

Abstract
Modulation of protein fate decision and protein homeostasis plays a significant role in altering the protein level, which acts as an orientation to develop drugs with new mechanisms. The molecular chaperones exert significant biological functions on modulation of protein fate decision and protein homeostasis under constantly changing environmental conditions through extensive protein–protein interactions (PPIs) with their client proteins. With the help of molecular chaperone machinery, the processes of protein folding, trafficking, quality control and degradation of client proteins could be arranged properly. The core members of molecular chaperones, including heat shock proteins (HSPs) family and their co-chaperones, are emerging as potential drug targets since they are involved in numerous disease conditions. Development of small molecule modulators targeting not only chaperones themselves but also the PPIs among chaperones, co-chaperones and clients is attracting more and more attention. These modulators are widely used as chemical tools to study chaperone networks as well as potential drug candidates for a broader set of diseases. Here, we reviewed the key checkpoints of molecular chaperone machinery HSPs as well as their co-chaperones to discuss the small molecules targeting on them for modulation of protein fate decision.
Key words: Molecular chaperone, Heat shock protein family, Small molecule inhibitors, Protein fate, Protein–protein interaction
Graphical abstract
This review summarizes the current states on modulation of molecular chaperone by small molecules, focusing on the protein fate decision by comprehensive strategies and different mechanisms.

1. Introduction
Targeting protein fate decision is a rational way to design drugs by modulating protein levels in various diseases. Molecular chaperones are one kind of significant proteins to regulate diverse downstream effectors. Abnormal expression of molecular chaperones could be regarded as potential biomarkers of multiple diseases. Molecular chaperones are obbligato for organisms to maintain homeostasis under stressed or changing environmental conditions1. In the human genome, approximately 150 genes are involved in molecular chaperones, such as heat shock proteins (HSPs) family and their associated proteins (co-chaperones), T-complex polypeptide 1 (TCP-1) ring complex, protein-disulfide isomerases, peptidyl-prolyl cis–trans isomerases, calnexin/calreticulin, etc.2,3 In fact, the molecular chaperone family is encoded by ∼170 genes to constitute a large number of proteins. Among them, HSPs are regarded as majority members of molecular chaperone which are categorized by their molecular weight, including HSP110, HSP90, HSP70, HSP60/40 and small HSPs (sHSPs). To date, HSP90 is the most studied molecular chaperone which is widely expressed in all cells and conserved in all eukaryotes, including HSP90α/β in cytosol, glucose-regulated protein 94 (GRP94) and tumor necrosis factor receptor-associated protein 1 (TRAP1) in the endoplasmic reticulum and mitochondria respectively. All these chaperones synergistically serve to achieve the balance of proteostasis, leading client proteins to avoid misfolded and/or aggregated4. The most essential function of molecular chaperones is to ensure the client proteins to achieve proteostasis under the diverse changes that affect proteins (such as a sudden increase of temperature, “heat shock”), thus leading to cellular homeostasis5. Molecular chaperones interact with other proteins (also known as client proteins or clients) to help them acquiring correctly folded forms, and only dissociate until the final active structures are accomplished (Fig. 1)6. Most chaperones work with the help of different co-chaperones to achieve different functions. They are directly related to important physical functions including anti-aggregation, intracellular trafficking, maintaining in metastable state and degradation. Based on the fundamental functions of molecular chaperones, which mainly include assisting and folding of enzymes with an inter connection of molecular chaperones and co-chaperones, disorders of molecular chaperone can be found in diverse diseases. Currently, directly inhibition of molecular chaperone and disruption of PPIs between chaperone and co-chaperones by small molecules have been identified as efficacious ways to modulate protein fate decision. Here, we discussed the major molecular chaperones, HSPs and their co-chaperones, which were discovered for their specific and elevated expression under the heat shock response. In this review, we briefly introduced the biological functions of HSPs and PPIs with co-chaperones to emphasize the development of small molecules targeting them.
Figure 1.

Constitution, brief regulation process, main function of molecular chaperone machinery and inhibition strategies by small molecule inhibitors.
2. Targeting HSPs family and small molecule inhibitors
2.1. Large HSPs
Large HSPs are mainly composed of HSP110 (or HSP105, which is induced by heat shock response) and GRP170 (retained in ER, which is induced by glucose deprivation)7. The major function of HSP110 is to recognize the denatured proteins and turn them to soluble and stable states. Although this function could be achieved by HSP70, HSP110 exhibits a more efficient pattern by interacting with other HSPs including HSP70 and HSP27. Considering a possible relationship of large HSPs (HSP110 and GRP170) to cancer, which concludes their immunostimulatory potency to target protein antigens for enhancement of antigen-based cancer vaccines, they appeal to be interesting targets. GRP170 shows similar structure and function of HSP110. The structure of HSP110 reveals comparability to Escherichia coli DnaK, which contains an N-terminal domain (ATPase domain, domain A), a peptide binding domain (domain B) and a C-terminal domain (domain H). A long acidic loop (domain L) exists for linking the peptide-binding domain and the C-terminal domain. In various cancers, HSP110 is overexpressed with diverse functions, providing therapeutic potentials in non-Hodgkin, lymphoma, melanoma and colorectal cancer. Currently, although there is no small molecule reported to regulate HSP110 or GRP170, considering their high chaperoning and immunological activity, large HSPs have been applicated to prepare vaccines for cancer therapy7, 8, 9, 10, 11, 12.
2.2. HSP90
HSP90 family is one of the most widely studied molecular chaperones in HSPs family, which comprises HSP90α and HSP90β in cytosol, glucose regulated protein 94 (GRP94) in endoplasmic reticulum and TNF receptor-associated protein 1 (TRAP1) in mitochondrion (Fig. 2A)13. HSP90 works through a homodimer and its dimerization process is important for its function in vivo14. One HSP90 monomer is constituted by an N-terminal domain (NTD) functioning as an ATP binding site, a middle domain (MD) for client binding and a C-terminal domain (CTD) for its dimerization15. Specifically, CTD involves a Met-Glu-Glu-Val-Asp (MEEVD) motif for interaction with tetratricopeptide repeat domain (TPR) containing co-chaperones16. NTD of HSP90 remains an open state in the absence of ATP and turns to a closed-state when ATP is hydrolyzed via binding clients and intermediate steps17. The client proteins of HSP90 are expanding rapidly and are consisted of conformationally labile signaling transducers which involve growth control, cell survival and development processes18. As a typical molecular chaperone, HSP90 helps diverse proteins for folding, maturation and degradation19. One representative client of HSP90 is the signaling of steroid hormone receptors (SHRs), which belongs to eukaryotic transcription factors. SHRs form large protein complexes which include the major components of HSP90 and its co-chaperones20. Another large class of HSP90 clients are protein kinases, including notorious oncogenes (RAF-1, CDK4/6, AKT, Src, c-Met, BCR-ABL, VEGF, HER2, etc.) HSP90 is essential for the stability and function of a number of oncogenic proteins, such as signaling kinases, steroid hormone receptors, telomerase, and many others that directly contribute to the hallmarks of cancer, making HSP90 as an attracting anti-cancer target over the past decade21,22. In addition, HSF-1 pathway could be activated by HSP90 inhibition, which was regarded as an inducer of anti-inflammatory and immunosuppressive genes, so blockade of HSP90 might provide a potential molecular target for autoimmune diseases23. Given the essential roles played by HSP90, inhibiting HSP90 by small molecules is therefore an attractive therapeutic strategy and several clinical trials are ongoing.
Figure 2.

Structure and biological function of HSP90. (A) Crystal structures of HSP90 (PDB:2CG9), GRP94 (PDB:5ULS) and TRAP1 (PDB: 4IPE). (B) Co-chaperones (including TPR proteins and non-TPR proteins), client proteins and processors in HSP90 chaperone cycle.
2.2.1. Functions and chaperone cycles of HSP90
HSP90 is important to maintain the protein homeostasis by regulating the active conformations of client proteins that widely take part in many biological processes, such as cell cycle, signal transduction, immune response, viral infections, and cancer development (Fig. 2B)24. HSP90-mediated protein folding cycle operates with the help of ATP and a number of co-chaperones24. Over 20 co-chaperones are known to form the multiprotein complexes with HSP90 to make the cycle proceed smoothly24. Initially, the complex of HSP70/HSP40/ADP is in charge for identifying nascent polypeptides, which could be stabilized by HSP70-interacting protein (HIP). Subsequently, as an adaptor protein, HOP/Sti1 (HSP90–HSP70 organizing protein) delivers the complex of HSP70/HSP40/ADP/client to HSP90. CDC37 (cell-division-cycle 37 homologue) is responsible for the fate of protein kinases. With the help of Cdc37 and Hop, client kinases are loaded onto HSP90. Next, various co-chaperones such as immunophilins (FKBP51, FKBP52) and partner proteins are recruited to complexes to form an activated HSP90 homodimer following concomitant release of HSP70, HIP, and HOP. With the binding of ATP at HSP90 N-terminal pocket, an “open” HSP90 complex changed into a “closed” conformation briefly. Co-chaperone P23 and AHA 1 (activator of HSP90 ATPase homologue 1) are recruited to the MD of HSP90 monomer, resulting in the hydrolysis of ATP and promoting the folding and maturation of clients24. The chaperone cycle is a complex process that requires the co-operation of different co-chaperones and co-activators that work with HSP90 to control the fate of client proteins. Small molecule inhibitors targeting HSP90 can interfere the different stage of the chaperone cycle, which prevents the normal work of HSP90, and ultimately, leads to the degradation of diverse clients via the ubiquitin–proteasome pathway.
2.2.2. HSP90 inhibitors
2.2.2.1. Pan-HSP90 N-terminal inhibitors
Pan-HSP90 N-terminal inhibitors competitively occupy the ATP pocket to block ATP hydrolysis of HSP90 and subsequently impair the maturation of client proteins. The inhibitors with similar affinity to all four isoforms (HSP90α, HSP90β, GRP94 and TRAP1) are named pan-HSP90 N-terminal inhibitors25. So far, more than 20 distinct N-terminal HSP90 inhibitors have entered into clinical trials and more compounds have been authorized to preclinical studies26. Briefly, these inhibitors can be divided into four classes according to their chemical scaffolds including ansamycin-based, resorcinol-based, purine-based and benzamide-based inhibitors27. Among them, geldanamycin (GDA), which was isolated from Streptomyces hygroscopicus, was identified as the first prominent HSP90 inhibitor that exhibited potent antitumor activity28. Analogues of GDA such as IPI-504 was the first-generation of HSP90 inhibitors29, 30, 31. To improve the drug-like properties of geldanamycin analogues, the second-generation of synthetic small molecule inhibitors are developed, such as purine derivatives (PU-H71), resorcinol derivatives (AT13387) and benzamide derivatives (SNX-2112)32, 33, 34. Among them, NVP-AUY922 (Luminespib, in phase II clinical trial), CUDC-305 (Debio 0932, in phase I clinical trial) and STA-9090 (Ganetespib, in phase II clinical trial) are currently important and representative HSP90 inhibitors with diverse researches on going in clinical trials, including multi-types of cancer35, 36, 37. In 2016, our group38 also discovered a promising and safe HSP90 inhibitor, DDO-5543 (compound 73), with potent antitumor effect in an HCT116 xenograft model. The current state of these inhibitors was comprehensively reviewed as previous reported25, 26, 27,39.
2.2.2.2. HSP90 C-terminal inhibitors
Based on the unique structure of HSP90, the function of C-terminal domain and the inhibition strategies targeting on it are raising more and more attention. Some natural products and their derivatives were reported to exhibit promising antitumor effect through targeting HSP90 C-terminal domain40. Novobiocin was reported as the first HSP90 C-terminal inhibitor with moderate antitumor activity. A comprehensive structure–activity relationship (SAR) study was conducted to improve the activity by Blagg group41 and many potent derivatives were reported to exhibit potent anti-proliferative activities against various cancer cell lines, such as KU-135 and KU675. A novobiocin based rapid overlay of chemical structures (ROCS) model was performed by our group42 and a new scaffold containing aminoquinoline was identified as HSP90 C-terminal inhibitor with potent tumor growth inhibition and anti-metastasis effect, leading DDO-5713 as a potent antitumor agent. Most importantly, unlike the N-terminal inhibitors, these C-terminal inhibitors did not induce heat shock response. Thus, C-terminal inhibitors provide new options to control the client protein with more efficacy and safety.
2.2.2.3. HSP90α/β selective inhibitors and HSP90β selective inhibitors
The pan-HSP90 inhibitors cannot induce the extended α-helix conformation, so they exhibit similar activity against HSP90, GRP94 and TRAP143. While, based on the sequential difference of HSP90α/β, structural insights for the design of inhibitors with improved HSP90α/β selectivity have been provided44. TAS-116, an HSP90α/β selective inhibitor which was developed through fragment-based drug design by Taiho Pharmaceutical Co., Ltd., has been entered into clinical trials. TAS-116 possessed excellent HSP90α/β selective inhibition activity with Ki values of 34.7, 21.3, >50 and > 50 μmol/L for HSP90α, HSP90β, GRP94, and TRAP1, respectively45. HSP90β is considered to be constitutively expressed in the cytoplasm, while HSP90α is an inducible form that is overexpressed during cellular stress43. HSP90α and HSP90β share ∼95% identity in the ATP binding pockets and the subtle difference is that only two amino acids differ between the α and β isoforms, making it difficult to develop of HSP90α- or HSP90β-selective inhibitors. In the co-crystal structure of HSP90β with radicicol, Ala52 and Leu91 make a sub-pocket that could tolerate a reasonable substituent at the 3-position of resorcinol, while there is an unfavorable steric effect at the same site of HSP90α. Thus, SAR at the 3-position of resorcinol was studied, leading to a benzoisoxazole scaffold (KUNB31) that could selectively inhibit the HSP90β isoform with >50-fold selectivity. Unlike pan-HSP90 N-terminal inhibitors, selective inhibition of HSP90β does not influence other isoforms of HSP90, suggesting an alternative and promising mechanism for the therapy of some tumor driven by HSP90β-dependent client proteins46.
