

Abstract
Acute kidney injury (AKI) is common in critically ill children and adults, and sepsis-associated AKI (SA-AKI) is the most frequent cause of AKI in the ICU. To date, no mechanistically targeted therapeutic interventions have been identified. High-throughput “omic” technologies (e.g. genomics, proteomics, metabolomics, etc.) offer a new angle of approach to achieve this end. In this review, we provide an update on the current understanding of SA-AKI pathophysiology. Omic technologies themselves are briefly discussed to facilitate interpretation of studies using them. We next summarize the body of SA-AKI research to date that has employed omic technologies. Importantly, omic studies are helping to elucidate a pathophysiology of SA-AKI centered around cellular stress responses, metabolic changes, and dysregulation of energy production that underlie its clinical features. Finally, we propose opportunities for future research using clinically relevant animal models, integrating multiple omic technologies, and ultimately progressing to translational human studies focusing therapeutic strategies on targeted disease mechanisms.
Keywords: Sepsis, Acute Kidney Injury, Genomics, Transcriptomics, Proteomics, Metabolomics
Introduction
Acute kidney injury (AKI) is common in critically ill children and adults [1, 2]. Sepsis is the most frequent etiology of AKI in the ICU, causing 40–50% of all cases in the critical care setting [3–5]. Compared to AKI from other etiologies, sepsis-associated AKI (SA-AKI) leads to more pronounced oliguria, greater fluid overload, increased hospital length of stay and cost, and increased risk of chronic kidney disease [2, 4–9]. Though definitions of sepsis and AKI vary between research populations, multiple studies have shown that the intersection of sepsis and AKI increases mortality above either condition alone. One pediatric study reported that children with severe sepsis who develop AKI have a mortality of 17% compared with only 8% in those who never develop AKI [10]. Another found that among those with septic shock, severe persistent AKI increased mortality from 6% to 38% [11]. Similarly, several adult studies show a clinically important impact of AKI on sepsis survival [3, 5, 12].
To date, no mechanistically targeted therapeutic interventions have been identified for sepsis, much less for SA-AKI. Current treatment relies wholly upon supportive care and renal replacement technologies. While preventive measures are important for the critically ill, many patients with severe sepsis demonstrate AKI on presentation [10, 13]. Given the complexity of this condition, it is unlikely that single target, single therapy approaches will yield the clinical improvements we hope to attain. While the use of biomarkers to stratify and endotype patients is an important part of this effort, it is insufficient to achieve the breakthroughs necessary. The organismal, tissue, and cellular derangements that underpin the endotypes must be understood to employ specific, mechanistic interventions advantageously. High-throughput technologies and computational integration of multidimensional data offer a new angle of approach, providing detailed understanding of disease mechanisms and the possibility to personalize therapy on an individual basis.
In this review, we explore the current understanding of SA-AKI pathophysiology. We then introduce various “omic” technologies, focusing primarily on genomics, transcriptomics, proteomics, and metabolomics, and discuss their application to date in SA-AKI research. We identify multiple opportunities for future investigation in this clinically important area.
SA-AKI Mechanisms
Clinically, SA-AKI is characterized by oliguria and decreased renal solute clearance [14]. These often lead to electrolyte and acid base disturbances, fluid overload, and toxic accumulation of metabolites and medications dependent upon renal elimination. Similar to sepsis, it is probable that many etiologic subtypes of SA-AKI exist with disparate underlying mechanisms, which may contribute singularly or in combination to its pathophysiology. Early hypotheses posited that poor global renal perfusion and oxygenation induced acute tubular necrosis, but frank tubular necrosis is seen in only a small minority of cases on biopsy tissue [15–22].
Generalized, dysregulated inflammation is one of the defining features of sepsis pathophysiology and the primary cause of many of its downstream complications, including kidney injury. This occurs via multiple mechanisms. Circulating inflammatory mediators change the vascular endothelium, resulting in endothelial dysfunction. Inflammatory cytokines stimulate endothelial cell production of nitric oxide, resulting in vascular dilatation and abrogation of autoregulation. Tight cellular junctions loosen and intravascular volume escapes into the tissues, resulting in relative intravascular hypovolemia. Additionally, the activated endothelium contributes to a prothrombotic state that can lead to microvascular thrombosis [23, 24].
Sepsis-induced changes in vascular function and tone can lead to decreased renal blood flow (RBF), resulting in insufficient delivery of oxygen and metabolic substrates as well as inadequate clearance of cellular byproducts (e.g. lactate). The relationship between systemic blood pressure, RBF, and kidney oxygenation, however, is complicated. While many reports describe sepsis-induced changes in macroscopic RBF and microcirculation, and others report clinical associations between hypotension and AKI, no direct correlation between RBF and GFR has been identified, and vasopressor administration has not consistently improved kidney-related outcomes in sepsis [9, 25–27]. Furthermore, several studies demonstrate that SA-AKI can occur with preserved renal perfusion and oxygenation [20, 28–30]. Therefore, while micro and macro-circulatory deficits have the potential to induce SA-AKI, it is inaccurate to ascribe all its pathophysiologic features to this mechanism.
While some degree of immune activation may be beneficial in sepsis, given IL-10 blockade and regulatory T cell depletion has resulted in renoprotection in an experimental model [31], the hyperinflammatory milieu of the host environment in sepsis could more likely lead to direct kidney damage. Infiltrating leukocytes migrating through activated endothelia can cause tissue injury. Reactive oxygen and nitrogen species (ROS, RNS), elaborated by activated macrophages and neutrophils, might contribute to external oxidative cellular stress. Errant hyper- timulated T and NK cells could possibly induce cell-mediated killing of uninfected host targets. These direct tissue toxicities, however, are not well described in the scientific literature. Recent research has illuminated a new paradigm for understanding sepsis-related organ system dysfunction [32]. We will discuss these mechanisms as they relate specifically to SA-AKI.
Of the more than two dozen cell types in the kidney, the renal tubular epithelial cells (RTECs) perform the lion share of work in support of kidney function. These are the most metabolically active cells in the kidney, generating vast amounts of ATP to support energy expenditures involved in tubular reabsorption and secretion that underpin their maintenance of homeostasis. Because of the metabolic requirements for this work, RTECs are exquisitely sensitive to sepsis-associated injury and appear to be the cell type most injured by this condition. While RTECs are susceptible to damage caused by hypoperfusion and dysoxia as discussed above, it has become apparent that programmed cellular responses to stress and danger signals mediate their decreased function in sepsis [33].
Similar to leukocytes, RTECs express pattern recognition and cytokine receptors that detect pathogen associated and tissue-damage associated molecules. These activate intracellular stress response pathways that reprioritize energy-demanding cellular functions along a specific hierarchy, sacrificing specialized functions associated with reabsorption and secretion to preserve only those activities that maintain viability. It has been hypothesized that RTECs serve as a sensor in an afferent feedback loop that intentionally reduces GFR to limit tubular energy expenditure during times of precarious physiology. This leads to loss of global kidney function but prevents cellular demise [34, 35].
As part of this protective maneuver, RTECs undergo cell-cycle arrest and enter a state of senescence where the majority of protein translation is shut down as the cell activates only those programs requisite for life [36–38]. This response may involve a kind of “dedifferentiation,” where cells assume more primitive transcriptional and translational postures to adapt to stress [33, 39]. RTECs shift their substrate utilization from fatty acids to carbohydrates and prioritize aerobic glycolysis over oxidative phosphorylation to generate anabolic precursors necessary for the stress response [37]. Autophagy, a program whereby a cell digests its own internal structures, is activated to provide energy during a period of potential exogenous substrate deficit and to recycle damaged cellular components. Several reports associate renal dysfunction with decreased autophagy [40–42].
