

Abstract
Lysosome is a ubiquitous acidic organelle fundamental for the turnover of unwanted cellular molecules, particles, and organelles. Currently, the pivotal role of lysosome in regulating cell death is drawing great attention. Over the past decades, we largely focused on how lysosome influences apoptosis and autophagic cell death. However, extensive studies showed that lysosome is also prerequisite for the execution of regulated necrosis (RN). Different types of RN have been uncovered, among which, necroptosis, ferroptosis, and pyroptosis are under the most intensive investigation. It becomes a hot topic nowadays to target RN as a therapeutic intervention, since it is important in many patho/physiological settings and contributing to numerous diseases. It is promising to target lysosome to control the occurrence of RN thus altering the outcomes of diseases. Therefore, we aim to give an introduction about the common factors influencing lysosomal stability and then summarize the current knowledge on the role of lysosome in the execution of RN, especially in that of necroptosis, ferroptosis, and pyroptosis.
KEY WORDS: Ferroptosis, Lysosome, Necroptosis, Pyroptosis, Regulated necrosis
Graphical abstract
As a versatile organelle, lysosome dominates the occurrence of regulated necrosis, especially necroptosis, ferroptosis and pyroptosis. Lysosomal reagents or genetic modifications can influence lysosomal stability and its function. Targeting lysosome is promising to enhance the prognosis of relevant diseases by controlling the process of regulated necrosis.
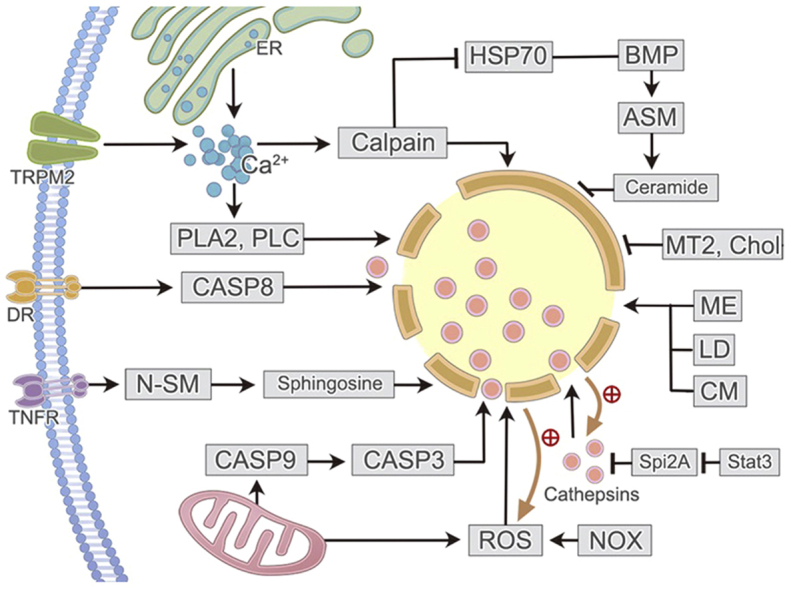
1. Introduction
Lysosome is a widespread intracellular organelle critical for cell homeostasis. It is once regarded as a simple housekeeping organelle owing to its critical impact on the turnover of the intracellular proteins and organelles1. However, more and more functions of lysosome have been explored nowadays, indicating that lysosome is a command-and-control center for cellular signaling, metabolism, and quality control2,3. Furthermore, it is generally acknowledged nowadays that lysosome plays key roles in cell death both in physiological and pathological conditions. Unlike the previous cognition, a novel method about the classification of cell death recommended by Nomenclature Committee on Cell Death (NCCD) has been commonly accepted4,5 (Fig. 1). Apart from accidental cell death (ACD), at least 20 different types of programmed cell death have been enumerated, including apoptosis, autophagic cell death, and regulated necrosis (RN)4, 5, 6, 7, 8, 9, 10. In the past, the investigations concerning lysosome and cell death largely focused on apoptosis and autophagic cell death3,11, 12, 13. Nonetheless, tremendous studies gradually verified the capability of lysosomal alterations in dominating the outcomes of a new type of cell death: RN.
Figure 1.

Classification of cell death. Cell death can be divided into ACD and programmed cell death (PCD). PCD includes apoptosis, autophagic cell death, and RN. RN comprises necroptosis, ferroptosis, pyroptosis, parthanatos, oxytosis, NETosis, pyronecrosis, cyclophilin D-mediated necrosis, and MPT-driven necrosis, etc.
Instead of being accidental and uncontrolled, the occurrence of necrosis is now believed to be highly regulated in contrast to ACD5. RN, sometimes also called programmed necrosis, consists of a series of subtypes of cell death, including necroptosis14, parthanatos15, ferroptosis16, cyclophilin D-mediated necrosis6, oxytosis17, NETosis (neutrophil extracellular trap, NET)18, pyronecrosis19, pyroptosis20, mitochondrial permeability transition (MPT)-driven necrosis21, poly-[adenosine diphosphate (ADP)-ribose] polymerase (PARP)-mediated cell death9. Lysosome takes part in different types of RN in different manners. Triggered by various inducers, lysosomal destabilization with ensuing lysosomal contents (cathepsins) release is observed both in necroptosis and pyroptosis, although they activate different signaling pathways22,23. Nevertheless, lysosomal membrane permeabilization (LMP) is not the only way for lysosome to influence RN. In ferroptosis, lysosome mainly functions in an autophagy-dependent manner including ferrintinophagy, clockophagy, and chaperon-mediated autophagy (CMA), exacerbating oxidative injury by promoting production of iron-dependent reactive oxygen species (ROS)24,25. In spite of this, other roles of lysosome in controlling RN are emerging.
Due to apoptosis defects and drug resistance, RN is considered as a novel target to overcome many diseases, especially cancers which show apoptosis- and therapy-resistance. Targeting lysosome may be a valuable approach for therapy, either by triggering cell death or by promoting therapeutic drug release that target the endocytic pathway23. Here we aim to give an introduction upon some common factors that influence the stability of lysosome and shed light on the current knowledge of how lysosome participates in the process of RN, especially in the three major forms of RN: necroptosis, ferroptosis, and pyroptosis. Table 1 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78 summarizes the key lysosomal factors that are involved in RN. Finally, we describe how interfering with lysosome can end up with enhanced prognosis of diseases by affecting RN.
Table 1.
Key lysosomal factors that are involved in regulated necrosis.
| Type of regulated necrosis | Key lysosomal factor |
|---|---|
|
|
|
|
|
|
| Parthanatos | The occurrence of parthanatos may be accompanied by the activation of calpains, LMP, and cathepsin B/D release74,75. |
| NETosis | NETs contains lysosomal enzymes. And instability of lysosome can cause the immediate formation of NETs and promote NETosis18,76,77. |
| Cyclophilin D-mediated necrosis | Mitochondrial ROS induced generation of lysosomal ceramide activates cytosolic protein BAX which triggers a cascade of reaction with ultimate necrosis78. |
2. Lysosomal stability
The functions of lysosome mainly depend on the hydrolases it contains, of which cathepsins B and D are the most important ones that can retain their activity both in the acid environment of lysosome and in the physiological pH value of cytosol26. LMP with subsequent hydrolases release is an essential stage during the occurrence of RN in most cases. Current knowledge suggests that LMP is either an initiator or a proteolytic amplifier that causes the final executioner phase of RN6. Although the underlying mechanisms by which the lysosomal lipid membrane gets altered remains obscure, several various pathways have been revealed to induce LMP79.
Firstly, mitochondria-originated, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-generated ROS production causes lipid peroxidation (LPO) in lysosomal membrane thus promoting LMP. Secondly, increased cellular Ca2+ concentration owing to Ca2+ influx from outside or elevated Ca2+ release from inside stores, activates calcium-dependent enzymes (calpains and phospholipase in particular) and the phosphatidylinositol 3 kinase (PI3K) pathway to influence LMP. Thirdly, activated caspase-3 and -8 are closely related to LMP. Fourthly, tumor necrosis factor (TNF) induces LMP by activating neutral sphingomyelinase (N-SM), an initiator of sphingosine generation that inserts into lysosomal membrane. Fifthly, STAT3 promotes LMP through negatively regulating endogenous inhibitors of cathepsins, serine protease inhibitor 2A (Spi2A), or controlling the formation of lysosomal vacuoles that contain triglyceride. Triglyceride is metabolized to free fatty acids, resulting in destroyed lysosomal membrane with ensuing leakage of contents thus promoting lysosome-mediated necrotic cell death. Sixthly, the activation of P53 and Bax are also reported to induce LMP26,27,79, 80, 81, 82, 83, 84, 85, 86, 87. Finally, several synthetic compounds, like methyl esters and lysosomotropic detergents can also cause LMP23. On the contrary, cholesterol accumulation, metallothionein 2 (MT2) and heat shock protein 70 (HSP70) are well-known protectors of lysosomal membrane79,88.
HSP70, a highly conserved molecular chaperone located in lysosomal membrane lipids, is reported to inhibit LMP and prevent cell death in HSP70–bis-monoacylglycero phosphate (BMP)–acid sphingomyelinase (ASM)–ceramide pathway89, 90, 91. HSP70 can bind to an endolysosomal phospholipid, BMP, enhancing the activity of ASM89,91,92. By binding to BMP, ASM promotes the production of ceramide which contributes to updated lysosomal membrane composition and increased membrane volume91,93, 94, 95. Downregulation of this pathway by inhibiting HSP70 or ASM could end up with destabilized lysosomal membranes and increased RN in cancer and neuronal cells89,91,92,96, 97, 98. On the contrary, upregulation or administration of HSP70 inhibits cell death and promotes neuroprotection99,100. It is noteworthy that calpains, another important inducer of LMP, can mediate the cleavage of oxidized HSP70 in hippocampal region of brain then induce lysosomal cell death and neurodegeneration89,101,102.
3. Lysosome and necroptosis
Necroptosis is defined as a programmed form of lytic cell death in which receptor-interacting protein kinase 3 (RIPK3) activation leads to subsequent activation of the mixed lineage kinase domain-like protein (MLKL) and acute permeabilization of the plasma membrane103. As a prototype of RN6, necroptosis shows morphological features similar to necrosis, namely ACD104. Therefore, it becomes hampered to distinguish necroptosis from ACD morphologically. Nevertheless, the discovery of MLKL which participates in the late event of necroptosis helps us better identify molecules that solely mediates necroptosis, thus providing probes for better assessing the role of necroptosis103. Unlike apoptosis, in which dying cells are cleared by phagocytes nearby before plasma membrane altered105, cell death in necroptosis causes cell-membrane rupture with subsequent release of intracellular components that can stimulate an innate immune response106.
3.1. The molecular mechanisms of necroptosis
When first being observed in 1990s, necroptosis was discovered to be a kind of TNF-induced necrotic cell death negatively regulated by caspase-1 and -85. To date, except for TNF, an array of other stimuli has been discovered to induce necroptosis as well, followed by a set of well-understood signally pathway. Those identified stimuli include CD95 ligand [CD95L, also known as FAS ligand (FASL)], tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), tumor necrosis factor-related weak inducer of apoptosis (TWEAK), genotoxic stress, polyclonal stimulation of T-cell receptors, DNA-dependent activator of interferon regulatory factors (DAI), anticancer drugs, pathogen-associated molecular patterns (PAMPs), RIG-I-like receptors (RLRs), lipopolysaccharide (LPS), interferons (IFNs), and smac mimetic, etc.6,107 However, death receptor-induced necroptosis, especially TNF-induced necroptosis, is still the best-understood among all these triggers in various backgrounds. Intriguingly, necroptosis can also be triggered in a receptor-independent manner108.
The molecular mechanism of death receptor-induced necroptosis is a representative of all the triggers. Furthermore, TNF is the most frequently used death receptor activator to study nectoptotic cell death. However, TNF can induce not only necroptosis, but also caspase-dependent apoptosis6,109. In the presence of caspase-8, TNF tends to induce apoptosis since caspase-8 inhibits the function of RIPK110 while inactive caspase-8 contributes to necroptosis111. Thus, it is of vital importance to eliminate the disturbance of apoptosis while studying necroptosis. Notably, caspase-8 can be inhibited by Z-VAD-fmk (a pan-caspase inhibitor), FAS-associated death domain-like interleukin-1β-converting enzyme (FLICE)-like inhibitory protein (FLIP), caspase-8 knockout or FAS-associated death domain (Fadd) knockout, thus inhibiting apoptosis79,112.
Under the circumstance of caspase-8 elimination, upon binding to death receptors on the membrane, TNF receptor 1 (TNFR1) signaling complex (TNF-RSC, also called complex I) recruits RIPK1 together with some other signaling molecules within minutes, forming a super-molecular complex that allows RIPK1 to recruit and activate its homologue RIPK3 by phosphorylating it6. It is observed that RIPK3, as an energy metabolism regulator related to the regulation of metabolic enzymes-glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), glutamate dehydrogenase 1 (GLUD1) and subsequent ROS production, plays an essential role in the switch between apoptosis and necroptosis111. The complex I is reported to activate RIPK1 via transforming growth factor-β-activated kinase 1 (TAK1)-mediated phosphorylation or regulating the ubiquitination status of RIPK1113, 114, 115, 116. Ubiquitylation and deubiquitylation of RIPK1 critically regulates the life and death of cells117. When caspase is inhibited, non-ubiquitinated RIPK1 dissociates from the complex I to the cytoplasm and gets phosphorylated26,118. Later on, activated RIPK1 interacts with RIPK3 stably to form a heterodimeric filamentous complex called necrosome, the process of which is mediated by RIP homotypic interaction motifs (RHIMs) of RIPK1 and RIPK3119,120. Ripk3 knockout in animals results in resistance to necrosis and absent in inflammation121.
MLKL, a critical downstream effector of RIPK3, is phosphorylated by RIPK3 and recruited to necrosome through its interaction with RIPK3, ending up with membrane permeability and cell death122,123. Still, how exactly phosphorylated MLKL leads to necropoptic cell death remains controversial. Chen et al.124 asserted that activated MLKL will tetramerize and translocate to lipid rafts of plasma membrane, causing Na+ influx afterwards, which increases osmotic pressure and results in membrane rupture eventually. Conversely, Cai et al.122 and Schappe et al.125 declared that MLKL trimerizes and translocates to plasma membrane, thereby promoting transient receptor potential melastatin-like 7 (TRPM7)-dependent Ca2+ influx rather than sodium influx, which is an early event of TNF-induced necroptosis. The oligomerization and/or translocation of MLKL to the plasma membrane relies on HSP90126.
Generally speaking, lysosome can trigger the progression of necroptosis in two pathways: (1) LMP induced by Ca2+-dependent enzymes and/or ROS promotes the release of cathepsins B and D into cytoplasm which finally causes necroptosis; (2) lysosome can regulate the key signaling molecules of necroptosis, RIPK1 and RIPK3 (Fig. 2).
Figure 2.

Lysosome and necroptosis. Necroptosis occurs in the context of caspase-8 inhibition (Z-VAD-fmk, FLIP, Casp8 knockout, or Fadd knockout). Activated receptors on the membrane attract and activate RIPK1, which consecutively recruits and phosphorylates RIPK3 and MLKL. Phosphorylated MLKL translocates to plasma membrane, ending up with Na+ or Ca2+ influx. ER stress, as a result of RIPK3 and MLKL phosphorylation, also causes Ca2+ dysregulation. Increased Ca2+ concentration activates Ca2+ dependent enzymes in cytosol, particularly calpains and cPLA2, which can later cause LMP. In addition, mitochondria- and NOX-dependent ROS production is another source of LMP inducer. The generation of ROS relies on necrosomal RIPK3 and it in turn facilitates RIPK1 autophosphorylation. Subsequently, disturbed lysosome releases acid hydrolases (especially cathepsins B and D) into cytoplasm, resulting in plasma membrane permeability and eventual necroptosis. Besides, lysosome is discovered to be essential for the post-translation of RIPK1 and RIPK3 which guarantees the occurrence of necroptosis. Cat: cathepsin; Casp: caspase.
3.2. LMP with subsequent cathepsins release triggers necroptosis
Like what's observed in apoptosis, lysosomal membrane damage like LMP with subsequent release of hydrolytic enzymes into cytosol occurs during the process of necroptosis22,127. A variety of cell death stimuli can lead to LMP, causing cathepsins release from lysosomal lumen into cytoplasm, thus inducing caspase-dependent or caspase-independent cell death36,96,128,129. Vanden Berghe et al.29 demonstrated that lysosomal damage is a late event in TNF-induced necroptosis, whereas inhibition of lysosomal acidification led to decrease in the level of TNF-induced necroptosis, although it has no difference or contrarily cause increase in the level of TRAIL-induced necroptosis130. It is described that ATPase H+ transporting V1 subunit G2 (ATP6V1G2, H+-ATPase), a ubiquitous multi-subunit pump prerequisite for lysosomal acidification as well as activation of hydrolytic enzymes inside, is a hit required for necroptosis128. Besides, it is reported that granulysin, a saposin-like protein in human natural killer (NK) and cytotoxic T lymphocyte cells, can cause the disruption of lysosomal membranes in tumor cells and release lysosomal contents, especially cathepsin B, resulting in necroptotic cell death with single strand DNA (ssDNA) nicks131. Likewise, polyinosinic-polycytidylic acid (poly IC) can induce lysosomal destabilization and cathepsin D release into cytoplasm and then promote RLRs-induced necroptosis in dendritic cells. This may contribute to the cleavage of caspase-8 by cathepsin D and ubiquitination of RIPK1 within a cathepsin D–caspase-8–IPS-1–RIP-1 complex (IPS-1: an adaptor molecule for RLRs) formed in mitochondria after cathepsin D release132.