2.2.2.4. GRP94 inhibitors
Glucose-regulated protein 94 (GRP94), also known as gp96, is a molecular chaperone in the lumen of the endoplasmic reticulum (ER), which belongs to the HSP90 family. GRP94 is one of the key downstream chaperones of the ER unfolded protein response (UPR), which is an essential adaptive intracellular signaling pathway to restore protein homeostasis when cells are subjected to the burden of unfolded proteins in the ER47. GRP94 has emerged as a potential therapeutic target for a host of diseases, including cancer, primary open-angle glaucoma and autoimmune diseases51, 52, 53. GRP94 controls the maturation and secretion of a variety of cancer-associated proteins, including TLR receptors, integrins, LDL receptor related protein 6 (LRP6), glycoprotein A repetitions predominant (GARP), glycoprotein Ib–IX–IV, HER2 and insulin-like growth factors (IGFs)54. Like other HSP90 isoforms, GRP94 exists as obligatory homodimers with each monomer consisting of the following three major functional domains: NTD, MD, and CTD. The GRP94 N-terminal ATP-binding pocket shares 85% identity to other HSP90 isoforms. The helix 1-4-5 of GRP94 subdomain contains a five-amino acids (QEDGE) insertion, resulting in a conformational change within the N-terminal ATP-binding pocket that exposes two different hydrophobic clefts adjacent to the adenine binding region of the N-terminal ATP-binding pocket, termed sites two and three pockets, respectively55. Currently, design of GRP94-selective inhibitors is based on the identification and exploitation of these two cavities. At present, the reported GRP94 inhibitors can be chemically classified into three classes: resorcinol derivatives, purine derivatives and benzamide derivatives47. In 2018, our group48 indicated that the “Phe199 shift” effect induced by the ligand is the structural basis for the selective inhibition of GRP94. By analyzing the binding modes of three classes of scaffolds, we found that the benzamide-scaffold is suitable to design new GRP94-selective inhibitors since the 3-position of the carbamoyl group points to Phe199. On the basis of these observations, we further introduced a rigid and hydrophobic phenyl ring at the 3-position of the carbamoyl group, which ultimately led to the discovery of DDO-5813 (compound 54). In a label-free biolayer interferometry (BLI) assay, DDO-5813 bound to GRP94 with a Kd value of 19.6 nmol/L and to HSP90α with a Kd value of 20.6 μmol/L In a DSS-induced mouse model of ulcerative colitis (UC), treatment with DDO-5813 could ameliorate the inflammatory symptoms and reduce the expression of inflammatory cytokines in the colon and serum. GRP94 isoform-selective inhibitors only regulate the GRP94-specific clients, endowing them with more favorable safety profiles and clinical potentials48.
2.2.2.5. TRAP1 inhibitors
As the mitochondrial paralog of HSP90, TNF receptor-associated protein 1 (TRAP1) was firstly identified by screening for proteins that bind to the intracellular domain of the type one receptor for tumor necrosis factor (TNFR-1IC) in the yeast based on two hybrid technologies56. With an improved understanding of the organelle architecture in mitochondria, the global role of TRAP1 in mitochondrial physiology, mitochondrial respiration and aerobic glycolysis, organelle-compartmentalized protein folding, and oxidative stress became increasingly clear57,58. Dysfunction of TRAP1 has been noticed in cancer and neurodegenerative diseases58,59. TRAP1 is induced in the different types of cancer and is involved in the regulation of metabolic switch in the mitochondria of tumor cells through the regulation of maturation and secretion of selective proteins, such as cyclophilin D, mitochondrial c-Src and sorcin60, 61, 62, 63. Targeting the TRAP1 system definitely influences mitochondrial physiology, impacts both glycolysis and oxidative stress, and could provide a novel anticancer approach for humans59. In 2009, the first TRAP1 inhibitor gamitrinibs (G-G1−G-G4) was reported by Byoung Heon Kang and coworkers49. Gamitrinibs are a series of combinatorial molecules, containing a backbone of pan-HSP90 inhibitor 17-AAG, a linker, and a mitochondrial-targeted moiety, either provided by one–four tandem repeats of cyclic guanidinium (Gamitrinib-G1–G4) or triphenylphosphonium (Gamitrinib–TPP–OH). Gamitrinibs could accumulate in the mitochondria of human tumor cell lines and inhibit HSP90 ATPase activity49. SMTIN-P01, a conjugate of PU-H71 and TPP, was another mitochondria-accumulating HSP90 inhibitor, showing an improved cytotoxicity to cancer cells. The active site of lid structure (Leu172-Phe201) in TRAP1 was disordered and its conserved residues Asn171 and Gly202 showed different configurations. Thus, it is important to generate the interactions with the disordered TRAP1 active site lid to improve the TRAP1 selectivity57. DN401 was the first TRAP1 inhibitor without using the mitochondrial delivery vehicle, discovered by comparison of crystal structures of TRAP1 and HSP90 complexed with HSP90 inhibitor BIIB02150. TRAP1 selective inhibitors may overcome the heat shock response effect of pan-HSP90 inhibitors. Development of TRAP1 selective inhibitor could control and regulate the fate of subcellular client proteins and subsequently generate a potent anti-tumor activity with a novel mode of action. All the representative HSP90 inhibitors were summarized in Table 126, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50.
Table 1.
Representative HSP90 inhibitors with different mechanisms.
| Name | Year | Structure | Mechanism | Activity | Ref. |
|---|---|---|---|---|---|
| Radicicol | 1998 | 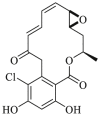 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.019 μmol/L (FP) |
26, 27 |
| GDA | 1997 | 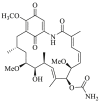 |
Pan-HSP90 N-terminal ATP inhibitors |
HSP90 all isoforms: IC50 = 0.005 μmol/L (FP) |
28 |
| IPI-504 | 2017 |  |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.064 μmol/L (FP) |
29, 30, 31 |
| AT13387 (onalespib) | 2010 |  |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.7 nmol/L (FP) |
32 |
| PU-H71 | 2005 | 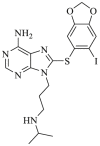 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.009 μmol/L (FP) |
33 |
| SNX-2112 | 2009 | 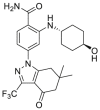 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.003 μmol/L (FP) |
34 |
| NVP-AUY922 (luminespib) | 2008 | 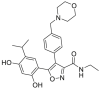 |
Pan-HSP90-N-terninal ATP inhibitors |
HSP90α, IC50 = 0.008 μmol/L HSP90β, IC50 = 0.021 μmol/L GRP94, IC50 = 0.535 μmol/L TRAP1, IC50 = 0.085 μmol/L |
35 |
| CUDC-305 (debio 0932) | 2009 | 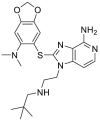 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90α, IC50 = 0.01 μmol/L HSP90β, IC50 = 0.01 μmol/L |
36 |
| STA-9090 (ganetespib) | 2011 | 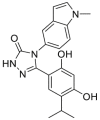 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50<0.01 μmol/L |
37 |
| DDO-5543 (73) | 2016 | 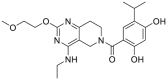 |
Pan-HSP90 N-terminal ATP inhibitors | HSP90 all isoforms: IC50 = 0.032 μmol/L (FP) |
38, 39 |
| Novobiocin | 2000 | 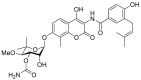 |
HSP90 C-terminal ATP inhibitors | Cellular activity: IC50 = 256 μmol/L |
40, 41 |
| DDO-5713 (69) | 2017 | 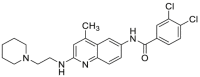 |
HSP90 C-terminal ATP inhibitors | Cell activity: IC50 = 1.19 μmol/L |
42 |
| SNX0723 | 2014 | 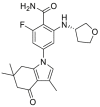 |
HSP90α/β selective inhibitor | HSP90α, Ki = 0.003 μmol/L HSP90β, Ki = 0.004 μmol/L GRP94, Ki = 0.375 μmol/L TRAP1, Ki = 1.195 μmol/L |
43, 44 |
| TAS-116 | 2015 | 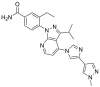 |
HSP90α/β selective inhibitor | HSP90α, Ki = 0.035 μmol/L HSP90β, Ki = 0.021 μmol/L GRP94 Ki > 50 μmol/L Trap1 Ki > 50 μmol/L |
45 |
| KUNB31 | 2018 | 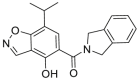 |
HSP90β selective inhibitor | HSP90α, Kd = 9.55 μmol/L HSP90β, Kd = 0.18 μmol/L GRP94, Kd = 8.48 μmol/L |
46 |
| PU-W13 | 2015 | 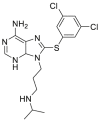 |
GRP94 selective inhibitor | GRP94: IC50 = 0.22 μmol/L HSP90α: IC50 = 2.73 μmol/L |
47 |
| KUNG29 | 2017 | 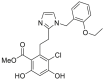 |
GRP94 selective inhibitor | GRP94: IC50 = 0.20 μmol/L HSP90α: IC50 = 8.10 μmol/L |
47 |
| KUNG94 | 2017 | 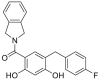 |
GRP94 selective inhibitor | GRP94: IC50 = 0.008 μmol/L HSP90α: IC50 = 0.077 μmol/L |
47 |
| ACO1 | 2017 | 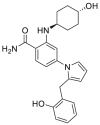 |
GRP94 selective inhibitor | GRP94: IC50 = 0.44 μmol/L HSP90α: no inhibition |
47 |
| SMTIN-P01 | 2015 | 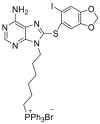 |
TRAP1 selective inhibitor | Cell activity: better than PU-H71 (data unavailable) | 47 |
| DDO-5813 (54) | 2018 | 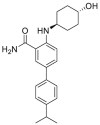 |
GRP94 selective inhibitor | GRP94: IC50 = 0.002 μmol/L HSP90α: no inhibition |
48 |
| Gamitrinibs | 2009 | 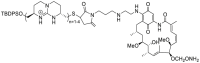 |
TRAP1 one selective inhibitor | Cell activity: IC50 = 4.0 μmol/L (A431) |
49 |
| DN401 | 2017 | 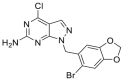 |
TRAP1 selective inhibitor | TRAP1: IC50 = 0.079 μmol/L HSP90α: IC50 = 0.698 μmol/L |
50 |
2.3. HSP70
2.3.1. Functions and chaperone cycle of HSP70
Besides HSP90, HSPs family contains another widely expressed and well-studied chaperone, HSP70, which is consisted of multiple homologous chaperone proteins. In normal cells, HSP70 mainly resides in the cytoplasm64. Other specific HSP70 proteins are localized in different organelle whereas GRP78 (also called as BIP or HSP70-5) is found in endoplasmic reticulum (ER) and HSP70-9 (also called as mtHSP70, mortalin or GRP75) is found in mitochondria65. Other known members including HSP70-1a, HSP70-1b, HSP70-2 and heat shock cognate protein 70 (HSC70) are mainly located in the cytosol and nucleus66. HSP70 is highly conserved and shares a similar domain architecture, which contains an N-terminal nucleotide binding domain (NBD) connected with a C-terminal substrate-binding domain (SBD). The NBD is further divided into two subdomains Ⅰ and Ⅱ, which works as an ATP binding domain through a nucleotide-binding cassette67,68. The SBD is composed of a ∼10 kDa α-helix and a ∼15 kDa β-sandwich structure (Fig. 3A)69. Protein–protein interactions including J domain with NBD of HSP70 enhance ATP turnover and distinct cochaperones as well as the nucleotide exchange factors (NEFs) contribute to the completion of ATPase cycle, which all are important regulatory factors and obbligato for diverse chaperone functions of HSP7070,71. Interestingly, similar with the MEEVD sequence on HSP90, the C-terminus of HSP70 shares an EEVD sequence for the binding of tetratricopeptide repeat (TPR)-containing co-chaperones72. In normal cells, HSP70 remains at low levels but its expression could be rapidly increased by the induction of transcription factor heat shock factor 1 (HSF-1) under cellular stress (including the stress of oncogenesis). The main clients of HSP70 are linear peptides, including newly synthesized proteins or unfolded intermediates (such as clathrin, transcriptional activation complex, nuclear hormone receptors, etc.)73,74. During normal cell growth, HSP70 also exhibits multiple functions including (1) folding the unfolded proteins75,76, (2) transporting proteins and vesicles in the subcellular fraction77, (3) forming complex with other chaperones78, and (4) degradation of mis-folded proteins (Fig. 3B)79. Due to the important activities of HSP70, a wide range of diseases have been implicated by genetic and biochemical studies, including cancer and apoptosis, protein misfolding and neurodegenerative disease and infectious disease and immunity (Fig. 3C)80, 81, 82.
Figure 3.

Structure, functional cycle and cancer-related pathway of HSP70. (A) The crystal structure of HSP70 containing NBD, SBD and TPR binding domain. (B) Functional cycles of HSP70. With the help of HSP40, unfolded polypeptide substrate firstly binding to HSP70 with an ATP bound state (this process exhibits a low affinity for polypeptide). After ATP hydrolyzed, substrates exhibit high affinity with an ADP bound state, before NEF induced ADP release. During this process, substrates are fully folded and released. (C) Roles and functions of HSP70 in cancer-related pathways as a potential therapeutic strategy, including inhibition of apoptosis, control of oncogene-induced cell senescence, stabilization of lysosome function and regulation of autophagy by HSP70, and regulation of HSP90 client proteins as a co-chaperone.
2.3.2. HSP70 inhibitors
Unlike HSP90 (weak ATPase), which has abundant ATPase inhibitors with multiple chemical types, the discovery of HSP70 ATPase inhibitors meets strong resistance. Although many reviews and researches have demonstrated HSP70 as a promising target for cancer therapy, there is no drug-like compound that has been identified yet83. This difficulty may be caused by the high affinity and abundance of its endogenous nucleotide substrates. Currently, small molecules are only used as chemical probes to investigate modulation mechanism by targeting HSP70 including directly blocking its ATPases activity, targeting with an allosteric modulation mechanism and disrupting the PPIs of HSP70 and its co-chaperones84.
2.3.2.1. ATPase inhibitors
In 2011, Massey et al.85,86 discovered VER-155008 as a potent HSP70 ATPase inhibitor based on co-crystal structures through structure-based drug design and optimization, which confirmed that VER-155008 could compete nucleotide for binding HSP70 and exhibited anti-proliferative activity in HCT116 cells. Of the HSP70 inhibitors so far described, VER-155008 was the only one to exhibit clearly binding modes and SARs as an ATP mimetic. Notably, VER-155008 exhibited therapeutic potentials in treatment of cancer87, 88, 89, viral diseases90 and Alzheimer's disease91. Considering the clear binding modes of VER-155008, it has also been designed as an irreversible inhibitor by covalently binding to Lys56 of HSP70, exhibiting as another effective strategy92. Another typical HSP70 ATPase inhibitor, apoptozole, was discovered in 2015 by immobilized pull down assay93. Apoptozole specifically binds to HSP70 but no other types of heat shock proteins, resulting in apoptotic phenotypes in cancer cells through directly inhibiting HSP70 ATPase. Interestingly, apoptozole acts as an ATPase inhibitor without an ATP mimetic structure (Table 2)85, 86, 87, 88, 89, 90, 91, 92, 93.
