

Abstract
目的
遗传变异中的单体型扩增因具有潜在的选择优势和克隆演变敏感性, 成为寻找易感癌基因的一个重要标志。本文充分考虑单体型扩增状态的影响因素, 有效实现稀有变异关联分析。
方法
通过等位基因变异频率估计单体型扩增状态。首先采用置换检验, 基于等位基因变异频率实现候选变异位点的聚类。再应用似然聚类方法, 确定隐马尔科夫随机场模型中的邻域系统。此外, 引入一个威尔逊区间和错误识别率的组合过滤机制, 进一步提高变异位点识别精度。最后将候选集与单体型扩增状态合并到加权虚拟位点中用于关联分析。
结果
通过仿真实验, 分别对不同次等位基因变异频率的Ⅰ型错误率比较分析, 发现Ⅰ型错误率基本稳定在2%以内。与其他5种关联分析方法分别进行Ⅰ型和Ⅱ错误率比较分析, Ⅰ型与Ⅱ型错误率均控制在2%以内, 显示出其显著优势及较好的统计能力。
结论
本研究提出的针对单体型扩增区域的肿瘤易感变异关联分析方法能够较为精确的识别单体型扩增区域的肿瘤易感变异, 具有良好的健壮性与稳定性, 可为临床诊断提供决策支持。
Keywords: 肿瘤基因组学, 疾病关联分析, 稀有变异, 单体型扩增
Abstract
Objective
Haplotype amplification on germline variants is suggested to imply potential selective advantages and clonal expansion susceptibility and has become an important signature for seeking cancer susceptibility gene.Here we propose an improved association method that fully considers the haplotype amplification status.
Methods
The haplotype amplification status was estimated by the variant allelic frequencies.We adopted a permutation test on variant allelic frequencies to divide the candidate variants into multiple groups.A likelihood clustering method was then applied to establish the neighborhood system of the hidden Markov random field framework.A filtering pipeline was introduced into the proposed method to further refine the candidate variants, including a Wilson's interval filter and a false discovery rate controller.The final candidate set along with the haplotype amplification status was collapsed into the weighted virtual sites for association tests.
Results
Through simulated tests on a series of datasets, we compared the type Ⅰ error rates of different minor allele frequencies, which stably fell within 2%, suggesting good robustness of the algorithm.In addition, we compared another 5 published association approaches for Type-Ⅰ and Type-Ⅱ error rates with the proposed method, which resulted in the error rates all within 2%, demonstrating significant advantages and a good statistical ability of the proposed method.
Conclusion
The proposed method can accurately identify tumor susceptibility variants in haplotype amplification area with good robustness and stability.
Keywords: cancer genomics, variant association method, rare variants, haplotype amplification