Sharapova et al.26 declared that activated MLKL contributes to an increase of intracellular Ca2+ concentration which activates Ca2+-dependent enzymes, including calpains, phospholipase (e.g., cytosolic phospholipase A2, cPLA2), and protease family members in FAS-dependent necroptosis of K562 cells. Inhibition of calpains and cPLA2 results in decreased cell death26. It is found that activation of calpains is also an essential stage in TNFR1-mediated necroptosis of tumor cells133. Calpains participate in the process of necroptosis via interfering with lysosome and subsequent cathepsins release27,28. Activated calpains cause LMP and release of cathepsins B and D, resulting in mitochondrial membrane depolarization, ROS production, and final cell death27. Fifteen members of the calpains family have been identified up to now, of which the best two characterized distinct, heterodimeric subtypes are μ- and m-calpains28. The localization of activated μ-calpains on lysosomal membrane was observed to be prior to the release of cathepsins while inhibition of calpains by inhibitors causes reduced necroptosis134,135. And it has been proven directly that calpains induce lysosomal rupture by the results of treating isolated lysosome with calpains136.
Calpains are a group of Ca2+-dependent proteases that are widely expressed in different tissues and organisms28. When activated, these enzymes truncate specific proteins by limited proteolysis to modify their structure and activity unlike the overall protein degradation mediated by proteasomes or lysosome137. Inactivated calpains exist in the cytoplasm whereas activated ones translocate to different cellular areas including nuclei, mitochondria, and lysosome, etc., and function distinctly28,138. Later on, in the presence of phospholipids, activated calpains cleave substrate proteins on membranes or in the cytoplasm when released from membranes, which may explain how activated calpains end up with LMP in cells undergoing necroptosis138,139. Intriguingly, calpains, with the downstream STAT3, are reported to regulate the expression of RIPK3 and phosphorylation of MLKL then induce endoplasmic reticulum (ER) stress and mitochondrial calcium dysregulation during ischemic/reperfusion induced lung injury which offer a positive feedback and amplify the function of calpains135. Moreover, upon initiation of necroptosis, activated calpains can also lead to phingomyelinase-mediated ceramide generation, and activation of c-Jun N-terminal kinase (JNK) besides inducing LMP140.
Latest data also indicate the involvement of ROS in necroptosis in a context- and cell type-dependent manner. Although the significance of ROS in the induction of necroptosis have been proven in both in vitro and in vivo experiments, either in a direct or indirect method of inhibiting ROS generation30,141,142. However, He et al.121 conversely showed that elimination of ROS makes no difference to necroptosis. As shown in recent study, the enhancement of ROS generation during necroptosis mainly lies in three distinct pathways which is mitochondria- and NADPH oxidase (NOX)-dependent, rather than Fenton reaction-reliant. The first is cathepsins-dependent mitochondria membrane potential reduction. The second is physical interactions between RIPK3 and enzymes of glycolysis which lead to abnormal energy metabolism and increase ROS production. The third is the interaction of TNFR and cell surface NOX enzymes which provides a significant source of death-inducing ROS in TNF-stimulated necroptosis26,29,111,143,144. Regardless of the sources of ROS, the downstream pathway between ROS and necroptosis has not been fully understood yet.
Current studies with respect to the relationship between ROS and necroptosis focus on the positive feedback circle which indicates that the production of ROS relies on necrosomal RIPK3, whereas ROS facilitates RIPK1 autophosphorylation on serine residue 161 and promotes the recruitment of RIPK3, thus enhancing the formation of necrosome and the occurrence of necroptosis111,142,145, 146, 147. Although ROS is a critical inducer of LMP, the direct evidence of ROS interfering with lysosome during ROS-induced necroptosis is still insufficient. Researchers found that lysosomal damage is a cPLA2-and ROS-dependent event and there is a steady increase of ROS generation in cells with necroptotic features which is followed by mitochondrial hyperpolarization, LMP, oxidative burst, and eventually plasma membrane permeabilization (PMP)29. Cai et al.30 demonstrated that ROS-mediated LMP is involved in bupivacaine-induced necroptosis in rabbit intervertebral disc cells as well. Similarly, RIPK1-induced mitochondrial-ROS generation is followed by LMP and mitochondrial dysfunction as a result of a positive feedback loop between Ca2+ and JNK, culminating in necroptosis of A549 and H1299 cells31,32. Nevertheless, it is worthwhile to identify the specific types of ROS involved in necroptosis and how they participate in the occurrence of necroptosis.
ROS and Ca2+ trigger LMP synergistically rather than respectively. Upregulated-RIPK3 in necroptosis evokes endoplasmic reticulum stress which increases cellular Ca2+ concentration and the expression of xanthine oxidase leading to an enhanced ROS generation148. P2X7, an ATP-gated, nonselective cation channel, is possibly related to the execution of necroptosis after hemorrhagic stroke by stimulating Ca2+ and ROS rise in cytoplasm146,149. In post-ischemic neuronal necrosis, lysosomal rupture is induced in two different pathways at different times150. For one thing, in previous study of ischemic brain model of monkey, Yamashima et al.33,151,152 formulated the “calpains–cathepsin” hypothesis to illustrate the execution of programmed neuronal necrosis affecting lysosomal membrane. μ-Calpains at the lysosomal membrane of cornu ammonis 1 (CA1) neurons gets activated when Ca2+ homeostasis is broken during transient brain ischemia. The activated form of μ-calpains was localized at the vacuolated or disrupted membrane of lysosome causing lysosomal membrane disruption and ensuing spillage of cathepsins from lysosome33,151. For another, during reperfusion, ROS oxidizes membrane fatty acids (e.g., linoleic and arachidonic acid) to generates 4-hydroxy-2-nonenal which can cause carbonylation of HSP70150. Oxidative stress-induced carbonylated HSP70 at the lysosomal membrane is efficiently cleaved by activated μ-calpains followed by lysosomal rupture/permeabilization102,150.
Lysosome contains around 70 kinds of broad-spectrum enzymes which can be released into cytosol as a consequence of lysosomal membrane damage. However, only cathepsins B and D display their enzymatic activity at physiological pH values26. The mechanisms of necroptotic cell death signaling owing to lysosomal activation have not been thoroughly illustrated up to now, but the release of lysosomal enzymes into cytoplasm is regarded as an indispensable event. That means in most cases, cathepsins are released into cytoplasm from lysosome after LMP, and display their hydrolytic functions which directly lead to cell death37, 38, 39. Leaked cathepsins interact with cytosolic signaling proteins, gradually self-digest certain cellular constituents, enhance LMP, induce mitochondrial membrane permeabilization (MMP), and finally cause cell death27,33, 34, 35, 36. Cathepsins can induce MMP by directly interacting with membrane proteins or by indirectly processing the cytosolic proteins Bid, ending up with pores formation in the outer membrane of mitochondria26,27,131.
Cathepsins B and D are the major functional contents released from impaired lysosome during the process of cell death30,132. Cytotoxin-1-induced necroptosis in leukemia cells is relevant to LMP and cathepsin B release153. And the formation of necrosome, phosphorylation and recruitment of MLKL with the ensuing cathepsin D activation, augmented the necroptotic pathway154. Using specific inhibitors of cathepsins B and D respectively can only partially reduce the execution of necroptosis whereas simultaneous pretreatment with both inhibitors completely blocked this type of cell death26,27,155. Ripk1 knockdown decreased not only RIPK1–RIPK3 necrosome formation, but also LMP and the levels of active cathepsin B, thus reducing necroptosis in ischemic stroke model156. Similarly, administration of necrostatin-1 (Nec-1, an inhibitor of necroptosis) decreased the release of cathepsin B from lysosomes, which would explain at least partly why Nec-1 could protect hippocampal neuronal cells from necroptosis induced by ischemia/reperfusion injury157. The neuroprotective effect of Nec-1 also involves cathepsin D inhibition in its underlying mechanism158.
Apart from cathepsins B and D, cathepsin L is also reported to be involved in certain context. Docosahexaenoic acid (DHA) attenuates TNF-induced necroptosis in L929 cells by reducing ROS generation, ceramide production, lysosomal dysfunction, and cathepsin L activation159. It is noteworthy that a positive feedback loop exists between LMP and cathepsins as well. As mentioned before in this paper, cathepsins are important inducers of LMP. However, the process of necroptosis is not always cathepsin-dependent. McComb et al.160 demonstrated that cathepsins B and S directly cleaved RIPK1 and limited macrophage necroptosis. It is also reported that in L929Ts fibrosarcoma cells (a modified L929 subline sensitive to TRAIL), inhibitors of cathepsin B (Ca074Me), cathepsin L (zFF-fmk), or co-inhibitors of cathepsin B/L (zFA-fmk), failed to protect cells from TNF-induced necroptosis161,162. Therefore, the attendance of cathepsins in necroptosis is cell type- and stimuli-dependent.
3.3. Lysosomal regulation of RIPK1 and RIPK3
Recently, we discovered that lysosome also plays a central role in maintaining normal homeostasis of RIPKs (RIPK1 and RIPK3) through post-translational modification in TNF-induced necroptosis. It is demonstrated that CHIP (also known as STUB1) is a direct E3 ligase which negatively regulates RIPKs by ubiquitylating them, thus inhibiting necroptosis in the mice model. Notably, ubiquitylated RIPKs co-localize to lysosome with CHIP and get degraded in a lysosome-dependent pathway other than proteasome-dependent pathway independent of HSP9040,41. On the contrary, lysosomal dysfunction after spinal cord injury may lead to decreased lysosomal localization and degradation of RIPKs, inducing rapid accumulation of RIPKs and MLKL in neurons, both in vitro and in vivo, thus inducing necroptosis that contributes to neuronal and glial cell death. Interestingly, inhibited lysosome may be enough to trigger necroptosis alone. In PC12 cells treated with Baf-A1 (bafilomycin A1, an inhibitor of lysosome's functions) and pan-caspase inhibitor, necroptotic cell death occurs even without the stimuli TNF163. Then, how did accumulated necroptotic mediators get activated without the stimuli? It is once demonstrated that RIPK1 is connected with autocrine production of TNF164. But whether this has happened in the context of lysosomal inhibition remains to be affirmed.
3.4. Others
Apart from what mentioned above, some various functions of lysosome during the initiation of necroptosis have been announced as well. Firstly, it is observed that necroptosis is usually accompanied by autophagy. Cells undergoing necroptosis show an increase in the number and size of lysosome165. RIPK3 overexpression in combination with chloroquine (CQ, which causes accumulation of vesicles but inhibits autophagy) enhanced LMP through the increase in autophagic flux and induced necroptosis in colon cancer cells166. Secondly, as discussed before, phosphorylated MLKL translocates to plasma membrane, then causes membrane rupture and the subsequent cell death. Lately, it is discovered that lysosomal integrity takes part in the membrane repair system of plasma membrane, which consists of lysosomal degradation of endocytic MLKL and the exocytosis of MLKL on plasma membrane, thus suppressing the occurrence of necroptosis42. Thirdly, lysosomal dysfunction plays an important role in the occurrence of necroptosis. In Parkinson's disease models, high levels of ROS may contribute to the activation of necroptosis in vitro with a decreased number of active lysosome and intra-lysosomal cathepsin D, increasing amount of total P62 protein which is degraded by lysosome, as well as the downregulated lysosomal genes and master regulator transcription factor EB (TFEB)167. However, 2-methoxy-6-acetyl-7-methyljuglone induced necroptosis in human colon cancer cells is reported to be independent of TNFα, MLKL, and LMP, instead dependent on cytosolic calcium accumulation-triggered JNK activation and mitochondrial ROS production168. Therefore, the role of lysosome in the execution of necroptosis may be stimuli type-dependent.
4. Lysosome and ferroptosis
Ferroptosis is a type of iron- and ROS-dependent, oxidative form of RN, which shows morphological, biochemical, and genetic distinction from other types of cell death like apoptosis and necroptosis, etc.5,16,169. It was first observed in 2003 to selectively kill oncogenic Ras mutated cells and was first formally termed by Dixon et al.16 in 2012 using erastin and RAS-selective lethal small molecule 3 (RSL3)170. The morphology of ferroptotic cells is characterized by smaller mitochondrial membrane densities with reduced/devoid crista and ruptured outer mitochondrial membrane171.
4.1. The molecular mechanisms of ferroptosis
Ferroptosis is initiated as a consequence of the imbalance between iron accumulation-induced ROS generation and antioxidant defense system that prevents LPO. Normally, on the one hand, as the pivotal protective mechanism against peroxidation damage, glutathione peroxidase 4 (GPX4) is activated relying on cystine (Cys2) import that is mediated mainly by system xc− (the glutamate/cystine antiporter) or by other routes like transsulfuration pathway169,172,173. When translocated into cells, Cys2 is reduced into cysteine which is used to synthesize glutathione (GSH), the recycling of which requires NADPH174. Subsequently, GPX4 uses GSH to eliminate phospholipid hydroperoxides (PLOOH) by converting it into non-toxic lipid alcohols (L-OH), so that iron can no longer react, thus reducing ROS accumulation5,169,175. In addition to GPX4, some other proteins including the transcription factor nuclear factor erythroid 2 like 2 (NFE2L2) and certain HSPs, like HSP25, HSP beta 1(HSPB1), and HSP family A member 5 (HSPA5) play antioxidant roles as well5,176. On the other hand, as for oxidative damage, it is demonstrated that lysophosphatidylcholine acyltransferase 3 (LPCAT3), acyl-CoA synthetase long-chain family member 4 (ACSL4), and arachidonate lipoxygenases (ALOXs, like ALOX12 and ALOX15) are involved in the catalytic process during the oxidation of polyunsaturated fatty acids (PUFAs) in lipid, which is critical for lethal lipid ROS accumulation5,173,177. Although it is a consensus that iron is indispensable in the process of ferroptosis, the precise role of iron still remains elusive. One tentative speculation is that iron regulates ferroptosis by influencing ROS accumulation via iron-dependent enzymatic and non-enzymatic (Fenton reaction) mechanisms5. The former refers to enzymes containing active iron site, like lipoxygenase (LOX) family enzymes which catalyzes the peroxidation of phospholipid PUFAs and hypoxia-inducible factor prolyl-4-hydroxylase isoform 1 (PHD1, also known as EGLN2)169,178.
A plenty of reagents and genes have been discovered to induce or inhibit ferroptosis with distinct targets mentioned above to break the balance between oxidative damage and antioxidant system179, among which the Raf kinase inhibitor sorafenib has already become a clinically-approved inducer of ferroptosis180. First of all, inactivation of GPX4 is the central stage. Regularly, we can use system xc− inhibitors (e.g., erastin, sulfasalazine, sorafenib, and high extracellular glutamate) to deplete GSH thus reduce the activity of GPX4, which are referred to as class I ferroptosis inducers. What's more, we can also directly inhibit GPX4 utilizing its inhibitors such as (1S,3R)-RSL3, ML162, FINO2, or knockdown of Gpx4 which causes overwhelming LPO and the eventual ferroptosis. And GPX4 inhibitors are regarded as class II ferroptosis inducers5,169,173,181,182. Of note, it is also observed that GPX4 depletion improves sensitivity to apoptosis, necroptosis, and pyroptosis, suggesting that LPO can accelerate multiple distinct RN modalities5,108,183, 184, 185. In comparison, vitamin E, ferrostatin-1, and coenzyme Q10, etc. can eliminate LPO and inhibit ferroptosis5,16,179. Nowadays, iron overload is considered to promote ferroptosis via stimulating ROS accumulation. Increasing iron concentration by using iron chloride, hemoglobin, and ferric ammonium citrate, etc. is reported to sensitize cells to ferroptosis whilst iron chelators, like deferoxamine (DFO), cyclipirox (CPX), or deferiprone can depletes iron and prevents iron-dependent lipid ROS accumulation and ferroptosis5,16,186. In addition, it is lately discovered that IFN-γ produced by CD8+ T cells in immunotherapy can induce ferroptosis to kill cancer cells187,188. Furthermore, P53 is found out to suppress tumor in a way of inhibiting cystine uptake and sensitizing cells to ferroptosis189.