Table 2.
Structures, mechanisms and activities of HSP70 inhibitors.
| Name | Year | Structure | Mechanism | In vitro assay | In vivo efficacy | Ref. |
|---|---|---|---|---|---|---|
| VER-155008 | 2011 |  |
ATPase inhibition | Kd = 0.08 μmol/L | Yesa | 85, 86, 87, 88, 89, 90, 91, 92 |
| Apoptozole | 2015 | 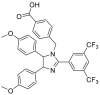 |
ATPase inhibition | Low μmol/L | Yes | 93 |
| MAL3-101 | 2004 | 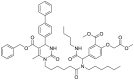 |
Disrupt HSP70–HSP40 (allosteric) | Mid to high μmol/L | Yes | 94, 95, 96, 97 |
| JG-231 | 2000 |  |
Disrupt HSP70–BAG (allosteric) | EC50 = 0.03 μmol/L | Yes | 98, 99 |
| YK5 | 2014 | 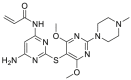 |
Allosteric on NBD | Low to Mid μmol/L | NAb | 100 |
| HS-72 | 2014 | 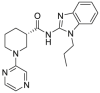 |
Allosteric on SBD | Mid μmol/L | Yes | 101 |
| PES | 2009 | 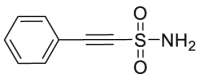 |
Allosteric on SBD | Not reported | Yes | 102 |
| Novolactone | 2015 | 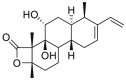 |
Covalently and allosteric on SBD | IC50 = 0.25 μmol/L | NA | 103 |
Effective in vivo.
No activity.
2.3.2.2. Allosteric and PPI inhibitors
Targeting the network of HSP70 PPIs is another way to achieve HSP70 inhibition. Dihydropyrimidines scaffold compounds, represented by MAL3-101, were identified as the first evidence to bind the interface between HSP70 and JDPs, leading to down-regulation of the HSP70 biomarkers (Table 2)94, 95, 96. Although the potency of such series compounds remained moderate (EC50 values remain micromolar level), given the important biological function of JDPs to HSP70, these compounds were valuable for further optimization97. Despite HSP70–JDPs, HSP70–BAG PPIs are important for controlling the release of clients from the complex, which include NEFs as one of the major categories98. A compound scaffold of rhodacyanine-benzothiazoles (such as JG-231) exhibited the potency to block NEF binding to HSP70, which was expected to increase the binding of clients in the chaperone complex, leading to a favorable degradation process in specific cases99. These compounds were firstly discovered by Wadhwa et al.47 through phenotypic anticancer screens and their targets were identified as HSP70 by pull down assay. Using these structures, further NMR studies revealed a novel allosteric pocket on HSP70 for blocking its interaction with BAG proteins, which was caused by a conformational change104,105. For the significant role of HSP70–BAG PPIs, JG-231 was used as a small molecule probe in diverse diseases including tau homeostasis106, dengue viral replication107, castration-resistant prostate cancer108 and breast cancer109. Other allosteric inhibitors include compound YK-5 and its analogs, which bind to a distinct allosteric site on NBD of HSP70100. In addition, diverse and different allosteric HSP70 inhibitors were identified for binding at SBD of HSP70 through phenotypic screens and assays of structural biology. Among them, HS-72 was discovered in a screen for nucleotide-binding molecules and was confirmed as a potential HSP70 inhibitor by pull-down assays101. 2-Phenylethynesulfonamide (PES) was discovered in phenotypic screens and identified as a HSP70 inhibitor through binding to an allosteric site on SBD, which was confirmed by mutagenesis102. Novolactone was also an allosteric HSP70 inhibitor, bound to SBD of HSP70 through a highly conserved covalent interaction103. Interestingly, all these inhibitors held different binding sites on HSP70 to exhibit diverse mechanisms of action which were expected for further investigation110.
2.4. HSP60
2.4.1. Structures, chaperone cycles and inhibitors of HSP60
HSP60 (also known as chaperonin 60 or Cpn60, named GroEL in E. coli; CCT in mammals; thermosome in archaea) is located in cytoplasm and mitochondria under a normal physiological condition, which mainly functions as an oligomer to bind unfolding client proteins for further interacting with the large ATP-controlled HSPs111,112. According to its canonical structure, HSP60 is consisted of three domains including apical domain, intermediates and equatorial domain (Fig. 4A and B). HSP60 exhibits its function through a ring structure containing seven subunits and functional PPIs for lid-shaped cochaperones (GroES in bacteria, HSP10 in mitochondria, and Cpn10/Cpn20 in chloroplasts)113. The reaction cycle is indicated in Fig. 4C. Under normal conditions, HSP60 could associate with diverse proteins, such as Y-box-binding protein one and fibrous structural protein Keratin 23,114,115. HSP60 exhibits induced expression by cancer cells and participates in transformation, promotion of angiogenesis and metastasis116,117. HSP60 can enhance anti-apoptotic effects and antagonize cellular stress induced by chemotherapeutic agents, through binding to and modulating the intracellular protein clusterin as well as interacting with cyclophilin D in the mitochondrial permeability transition pore112,118. Direct inhibition of HSP60 results in cyclophilin D-dependent mitochondrial permeability transition, caspase-dependent apoptosis, and suppression of tumor growth119. Numerous cancer factors are correlated with HSP60, such as insulinlike growth factor binding protein 7 (IGFBP7) in colorectal cancer cells and IκB kinase (IKK) in human cervical cancer HeLa cells120,121. Through the interaction with β-catenin, HSP60 promotes tumor metastasis and overexpresses in many cancer cells122. In addition, HSP60 has been found up-regulated in multiple human cancers, which make it a potential target for the diagnosis and prognosis of cancer123.
Figure 4.

Structure and functional cycle of HSP60. (A) The crystal structure of HSP60 oligomer (PDB: 1SS8). The client binding site is indicated as red dots. (B) One subunit of HSP60, including apical domain, intermediate domain and equatorial domain. (C) HSP60–HSP10 reaction cycle. Unfolded intermediates firstly bound to HSP60 ring of the asymmetrical HSP60–HSP10 complex. Subsequently, binding of seven ATP to each of HSP60 subunit and HSP10 in the cis complex resulted in a conformational change in the apical domain to release the HSP10 from the trans ring. The newly encapsulated complex hydrolyzed seven ATP molecules and the binding of HSP10 to the trans ring led the bound-state HSP10 to dissociate from the cis ring and release the folded proteins.
Currently, HSP60 inhibitors, including BF844124, phenoxyacetanilide125, KHS-101126, suvanine127, epolactaene128 and myrtucommulone129, were discovered by phenotypic screens and identified through pull-down assays, leading to an unclear binding mechanism of these compounds (Table 3)124, 125, 126, 127, 128, 129. Although these compounds exhibited limited selectivity in cells, HSP60 could be regarded as an identified target for them. To date, no small molecule targeting HSP60 has been discovered as specific inhibitors and no evidence of medicinal chemistry work has been performed for further study of HSP60 inhibition. Given the significance of HSP60 in mitochondrial protein quality control, more investments are needed to identify the chemical probes of specific HSP60 inhibitors for study this system130.
Table 3.
Chemical structures, regulation mechanisms and in vitro/in vivo activities of potential HSP60 inhibitors.
| Name | Year | Structure | Mechanism | In vitro assay | In vivo efficacy | Ref. |
|---|---|---|---|---|---|---|
| BF844 | 2016 | 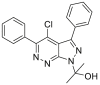 |
Unknown | – | NDa | 124 |
| Phenoxyacetanilide | 2010 | 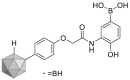 |
Directly binding to HSP60 | IC50 = 0.74 μmol/L | NDa | 125 |
| KHS-101 | 2018 | 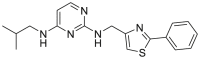 |
Unknown | – | NDa | 126 |
| Suvanine | 2012 | 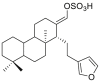 |
Directly binding to HSP60 | IC50 = 0.6 μmol/L | NDa | 127 |
| Epolactaene | 2005 | 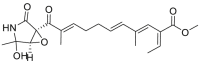 |
Covalently binding to HSP60 Cys442 | Low μmol/L | NDa | 128 |
| Myrtucommulone | 2017 | 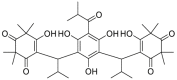 |
Directly binding to HSP60 | Micromolar level | NDa | 129 |
Not determined. –Not applicable.
2.5. sHSPs
Different with HSP90, HSP70 and HSP60, although sHSPs are less studied, their functions reveal diversity131,132. sHSPs are usually defined as those HSPs with subunit molecular masses of 12–43 kDa and all of them are ATP-independent. They are consisted of a less conserved NTD and a CTE with a highly conserved region of 80–100 amino acids (also known as “α-crystallin domain” (ACD))133, 134, 135.
2.5.1. Chaperone cycles and inhibitors of HSP40
HSP40 (DNAJ family) proteins are defined as homologs of bacterial DnaJ HSPs, which contains a “J” domain for interacting with HSP70, leading to be known as HSP70 co-chaperones as well136. The main function of HSP40 lies in stimulating the ATPase activity of HSP70 to regulate the protein folding, translation, translocation and degradation. Although HSP40 is not as well-known as HSP90 and HSP70, the major family of HSP40 reveals the largest number of 49 coded by human genome. As shown in Fig. 5A, HSP40 could be divided into three DNAJ classes including DNAJA (Type Ⅰ), DNAJB (Type Ⅱ), and DNAJC (Type Ⅲ)137. Due to the diverse constructions and precise functions of HSP40, it exhibits a high correlation to cancer development. Interestingly, HSP40 family proteins exhibit dual characters for playing a role as both anticancer and pro-cancer processes. For example, DNAJA3 (also known as Tid1) binds to HSP70 to directly interact with P53, leading to mitochondrial translocation of the complex and the induction of intrinsic apoptosis in breast cancer MCF-7 cells as a regulator of P53-mediated apoptosis138. DNAJB4 (also known as HLJ1) also works as a tumor suppressor to inhibit the proliferation, cell growth and invasion of lung cancer cells. High DNAJB4 level result in decelerating of cell-cycle progression of lung cancer through the STAT1/P21 pathway139,140. On the contrary, there was evidence that DNAJC6 increased the progression of hepatocellular carcinoma by enhancing EMT and DNAJB8 promoted the expression of cancer stem-like cells for elevation of tumor-initiating ability of renal cancer cells141,142. Overexpression of HSP40 family proteins has been found in gastric, colorectal, cervical, and lung cancers and involved in the therapeutic effects of chemical agents12,143,144.
Figure 5.

The structure and small molecule inhibitors of HSP40. (A) The classification of the functional domains of HSP40 (DNAJ). HSP40 could be divided into three subclasses (DNAJA, DNAJB and DNAJC). Each of them is partly consisted of J domain, glycine/phenylalanine-rich region (G/F), cysteine-repeat motif (Cys-rich) and a fully characterized C-terminal domain. (B) Chemical structures of small molecules involving the function of HSP40 inhibition.
Currently, there is no specific HSP40 inhibitor to be reported to achieve a direct HSP40 binding mechanism for regulation of HSP70–HSP40 chaperone cycle. While, HSP40 proteins are reported to be involved in multiple effects of chemotherapeutic agents. Few small molecule inhibitors have been identified to mediate the biological effects with HSP40 involved (Fig. 5B). KNK437 exhibits antitumor effects through a HSP27, HSP40, HSP72 and HSP110 inhibition manner in human colon cancer cells145. BMS-690514 is identified as a potent inhibitor targeting EGFR, HER and VEGFR, which also exerts its anti-tumor effect through inhibiting the expression level of HSP40 and other HSPs in non-small cell lung cancer (NSCLC) cells146. R115777 (tipifamib) is a farnesyltransferase inhibitor with biological effects including anti-tumor growth, survival inhibition and angiogenesis pathway in human breast cancer cells by inhibiting the multiple significant factors (including HDJ-2)147. Thus, development of small molecules targeting HSP40 remains an uncovered gold mine to be further explored.
2.5.2. Other sHSPs
Some members of the sHSPs, including HSP27, αA- and αB-crystallin, can form large oligomeric species. For their genome diversity, only HSP27 was discussed in this review. HSP27 can be regulated by phosphorylation at Ser15, Ser78, and Ser82 when induced in response to stress148. Current evidence indicated that HSP27 could interact with β-catenin, histone deacetylases 6 (HDAC6), signal transducer and activator of transcription 2 (STAT2) and procaspase-3 for development of various diseases including neurodegenerative diseases, ischemia, atherosclerosis, and cancer149, 150, 151. In addition, HSP27 is found up-regulated in a variety of cancers and could be regarded as a biomarker in cancer diagnosis and prognosis12. Currently, only OGX-427 (apatorsen), an antisense oligonucleotide in phase Ⅱ clinical trials, was reported to inhibit HSP27. OGX-427 exhibited the potential therapeutic effects to reduce tumor metastasis in a murine model of prostate cancer and showed efficiency in patients with metastatic prostate cancer for decreasing the number of circulation tumor cells in a phase Ⅰ trial152.