分子变异识别是基因组疾病研究的重点问题。高通量测序技术和大规模的肿瘤基因组计划,如TCGA[1]和ICGC[2],为成功检测肿瘤基因组中的易感变异提供了研究空间。全基因组关联研究(GWAS)已经成功地识别出许多与给定结果相关的常见遗传变异[3],但其对疾病风险解释不足。已有研究在遗传因素导致疾病风险的几个假设[4-7]中提出稀有变异更可能会导致疾病风险。现有的稀有变异检测方法试图从多角度运用多种策略来提高识别精度。第一,针对单一稀有变异较低的次等位基因变异频率(MAF)统计效力不足的问题,采用聚集多个稀有变异位点的合并方法[8],如Sum test[9]、CAST[10]、RareCover[11]等;以及对变异位点加权的统计检验方法,如RWAS[12]、LRT[13]、BUR和LiMB[14]等,从而提高MAF的统计显著性。第二,若将非致病位点或不同效力的稀有变异位点合并会引入噪声、降低统计能力,由此进一步考虑了稀有变异位点影响效应问题,如Seq-aSum-VS[3]方法基于关联方向将稀有变异分为正相关、负相关和无关3类;有研究提出了利用先验信息确定稀有变异是否应该被聚合的方法[15]。第三,稀有变异是稀疏数据[16],仅有一小部分的变异与疾病相关,绝大多数都是中性的,由此CCRS[17]和PMD[18]等方法引入降维机制提高运算性能。
上述方法仅考虑了变异位点的自身特性,忽略了其它因素的影响问题,如变异位点所处的结构性变异对疾病关联性的影响。然而,肿瘤不是仅由单核苷酸多态性位点所引起,而是遗传变异与体细胞变异间相互作用产生的结果[19]。研究表明,具有易感性的遗传变异在加性遗传模型中可能存在选择优势,如杂合缺失和单体型扩增,尤其是单体型扩增引起的体细胞拷贝数变异区域隐藏着易感的致病位点[20]。在一个典型的肿瘤样本中,研究发现癌基因平均有17%的扩增,且有33%的功能区被证实由扩增而激活[21]。目前已知7p、8q、13q和20q等多个区域的拷贝数目变异出现在多种人类实体肿瘤中[22],20q尤其是人类20号染色体长臂13区的扩增是多个肿瘤相关染色体变化的热点区域,存在于卵巢癌、胃癌、乳腺癌、肺癌、结肠直肠癌等多种肿瘤中[23-26]。因此,从海量变异基因数据中检测单体型扩增区域的变异位点与复杂疾病之间的关联关系,为提高复杂疾病的早期诊断率和发现新的个性化诊疗方法具有重要参考价值。单体型扩增区域常覆盖多个变异位点,将基因型数据与单体型扩增引起的等位基因变异频率数据联合考虑,不仅能够增加变异间相互作用的关联性,而且减少了合并无关变异位点所带来的统计效力降低的问题,并可减小Ⅰ型和Ⅱ型的错误识别率,有助于更好地理解肿瘤发生与演变机制[27]。
针对单体型扩增与稀有变异的联合作用关系,本文提出了一种基于隐马尔科夫随机场模型的肿瘤易感变异关联分析方法,RareProb-M,识别单体型扩增区域的致病变异位点。首先在初始化模块中,运用置换检验方法对变异等位基因频率集合进行致病状态的初始化,并应用隐马尔科夫模型初始化区域状态;然后在检测模块中,通过隐马尔可夫随机场模型来检测致病变异位点和显著区域;最后在优化模块中,采用威尔逊区间和错误发现率优化致病变异位点检测结果,获得显著的致病变异位点。仿真实验表明,在不同的参数设置下,本文所提方法优于现有其他同类方法。
1. 算法模型
1.1. 问题描述
在单体型扩增检测的基础上,实现病例―对照样本测序数据集合与复杂疾病之间的关联分析。单体型扩增不仅引起对应区域的读段厚度变化,也会改变等位基因变异频率。假定病例组与对照组的样本量各为N/2,有M个稀有变异。令G ={g1, …, gi, …, gM}表示每个样本中M个变异位点的基因型集合。其中,gi = 0(1 ≤ i ≤ M)表示变异位点si的两条单体型均为野生型等位基因,gi = 1表示当前位点至少有一条单体型发生了变异。在单体型扩增区域中,某一位点的两条单体型均发生变异的情况较为罕见,在此不做考虑。此外,给定M个变异位点对应的等位基因变异频率,V ={v1, …, vi, …, vM} (vi ∈[0, 1])。本文通过变异位点的变异频率来检测其与肿瘤的相关性。样本表型为二分变量,用向量P表示,pj = 'A'与pj = 'C'分别表示来自于病例组和对照组样本的第j个个体。
本文采用隐马尔科夫随机场模型(HMRF)检测肿瘤易感变异。基于在相同单体型扩增区域中的稀有变异对复杂性状具有相似影响的假设,模型为每个位点引入区域状态和致病状态。通常认为致病显著区比背景区中的变异位点具有更高的致病概率。令向量R表示区域状态,ri = 0/1分别表示第i个变异位点处于背景区或致病显著区;向量X表示致病状态,xi = 0/1分别表示第i个变异位点为非致病或致病状态。
在HMRF模型中,有两种输入数据,即每个位点的基因型数据及等位基因变异频率;有两种概率,发射概率建立基因型、表型和隐状态之间的联系,转移概率建立致病/非致病隐状态与区域隐状态之间的联系。隐状态间的相互作用关系通过构建邻居系统来实现。RareProb-M方法划分为初始化、检测和优化3个模块,总体框架如图 1。①为初始模块,对等位基因变异频率集合进行置换检验,根据产生的p值列表初始化致病/非致病状态,并计算聚合似然。由于两个隐状态间具有依赖关系,将初始化的致病/非致病状态作为马尔科夫模型的输入,实现区域隐状态的初始化;②为检测模块,将基因型数据计算得到的次等位基因频率、初始的隐状态结果及邻居系统作为隐马尔科夫随机场模型的输入,采用条件迭代模型实现致病位点的检测;③为优化模块,为了提高模型精度并降低Ⅰ型与Ⅱ型的错误率,通过威尔逊区间、错误发现率及显著检验策略联合筛选显著性致病位点。
1.