Mitochondrial changes in function and structure can also occur in kidneys of organisms exposed to sepsis. Multiple studies have indicated that mitochondrial energy production and its attendant oxygen consumption decrease in sepsis commensurate with any decrease in renal oxygen delivery, which is likely an adaptive phenomenon [43, 44]. Dysfunction of the electron transport chain can result in excessive ROS generation, imposing oxidative stress on the cell. Subsequent mitochondrial membrane permeability can result in both dissipation of the electrochemical gradient needed to generate ATP and mitochondrial swelling that may result in apoptosis [43, 45, 46].
As noted above, however, many histologic studies of SA-AKI show a paucity of necrosis or even apoptosis in the kidney despite severe renal dysfunction [15, 19, 20, 22, 47]. Additionally, as sepsis improves in survivors, recovery of kidney function occurs quicker than would be expected from cellular regeneration, indicating that renal tissues merely emerge from a functionally senescent state as sepsis-related stress stimuli dissipate [48]. This has led some investigators to conceptually reframe “acute renal failure” as “acute renal success,” in that at least some of the aforementioned mechanisms mediate adaptive responses that temporarily sacrifice normal kidney function during sepsis to preserve long term viability [49, 50]. The Figure highlights the interplay between these varied yet interconnected disease mechanisms.
Omic Technologies
Recent years have seen rapid increase in research using omic technologies, ranging from genomics to metabolomics, to elucidate biological complexity in health and disease. These high-dimensional approaches study biological molecules on a huge scale, requiring sensitive instruments and significant computational resources.
The term “genomics” was coined over 30 years ago around the inception of the Human Genome Project to distinguish genetics, the study of individual heritable traits at relatively few specific loci, from the study scientists were undertaking to investigate differences at very many loci across an organism’s entire genetic code. The objective of genomic research is to identify individual genetic variations that confer susceptibility to a specific disease or condition. Genetic variations include single nucleotide polymorphisms (SNPs), sites of variability where only one base pair is changed in the genetic sequence, copy number variants (CNVs); where segments of the genome are duplicated and the replicate count differs between individuals; and insertion/deletion mutations that can disrupt an entire gene. There are currently three primary approaches to pursue genomic studies: 1) Whole genome sequencing allows the identification of potentially disease-causing variants anywhere in the genetic code including regulatory and other untranslated regions. 2) Whole exome sequencing focuses only on the roughly 2% of the genome that is translated into protein, providing a quicker and less expensive technology, but one that may miss important disease-related variants. 3) DNA microarrays rely on nucleic acid hybridization rather than sequencing to detect the presence of SNPs and CNVs. Genome-wide association studies rely on this technology to correlate diseases with possible genetic mutations.
While genomics focuses on static DNA sequences, transcriptomics evaluates dynamic gene expression patterns by quantifying RNA transcripts to identify genes and gene networks activated or repressed under specific conditions. Transcriptomics has historically relied primarily on microarray technology where probe sets were developed to hybridize with specific targets to quantify gene expression. As sequencing has become more available and affordable, many researchers now employ RNA sequencing to study whole genome gene expression. RNA sequencing can be highly sensitive and offers significant dynamic range relative to microarray dependent approaches. It can also detect splice variants, gene fusions, and unanticipated transcripts potentially missed on microarrays, which require a priori knowledge of possible targets to develop the probes used to detect them. Usually, transcriptomic studies are run on bulk RNA samples derived from many cells or whole tissue, but the advent of single-cell RNA sequencing allows investigators the opportunity to study gene expression at the individual cell level. This technology offers advantages in studying gene expression in any organ, but is tremendously important in the kidney, which has a large number of different cell types, with radically different functions, physically interspersed among one another. Separating these cell types to evaluate their unique transcriptional programs is extremely challenging using traditional, whole-tissue RNA extraction techniques. Identifying cell type-specific transcriptional changes could help investigators discover new potentially targetable disease mechanisms.
Due to significant regulation of mRNA translation and cellular protein turnover, specific protein levels cannot necessarily be inferred from gene expression levels. Proteomics attempts to simultaneously quantify large numbers of proteins in biological samples. Researchers employ a variety of technologies: 1) 2-dimensional gel electrophoresis, 2) liquid chromatography, and 3) mass spectrometry [51]. Working with and identifying proteins is more difficult than nucleic acids and genes, leading to far fewer numbers of proteins quantified in proteomic studies.
Metabolomics is the measurement of molecules smaller than 1000 Daltons, which are consumed or produced by cellular metabolism [52]. Proteins are subject to posttranslational modification and regulation; thus, metabolomics has the potential to more accurately describe the cellular processes active in any set of conditions. Metabolomic studies employ two primary modalities to detect metabolites: nuclear magnetic resonance (NMR) and liquid chromatography/mass spectrometry (LC/MS). NMR requires relatively larger biological sample sizes and does not identify as many metabolites, but more accurately quantifies metabolite abundance with better reproducibility. LC/MS has much higher sensitivity and can quantify many more metabolites than NMR, though with less accuracy and reproducibility.
Each of these technologies independently offers a potential wealth of information to investigators, but many studies are now seeking to integrate findings from these various approaches to provide a more holistic understanding of cellular processes in disease states. The statistical analyses and computer algorithms necessary to accomplish this “multi-omic” integration are areas of active research.
Genomics, Transcriptomics, Proteomics, and Metabolomics in SA-AKI
The Table summarizes the articles discussed below, specifically pointing out experimental methods and omic technology employed, as well as major findings.
Genomics
Only one study to date has used a large-scale genomic approach to identify genetic variability associated with SA-AKI. This research relied on microarray technology, which is biased toward known variants in specific diseases, to identify SNPs. Although CNVs and other forms of genetic variability have recently been associated with various kinds of kidney injury [53–55], none of these have been explored in SA-AKI. Additionally, no studies in SA-AKI have yet employed DNA sequencing genomic technologies.
Using a DNA microarray, which tested for 48,742 markers across 2,100 genes previously associated with cardiovascular disease, Frank et al. genotyped 887 patients admitted to the ICU with septic shock, of which roughly 50% developed SA-AKI [56]. They identified 5 SNPs associated with SA-AKI involving the BCL2, SERPINA, and SIK3 genes. The BCL2 protein regulates apoptosis. The SERPINA genes code for serine protease inhibitors involved in apoptosis, inflammation, and tumorigenesis. The SIK3 protein regulates cellular metabolism via mTOR signaling. In a follow-up, prospective confirmation study, Vilander et al. and the FINNAKI study group genotyped just these 5 SNPs in 653 patients with sepsis and found that only SERPINA4 and 5 were associated with SA-AKI after controlling for confounding variables [57].
A recent review of genomic research in AKI of any etiology showed that of 35 studies, only two leveraged genomic technologies in an unbiased fashion. The remaining studies were smaller, targeted investigations of previously identified variants. The review authors concluded that research to date has failed to identify consistent variants contributing to AKI susceptibility [58]. This review highlights an opportunity to apply genomics to the study of SA-AKI to identify genetic determinants of disease and generate new mechanistic hypotheses. This technology, however, requires either very large numbers of patients, or the identification of a small cohort of patients in unique clinical scenarios at especially high risk. These considerations make the conduct of such investigations challenging and expensive.