3. HSP90 and its co-chaperones
3.1. Targeting HSP90–CDC37 PPI
3.1.1. The structure of HSP90–CDC37 PPI and its regulation mechanisms for protein kinases
CDC37 (also referred to P50) is a typical molecular chaperone to physically stabilize the catalytic domains of protein kinases, leading to a wide spectrum regulation of phosphorylation of protein kinases153. In normal cells, CDC37 recognizes the newly synthesized protein kinases and interacts with specific kinase binding domains to promote their maturation and recruitment to HSP90. Due to the specificity to protein kinases, CDC37 has been regarded as a “kinases guard” and accelerates cell proliferation observed in many types of cancer through promoting the activities of a wide-spectrum of protein kinases, but including one exception of the androgen receptor (AR) as a rare non-kinase CDC37 client154, 155, 156, 157. Early studies have identified the structure of CDC37, including an N-terminal domain for kinase interacting (including a large number of protein kinases to be dysregulated in cancer, such as RAF-1, AKT, EGFR, FGFR, CDK4, etc.), a middle domain for HSP90 binding and a C-terminal domain with unknow properties158. CDC37 acts as a linker and accelerator between HSP90 and its protein kinase clients. Currently, CDC37 is well-defined as a co-chaperone to mediate the maturation and stabilization of protein kinases by forming complex with HSP90, although there is evidence showing that CDC37 could achieve its function without HSP90 (Fig. 6A)159. Structural evidence identified the mechanism of HSP90–CDC37 binding process and its functions for trapping and stabilizing unfolded kinases (Fig. 6B)160. Due to its significant roles in maturation of protein kinases, elevated level of CDC37 has been found in diverse clinical cancers, including prostate carcinoma, anaplastic large cell lymphoma, acute myelocytic leukemia, hepatocellular carcinoma and multiple myeloma161, 162, 163. Considering most normal tissues can tolerate the absence of CDC37 while similar abundance of HSP90 exist in both normal tissues and malignant cells, a potential therapeutic window is available for targeting HSP90–CDC37 complex or CDC37 itself as therapeutic targets (Fig. 6C)164,165.
Figure 6.

Structure and regulation mechanism of HSP90–CDC37 PPI. (A) The solution NMR structure of HSP90 N-terminal and CDC37-M domain (PDB:2K5B). (B) The co-crystal structure of HSP90–CDC37–CDK4 complex (PDB:5FWL). (C) The regulation mechanisms of HSP90–CDC37 PPI for controlling protein kinases from the unfolded to folded ones.
3.1.2. Small molecule inhibitors targeting HSP90–CDC37 PPI
Disrupting HSP90–CDC37 PPI could selectively downregulate the kinase clients of HSP90, which provides a specific horizon for cancer therapy22. Considering the complexity and dynamic process of HSP90–CDC37 PPI, as well as large binding surface, rational design of inhibitors targeting HSP90–CDC37 PPI exit numerous obstacles. During the past few years, several kinds of inhibitors with the potency to disrupt HSP90–CDC37 PPI have been reported successively166, as shown in Table 4167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179. Until 2019, there was no specific HSP90–CDC37 PPI inhibitor with clear binding mode and SAR has been discovered to confirm the accurate binding sites for small molecule inhibitors. Recently, our group discovered DDO-5936 as a first identified small molecule inhibitor of HSP90–CDC37 PPI through directly binding to a novel binding site on HSP90, resulting in selective downregulation of HSP90 kinase clients in HCT116 cells. This evidence not only provided a novel binding site on HSP90 and a specific HSP90–CDC37 PPI small molecule inhibitor, but also established a feasible way for the rational design of HSP90–CDC37 inhibitors.
Table 4.
Small molecules inhibitors targeting HSP90–CDC37 PPI.
| Name | Year | Structure | Mechanism | In vitro assay | In vivo efficacy | Ref. |
|---|---|---|---|---|---|---|
| Celastrol | 2008 | 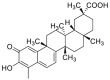 |
Covalently binding to CDC37 | Low | Yesa | 167, 168, 169, 170, 171, 172 |
| Withaferin A (WA) | 2011 | 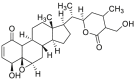 |
Covalently binding to HSP90 C-terminus | Low | Yes | 173, 174 |
| Kongensin A (KA) | 2016 | 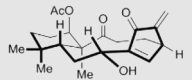 |
Covalently binding to HSP90-M domain | Low | NDb | 175 |
| FW-04-804 | 2014 | 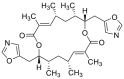 |
Binding to HSP90 N-terminus | Low | Yes | 176 |
| DCZ3112 | 2018 | 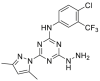 |
Binding to HSP90 N-terminus | Kd = 4.98 μmol/L | Yes | 177 |
| VS-8 | 2017 | Binding to HSP90 N-terminus | Kd = 40.4 μmol/L | ND | 178 | |
| DDO-5936 | 2019 | 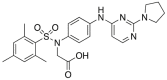 |
Binding to HSP90 N-terminus | Kd = 3.86 μmol/L | Yes | 179 |
Effective in vivo.
Not determined.
Here, based on their binding mechanisms, all of the modulators with potency to disrupt HSP90–CDC37 PPI were divided into covalent inhibitors targeting HSP90 or CDC37 and allosteric inhibitors targeting HSP90 N-terminus.
3.1.2.1. Covalent inhibitors targeting HSP90 or CDC37
Celastrol is one of the well-studied natural products with potential anti-tumor efficiency167. Its ability to downregulate the multiple signaling factors, especially kinase proteins, of HSP90 leads to make HSP90 and its co-chaperone CDC37 as one of feasible targets168. It is worthy note that celastrol could be regarded as the first evidence to disrupt HSP90–CDC37 PPI in cells by a small molecule inhibitor, although celastrol exhibit multiple targets and this consequence might be a result of diverse biological functions169. Currently, celastrol has been measured to bind to HSP90 N-terminus and C-terminus by molecular docking and fingerprinting assay, separately170,171. While, more convincing evidence showed a mechanism that celastrol covalently bounds to CDC37 through 1H–15N HSQC spectrum, thus leading to a disruption of HSP90–CDC37 PPI168. This also explained the result that celastrol inhibits HSP90 pathway through a HSP90–CDC37 PPI inhibition manner instead of binding to ATP pocket like traditional HSP90 inhibitors, leading to its potent anti-cancer effect. Based on the chemical structure of celastrol, our group also performed a SAR study, leading to CEL20 with improved activity and drug-like property172. Withaferin A (WA), is also a bioactive compound to show anti-proliferative activity by blocking HSP90 chaperone machine with a non-ATP inhibition manner. Subsequent assays discovered that WA covalently bound to HSP90 C-terminus to inhibit HSP90–CDC37 PPI through inducing the conformational change of HSP90173. In addition, another natural product Kongensin A (KA) was identified as a non-canonical HSP90 inhibitor by covalently binding to an uncharacterized cysteine 420 in the HSP90 MD, which exhibited the potency to disrupt HSP90–CDC37 PPI174. This was also the first evidence to inhibit HSP90–CDC37 PPI by covalently binding to HSP90 MD. Although these natural inhibitors could not be regarded as specific small molecule inhibitors targeting HSP90–CDC37 PPI, they provided valuable evidence to study novel mechanisms through modulating the PPIs of HSP90 and its co-chaperones instead of directly inhibiting HSP90 ATPase activity.
3.1.2.2. Allosteric inhibitors targeting HSP90 N-terminus
FW-04-804 was discovered by a chemoproteomics method to identify clinical candidates targeting HSP90. FW-04-804 disrupted HSP90–CDC37 PPI in cells and inhibited the proliferation of cancer cells by decreasing the levels of HSP90 kinase clients without ATPase inhibition. Molecular docking predicted HSP90 N-terminus as the potential binding site of FW-04-804,175. Besides randomly discovered natural products, with the development of synthetic small molecules of HSP90 ATPase inhibitors, DCZ3112 (a derivative of triazine scaffold compounds targeting HSP90 N-terminus) was found to inhibit HSP90–CDC37 PPI through directly binding to HSP90 N-terminus. Interestingly, DCZ3112 exhibited no HSP90 ATPase inhibition ability but predominantly inhibited the proliferation of HER2 positive cell lines176. Currently, VS-8 was discovered by our group through a structure-based virtual screening to simulate the key binding pattern between HSP90–CDC37177. Based on the predicted binding modes of CDC37-derived peptides, potential significant pharmacophores were established to in silico screen compound libraries, resulting in VS-8 with moderate binding affinity to HSP90 N-terminus and the ability of HSP90–CDC37 PPI inhibition in vitro (Table 4)178. Notably, a recently reported small molecule inhibitor, DDO-5936, which was also discovered by our group, showed a clear binding mode with specificity targeting HSP90–CDC37 PPI. DDO-5936 was identified to bind HSP90 with micromolar affinity in vitro and disrupt the interaction of HSP90–CDC37 through binding to Glu47 of HSP90, which was one binding determinants of HSP90–CDC37 PPI. DDO-5936 could be regarded as a first evidence to achieve therapeutic potency in colorectal cancer through a specific inhibition manner of HSP90–CDC37 PPI179. Currently, the small molecule inhibitors targeting HSP90–CDC37 PPI exhibit a feasible strategy to achieve specific modulation of kinase clients and show a promising direction for drug design based on regulation of chaperone cycles, although the binding affinity is moderate and SAR targeting on the binding interface of HSP90–CDC37 remains unclear.
3.2. Targeting the HSP90–HOP–HSP70 PPIs
3.2.1. Structures and regulation mechanisms of HSP90–HOP–HSP70 PPIs
HSP90-organizing protein (HOP, also known as Stil in yeast), is one of the best-characterized co-chaperones containing TPR domain (consisting a repeat of a 34-residue TPR motif), which simultaneously binds to HSP90 and HSP70, and functions as an adaptor to transfer client proteins from HSP70 to HSP90180,181. This transfer process is regulated by phosphorylation, which also inhibits the interaction between HSP70 and HOP182. HOP acts as a non-competitive inhibitor of HSP90 to inhibit its ATPase activity which maintains HSP90 in an open status183. Structurally, HOP contains three TPR domains and two Asp–Pro (DP)-rich domains with the following order: TPR1-DP1-TPR2A-TPR2B-DP2. Among them, it is sufficient to bind HSP90 and HSP70, as well as inhibiting the ATPase activity of HSP90, with only TPR2A-TPR2B module184. In addition, there is evidence to show that HOP selectively binds to the ADP-bound state of HSP70 by TRP1 and TPR2B, and TPR2A preferably binds to HSP90185, 186, 187. HOP promotes the conformational changes of HSP90 through binding to its MD-CTD region, leading to a stabilized and open conformation of HSP90 for subsequent client loading by HSP70 and N-terminus dimerization for ATP hydrolysis188. Thus, HOP acts as a bridge between HSP90 and HSP70 to achieve function of promoting clients folding, which has been found especially for recruiting the steroid hormone receptor clients (Fig. 7C)189. Disrupting the PPIs between HSP70–HOP or HSP90–HOP might achieve a specific modulation mechanism instead of ATPase inhibition.
Figure 7.

The structure, regulation mechanism and small molecules targeting HSP70–HOP–HSP90 chaperone. (A) Structures of different parts of HOP. TRP1 (tetratricopeptide repeat domain 1, PDB:3ESK), DP1 (aspartate-proline motif domain, PDB:2LLV), TPR2AB (tetratricopeptide repeat domains 2A and B, PDB:3UQ3), DP2 (aspartate-proline motif domain, PDB:2LLW). (B) HOP domain structures of different model organisms. No. 1 indicated the N-terminus and total number of amino acids in the proteins were indicated at the C-terminus. (C) Regulation mechanism of HSP90–HOP–HSP70 chaperone complex. HOP's TPR1 domain binds to SBD of HSP70 and TPR2AB domain binds to CTD-MD of HSP90. Three helices are involved in the interaction of binding mode, which contains 1A, 2A and 3A. (D) Chemical structures of modulators (C1 and SY8) targeting HSP70–HOP.
3.2.2. Inhibitors targeting HSP70–HOP PPI
Currently, two compounds (C1 and SY8) have been reported to directly targeting HSP70–HOP interaction (Fig. 7D). Through a rational design using the crystal structure of HOP's TRP1 domain, C1 and SY8 are expected to regulate the interaction between HSP70 and HOP based on the sequence of the interacting interface. HOP's TRP1 domain is consisted of 7 α-helix and three of them are interacting with the C-terminus of HSP70 (helix 1A, 2A and 3A)190. Interestingly, C1 and SY8 exhibit a totally different regulation mechanism although they are all designed from the critical residues on the interacting helix. C1 is designed from the helix 3A, which functions to stabilize the binding between HSP70 and HOP, and acts as an effective modulator by trapping the HSP70 to HOP as complex, finally leading to disruption of the protein folding system. On the contrary, SY8 acts as a typical peptide inhibitor to directly disrupt the interaction between HSP70 and HOP. This is mainly because of the longer size of SY8, compared to C1, which make the binding process of SY8 extending outside the region of C1191. Although the current modulators have not been comprehensively evaluated in vitro and in vivo for their potential therapeutic applications, this novel modulation mechanism between HSP70 and HOP established a new insight for the dynamic system of HSP90 chaperone machine.
3.3. HSP90–AHA1
3.3.1. AHA1 accelerates the ATPase activity of HSP90
It is known that HSP90 itself is an ATPase with low enzymatic activity, indicating a weak binding affinity between ATP and HSP9015,192. To facilitate the ATPase activity of HSP90, its co-chaperone AHA1 (also known as activator of HSP90 ATPase) acts as the most potent stimulator of the low ATPase activity of HSP90193,194. In mammalian cells, AHA1 helps HSP90 to activate kinases and fold membrane proteins195. Structurally, AHA1 is consisted of two major domains including a N-terminus domain with 156 residues and a C-terminus domain with similar size, connecting by a loose structured linker (Fig. 8B). It has been certified that N-terminus domain of AHA1 interacts with the MD of HSP90 and leads to a conformation rearrangement in the NBD of HSP90196. The C-terminus domain of AHA1 interacts with the NBD dimerized region of HSP90 dimer197. Both two domains are important for ATPase stimulation of HSP90 and one AHA1 molecule per HSP90 dimer is sufficient to complete the whole process of ATPase of HSP90 (Fig. 8A). However, the detailed working mechanism of HSP90–AHA1 is currently not fully understood. For the importance of AHA1 in the HSP90 chaperone cycle, it is involved in the interaction of HSP90 with specific client proteins including protein kinases and steroid hormone receptors193,198. In addition, HSP90–AHA1 complex plays significant roles in the cystic fibrosis transmembrane conductance regulator (CFTR), especially for the quality control process of CFTR mutants. Considering the significant role of Aha1, it has been regarded as an attractively therapeutic strategy by specifically targeting HSP90–AHA1 interaction196,199,200.
Figure 8.

The regulation mechanisms, structures and small molecule inhibitors of HSP90–AHA1 chaperone complex. (A) The biological function of HSP90–AHA1 relies on accelerating the ATPase activity of HSP90. (B) Co-crystal structure of HSP90–AHA1 chaperone complex (PDB:1USU). (C) Small molecule inhibitors with the potency to modulate HSP90–AHA1 chaperone complex.