RareProb-M方法总体框架
Diagram of RareProb-M framework.
1.2. 初始化模块
1.2.1. 置换检验初始化致病/非致病状态
置换检验适用于总体分布未知且经典统计检验很难分析的假设问题。在此采用置换检验计算病例组与对照组中每个位点变异频率之间差异的统计量,构造经验抽样分布,求出p值进行推断。为了有效地检验变异位点si和肿瘤易感性之间的关联关系,定义H0为原假设,即无关联且位点si为非致病位点;H1为备择假设,即位点si为致病位点。设置显著水平α = 0.001。观测样本的统计量Tobs为,
 |
1 |
其中,A与C分别为病例组样本和对照组样本,vsi和vsj分别为病例组样本和对照组样本中位点的变异频率。
然后混合两组样本,随机抽取N/2个样本作为新的病例组样本A',余下的作为新的对照组样本C',按照公式(1)重新生成新的观测样本统计量Tper。计算并记录混合样本对两组所有可能划分形式下的样本均值差异的统计量,假设置换次数为K,根据K个样本统计量Tper和Tobs获得原假设下差值的正确分布。统计检验第i个位点的p值为,
 |
2 |
其中,I(·)为指示函数。
由此,获得了一个包含所有变异位点的p值列表,对致病/非致病状态向量X进行初始化,
 |
3 |
1.2.2. 隐马尔科夫模型初始化区域状态向量
建立一个简单的马尔科夫模型初始化区域状态向量R,致病/非致病状态向量X作为观察序列,T表示状态转移矩阵,若前一个变异位点si-1的区域状态为Rsi-1,当前变异位点si的区域状态为Rsi,则转移矩阵为,
 若变异位点si的区域状态为Rsi,不平衡致病状态为Xsi,则发射矩阵B为,
若变异位点si的区域状态为Rsi,不平衡致病状态为Xsi,则发射矩阵B为,
 在此采用前向—后向算法学习和更新参数,由维特比算法获得初始化的区域状态向量R。
在此采用前向—后向算法学习和更新参数,由维特比算法获得初始化的区域状态向量R。
1.2.3. 构建邻居系统
隐马尔科夫随机场模型描述了隐状态之间的依赖关系。在此采用文献[28]中的方法计算配对位点的聚合似然值。对于变异位点si和si′,聚合似然值为,
 |
4 |
其中,psi与psi′为变异位点对应的p值。ω(si, si′)值越大表示两个位点间的相似度越高。
为了进一步提高聚类的聚合性能,采用似然校正策略,在此使用两种聚类方法,一种是文献[29]中所用的RAPID方法,另一种是K均值法。对病例组样本采用RAPID方法,定义向量ls =[ls(s1), …, ls(si), …, ls(sn)]将所有个体变异位点的基因型数据映射到实数空间的一个向量上,使其与超平面上的一个点相对应。
 |
5 |
其中, gsi∈{0, 1}, rsi =∑(I(gsi=1)+I(gsi=0)), I (·)为指示函数。定义向量lsi与lsi′间的距离, dist (lsi, lsi′)=min (‖lsi -lsi′ ‖, ‖lsi +lsi′‖),通过距离相似性进行聚类。K均值方法实现病例组样本的变异频率聚类,每个位点视为向量的一列,即Vsi= [v1(si), …, vj(si), …, vN(si)]T(1 ≤ i ≤ M),其中vj(si)表示第j个个体在变异位点si处的变异频率。使用欧几里德距离计算变异位点的相似性,
 |
6 |
根据聚类结果,如果si和si′属于同类,ω(si, si′)保持不变,否则ω(si, si′) = ω(si, si′)/2。
1.3. 检测模块
在HMRF模型中, 有两个隐状态X和R。通过马尔科夫自动逻辑回归模型计算邻居系统中Xsi和Rsi的条件概率。
 |
7 |
其中,n(si)表示si的邻居,ΦX ={γ, η}。
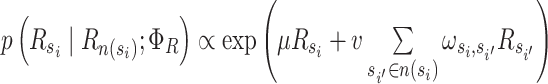 |
8 |
其中, ΦR={μ, ν}。参数γ和μ分别表示Xsi和Rsi自身影响概率, η和ν分别表示Xsi和Rsi受邻居影响的概率。
