

Abstract
A molecular level conceptualization of the pathogenesis of Alzheimer's disease (AD) remains elusive with many competing hypotheses, particularly via proteopathic and immunopathic mechanisms. However, these need not be competitive. If amyloid beta (Aβ) is regarded as an “early responder cytokine,” then proteopathic considerations become encompassed within an overarching hybrid proteopathic‐immunopathic mechanism. As argued in this commentary, Aβ is in fact a molecular constituent of the innate immune system. Aβ is an antimicrobial peptide (AMP) functioning not only as a killer peptide, but also as a modulatory immunopeptide. Aβ satisfies the definition of a cytokine, exhibiting interdependency with other cytokines. Aβ also satisfies the functional definition of a chemokine, existing within the AMP‐chemokine spectrum. Aβ, like conventional cytokines, both binds to and is released by microglial cells. Finally, Aβ interacts with the complement and Toll‐like receptor systems analogously to established cytokines. Aβ may thus be regarded as an effector molecule of innate immunity.
Keywords: Alzheimer's disease, amyloid beta, antimicrobial peptide, chemokine, cytokine, immunopathy, microglia
1. INTRODUCTION
A molecular level conceptualization of the pathogenesis of Alzheimer's disease (AD) remains elusive and hotly debated. 1 There are many different mechanistic proposals including proteopathy, tauopathy, immunopathy, gliopathy, mitochondriopathy, synapopathy—each with passionate supporters and detractors. In recent decades, the two leading contenders have been the amyloid protein misfolding hypothesis (proteopathy), and the innate immunotoxicity hypothesis (immunopathy). However, arising from multiple much‐publicized failures of amyloid‐targeting therapies, some researchers have ardently declared “the amyloid hypothesis is dead,” proclaiming the need to focus exclusively on other avenues such as immunopathy. But this need not be an “either‐or” situation; indeed, if amyloid beta (Aβ) is regarded as simply an early responder cytokine, then proteopathic considerations become encompassed within an overarching hybrid proteopathic‐immunopathic mechanism, embracing the comprehensive role of innate immunity in the etiopathogenesis of AD.
2. INNATE IMMUNE SYSTEM
Although phylogenetically ancient, the innate immune system is complex, presenting a multi‐tiered biomolecular defense strategy hierarchically consisting of tightly homeostatically regulated peptides (antimicrobial peptides), small proteins (cytokines, chemokines, interleukins, interferons), and cellular components (macrophages/microglia, innate lymphoid cells, neutrophils) cooperatively functioning in a concerted fashion. 2 This biochemical defense network is initiated, coordinated, and harmonized by two additional signaling systems (plasma‐based complement system, membrane‐based Toll‐like receptor system), which initially present instigating pathological processes to trigger the innate immune system, then integrate the various components of innate immunity with each other, and finally facilitate an innate‐to‐adaptive immune system hand‐off.
3. IS Aβ AN INNATE IMMUNITY EFFECTOR MOLECULE?
Despite decades of study, Aβ remains an enigmatic and poorly understood molecule. Its presence, either as a consequence or cause, is nonetheless central to the recognized pathology of AD. However, given the existence of specific enzymatic pathways for the biosynthesis of Aβ, coupled with the presence of selective uptake, breakdown, and clearance routes, it seems plausible that Aβ has a normal physiological role and is not merely a toxic molecule leading to dementia. Although multiple physiological roles for Aβ have been proposed, including regulation of cholesterol transport, protection against oxidative stress, and activation of key kinase enzymes, none of these capture the breadth of Aβ’s involvement in AD. 3 Recognizing the possibility of Aβ as an innate immunopeptide (antimicrobial peptide/cytokine/chemokine) would be consistent with Aβ’s multi‐faceted role in AD (see Figure 1).
FIGURE 1.