3.3.2. Inhibitors targeting HSP90-AHA1 chaperone complex
Interestingly, in 2017, several small molecule inhibitors were reported to inhibit the HSP90–AHA1 chaperone complex (Fig. 8C). HAM-1 was discovered by a FRET-based assay to obtain a desired compound targeting HSP90–AHA1 chaperone complex. Although NMR spectroscopy revealed a HSP90 N–terminal binding mechanism, instead of a direct PPI inhibiting manner, HAM-1 could affect the activation and processing of HSP90–AHA1-based client proteins in vivo201. By screening 14,440 druglike compounds, another chemical scaffold was found to disrupt HSP90–AHA1 complex using an amplified luminescence proximity homogeneous assay. Compounds A12 and A16 were identified with promising HSP90–AHA1 inhibitory effect to restore chloride channel activity in CFTRΔF508 mutant cells to be lead compounds with further development for cystic fibrosis patients202. In addition, it has been identified that a quercetin derivative, TL-2-8, induced significant cell death and immature mitophagy in breast cancer cells in vitro and in vivo by dose-dependently inhibiting the expression of AHA1203. All the above evidence revealed a feasible targeting strategy to inhibit HSP90–AHA1 chaperone complex for potential therapeutic effects.
3.4. HSP90–P23
P23/SBA1 is an important co-chaperone of HSP90, which stabilizes the closed conformation of HSP90 by binding to a dimerized region on NTDs of HSP90, playing a role in the late stage of the chaperone cycle204. P23/SBA1 is consisted of a folded Chord and SGT1 (CS) domain for binding to HSP90 NTD and an unstructured tail for its chaperone activity205, 206, 207, 208. The crystal structure of a full-length and closed state of HSP90–P23–ATP complex provided an insight into the structural information of HSP90 and the rearrangement process of NTD dimerization204. Functionally, P23/SBA1 is responsible for reducing the ATPase activity of HSP90 to regulate the progression of the reaction cycle209,210. The involvement of chaperone functions of HSP90–P23 interaction is important for a broad range of processes, including chromatin remodelling and ribosome biogenesis, which modulates the genome-wide protein–DNA binding dynamics211, 212, 213. Currently, no specific small molecule inhibitor was identified and discovered to selectively regulate HSP90–P23 chaperone complex. While, several natural products were found to impair the association of HSP90–P23 chaperone complex by directly binding to the different regions of HSP90 with various mechanisms, including EGCG, geldanamycin, celastrol and gedunin171,214, 215, 216. Silencing P23 in yeast results in improved sensitivity of structurally dissimilar natural inhibitors including geldanamycin and radicicol, while the overexpression of P23 is found in cancer to protect cells from these inhibitors217,218. Considering the difficulty of targeting HSP90–P23 PPI, it might be challenging to design small molecules to specifically regulate HSP90–P23 chaperone complex219.
3.5. PP5
The serine/threonine protein phosphatase 5 (PP5/Ppt1) regulates diverse cellular network in signaling pathway and functions as a TRP-containing co-chaperone to regulate HSP90 conformational cycle through a dephosphorylation mechanism220. It is known that phosphorylation of HSP90 influences on its conformational dynamics and plays significant roles in the maturation of clients221. PP5 modulates the dephosphorylation process of HSP90 as well as its co-chaperone CDC37, leading to autoinhibition of itself when forming a complex with HSP90222. Modulating the function of PP5 also affect the process of chaperone cycle.
4. Future directions and conclusions
For the past 30 years, much of the work on molecular chaperone has been performed to study the process of protein folding and stabilization. HSPs family and their co-chaperones attracted attentions due to the fundamental role for maintain homeostasis under stressed or changing environmental conditions. Targeting molecular chaperone machinery is becoming a feasible way to modulate the content of diverse proteins in different signaling pathways. Among them, HSP90 could be regarded as the most functional chaperone which yielded more than 20 inhibitors to enter the clinical trials. Although directly inhibiting ATPase of HSP90 could exhibit therapeutic potential against multiple cancer cells, the clinical data of recent 10 years of HSP90 inhibitor showed this is not the case, leading to no HSP90 inhibitor approved to market. For one hand, HSP90 is ubiquitously expressed (about 1%–3% of total cellular proteins) in both normal and cancer cells, which breaks a principle that ideal drug targets exhibit high correlation with malignant cells and be of low expression in normal cell and tissues, providing a possible therapeutic window for small molecule drugs223. For another hand, directly inhibiting HSP90 ATPase totally disrupts ATP hydrolysis, leading to a non-selective degradation of all the clients of HSP90, which might contribute to the toxicity of current HSP90 inhibitors.
Targeting HSP90 ATPase could be regarded as a direct way to totally inhibit the function of HSP90, thus to result in client degradation for potential therapeutic utilities. However, due to the non-selective degradation of diverse kinds of client proteins, HSP90 ATPase inhibition revealing toxicity with heat shock response in high doses and inefficacy in low doses, indicating a non-ideal therapeutic window and a tough issue in clinical trials. Recently, development of HSP90 inhibitors turns its emphasis from ATPase inhibition to other HSPs, HSP90 isoforms and protein–protein interactions between co-chaperones. All these strategies aim at improving the specificity by different regulation mechanisms to achieve therapeutic potentials with low toxicity. Currently, there are abundant data demonstrating that not only HSP90 itself, but also its different isoforms (including HSP90α, HSP90β, GRP94 and TRAP1), other components of HSPs family (including HSP70, HSP60, HSP40 and sHSPs) and the interactions between various co-chaperones play significant roles in cancer cells. As we reviewed, each of the strategies targeting different checkpoint of molecule chaperone might achieve specific therapeutic applications. Except directly inhibition of HSP90 ATPase, which results in the downregulation of all the client proteins, selective modulation of the specific molecular chaperone or disruption of the PPIs between co-chaperones achieves a more specific way to regulate certain client proteins. Based on current foundation, development of small molecules targeting molecular chaperone machinery by different mechanisms could be regarded as advantageous ways to modulate protein fate decision.
Acknowledgments
We gratefully acknowledged the financial support by National Natural Science Foundation of China (81773639, 81773581, 81872737 and 81930100); the Natural Science Foundation of Jiangsu Province (BK20190559, BK20160746 China); National Science & Technology Major Project ‘Key New Drug Creation and Manufacturing Program’ (Nos. 2018ZX09711002 and 2017ZX09302003, China); the Priority Academic Program Development of Jiangsu Higher Education Institutions; the Fundamental Research Funds for the Central Universities of China Pharmaceutical University (2632018ZD15, China); the Double First Class Innovation Team of China Pharmaceutical University (CPU2018GY02, China); Program for Outstanding Scientific and Technological Innovation Team of Jiangsu Higher Education and the Young Elite Scientists Sponsorship Program by CAST and the Jiangsu Qing Lan Project.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Zhengyu Jiang, Email: jiangzhengyucpu@163.com.
Qidong You, Email: youqd@163.com.
Author contributions
Qidong You and Zhengyu Jiang were responsible for the conception and design of the review. Lei Wang and Xiaoli Xu analyzed the literatures and summarized the results. Lei Wang drafted the manuscript. Qidong You and Zhengyu Jiang revised the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Hartl F.U., Bracher A., Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 2.Kampinga H.H., Hageman J., Vos M.J., Kubota H., Tanguay R.M., Bruford E.A. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandvold K.R., Morimoto R.I. The chemical biology of molecular chaperones—implications for modulation of proteostasis. J Mol Biol. 2015;427:2931–2947. doi: 10.1016/j.jmb.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powers E.T., Morimoto R.I., Dillin A., Kelly J.W., Balch W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 5.Rutherford S.L., Lindquist S. HSP90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 6.Koldewey P., Horowitz S., Bardwell J.C.A. Chaperone-client interactions: non-specificity engenders multifunctionality. J Biol Chem. 2017;292:120107. doi: 10.1074/jbc.R117.796862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X.Y., Subjeck J.R. High molecular weight stress proteins: identification, cloning and utilisation in cancer immunotherapy. Int J Hyperther. 2013;29:364–375. doi: 10.3109/02656736.2013.803607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuo D.M., Subjeck J., Wang X.Y. Unfolding the role of large heat shock proteins: new insights and therapeutic implications. Front Immunol. 2016;7 doi: 10.3389/fimmu.2016.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh H.J., Chen X., Subjeck J.R. HSPHSP110 protects heat-denatured proteins and confers cellular thermoresistance. J Biol Chem. 1997;272:31636–31640. doi: 10.1074/jbc.272.50.31636. [DOI] [PubMed] [Google Scholar]
- 10.Oh H.J., Easton D., Murawski M., Kaneko Y., Subjeck J.R. The chaperoning activity of HSP110—identification of functional domains by use of targeted deletions. J Biol Chem. 1999;274:15712–15718. doi: 10.1074/jbc.274.22.15712. [DOI] [PubMed] [Google Scholar]
- 11.Dorard C., de Thonel A., Collura A., Marisa L., Svrcek M., Lagrange A. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med. 2011;17:1283–1289. doi: 10.1038/nm.2457. [DOI] [PubMed] [Google Scholar]
- 12.Wu J., Liu T., Rios Z., Mei Q., Lin X., Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci. 2017;38:226–256. doi: 10.1016/j.tips.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 13.de Zwaan D.C., Freeman B.C. HSP90 manages the ends. Trends Biochem Sci. 2010;35:384–391. doi: 10.1016/j.tibs.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayer M.P., Le Breton L. HSP90: breaking the symmetry. Mol Cell. 2015;58:8–20. doi: 10.1016/j.molcel.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 15.Prodromou C., Roe S.M., O'Brien R., Ladbury J.E., Piper P.W., Pearl L.H. Identification and structural characterization of the ATP/ADP-binding site in the HSP90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 16.Wayne N., Bolon D.N. Dimerization of HSP90 is required for in vivo function. Design and analysis of monomers and dimers. J Biol Chem. 2007;282:35386–35395. doi: 10.1074/jbc.M703844200. [DOI] [PubMed] [Google Scholar]
- 17.Jahn M., Rehn A., Pelz B., Hellenkamp B., Richter K., Rief M. The charged linker of the molecular chaperone HSP90 modulates domain contacts and biological function. Proc Natl Acad Sci U S A. 2014;111:17881–17886. doi: 10.1073/pnas.1414073111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pratt W.B. The HSP90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med. 1998;217:420–434. doi: 10.3181/00379727-217-44252. [DOI] [PubMed] [Google Scholar]
- 19.Chiosis G., Dickey C.A., Johnson J.L. A global view of HSP90 functions. Nat Struct Mol Biol. 2013;20:1–4. doi: 10.1038/nsmb.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pratt W.B., Toft D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 21.Li Y., Zhang T., Schwartz S.J., Sun D. New developments in HSP90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updates. 2009;12:17–27. doi: 10.1016/j.drup.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitesell L., Lindquist S.L. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 23.Carpenter R.L., Gokmen-Polar Y. HSF1 as a cancer biomarker and therapeutic target. Curr Cancer Drug Targets. 2019;19:515–524. doi: 10.2174/1568009618666181018162117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trepel J., Mollapour M., Giaccone G., Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhat R., Tummalapalli S.R., Rotella D.P. Progress in the discovery and development of heat shock protein 90 (HSP90) inhibitors. J Med Chem. 2014;57:8718–8728. doi: 10.1021/jm500823a. [DOI] [PubMed] [Google Scholar]
- 26.Biamonte M.A., Van de Water R., Arndt J.W., Scannevin R.H., Perret D., Lee W.C. Heat shock protein 90: inhibitors in clinical trials. J Med Chem. 2010;53:3–17. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Carbonero R., Carnero A., Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14:e358–e369. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 28.Stebbins C.E., Russo A.A., Schneider C., Rosen N., Hartl F.U., Pavletich N.P. Crystal structure of an HSP90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 29.Jagannath S., Siegel D., Richardson P., Mazumder A., Sydor J., Goddard J. Phase I clinical trial of IPI-504, a novel, water-soluble HSP90 inhibitor, in patients with relapsed/refractory multiple myeloma (MM) Blood. 2005;106:719a–720a. [Google Scholar]
- 30.Mitsiades C.S., Mitsiades N., Rooney M., Negri J., Geer C.C., Pink M. IPI-504: a novel HSP90 inhibitor with in vitro and in vivo antitumor activity. Blood. 2004;104:660a. [Google Scholar]
- 31.Palombella V.J., Normant E., Ali J., Barrett J., Foley M., Gao Y. Anti-tumor activity of IPI-504, a novel HSP90 inhibitor in multiple myeloma. Blood. 2004;104:312b–313b. [Google Scholar]
- 32.Woodhead A.J., Angove H., Carr M.G., Chessari G., Congreve M., Coyle J.E. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone HSP90 by dragment based drug design. J Med Chem. 2010;53:5956–5969. doi: 10.1021/jm100060b. [DOI] [PubMed] [Google Scholar]
- 33.Varticovski L., Wright M.H., Caldas-Lopes E., Chiosis G., Robles A.I. Synergy of the purine-scaffold HSP90 inhibitor, PU-H71, with doxorubicin in non-Hodgkin's lymphoma cell lines. Blood. 2007;110:420a. [Google Scholar]
- 34.Steed R., Huang H., Fadden P., Rice J., Eaves J., Barbasz A. SNX-2112: a novel, selective, potent small molecule inhibitor of HSP90 with unique pharmacodynamic properties. EJC Suppl. 2006;4:165. [Google Scholar]