为了实现观察变量与隐状态之间的有效关联,HMRF模型中定义了两种概率,一是基因型向量G与不平衡性致病关联向量X之间的发射概率,p(G|X);另一是致病关联向量X与区域状态向量R两种隐状态之间的转移概率,p(X|R)。对于变异位点si,θsi和ρsi分别表示病例组与对照组中的致病风险率。令csA~Bin(N/2, θs)和csC ~Bin(N/2, ρs)表示病例组与对照组中的变异数目。条件迭代表示为,
 |
9 |
 |
10 |
其中,S/si表示除si外的所有变异位点。Xsi由 和
和 更新。Rsi与Xsi的更新方法相似。当模型收敛后,即可估计出不平衡致病状态
更新。Rsi与Xsi的更新方法相似。当模型收敛后,即可估计出不平衡致病状态 和显著致病区域
和显著致病区域 。
。
1.4. 优化模块
1.4.1. 威尔逊区间
在假设检验中,通常过滤掉p值不显著的变异位点[30],来提高错误发现率。在统计学上,二项分布的置信区间使用样本的估计比例且允许一定的抽样误差。然而在样本中单体型扩增区域引起读段深度的变化,二项式分布的置信水平会受采样深度影响,对位点变异频率的置信水平估计有偏差。因此,采用能够修正正态近似区间的威尔逊区间方法。令c是每个位点上发现的变异数目,c~Bin(N, 1/2),观察为 。各位点处等位基因变异频率的置信区间为,
。各位点处等位基因变异频率的置信区间为,
 |
11 |
其中,z是置信区间统计量,N是样本大小。通过置信区间做进一步筛选获得高质量的致病位点,若区间的上/下界p值 与差值满足条件
与差值满足条件 (ε默认值为12%),则移除不服从二项分布的位点。
(ε默认值为12%),则移除不服从二项分布的位点。
1.4.2. 错误发现率
在一个假设检验中,可通过选择一个拒绝的阈值控制Ⅰ类错误,而在多重假设检验时,出现Ⅰ类错误的概率会随着待检验数量的增加而增大。因此,需尽可能的平衡敏感度与特异度需求,检测出尽可能多的候选致病位点,同时将错误发现率控制在一个可接受的范围内。令Nt表示拒绝正确原假设的数目,Nr表示拒绝假设的总数目,Q为未观察的随机商。
 |
12 |
对于m重检验,定义p值列表P =(p(1), ..., p(m)),令p(0) ≡ 0,对p值进行排序,p(0) = 0 ≤ p(1) ≤ … ≤ p(m)。定义假设指示量H =(H(1), …, H(m)),若H(i) = 0表示原假设是正确的,否则H(i) = 1。我们拒绝p(i) ≤ T(P)的原假设,T为多重检验的阈值。最后,FDR ≤ α(α = 0.01)的变异位点为显著不平衡致病位点,即RareProb-M报出满足多重检验阈值的具有高显著性的致病位点。
1.4.3. 统计检验
通过FDR对不平衡致病向量X修正后,对该向量再次进行统计检验,计算其致病显著性并求出样本的总体p值。在此使用与文献[12]相同的z检验方法。对于变异位点si,病例组和对照组中的次等位基因频率值θs和ρs之间差值的统计量为,
 |
13 |
假设H0:θsi =ρsi, H1:θsi ≠ ρsi。变异位点统计量为,
 |
14 |
应用双侧检验获得总体p值。
总之,RareProb-M方法充分考虑单体型扩增区域中的基因型数据和变异频率数据,将两者联合起来检测变异与表型间的关联关系。采用HMRF模型估计候选的不平衡致病位点。此模型中,运用置换检验与聚类方法初始化隐状态向量,通过配对位点的聚合似然构建邻居系统计算致病状态与显著区域状态。
2. 实验结果与分析
在仿真实验中,首先生成基因型数据,参照文献[31]中的方法由群体归因危险度决定不平衡致病位点数目,再根据表型与基因型数据仿真变异频率。为了更好地体现RareProb-M方法性能优势,与其他几种方法做比较分析。主要比较这些方法识别的Ⅰ型和Ⅱ型错误率。实验中,调整变异数目M,不平衡致病位点数C和群体归因危险度PAR三个参数值。
2.1. 不同等位基因变异频率的I型错误率分析
Ⅰ型错误率在此定义为,从总的候选变异位点中漏识致病位点的概率。在仿真实验中,预置了100个变异位点,致病变异位点的变异数目(C)分别为50、60、70、80和90,群体归因危险度(PAR)变化值为0.02、0.03、0.04和0.05。在不同的参数下分别生成50组数据。当次等位基因频率(MAF)分别为0.4和0.01时,RareProb-M方法的Ⅰ型错误率如表 1所示。
1.