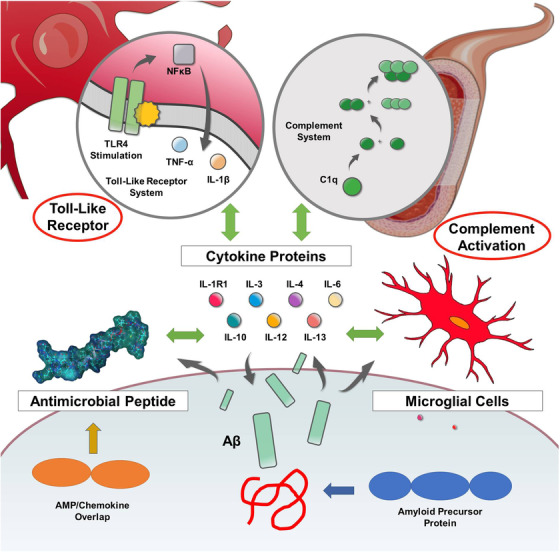
Amyloid beta (Aβ) as an early responder cytokine. The innate immune system is complex, presenting a multi‐tiered biomolecular defense strategy hierarchically consisting of peptides (antimicrobial peptides), small proteins (cytokines), and cellular components (microglia, lymphocytes) cooperatively functioning in a concerted fashion; this biochemical defense network is initiated, coordinated, and harmonized by two additional signaling systems (plasma‐based complement system, membrane‐based Toll‐like receptor system). Aβ interacts with all aspects of this interconnected innate immune system. Thus, Aβ may be regarded as an effector molecule of innate immunity and functions as an early responder cytokine
3.1. Aβ as an antimicrobial peptide
Antimicrobial peptides (AMPs; or host defense peptides) are the first responder molecules of the innate immune system, exhibiting dual functional roles: first as killer peptides destroying microbes, second as immunopeptides modulating the subsequent innate immune response. AMPs are potent, broad spectrum antimicrobials targeting enveloped viruses, bacteria, and fungi; they are also immunomodulatory, inhibiting pro‐inflammatory cytokine production, inducing chemokine release, and stimulating chemotaxis by acting as chemoattractants. To achieve certain immunomodulatory effects, AMPs bind to membrane‐bound glycosaminoglycans (GAGs), sometimes via BBXB (where B is any basic amino acid) peptidic motifs within their primary amino acid sequence. 4 , 5 In fulfilling these biochemical functions, many AMPs oligomerize and aggregate, having their actions modified by ambient concentrations of cholesterol and metallic ions (Zn2+, Cu2+). AMPs may themselves also act as cytokines/chemokines with some studies suggesting a significant AMP‐chemokine functional overlap (the AMP/cytokine differentiation may be an artefact of nomenclature, with many AMPs and cytokines existing on a common functional spectrum such that AMPs have cytokine‐like properties and cytokines have AMP‐like properties).
Aβ is an AMP. 6 Multiple in vitro and in vivo studies demonstrate that Aβ has potent, broad‐spectrum antimicrobial properties against viruses, bacteria, and fungi, enabled by the formation of oligomers that disrupt membranes and entrap pathogens; moreover, overexpression of Aβ confers increased resistance to infection from both bacteria and viruses. 7 Like AMPs, Aβ’s actions are influenced by cholesterol, Zn2+, and Cu2+. In fulfilling the dual functional roles of AMPs, Aβ is likewise an immunopeptide modulating innate immune responses and binding to GAGs via its H13HQK16 (BBXB) motif. 8
3.2. Aβ as a cytokine/chemokine
Cytokines, the molecular backbone of innate immunity, are a group of diverse small proteins (4‐20 kDa) which function as immunomodulatory signaling molecules regulating immunity and inflammation by influencing the maturation and responsiveness of designated cell populations and by inducing changes in gene expression that affect cellular function. 9 Cytokines are produced by a range of cells, including macrophages/microglia, lymphocytes, mast cells, endothelial cells, and various stromal cells. Most cytokines enhance or inhibit the action of other cytokines through a complex interdependency that involves pleotropism, redundancy, and synergism. Chemokines, the largest subgroup of cytokine, are chemoattractant proteins inducing cellular movement to loci of pathology; their release is stimulated by pathological pro‐inflammatory stimuli, such as interleukin‐1 (IL‐1), tumor necrosis factor‐α (TNF‐α), or lipopolysaccharide (LPS). Chemokines are characterized by their functional and structural characteristics: functionally, they stimulate the chemotactic migration of diverse cells, particularly leukocytes; structurally, they have cysteine residues in conserved locations to enforce a constrained 3‐dimensional conformation. Oligomerization and GAG binding are vital for the functions of multiple chemokines; 10 a conserved, BBXB motif is key to the immunomodulatory binding of some chemokines that recognize GAGs as co‐receptors. 11 Select chemokines also function as AMPs, giving rise to a broader spectrum class of AMP/chemokine hybrid proteins.