- 35.Eccles S.A., Massey A., Raynaud F.I., Sharp S.Y., Box G., Valenti M. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 36.Bao R., Lai C.J., Qu H., Wang D., Yin L., Zifcak B. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin Cancer Res. 2009;15:4046–4057. doi: 10.1158/1078-0432.CCR-09-0152. [DOI] [PubMed] [Google Scholar]
- 37.Shimamura T., Perera S.A., Foley K.P., Sang J., Rodig S.J., Inoue T. Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor, has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clin Cancer Res. 2012;18:4973–4985. doi: 10.1158/1078-0432.CCR-11-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang F., Wang H.J., Jin Y.H., Zhang Q., Wang Z.H., Jia J.M. Novel tetrahydropyrido[4,3-d]pyrimidines as potent inhibitors of chaperone heat shock protein 90. J Med Chem. 2016;59:10498–10519. doi: 10.1021/acs.jmedchem.6b00912. [DOI] [PubMed] [Google Scholar]
- 39.Li L., Wang L., You Q.D., Xu X.L. Heat shock protein 90 (HSP90) inhibitors: an update on achievements, challenges, and future directions. J Med Chem. 2020;63:1798–1822. doi: 10.1021/acs.jmedchem.9b00940. [DOI] [PubMed] [Google Scholar]
- 40.Weikl T., Muschler P., Richter K., Veit T., Reinstein J., Buchner J. C-terminal regions of HSP90 are important for trapping the nucleotide during the ATPase cycle. J Mol Biol. 2000;303:583–592. doi: 10.1006/jmbi.2000.4157. [DOI] [PubMed] [Google Scholar]
- 41.Yu X.M., Shen G., Neckers L., Blake H., Holzbeierlein J., Cronk B. HSP90 inhibitors identified from a library of novobiocin analogues. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 42.Jiang F., Guo A.P., Xu J.C., Wang H.J., Mo X.F., You Q.D. Identification and optimization of novel 6-acylamino-2-aminoquinolines as potent HSP90 C-terminal inhibitors. Eur J Med Chem. 2017;141:1–14. doi: 10.1016/j.ejmech.2017.07.080. [DOI] [PubMed] [Google Scholar]
- 43.Gewirth D.T. Paralog specific HSP90 inhibitors—a brief history and a bright future. Curr Top Med Chem. 2016;16:2779–2791. doi: 10.2174/1568026616666160413141154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ernst J.T., Liu M., Zuccola H., Neubert T., Beaumont K., Turnbull A. Correlation between chemotype-dependent binding conformations of HSP90alpha/beta and isoform selectivity-implications for the structure-based design of HSP90alpha/beta selective inhibitors for treating neurodegenerative diseases. Bioorg Med Chem Lett. 2014;24:204–208. doi: 10.1016/j.bmcl.2013.11.036. [DOI] [PubMed] [Google Scholar]
- 45.Ohkubo S., Kodama Y., Muraoka H., Hitotsumachi H., Yoshimura C., Kitade M. TAS-116, a highly selective inhibitor of heat shock protein 90alpha and beta, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Canc Therapeut. 2015;14:14–22. doi: 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- 46.Khandelwal A., Kent C.N., Balch M., Peng S., Mishra S.J., Deng J. Structure-guided design of an HSP90beta N-terminal isoform-selective inhibitor. Nat Commun. 2018;9:425. doi: 10.1038/s41467-017-02013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu B.X., Hong F., Zhang Y.L., Ansa-Addo E., Li Z.H. GRP94/gp96 in cancer: biology, structure, immunology, and drug development. Adv Cancer Res. 2016;129:165–190. doi: 10.1016/bs.acr.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Jiang F., Guo A.P., Xu J.C., You Q.D., Xu X.L. Discovery of a potent GRP94 selective inhibitor with anti-inflammatory efficacy in a mouse model of ulcerative colitis. J Med Chem. 2018;61:9513–9533. doi: 10.1021/acs.jmedchem.8b00800. [DOI] [PubMed] [Google Scholar]
- 49.Kang B.H., Plescia J., Song H.Y., Meli M., Colombo G., Beebe K. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial HSP90. J Clin Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park H.K., Jeong H., Ko E., Lee G., Lee J.E., Lee S.K. Paralog specificity determines subcellular distribution, action mechanism, and anticancer activity of TRAP1 inhibitors. J Med Chem. 2017;60:7569–7578. doi: 10.1021/acs.jmedchem.7b00978. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y.Y., Favoino E., Yu L., Ferrone C.R., Ferrone S., Wang X.H. Vol. 72. 2012. Abstract 4390: heat shock protein (HSP) GRP94-targeted combinatorial immunotherapy for pancreatic cancer; pp. AM2012–AM4390. (Proceedings of AACR 103rd annual meeting, 2012 mar 31–apr 4; Chicago, IL, USA. Cancer Res). [Google Scholar]
- 52.Michelakos T., Cai L., Goyal L., Zhu A.X., Tanabe K.K., Ferrone S. Glucose-regulated protein of 94kDa (GRP94)-targeted antibody-based combinatorial immunotherapy for intrahepatic cholangiocarcinoma. J Am Coll Surg. 2016;223:E136–E137. [Google Scholar]
- 53.Shen Y.L., Liu B., Zhang P.L. GRP94, a chaperone protein, represents a new target for treating multiple myeloma. Lab Invest. 2017;97:377a. [Google Scholar]
- 54.Mishra S.J., Ghosh S., Stothert A.R., Dickey C.A., Blagg B.S. Transformation of the non-selective aminocyclohexanol-based HSP90 inhibitor into a GRP94-seletive scaffold. ACS Chem Biol. 2017;12:244–253. doi: 10.1021/acschembio.6b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel H.J., Patel P.D., Ochiana S.O., Yan P., Sun W., Patel M.R. Structure–activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum HSP90 paralog GRP94. J Med Chem. 2015;58:3922–3943. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altieri D.C., Stein G.S., Lian J.B., Languino L.R. TRAP-1, the mitochondrial HSP90. Biochim Biophys Acta. 2012;1823:767–773. doi: 10.1016/j.bbamcr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Felts S.J., Owen B.A.L., Nguyen P., Trepel J., Donner D.B., Toft D.O. The HSP90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275:3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- 58.Lavery L.A., Partridge J.R., Ramelot T.A., Elnatan D., Kennedy M.A., Agard D.A. Structural asymmetry in the closed state of mitochondrial HSP90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol Cell. 2014;53:330–343. doi: 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee C.W., Park H.K., Jeong H., Lim J., Lee A.J., Cheon K.Y. Development of a mitochondria-targeted HSP90 inhibitor based on the crystal atructures of human TRAP1. J Am Chem Soc. 2015;137:4358–4367. doi: 10.1021/ja511893n. [DOI] [PubMed] [Google Scholar]
- 60.Rondanin R., Lettini G., Oliva P., Baruchello R., Costantini C., Trapella C. New TRAP1 and HSP90 chaperone inhibitors with cationic components: preliminary studies on mitochondrial targeting. Bioorg Med Chem Lett. 2018;28:2289–2293. doi: 10.1016/j.bmcl.2018.05.031. [DOI] [PubMed] [Google Scholar]
- 61.Vo V.T., Phan A.N., Hua T.N., Jeong Y., Kang B.H., Kim H.W. Vol. 77. 2017. Abstract 5434: tumor necrosis factor receptor-associated protein 1 (TRAP1) as a potential target for glutamine addicted cancer cells; pp. AM2017–AM5434. (Proceedings of AACR annual meeting 2017 April 1–5; Washington D.C., USA. Cancer Res). [Google Scholar]
- 62.Landriscina M., Condelli V., Maddalena F., Sisinni L., Piscazzi A., Lettini G. TRAP1 is a novel molecular target in BRAF-driven human colorectal carcinomas. Eur J Cancer. 2015;51:S394. [Google Scholar]
- 63.Lettini G., Maddalena F., Sisinni L., Condelli V., Matassa D.S., Costi M.P. TRAP1: a viable therapeutic target for future cancer treatments?. Expert Opin Ther Targets. 2017;21:805–815. doi: 10.1080/14728222.2017.1349755. [DOI] [PubMed] [Google Scholar]
- 64.Daugaard M., Rohde M., Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 65.Murphy M.E. The HSP70 family and cancer. Carcinogenesis. 2013;34:1181–1188. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stricher F., Macri C., Ruff M., Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9:1937–1954. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- 67.Bertelsen E.B., Chang L., Gestwicki J.E., Zuiderweg E.R. Solution conformation of wild-type E. coli HSP70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bork P., Sander C., Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and HSP70 heat shock proteins. Proc Natl Acad Sci U S A. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu X., Zhao X., Burkholder W.F., Gragerov A., Ogata C.M., Gottesman M.E. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bukau B., Weissman J., Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 71.Pierpaoli E.V., Sandmeier E., Baici A., Schonfeld H.J., Gisler S., Christen P. The power stroke of the DnaK/DnaJ/GRPE molecular chaperone system. J Mol Biol. 1997;269:757–768. doi: 10.1006/jmbi.1997.1072. [DOI] [PubMed] [Google Scholar]
- 72.Blatch G.L., Lassle M. The tetratricopeptide repeat: a structural motif mediating protein–protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 73.Erbse A., Mayer M.P., Bukau B. Mechanism of substrate recognition by HSP70 chaperones. Biochem Soc Trans. 2004;32:617–621. doi: 10.1042/BST0320617. [DOI] [PubMed] [Google Scholar]
- 74.Xing Y., Bocking T., Wolf M., Grigorieff N., Kirchhausen T., Harrison S.C. Structure of clathrin coat with bound Hsc70 and auxilin: mechanism of Hsc70-facilitated disassembly. EMBO J. 2010;29:655–665. doi: 10.1038/emboj.2009.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 76.Schaffitzel E., Rudiger S., Bukau B., Deuerling E. Functional dissection of trigger factor and DnaK: interactions with nascent polypeptides and thermally denatured proteins. Biol Chem. 2001;382:1235–1243. doi: 10.1515/BC.2001.154. [DOI] [PubMed] [Google Scholar]
- 77.Pratt W.B., Toft D.O. Regulation of signaling protein function and trafficking by the HSP90/HSP70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 78.Young J.C., Barral J.M., Ulrich Hartl F. More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci. 2003;28:541–547. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 79.Chiang H.L., Terlecky S.R., Plant C.P., Dice J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 80.Evans C.G., Chang L., Gestwicki J.E. Heat shock protein 70 (HSP70) as an emerging drug target. J Med Chem. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kirkegaard T., Roth A.G., Petersen N.H., Mahalka A.K., Olsen O.D., Moilanen I. HSP70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 82.Mosser D.D., Morimoto R.I. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907–2918. doi: 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- 83.Powers M.V., Jones K., Barillari C., Westwood I., van Montfort R.L., Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone?. Cell Cycle. 2010;9:1542–1550. doi: 10.4161/cc.9.8.11204. [DOI] [PubMed] [Google Scholar]
- 84.Assimon V.A., Gillies A.T., Rauch J.N., Gestwicki J.E. HSP70 protein complexes as drug targets. Curr Pharmaceut Des. 2013;19:404–417. doi: 10.2174/138161213804143699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Macias A.T., Williamson D.S., Allen N., Borgognoni J., Clay A., Daniels Z. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (GRP78) ATPase: insights into isoform selectivity. J Med Chem. 2011;54:4034–4041. doi: 10.1021/jm101625x. [DOI] [PubMed] [Google Scholar]
- 86.Williamson D.S., Borgognoni J., Clay A., Daniels Z., Dokurno P., Drysdale M.J. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J Med Chem. 2009;52:1510–1513. doi: 10.1021/jm801627a. [DOI] [PubMed] [Google Scholar]
- 87.Tang X., Tan L., Shi K., Peng J., Xiao Y., Li W. Gold nanorods together with HSP inhibitor-VER-155008 micelles for colon cancer mild-temperature photothermal therapy. Acta Pharm Sin B. 2018;8:587–601. doi: 10.1016/j.apsb.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kita K., Shiota M., Tanaka M., Otsuka A., Matsumoto M., Kato M. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci. 2017;108:1820–1827. doi: 10.1111/cas.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wen W., Liu W., Shao Y., Chen L. VER-155008, a small molecule inhibitor of HSP70 with potent anti-cancer activity on lung cancer cell lines. Exp Biol Med. 2014;239:638–645. doi: 10.1177/1535370214527899. [DOI] [PubMed] [Google Scholar]
- 90.Shan L.P., Chen X.H., Ling F., Zhu B., Wang G.X. Targeting heat shock protein 70 as an antiviral strategy against grass carp reovirus infection. Virus Res. 2018;247:1–9. doi: 10.1016/j.virusres.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 91.Yang X., Tohda C. Heat shock cognate 70 inhibitor, VER-155008, reduces memory deficits and axonal degeneration in a mouse model of Alzheimer's disease. Front Pharmacol. 2018;9:48. doi: 10.3389/fphar.2018.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pettinger J., Le Bihan Y.V., Widya M., van Montfort R.L., Jones K., Cheeseman M.D. An irreversible inhibitor of HSP72 that unexpectedly targets Lysine-56. Angew Chem Int Ed Engl. 2017;56:3536–3540. doi: 10.1002/anie.201611907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ko S.K., Kim J., Na D.C., Park S., Park S.H., Hyun J.Y. A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem Biol. 2015;22:391–403. doi: 10.1016/j.chembiol.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 94.Fewell S.W., Smith C.M., Lyon M.A., Dumitrescu T.P., Wipf P., Day B.W. Small molecule modulators of endogenous and co-chaperone-stimulated HSP70 ATPase activity. J Biol Chem. 2004;279:51131–51140. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- 95.Kampinga H.H., Craig E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huryn D.M., Brodsky J.L., Brummond K.M., Chambers P.G., Eyer B., Ireland A.W. Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (HSP70) modulators. Proc Natl Acad Sci U S A. 2011;108:6757–6762. doi: 10.1073/pnas.1015251108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sabnis A.J., Guerriero C.J., Olivas V., Sayana A., Shue J., Flanagan J. Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc Natl Acad Sci U S A. 2016;113:9015–9020. doi: 10.1073/pnas.1603883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gowda N.K.C., Kaimal J.M., Kityk R., Daniel C., Liebau J., Ohman M. Nucleotide exchange factors Fes1 and HSPBP1 mimic substrate to release misfolded proteins from HSP70. Nat Struct Mol Biol. 2018;25:83–89. doi: 10.1038/s41594-017-0008-2. [DOI] [PubMed] [Google Scholar]
- 99.Young Z.T., Rauch J.N., Assimon V.A., Jinwal U.K., Ahn M., Li X. Stabilizing the HSP70-Tau complex promotes turnover in models of tauopathy. Cell Chem Biol. 2016;23:992–1001. doi: 10.1016/j.chembiol.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abbas-Terki T., Donze O., Picard D. The molecular chaperone CDC37 is required for Ste11 function and pheromone-induced cell cycle arrest. FEBS Lett. 2000;467:111–116. doi: 10.1016/s0014-5793(00)01134-0. [DOI] [PubMed] [Google Scholar]
