = 17.4, 14.0, 5.4, 1H), 2.68–2.49 (m, 2H), 2.04 (ddt, J = 13.0, 5.6, 3.3, 1H). 13C NMR (100 MHz, DMSO-d6), δ 173.24, 170.50, 169.41, 167.76, 146.90, 136.69, 132.57, 117.92, 111.14, 109.73, 72.81, 70.32, 70.28, 70.25, 69.96, 69.37, 60.69, 55.37, 49.05, 42.20, 31.45, 22.62. MS (ESI), HR-MS Calcd. for C29H43N3O12 [M+H]+: 626.2847, Found: 626.2947.
19a: Yield 688.9 mg, 57%. 1H NMR (400 MHz, DMSO-d6), δ 11.09 (s, 1H), 7.84–7.71 (m, 2H), 7.58 (dd, J = 8.6, 7.1, 1H), 7.47 (d, J = 8.1, 2H), 7.21–6.99 (m, 2H), 6.59 (s, 1H), 5.05 (dd, J = 12.9, 5.4, 1H), 4.10 (dd, J = 5.5, 3.3, 2H), 3.75–3.42 (m, 14H), 2.96–2.82 (m, 1H), 2.64–2.52 (m, 2H), 2.41 (s, 3H), 2.06–1.96 (m, 1H). MS (ESI), Calcd. for C28H33N3O10S [M+H]+: 604.2, Found: 604.3.
19b: Yield 637 mg, 46%. 1H NMR (400 MHz, DMSO-d6), δ 11.09 (s, 1H), 7.87–7.72 (m, 2H), 7.59 (dd, J = 8.5, 7.0, 1H), 7.48 (d, J = 8.0, 2H), 7.15 (d, J = 8.6, 1H), 7.04 (d, J = 7.0, 1H), 6.60 (t, J = 5.9, 1H), 5.06 (dd, J = 12.9, 5.4, 1H), 4.18–4.02 (m, 2H), 3.71–3.43 (m, 22H), 2.89 (ddd, J = 17.3, 14.0, 5.4, 1H), 2.66–2.52 (m, 2H), 2.42 (s, 3H), 2.10–1.96 (m, 1H). MS (ESI), Calcd. for C32H41N3O12S [M+H]+: 692.3, Found: 692.4.
19c: Yield 639.85 mg, 41%. 1H NMR (400 MHz, DMSO-d6), δ 11.08 (s, 1H), 7.86–7.72 (m, 2H), 7.59 (dd, J = 8.6, 7.1, 1H), 7.48 (d, J = 8.0, 2H), 7.15 (d, J = 8.6, 1H), 7.04 (d, J = 7.0, 1H), 6.60 (t, J = 5.8, 1H), 5.05 (dd, J = 12.9, 5.4, 1H), 4.10 (dd, J = 5.6, 3.1, 2H), 3.66–3.47 (m, 30H), 2.94–2.83 (m, 1H), 2.55 (s, 2H), 2.42 (s, 3H), 2.09–1.96 (m, 1H). MS (ESI), Calcd. for C36H49N3O14S [M+H]+: 780.3, Found: 780.3.
19d: Yield 780.66 mg, 45%. 1H NMR (400 MHz, DMSO-d6), δ 11.09 (s, 1H), 7.84–7.75 (m, 2H), 7.59 (dd, J = 8.5, 7.1, 1H), 7.48 (d, J = 8.0, 2H), 7.15 (d, J = 8.6, 1H), 7.05 (d, J = 7.0, 1H), 6.60 (t, J = 5.8, 1H), 5.06 (dd, J = 12.9, 5.4, 1H), 4.12 (s, 2H), 3.66–3.49 (m, 38H), 2.89 (ddd, J = 17.5, 14.1, 5.4, 1H), 2.64–2.53 (m, 2H), 2.42 (s, 3H), 2.07–1.98 (m, 1H). MS (ESI), Calcd. for C40H57N3O16S [M+H]+: 867.3, Found: 867.5.
19c-CH3: Yield 762.53 mg, 48%. 1H NMR (400 MHz, DMSO-d6), δ 7.88–7.71 (m, 2H), 7.59 (dd, J = 8.5, 7.1, 1H), 7.48 (d, J = 8.0, 2H), 7.15 (d, J = 8.6, 1H), 7.04 (d, J = 7.0, 1H), 6.60 (t, J = 5.8, 1H), 5.05 (dd, J = 12.9, 5.4, 1H), 4.11 (dd, J = 5.6, 3.1, 2H), 3.66–3.47 (m, 30H), 3.05–2.99 (s, 3H), 2.94–2.83 (m, 1H), 2.55 (s, 2H), 2.42 (s, 3H), 2.09–1.96 (m, 1H). MS (ESI), Calcd. for C37H51N3O14S [M+H]+: 794.3, Found: 794.3.