Aβ satisfies the functional definition of a cytokine. Aβ is a small protein (4 kDa) which functions as immunomodulatory signaling molecule mediating intercellular interactions, and regulating immunity and inflammation. Via c‐jun‐n‐terminal‐kinase (JNK‐AP1) signaling pathways, Aβ elicits expression of monocyte chemoattractant protein‐1 (MCP‐1), growth related oncogene (GRO), IL‐1β, and IL‐6 inflammatory genes; 12 Aβ likewise impacts other gene expression pathways important for vesicle trafficking, cell adhesion, actin cytoskeleton dynamics, insulin signaling, and synaptophysin downregulation. 13 Aβ can be produced by numerous types of cells such as neurons, astrocytes, neuroblastoma cells, hepatoma cells, and fibroblasts. Aβ’s agonistic/antagonistic interactions with established cytokines display pleotropism, redundancy, and synergism. Although Aβ lacks the defining cystine disulfide loops of chemokines, it does exist functionally within the AMP‐chemokine overlap spectrum.
3.3. Aβ and microglia
Microglia are the cellular backbone of the innate immune system, being a specialized population of phagocytic macrophage‐like cells functioning as immunological sentinels capable of choreographing a potent inflammatory response. Microglia are both a source and a target of cytokines: activated microglia elicit the expression of pro‐inflammatory cytokines such as IL‐1β, IL‐6, and TNF‐α influencing the surrounding brain tissue. 14
Microglia are a source and target of neurotoxic Aβ, as they are for other cytokines. Microglia can secrete Aβ when appropriately stimulated by LPS, other cytokines, and even Aβ itself. Correspondingly, Aβ activation of microglia induces gene transcription including activation of many pro‐inflammatory cytokines, most notably IL‐1β, IL‐8, and matrix metalloproteinases (MMP), amplifying ongoing inflammation and neuronal loss. 15 Like other cytokines, Aβ also activates B‐lymphocytes and other leukocytes. 16
3.4. Aβ interactions with the complement cascade/Toll‐like receptor systems
The Toll‐like receptor (TLR) system of membrane‐spanning proteins initiates and integrates innate immune responses by recognizing specific molecular patterns. TLR engagement leads to the activation of nuclear factor‐κB (NF‐κB) and interferon regulatory factors, resulting in the upregulation of downstream target genes including an array of pro‐inflammatory cytokines, chemokines, and interferon‐responsive genes; in macrophages TLR signaling also induces autophagy. The concomitantly activated complement system of plasma proteins also traverses the histobiomolecular spectrum of innate immunity, triggering and expanding innate responses after detecting pathogen‐associated molecular patterns (PAMPs). Subsequent protease activation releases cytokines and establishes an amplifying cascade that also attracts macrophages and neutrophils; arising from subsequent antibody interactions, the complement system is ultimately active at the interface between the innate and adaptive immune systems.
In a manner analogous to other cytokines, Aβ influences and is influenced by the TLR and complement systems. In the early stages of AD, fibrillar Aβ can directly interact with TLR2 and TLR4 Toll‐like receptors to induce microglial Aβ phagocytosis; 17 similarly, TLR7, TLR8, and TLR9 can enhance microglial Aβ uptake at other stages of AD. 18 Within the complement system, Aβ binds C1q protein and activates the classical pathway, inducing complement‐mediated toxicity against neurons. Complement proteins are integral components of Aβ plaques and their accumulation occurs at the earliest stages of Aβ deposition. 19 , 20
4. Aβ IS AN INNATE IMMUNITY EFFECTOR MOLECULE
Aβ is a molecular constituent of the innate immune system. Aβ is an antimicrobial peptide functioning not only as a killer peptide, but also a modulatory immunopeptide. Aβ satisfies the definition of a cytokine and exhibits interdependency with other cytokines characterized by pleotropism, redundancy, and synergism. Aβ also satisfies the functional definition of a chemokine, but not the strict structural definition; Aβ exists within the AMP‐chemokine spectrum also exhibiting oligomerization and binding to GAGs via its HHQK(BBXB) motif. Aβ, like conventional cytokines, both binds to and is released by microglial cells. Finally, Aβ interfaces with the complement and Toll‐like receptor systems analogously to established cytokines (see Figure 1).