4.2. Lysosome in regulating iron homeostasis
Increasing evidence indicates the participation of lysosome in the process of ferroptosis nowadays46,190 (Fig. 3). What is the most focused at present is that lysosome regulates ferroptosis by interfering with iron metabolism. Iron metabolism mainly consists of three parts: iron uptake, storage, and efflux. Ferritin is the main storage form of iron. Iron uptake is mediated by transferrin and transferrin receptor on the plasma membrane to transport iron into cells. On the contrary, ferroportin is the only known iron efflux pump to export iron out of cells191,192. Ordinarily, these three parts achieve a balance so that intracellular iron maintains a stable concentration which guarantees its normal function. However, lysosome contributes to ferroptosis by modulating iron equilibria and causing ensuing burst of ROS expression in lysosome46,193. It has been revealed that elevated level of labile Fe(II) both in lysosome and ER are responsible for erastin-induced ferroptosis in HT1080 cells, which is consistent with the intracellular location of ROS generation during ferroptosis194.
Figure 3.

Lysosome and ferroptosis. Ferroptosis occurs owing to the imbalance between ROS generation and antioxidant defense system. Iron promotes ROS accumulation by enhancing the enzymetic activity of LOX or through Fenton reaction. But GPX4, coming from increased Cys2, reduces oxidative injury. Lysosome affects ferroptosis via regulating iron homeostasis, CMA (lysosomal degradation of GPX4) or clockophagy (lysosomal degradation of circadian clock protein ARNTL, which facilitates EGLN2 expression, thus destabilizing HIF1A and promoting LPO with subsequent ferroptosis). Lysosome increases the concentration of intracellular active iron, and NCOA4-dependent ferritinophagy is the most important form. Ferritin is sequestered into autophagosomes and then delivered to lysosome for degradation, liberating iron from ferritin. Increased labile iron concentration mediated by ferritinophagy in turn engages the IRP–IRE iron homeostatic system which leads to a continuous increase of ferritin synthesis, thus forming a positive feedback. Moreover, lysosome may also be related to increased transferrin and attenuated ferroportin-1 expression. Cys2: cystine; TFR1: transferrin (TF) receptor 1.
Lysosomal ROS production attributes to the low pH and high iron content within lysosome unlike that originated from mitochondria and NADPH5,195. Subsequent LMP may be involved in ferroptosis in response to iron overload and ROS accumulation which causes peroxidation of the lipid components in lysosomal membrane53,127,190. In this way, LMP results in heightened Fenton radical generation, cell membrane denaturation, and GSH consumption127. DFO are also shown to block ferroptosis via inhibiting oxidative stress-induced LMP196. In spite of this, some dissenting voice put forward that LMP is not observed in ferroptosis197. Likewise, the reaction of downstream effector including cathepsin B varies in diverse cell types. In breast cancer cells, cathepsin B is discovered to be irrelevant to the ferroptosis process50. On the contrary, Gao et al.190 presented that ferroptosis at least requires the activation of STAT3-mediated cathepsin B expression since its inhibition ended up with blocked ferroptosis in pancreatic ductal adenocarcinoma cells (PDACs). Therefore, the involvement of LMP–cathepsin axis in ferroptosis is context-dependent.
Among the organelles, the lysosome has the highest abundance of active catalytic Fe2+ probably owing to the acid environment inside lysosome where higher amounts of iron can be solubilized43. Carbonic anhydrase 9 inhibition causes intracellular iron overload co-localized both in lysosome and mitochondria in malignant mesothelioma cells, inducing oxidative stress and triggering LPO with final ferroptosis198. The lysosomal disrupter siramesine, combined with lapatinib (a dual tyrosine kinase inhibitor) or alone, induces a prompt rise in lysosomal pH followed by destabilization of lysosomal membrane, resulting in increased reactive iron and ROS production causing ferroptosis due to blocked iron transport and iron release by lysosomal disruption52,96,199,200. It is reported that lysosomal catalytic Fe2+ induced ferroptosis is critical for the efficacy of non-thermal plasma on killing oral squamous cell carcinoma201. Targeting the lysosomal pool of iron provides an unprecedented therapeutic strategy to eradicate cancer stem cells49. Moreover, the iron chelator DFO likely protects against ferroptosis by chelating iron accumulation in lysosome51. And DFO is observed to be eventually localized within the lysosome, further confirming the localization of active iron in lysosome196,202. Artesunate is also able to induce ferroptosis in a lysosomal iron-dependent manner, since it can be fully blocked by lysosomal DFO whereas increasing lysosomal free iron by co-treatment with iron-saturated, diferric holo-transferrin significantly increased ferroptosis in PDACs203,204.
But it is still elusive how lysosome enhances active iron to regulate ferroptosis. One possible explanation is through the connection with autophagy, so called ferritinophagy24,25. This process has been proven both in vitro and in vivo205 and in various types of cells including fibroblasts cells, cancer cells44, hepatic stellate cells25, and breast cancer cells205, etc. Moreover, some common inducers of ferroptosis, including erastin206, cysteine depletion207, artesunate (a water-soluble hemisuccinate derivative of artemisinin)25,208, salinomycin, and its synthetic derivative ironomycin49,205, are indicated to function via stimulating ferritinophagy with consequent iron overload. On the contrary, ascorbate can delay lysosomal ferritinophagy and stabilize ferritin, thus attenuate cell death by ferroptosis in relation to inflammatory pathology and anti-cancer therapy209. However, it is still debatable that whether another derivative of antimalarial drug artemisinin: dihydroartemisinin (DAT) induces ferroptosis by activating ferritinophagy210,211 or not206.
4.3. Ferritinophagy
Ferritinophagy refers to an autophagic process of ferritin being sequestered into autophagosomes and delivered to lysosome for degradation, which is essential for liberating active iron from ferritin and thus for maintaining cellular iron homeostasis47,48. This lysosomal degradation of ferritin is of great importance to maintain normal iron metabolism. Elevated intracellular iron concentrations can trigger ferroptosis by enhancing the production of lipid peroxides, which can be reverted by iron chelators, such as DFO46. However, the entry of ferritin into lysosome is independent of the presence of lysosome-associated membrane protein 2A (LAMP-2A), suggesting that ferritin entry has nothing to do with CMA212. Instead, the cargo receptor nuclear receptor coactivator 4 (NCOA4) is a significant regulator of ferritinophagy which binds to ferritin and selectively transports it into autophagosome, forming mature autophagosome that merges with the lysosome for degradation47. Activation of ferritinophagy followed by enhanced ferritin degradation is required for regulating ferroptosis213. For example, overexpression of NCOA4 increased ferritin degradation and promoted ferroptosis44. Contrarily, depletion of ferritinophagy either by knockdown of Ncoa4 or by inhibition of autophagy using specific lysosomal lumen alkalizer inhibitor-CQ blocked the accumulation of cellular labile (or free) iron and lipid ROS, thus eventually suppressing ferroptosis25,44,214.
Ferritin, composed of ferritin light polypeptide 1 (FTL1) and ferritin heavy polypeptide (FTH1), is the major intracellular iron storage protein215. It has been shown that ferritin expression and ferroptosis are negatively correlated44,45. Namely, increased ferritin expression limits ferroptosis and vice versa. That is because the initiation of ferroptosis at least partly relies on the autophagic degradation of ferritin in lysosome with subsequent increased labile iron levels and increased ROS, both of which are prerequisite for ferroptosis44,45. The lysosomal degradation of ferritin is mediated by lysosomal acid hydrolases like cathepsin B inside lysosome44,49. Lysosomal dysfunction that occurs in senescent cells causes ferritin accumulation and a deficiency in labile iron as a result of impaired ferritinophagy, inducing resistance to ferroptosis216. However, re-building the function of ferritinophagy with rapamycin averted the iron accumulation phenotype of senescent cells, yet still unable to sensitize senescent cells to ferroptosis. So, it is noteworthy that in those cells, lysosome is pivotal but not unique in regulating ferroptosis216. In contrast, activated ferritinophagy increases labile iron pool (LIP) and promotes ferroptosis213,214.
Notably, increased cellular labile iron concentration mediated by ferritinophagy can in turn engages the IRP–IRE iron homeostatic system (IRE: iron-responsive element) which leads to a continuous increase of ferritin synthesis, thus forming a positive feedback with continued free iron increase and ROS enhancement206. This efficient pathway has been observed, including but not limited to erastin- and DAT-induced ferroptosis206. Furthermore, unlike autophagy, NCOA4-mediated ferritinophagy can be done directly in lysosome rather than relying on autophagic vacuoles44. But inhibitor of autophagy can suppress this process44. The underlying mechanisms of this merit need further investigation.
Ferrintinophagy with subsequent release of free iron and ROS generation may underlie ferroptosis in a sum of disease such as neurodegenerative diseases (e.g., Alzheimer's, Parkinson's, and Huntington's diseases)217 and liver fibrosis25,213. A recent study proposed that the upregulated mRNA binding protein ELAVL1/HuR (ELAV like RNA binding protein 1) in activated hepatic satellite cells was shown to enhance autophagy and meanwhile promote ferritinophagy and ferroptosis, contributing to liver fibrosis213. Treatment with sorafenib or artesunate inhibited this pathway and ameliorated liver fibrosis25,213. Although these data require further confirmation, including the newly discovered HuR pathway, understanding the role of ferritinophagy in regulating ferroptosis may provide a promising insight into overcoming ferroptosis-related diseases.
Lysosome also has impact on iron metabolisms via regulating transferrin and ferroportin-1. Under normal conditions, lysosome stores iron imported by endocytosis of transferrin, a critical process for ferroptosis51. It is demonstrated that lysosome disrupting agent, siramesine, synergistic with lapatinib, is capable of increasing transferrin expression and conversely attenuating ferroportin-1 expression, leading to increased iron concentration and causes ferroptosis in breast cancer cells50. This result is further confirmed in their research again later in 201752 with increased transferrin and decreased ferritin partly due to increased autophagy. In contrast, transferrin receptor expression is observed to be reduced during impaired ferroptosis owing to the high mobility group box 1 (HMGB1) gene knockdown in HL-60 cells46.
4.4. Chaperon-mediated autophagy and clockophagy in ferroptosis
CMA may not relate to iron metabolism212, but it can affect ferroptosis through another pathway rather than affecting iron homeostasis. It is reported that the induction of ferroptosis is accompanied with an increase in the levels of LAMP-2A to promote HSP90–CMA which in turn, mediate the degradation of GPX4 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)53,54. On the contrary, using lysosome inhibitors (e.g., CQ, NH4Cl, and Baf-A1) or HSP90 inhibitor 2-amino-5-chloro-N,3-dimethylbenzamide (CDDO), or otherwise knockdown of Lamp-2a is able to restore GPX4 levels and reduces ferroptosis significantly54. Apart from regulating iron homeostasis or CMA, recently we found that lysosome can also affect ferroptosis by selective autophagic degradation of the core circadian clock protein aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL), named clockophagy55,56. The autophagy-mediated degradation of ARNTL can facilitate the expression of egl nine homolog 2 (EGLN2), thus destabilizing the pro-survival factor hypoxia-inducible factor 1 subunit α (HIF1A) and ultimately promote LPO and subsequent ferroptosis55,56.
4.5. Others
Interestingly, we observed that most ferrostatins (an inhibitor of ferroptosis) are located within lysosome, mitochondria, and the endoplasmic reticulum which blocks ferroptosis through inhibition of LPO51,197. What's more, the lysosome-located ferrostatins are observed with alleviated potency whereas utilizing lysosome disrupter CQ to prevent its accumulation in lysosome can increase the potency of ferrostatin-151. Therefore, it is not hard to understand that lysosome can enhance the sensitivity of cells to ferroptosis both in inductive or inhibitive environment. Moreover, the lysosomal acid environment is essential for the release of sorafenib from nanoparticles where it is loaded to inhibit GPX4 enzyme for ferroptosis initiation and enhances the efficacy of cancer treatment218. Similarly, iron-based nanoparticles could release ferrous (Fe2+) or ferric (Fe3+) ions inside acidic lysosome, thus rapidly increase the generation of ROS in tumor cells191. Ultrasmall nanoparticles, which reside in lysosome, can induce ferroptosis in nutrient-deprived cancer cells via a novel pathway and suppress tumor growth219. However, lysosome can sometimes entrap nanocarriers of ferroptosis inducers after internalization. On the contrary, nanoparticles modified to escape from lysosome and arrive at their subcellular targets can induce ferroptosis more efficiently and enhance the therapy efficacy in tumor cells220. Therefore, we come to a conclusion that the association of lysosome with nano-therapy of ferroptosis relies on the various types and targets of nanoparticles.
In summary, lysosome initiates ferroptosis by increasing iron-uptake (transferrin receptor) or decreasing iron efflux (ferroportin-1) and iron storage (ferritin), dependent/independent of NCOA4-mediated ferritin autophagy (ferritinophagy)198. Moreover, it is not hard to understand that ferroptosis is closely related to autophagy, referring to the involvement of ferritinophagy, clockophagy, and CMA.
5. Lysosome and pyroptosis
Pyroptosis is a lytic form of regulated cell death (RCD) that is driven by the activation of inflammasome and critically depends on the formation of plasma membrane pores mediated by gasdermin protein family5,7. It was first termed by Cookson and Brennan20 in 2001 to describe an atypical caspase-1-dependent cell death of macrophages infected with Salmonella or Shigella. But now we found that bacterial infection is far from the only inducer of this type of cell death. The assembly of inflammasome can also be activated in the context of tissue injury, metabolism in macrophages, monocytes, and other cells69. Mostly, although involving different caspase sub-types221, pyroptosis is considered as a kind of caspase-dependent cell death103 like apoptosis, yet pyroptosis simultaneously shares morphological features with both necrosis and apoptosis222. Pyroptosis is characterized by the absence of DNA fragmentation, instead by the presence of a specific form of chromatin condensation coupled to cellular swelling and PMP as a result of the membrane pores formation, forming an apoptotic body-like cell protrusions (termed pyroptotic bodies)223,224.
5.1. The molecular mechanisms of pyroptosis
Gasdermin D (GSDMD) to pyroptosis is exactly what MLKL to necroptosis. That is to say, GSDMD is the key effector for inducing pyroptosis, which is cleaved and activated by the inflammatory caspase-1 and -11 (along with its human homologs caspase-4 and -5)69,224,225. When activated, GSDMD generates C (GSDMD-C) and N-terminal (GSDMD-N) fragments of GSDMD, the latter of which forms oligomer and is translocated to plasma membrane to form non-selective pores on it, coordinating membrane lysis and the release of inflammatory cytokines, interleukin-1β (IL-1β), and IL-18 included69,223,224,226. In contrast, GSDMD-C is thought to fold back on and inhibit GSDMD-N, which is removed by the cleavage of activated inflammasome227. Except GSDMD, it is discovered that other gasdermin family members like gasdermin A (GSDMA), GSDMA3, and gasdermin E (GSDME, also called DFNA5) share the GSDMD-N domains and show similar pore-forming activity227,228. Pyroptosis can be pharmacologically inhibited by a direct inhibitor of GSDMD, necrosulfonamide, which offers a new insight into artificial manipulation of this type of cell death223.
Caspase-1 is stimulated by canonical inflammasome to activate GSDMD. It is a multiprotein complexes constituted by pro-caspase-1, pattern recognition receptors (PRRs), and the apoptosis speck-like adaptor protein (ASC)149. The PRRs are composed of two sub-types. The first type refers to nucleotide-binding oligomerization domain (NOD)-like receptors (NLR) family pyrin domain (NLRP) containing 1–3, 6, and 7 (NLRP1–3, NLRP6, andNLRP7), NLR family caspase recruitment domain (CARD) domain-containing 4 (NLRC4) among which NLRP3 is the most intensively investigated one. And the second type is non-NLR, such as absent in melanoma 2 (AIM2) and tripartite motif-containing (TRIM)69,229,230. They act as a sensor that NLR recognizes a wide variety of danger-associated molecular patterns (DAMPs) and PAMPs whereas non-NLR including AIM2 recognizes double-stranded DNA from bacteria or host cells69,229,231,232. ASC, however, functions as a bridge between the upstream sensor and pro-caspase-1, which contains two death-fold domains including pyrin domain (PYD) and CARD69. When this inflammasome is activated, it would in turn activate pro-caspase-1 into mature caspase-1 which triggers pyroptosis and cleaves pro-IL-1β and pro-IL-18 into active IL-1β and IL-18, respectively233. Recently, some studies reported that during the pyroptotic process, caspase-1 could be activated by both inflammasome and pyroptosome (composed of oligomerized ASC dimers)72.
On the contrary, the other activator of GSDMD, caspase-11 (as well as its homologs caspase-4 and -5 in human) is stimulated by non-canonical inflammasome which is activated by cytosolic LPS from invading Gram-negative bacteria69,234. HMGB1 and outer membrane vesicles (OMVs) produced by Gram-negative bacteria are critical for the delivery of extracellular LPS for caspase-11 dependent pyroptosis70,235,236. However, the canonical and non-canonical pathways are not absolutely isolated. Instead, there are some amplifying interconnections between canonical (NLRP3- and AIM2-dependent) and non-canonical pathways237,238. Non-canonical caspase-11 can induce activation of canonical NLRP3 by activating downstream pennexin 1 channel and P2X7 receptor causing potassium efflux229,239,240. Similar to necroptosis, endosomal sorting complexes required for transport III (ESCRT-III) dependent membrane repair system negatively regulates pyroptosis upon GSDMD activation241.