4.1.1.4. Synthesis of compound 2
The synthetic procedure of compound 2 was same as compound 1.
2a: Yield 71.12 mg, 45%, m.p. 147.7–151.1 °C, HPLC >95%. 1H NMR (400 MHz, DMSO-d6), δ 11.48 (s, 1H), 8.85 (s, 1H), 7.62–7.45 (m, 1H), 7.29–6.96 (m, 4H), 6.77 (ddd, J = 8.8, 4.1, 2.7, 1H), 6.59 (t, J = 5.8, 1H), 6.19 (d, J = 5.1, 1H), 5.05 (dd, J = 12.9, 5.4, 1H), 4.32 (q, J = 7.1, 0.5H), 4.03 (q, J = 7.1, 0.5H), 3.72–3.39 (m, 12H), 3.27 (d, J = 5.4, 2H), 2.88 (ddd, J = 17.7, 13.8, 5.3, 1H), 2.73 (d, J = 27.6, 4H), 2.51 (d, J = 1.9, 3H), 2.03 (s, 1H). 13C NMR (100 MHz, DMSO-d6), δ 173.26, 170.53, 169.41, 167.76, 156.15, 146.89, 140.38, 139.82, 138.52, 136.69, 132.57, 129.54, 129.21, 125.22, 121.97, 117.91, 116.51, 111.16, 109.74, 70.26, 70.10, 69.36, 49.04, 48.67, 47.73, 31.45. HR-MS Calcd. for C32H37N9O9 [M+H]+: 790.1882, Found: 790.2073.
2b: Yield 80.79 mg, 46%, m.p. 138.2–141.5 °C, HPLC >95%. 1H NMR (400 MHz, CD3OD), δ 7.52 (dd, J = 8.6, 7.1, 1H), 7.12 (dd, J = 6.0, 2.7, 1H), 7.08–6.98 (m, 3H), 6.85 (ddd, J = 8.8, 4.1, 2.7, 1H), 5.03 (dd, J = 12.4, 5.5, 0.5H), 4.80–4.69 (m, 0.5H), 3.70 (t, J = 5.3, 2H), 3.66–3.54 (m, 18H), 3.47 (t, J = 5.6, 4H), 2.96 (t, J = 6.1, 2H), 2.91–2.63 (m, 5H), 2.09 (ddd, J = 12.6, 5.1, 2.8, 1H). 13C NMR (100 MHz, CD3OD), δ 173.28, 170.19, 169.30, 167.90, 155.73, 154.04, 146.73, 140.60, 139.47, 137.62, 135.83, 135.70, 132.46, 126.44, 122.56, 122.49, 116.88, 116.70, 115.51, 110.69, 110.59, 109.89, 107.51, 107.29, 70.23, 70.16, 70.09, 70.06, 69.85, 69,23, 68.46, 42.72, 41.87, 30.81, 30.34, 22.34. HR-MS Calcd. for C36H45BrFN9O11 [M+H]+: 878.2414, Found: 878.2639.
2c: Yield 71.51 mg, 37%, m.p. 123.1–126.5 °C, HPLC >95%. 1H NMR (400 MHz, CD3OD), δ 7.53 (dd, J = 8.6, 7.0, 1H), 7.18–6.99 (m, 4H), 6.85 (ddd, J = 8.8, 4.1, 2.7, 1H), 5.04 (dd, J = 12.5, 5.5, 0.5H), 4.74 (t, J = 7.8, 0.5H), 3.72–3.43 (m, 32H), 2.96–2.60 (m, 7H), 2.10 (ddd, J = 12.6, 5.2, 2.8, 1H). 13C NMR (100 MHz, CD3OD), δ 173.27, 170.16, 169.28, 167.91, 155.73, 146.82, 140.58, 139.47, 137.66, 135.82, 135.68, 132.49, 126.39, 122.51, 122.44, 116.89, 116.70, 115.51, 115.27, 110.65, 110.55, 109.91, 107.50, 107.28, 70.26, 70.23, 70.21, 70.16, 70.13, 70.10, 69.89, 69.25, 69.12, 43.17, 41.89, 30.81, 29.34, 22.34. HR-MS Calcd. for C40H53BrFN9O13 [M+H]+: 966.2930, Found: 966.3238.