不同次等位基因频率下的Ⅰ型错误率
Type-Ⅰ error for different MAF
| PAR | C | MAF | |
| 0.4 | 0.01 | ||
| 0.02 | |||
| 50 | 1.23% | 0.27% | |
| 60 | 1.30% | 0.00% | |
| 70 | 0.96% | 0.24% | |
| 80 | 1.48% | 0.13% | |
| 90 | 1.17% | 0.33% | |
| 0.03 | |||
| 50 | 1.02% | 0.47% | |
| 60 | 1.96% | 0.46% | |
| 70 | 1.09% | 0.33% | |
| 80 | 1.53% | 0.46% | |
| 90 | 1.30% | 0.70% | |
| 0.04 | |||
| 50 | 1.01% | 0.23% | |
| 60 | 1.37% | 0.34% | |
| 70 | 2.00% | 0.23% | |
| 80 | 2.09% | 0.50% | |
| 90 | 1.22% | 1.28% | |
| 0.05 | |||
| 50 | 0.73% | 0.20% | |
| 60 | 1.57% | 0.48% | |
| 70 | 1.50% | 0.15% | |
| 80 | 1.79% | 0.23% | |
| 90 | 1.48% | 0.54% | |
从表中结果可见,RareProb-M方法对常见变异(MAF=0.4)和稀有变异(MAF=0.01)都有较低的Ⅰ型错误率。在不同的PAR和致病位点数下,Ⅰ型错误率基本保持稳定,表明本方法具有较好的健壮性和稳定性。
2.2. 不同方法的Ⅰ型错误率比较分析
在本实验中,设置总的位点数为100;致病位点数目分别为50、60、70、80和90;PAR的变化范围[0.02, 0.05];次等位基因变异频率为0.01。每组参数下分别对50组数据集进行比较实验,结果取其均值,对RareProb- M、RareProb[32]、RareProb- C[33]、LBL、LiMB和CCRS 6种方法进行Ⅰ型错误率比较分析(图 2)。其中,LiMB和CCRS两种方法无法在线获得对应程序,所以根据前期的工作基础,依据对原文的理解重新编程实现方法思想,并做了相应的结果统计,可能会存在一定的偏差。
2.

不同方法的Ⅰ型错误率比较
Comparison of Type-Ⅰ error rates of different approaches.A:PAR=0.02;B:PAR=0.03;C:PAR=0.04;D:PAR= 0.05.
图 2A~D分别表示各种方法在群体归因危险度PAR从0.02到0.05的不同取值下,识别致病位点从50变化到90的Ⅰ型错误率情况。从图中可见,LiMB和CCRS两种方法在不同的PAR值下均有较高的Ⅰ类错误率。相比之下,其他4种方法的Ⅰ型错误率随着PAR值和致病位点的增加略有增高,但基本控制在20%以内。在不同的PAR下RareProb-M方法的Ⅰ类错误率均很低,说明致病位点数目对其影响较小。表明RareProb-M对单体型扩增区域的致病位点比其他方法具有更好的识别性能。
2.3. 不同方法的Ⅱ型错误率比较分析
Ⅱ型错误率在此定义为,对于候选变异位点,将非致病位点错选为致病位点的概率。将RareProb-M方法与其他5种方法进行比较,PAR的取值范围[0.02, 0.05],致病变异位点数目的取值范围[50, 90],次等位基因变异频率为0.01。
结果显示,RareProb-M方法均在各种情况下具有较低的Ⅱ型错误率,RareProc和RareProb-M方法的Ⅱ型错误率也能控制在5%以内,基本不受致病位点数目的影响(表 2)。LBL方法随着致病位点数的增加呈下降趋势,且随着PAR的升高也呈下降趋势。CCRS与LiMB两种方法基本不受致病位点数目的影响,CCRS的Ⅱ型错误率随着PAR的升高而升高,LiMB在各种情况下均表现出稳定的Ⅱ型错误率。
2.