- 101.Howe M.K., Bodoor K., Carlson D.A., Hughes P.F., AlwarawrahY, Loiselle D.R. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem Biol. 2014;21:1648–1659. doi: 10.1016/j.chembiol.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leu J.I., Pimkina J., Frank A., Murphy M.E., George D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hassan A.Q., Kirby C.A., Zhou W., Schuhmann T., Kityk R., Kipp D.R. The novolactone natural product disrupts the allosteric regulation of HSP70. Chem Biol. 2015;22:87–97. doi: 10.1016/j.chembiol.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 104.Wadhwa R., Sugihara T., Yoshida A., Nomura H., Reddel R.R., Simpson R. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the HSP70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–6821. [PubMed] [Google Scholar]
- 105.Rousaki A., Miyata Y., Jinwal U.K., Dickey C.A., Gestwicki J.E., Zuiderweg E.R. Allosteric drugs: the interaction of antitumor compound MKT-077 with human HSP70 chaperones. J Mol Biol. 2011;411:614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Abisambra J., Jinwal U.K., Miyata Y., Rogers J., Blair L., Li X. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatr. 2013;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Taguwa S., Maringer K., Li X., Bernal-Rubio D., Rauch J.N., Gestwicki J.E. Defining HSP70 subnetworks in Dengue virus replication reveals key vulnerability in flavivirus infection. Cell. 2015;163:1108–1123. doi: 10.1016/j.cell.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moses M.A., Kim Y.S., Rivera-Marquez G.M., Oshima N., Watson M.J., Beebe K.E. Targeting the HSP40/HSP70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res. 2018;78:4022–4035. doi: 10.1158/0008-5472.CAN-17-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Colvin T.A., Gabai V.L., Gong J., Calderwood S.K., Li H., Gummuluru S. HSP70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014;74:4731–4740. doi: 10.1158/0008-5472.CAN-14-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li X., Shao H., Taylor I.R., Gestwicki J.E. Targeting allosteric control mechanisms in heat shock protein 70 (HSP70) Curr Top Med Chem. 2016;16:2729–2740. doi: 10.2174/1568026616666160413140911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jego G., Hazoume A., Seigneuric R., Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332:275–285. doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 112.Pace A., Barone G., Lauria A., Martorana A., Piccionello A.P., Pierro P. HSP60, a novel target for antitumor therapy: structure-function features and prospective drugs design. Curr Pharmaceut Des. 2013;19:2757–2764. doi: 10.2174/1381612811319150011. [DOI] [PubMed] [Google Scholar]
- 113.Sigler P.B., Xu Z.H., Rye H.S., Burston S.G., Fenton W.A., Horwich A.L. Structure and function in GroEL-mediated protein folding. Annu Rev Biochem. 1998;67:581–608. doi: 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]
- 114.Ohashi S., Atsumi M., Kobayashi S. HSP60 interacts with YB-1 and affects its polysome association and subcellular localization. Biochem Biophys Res Commun. 2009;385:545–550. doi: 10.1016/j.bbrc.2009.05.094. [DOI] [PubMed] [Google Scholar]
- 115.Liffers S.T., Maghnouj A., Munding J.B., Jackstadt R., Herbrand U., Schulenborg T. Keratin 23, a novel DPC4/Smad4 target gene which binds 14-3-3epsilon. BMC Cancer. 2011;11:137. doi: 10.1186/1471-2407-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lin C.S., He P.J., Hsu W.T., Wu M.S., Wu C.J., Shen H.W. Helicobacter pylori-derived heat shock protein 60 enhances angiogenesis via a CXCR2-mediated signaling pathway. Biochem Biophys Res Commun. 2010;397:283–289. doi: 10.1016/j.bbrc.2010.05.101. [DOI] [PubMed] [Google Scholar]
- 117.Tsai Y.P., Teng S.C., Wu K.J. Direct regulation of HSP60 expression by c-MYC induces transformation. FEBS Lett. 2008;582:4083–4088. doi: 10.1016/j.febslet.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 118.Chaiwatanasirikul K.A., Sala A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011;2:e219. doi: 10.1038/cddis.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ghosh J.C., Siegelin M.D., Dohi T., Altieri D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010;70:8988–8993. doi: 10.1158/0008-5472.CAN-10-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ruan W., Wang Y., Ma Y., Xing X., Lin J., Cui J. HSP60, a protein downregulated by IGFBP7 in colorectal carcinoma. J Exp Clin Cancer Res. 2010;29:41. doi: 10.1186/1756-9966-29-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chun J.N., Choi B., Lee K.W., Lee D.J., Kang D.H., Lee J.Y. Cytosolic HSP60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tsai Y.P., Yang M.H., Huang C.H., Chang S.Y., Chen P.M., Liu C.J. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis. 2009;30:1049–1057. doi: 10.1093/carcin/bgp087. [DOI] [PubMed] [Google Scholar]
- 123.Lianos G.D., Alexiou G.A., Mangano A., Mangano A., Rausei S., Boni L. The role of heat shock proteins in cancer. Cancer Lett. 2015;360:114–118. doi: 10.1016/j.canlet.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 124.Alagramam K.N., Gopal S.R., Geng R., Chen D.H., Nemet I., Lee R. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat Chem Biol. 2016;12:444–451. doi: 10.1038/nchembio.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ban H.S., Shimizu K., Minegishi H., Nakamura H. Identification of HSP60 as a primary target of o-carboranylphenoxyacetanilide, an HIF-1alpha inhibitor. J Am Chem Soc. 2010;132:11870–11871. doi: 10.1021/ja104739t. [DOI] [PubMed] [Google Scholar]
- 126.Polson E.S., Kuchler V.B., Abbosh C., Ross E.M., Mathew R.K., Beard H.A. KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci Transl Med. 2018;10 doi: 10.1126/scitranslmed.aar2718. eaar2718. [DOI] [PubMed] [Google Scholar]
- 127.Cassiano C., Monti M.C., Festa C., Zampella A., Riccio R., Casapullo A. Chemical proteomics reveals heat shock protein 60 to be the main cellular target of the marine bioactive sesterterpene suvanine. Chembiochem. 2012;13:1953–1958. doi: 10.1002/cbic.201200291. [DOI] [PubMed] [Google Scholar]
- 128.Nagumo Y., Kakeya H., Shoji M., Hayashi Y., Dohmae N., Osada H. Epolactaene binds human HSP60 Cys442 resulting in the inhibition of chaperone activity. Biochem J. 2005;387:835–840. doi: 10.1042/BJ20041355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wiechmann K., Muller H., Konig S., Wielsch N., Svatos A., Jauch J. Mitochondrial chaperonin HSP60 is the apoptosis-related target for myrtucommulone. Cell Chem Biol. 2017;24:614–623. doi: 10.1016/j.chembiol.2017.04.008. e6. [DOI] [PubMed] [Google Scholar]
- 130.Cappello F., Conway de Macario E., Marasa L., Zummo G., Macario A.J. HSP60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol Ther. 2008;7:801–809. doi: 10.4161/cbt.7.6.6281. [DOI] [PubMed] [Google Scholar]
- 131.Nahleh Z., Tfayli A., Najm A., El Sayed A., Nahle Z. Heat shock proteins in cancer: targeting the 'chaperones'. Future Med Chem. 2012;4:927–935. doi: 10.4155/fmc.12.50. [DOI] [PubMed] [Google Scholar]
- 132.Chatterjee S., Burns T.F. Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci. 2017;18:E1978. doi: 10.3390/ijms18091978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kappe G., Franck E., Verschuure P., Boelens W.C., Leunissen J.A., de Jong W.W. The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HSPB1-10. Cell Stress Chaperones. 2003;8:53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kriehuber T., Rattei T., Weinmaier T., Bepperling A., Haslbeck M., Buchner J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010;24:3633–3642. doi: 10.1096/fj.10-156992. [DOI] [PubMed] [Google Scholar]
- 135.Franck E., Madsen O., van Rheede T., Ricard G., Huynen M.A., de Jong W.W. Evolutionary diversity of vertebrate small heat shock proteins. J Mol Evol. 2004;59:792–805. doi: 10.1007/s00239-004-0013-z. [DOI] [PubMed] [Google Scholar]
- 136.Hata M., Ohtsuka K. Characterization of HSE sequences in human HSP40 gene: structural and promoter analysis. Biochim Biophys Acta. 1998;1397:43–55. doi: 10.1016/s0167-4781(97)00208-x. [DOI] [PubMed] [Google Scholar]
- 137.Sterrenberg J.N., Blatch G.L., Edkins A.L. Human DNAJ in cancer and stem cells. Cancer Lett. 2011;312:129–142. doi: 10.1016/j.canlet.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 138.Trinh D.L., Elwi A.N., Kim S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget. 2010;1:396–404. doi: 10.18632/oncotarget.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang C.C., Tsai M.F., Hong T.M., Chang G.C., Chen C.Y., Yang W.M. The transcriptional factor YY1 upregulates the novel invasion suppressor HLJ1 expression and inhibits cancer cell invasion. Oncogene. 2005;24:4081–4093. doi: 10.1038/sj.onc.1208573. [DOI] [PubMed] [Google Scholar]
- 140.Zhang L., Cai X., Chen K., Wang Z., Wang L., Ren M. Hepatitis B virus protein up-regulated HLJ1 expression via the transcription factor YY1 in human hepatocarcinoma cells. Virus Res. 2011;157:76–81. doi: 10.1016/j.virusres.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 141.Yang T., Li X.N., Li X.G., Li M., Gao P.Z. DNAJC6 promotes hepatocellular carcinoma progression through induction of epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;455:298–304. doi: 10.1016/j.bbrc.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 142.Nishizawa S., Hirohashi Y., Torigoe T., Takahashi A., Tamura Y., Mori T. HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res. 2012;72:2844–2854. doi: 10.1158/0008-5472.CAN-11-3062. [DOI] [PubMed] [Google Scholar]
- 143.Castle P.E., Ashfaq R., Ansari F., Muller C.Y. Immunohistochemical evaluation of heat shock proteins in normal and preinvasive lesions of the cervix. Cancer Lett. 2005;229:245–252. doi: 10.1016/j.canlet.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 144.Isomoto H., Oka M., Yano Y., Kanazawa Y., Soda H., Terada R. Expression of heat shock protein (HSP) 70 and HSP 40 in gastric cancer. Cancer Lett. 2003;198:219–228. doi: 10.1016/s0304-3835(03)00305-7. [DOI] [PubMed] [Google Scholar]
- 145.Yokota S., Kitahara M., Nagata K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000;60:2942–2948. [PubMed] [Google Scholar]
- 146.de La Motte Rouge T., Galluzzi L., Olaussen K.A., Zermati Y., Tasdemir E., Robert T. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res. 2007;67:6253–6262. doi: 10.1158/0008-5472.CAN-07-0538. [DOI] [PubMed] [Google Scholar]
- 147.Izbicka E., Campos D., Carrizales G., Patnaik A. Biomarkers of anticancer activity of R115777 (Tipifarnib, Zarnestra) in human breast cancer models in vitro. Anticancer Res. 2005;25:3215–3223. [PubMed] [Google Scholar]
- 148.Vidyasagar A., Wilson N.A., Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair. 2012;5:7. doi: 10.1186/1755-1536-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cayado-Gutierrez N., Moncalero V.L., Rosales E.M., Beron W., Salvatierra E.E., Alvarez-Olmedo D. Downregulation of HSP27 (HSPB1) in MCF-7 human breast cancer cells induces upregulation of PTEN. Cell Stress Chaperones. 2013;18:243–249. doi: 10.1007/s12192-012-0367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghayour-Mobarhan M., Saber H., Ferns G.A. The potential role of heat shock protein 27 in cardiovascular disease. Clin Chim Acta. 2012;413:15–24. doi: 10.1016/j.cca.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 151.Abisambra J.F., Blair L.J., Hill S.E., Jones J.R., Kraft C., Rogers J. Phosphorylation dynamics regulate HSP27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 2010;30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shiota M., Bishop J.L., Nip K.M., Zardan A., Takeuchi A., Cordonnier T. HSP27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013;73:3109–3119. doi: 10.1158/0008-5472.CAN-12-3979. [DOI] [PubMed] [Google Scholar]
- 153.Gray P.J., Jr., Prince T., Cheng J., Stevenson M.A., Calderwood S.K. Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer. 2008;8:491–495. doi: 10.1038/nrc2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Caplan A.J., Ma'ayan A., Willis I.M. Multiple kinases and system robustness: a link between CDC37 and genome integrity. Cell Cycle. 2007;6:3145–3147. doi: 10.4161/cc.6.24.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pearl L.H. HSP90 and CDC37—a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 156.Caplan A.J., Mandal A.K., Theodoraki M.A. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 157.Robzyk K., Oen H., Buchanan G., Butler L.M., Tilley W.D., Mandal A.K. Uncoupling of hormone-dependence from chaperone-dependence in the L701H mutation of the androgen receptor. Mol Cell Endocrinol. 2007;268:67–74. doi: 10.1016/j.mce.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shao J., Irwin A., Hartson S.D., Matts R.L. Functional dissection of CDC37: characterization of domain structure and amino acid residues critical for protein kinase binding. Biochemistry. 2003;42:12577–12588. doi: 10.1021/bi035138j. [DOI] [PubMed] [Google Scholar]
- 159.Lee P., Rao J., Fliss A., Yang E., Garrett S., Caplan A.J. The CDC37 protein kinase-binding domain is sufficient for protein kinase activity and cell viability. J Cell Biol. 2002;159:1051–1059. doi: 10.1083/jcb.200210121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Verba K.A., Wang R.Y.R., Arakawa A., Liu Y.X., Shirouzu M., Yokoyama S. Atomic structure of HSP90-CDC37-Cdk4 reveals that HSP90 traps and stabilizes an unfolded kinase. Science. 2016;352:1542–1547. doi: 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Feo F., de Miglio M.R., Simile M.M., Muroni M.R., Calvisi D.F., Frau M. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta. 2006;1765:126–147. doi: 10.1016/j.bbcan.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 162.Casas S., Ollila J., Aventin A., Vihinen M., Sierra J., Knuutila S. Changes in apoptosis-related pathways in acute myelocytic leukemia. Cancer Genet Cytogenet. 2003;146:89–101. doi: 10.1016/s0165-4608(03)00102-x. [DOI] [PubMed] [Google Scholar]