Transcriptomics
One of the first efforts to characterize genome-wide expression patterns in SA-AKI provided important insights into the metabolic nature of the cellular response. Using an LPS model of sepsis and microarray technology, Tran et al. showed that changes in gene expression pathways associated with cellular metabolism and mitochondrial function were most enriched in the sick mice [47]. They found PGC1-α, a transcriptional regulator governing fatty acid oxidation and mitochondrial biogenesis, to be central to renal recovery. Ferreyra et el. conducted a microarray study after injecting mice with staphylococcal enterotoxin and evaluated gene expression across multiple tissues looking for expression patterns shared between them. They identified genes related to the innate immune response, apoptosis, DNA damage and metabolic stress consistently upregulated between organs, though they did not discuss changes specific to the kidneys [59]. Hultström et al. reanalyzed and compared the publicly available gene expression data of 6 different models of AKI, including the aforementioned study as one of two sepsis models. They confirmed the findings of Tran et al. that mitochondrial genes were highly regulated in sepsis and showed additionally in the data from Ferreyra et al. that genes related to mesenchymal development, protein degradation, and oxidative stress were highly enriched. Of the transcriptomic features shared with other models of AKI, activation of MYC, an oncogene related to epithelial-to-mesenchymal transition, was the most prominent, indicating that injury could induce a state of cellular dedifferentiation [39].
Taking a slightly different approach, other investigators have analyzed gene expression in whole blood-derived RNA, effectively assessing the circulating leukocyte transcriptome to study associations with SA-AKI. Seeking to identify children at risk for developing SA-AKI, Basu et al. used gene expression microarrays to identify 21 genes, primarily related to immune signaling and function, whose elevated expression predicted SA-AKI with a high degree of accuracy [60]. The authors subsequently used these data to build a successful SA-AKI biomarker panel [61]. In another human study, Ge et al. used microarrays targeting whole-blood derived miRNAs, untranslated RNA molecules with transcriptional regulatory functions, to compare patients with SA-AKI to those with sepsis that did not develop AKI. They found that miRNAs regulating aspects of kidney development, response to oxidative stress, and cellular metabolism (including mTOR and PGC1-α) were enriched in their data set [62]. Considering that miRNA can be packaged in exosomes for cell-to-cell communications, these could have direct influence on renal stress responses in SA-AKI.
Single-cell RNA sequencing technology has not yet been employed in SA-AKI research.
Proteomics
High-throughput protein analyses have been conducted on a variety of tissue, blood, and urine samples. In a mouse cecal ligation and puncture model of sepsis, Wu et al. performed 2D-gel electrophoresis on tissue protein extract followed by mass spectrometry analysis to identify changes associated with SA-AKI [63]. They found phosphorylated myosin regulatory light chain 12B, a protein important to cytoskeletal integrity, increased in the septic kidney. Using a similar proteomic approach with a rat model of SA-AKI, Hinkelbein et al. found upregulation of myosin and cytochrome C oxidase proteins in kidney tissue and downregulation of major urinary protein 5, whose functions associate with cell adhesion and migration, vascular smooth muscle tone, mitochondrial energy production, and fatty acid and tubular transport [64]. Matejovic et al. identified 21 differentially regulated renal cortical proteins in a pig sepsis model that fell into 3 broad functional groups: 1) protein chaperones, 2) endoplasmic reticulum and oxidative stress responses, and 3) cellular energy metabolism [65]. These researchers highlighted a particular protein increased in sepsis, Na/H exchange regulatory cofactor 3, which regulates the activity of other renal membrane transporters and likely facilitates an adaptive reduction in GFR to reduce RTEC resorptive energy expenditure.
Evaluating the plasma proteome using a pig model, Thongboonkerd et al. identified 27 proteins whose levels changed in sepsis globally. These protein functions related to inflammation, oxidative stress response, and cytoskeletal components [66]. Hashida et al. studied 20 patients with AKI on ICU admission who received continuous renal replacement therapy, 10 of which had sepsis [67]. Protein was extracted from hemofilter adsorbates and analyzed by LC/MS to identify over 350 proteins. Functional annotations for these proteins fell into metabolic, stress response, and immune response categories. Patients with sepsis had more proteins in the immune system process and biological adhesion functional annotations than did those without sepsis. Gong et al. studied serum proteome changes in patients with SA-AKI over the course of their first 72 hours of continuous renal replacement therapy, noting changes consistent with resolution of inflammation, dysfunctional coagulation, and oxidative stress [68].
Urine proteomic studies have provided evidence for disrupted glomerular filtration, increased extracellular matrix remodeling, inflammation, and oxidative stress responses in SA-AKI. Multiple researchers have identified increased serum proteins in the urine, including albumin, β2-microglobulin, and α−2u-globulin indicating loss of glomerular membrane integrity [69–71]. Increased urinary levels of collagen and fibrinogen fragments and changes in brush border enzymes involved in extracellular matrix remodeling (aminopeptidases, meprin, chitinase-like proteins) point toward increased turnover [69, 71, 72]. Maddens et al. found inflammation-related proteins neutrophil gelatinase-associated lipocalin and protein S100-A8 in their data set as well as the antioxidant thioredoxin. This group also reported urinary increases in Na/H exchange regulatory cofactor 3, noted above, augmenting the potential biologic importance of the signal [72].
Metabolomics
Metabolomics is the youngest omic technology, and only in the past few years have investigators begun to apply metabolomic analyses to SA-AKI. In one of the first approaches, Waltz et al. used GC/MS and LC/MS to detect over 300 metabolites in kidney tissue using a mouse model of sepsis [73]. They found that glucose and glycolytic intermediates increased with evidence of carbohydrate shunting through the sorbitol pathway. TCA cycle intermediates were decreased, indicating reduced mitochondrial function. Metabolites associated with inflammation were increased while antioxidants decreased in the septic tissue indicating oxidative stress. Additionally, renal tissue osmolyte levels were increased, which could be an adaptive change to help concentrate urine and retain fluid in the setting of critical illness. Izquierdo-Garcia et al. conducted a robust analysis of SA-AKI in a swine E. coli infusion model of sepsis [74]. They analyzed kidney tissue, plasma, and urine metabolomics with NMR and found metabolite changes consistent with cellular energy dysregulation. Specifically, plasma elevations of lactate, pyruvate and alanine imply decreased acetyl-CoA incorporation into the TCA cycle. Additionally, changes in nicotinuric acid, phenylacetylglycine, and isovaleroglycine concentration implicate alterations in fatty acid metabolism, as acylglycine excretion associates with mitochondrial beta-oxidation dysfunction. This group also found dysregulated osmolyte levels, with betaine and myo-inositol lower in both plasma and urine. The authors do not mention, however, whether any osmolytes were identified in the spectra collected from tissue samples, which would allow direct comparison between studies.
Two groups have used metabolomic analyses to guide investigations into treatments of SA-AKI. Using NMR based metabolomics to analyze kidney tissue and serum, Li et al. studied the effect of Chinese herbs in a mouse LPS model of SA-AKI [75]. They found evidence of energy deficiency and oxidative stress ameliorated by the herbal treatment. Rodrigues et al. evaluated the effect of gingerol compounds on SA-AKI, employing urinary NMR metabolomics in a rat model to show that sepsis induced oxidative and nitrosative stress in the kidney. Therapeutic administration of gingerol increased the antioxidant and anti-inflammatory compounds dimethylamine and methylsulfonylmethane in the urine and was associated with decreased renal dysfunction and improved overall survival [76].