5.2. The role of lysosome in pyroptosis
The role of lysosome in the execution of pyroptosis has been briefly introduced by Repnik et al.23. Thus, here we would supplement with most recent findings (Fig. 4). Similar to that of necroptosis, LMP with subsequent cathepsins release, particularly cathepsin B, is a central regulator of inflammasome-dependent pyroptosis. This has been proven in numerous experiments on non-epithelial cells, especially in macrophages exposed to infectious agents59,64,65. Activation of pyroptosis during lysosomal damage could be alleviated through inhibition of lysosomal acidification or cathepsins. Lysosomal destabilization induced by dipeptidyl methyl ester Leu-Leu-OMe (LLME) triggers caspase-1 mediated pyroptosis whilst blocking lysosomal disruption abrogates the cytotoxicity of LLME242. Moreover, overexpression of cathepsin B by ablation of cystatin C (endogenous inhibitor of cathepsin B) significantly exacerbates, whereas the cathepsin B inhibition either by genetically knockout of cathepsin B or cathepsin B-selective inhibitor Ca074Me attenuates caspase-1 dependent pyroptosis60,62,243,244. Functional lysosomal cathepsins are important for downstream signaling since Baf-A1 (a lysosomal disrupter) prevents the activation of caspase-1245. In tamoxifen-treated retinal pigment epithelial cells, blockade of the activity of cathepsins B and L leads to a significant decrease not only in apoptosis and necroptosis, but also in pyroptosis246.
Figure 4.

Lysosome and pyroptosis. In pyroptosis, lysosomal rupture can be initiated by various crystalline materials, nano-particles, chemical compounds, rare earth oxide, and maybe ROS. Subsequent CatB release and K+ efflux activate NLRP3 inflammasome. But in pyroptosis induced by the endocytosis of HMGB1, lysosome destabilization and CatB release are necessary for pyroptosome formation. Activated NLRP3 inflammasome or pyroptosome stimulates caspase-1. CatB can be inhibited by Ca074Me, endogenous cystatin C, or genetic knockout of CatB. Lysosome destabilization caused by HMGB1 is also indispensable for LPS-induced activation of non-canonical inflammasome and caspase-11, since it allows LPS internalized through HMGB1–RAGE signaling pathway to escape from lysosomal degradation. Activated caspase-1 and -11 drive the cleavage of pro-IL-1β/18 and GSDMD, coordinating membrane lysis, mature IL-1β/18 release and ultimate pyroptosis. CatB: cathepsin B.
Lysosomal rupture is one of the common stimulators of NLRP3 inflammasome which results in direct activation of caspase-1247. In macrophages, various crystalline materials (e.g., silica or alum) or chemical compounds (e.g., LLME) can cause LMP and cathepsin B release, which is an important mechanism of NLRP3 inflammasome and subsequent caspase-1 activation and then pyroptosis23,245. In addition, NLRP3-mediated pyroptosis was observed after viral infection or 27-hydroxycholesterol-, cholesterol crystals-, rare earth oxide-induced lysosomal damage in other cell types, like nerve cells and Kupffer cells39,57, 58, 59. Nanomaterials, like nano-silver and nano rare earth metal oxide, are capable of stimulating inflammasome activation owing to lysosomal disruption which is closely related to the morphological characteristics of nanoparticles for needle-shaped nanoparticles are more prone to pierce lysosomal membrane248, 249, 250, 251.
Cathepsin B's release, as a result of lysosomal disruption, attributes to NLRP3 activation. Unlike necroptosis, cathepsins function as an upstream initiator of pyroptotic cell death rather than an executor. In Table 2, a comparison about the functions of cathepsins in necroptosis and pyroptosis is summarized. Nano-therapy based on magnetic intra-lysosomal hyperthermia involves an enhanced production of ROS through lysosomal Fenton reaction which subsequently induced the release of LPO, LMP, and cathepsin B that in turn activates caspase-1 (either by directly or indirectly activating upstream inflammasome), and ultimately induces pyroptosis in pancreatic endocrine, pancreatic exocrine, and gastric cancer cells without IL-1β secretion252. Cathepsin B binds to NLRP3 as verified by co-precipitation using either an anti-NLRP3 or anti-cathepsin B antibody60. Cathepsin B may activate NLRP3 inflammasome through mitogen-activated protein kinase (MAPK) and TAK1–JNK pathway61. This mechanism of activating NLRP3 accounts for the cytotoxicity of cathepsin B to coxsackie virus B3-induced myocarditis243. In Kawasaki disease, cathepsin B's release induced endothelial cell pyroptosis by activating NLRP3 inflammasome253. 27-Hydroxycholesterol, which initiates pyroptosis procedures in neurons and causes neurodegenerative diseases, functions through inducing LMP and cathepsin B release, leading to NLRP3 inflammasome activation as well59,254,255. We also found that multi-walled carbon nanotubes induced pyroptosis and inflammasome activation in primary human bronchial epithelial cells can be distinctly decreased by cathepsin B inhibitor Ca074Me256. Uptake of boron nitride nanotubes caused lysosomal destabilization, cathepsin B increase, inflammasome activation, and pyroptosis both in vitro and in vivo257.
Table 2.
The functions of cathepsins in necroptosis and pyroptosis.
| Content | Necroptosis | Pyroptosis |
|---|---|---|
| Similarities |
|
|
| Types involved | Mainly: B and D; others: L | Mainly: B; others: D, L, and X |
| Role | Downstream executor | Upstream trigger |
| Functions | Interact with cytosolic signaling proteins, gradually self-digest certain cellular constituents, enhance LMP, induce mitochondrial and plasma membrane permeabilization, and finally cause cell death. |
|
However, some studies revealed that the execution of pyroptotic cell death stimulated by particle and pyroptosis inducers like nigericin and ATP relies on multiple cathepsins rather than just cathepsin B. Because cathepsin B's deficiency does not prevent the induction of pyroptosis under this circumstance59,62,63. This includes another type of cathepsin, cathepsin X59,63. Moreover, inhibition of cathepsins B and L suppresses inflammasome activation and significantly reduced pyroptotic cell death in primed retinal pigment epithelial cells which revealed the involvement of both cathepsins in pyroptosis258. Similarly, the death of retinal pigment epithelium cells activated by all-trans retinal is related to cathepsins B and D, as well as NLRP3 inflammasome activation259. But the cytosolic flagellin-induced cathepsin B release in macrophage may induce cell death by activating neuronal apoptosis inhibitory protein 5 (NAIP5)/NLRC4 inflammasome rather than NLRP3 inflammasome64.
In addition to the cathepsin release, ROS production and K+ efflux are common stimulators for inflammasome activation as well66, 67, 68. When exposed to excess all-trans retinal, both ROS (including mitochondria ROS) and cathepsins released from lysosome contribute to NLRP3 inflammasome activation and the subsequent pyroptosis in human ARPE-19 cells259. Notably, ROS appears to be upstream of lysosomal damage in the context of iron oxide nanoparticles-induced inflammasome activation, as ROS inhibitor significantly reduced lysosomal damage, while cathepsin B's inhibitor shows no difference to ROS production251. Moreover, polymers induce lysosomal rupture followed by the concurrent K+ efflux from the cells, which leads to subsequent pyroptosis as observed by the propidium iodide stain260. Cytosolic DNA-induced IL-1β maturation and pyroptosis are essential for the anti-viral immunity in human myeloid cells, during which activated stimulator of interferon genes (STING, a component of DNA detection axis) would translocate to lysosome and trigger LMP, initiating K+ efflux upstream of NLRP3 inflammasome activation261. Some other studies indicated that lysosome rupture induces Ca2+ release from the lysosome to the cytosol and then promotes complete NLRP3 inflammasome activation through the oligomerization of ASC via TAK1–JNK pathway61. However, it is reported that only low level of lysosome disruption activates NLRP3 inflammasome initiating by K+ efflux nevertheless extensive lysosome rupture is inhibitory262. Variously, Shigella infection-induced pyroptosis in macrophage influences K+ metabolism in a unique manner. This infection can cause the internalization of the invasion plasmid antigen B channels which permit K+ influx within endolysosomal compartments followed by vacuolar destabilization, endolysosomal leakage, and the ultimate caspase-1-dependent pyroptosis263.
Therefore, it is conceivable that lysosomal rupture can regulate the occurrence of pyroptosis via cathepsins release, ROS pathway or by reducing the K+ concentration in cytoplasma. At first, it is believed that lysosomal rupture is an upstream activator of NLRP3 inflammation independent of caspase-11234,245,264. Recently, there are a few dissenting voice claims that lysosomal rupture occurs either upstream or downstream of inflammasome activation. They presented that NLRP1b/NLRP3 inflammasome ignited by pyroptosis agonists was prior to lysosomal disruptsion64,265. Furthermore, it is postulated that activated caspase-1 contributes to lysosomal acidification in systemic lupus erythematosus266. Therefore, further investigation requires to be done to clarify the relationship between lysosomal rupture and NLRP3 activation.
Lysosomal permeabilization also takes part in the activation of non-canonical inflammasome by allowing LPS stimulation. Deng et al.70 demonstrated that extracellular LPS molecules are internalized into the acidic lysosome of macrophages and endothelial cells by physically binding to hepatocyte-released HMGB1 through HMGB1-RAGE (receptor for advanced glycation end-products) signaling pathway. HMGB1 is a nuclear protein which can activate multiple cell surface receptors including RAGE or Toll-like receptors to initiate innate immune response when released into extracellular fluid267. When internalized, HMGB1 can directly permeabilize the phospholipid bilayer of the lysosome under acidic conditions, thus allowing LPS to escape from lysosomal degradation and then leak out into its key pathogenic cytosolic receptor, caspase-11, which culminates in the activation of non-canonical inflammasome and pyroptosis in endotoxemia69,70. Inhibition of HMGB1/RAGE mediated endocytosis prevented caspase-11 dependent pyroptosis in endotoxemia and bacterial sepsis70,268. Cathepsin G mediated caspase-4 activation by a bacterial surface protein in human gingival fibroblasts and induced pyroptosis269. Nevertheless, by inducing lysosomal rupture with ensuing lysosomal contents release including cathepsin D, HMGB1 can also activate NLRP3 inflammasome independent of caspase-11, subsequently inducing caspase-1 activation through Toll-like receptors70,72.
However, we should notice that in pyroptosis induced by the endocytosis of HMGB1, lysosome destabilization and cathepsin B release are necessary for pyroptosome formation. Acting through the RAGE and dynamin-dependent signaling, the endocytosis of HMGB1 triggers a cascade of intracellular molecular events, including lysosomal rupture with cathpsin B release followed by pyroptosome assembly and NLRP3 inflammasome-independent caspase-1 activation and pyroptosis both in vitro and in vivo71, 72, 73,270. Furthermore, these events are reported to be indispensable for LPS-induced pyroptosis. Yet it is indicated that HMGB1 endocytosis and subsequent cathepsin B activation is only required for late-phase of caspase-1 activation unlike that NLRP3 inflammasome accounts for the early-phase73.
To sum up, lysosomal rupture triggers the execution of pyroptosis either by activating NLRP inflammasome or by promoting pyroptosome assembly.
6. Lysosome and other forms of RN
6.1. Parthanatos
Parthanatos is a form of RN characterized by hyperactivation of the nuclear protein PARP1 and consequent apoptosis inducing factor (AIF)- or macrophage migration inhibitory factor (MIF)-dependent DNA degradation7. PARP1 is essential for restoring cellular homeostasis by taking part in DNA repair, chromosome stability, and the inflammatory response6,10. But overactivated PARP1 stimulated by a wide array of stimuli such as DNA damage, oxidative stress, hypoxia, hypoglycemia, or inflammatory cues ends up with parthanatotic cell death by transferring PARP1 from nicotinamide adenine dinucleotide (NAD+) and binding to AIF or hexokinase1 (HK1), which mediates the release of AIF from mitochondria into cytosol and its translocation into the nucleus where it promotes large-scale DNA fragmentation and chromatin condensation, leading to cell death5,7,10. Through binding to PARP1, HK1 inhibits glycolysis to cause the energy depletion and precipitate parthanatos271. Recently, MIF emerges as the main nuclease that binds to AIF and produces large DNA fragment to induce pathanatos272. On the contrary, PARP1 inhibition by pharmacological or genetic modification provides cytoprotective effects in multiple animal models of diseases10.
The relationship of lysosome and parthanatos is largely unknown. Porte Alcon et al.74 discovered that the occurrence of parthanatos in microglial cell death triggered by manganese is accompanied by LMP and cathepsin B/D release. Inhibition of both cathepsins B and D results in partial decrease of cell death combined pretreatment retinal photoreceptor with calpains and cathepsin inhibitors, rather than cathepsin inhibitors alone, reduced parthanototic cell death75.
6.2. NETosis
NETosis, also called NETotic cell death, is a ROS-dependent modality of RN which is driven by NETs release in response to infection or injury in cells of hematopoietic derivation like neutrophils7,273. ROS generation in NETosis is dependent on NOX activation and can promote the translocation of granular enzymes including elastase, neutrophil expressed (ELANE), myeloperoxidase, and peptides of the cathelecidin family from cytosol to nucleus274,275. Later on, the myeloperoxidase-dependent proteolytic activity of ELANE gets promoted, which culminates in histone citrullination, chromatin decondensation, extrusion of chromatin fibers along with cytoplasmic and nuclear components, ultimately resulting in plasma membrane rupture and necrosis5,276,277.
It has been demonstrated that NETs contains unfolded chromatin and lysosomal enzymes18. Moreover, instability of the lysosomal compartment and fast damage of plasma membranes in various cell types and tissues cause the immediate formation of NETs in the context of 10–40 nm sized nanoparticles76. Similarly, it is observed that LLME induced crystal-triggered NETosis in a dose-dependent fashion, suggesting that lysosomal disruption is involved in the execution of NETosis as well77.
6.3. Cyclophilin D-mediated necrosis
Cyclophilin D-mediated necrosis is another form of RN that relies on the cyclophilin D-dependent formation of mitochondrial permeability transition pore10. Most recently, researchers found that cyclophilin D-mediated necrosis in tuberculosis-infected macrophage relies on a mitochondrial–lysosomal–endoplasmic reticulum circuit78. During this process, mitochondrial ROS induced generation of lysosomal ceramide is a critical activator of the cytosolic protein BAX which triggers a cascade of reaction with ultimate necrosis.
Nevertheless, the evidence indicating a direct connection between lysosome and parthanatos, cyclophilin D-mediated necrosis, and NETosis is far from sufficient. And up to now, no direct proof has referred to the connection between lysosome and oxytosis or MPT-driven necrosis. Therefore, a broad area about the relationship between lysosome and RN can be explored in the future.
7. Physiological and pathological significance
Cell death is essential for living organisms and is a hallmark of numerous disorders. It also has a significant influence on the treatment of certain diseases, for example, cancer278. In general, RN also occurs both under physiological and pathological conditions. On the one hand, it is of great importance for embryogenesis, lymphocyte function, and the involution of post-lactation mammary gland85,87,279. On the other hand, RN is a genetically determined process that contributes to plenty of human diseases. Different types of RN may account for various pathological injuries either simultaneously or respectively. For example, during ischemic brain injuries and neurodegenerative diseases, RN rather than apoptosis seems to compose the major cell death mode152. Analogously, necroptosis, mitochondrial-mediated necrosis, ferroptosis, parthanatos, and pyroptosis are probably the main contributors of renal and myocardial ischemic-reperfusion injury and renal transplantation8,54,280,281.
It is noteworthy that the pathways of RN can be therapeutically targeted10,16,282,283. What's more, targeting lysosome may provide a novel insight into controlling RN thus further improve the outcomes of diseases. A large number of experiments have confirmed that reagents or genetic modifications of lysosome and relevant factors showed positive efficacy in controlling RN, and some of them can significantly enhance the prognosis of various diseases. Numerous lysosomotropic agents showed positive efficacy in treating various cancers including breast cancers, lymphoma, leukemia, and those showing resistance to traditional forms of treatment. Many of those agents are U.S. Food and Drug Administration (FDA)-approved drugs already or under clinical trials284,285. Cathepsins and calpains are promising therapeutic targets, especially for cancers and neurological diseases. Inhibitors of cathepsins have reached clinical trials in osteoporosis and cancer. E-64, an inhibitor of both calpains and cathepsins, is in clinical trials of Alzheimer's disease286. What's challenging now is that clinical trials targeting this complex regulating network of lysosome in RN is still insufficient. Here we would describe the known human diseases related to each kind of regulated necrosis and the potential roles of lysosome in these diseases via dominating the occurrence of RN.