2d: Yield 69.58 mg, 33%, m.p. 93.1–96.4 °C, HPLC >95%. 1H NMR (400 MHz, CD3OD), δ 7.53 (ddd, J = 9.8, 7.1, 2.8, 1H), 7.19–6.99 (m, 4H), 6.85 (ddd, J = 8.7, 4.1, 2.7, 1H), 5.04 (dd, J = 12.5, 5.5, 0.5H), 4.75 (d, J = 7.8, 0.5H), 3.71 (t, J = 5.3, 2H), 3.66–3.57 (m, 34H), 3.46 (dt, J = 17.3, 5.7, 4H), 2.98–2.59 (m, 7H), 2.10 (ddd, J = 12.6, 5.1, 2.8, 1H). 13C NMR (100 MHz, CD3OD), δ 173.26, 167.90, 155.73, 146.82, 140.57, 139.48, 137.67, 135.83, 135.69, 132.49, 126.37, 122.50, 122.43, 116.90, 116.70, 115.52, 110.66, 110.55, 109.91, 107.51, 107.28, 70.27, 70.23, 70.21, 70.17, 70.14, 70.13, 70.11, 69.89, 69.24, 69.09, 43.15, 41.90, 30.82, 30.34, 29.35, 22.34. HR-MS Calcd. for C44H61BrFN9O15 [M+H]+: 1054.3455, Found: 1054.3734.
2c-CH3: Yield 80.38 mg, 41%, m.p. 132.7–135.4 °C, HPLC >95%. 1H NMR (400 MHz, DMSO-d6), δ 7.59 (dd, J = 8.6, 7.1, 1H), 7.21–7.10 (m, 3H), 7.05 (d, J = 7.1, 1H), 6.79 (dt, J = 8.9, 3.4, 1H), 5.13 (dd, J = 13.0, 5.4, 1H), 3.63 (t, J = 5.4, 2H), 3.53–3.40 (m, 28H), 3.27 (t, J = 6.1, 2H), 3.03 (s, 3H), 2.99–2.89 (m, 1H), 2.77 (dt, J = 12.2, 3.9, 3H), 2.68 (t, J = 5.7, 2H), 2.57 (td, J = 13.3, 4.6, 1H), 2.11–2.01 (m, 1H). 13C NMR (100 MHz, DMSO-d6), δ 172.24, 170.78, 170.25, 169.38, 167.72, 156,16, 156.10, 155.41, 153.03, 1146.82, 140.33, 139.71, 138.41, 138.38, 136.69, 132.55, 125.25, 121.92, 121.85, 117.89, 116.25, 111.15, 109.72, 109.69, 107.59, 107.37, 70.46, 70.31, 70.25, 70.09, 69.35, 60.21, 55.34, 49.61, 48.71, 47.79, 44.18, 44.08, 42.19, 42.09, 31.57, 27.04, 21.85, 21.76, 21.19, 14.53. HR-MS Calcd. for C41H55BrFN9O13 [M+H]+: 980.3087, Found: 980.3167.
4.2. Pharmacology
4.2.1. Cell lines
HeLa cell (#CCL-2) used in the study was obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were propagated in DMEM medium containing 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics (penicillin and streptomycin) in 5% CO2 at 37 °C.
4.2.2. Cell toxicity assay
The cell viability of IDO1 PROTACs treated cells was assessed by MTT assay. Briefly, the exponentially growing cells (2–6 × 103 cells/well) were plated in 96-well plates (100 μL/well) and incubated for 24 h. Then the cells were treated with gradient concentrations of IDO1 PROTACs. After treatment for 48 h, 20 μL of 5 mg/mL MTT was added to each well, and the plates were incubated at 37 °C for additional 2–4 h. The medium was removed and the purple colored precipitates of formazan were dissolved in 150 μL of DMSO. The color absorbance was recorded at 492 nm using a Spectra MAX M5 microplate spectrophotometer (Molecular Devices, CA, USA). Data are expressed as the mean ± SD (n = 3).
4.3. Determination of inhibitor activity in HeLa cell-based IDO1/kynurenine assay
This experiment was conducted according to previous report34.