Integration
While the studies reviewed here have differed significantly from each other, consistent pathophysiologic themes in SA-AKI have emerged that support those elucidated by more traditional research approaches; namely, increased inflammatory signaling, cellular metabolic changes, oxidative stress, and osmolyte derangement. The research reviewed to this point has focused on single-omic studies. While several investigations into both sepsis and AKI separately have made important discoveries by integrating multiple omic data sets [77–80], only two have pursued this experimental approach for SA-AKI specifically. A team lead by Hato and Dagher recently published both studies.
In the first, the authors used transcriptomics, proteomics, and metabolomics to demonstrate that endotoxin preconditioning prevents severe AKI in mouse models of sepsis by activating protective stress response programs and metabolic pathways in both tissue resident macrophages and proximal RTECs [81]. Of particular interest, they showed that the anti-inflammatory, antimicrobial metabolite itaconate had increased abundance in the proximal RTECs of preconditioned mice and likely contributed to their protected state. The integral role of itaconate in macrophage activation and function was recently discovered by another group using combined transcriptomics and metabolomics [82]. Hato’s work is the first to report this metabolite in the kidney.
In their second study, Hato et al. employed transcriptomics, metabolomics and cutting-edge nascent proteomic and Ribo-seq translatomic technologies to show that sepsis induces profound changes in the RTEC translational program via activation of the Eif2 integrated stress response system, previously understood to be triggered principally by viral pathogens [38]. Protein translation was downregulated for most genes, but for those related to specific aspects of metabolism (including itaconate production) and stress response, translation was upregulated. They discovered new upstream open reading frames in the 5’ untranslated region of many mRNA occupied by ribosomes in sepsis, which likely contributed to translational regulation. The authors proposed that this global translation shutdown is likely an important contributor to sepsis-associated organ failure, including SA-AKI.
These two studies identify multiple potential therapeutic targets that could improve patient centered outcomes in SA-AKI. Administration of metabolites such as itaconate may enhance cellular stress response programs that facilitate resistance to injury and accelerate recovery. Inhibition of Eif2 signaling during the resolution phase of sepsis might help reverse persistent SA-AKI.
Limitations
Omic technologies are powerful but have important limitations. In the research reviewed here, rarely did any of the studies identify the same genes, proteins, or metabolites contributing to SA-AKI pathophysiology. This is likely due primarily to lack of standardization. Each study used different experimental models of sepsis, evaluated different kinds of samples, and employed variable analytical approaches. Even within a specific technology, distinct protocols for sample processing and analysis can render divergent results. Additionally, the techniques employed for aligning nucleic acid sequencing reads to genes or assigning protein and metabolite identifications to NMR and LC/MS spectral features can introduce further variability between studies. Even once investigators have significant findings in hand, their clinical utility may be difficult to discern when lesser-known genes, proteins, metabolites and pathways are identified by the analysis.
Another important drawback to employing omic technologies in SA-AKI research is that the equipment, technical expertise and reagents required to conduct these high-throughput analyses have historically been quite expensive. The data science necessary to integrate output and draw clinically relevant conclusions from the mass of information generated has also been a considerable roadblock to their widespread adoption.
As these technologies mature, however, standardization of experimental design and analytical approach will facilitate generalization of findings. This has already happened, to a large extent, in the genomic and transcriptomic arenas that emerged first. Furthermore, databases that catalogue gene, protein and metabolite functions and organize them in coherent, biologically relevant pathways are ever improving (Ingenuity Pathway Analysis, Toppgene, Kyoto Encyclopedia of Genes and Genomes, The Human Metabolome Database), which facilitates the clinical application of the research. With ever increasing adoption of these methods and improvement in the fundamental technologies, economies of scale are driving down costs. Finally, free, open-source data analysis platforms such as Python and R, which have robust documentation, tutorials, and user communities, are making the data science much more accessible to interested researchers.
Translating Omics to Bedside Therapeutics
Employing omic technologies to elucidate fundamental pathophysiologies in SA-AKI has the important potential to bring new clinical therapies to the bedside. For this translation to occur, however, future investigation in SA-AKI should pursue mechanistic discoveries in clinically relevant animal models, which allow study of tissues and cells currently inaccessible to human research. Studies should be planned and sufficiently funded to integrate findings from multiple omic technologies to give a more complete understanding of the pathophysiology. Multidisciplinary teams should be assembled with expertise in bench research, clinical studies, and highly-dimensional data analysis.
After identifying potential SA-AKI mechanistic targets in model organisms, research should progress toward detecting non-invasive markers of these processes in clinically accessible biological samples, such as blood and urine. These “mechanistic biomarkers” would reveal pathophysiological processes at play in tissues not sampled in humans, and facilitate identification of deranged etiologic processes and tracking of response to novel treatments.
Therapeutic strategies based on underlying pathophysiologic mechanisms rather than clinical presentation should be studied first in models and then translated to patients most at risk of poor outcome. For example, elevation of serum creatinine levels in a patient with septic shock reveals nothing to clinicians about the underlying cause of renal failure, just that it is present. But posit that recent, repeatable findings in multiple animal models of sepsis indicate through gene expression, protein abundance, and cellular metabolite levels that decreased RTEC fatty acid oxidation is directly related to severe SA-AKI. Furthermore, studies show that this derangement is accompanied by decreased serum free carnitine levels and increased levels of certain acyl-carnitine species in blood and urine in both model organisms and humans. Finding these markers elevated in a patient would indicate potential candidacy for a drug that increases fatty acid oxidation to restore renal function. If, however, these specific markers were not present, but rather others indicative of microvascular thrombosis that compromised renal circulation, then this patient could benefit more from a therapy designed to inhibit capillary thrombin deposition or to enhance thrombolysis. This is the desired outcome of mechanism-driven multi-omic research, truly personalized medicine targeting the unique pathophysiologic signature of each critically ill patient.
Conclusions
SA-AKI is a prevalent condition in our ICUs that increases patient morbidity and mortality. While supportive therapies promote survival in those for whom the condition resolves, no mechanistic therapies exist to alter the underlying pathophysiology. Our understanding of the basic mechanisms of disease has evolved over recent years as new studies show that changes in cellular gene expression, protein abundance, and metabolism affect kidney function in sepsis. While some work has been done, the potential of new omic technologies to elucidate these mechanisms has not yet been realized. The proposed approach to future research in SA-AKI will build on recent advances in both basic and clinical research and take advantage of important new omic technologies to improve outcomes for some of our sickest patients.
Figure.