7.1. Necroptosis
As an alternative cell death pathway in apoptosis-defective cells, necroptosis is implicated in a wide range of pathological cell death paradigms, including infectious diseases, ischemic–reperfusion injury in brain, inflammatory diseases, tumorigenesis, myocardial infarctions, acute lung/kidney/liver injury, amyotrophic lateral sclerosis, and chemotherapy-induced cell death, etc.118,287, 288, 289 Notably, necroptotic cell death pathway becomes a novel therapeutic target in the treatment of many diseases nowadays, particularly in the treatment of apoptosis- and therapy-resistant cancers such as breast, colon, and liver cancers79,85,287,290,291. Many cancers contain mutations that inactivate apoptotic pathway and cause drug-resistance. However, the idea that tumors may be sensitive to RN can hopefully offer another direction for overcoming the resistance292. Researches regarding the necroptosis in cancer therapy mainly focus on two aspects. The first is that the induction of necroptosis can eliminate therapy-resistant cancer cells or enhance surveillance and protective immunity against cancer. On the contrary, inhibition of necroptosis reduces cancer-promoting inflammation287. The inhibitor of RIPK1, benzoxazepinone, has already reached phase 2 clinical trial293. RIPK3 is now considered as a potential therapeutic target for Gaucher's disease, a lysosomal storage disease with insufficient lysosomal enzyme β-glucocerebrosidase294. Table 3 summarized the reagents or genetic modifications of lysosome and relevant factors which could affect RN and change the outcomes of various diseases.
Table 3.
Lysosomal reagents or genetic modifications in regulating RN and relevant diseases.
| Name | Target | State | Diseases/model | Result | Effect on injury |
|---|---|---|---|---|---|
| Desipramine | (−) ASM | In vitro | TB infection | Reduced susceptibility to necroptosis | Reduce bacterial burdens, making zebrafish hyperresistant295 |
| ALLN | (−) Calpain | In vitro and in vivo | IRI in hippocampal neurons | (−) Necrosis | Neuroprotection101 |
| In vitro | IRI in lung | (−) Necroptosis | (−) Cell death after IRI in lung135 | ||
| Calpastatin overexpression | Endogenous calpain inhibitor | In vitro and in vivo | (−) Caspase-independent neuronal cell death | Neuroprotection296 | |
| DPI | (−) NOX | In vitro | Human umbilical vein endothelial cells | (−) Apoptosis and necroptosis | ND297 |
| GKT137831 Nox4 siRNA | (−) NOX4 | In vitro | (−) Apoptosis and necroptosis | ND297 | |
| N-Acetyl-l-cysteine | (−) ROS | In vitro | Bupivacaine-induced cytotoxicity of IVD cells | (−) LMP and necroptosis | Reduced cytotoxity30 |
| CDDO | (−) HSP90 | In vitro | (−) Ferroptosis and necroptosis | ND54 | |
| CQ, NH4Cl, and Baf-A1 | Lysosome inhibitor | In vitro | (−) Ferroptosis | ND54 | |
| Baf-A1 and CQ | Lysosome inhibitor | In vitro | In certain cancer cells (e.g., HT1080 and BJeLR) | Can't modulate ferroptosis | ND16 |
| In vitro | MEFs and HT1080 cells | (−) Ferroptosis | ND214 | ||
| Baf-A1 | Lysosome inhibitor | In vitro | PDAC | (−) Ferroptosis | ND190 |
| In vitro and in vivo | Cytosolic flagellin stimulated macrophages | (−) Pyroptosis | ND64 | ||
| Lysosomal lumen alkalizer-CQ | Lysosome inhibitor | In vitro and in vivo | Liver fibrosis | Impair ferritinophagy and ferroptosis | Inhibited anti-fibrosis function of artesunate25 |
| LLME | Lysosomal disrupter | In vitro | AMD | (+) Pyroptosis | ND242 |
| In vitro | Acutely inflamed joints of gout | (+) NETosis | ND77 | ||
| In vitro and in vivo | murine BMDC | (+) Pyroptosis | ND262 | ||
| Ca074Me | (−) Cat B | In vivo | Ischemic injury of hippocampus | Inhibit 67% of programmed necrosis | Neuroprotection33 |
| In vitro and in vivo | endotoxemia | (−) Pyroptosis | ND73 | ||
| In vitro | SH-SY5Y and C6 cells | Partially prevented pyroptosis | ND254 | ||
| in vitro and in vivo | CLP sepsis | (−) Pyroptosis | Attenuate inflammatory responses71 | ||
| In vitro | KUP5 cells and macrophages | (−) Pyroptosis | ND57 | ||
| In vitro | (−) Necroptosis | ND298 | |||
| In vitro | Promyelocytic leukemia cells | (−) Necroptosis | ND299 | ||
| In vitro | PDAC | (−) Ferroptosis | ND190 | ||
| In vitro | Cancer stem cells | (−) Ferroptosis | ND49 | ||
| Genetic knockout of CatB | (−) Cat B | In vitro and in vivo | Coxsackievirus B3-induced viral myocarditis | (−) Pyroptosis | Attenuate243 |
| Genetic deletion of cystatin C | (+) Cat B | In vitro and in vivo | (+) Pyroptosis | Aggravate243 | |
| Ca074Me and pepstatin A | (−) Cats B and D | In vitro | Bupivacaine-induced cytotoxicity of IVD cells | (−) Necroptosis | Reduced cytotoxity30 |
| In vitro | ARPE-19 cells | (−) Apoptosis and pyroptosis | ND259 | ||
| In vitro | Mn2+ exposed BV-2 cells | Prevented a ∼13% parthanatos | Alleviates toxicity74 | ||
| In vitro and in vivo | Cytosolic flagellin stimulated macrophages | Halved pyroptosis | ND64 | ||
| Ca074Me/pepstatin A alone | (−) Cat B or D | In vitro and in vivo | Cytosolic flagellin stimulated macrophages | No effect on pyroptosis | ND64 |
| Ca074Me and Z-FY(t-Bu)FMK | (−) Cats B and L | In vitro | Tamoxifen treated human RPE cells | (−) Apoptosis, necroptosis, pyroptosis | ND246 |
(−): Inhibit; (+): promote; CAD: cationic amphiphilic drugs; MDR: multidrug resistance; TB: tuberculosis; DPI: diphenyleneiodonium; IRI: ischemia–reperfusion injury; IVD: intervertebral disc; Cat: cathepsin; (A)RPE cells: (acute) retinal pigment epithelial cells; CLP: cecal ligation and puncture; AMD: age-related macular degeneration; BMDC: bone marrow-derived dendritic cells; ND: not determined.
In a zebrafish model of tuberculosis infection, necroptosis of macrophage releases residual bacteria into permissive extracellular milieu and causes continuous infection. However, desipramine, a tricyclic antidepressant that promotes the degradation of ASM and inhibits ceramide production which is an essential step for LMP, decreased the susceptibility of macrophage to necroptosis and reduced bacterial burdens, making zebrafish hyperresistant to infection295. Using N-acetyl-Leu-Leu-norleucinal (ALLN) to block calpain (another inducer of LMP) inhibited the occurrence of necroptosis which occurs after ischemia–reperfusion injury in lung135. Similarly, overexpression of endogenous calpain inhibitor, calpastatin, inhibited caspase-independent neuronal cell death and promoted neuroprotection in calpastatin mutant mice296. Diphenyleneiodonium (DPI), GKT137831 or Nox4 siRNA inhibited the function of NOX, reduced the production of ROS, and inhibited the execution of both apoptosis and necroptosis in vitro297. Inhibition of ROS directly by N-acetyl-l-cysteine effectively blocked bupivacaine-induced LMP and necroptosis, reducing cytotoxicity to intervertebral disc (IVD) cells30.
CDDO, an inhibitor of lysosomal membrane protector HSP90, effectively blocked necroptosis of HT-22, 661W, HT29, and human leukemia Fadd-deficient Jurkat cells by inhibiting the activation of RIPK154. The activity of the lysosomal protease cathepsin B was observed to be increased on Days 3–5 after ischemia injury in hippocampus of monkeys. However, blockade of lysosomal activity rescued neurons from death which showed mild central chromatolysis. A specific inhibitor of cathepsin B, Ca074Me, saved around 67% of CA1 neurons from programmed necrosis and showed a promising capability of neuroprotection after ischemic injury33. Similarly, Ca074Me promoted the survival of U937 cells and promyelocytic leukemia cells dying from necroptosis298,299. Apart from cathepsin B, other cathepsins also count in the process of necroptosis. Ca074Me along with pepstatin A (cathepsin D inhibitor) prevented necroptotic cell death and reduced bupivacaine-induced cytotoxicity of IVD cells30. What's more, Ca074Me combined with cathepsin L inhibitor, Z-FY(t-Bu)FMK, blocked apoptosis, necroptosis, and pyroptosis which occur in tamoxifen-treated human retinal pigment epithelial cells246.
7.2. Ferroptosis
Ferroptotic cell death is discovered to be involved in many diseases including cancers50,190, neurodegenerative disorders300, acute renal injury, renal IRI301, and infectious diseases, etc.44 It is noteworthy that pharmacological inhibition or inducement of this type of cell death may be a novel therapeutic approach for those diseases187,188,206,302. In some cases, the metabolic reprogramming in cancer cells heightens their sensitivity to ferroptosis, therefore opening up a novel insight for overcoming tumors, particularly the therapy-resistant ones171,302, 303, 304. Sorafenib, a multikinase inhibitor drug for the treatment of kidney/liver/thyroid cancer, has already become a clinically-approved inducer of ferroptosis10,180. Moreover, it is reported that ferroptosis, which is triggered by CD8+ T cells via releasing IFN-γ, can boost the effectiveness of immune checkpoint inhibitors305.
Similar to necroptosis, CDDO could also inhibit ferroptosis of HT-22, 661W, and human HT-1080 cells54. What's more, lysosome pathway inhibitor CQ, NH4Cl, or Baf-A1 inhibited GPX4 degradation and subsequent ferroptosis induced by glutamate or erastin in vitro54. Although Baf-A1 and CQ could block ferroptosis in various cell lines, like MEFs, HT1080 cells, and PDAC, they failed to modulate ferroptosis in certain cancer cells (e.g., HT1080 and BJeLR)16,190,214. Artesunate alleviates liver fibrosis by triggering activated hepatic stellate cell (HSC) ferroptosis. However, the lysosomal lumen alkalizer CQ, which disturbs the fusion of lysosomes with autophagosomes, impaired ferritinophagy and ferroptosis both in vitro and in vivo, thus inhibiting anti-fibrosis function of artesunate25. The inhibitor of cathepsin B, Ca074Me, could block ferroptosis in PDAC and cancer stem cells as well49,190.
7.3. Pyroptosis
Pyroptosis is crucial for immune defenses (e.g., against infection) and numerous diseases including hemorrhagic stroke and sepsis71,149,227,230,306. Of note, it is reported that except apoptosis, pyroptosis accounts for over 95% of quiescent CD4+ T cells death in HIV-1 infection307. Interestingly, chemotherapy drugs can induce pyroptosis through caspase-3 cleavage of GSDME228.
The usage of lysosomal inhibitor Baf-A1 showed a significant inhibitory effect on cytosolic flagellin-induced pyroptosis in macrophages64. The lysosomotropic agent LLME which is used to disrupt lysosome and induce lysosomal destabilization, triggered pyroptosis in ARPE-19 cells of age-related macular degeneration patients and murine bone marrow-derived dendritic cells242,262. Suppression of cathepsin B with its inhibitor Ca074Me prevented pyroptosis in bone marrow-derived macrophages of endotoxemia73, in SH-SY5Y and C6 cells (partially prevented)254, in macrophages of a mouse sepsis model and attenuate inflammatory responses71 and in Kupffer cells57. Analogously, genetic knockout of cathepsin B also inhibited pyroptosis and attenuated coxsackievirus B3-induced myocarditis. But genetic deletion of cystatin C (endogenous inhibitor of cathepsin B) contrarily promoted cell death and aggravated myocarditis243. However, in cytosolic flagellin stimulated macrophages, pyroptosis seems to be promoted by both cathepsins B and D since inhibiting both cathepsins simultaneously with Ca074Me and pepstatin A can inhibit pyroptosis. Ca074Me or pepstatin A alone failed to have effect on pyroptosis64.
7.4. Other forms of regulated necrosis
Other regulated forms of necrosis have been involved in various pathological conditions and provide a potential for drug development. For instance, parthanatos has been studied mostly under the background of neuronal damage and neurodegeneration such as Parkinson disease10,308. Plenty of pathological conditions are reported to attribute to parthanatos, including neurodegeneration, some cardiovascular and renal disorders, diabetes, and cerebral ischemia7. NETosis reportedly contributes to a wide range of human pathologies, including diabetes, cancers, allergic asthma systemic, autoimmune diseases, and renal diseases etc.309, 310, 311, 312, 313 Another type of cell death, cyclophilin D-mediated necrosis, has been reported to be involved in cardiac and renal IRI314, muscular dystrophy315, thrombosis316, and diabetes317. Cathepsin inhibitors Ca074Me and pepstatin A prevented around 13% parthanatos and reduced the cytotoxicity of Mn2+ to BV-2 cells74. Lysosomal disrupter LLME induced NETosis in a dose-dependent manner in vitro77.
8. Conclusions, perspective, and challenges
The role of lysosome in regulating necroptosis, ferroptosis, and pyroptosis shares both similarities and differences. The release of hydrolytic enzymes as a result of LMP, especially cathepsin B, is closely involved in the occurrence of both necroptosis and pyroptosis. However, in cells undergoing necroptosis, LMP, and cathepsins release, triggered by various inducers like Ca2+ and ROS, are more prone to be a frequent downstream occurrence of MLKL and can directly cause cell death. In contrast, LMP and cathepsin B release is the initiator of pyroptotic cell death. In pyroptosis, cathepsin B functions by promoting the assembly of the pivotal complex, canonical inflammasome, or pyroptosome, thus activates caspase-1 and initiates pyroptotic cell death. Diversely, the role of lysosome in ferroptosis is rather different. Lysosome acts in an autophagy-dependent manner to control the degradation of ferritin and cause iron accumulation, namely ferrintinophagy. It is noteworthy that the roles of lysosome in regulating RN are far from just these. Instead, some other functions are surfacing, including lysosomal post-translation of RIPK1 and RIPK3 in necroptosis, CMA-dependent degradation of GPX4, or clockophagy in ferroptosis and lysosome-dependent delivery system of LPS in pyroptosis, etc. Utilizing agents or genetic methods to affect lysosome or lysosomal molecules can vigorously dominate the fate of cells and even influence the outcomes of diseases. Therefore, targeting lysosome and relevant events may provide a promising therapeutic strategy to alter the hallmarks of RN, thus further improve the prognosis of diseases, especially of cancers which show resistance to apoptosis and traditional treatments.
In the past, the researches concerning RN have been principally biased towards necroptosis, ferroptosis, and pyroptosis, and consequently lysosome was supposedly thought to be responsible for these three types of RN mainly. Therefore, the evidence for lysosomal involvement in other forms of RN like parthanatos, PARP-mediated cell death, etc., is still insufficient. However, we now noticed that other forms of RN also play an essential role in many physiological and pathological situations, hence it is necessary to expand our study to uncover the role of lysosome in a wider range of RN. Moreover, due to the diversity and abundance of cathepsins in lysosome, as well as the complexity of stimuli and molecular mechanisms in necroptosis and pyroptosis, the functions of different types of cathepsins in these two forms of cell death remain ambiguous, although most studies agreed that cathepsins promote these processes. Some results are even contradictory in different context. Considering that cathepsins are validated targets for the treatment of human diseases, it's worthwhile to further investigate the complicated interactions of cathepsins and RN. Finally, lysosomal factors are emerging as therapeutic targets for many diseases. Strategies that alter the status of lysosome provided a method of dominating the occurrence of RN and showed therapeutic effects in certain diseases. Future studies should explore the therapeutic effects of lysosome through interfering with RN in different diseases background.
Acknowledgments
This work is supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001, China), the Development Program of China (No. 2016YFA0201402), and by the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No. 2019JDJQ008).
Author contributions
Aqu Alu wrote the initial manuscript and made the tables. Xuejiao Han contributed writing materials and new ideas. Xuelei Ma created the figures and revised the manuscript. Min Wu,Yuquan Wei and Xiawei Wei revised the manuscript as well and approved the final version. All authors have read and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Author contributions
Aqu Alu wrote the initial manuscript and made the tables. Xuejiao Han contributed writing materials and new ideas. Xuelei Ma created the figures and revised the manuscript. Min Wu, Yuquan Wei and Xiawei Wei revised the manuscript as well and approved the final version. All authors have read and approved the final manuscript.