4.4. QPCR
HeLa cells were treated with different concentration of IFN-γ for 24 h, then medium was discarded and the cells were washed 3 times with PBS. 700 μL of QIAzol lysate (Axygen, Tewksbury, MA, USA) were added collected HeLa cells, then the total RNA was extracted referring to the instructions to obtain 30 μL of RNA samples. The genomic DNA was removed, and 1 μg of RNA was used for the next step. The cDNA was obtained by reverse transcription referring to the reverse transcription kit (Takara, Kyoto, Japan) instructions. The Real Time PCR reaction was then performed using SYBR Premix Ex TaqTM II (Takara). The IDO1 mRNA level of the control group was set as baseline, and the expression of IDO1 mRNA in HeLa cells induced by different concentrations of IFN-γ was calculated. The primers sequences used were as follows: IDO1 forward: 5ʹ-CGGGAAGGAAATGAATGGGC-3ʹ, reverse: 5ʹ-GCATCACCCGGAGGAGAAAT-3ʹ; GAPDH forward primer: 5ʹ-AGGGTTCTGGGAAGACCCAA-3ʹ, revers primer: 5ʹ-ATGTCCTCCACCAGCAGTCT-3ʹ.
4.5. Western blot assay
HeLa cells were treated with IDO1 PROTACs in designed concentration for 24 h. The cells were washed twice with cold PBS and lysed in RIPA buffer. The protein concentrations were measured using the Coomassie brilliant blue G-250 method and equalized before loading. Equal amounts of protein from each sample was subjected to sodium dodecyl sulfate-polyacrylamidegel electrophoresis (SDS-PAGE) gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Bioscience, Piscataway, NJ, USA). Then, the membranes were blocked for 2 h at 37 °C and incubated with IDO1 antibodies (Abcam, Cambridge, UK) overnight at 4 °C. After incubation with the relevant secondary antibodies, the reactive bands were identified using an enhanced chemiluminescence kit (Amersham).
4.6. Immunofluorescence
HeLa cells (1–2 × 104 cells/well) were plated in 24-well plates that pre-placed slides and incubated for 24 h. Then the cells were treated with 2c (5 μmol/L) and IFN-γ (5 ng/mL) for 24 h, cells were washed three times with PBS and fixed with 4% paraformaldehyde for 15 min. Then cells were washed three times with PBS and permeabilized with 0.2% Triton X-100 for 20 min, washed three times with PBS and blocked with goat serum for 30 min at 37 °C. Discarded the blocking solution and incubated with IDO1 antibodies (Abcam) in wet box overnight at 4 °C. Slides were washed three times with PBST and incubated with corresponding fluorescent second antibodies for 1 h at room temperature in wet box. Then, the slides were incubated with DAPI for 5 min to stain nucleus, slides were sealed with a seal liquid containing anti-fluorescence quencher after washed four times with PBST, cells images were obtained with a LSM 880 confocal microscope (Zeiss, Jena, Germany).
4.7. Real-time cell analysis
The HeLa cells and HER-2 CAR-T cells used here were confirmed HER-2 positive by flow cytometry. HeLa cells (8 × 103 cells/well) were plated in 96-well real-time microelectronic detection plates (E-Plate®96, ACEA, San Diego, CA, USA) for 24 h. Then the cells were treated with 2c (5 μmol/L) and IFN-γ (5 ng/mL) for 24 h, replaced the medium and HER-2 CAR-T cells (4 × 104 cells/well) were added. The cell index–time plot was recorded by xCElligence RTCA MP (ACEA).
Author contributions
Yongmei Xie, Wei Wang, Xifei Yang, Gongping Liu and Mingxing Hu participated in research design. Mingxing Hu, Weilin Zhou, Yijie Wang and Dongping Yao conducted experiments. Yongmei Xie, Tinghong Ye, Yuqin Yao, Bin Chen and Mingxing Hu performed data analysis and contributed to the writing of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Institute of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.02.010.
Contributor Information
Wei Wang, Email: weiwang@scu.edu.cn.