- 163.Thompson M.A., Stumph J., Henrickson S.E., Rosenwald A., Wang Q., Olson S. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36:494–504. doi: 10.1016/j.humpath.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 164.Stepanova L., Yang G., DeMayo F., Wheeler T.M., Finegold M., Thompson T.C. Induction of human CDC37 in prostate cancer correlates with the ability of targeted CDC37 expression to promote prostatic hyperplasia. Oncogene. 2000;19:2186–2193. doi: 10.1038/sj.onc.1203561. [DOI] [PubMed] [Google Scholar]
- 165.Waza M., Adachi H., Katsuno M., Minamiyama M., Tanaka F., Doyu M. Modulation of HSP90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med (Berl) 2006;84:635–646. doi: 10.1007/s00109-006-0066-0. [DOI] [PubMed] [Google Scholar]
- 166.Wang L., Li L., Gu K., Xu X.L., Sun Y., You Q.D. Targeting HSP90-CDC37: a promising therapeutic strategy by inhibiting HSP90 chaperone function. Curr Drug Targets. 2017;18:1572–1585. doi: 10.2174/1389450117666160527125522. [DOI] [PubMed] [Google Scholar]
- 167.Boridy S., Le P.U., Petrecca K., Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014;5:e1216. doi: 10.1038/cddis.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sreeramulu S., Gande S.L., Gobel M., Schwalbe H. Molecular mechanism of inhibition of the human protein complex HSP90–CDC37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl. 2009;48:5853–5855. doi: 10.1002/anie.200900929. [DOI] [PubMed] [Google Scholar]
- 169.Kannaiyan R., Shanmugam M.K., Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 170.Zhang T., Li Y., Yu Y., Zou P., Jiang Y., Sun D. Characterization of celastrol to inhibit HSP90 and CDC37 interaction. J Biol Chem. 2009;284:35381–35389. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C.G. A novel HSP90 inhibitor to disrupt HSP90/CDC37 complex against pancreatic cancer cells. Mol Canc Therapeut. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 172.Jiang F., Wang H.J., Bao Q.C., Wang L., Jin Y.H., Zhang Q. Optimization and biological evaluation of celastrol derivatives as HSP90–CDC37 interaction disruptors with improved druglike properties. Biorg Med Chem. 2016;24:5431–5439. doi: 10.1016/j.bmc.2016.08.070. [DOI] [PubMed] [Google Scholar]
- 173.Grover A., Shandilya A., Agrawal V., Pratik P., Bhasme D., Bisaria V.S. HSP90/CDC37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinf. 2011;12(1):S30. doi: 10.1186/1471-2105-12-S1-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li D., Li C., Li L., Chen S., Wang L., Li Q. Natural product kongensin A is a non-canonical HSP90 inhibitor that blocks RIP3-dependent necroptosis. Cell Chem Biol. 2016;23:257–266. doi: 10.1016/j.chembiol.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 175.Huang W., Ye M., Zhang L.R., Wu Q.D., Zhang M., Xu J.H. FW-04-806 inhibits proliferation and induces apoptosis in human breast cancer cells by binding to N-terminus of HSP90 and disrupting HSP90–CDC37 complex formation. Mol Cancer. 2014;13:150. doi: 10.1186/1476-4598-13-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chen X., Liu P., Wang Q., Li Y., Fu L., Fu H. DCZ3112, a novel HSP90 inhibitor, exerts potent antitumor activity against HER2-positive breast cancer through disruption of HSP90–CDC37 interaction. Cancer Lett. 2018;434:70–80. doi: 10.1016/j.canlet.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 177.Wang L., Li L., Zhou Z.H., Jiang Z.Y., You Q.D., Xu X.L. Structure-based virtual screening and optimization of modulators targeting HSP90-CDC37 interaction. Eur J Med Chem. 2017;136:63–73. doi: 10.1016/j.ejmech.2017.04.074. [DOI] [PubMed] [Google Scholar]
- 178.Wang L., Li L., Fu W.T., Jiang Z.Y., You Q.D., Xu X.L. Optimization and bioevaluation of CDC37-derived peptides: an insight into HSP90-CDC37 protein–protein interaction modulators. Bioorg Med Chem. 2017;25:233–240. doi: 10.1016/j.bmc.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 179.Wang L., Zhang L., Li L., Jiang J., Zheng Z., Shang J. Small-molecule inhibitor targeting the HSP90-CDC37 protein–protein interaction in colorectal cancer. Sci Adv. 2019 doi: 10.1126/sciadv.aax2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wegele H., Wandinger S.K., Schmid A.B., Reinstein J., Buchner J. Substrate transfer from the chaperone HSP70 to HSP90. J Mol Biol. 2006;356:802–811. doi: 10.1016/j.jmb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 181.Li J., Richter K., Buchner J. Mixed HSP90-cochaperone complexes are important for the progression of the reaction cycle. Nat Struct Mol Biol. 2011;18:61–66. doi: 10.1038/nsmb.1965. [DOI] [PubMed] [Google Scholar]
- 182.Rohl A., Tippel F., Bender E., Schmid A.B., Richter K., Madl T. Hop/Sti1 phosphorylation inhibits its co-chaperone function. EMBO Rep. 2015;16:240–249. doi: 10.15252/embr.201439198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Johnson B.D., Schumacher R.J., Ross E.D., Toft D.O. Hop modulates HSP70/HSP90 interactions in protein folding. J Biol Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- 184.Schmid A.B., Lagleder S., Grawert M.A., Rohl A., Hagn F., Wandinger S.K. The architecture of functional modules in the HSP90 co-chaperone Sti1/Hop. EMBO J. 2012;31:1506–1517. doi: 10.1038/emboj.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Flom G., Behal R.H., Rosen L., Cole D.G., Johnson J.L. Definition of the minimal fragments of Sti1 required for dimerization, interaction with HSP70 and HSP90 and in vivo functions. Biochem J. 2007;404:159–167. doi: 10.1042/BJ20070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Carrigan P.E., Nelson G.M., Roberts P.J., Stoffer J., Riggs D.L., Smith D.F. Multiple domains of the co-chaperone Hop are important for HSP70 binding. J Biol Chem. 2004;279:16185–16193. doi: 10.1074/jbc.M314130200. [DOI] [PubMed] [Google Scholar]
- 187.Hernandez M.P., Sullivan W.P., Toft D.O. The assembly and intermolecular properties of the HSP70-Hop-HSP90 molecular chaperone complex. J Biol Chem. 2002;277:38294–38304. doi: 10.1074/jbc.M206566200. [DOI] [PubMed] [Google Scholar]
- 188.Southworth D.R., Agard D.A. Client-loading conformation of the HSP90 molecular chaperone revealed in the cryo-EM structure of the human HSP90:Hop complex. Mol Cell. 2011;42:771–781. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Onuoha S.C., Coulstock E.T., Grossmann J.G., Jackson S.E. Structural studies on the co-chaperone Hop and its complexes with HSP90. J Mol Biol. 2008;379:732–744. doi: 10.1016/j.jmb.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 190.Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H. Structure of TPR domain-peptide complexes: critical elements in the assembly of the HSP70-HSP90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 191.Zaiter S.S., Huo Y.T., Tiew F.Y., Gestwicki J.E., McAlpine S.R. Designing de novo small molecules that control heat shock protein 70 (HSP70) and heat shock organizing protein (HOP) within the chaperone protein-folding machinery. J Med Chem. 2019;62:742–761. doi: 10.1021/acs.jmedchem.8b01436. [DOI] [PubMed] [Google Scholar]
- 192.Scheibel T., Neuhofen S., Weikl T., Mayr C., Reinstein J., Vogel P.D. ATP-binding properties of human HSP90. J Biol Chem. 1997;272:18608–18613. doi: 10.1074/jbc.272.30.18608. [DOI] [PubMed] [Google Scholar]
- 193.Meyer P., Prodromou C., Liao C., Hu B., Roe S.M., Vaughan C.K. Structural basis for recruitment of the ATPase activator Aha1 to the HSP90 chaperone machinery. EMBO J. 2004;23:1402–1410. doi: 10.1038/sj.emboj.7600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Horvat N.K., Armstrong H., Lee B.L., Mercier R., Wolmarans A., Knowles J. A mutation in the catalytic loop of HSP90 specifically impairs ATPase stimulation by Aha1p, but not Hch1p. J Mol Biol. 2014;426:2379–2392. doi: 10.1016/j.jmb.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 195.Holmes J.L., Sharp S.Y., Hobbs S., Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68:1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- 196.Koulov A.V., LaPointe P., Lu B., Razvi A., Coppinger J., Dong M.Q. Biological and structural basis for Aha1 regulation of HSP90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol Biol Cell. 2010;21:871–884. doi: 10.1091/mbc.E09-12-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H. Asymmetric activation of the HSP90 dimer by its cochaperone aha1. Mol Cell. 2010;37:344–354. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 198.Harst A., Lin H., Obermann W.M. Aha1 competes with Hop, p50 and P23 for binding to the molecular chaperone HSP90 and contributes to kinase and hormone receptor activation. Biochem J. 2005;387:789–796. doi: 10.1042/BJ20041283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang X., Venable J., LaPointe P., Hutt D.M., Koulov A.V., Coppinger J. HSP90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 200.Loo M.A., Jensen T.J., Cui L., Hou Y., Chang X.B., Riordan J.R. Perturbation of HSP90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stiegler S.C., Rubbelke M., Korotkov V.S., Weiwad M., John C., Fischer G. A chemical compound inhibiting the Aha1-HSP90 chaperone complex. J Biol Chem. 2017;292:17073–17083. doi: 10.1074/jbc.M117.797829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ihrig V., Obermann W.M.J. Identifying inhibitors of the HSP90-Aha1 protein complex, a potential target to drug cystic fibrosis, by alpha technology. SLAS Discov. 2017;22:923–928. doi: 10.1177/2472555216688312. [DOI] [PubMed] [Google Scholar]
- 203.Liu H.J., Jiang X.X., Guo Y.Z., Sun F.H., Kou X.H., Bao Y. The flavonoid TL-2-8 induces cell death and immature mitophagy in breast cancer cells via abrogating the function of the AHA1/HSP90 complex. Acta Pharmacol Sin. 2017;38:1381–1393. doi: 10.1038/aps.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ali M.M., Roe S.M., Vaughan C.K., Meyer P., Panaretou B., Piper P.W. Crystal structure of an HSP90-nucleotide-P23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Weaver A.J., Sullivan W.P., Felts S.J., Owen B.A., Toft D.O. Crystal structure and activity of human P23, a heat shock protein 90 co-chaperone. J Biol Chem. 2000;275:23045–23052. doi: 10.1074/jbc.M003410200. [DOI] [PubMed] [Google Scholar]
- 206.Weikl T., Abelmann K., Buchner J. An unstructured C-terminal region of the HSP90 co-chaperone P23 is important for its chaperone function. J Mol Biol. 1999;293:685–691. doi: 10.1006/jmbi.1999.3172. [DOI] [PubMed] [Google Scholar]
- 207.Bose S., Weikl T., Bugl H., Buchner J. Chaperone function of HSP90-associated proteins. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- 208.Freeman B.C., Toft O., Morimoto R.I. Molecular chaperone machines: chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein P23. Science. 1996;274:1718–1720. doi: 10.1126/science.274.5293.1718. [DOI] [PubMed] [Google Scholar]
- 209.Johnson J.L., Toft D.O. A novel chaperone complex for steroid receptors involving heat shock proteins, immunophilins, and P23. J Biol Chem. 1994;269:24989–24993. [PubMed] [Google Scholar]
- 210.Richter K., Walter S., Buchner J. The Co-chaperone Sba1 connects the ATPase reaction of HSP90 to the progression of the chaperone cycle. J Mol Biol. 2004;342:1403–1413. doi: 10.1016/j.jmb.2004.07.064. [DOI] [PubMed] [Google Scholar]
- 211.Echtenkamp F.J., Gvozdenov Z., Adkins N.L., Zhang Y., Lynch-Day M., Watanabe S. HSP90 and P23 molecular chaperones control chromatin architecture by maintaining the functional pool of the RSC chromatin remodeler. Mol Cell. 2016;64:888–899. doi: 10.1016/j.molcel.2016.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Echtenkamp F.J., Zelin E., Oxelmark E., Woo J.I., Andrews B.J., Garabedian M. Global functional map of the P23 molecular chaperone reveals an extensive cellular network. Mol Cell. 2011;43:229–241. doi: 10.1016/j.molcel.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zelin E., Zhang Y., Toogun O.A., Zhong S., Freeman B.C. The P23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol Cell. 2012;48:459–470. doi: 10.1016/j.molcel.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chadli A., Felts S.J., Wang Q., Sullivan W.P., Botuyan M.V., Fauq A. Celastrol inhibits HSP90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone P23. J Biol Chem. 2010;285:4224–4231. doi: 10.1074/jbc.M109.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Patwardhan C.A., Fauq A., Peterson L.B., Miller C., Blagg B.S., Chadli A. Gedunin inactivates the co-chaperone P23 protein causing cancer cell death by apoptosis. J Biol Chem. 2013;288:7313–7325. doi: 10.1074/jbc.M112.427328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li Y., Zhang T., Jiang Y., Lee H., Schwartz S.J., Sun D. (–)-Epigallocatechin-3-gallate inhibits HSP90 function by impairing HSP90 association with cochaperones in pancreatic cancer cell line Mia Paca-2. Mol Pharm. 2009;6:1152–1159. doi: 10.1021/mp900037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.McDowell C.L., Bryan Sutton R., Obermann W.M. Expression of HSP90 chaperone proteins in human tumor tissue. Int J Biol Macromol. 2009;45:310–314. doi: 10.1016/j.ijbiomac.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 218.Forafonov F., Toogun O.A., Grad I., Suslova E., Freeman B.C., Picard D. P23/Sba1p protects against HSP90 inhibitors independently of its intrinsic chaperone activity. Mol Cell Biol. 2008;28:3446–3456. doi: 10.1128/MCB.02246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gestwicki J.E., Shao H. Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem. 2019;294:2151–2161. doi: 10.1074/jbc.TM118.002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wandinger S.K., Suhre M.H., Wegele H., Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone HSP90. EMBO J. 2006;25:367–376. doi: 10.1038/sj.emboj.7600930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Soroka J., Wandinger S.K., Mausbacher N., Schreiber T., Richter K., Daub H. Conformational switching of the molecular chaperone HSP90 via regulated phosphorylation. Mol Cell. 2012;45:517–528. doi: 10.1016/j.molcel.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 222.Vaughan C.K., Mollapour M., Smith J.R., Truman A., Hu B., Good V.M. HSP90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of CDC37. Mol Cell. 2008;31:886–895. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ross J.S., Schenkein D.P., Pietrusko R., Rolfe M., Linette G.P., Stec J. Targeted therapies for cancer 2004. Am J Clin Pathol. 2004;122:598–609. doi: 10.1309/5CWP-U41A-FR1V-YM3F. [DOI] [PubMed] [Google Scholar]