References
- 1.Appelqvist H., Wäster P., Kågedal K., Öllinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–226. doi: 10.1093/jmcb/mjt022. [DOI] [PubMed] [Google Scholar]
- 2.Lim C.Y., Zoncu R. The lysosome as a command-and-control center for cellular metabolism. J Cell Biol. 2016;214:653–664. doi: 10.1083/jcb.201607005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence R.E., Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21:133–142. doi: 10.1038/s41556-018-0244-7. [DOI] [PubMed] [Google Scholar]
- 4.Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E.S., Baehrecke E.H. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang D., Kang R., Berghe T.V., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Re D.P., Amgalan D., Linkermann A., Liu Q., Kitsis R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–1817. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Degterev A., Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 10.Conrad M., Angeli J.P., Vandenabeele P., Stockwell B.R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberle C., Huai J., Reinheckel T., Tacke M., Rassner M., Ekert P.G. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17:1167–1178. doi: 10.1038/cdd.2009.214. [DOI] [PubMed] [Google Scholar]
- 12.Erdal H., Berndtsson M., Castro J., Brunk U., Shoshan M.C., Linder S. Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc Natl Acad Sci U S A. 2005;102:192–197. doi: 10.1073/pnas.0408592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Sintes R., Ledesma M.D., Boya P. Lysosomal cell death mechanisms in aging. Ageing Res Rev. 2016;32:150–168. doi: 10.1016/j.arr.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 15.David K.K., Andrabi S.A., Dawson T.M., Dawson V.L. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan S., Schubert D., Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 18.Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 19.Willingham S.B., Bergstralh D.T., O'Connor W., Morrison A.C., Taxman D.J., Duncan J.A. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–159. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cookson B.T., Brennan M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 21.Cantoni O., Tommasini I., Cerioni L., Palomba L., Carloni E., Guidarelli A. Survival pathways triggered by peroxynitrite in cells belonging to the monocyte/macrophage lineage. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:118–123. doi: 10.1016/j.cbpb.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 22.Serrano-Puebla A., Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann N Y Acad Sci. 2016;1371:30–44. doi: 10.1111/nyas.12966. [DOI] [PubMed] [Google Scholar]
- 23.Repnik U., Hafner Česen M., Turk B. Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion. 2014;19 Pt:49–57. doi: 10.1016/j.mito.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Skoupilová H., Michalová E., Hrstka R. Ferroptosis as a new type of cell death and its role in cancer treatment. Klin Onkol. 2018;31:21–26. doi: 10.14735/amko20182S21. [DOI] [PubMed] [Google Scholar]
- 25.Kong Z., Liu R., Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2019;109:2043–2053. doi: 10.1016/j.biopha.2018.11.030. [DOI] [PubMed] [Google Scholar]
- 26.Sharapova T.N., Romanova E.A., Sashchenko L.P., Yashin D.V. FasL on the surface of Tag7 (PGRP-S)-activated lymphocytes induces necroptosis in HLA-negative tumor cells with the involvement of lysosomes and mitochondria. Biochimie. 2018;152:174–180. doi: 10.1016/j.biochi.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Yashin D.V., Romanova E.A., Ivanova O.K., Sashchenko L.P. The Tag7–Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie. 2016;123:32–36. doi: 10.1016/j.biochi.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Cheng S.Y., Wang S.C., Lei M., Wang Z., Xiong K. Regulatory role of calpain in neuronal death. Neural Regen Res. 2018;13:556–562. doi: 10.4103/1673-5374.228762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanden Berghe T., Vanlangenakker N., Parthoens E., Deckers W., Devos M., Festjens N. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 30.Cai X., Liu Y., Hu Y., Liu X., Jiang H., Yang S. ROS-mediated lysosomal membrane permeabilization is involved in bupivacaine-induced death of rabbit intervertebral disc cells. Redox Biol. 2018;18:65–76. doi: 10.1016/j.redox.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun W., Yu J., Gao H., Wu X., Wang S., Hou Y. Inhibition of lung cancer by 2-methoxy-6-acetyl-7-methyljuglone through induction of necroptosis by targeting receptor-interacting protein 1. Antioxid Redox Signal. 2019;31:93–108. doi: 10.1089/ars.2017.7376. [DOI] [PubMed] [Google Scholar]
- 32.Ma Y.M., Peng Y.M., Zhu Q.H., Gao A.H., Chao B., He Q.J. Novel CHOP activator LGH00168 induces necroptosis in A549 human cancer cells via ROS-mediated ER stress and NF-kappaB inhibition. Acta Pharmacol Sin. 2016;37:1381–1390. doi: 10.1038/aps.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashima T., Kohda Y., Tsuchiya K., Ueno T., Yamashita J., Yoshioka T. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain–cathepsin hypothesis’. Eur J Neurosci. 1998;10:1723–1733. doi: 10.1046/j.1460-9568.1998.00184.x. [DOI] [PubMed] [Google Scholar]
- 34.Boya P., Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 35.Droga-Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of Bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283:19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 36.Guicciardi M.E., Leist M., Gores G.J. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 37.Aljabri A., Vijayan V., Stankov M., Nikolin C., Figueiredo C., Blasczyk R. HLA class II antibodies induce necrotic cell death in human endothelial cells via a lysosomal membrane permeabilization-mediated pathway. Cell Death Dis. 2019;10:235. doi: 10.1038/s41419-019-1319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulay S.R., Honarpisheh M.M., Foresto-Neto O., Shi C., Desai J., Zhao Z.B. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol. 2019;30:1857–1869. doi: 10.1681/ASN.2018121218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aits S., Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–1912. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 40.Seo J., Lee E.W., Sung H., Seong D., Dondelinger Y., Shin J. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol. 2016;18:291–302. doi: 10.1038/ncb3314. [DOI] [PubMed] [Google Scholar]
- 41.Seo J., Lee E.W., Song J. New role of E3 ubiquitin ligase in the regulation of necroptosis. BMB Rep. 2016;49:247–248. doi: 10.5483/BMBRep.2016.49.5.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan W., Guo J., Gao B., Zhang W., Ling L., Xu T. Flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci Signal. 2019;12 doi: 10.1126/scisignal.aaw3423. [DOI] [PubMed] [Google Scholar]
- 43.Toyokuni S., Ito F., Yamashita K., Okazaki Y., Akatsuka S. Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Radic Biol Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 44.Hou W., Xie Y., Song X., Sun X., Lotze M.T., Zeh H.J., 3rd Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye F., Chai W., Xie M., Yang M., Yu Y., Cao L. HMGB1 regulates erastin-induced ferroptosis via RAS–JNK/p38 signaling in HL-60/NRASQ61L cells. Am J Cancer Res. 2019;9:730–739. [PMC free article] [PubMed] [Google Scholar]
- 47.Mancias J.D., Wang X., Gygi S.P., Harper J.W., Kimmelman A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Mikhael M., Xu D., Li Y., Soe-Lin S., Ning B. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid Redox Signal. 2010;13:999–1009. doi: 10.1089/ars.2010.3129. [DOI] [PubMed] [Google Scholar]
- 49.Hamaï A., Cañeque T., Müller S., Mai T.T., Hienzsch A., Ginestier C. An iron hand over cancer stem cells. Autophagy. 2017;13:1465–1466. doi: 10.1080/15548627.2017.1327104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma S., Henson E.S., Chen Y., Gibson S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaschler M.M., Hu F., Feng H., Linkermann A., Min W., Stockwell B.R. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13:1013–1020. doi: 10.1021/acschembio.8b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma S., Dielschneider R.F., Henson E.S., Xiao W., Choquette T.R., Blankstein A.R. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12 doi: 10.1371/journal.pone.0182921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou B., Liu J., Kang R., Klionsky D.J., Kroemer G., Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. doi: 10.1016/j.semcancer.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Wu Z., Geng Y., Lu X., Shi Y., Wu G., Zhang M. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116:2996–3005. doi: 10.1073/pnas.1819728116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J., Yang M., Kang R., Klionsky D.J., Tang D. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15:2033–2035. doi: 10.1080/15548627.2019.1659623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang M., Chen P., Liu J., Zhu S., Kroemer G., Klionsky D.J. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5 doi: 10.1126/sciadv.aaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mirshafiee V., Sun B., Chang C.H., Liao Y.P., Jiang W., Jiang J. Toxicological profiling of metal oxide nanoparticles in liver context reveals pyroptosis in Kupffer cells and macrophages versus apoptosis in hepatocytes. ACS Nano. 2018;12:3836–3852. doi: 10.1021/acsnano.8b01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duewell P., Kono H., Rayner K.J., Sirois C.M., Vladimer G., Bauernfeind F.G. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orlowski G.M., Sharma S., Colbert J.D., Bogyo M., Robertson S.A., Kataoka H. Frontline Science: multiple cathepsins promote inflammasome-independent, particle-induced cell death during NLRP3-dependent IL-1beta activation. J Leukoc Biol. 2017;102:7–17. doi: 10.1189/jlb.3HI0316-152R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bruchard M., Mignot G., Derangère V., Chalmin F., Chevriaux A., Végran F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the NLRP3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 61.Okada M., Matsuzawa A., Yoshimura A., Ichijo H. The lysosome rupture-activated TAK1–JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. 2014;289:32926–32936. doi: 10.1074/jbc.M114.579961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brojatsch J., Lima H., Jr., Palliser D., Jacobson L.S., Muehlbauer S.M., Furtado R. Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lysosome-destabilizing agents. Cell Cycle. 2015;14:964–972. doi: 10.4161/15384101.2014.991194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orlowski G.M., Colbert J.D., Sharma S., Bogyo M., Robertson S.A., Rock K.L. Multiple cathepsins promote pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J Immunol. 2015;195:1685–1697. doi: 10.4049/jimmunol.1500509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lage S.L., Buzzo C.L., Amaral E.P., Matteucci K.C., Massis L.M., Icimoto M.Y. Cytosolic flagellin-induced lysosomal pathway regulates inflammasome-dependent and -independent macrophage responses. Proc Natl Acad Sci U S A. 2013;110:E3321–E3330. doi: 10.1073/pnas.1305316110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galluzzi L., Vitale I., Abrams J.M., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H., Wu S., Mao L., Lei G., Zhang L., Lu A. Human pathogenic fungus Trichophyton schoenleinii activates the NLRP3 inflammasome. Protein Cell. 2013;4:529–538. doi: 10.1007/s13238-013-2127-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tschopp J., Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production?. Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 68.Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B.L., Rajendiran T.M., Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Broz P., Dixit V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 70.Deng M., Tang Y., Li W., Wang X., Zhang R., Zhang X. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49:740–753.e7. doi: 10.1016/j.immuni.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L., Zhao Y., Lai D., Zhang P., Yang Y., Li Y. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9:597. doi: 10.1038/s41419-018-0538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang J., Zhao Y., Zhang P., Li Y., Yang Y., Yang Y. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis. 2016;7:e2363. doi: 10.1038/cddis.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu J., Jiang Y., Wang J., Shi X., Liu Q., Liu Z. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–1239. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porte Alcon S., Gorojod R.M., Kotler M.L. Regulated necrosis orchestrates microglial cell death in manganese-induced toxicity. Neuroscience. 2018;393:206–225. doi: 10.1016/j.neuroscience.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 75.Prado Spalm F.H., Vera M.S., Dibo M.J., Simón M.V., Politi L.E., Rotstein N.P. Ceramide induces the death of retina photoreceptors through activation of parthanatos. Mol Neurobiol. 2019;56:4760–4777. doi: 10.1007/s12035-018-1402-4. [DOI] [PubMed] [Google Scholar]
- 76.Muñoz L.E., Bilyy R., Biermann M.H., Kienhöfer D., Maueröder C., Hahn J. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci U S A. 2016;113:E5856–E5865. doi: 10.1073/pnas.1602230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatfield S.M., Grebe K., Whitehead L.W., Rogers K.L., Nebl T., Murphy J.M. Monosodium urate crystals generate nuclease-resistant neutrophil extracellular traps via a distinct molecular pathway. J Immunol. 2018;200:1802–1816. doi: 10.4049/jimmunol.1701382. [DOI] [PubMed] [Google Scholar]
- 78.Roca F.J., Whitworth L.J., Redmond S., Jones A.A., Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial–lysosomal–endoplasmic reticulum circuit. Cell. 2019;178:1344–1361.e11. doi: 10.1016/j.cell.2019.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kreuzaler P., Watson C.J. Killing a cancer: what are the alternatives?. Nat Rev Cancer. 2012;12:411–424. doi: 10.1038/nrc3264. [DOI] [PubMed] [Google Scholar]
- 80.Putney J.W., Jr. Excitement about calcium signaling in inexcitable cells. Science. 1993;262:676–678. doi: 10.1126/science.8235587. [DOI] [PubMed] [Google Scholar]
- 81.Ghosh A., Greenberg M.E. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 82.Rodríguez-Muela N., Hernández-Pinto A.M., Serrano-Puebla A., García-Ledo L., Latorre S.H., de la Rosa E.J. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015;22:476–487. doi: 10.1038/cdd.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarkar C., Jones J.W., Hegdekar N., Thayer J.A., Kumar A., Faden A.I. PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy. 2020;16:466–485. doi: 10.1080/15548627.2019.1628538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noutsopoulos D., Markopoulos G., Vartholomatos G., Kolettas E., Kolaitis N., Tzavaras T. Vl30 retrotransposition signals activation of a caspase-independent and p53-dependent death pathway associated with mitochondrial and lysosomal damage. Cell Res. 2010;20:553–562. doi: 10.1038/cr.2010.48. [DOI] [PubMed] [Google Scholar]
- 85.Kreuzaler P.A., Staniszewska A.D., Li W., Omidvar N., Kedjouar B., Turkson J. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13:303–309. doi: 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- 86.Sargeant T.J., Lloyd-Lewis B., Resemann H.K., Ramos Montoya A., Skepper J., Watson C.J. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat Cell Biol. 2014;16:1057–1068. doi: 10.1038/ncb3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luke C.J., Silverman G.A. Necrotic cell death: harnessing the dark side of the force in mammary gland involution. Nat Cell Biol. 2011;13:197–199. doi: 10.1038/ncb0311-197. [DOI] [PubMed] [Google Scholar]
- 88.Appelqvist H., Nilsson C., Garner B., Brown A.J., Kågedal K., Ollinger K. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol. 2011;178:629–639. doi: 10.1016/j.ajpath.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu H., Yoshimoto T., Yamashima T. Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem. 2014;289:27432–27443. doi: 10.1074/jbc.M114.560334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nylandsted J., Gyrd-Hansen M., Danielewicz A., Fehrenbacher N., Lademann U., Høyer-Hansen M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kirkegaard T., Roth A.G., Petersen N.H., Mahalka A.K., Olsen O.D., Moilanen I. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 92.Saftig P., Sandhoff K. Cancer: killing from the inside. Nature. 2013;502:312–313. doi: 10.1038/nature12692. [DOI] [PubMed] [Google Scholar]
- 93.Kolter T., Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 94.Zeidan Y.H., Hannun Y.A. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med. 2010;10:454–466. doi: 10.2174/156652410791608225. [DOI] [PubMed] [Google Scholar]
- 95.Goñi F.M., Alonso A. Sphingomyelinases: enzymology and membrane activity. FEBS Lett. 2002;531:38–46. doi: 10.1016/s0014-5793(02)03482-8. [DOI] [PubMed] [Google Scholar]
- 96.Petersen N.H., Olsen O.D., Groth-Pedersen L., Ellegaard A.M., Bilgin M., Redmer S. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393. doi: 10.1016/j.ccr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 97.Leu J.I., Pimkina J., Frank A., Murphy M.E., George D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Calderwood S.K., Khaleque M.A., Sawyer D.B., Ciocca D.R. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–172. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 99.Tidwell J.L., Houenou L.J., Tytell M. Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress Chaperones. 2004;9:88–98. doi: 10.1379/CSC-9R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu H., Yoshimoto T., Imajo-Ohmi S., Dazortsava M., Mathivanan A., Yamashima T. Why are hippocampal CA1 neurons vulnerable but motor cortex neurons resistant to transient ischemia?. J Neurochem. 2012;120:574–585. doi: 10.1111/j.1471-4159.2011.07550.x. [DOI] [PubMed] [Google Scholar]
- 101.Sahara S., Yamashima T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys Res Commun. 2010;393:806–811. doi: 10.1016/j.bbrc.2010.02.087. [DOI] [PubMed] [Google Scholar]