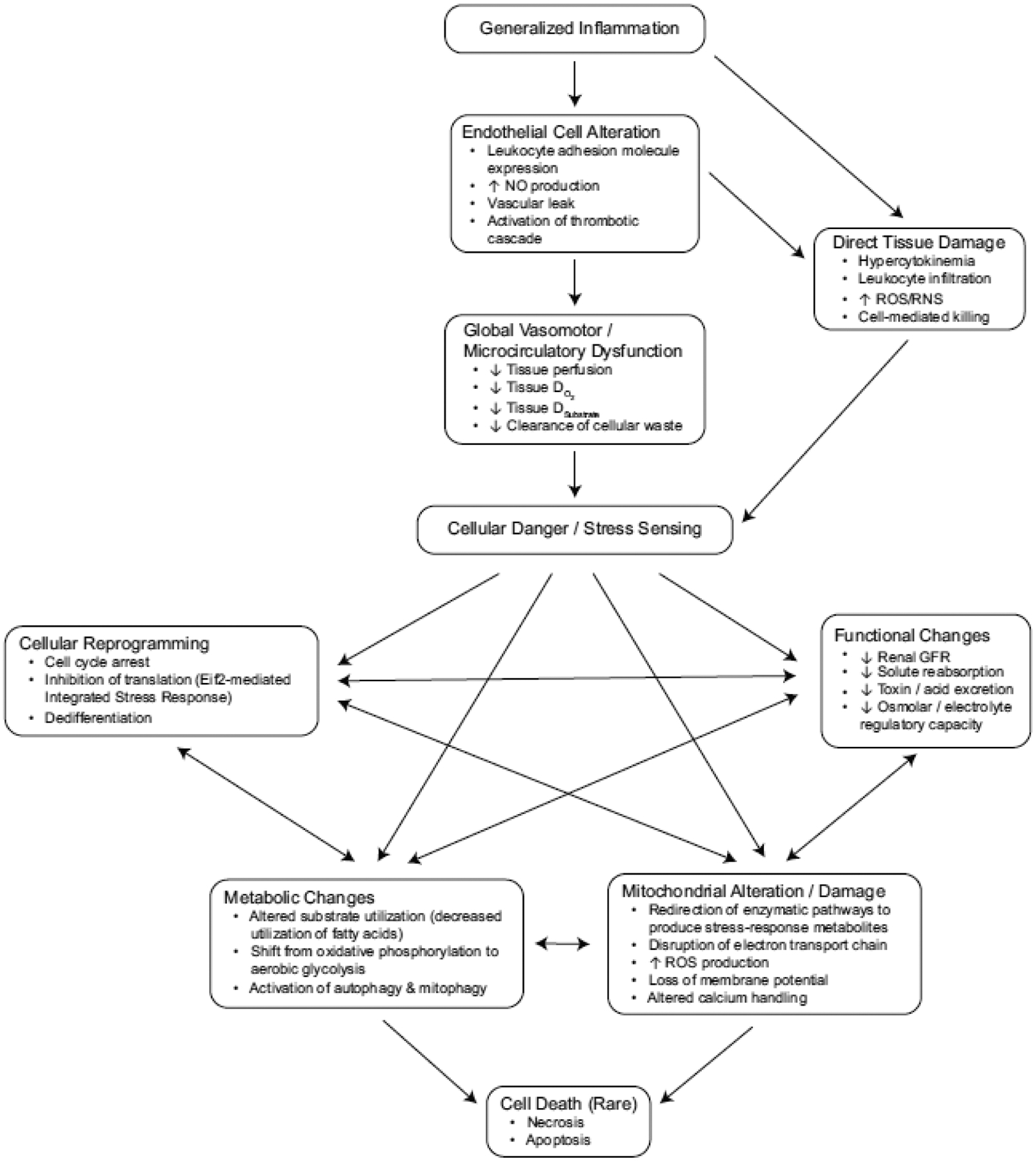
Concept map of multiple proposed SA-AKI mechanisms. NO: nitric oxide. ROS: reactive oxygen species. RNS: reactive nitrogen species. DO2: oxygen delivery. Dsubstrate: substrate delivery
Table.
Brief summary of each “omic” manuscript, including type of omic technology utilized, study population, and major findings. Ox-phos: oxidative phosphorylation. PsA: pseudomonas aeruginosa. 2DE: 2D gel electrophoresis. DIGE: 2D difference gel electrophoresis. CE: capillary electrophoresis. MS: mass spectrometry. MALDI: matrix-assisted laser desorption/ionization. A1AT: alpha-1 antitrypsin. SDS PAGE: SDS-polyacrylamide gel electrophoresis. GC: gas chromatography. LC: liquid chromatography. Ribo-seq: ribosomal profiling technique
| Author | Omic Technology | Methods/ Models | Major Findings |
|---|---|---|---|
| Frank [53] | Genomics-DNA microarray | 887 critically ill patients with septic shock | 5 SNPs were associated with SA-AKI: BCL2, SERPINA, SIK3 genes. |
| Vilander [54] | Genomics-SNP genotyping | 837 septic patients, 627 with septic shock | SNPs in SERPINA4,5 but not BCL2 nor SIK3, associated with SA-AKI after controlling for confounders. |
| Tran [47] | Transcriptomics- Gene expression microarray | Global and tubule-specific PGC-1a knockout mice exposed to endotoxemia | Ox-phos genes were selectively suppressed during SA-AKI, and PGC1a was proportionally suppressed. PGC1a KO mice suffered persistent kidney injury after sepsis. |
| Basu [57] | Transcriptomics- Gene expression microarray | 179 critically ill children with septic shock | Elevated expression of 21 genes predicted SA-AKI. The genes mainly related to immune signaling and function. |
| Ferreyra [56] | Transcriptomics- Gene expression microarray | Mice exposed to intranasal staph enterotoxin B | Global gene-expression changes found a host-wide Interferon- response. |
| Ge [59] | Transcriptomics- Gene expression microarray | Whole-blood derived miRNAs from septic patients with/ without AKI | 2 miRNAs that regulate AKT, NOX5 expression (involved in oxidative stress, mitochondrial dysfunction) significantly overexpressed in SA-AKI. |
| Hultstrom [39] | Transcriptomics- Gene expression microarrays | Microarray studies of renal gene expression after AKI in 6 different models | 5254 differentially expressed genes. 2/3 found in only 1 model. 4 genes in all 6 models. Pathway analysis showed MYC to be a central connection in AKI. |
| Holly [66] | Urine Proteomics- DIGE, MS | Rat CLP sepsis | Meprin-1-alpha, a brush-border enzyme, was changed in rats with septic AKI, and an inhibitor of this enzyme prevented acute renal failure in aged mice. |
| Gong [65] | Serum proteomics- 2DE, MS | 20 patients with severe sepsis receiving CVVH | 10 proteins differentially expressed in pts getting CVVH, 7 proteins decreased in serum, 3 increased. |
| Thongboonkerd [63] | Plasma proteomics- 2DE, MS | Intraperitoneal injection of feces into pigs | 27 unique proteins whose levels changed in sepsis, related to inflammation, oxidative/nitrosative stress, and cytoskeleton. |
| Metzger [67] | Urine Proteomics- CE-MS | Critically ill patients | 6 proteins associated with AKI--albumin, A1AT, b2- microglobulin upregulated; collagen, fibrinogen downregulated. |
| Maddens [69] | Urine, plasma, tissue proteomics- gel free technique | Mice after uterine ligation and E Coli inoculation | Urinary chitinase3-like proteins and acidic mammalian chitinase were only detected in septic mice with AKI. |
| Wu [60] | Tissue Proteomics- DIGE, MS | Mouse CLP sepsis | Phosphorylated myosin regulatory light chain 12B was upregulated in septic AKI, which is important for cytoskeletal integrity. |
| Carrick [68] | Urinary proteomics- MALDI-MS | 95 septic patients | 39 urinary peptides were markers of AKI; proteins included b-2-microglobulin, collagen, fibrinogen,A1AT. |
| Matejovic [62] | Tissue proteomics- 2DE, MS | Continuous infusion of PsA in pigs and biopsies of renal cortical tissue. | 21 proteins were differentially regulated in septic AKI, specifically Na/H exchange regulatory cofactor 3. |
| Hashida [64] | Proteomics from hemofilter adsorbates- SDS PAGE, MS | 20 critically ill patients with AKI requiring CRRT. | 3 proteins, including carbonic anhydrase 1 and leucine-rich alpha-glycoprotein, were in all septic patients with AKI but not in non-septic patients. |
| Hinkelbein [61] | Tissue proteomics- DIGE, MS | Rat CLP sepsis | Myosin and cytochrome C oxidase proteins upregulated; major urinary protein 5 downregulated in SA-AKI. These proteins correlate with mitochondrial energy production and electron transport. |
| Waltz [70] | Tissue (whole kidney) metabolomics- LC/MS | Mice CLP sepsis | Sepsis resulted in increased glucose and glycolytic intermediates, decreased TCA cycle intermediates, increased renal osmolytes (pinitol, urea, taurine), and metabolites associated with inflammation; decreased antioxidants (ascorbate, a-tocopherol, erogthioneine). |
| Li [72] | Tissue and serum metabolomics-NMR | Mice after intraperitoneal injection of LPS | Decreased antioxidants/regulatory osmolytes (betaine, taurine), membrane repair metabolites (choline, phosphocholine, ethanolamine). Decreased glucose, lactate, alanine, citrate and a-oxoglutarate, increased ketone bodies, creatine, creatinine, and ATP breakdown products (adenosine, inosine), marker of energy deficiency. Oxidative stress, energy metabolism pathways most significantly affected. |
| Rodrigues [73] | Urine metabolomics-NMR | Rat CLP sepsis | Elevated urinary creatine, allantoin, and dimethylglycine in CLP compared to sham mice. |
| Izquierdo-Garcia [71] | Tissue, plasma, and urine metabolomics- NMR | Pigs infused with E Coli | Metabolic differences between sepsis and controls--Tissue: increased lactate, nicotinuric acid; decreased value, aspartate, glucose, threonine. Serum: increased lactate, pyruvate, alanine, glutamine; decreased glucose, betaine. Urine: increased ascorbic acid, isovaleroglycine, aminoadipic acid, N-acetylglutamine, N-acetylaspartate, and decreased myoinositol, phenylacetylglycine. Altogether signified alterations of cellular energy pathways, specifically fatty acid metabolism. |
| Hato [74] | Unbiased transcriptomics, tissue metabolomics- DIGE, MS; GC/MS, LC/MS | Endotoxin preconditioning in CLP mouse model | There are benefits of preconditioning from molecules involved in antibacterial defense, redox balance, and tissue healing, specifically increased itaconate in proximal RTECs, that protect the kidney. |