5. CONCLUSIONS
Within the context of AD disease pathogenesis, Aβ may be regarded as an early responder cytokine. This possibility simplifies the complex collaborative connections between Aβ and the established anti/pro‐inflammatory cytokines of innate immunity, and enables incorporation of amyloid misfolding into the evolving immunopathic disease mechanism hypotheses of AD.
FUNDING INFORMATION
DFW acknowledges salary support from a Canada Research Chair, Tier 1.
CONFLICTS OF INTEREST
The author declares no conflicts of interest.
Weaver DF. Amyloid beta is an early responder cytokine and immunopeptide of the innate immune system. Alzheimer's Dement. 2020;6:e12100 10.1002/trc2.12100
REFERENCES
- 1. Gray SC, Kinghorn KJ, Woodling NS. Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen Res. 2020;15:1208‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang H, Shen Y, Chuang H, Chiu C, Ye Y, Zhao L. Neuroinflammation in Alzheimer's disease: microglia, molecular participants and therapeutic choices. Curr Alzheimer Res. 2019;16:659‐674. [DOI] [PubMed] [Google Scholar]
- 3. Dawkins E, Small DH. Insights into the physiological function of the β‐amyloid precursor protein: beyond Alzheimer's disease. J Neurochem. 2014;129:756‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herrera R, Morris M, Rosbe K, Feng Z, Weinberg A, Tugizov S. Human beta‐defensins‐2 and ‐3 co‐internalize with human immunodeficiency virus via heparan sulfate proteoglycans and reduce infectivity of intracellular virions in tonsil epithelial cells. Virology. 2016;487:172‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silva de Paula V, Valente AP. A dynamic overview of antimicrobial peptides and their complexes. Molecules. 2018;23:2040‐2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14:1602‐1614. [DOI] [PubMed] [Google Scholar]
- 7. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer's disease‐associated β‐Amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 2018;99:56‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giulian D, Haverkamp LJ, Yu J, et al. The HHQK domain of beta‐amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem. 1998;273:29719‐29726. [DOI] [PubMed] [Google Scholar]
- 9. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer's disease. Immunol Cell Biol. 2020;98:28‐41. [DOI] [PubMed] [Google Scholar]
- 10. Sepuru KM, Rajarathnam K. Structural basis of chemokine interactions with heparan sulfate, chondroitin sulfate, and dermatan sulfate. J Biol Chem. 2019;294:15650‐15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Proudfoot AE, Fritchley S, Borlat F, et al. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J Biol Chem. 2001;276:10620‐10626. [DOI] [PubMed] [Google Scholar]
- 12. Vukic V, Callaghan D, Walker D, et al. Expression of inflammatory genes induced by beta‐amyloid peptides in human brain endothelial cells and in Alzheimer's brain is mediated by the JNK‐AP1 signaling pathway. Neurobiol Dis. 2009;34:95‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sebollela A, Freitas‐Correa L, Oliveira FF, et al. Amyloid‐β oligomers induce differential gene expression in adult human brain slices. J Biol Chem. 2012;287:7436‐7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bitting L, Naidu A, Cordell B, Murphy GM. Beta‐amyloid peptide secretion by a microglial cell line is induced by beta‐amyloid‐(25‐35) and lipopolysaccharide. J Biol Chem. 1996;271:16084‐16089. [DOI] [PubMed] [Google Scholar]
- 15. Hussain AA, Lee Y, Zhang JJ, Francis PT, Marshall J. Disturbed matrix metalloproteinase pathway in both age‐related macular degeneration and Alzheimer's disease. J Neurodegener Dis. 2017;2017:4810232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurnellas MP, Ghosn EE, Schartner JM, et al. Amyloid fibrils activate B‐1a lymphocytes to ameliorate inflammatory brain disease. Proc Natl Acad Sci USA. 2015;112:15016‐15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll‐like receptors 2 and 4 are required for fibrillar Aβ‐stimulated microglial activation. J Neurosci. 2009;29:11982‐11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landreth GE, Reed‐Geaghan EG. Toll‐like receptors in Alzheimer's disease. Curr Top Microbiol Immunol. 2009;336:137‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bradt B, Kolb W, Cooper N. Complement‐dependent proinflammatory properties of the Alzheimer's disease beta‐peptide. J Exp Med. 1998;188:431‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bergamaschini L, Donarini C, Gobbo G, Parnetti L, Gallai V. Activation of complement and contact system in Alzheimer's disease. Mech Ageing Dev. 2001;122:1971‐1983. [DOI] [PubMed] [Google Scholar]