- 102.Yamashima T., Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 103.Wallach D., Kang T.B., Dillon C.P., Green D.R. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 104.Cai Z., Zhang A., Choksi S., Li W., Li T., Zhang X.M. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26:886–900. doi: 10.1038/cr.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cullen S.P., Henry C.M., Kearney C.J., Logue S.E., Feoktistova M., Tynan G.A. Fas/CD95-induced chemokines can serve as "find-me" signals for apoptotic cells. Mol Cell. 2013;49:1034–1048. doi: 10.1016/j.molcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 106.Newton K., Manning G. Necroptosis and inflammation. Annu Rev Biochem. 2016;85:743–763. doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- 107.Rohde K., Kleinesudeik L., Roesler S., Löwe O., Heidler J., Schröder K. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 2017;24:83–97. doi: 10.1038/cdd.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ö Canli, Alankuş Y.B., Grootjans S., Vegi N., Hültner L., Hoppe P.S. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ashkenazi A., Dixit V.M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 110.Lin Y., Devin A., Rodriguez Y., Liu Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C. Rip3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 112.Cowburn A.S., White J.F., Deighton J., Walmsley S.R., Chilvers E.R. z-VAD-fmk augmentation of TNF alpha-stimulated neutrophil apoptosis is compound specific and does not involve the generation of reactive oxygen species. Blood. 2005;105:2970–2972. doi: 10.1182/blood-2004-07-2870. [DOI] [PubMed] [Google Scholar]
- 113.Galluzzi L., Kepp O., Kroemer G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014;24:139–140. doi: 10.1038/cr.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Geng J., Ito Y., Shi L., Amin P., Chu J., Ouchida A.T. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;8:359. doi: 10.1038/s41467-017-00406-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peltzer N., Darding M., Walczak H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 2016;26:445–461. doi: 10.1016/j.tcb.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 116.Bertrand M.J., Milutinovic S., Dickson K.M., Ho W.C., Boudreault A., Durkin J. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote rip1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 117.Vucic D., Dixit V.M., Wertz I.E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- 118.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Papatriantafyllou M. Cell signalling: a necrosome build-up. Nat Rev Mol Cell Biol. 2012;13:540. doi: 10.1038/nrm3415. [DOI] [PubMed] [Google Scholar]
- 120.Li J., McQuade T., Siemer A.B., Napetschnig J., Moriwaki K., Hsiao Y.S. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.He S., Wang L., Miao L., Wang T., Du F., Zhao L. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 122.Cai Z., Jitkaew S., Zhao J., Chiang H.C., Choksi S., Liu J. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sun L., Wang H., Wang Z., He S., Chen S., Liao D. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 124.Chen X., Li W., Ren J., Huang D., He W.T., Song Y. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schappe M.S., Szteyn K., Stremska M.E., Mendu S.K., Downs T.K., Seegren P.V. Chanzyme TRPM7 mediates the Ca2+ influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation. Immunity. 2018;48:59–74.e5. doi: 10.1016/j.immuni.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacobsen A.V., Lowes K.N., Tanzer M.C., Lucet I.S., Hildebrand J.M., Petrie E.J. Hsp90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2015.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ferri K.F., Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–E263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 128.Hitomi J., Christofferson D.E., Ng A., Yao J., Degterev A., Xavier R.J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Z., Cheng X., Meng Q., Wang P., Shu B., Hu Q. Azadirachtin induced apoptosis involves lysosomal membrane permeabilization and cathepsin L release in Spodoptera frugiperda Sf9 cells. Int J Biochem Cell Biol. 2015;64:126–135. doi: 10.1016/j.biocel.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 130.Sosna J., Philipp S., Fuchslocher Chico J., Saggau C., Fritsch J., Föll A. Differences and similarities in TRAIL and tumor necrosis factor mediated necroptotic signaling in cancer cells. Mol Cell Biol. 2016;36:2626–2644. doi: 10.1128/MCB.00941-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang H., Zhong C., Shi L., Guo Y., Fan Z. Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondria through processing of Bid leading to necroptosis. J Immunol. 2009;182:6993–7000. doi: 10.4049/jimmunol.0802502. [DOI] [PubMed] [Google Scholar]
- 132.Zou J., Kawai T., Tsuchida T., Kozaki T., Tanaka H., Shin K.S. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity. 2013;38:717–728. doi: 10.1016/j.immuni.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 133.Yashin D.V., Ivanova O.K., Soshnikova N.V., Sheludchenkov A.A., Romanova E.A., Dukhanina E.A. Tag7 (PGLYRP1) in complex with Hsp70 induces alternative cytotoxic processes in tumor cells via TNFR1 receptor. J Biol Chem. 2015;290:21724–21731. doi: 10.1074/jbc.M115.639732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamashima T., Tonchev A.B., Tsukada T., Saido T.C., ImajohOhmi S., Momoi T. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791–800. doi: 10.1002/hipo.10127. [DOI] [PubMed] [Google Scholar]
- 135.Kim H., Zamel R., Bai X.H., Lu C., Keshavjee S., Keshavjee S. Ischemia–reperfusion induces death receptor-independent necroptosis via calpain–STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol. 2018;315:L595–L608. doi: 10.1152/ajplung.00069.2018. [DOI] [PubMed] [Google Scholar]
- 136.Yap Y.W., Whiteman M., Bay B.H., Li Y., Sheu F.S., Qi R.Z. Hypochlorous acid induces apoptosis of cultured cortical neurons through activation of calpains and rupture of lysosomes. J Neurochem. 2006;98:1597–1609. doi: 10.1111/j.1471-4159.2006.03996.x. [DOI] [PubMed] [Google Scholar]
- 137.Curcio M., Salazar I.L., Mele M., Canzoniero L.M., Duarte C.B. Calpains and neuronal damage in the ischemic brain: the swiss knife in synaptic injury. Prog Neurobiol. 2016;143:1–35. doi: 10.1016/j.pneurobio.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 138.Suzuki S., Murotomi K., Nakajima Y., Kawai K., Ohta K., Warita K. Development of an artificial calcium-dependent transcription factor to detect sustained intracellular calcium elevation. ACS Synth Biol. 2014;3:717–722. doi: 10.1021/sb500070c. [DOI] [PubMed] [Google Scholar]
- 139.Singh R., Brewer M.K., Mashburn C.B., Lou D., Bondada V., Graham B. Calpain 5 is highly expressed in the central nervous system (CNS), carries dual nuclear localization signals, and is associated with nuclear promyelocytic leukemia protein bodies. J Biol Chem. 2014;289:19383–19394. doi: 10.1074/jbc.M114.575159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Andón F.T., Fadeel B. Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc Chem Res. 2013;46:733–742. doi: 10.1021/ar300020b. [DOI] [PubMed] [Google Scholar]
- 141.Vanlangenakker N., Vanden Berghe T., Bogaert P., Laukens B., Zobel K., Deshayes K. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schenk B., Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene. 2015;34:5796–5806. doi: 10.1038/onc.2015.35. [DOI] [PubMed] [Google Scholar]
- 143.Yazdanpanah B., Wiegmann K., Tchikov V., Krut O., Pongratz C., Schramm M. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 144.Kim Y.S., Morgan M.J., Choksi S., Liu Z.G. TNF induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 145.Zhang Y., Su S.S., Zhao S., Yang Z., Zhong C.Q., Chen X. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 147.Han J., Zhong C.Q., Zhang D.W. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–1149. doi: 10.1038/ni.2159. [DOI] [PubMed] [Google Scholar]
- 148.Zhu P., Hu S., Jin Q., Li D., Tian F., Toan S. RIPK3 promotes ER stress induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhao H., Chen Y., Feng H. P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol. 2018;16:1282–1295. doi: 10.2174/1570159X16666180516094500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yamashima T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J Neurochem. 2012;120:477–494. doi: 10.1111/j.1471-4159.2011.07596.x. [DOI] [PubMed] [Google Scholar]
- 151.Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–295. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 152.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved ‘calpain–cathepsin cascade’ from nematodes to primates. Cell Calcium. 2004;36:285–293. doi: 10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 153.Liu Y., Ming W., Wang Y., Liu S., Qiu Y., Xiang Y. Cytotoxin 1 from Naja atra Cantor venom induced necroptosis of leukemia cells. Toxicon. 2019;165:110–115. doi: 10.1016/j.toxicon.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 154.Wang W., Wang W.H., Azadzoi K.M., Su N., Dai P., Sun J. Activation of innate antiviral immune response via double-stranded RNA-dependent RLR receptor-mediated necroptosis. Sci Rep. 2016;6:22550. doi: 10.1038/srep22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Moriwaki K., Bertin J., Gough P.J., Orlowski G.M., Chan F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636. doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ni Y., Gu W.W., Liu Z.H., Zhu Y.M., Rong J.G., Kent T.A. RIP1K contributes to neuronal and astrocytic cell death in ischemic stroke via activating autophagic–lysosomal pathway. Neuroscience. 2018;371:60–74. doi: 10.1016/j.neuroscience.2017.10.038. [DOI] [PubMed] [Google Scholar]
- 157.Yin B., Xu Y., Wei R.L., He F., Luo B.Y., Wang J.Y. Inhibition of receptor-interacting protein 3 upregulation and nuclear translocation involved in Necrostatin-1 protection against hippocampal neuronal programmed necrosis induced by ischemia/reperfusion injury. Brain Res. 2015;1609:63–71. doi: 10.1016/j.brainres.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 158.Jantas D., Chwastek J., Grygier B., Lasoń W. Neuroprotective effects of Necrostatin-1 against oxidative stress-induced cell damage: an involvement of cathepsin D inhibition. Neurotox Res. 2020;37:525–542. doi: 10.1007/s12640-020-00164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pacheco F.J., Almaguel F.G., Evans W., Rios-Colon L., Filippov V., Leoh L.S. Docosahexanoic acid antagonizes TNF-alpha-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflamm Res. 2014;63:859–871. doi: 10.1007/s00011-014-0760-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.McComb S., Shutinoski B., Thurston S., Cessford E., Kumar K., Sad S. Cathepsins limit macrophage necroptosis through cleavage of RIP1 kinase. J Immunol. 2014;192:5671–5678. doi: 10.4049/jimmunol.1303380. [DOI] [PubMed] [Google Scholar]
- 161.Sosna J., Voigt S., Mathieu S., Kabelitz D., Trad A., Janssen O. The proteases HtrA2/Omi and UCH-L1 regulate TNF-induced necroptosis. Cell Commun Signal. 2013;11:76. [Google Scholar]
- 162.Sosna J., Voigt S., Mathieu S., Lange A., Thon L., Davarnia P. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. 2014;71:331–348. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu S., Li Y., Choi H.M.C., Sarkar C., Koh E.Y., Wu J. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018;9:476. doi: 10.1038/s41419-018-0469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Christofferson D.E., Li Y., Hitomi J., Zhou W., Upperman C., Zhu H. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012;3 doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Asare N., Lagadic-Gossmann D., Holme J.A. 3-Nitrofluoranthene (3-NF)-induced apoptosis and programmed necrosis. Autophagy. 2009;5:751–752. doi: 10.4161/auto.5.5.8790. [DOI] [PubMed] [Google Scholar]
- 166.Hou X., Yang C., Zhang L., Hu T., Sun D., Cao H. Killing colon cancer cells through PCD pathways by a novel hyaluronic acid-modified shell–core nanoparticle loaded with RIP3 in combination with chloroquine. Biomaterials. 2017;124:195–210. doi: 10.1016/j.biomaterials.2016.12.032. [DOI] [PubMed] [Google Scholar]
- 167.Iannielli A., Bido S., Folladori L., Segnali A., Cancellieri C., Maresca A. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson's disease models. Cell Rep. 2018;22:2066–2079. doi: 10.1016/j.celrep.2018.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sun W., Wu X., Gao H., Yu J., Zhao W., Lu J.J. Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Radic Biol Med. 2017;108:433–444. doi: 10.1016/j.freeradbiomed.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 169.Dixon S.J. Ferroptosis: bug or feature?. Immunol Rev. 2017;277:150–157. doi: 10.1111/imr.12533. [DOI] [PubMed] [Google Scholar]
- 170.Dolma S., Lessnick S.L., Hahn W.C., Stockwell B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 171.Sang M., Luo R., Bai Y., Dou J., Zhang Z., Liu F. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics. 2019;9:6209–6223. doi: 10.7150/thno.36283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hayano M., Yang W.S., Corn C.K., Pagano N.C., Stockwell B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–278. doi: 10.1038/cdd.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Green D.R. The coming decade of cell death research: five riddles. Cell. 2019;177:1094–1107. doi: 10.1016/j.cell.2019.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rennekamp A.J. The ferrous awakens. Cell. 2017;171:1225–1227. doi: 10.1016/j.cell.2017.11.029. [DOI] [PubMed] [Google Scholar]
- 176.Sun X., Ou Z., Xie M., Kang R., Fan Y., Niu X. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–5625. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chu B., Kon N., Chen D., Li T., Liu T., Jiang L. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–591. doi: 10.1038/s41556-019-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dixon S.J., Stockwell B.R. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 179.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Louandre C., Marcq I., Bouhlal H., Lachaier E., Godin C., Saidak Z. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971–977. doi: 10.1016/j.canlet.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 181.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xie Y., Hou W., Song X., Yu Y., Huang J., Sun X. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Seiler A., Schneider M., Förster H., Roth S., Wirth E.K., Culmsee C. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metabol. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 184.Ran Q., Van Remmen H., Gu M., Qi W., Roberts L.J., 2nd, Prolla T. Embryonic fibroblasts from Gpx4+/– mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic Biol Med. 2003;35:1101–1109. doi: 10.1016/s0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- 185.Kang R., Zeng L., Zhu S., Xie Y., Liu J., Wen Q. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108.e4. doi: 10.1016/j.chom.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Friedmann Angeli J.P., Schneider M., Proneth B., Tyurina Y.Y., Tyurin V.A., Hammond V.J. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zitvogel L., Kroemer G. Interferon-gamma induces cancer cell ferroptosis. Cell Res. 2019;29:692–693. doi: 10.1038/s41422-019-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang W., Green M., Choi J.E., Gijón M., Kennedy P.D., Johnson J.K. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gao H., Bai Y., Jia Y., Zhao Y., Kang R., Tang D. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. 2018;503:1550–1556. doi: 10.1016/j.bbrc.2018.07.078. [DOI] [PubMed] [Google Scholar]
- 191.Wang S., Luo J., Zhang Z., Dong D., Shen Y., Fang Y. Iron and magnetic: new research direction of the ferroptosis-based cancer therapy. Am J Cancer Res. 2018;8:1933–1946. [PMC free article] [PubMed] [Google Scholar]
- 192.Gkouvatsos K., Papanikolaou G., Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Biophys Acta. 2012;1820:188–202. doi: 10.1016/j.bbagen.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 193.Torii S., Shintoku R., Kubota C., Yaegashi M., Torii R., Sasaki M. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769–777. doi: 10.1042/BJ20150658. [DOI] [PubMed] [Google Scholar]
- 194.Hirayama T., Miki A., Nagasawa H. Organelle-specific analysis of labile Fe(II) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics. 2019;11:111–117. doi: 10.1039/c8mt00212f. [DOI] [PubMed] [Google Scholar]
- 195.Kurz T., Eaton J.W., Brunk U.T. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011;43:1686–1697. doi: 10.1016/j.biocel.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 196.Homma T., Fujii J. Application of glutathione as anti-oxidative and anti-aging drugs. Curr Drug Metabol. 2015;16:560–571. doi: 10.2174/1389200216666151015114515. [DOI] [PubMed] [Google Scholar]
- 197.Skouta R., Dixon S.J., Wang J., Dunn D.E., Orman M., Shimada K. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li Z., Jiang L., Chew S.H., Hirayama T., Sekido Y., Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol. 2019;26:101297. doi: 10.1016/j.redox.2019.101297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wood E.R., Truesdale A.T., McDonald O.B., Yuan D., Hassell A., Dickerson S.H. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 200.Ostenfeld M.S., Fehrenbacher N., Høyer-Hansen M., Thomsen C., Farkas T., Jäättelä M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65:8975–8983. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- 201.Sato K., Shi L., Ito F., Ohara Y., Motooka Y., Tanaka H. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe(II)-dependent manner. J Clin Biochem Nutr. 2019;65:8–15. doi: 10.3164/jcbn.18-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Horackova M., Ponka P., Byczko Z. The antioxidant effects of a novel iron chelator salicylaldehyde isonicotinoyl hydrazone in the prevention of H2O2 injury in adult cardiomyocytes. Cardiovasc Res. 2000;47:529–536. doi: 10.1016/s0008-6363(00)00088-2. [DOI] [PubMed] [Google Scholar]
- 203.Eling N., Reuter L., Hazin J., Hamacher-Brady A., Brady N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–532. doi: 10.18632/oncoscience.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kasukabe T., Honma Y., Okabe-Kado J., Higuchi Y., Kato N., Kumakura S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. 2016;36:968–976. doi: 10.3892/or.2016.4867. [DOI] [PubMed] [Google Scholar]
- 205.Mai T.T., Hamaï A., Hienzsch A., Cañeque T., Müller S., Wicinski J. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025–1033. doi: 10.1038/nchem.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chen G.Q., Benthani F.A., Wu J., Liang D., Bian Z.X., Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242–254. doi: 10.1038/s41418-019-0352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Latunde-Dada G.O. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 208.Guo Z. Artemisinin anti-malarial drugs in China. Acta Pharm Sin B. 2016;6:115–124. doi: 10.1016/j.apsb.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Badu-Boateng C., Naftalin R.J. Ascorbate and ferritin interactions: consequences for iron release in vitro and in vivo and implications for inflammation. Free Radic Biol Med. 2019;133:75–87. doi: 10.1016/j.freeradbiomed.2018.09.041. [DOI] [PubMed] [Google Scholar]