| Hato [38] | Transcriptomics (RNAseq), proteomics (nascent), metabolomics (GC/MS, LC/MS) Ribo-seq translatomics | Mice injected with LPS. Transcriptomics performed on S1 and S2/S3 kidney segments after laser and manual microdissection. | Activation of Eif2ak2/Eif2a axis is the key mediator of translation initiation block in late-phase sepsis in RTECs. Reversal of this axis mitigated kidney injury and this global translational shutdown is likely an important contributor to SA-AKI. |
Acknowledgements
The authors would like to thank Dr. Prasad Devarajan and Dr. Hector Wong for helping conceive and develop this work and for their insightful editorial input.
Funding
SWS receives grant support from the National Heart, Lung, and Blood Institute, National Institutes of Health (1K08HL133377-01A1).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
No author has any conflict of interest, financial or otherwise, relative to this manuscript.
Conflicts of interest/Competing interests: No author has any conflict of interest, financial or otherwise, relative to this manuscript.
Availability of data and material: Not applicable
Code availability: Not applicable
References
- 1.Kaddourah A, Basu RK, Bagshaw SM, et al. (2017) Epidemiology of Acute Kidney Injury in Critically Ill Children and Young Adults. N Engl J Med 376:11–20. 10.1056/NEJMoa1611391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouchard J, Acharya A, Cerda J, et al. (2015) A Prospective International Multicenter Study of AKI in the Intensive Care Unit. Clin J Am Soc Nephrol CJASN 10:1324–1331. 10.2215/CJN.04360514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagshaw SM, Uchino S, Bellomo R, et al. (2007) Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol CJASN 2:431–439. 10.2215/CJN.03681106 [DOI] [PubMed] [Google Scholar]
- 4.Hoste EAJ, Lameire NH, Vanholder RC, et al. (2003) Acute renal failure in patients with sepsis in a surgical ICU: predictive factors, incidence, comorbidity, and outcome. J Am Soc Nephrol JASN 14:1022–1030 [DOI] [PubMed] [Google Scholar]
- 5.Bagshaw SM, George C, Bellomo R, ANZICS Database Management Committee (2008) Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care Lond Engl 12:R47. 10.1186/cc6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzgerald JC, Basu RK, Akcan-Arikan A, et al. (2016) Acute Kidney Injury in Pediatric Severe Sepsis: An Independent Risk Factor for Death and New Disability. Crit Care Med 44:2241–2250. 10.1097/CCM.0000000000002007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss SL, Balamuth F, Thurm CW, et al. (2019) Major Adverse Kidney Events in Pediatric Sepsis. Clin J Am Soc Nephrol CJASN 14:664–672. 10.2215/CJN.12201018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alobaidi R, Basu RK, Goldstein SL, Bagshaw SM (2015) Sepsis-associated acute kidney injury. Semin Nephrol 35:2–11. 10.1016/j.semnephrol.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellomo R, Kellum JA, Ronco C, et al. (2017) Acute kidney injury in sepsis. Intensive Care Med 43:816–828. 10.1007/s00134-017-4755-7 [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald JC, Ross ME, Thomas NJ, et al. (2018) Risk factors and inpatient outcomes associated with acute kidney injury at pediatric severe sepsis presentation. Pediatr Nephrol Berl Ger 33:1781–1790. 10.1007/s00467-018-3981-8 [DOI] [PubMed] [Google Scholar]
- 11.Stanski NL, Stenson EK, Cvijanovich NZ, et al. (2020) PERSEVERE Biomarkers Predict Severe Acute Kidney Injury and Renal Recovery in Pediatric Septic Shock. Am J Respir Crit Care Med. 10.1164/rccm.201911-2187OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poukkanen M, Vaara ST, Pettilä V, et al. (2013) Acute kidney injury in patients with severe sepsis in Finnish Intensive Care Units. Acta Anaesthesiol Scand 57:863–872. 10.1111/aas.12133 [DOI] [PubMed] [Google Scholar]
- 13.Kellum JA, Chawla LS, Keener C, et al. (2016) The Effects of Alternative Resuscitation Strategies on Acute Kidney Injury in Patients with Septic Shock. Am J Respir Crit Care Med 193:281–287. 10.1164/rccm.201505-0995OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.KDIGO AKI Writing Group (2012) KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl 2:1–138 [Google Scholar]
- 15.otchkiss RS, Swanson PE, Freeman BD, et al. (1999) Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27:1230–1251 [DOI] [PubMed] [Google Scholar]
- 16.Kosaka J, Lankadeva YR, May CN, Bellomo R (2016) Histopathology of Septic Acute Kidney Injury: A Systematic Review of Experimental Data. Crit Care Med 44:e897–e903. 10.1097/CCM.0000000000001735 [DOI] [PubMed] [Google Scholar]
- 17.Lerolle N, Nochy D, Guérot E, et al. (2010) Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med 36:471–478. 10.1007/s00134-009-1723-x [DOI] [PubMed] [Google Scholar]
- 18.Aslan A, van den Heuvel MC, Stegeman CA, et al. (2018) Kidney histopathology in lethal human sepsis. Crit Care Lond Engl 22:359. 10.1186/s13054-018-2287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takasu O, Gaut JP, Watanabe E, et al. (2013) Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 187:509–517. 10.1164/rccm.201211-1983OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiden MJ, Otto S, Brealey JK, et al. (2016) Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am J Respir Crit Care Med 194:692–700. 10.1164/rccm.201511-2285OC [DOI] [PubMed] [Google Scholar]
- 21.Langenberg C, Bagshaw SM, May CN, Bellomo R (2008) The histopathology of septic acute kidney injury: a systematic review. Crit Care Lond Engl 12:R38. 10.1186/cc6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langenberg C, Gobe G, Hood S, et al. (2014) Renal histopathology during experimental septic acute kidney injury and recovery. Crit Care Med 42:e58–67. 10.1097/CCM.0b013e3182a639da [DOI] [PubMed] [Google Scholar]
- 23.Verma SK, Molitoris BA (2015) Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin Nephrol 35:96–107. 10.1016/j.semnephrol.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ince C, Mayeux PR, Nguyen T, et al. (2016) The Endothelium in Sepsis. Shock Augusta Ga 45:259–270. 10.1097/SHK.0000000000000473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Post EH, Kellum JA, Bellomo R, Vincent J-L (2017) Renal perfusion in sepsis: from macro- to microcirculation. Kidney Int 91:45–60. 10.1016/j.kint.2016.07.032 [DOI] [PubMed] [Google Scholar]