与其他方法的Ⅱ型错误率比较
Comparison of Type-Ⅱ error rates of different approaches
| Causal | PAR | Approach | |||||
| RareProb-M | RareProb | RareProb-C | LBL | LiMB | CCRS | ||
| 50 | |||||||
| 0.02 | 0.56% | 3.16% | 1.96% | 58.67% | 23.39% | 25.35% | |
| 0.03 | 0.28% | 2.52% | 2.28% | 43.03% | 23.12% | 38.2 | |
| 0.04 | 0.58% | 3.36% | 2.24% | 27.13% | 22.90% | 51.65% | |
| 0.05 | 0.54% | 4.52% | 2.96% | 18.59% | 26.16% | 66.10% | |
| 60 | |||||||
| 0.02 | 0.73% | 2.17% | 1.80% | 49.69% | 28.31% | 27.07% | |
| 0.03 | 0.55% | 2.97% | 2.43% | 29.96% | 26.61% | 37.71% | |
| 0.04 | 0.62% | 4.03% | 2.63% | 16.03% | 21.63% | 52.05% | |
| 0.05 | 0.57% | 4.87% | 4.07% | 12.71% | 26.14% | 65.95% | |
| 70 | |||||||
| 0.02 | 0.50% | 2.49% | 2.06% | 42.00% | 26.19% | 24.81% | |
| 0.03 | 0.51% | 2.71% | 1.63% | 22.76% | 26.20% | 36.60% | |
| 0.04 | 0.40% | 2.89% | 1.03% | 9.23% | 25.29% | 50.24% | |
| 0.05 | 0.47% | 2.23% | 1.60% | 7.83% | 24.93% | 64.48% | |
| 80 | |||||||
| 0.02 | 0.35% | 2.90% | 1.75% | 35.16% | 25.35% | 25.14% | |
| 0.03 | 0.33% | 2.90% | 1.48% | 13.20% | 26.78% | 36.24% | |
| 0.04 | 0.39% | 3.85% | 1.10% | 4.82% | 19.20% | 49.92% | |
| 0.05 | 0.55% | 1.78% | 0.68% | 3.52% | 21.43% | 62.93% | |
| 90 | |||||||
| 0.02 | 0.52% | 2.36% | 1.13% | 32.50% | 24.30% | 23.36% | |
| 0.03 | 0.48% | 2.60% | 0.84% | 7.84% | 24.71% | 35.38% | |
| 0.04 | 0.47% | 3.02% | 1.36% | 1.67% | 22.79% | 46.18% | |
| 0.05 | 0.54% | 3.22% | 1.80% | 0.95% | 22.81% | 59.35% | |
从Ⅰ型错误率和Ⅱ型错误率综合来看,RareProb-M方法对单体型扩增区域的致病位点识别性能最优。
3. 讨论
本文提出了一种单体型扩增与复杂疾病间的关联分析方法,RareProb-M,此方法是RareProb方法的扩展,两种方法均基于位置相近的变异位点对性状影响相似的假设,引入背景区和显著致病区的概念,不是简单地将变异位点聚合在一起,而是将遗传效应的因素考虑其中。RareProb-M方法与RareProb方法不同之处,其一,不依赖于候选变异位点的先验信息,而是采用了置换检验和马尔科夫模型组合策略,实现了隐状态向量的初始化;其二,通过聚类构建邻居系统,并应用隐马尔科夫随机场模型迭代更新状态向量;其三,RareProb等方法仅是基于基因型数据检测稀有变异与表型间的关联关系,对于大量的低频等位基因检测是困难的,但RareProb-M方法考虑了单体型扩增因素,在各变异位点间可获得更合理可靠的关联因素。对于RareProb-M方法,优化模块主要通过威尔逊区间对样本置信区间实现不断校正,并使用错误发现率控制Ⅰ型和Ⅱ型错误率,从而提高检测精度。在获得高质量的候选致病位点后,采用重复抽样,逐步控制方法计算p值。总之,优化模块通过统计方法有效地提高了检测精度,并控制了Ⅰ型和Ⅱ型错误率。
此外,运用空模型检验收集Ⅰ型错误率的数据集水平。在数据集水平上的Ⅰ型错误率是衡量一个非显著数据集(由非致病变异位点组成)被误认为是显著致病数据集的比率。随机产生1 000 000个数据集,每个数据集包括1000个样本,每个样本中包含100个变异位点。预设每个样本中的位点变异概率为0.005。每个样本被指派为病例组或对照组的概率相同。在100万个数据集中,RareProb-M仅报出85个显著数据集,显示了强大的可靠性。
在下一步研究中,在基因型数据的基础上,考虑性别、年龄、吸烟史等各种表型数据的影响因素,实现多表型与基因型对复杂疾病的综合性关联分析;在聚合变异位点进行统计分析时,要充分考虑变异位点正负效果的影响因素,加强候选致病位点聚合效果;联合降维策略有效地实现高维数据检测分析。
Funding Statement
辽宁省自然科学基金计划项目(20180550161, 20180550855, 2019-ZD-0604)
References
- 1.National Caner Institute. The cancer genome atlas program[EB/ OL]. 2018. <a href="http://cancergenome.nih.gov" target="_blank">http://cancergenome.nih.gov</a>.
- 2.International cancer genome consortium. No cancer therapy is developed today without the genomic knowledge that ICGC provided to the world[EB/OL]. 2018. <a href="http://icgc.org" target="_blank">http://icgc.org</a>
- 3.Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet Epidemiol. 2011;35(7):606–19. doi: 10.1002/gepi.20609. [Basu S, Pan W. Comparison of statistical tests for disease association with rare variants[J]. Genet Epidemiol, 2011, 35(7): 606-19.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53. doi: 10.1038/nature08494. [Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases[J]. Nature, 2009, 461(7265): 747-53.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuk O, Hechter E, Sunyaev SR, et al. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci USA. 2012;109(4):1193–8. doi: 10.1073/pnas.1119675109. [Zuk O, Hechter E, Sunyaev SR, et al. The mystery of missing heritability: Genetic interactions create phantom heritability[J]. Proc Natl Acad Sci USA, 2012, 109(4): 1193-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SH, Wray NR, Goddard ME, et al. Estimating missing heritability for disease from genome-wide association studies. http://www.mendeley.com/catalog/supp-estimating-missing-heritability-disease-genomewide-association-studies/ Am J Hum Genet. 2011;88(3):294–305. doi: 10.1016/j.ajhg.2011.02.002. [Lee SH, Wray NR, Goddard ME, et al. Estimating missing heritability for disease from genome-wide association studies[J]. Am J Hum Genet, 2011, 88(3): 294-305.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slatkin M. Epigenetic inheritance and the missing heritability problem. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=d787e45a6354fc353bdb39da20d38a28. Genetics. 2009;182(3):845–50. doi: 10.1534/genetics.109.102798. [Slatkin M. Epigenetic inheritance and the missing heritability problem[J]. Genetics, 2009, 182(3): 845-50.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng G, Luo L, Siu H, et al. Gene and pathway-based second-wave analysis of genome-wide association studies. http://carcin.oxfordjournals.org/external-ref?access_num=10.1038/ejhg.2009.115&link_type=DOI. Eur J Hum Genet. 2010;18(1):111–7. doi: 10.1038/ejhg.2009.115. [Peng G, Luo L, Siu H, et al. Gene and pathway-based second-wave analysis of genome-wide association studies[J]. Eur J Hum Genet, 2010, 18(1): 111-7.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan W. Asymptotic tests of association with multiple SNPS in linkage disequilibrium. Genet Epidemiol. 2009;33(6):497–507. doi: 10.1002/gepi.20402. [Pan W. Asymptotic tests of association with multiple SNPS in linkage disequilibrium[J]. Genet Epidemiol, 2009, 33(6): 497-507.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=dab3558313363aa6910a52bed0bda789. Mutat Res. 2007;615(1/2):28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST)[J]. Mutat Res, 2007, 615(1/2): 28-56.] [DOI] [PubMed] [Google Scholar]
- 11.Bhatia G, Bansal V, Harismendy O, et al. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput Biol. 2010;6(10):e1000954–9. doi: 10.1371/journal.pcbi.1000954. [Bhatia G, Bansal V, Harismendy O, et al. A covering method for detecting genetic associations between rare variants and common phenotypes[J]. PLoS Comput Biol, 2010, 6(10): e1000954-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sul JH, Han B, He D, et al. An optimal weighted aggregated association test for identification of rare variants involved in common diseases. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=e1c8287ec53c03fc99a24d1b0c14ecca. Genetics. 2011;188(1):181–8. doi: 10.1534/genetics.110.125070. [Sul JH, Han B, He D, et al. An optimal weighted aggregated association test for identification of rare variants involved in common diseases[J]. Genetics, 2011, 188(1): 181-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sul JH, Han B, Eskin E. Increasing power of groupwise association test with likelihood ratio test. J Comput Biol. 2011;18(11):1611–24. doi: 10.1089/cmb.2011.0161. [Sul JH, Han B, Eskin E. Increasing power of groupwise association test with likelihood ratio test[J]. J Comput Biol, 2011, 18(11): 1611-24.