- 210.Du J., Wang T., Li Y., Zhou Y., Wang X., Yu X. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–369. doi: 10.1016/j.freeradbiomed.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 211.Lin R., Zhang Z., Chen L., Zhou Y., Zou P., Feng C. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381:165–175. doi: 10.1016/j.canlet.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 212.Dowdle W.E., Nyfeler B., Nagel J., Elling R.A., Liu S., Triantafellow E. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 213.Zhang Z.L., Yao Z., Wang L., Ding H., Shao J.J., Chen A.P. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–2103. doi: 10.1080/15548627.2018.1503146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gao M., Monian P., Pan Q., Zhang W., Xiang J., Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wish J.B. Assessing iron status: beyond serum ferritin and transferrin saturation. Clin J Am Soc Nephrol. 2006;1 Suppl 1:S4–S8. doi: 10.2215/CJN.01490506. [DOI] [PubMed] [Google Scholar]
- 216.Masaldan S., Clatworthy S.A.S., Gamell C., Meggyesy P.M., Rigopoulos A.T., Haupt S. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100–115. doi: 10.1016/j.redox.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Biasiotto G., Di Lorenzo D., Archetti S., Zanella I. Iron and neurodegeneration: is ferritinophagy the link?. Mol Neurobiol. 2016;53:5542–5574. doi: 10.1007/s12035-015-9473-y. [DOI] [PubMed] [Google Scholar]
- 218.Liu T., Liu W., Zhang M., Yu W., Gao F., Li C. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. 2018;12:12181–12192. doi: 10.1021/acsnano.8b05860. [DOI] [PubMed] [Google Scholar]
- 219.Kim S.E., Zhang L., Ma K., Riegman M., Chen F., Ingold I. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–985. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bao W., Liu X., Lv Y., Lu G.H., Li F., Zhang F. Nanolongan with multiple on-demand conversions for ferroptosis–apoptosis combined anticancer therapy. ACS Nano. 2019;13:260–273. doi: 10.1021/acsnano.8b05602. [DOI] [PubMed] [Google Scholar]
- 221.Yagami T., Yamamoto Y., Koma H. Pathophysiological roles of intracellular proteases in neuronal development and neurological diseases. Mol Neurobiol. 2019;56:3090–3112. doi: 10.1007/s12035-018-1277-4. [DOI] [PubMed] [Google Scholar]
- 222.Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B.T., Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rathkey J.K., Zhao J., Liu Z., Chen Y., Yang J., Kondolf H.C. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3 doi: 10.1126/sciimmunol.aat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chen X., He W.T., Hu L., Li J., Fang Y., Wang X. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from mlkl channel-mediated necroptosis. Cell Res. 2016;26:1007–1020. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 226.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 228.Wang Y., Gao W., Shi X., Ding J., Liu W., He H. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 229.He Y., Zeng M.Y., Yang D., Motro B., Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Jorgensen I., Rayamajhi M., Miao E.A. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fernandes-Alnemri T., Yu J.W., Juliana C., Solorzano L., Kang S., Wu J. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rathinam V.A., Jiang Z., Waggoner S.N., Sharma S., Cole L.E., Waggoner L. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Latz E., Xiao T.S., Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 235.Kim H.M., Kim Y.M. HMGB1: LPS delivery vehicle for caspase-11-mediated pyroptosis. Immunity. 2018;49:582–584. doi: 10.1016/j.immuni.2018.09.021. [DOI] [PubMed] [Google Scholar]
- 236.Vanaja S.K., Russo A.J., Behl B., Banerjee I., Yankova M., Deshmukh S.D. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. 2016;165:1106–1119. doi: 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Man S.M., Karki R., Sasai M., Place D.E., Kesavardhana S., Temirov J. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell. 2016;167:382–396.e17. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rathinam V.A., Vanaja S.K., Waggoner L., Sokolovska A., Becker C., Stuart L.M. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yang D., He Y., Muñoz-Planillo R., Liu Q., Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43:923–932. doi: 10.1016/j.immuni.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rühl S., Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ Eur J Immunol. 2015;45:2927–2936. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 241.Rühl S., Shkarina K., Demarco B., Heilig R., Santos J.C., Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362:956–960. doi: 10.1126/science.aar7607. [DOI] [PubMed] [Google Scholar]
- 242.Tseng W.A., Thein T., Kinnunen K., Lashkari K., Gregory M.S., D'Amore P.A. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54:110–120. doi: 10.1167/iovs.12-10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wang Y., Jia L., Shen J., Wang Y., Fu Z., Su S.A. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1006872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Masters S.L., Dunne A., Subramanian S.L., Hull R.L., Tannahill G.M., Sharp F.A. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hornung V., Bauernfeind F., Halle A., Samstad E.O., Kono H., Rock K.L. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kim L.A., Amarnani D., Gnanaguru G., Tseng W.A., Vavvas D.G., D'Amore P.A. Tamoxifen toxicity in cultured retinal pigment epithelial cells is mediated by concurrent regulated cell death mechanisms. Invest Ophthalmol Vis Sci. 2014;55:4747–4758. doi: 10.1167/iovs.13-13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 248.Mishra A.R., Zheng J., Tang X., Goering P.L. Silver nanoparticle-induced autophagic–lysosomal disruption and NLRP3-inflammasome activation in HepG2 cells is size-dependent. Toxicol Sci. 2016;150:473–487. doi: 10.1093/toxsci/kfw011. [DOI] [PubMed] [Google Scholar]
- 249.Li R., Ji Z., Qin H., Kang X., Sun B., Wang M. Interference in autophagosome fusion by rare earth nanoparticles disrupts autophagic flux and regulation of an interleukin-1beta producing inflammasome. ACS Nano. 2014;8:10280–10292. doi: 10.1021/nn505002w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sun B., Wang X., Ji Z., Li R., Xia T. NLRP3 inflammasome activation induced by engineered nanomaterials. Small. 2013;9:1595–1607. doi: 10.1002/smll.201201962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Liu L., Sha R., Yang L., Zhao X., Zhu Y., Gao J. Impact of morphology on iron oxide nanoparticles-induced inflammasome activation in macrophages. ACS Appl Mater Interfaces. 2018;10:41197–41206. doi: 10.1021/acsami.8b17474. [DOI] [PubMed] [Google Scholar]
- 252.Clerc P., Jeanjean P., Hallali N., Gougeon M., Pipy B., Carrey J. Targeted magnetic intra-lysosomal hyperthermia produces lysosomal reactive oxygen species and causes caspase-1 dependent cell death. J Control Release. 2018;270:120–134. doi: 10.1016/j.jconrel.2017.11.050. [DOI] [PubMed] [Google Scholar]
- 253.Jia C., Zhang J., Chen H., Zhuge Y., Chen H., Qian F. Endothelial cell pyroptosis plays an important role in kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10:778. doi: 10.1038/s41419-019-2021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Chen S., Zhou C., Yu H., Tao L., An Y., Zhang X. 27-Hydroxycholesterol contributes to lysosomal membrane permeabilization-mediated pyroptosis in co-cultured SH-SY5Y cells and C6 cells. Front Mol Neurosci. 2019;12:14. doi: 10.3389/fnmol.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gorojod R.M., Alaimo A., Porte Alcon S., Pomilio C., Saravia F., Kotler M.L. The autophagic–lysosomal pathway determines the fate of glial cells under manganese-induced oxidative stress conditions. Free Radic Biol Med. 2015;87:237–251. doi: 10.1016/j.freeradbiomed.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 256.Hussain S., Sangtian S., Anderson S.M., Snyder R.J., Marshburn J.D., Rice A.B. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part Fibre Toxicol. 2014;11:28. doi: 10.1186/1743-8977-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kodali V.K., Roberts J.R., Shoeb M., Wolfarth M.G., Bishop L., Eye T. Acute in vitro and in vivo toxicity of a commercial grade boron nitride nanotube mixture. Nanotoxicology. 2017;11:1040–1058. doi: 10.1080/17435390.2017.1390177. [DOI] [PubMed] [Google Scholar]
- 258.Brandstetter C., Patt J., Holz F.G., Krohne T.U. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J Photochem Photobiol B. 2016;161:177–183. doi: 10.1016/j.jphotobiol.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 259.Liao Y., Zhang H., He D., Wang Y., Cai B., Chen J. Retinal pigment epithelium cell death is associated with NLRP3 inflammasome activation by all-trans retinal. Invest Ophthalmol Vis Sci. 2019;60:3034–3045. doi: 10.1167/iovs.18-26360. [DOI] [PubMed] [Google Scholar]
- 260.Manna S., Howitz W.J., Oldenhuis N.J., Eldredge A.C., Shen J., Nihesh F.N. Immunomodulation of the NLRP3 inflammasome through structure-based activator design and functional regulation via lysosomal rupture. ACS Cent Sci. 2018;4:982–995. doi: 10.1021/acscentsci.8b00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171:1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Katsnelson M.A., Lozada-Soto K.M., Russo H.M., Miller B.A., Dubyak G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol. 2016;311:C83–C100. doi: 10.1152/ajpcell.00298.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Senerovic L., Tsunoda S.P., Goosmann C., Brinkmann V., Zychlinsky A., Meissner F. Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 2012;3:e384. doi: 10.1038/cddis.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Zitvogel L., Kepp O., Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 265.Lima H., Jr., Jacobson L.S., Goldberg M.F., Chandran K., Diaz-Griffero F., Lisanti M.P. Role of lysosome rupture in controlling NLRP3 signaling and necrotic cell death. Cell Cycle. 2013;12:1868–1878. doi: 10.4161/cc.24903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Monteith A.J., Vincent H.A., Kang S., Li P., Claiborne T.M., Rajfur Z. mTORC2 activity disrupts lysosome acidification in systemic lupus erythematosus by impairing caspase-1 cleavage of Rab39a. J Immunol. 2018;201:371–382. doi: 10.4049/jimmunol.1701712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Chen Q., Guan X., Zuo X., Wang J., Yin W. The role of high mobility group box 1 (HMGB1) in the pathogenesis of kidney diseases. Acta Pharm Sin B. 2016;6:183–188. doi: 10.1016/j.apsb.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yang H., Liu H., Zeng Q., Imperato G.H., Addorisio M.E., Li J. Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol Med. 2019;25:13. doi: 10.1186/s10020-019-0081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Jun H.K., Jung Y.J., Ji S., An S.J., Choi B.K. Caspase-4 activation by a bacterial surface protein is mediated by cathepsin G in human gingival fibroblasts. Cell Death Differ. 2018;25:380–391. doi: 10.1038/cdd.2017.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Luan Z.G., Zhang H., Yang P.T., Ma X.C., Zhang C., Guo R.X. HMGB1 activates nuclear factor-kappaB signaling by RAGE and increases the production of TNF-alpha in human umbilical vein endothelial cells. Immunobiology. 2010;215:956–962. doi: 10.1016/j.imbio.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 271.Andrabi S.A., Umanah G.K., Chang C., Stevens D.A., Karuppagounder S.S., Gagné J.P. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wang Y., An R., Umanah G.K., Park H., Nambiar K., Eacker S.M. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;354:aad6872. doi: 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Yipp B.G., Kubes P. NETosis: how vital is it?. Blood. 2013;122:2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 274.Remijsen Q., Vanden Berghe T., Wirawan E., Asselbergh B., Parthoens E., de Rycke R. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Papayannopoulos V., Metzler K.D., Hakkim A., Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Yipp B.G., Petri B., Salina D., Jenne C.N., Scott B.N., Zbytnuik L.D. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhang D., Gao M., Jin Q., Ni Y., Zhang J. Updated developments on molecular imaging and therapeutic strategies directed against necrosis. Acta Pharm Sin B. 2019;9:455–468. doi: 10.1016/j.apsb.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Zhang H., Zhou X., McQuade T., Li J., Chan F.K., Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sarhan M., von Mässenhausen A., Hugo C., Oberbauer R., Linkermann A. Immunological consequences of kidney cell death. Cell Death Dis. 2018;9:114. doi: 10.1038/s41419-017-0057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Tonnus W., Linkermann A. The in vivo evidence for regulated necrosis. Immunol Rev. 2017;277:128–149. doi: 10.1111/imr.12551. [DOI] [PubMed] [Google Scholar]
- 282.Linkermann A., Green D.R. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Degterev A., Hitomi J., Germscheid M., Ch'en I.L., Korkina O., Teng X. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Dielschneider R.F., Henson E.S., Gibson S.B. Lysosomes as oxidative targets for cancer therapy. Oxid Med Cell Longev. 2017;2017:3749157. doi: 10.1155/2017/3749157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Domenech M., Marrero–Berrios I., Torres-Lugo M., Rinaldi C. Lysosomal membrane permeabilization by targeted magnetic nanoparticles in alternating magnetic fields. ACS Nano. 2013;7:5091–5101. doi: 10.1021/nn4007048. [DOI] [PubMed] [Google Scholar]
- 286.Siklos M., BenAissa M., Thatcher G.R. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B. 2015;5:506–519. doi: 10.1016/j.apsb.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Wegner K.W., Saleh D., Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci. 2017;38:202–225. doi: 10.1016/j.tips.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 289.Moreno-Gonzalez G., Vandenabeele P., Krysko D.V. Necroptosis: a novel cell death modality and its potential relevance for critical care medicine. Am J Respir Crit Care Med. 2016;194:415–428. doi: 10.1164/rccm.201510-2106CI. [DOI] [PubMed] [Google Scholar]
- 290.Fulda S. Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol. 2014;35:51–56. doi: 10.1016/j.semcdb.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 291.Groth-Pedersen L., Jäättelä M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013;332:265–274. doi: 10.1016/j.canlet.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 292.Edinger A.L., Thompson C.B. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 293.Harris P.A., King B.W., Bandyopadhyay D., Berger S.B., Campobasso N., Capriotti C.A. DNA-encoded library screening identifies benzo[b][1,4]oxazepin-4-ones as highly potent and monoselective receptor interacting protein 1 kinase inhibitors. J Med Chem. 2016;59:2163–2178. doi: 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- 294.Vitner E.B., Salomon R., Farfel-Becker T., Meshcheriakova A., Ali M., Klein A.D. RIPK3 as a potential therapeutic target for Gaucher's disease. Nat Med. 2014;20:204–208. doi: 10.1038/nm.3449. [DOI] [PubMed] [Google Scholar]
- 295.Roca F.J., Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Takano J., Tomioka M., Tsubuki S., Higuchi M., Iwata N., Itohara S. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 297.Zhao W., Feng H., Sun W., Liu K., Lu J.J., Chen X. Tert-Butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: roles of NOX4 and mitochondrion. Redox Biol. 2017;11:524–534. doi: 10.1016/j.redox.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Dunai Z.A., Imre G., Barna G., Korcsmaros T., Petak I., Bauer P.I. Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Mihalik R., Imre G., Petak I., Szende B., Kopper L. Cathepsin B-independent abrogation of cell death by CA-074-ome upstream of lysosomal breakdown. Cell Death Differ. 2004;11:1357–1360. doi: 10.1038/sj.cdd.4401493. [DOI] [PubMed] [Google Scholar]
- 300.Morris G., Walker A.J., Berk M., Maes M., Puri B.K. Cell death pathways: a novel therapeutic approach for neuroscientists. Mol Neurobiol. 2018;55:5767–5786. doi: 10.1007/s12035-017-0793-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Linkermann A., Skouta R., Himmerkus N., Mulay S.R., Dewitz C., de Zen F. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111:16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hangauer M.J., Viswanathan V.S., Ryan M.J., Bole D., Eaton J.K., Matov A. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–250. doi: 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Friedmann Angeli J.P., Krysko D.V., Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–414. doi: 10.1038/s41568-019-0149-1. [DOI] [PubMed] [Google Scholar]
- 305.No authors listed Immunotherapy activates unexpected cell death mechanism. Cancer Discov. 2019;9:OF2. doi: 10.1158/2159-8290.CD-NB2019-058. [DOI] [PubMed] [Google Scholar]
- 306.Zhu S., Ding S., Wang P., Wei Z., Pan W., Palm N.W. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature. 2017;546:667–670. doi: 10.1038/nature22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Doitsh G., Galloway N.L., Geng X., Yang Z., Monroe K.M., Zepeda O. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lee Y., Karuppagounder S.S., Shin J.H., Lee Y.I., Ko H.S., Swing D. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013;16:1392–1400. doi: 10.1038/nn.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Cedervall J., Zhang Y., Olsson A.K. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. 2016;76:4311–4315. doi: 10.1158/0008-5472.CAN-15-3051. [DOI] [PubMed] [Google Scholar]
- 310.Wong S.L., Demers M., Martinod K., Gallant M., Wang Y., Goldfine A.B. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Toussaint M., Jackson D.J., Swieboda D., Guedán A., Tsourouktsoglou T.D., Ching Y.M. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. 2017;23:681–691. doi: 10.1038/nm.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Gupta S., Kaplan M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12:402–413. doi: 10.1038/nrneph.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Albrengues J., Shields M.A., Ng D., Park C.G., Ambrico A., Poindexter M.E. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361 doi: 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Devalaraja-Narashimha K., Diener A.M., Padanilam B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. 2009;297:F749–F759. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 315.Millay D.P., Sargent M.A., Osinska H., Baines C.P., Barton E.R., Vuagniaux G. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Jobe S.M., Wilson K.M., Leo L., Raimondi A., Molkentin J.D., Lentz S.R. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111:1257–1265. doi: 10.1182/blood-2007-05-092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Fujimoto K., Chen Y., Polonsky K.S., Dorn G.W., 2nd Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci U S A. 2010;107:10214–10219. doi: 10.1073/pnas.0914209107. [DOI] [PMC free article] [PubMed] [Google Scholar]