- 26.Gordon AC, Mason AJ, Thirunavukkarasu N, et al. (2016) Effect of Early Vasopressin vs Norepinephrine on Kidney Failure in Patients With Septic Shock: The VANISH Randomized Clinical Trial. JAMA 316:509–518. 10.1001/jama.2016.10485 [DOI] [PubMed] [Google Scholar]
- 27.Ma S, Evans RG, Iguchi N, et al. (2019) Sepsis-induced acute kidney injury: A disease of the microcirculation. Microcirc N Y N 1994 26:e12483. 10.1111/micc.12483 [DOI] [PubMed] [Google Scholar]
- 28.Murugan R, Karajala-Subramanyam V, Lee M, et al. (2010) Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int 77:527–535. 10.1038/ki.2009.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langenberg C, Wan L, Egi M, et al. (2006) Renal blood flow in experimental septic acute renal failure. Kidney Int 69:1996–2002. 10.1038/sj.ki.5000440 [DOI] [PubMed] [Google Scholar]
- 30.Langenberg C, Bellomo R, May C, et al. (2005) Renal blood flow in sepsis. Crit Care Lond Engl 9:R363–374. 10.1186/cc3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S-Y, Lee Y-S, Choi H-M, et al. (2012) Distinct pathophysiologic mechanisms of septic acute kidney injury: Role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury. Crit Care Med 40:2997–3006. 10.1097/CCM.0b013e31825b912d [DOI] [PubMed] [Google Scholar]
- 32.Pool R, Gomez H, Kellum JA (2018) Mechanisms of Organ Dysfunction in Sepsis. Crit Care Clin 34:63–80. 10.1016/j.ccc.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emlet DR, Shaw AD, Kellum JA (2015) Sepsis-associated AKI: epithelial cell dysfunction. Semin Nephrol 35:85–95. 10.1016/j.semnephrol.2015.01.009 [DOI] [PubMed] [Google Scholar]
- 34.Gómez H, Kellum JA (2016) Sepsis-induced acute kidney injury. Curr Opin Crit Care 22:546–553. 10.1097/MCC.0000000000000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA (2019) Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int 96:1083–1099. 10.1016/j.kint.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez H, Ince C, De Backer D, et al. (2014) A Unified Theory of Sepsis-Induced Acute Kidney Injury: Inflammation, microcirculatory dysfunction, bioenergetics and the tubular cell adaptation to injury. Shock Augusta Ga 41:3–11. 10.1097/SHK.0000000000000052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gómez H, Kellum JA, Ronco C (2017) Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol 13:143–151. 10.1038/nrneph.2016.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hato T, Maier B, Syed F, et al. (2019) Bacterial sepsis triggers an antiviral response that causes translation shutdown. J Clin Invest 129:296–309. 10.1172/JCI123284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hultström M, Becirovic-Agic M, Jönsson S (2018) Comparison of acute kidney injury of different etiology reveals in-common mechanisms of tissue damage. Physiol Genomics 50:127–141. 10.1152/physiolgenomics.00037.2017 [DOI] [PubMed] [Google Scholar]
- 40.Kaushal GP, Shah SV (2016) Autophagy in acute kidney injury. Kidney Int 89:779–791. 10.1016/j.kint.2015.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sunahara S, Watanabe E, Hatano M, et al. (2018) Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep 8:1050. 10.1038/s41598-018-19350-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsiao H-W, Tsai K-L, Wang L-F, et al. (2012) The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock Augusta Ga 37:289–296. 10.1097/SHK.0b013e318240b52a [DOI] [PubMed] [Google Scholar]
- 43.Parikh SM, Yang Y, He L, et al. (2015) Mitochondrial Function and Disturbances in the Septic Kidney. Semin Nephrol 35:108–119. 10.1016/j.semnephrol.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singer M (2014) The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 5:66–72. 10.4161/viru.26907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parikh SM (2013) Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Curr Opin Crit Care 19:554–559. 10.1097/MCC.0000000000000038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun J, Zhang J, Tian J, et al. (2019) Mitochondria in Sepsis-Induced AKI. J Am Soc Nephrol JASN 30:1151–1161. 10.1681/ASN.2018111126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran M, Tam D, Bardia A, et al. (2011) PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121:4003–4014. 10.1172/JCI58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singer M, De Santis V, Vitale D, Jeffcoate W (2004) Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet Lond Engl 364:545–548. 10.1016/S0140-6736(04)16815-3 [DOI] [PubMed] [Google Scholar]
- 49.Thurau K, Boylan JW (1976) Acute renal success. The unexpected logic of oliguria in acute renal failure. Am J Med 61:308–315. 10.1016/0002-9343(76)90365-x [DOI] [PubMed] [Google Scholar]
- 50.Singer M (2017) Critical illness and flat batteries. Crit Care Lond Engl 21:309. 10.1186/s13054-017-1913-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma NK, Salomao R (2017) Sepsis Through the Eyes of Proteomics: The Progress in the Last Decade. Shock Augusta Ga 47:17–25. 10.1097/SHK.0000000000000698 [DOI] [PubMed] [Google Scholar]
- 52.Marx D, Metzger J, Pejchinovski M, et al. (2018) Proteomics and Metabolomics for AKI Diagnosis. Semin Nephrol 38:63–87. 10.1016/j.semnephrol.2017.09.007 [DOI] [PubMed] [Google Scholar]
- 53.Li M, Carey J, Cristiano S, et al. (2017) Genome-Wide Association of Copy Number Polymorphisms and Kidney Function. PloS One 12:e0170815. 10.1371/journal.pone.0170815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cañadas-Garre M, Anderson K, Cappa R, et al. (2019) Genetic Susceptibility to Chronic Kidney Disease - Some More Pieces for the Heritability Puzzle. Front Genet 10:453. 10.3389/fgene.2019.00453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verbitsky M, Westland R, Perez A, et al. (2019) The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 51:117–127. 10.1038/s41588-018-0281-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frank AJ, Sheu C-C, Zhao Y, et al. (2012) BCL2 genetic variants are associated with acute kidney injury in septic shock*. Crit Care Med 40:2116–2123. 10.1097/CCM.0b013e3182514bca [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vilander LM, Kaunisto MA, Vaara ST, et al. (2017) Genetic variants in SERPINA4 and SERPINA5, but not BCL2 and SIK3 are associated with acute kidney injury in critically ill patients with septic shock. Crit Care Lond Engl 21:47. 10.1186/s13054-017-1631-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larach DB, Engoren MC, Schmidt EM, Heung M (2018) Genetic variants and acute kidney injury: A review of the literature. J Crit Care 44:203–211. 10.1016/j.jcrc.2017.11.019 [DOI] [PubMed] [Google Scholar]