Yongmei Xie, Email: xieym@scu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Platten M., Wick W., Van den Eynde B.J. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Canc Res. 2012;72:5435–5440. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 2.Munn D.H., Zhou M., Attwood J.T., Bondarev I., Conway S.J., Marshall B. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 3.Yoshiki K., Boyd C.A.R., Spyropoulou I., Redman C.W.G., Osamu T., Takafumi K. Indoleamine 2,3-dioxygenase: distribution and function in the developing human placenta. J Reprod Immunol. 2004;61:87–98. doi: 10.1016/j.jri.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Godin-Ethier J., Hanafi L.A., Piccirillo C.A., Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Canc Res. 2011;17:6985–6991. doi: 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- 5.Zamanakou M., Germenis A.E., Karanikas V. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol Lett. 2007;1112:69–75. doi: 10.1016/j.imlet.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Prendergast G.C., Smith C., Thomas S., Mandik-Nayak L., Laury-Kleintop L., Metz R. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–735. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz J.B., Muller A.J., Prendergast G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2010;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 8.Mellor A.L., Munn D.H. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 9.Rohrig U.F., Majjigapu S.R., Vogel P., Zoete V., Michielin O. Challenges and opportunities in the discovery of new therapeutics targeting the kynurenine pathway. J Med Chem. 2015;58:9421–9437. doi: 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. PROTACs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan S.G., Andrea T., Xavier L., Kwok-Ho C., Wenzhang C., Douglas J.L., Michael Z., Alessio C. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter G.E., Dennis L.B., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farnaby W., Koegl M., Roy M.J., Whitworth C., Diers E., Trainor N. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15:672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Gonzalez A., Kim K.B., Crews C.M., Deshaies R.J., Sakamoto K.M. Development of PROTACs to target the estrogen receptor for ubiquitination and degradation in breast cancer cells. Canc Res. 2006;66:574. [Google Scholar]
- 16.Ohh M., Park C.W., Ivan M., Hoffman M.A., Kim T.Y., Huang L.E. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 17.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 18.Galdeano C., Gadd M.S., Soares P., Scaffidi S., Molle I.V., Birced I. Structure-guided design and optimization of small molecules targeting the protein–protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y.J., Angelo A., Denzil B., Wang S.M. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58:1038–1052. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su S., Yang Z.M., Gao H.Y., Yang H.Y., Zhu S.B., An Z.X. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem. 2019;62:7575–7582. doi: 10.1021/acs.jmedchem.9b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai A.C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem Int Ed. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zoppi V., Hughes S.J., Maniaci C., Testa A., Gmaschitz T., Wieshofer C. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2019;62:699–726. doi: 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zengerle M., Chan K.H., Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H.Y., Lv W.X., He M., Deng H.T., Li H.T., Wu W. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem Commun. 2019;55:14848–14851. doi: 10.1039/c9cc08509b. [DOI] [PubMed] [Google Scholar]
- 25.Lu M.C., Liu T., Jiao Q., Ji J.N., Tao M.M., Liu Y.J. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Q.Y., Lan T.L., Su S., Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem Commun. 2019;55:369–372. doi: 10.1039/c8cc07813k. [DOI] [PubMed] [Google Scholar]
- 27.Toure M., Crews C.M. Small-molecule PROTACS: new approaches to protein degradation. Angew Chem. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson M., Crews C.M. PROteolysis TArgeting Chimeras (PROTACs)—past, present and future. Drug Discov Today. 2019;31:15–27. doi: 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Jiang X.Y., Feng F., Liu W.Y., Sun H.P. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2019;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z.Z., Lin Y.X., Song H., Qin X.J., Yu Z.X., Zhang Z. First small-molecule PROTACs for G protein-coupled receptors: inducing α1A-adrenergic receptor degradation. Acta Pharm Sin B. 2020;10:1669–1679. doi: 10.1016/j.apsb.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bondeson D.P., Mares A., Smith I.E., Ko E., Campos S., Miah A.H. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25:78–87. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018;25:67–77. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yue E.W., Sparks R., Polam P., Modi D., Douty B., Wayland B. INCB24360 (Epa), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett. 2017;8:486–491. doi: 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Long G.V., Dummer R., Hamid O., Gajewski T.F., Caglevic C., Dalle S. Epa plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–1097. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 36.Lewis-Ballester A., Pham K.N., Batabyal D., Karkashon S., Bonanno J.B., Poulos T.L. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat Commun. 2017;8:1693. doi: 10.1038/s41467-017-01725-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edmondson S.D., Yang B., Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29:1555–1564. doi: 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 38.Yang J.L., Li Y.B., Aguilar A., Zhao M.L., Yang C.Y., Wang S.M. Simple structural modifications converting a bona fide MDM2 PROTAC degrader into a molecular glue molecule: a cautionary tale in the design of PROTAC degraders. J Med Chem. 2019;62:9471–9487. doi: 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matyskiela M.E., Lu G., Ito T., Pagarigan B., Lu C.C., Miller K. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 40.Nowak R.P., DeAngelo S.L., Buckley D., He Z.X., Donovan K.A., An J. Plasticity in binding confers selectivity in ligand induced protein degradation. Nat Chem Biol. 2018;14:706–714. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.