- 59.Ferreyra GA, Elinoff JM, Demirkale CY, et al. (2014) Late multiple organ surge in interferon-regulated target genes characterizes staphylococcal enterotoxin B lethality. PloS One 9:e88756. 10.1371/journal.pone.0088756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basu RK, Standage SW, Cvijanovich NZ, et al. (2011) Identification of candidate serum biomarkers for severe septic shock-associated kidney injury via microarray. Crit Care Lond Engl 15:R273. 10.1186/cc10554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong HR, Cvijanovich NZ, Anas N, et al. (2015) A Multibiomarker-Based Model for Estimating the Risk of Septic Acute Kidney Injury. Crit Care Med 43:1646–1653. 10.1097/CCM.0000000000001079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ge Q-M, Huang C-M, Zhu X-Y, et al. (2017) Differentially expressed miRNAs in sepsis-induced acute kidney injury target oxidative stress and mitochondrial dysfunction pathways. PloS One 12:e0173292. 10.1371/journal.pone.0173292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu F, Dong X-J, Li Y-Y, et al. (2015) Identification of phosphorylated MYL12B as a potential plasma biomarker for septic acute kidney injury using a quantitative proteomic approach. Int J Clin Exp Pathol 8:14409–14416 [PMC free article] [PubMed] [Google Scholar]
- 64.Hinkelbein J, Böhm L, Braunecker S, et al. (2017) Decreased Tissue COX5B Expression and Mitochondrial Dysfunction during Sepsis-Induced Kidney Injury in Rats. Oxidative Med Cell Longev N Y 2017:. http://dx.doi.org.proxy.libraries.uc.edu/10.1155/2017/8498510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matejovic M, Tuma Z, Moravec J, et al. (2016) Renal Proteomic Responses to Severe Sepsis and Surgical Trauma: Dynamic Analysis of Porcine Tissue Biopsies. SHOCK 46:453–464. 10.1097/SHK.0000000000000613 [DOI] [PubMed] [Google Scholar]
- 66.Thongboonkerd V, Chiangjong W, Mares J, et al. (2009) Altered plasma proteome during an early phase of peritonitis-induced sepsis. Clin Sci 116:721–730. 10.1042/CS20080478 [DOI] [PubMed] [Google Scholar]
- 67.Hashida T, Nakada T-A, Satoh M, et al. (2017) Proteome analysis of hemofilter adsorbates to identify novel substances of sepsis: a pilot study. J Artif Organs Off J Jpn Soc Artif Organs 20:132–137. 10.1007/s10047-016-0936-3 [DOI] [PubMed] [Google Scholar]
- 68.Gong Y, Chen N, Wang F-Q, et al. (2009) Serum proteome alteration of severe sepsis in the treatment of continuous renal replacement therapy. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc 24:3108–3114. 10.1093/ndt/gfp231 [DOI] [PubMed] [Google Scholar]
- 69.Holly MK, Dear JW, Hu X, et al. (2006) Biomarker and drug-target discovery using proteomics in a new rat model of sepsis-induced acute renal failure. Kidney Int 70:496–506. 10.1038/sj.ki.5001575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Metzger J, Kirsch T, Schiffer E, et al. (2010) Urinary excretion of twenty peptides forms an early and accurate diagnostic pattern of acute kidney injury. Kidney Int 78:1252–1262. 10.1038/ki.2010.322 [DOI] [PubMed] [Google Scholar]
- 71.Carrick E, Vanmassenhove J, Glorieux G, et al. (2016) Development of a MALDI MS-based platform for early detection of acute kidney injury. Proteomics Clin Appl 10:732–742. 10.1002/prca.201500117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maddens B, Ghesquière B, Vanholder R, et al. (2012) Chitinase-like proteins are candidate biomarkers for sepsis-induced acute kidney injury. Mol Cell Proteomics MCP 11:M111.013094. 10.1074/mcp.M111.013094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waltz P, Carchman E, Gomez H, Zuckerbraun B (2016) Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res 202:8–12. 10.1016/j.jss.2015.12.011 [DOI] [PubMed] [Google Scholar]
- 74.Izquierdo-Garcia JL, Nin N, Cardinal-Fernandez P, et al. (2018) Identification of novel metabolomic biomarkers in an experimental model of septic acute kidney injury. Am J Physiol Renal Physiol. 10.1152/ajprenal.00315.2018 [DOI] [PubMed] [Google Scholar]
- 75.Li P, Liao S-T, Wang J-S, et al. (2017) Protection by Huang-Lian-Jie-Du decoction and its constituent herbs of lipopolysaccharide-induced acute kidney injury. FEBS Open Bio 7:221–236. 10.1002/2211-5463.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodrigues FA de P, Santos AD da C, de Medeiros PHQS, et al. (2018) Gingerol suppresses sepsis-induced acute kidney injury by modulating methylsulfonylmethane and dimethylamine production. Sci Rep 8:12154. 10.1038/s41598-018-30522-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langley RJ, Tsalik EL, van Velkinburgh JC, et al. (2013) An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med 5:195ra95. 10.1126/scitranslmed.3005893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langley RJ, Tipper JL, Bruse S, et al. (2014) Integrative “omic” analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am J Respir Crit Care Med 190:445–455. 10.1164/rccm.201404-0624OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cambiaghi A, Díaz R, Martinez JB, et al. (2018) An Innovative Approach for The Integration of Proteomics and Metabolomics Data In Severe Septic Shock Patients Stratified for Mortality. Sci Rep 8:6681. 10.1038/s41598-018-25035-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang H, van Dullemen LFA, Akhtar MZ, et al. (2018) Proteo-metabolomics reveals compensation between ischemic and non-injured contralateral kidneys after reperfusion. Sci Rep 8:8539. 10.1038/s41598-018-26804-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hato T, Zollman A, Plotkin Z, et al. (2018) Endotoxin Preconditioning Reprograms S1 Tubules and Macrophages to Protect the Kidney. J Am Soc Nephrol JASN 29:104–117. 10.1681/ASN.2017060624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jha AK, Huang SC-C, Sergushichev A, et al. (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42:419–430. 10.1016/j.immuni.2015.02.005 [DOI] [PubMed] [Google Scholar]