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coombes B, Basu S, Guha S, et al. Weighted score tests implementing model-averaging schemes in detection of rare variants in case-control studies. PLoS One. 2015;10(10):e0139355–63. doi: 10.1371/journal.pone.0139355. [Coombes B, Basu S, Guha S, et al. Weighted score tests implementing model-averaging schemes in detection of rare variants in case-control studies[J]. PLoS One, 2015, 10(10): e0139355-63.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants. PLoS One. 2010;5(11):e13584–93. doi: 10.1371/journal.pone.0013584. [Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants[J]. PLoS One, 2010, 5(11): e13584-93.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison AC, Voorman A, Johnson AD, et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat Genet. 2013;45(8):899–901. doi: 10.1038/ng.2671. [Morrison AC, Voorman A, Johnson AD, et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol[J]. Nat Genet, 2013, 45(8): 899-901.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yazdani A, Yazdani A, Boerwinkle E. Rare variants analysis using penalization methods for whole genome sequence data. BMC Bioinformatics. 2015;16:405–12. doi: 10.1186/s12859-015-0825-4. [Yazdani A, Yazdani A, Boerwinkle E. Rare variants analysis using penalization methods for whole genome sequence data[J]. BMC Bioinformatics, 2015, 16: 405-12.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10(3):515–34. doi: 10.1093/biostatistics/kxp008. [Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis[J]. Biostatistics, 2009, 10(3): 515-34.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basu S, Pan W, Oetting WS. A dimension reduction approach for modeling multi-locus interaction in case-control studies. Hum Hered. 2011;71(4):234–45. doi: 10.1159/000328842. [Basu S, Pan W, Oetting WS. A dimension reduction approach for modeling multi-locus interaction in case-control studies[J]. Hum Hered, 2011, 71(4): 234-45.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Xie MC, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015;6:10086–92. doi: 10.1038/ncomms10086. [Lu C, Xie MC, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types[J]. Nat Commun, 2015, 6: 10086-92.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers[J]. Nature, 2010, 463 (7283): 899-905.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albertson DG, Collins C, McCormick F, et al. Chromosome aberrations in solid tumors. Nat Genet. 2003;34(4):369–76. doi: 10.1038/ng1215. [Albertson DG, Collins C, McCormick F, et al. Chromosome aberrations in solid tumors[J]. Nat Genet, 2003, 34(4): 369-76.] [DOI] [PubMed] [Google Scholar]
- 23.Iwabuchi H, Sakamoto M, Sakunaga H, et al. Genetic analysis of benign, low-grade, and high-grade ovarian tumors. http://europepmc.org/abstract/MED/8521410. Cancer Res. 1995;55(24):6172–80. [Iwabuchi H, Sakamoto M, Sakunaga H, et al. Genetic analysis of benign, low-grade, and high-grade ovarian tumors[J]. Cancer Res, 1995, 55(24): 6172-80.] [PubMed] [Google Scholar]
- 24.Kimura Y, Noguchi T, Kawahara K, et al. Genetic alterations in 102 primary gastric cancers by comparative genomic hybridization: gain of 20q and loss of 18q are associated with tumor progression. Mod Pathol. 2004;17(11):1328–37. doi: 10.1038/modpathol.3800180. [Kimura Y, Noguchi T, Kawahara K, et al. Genetic alterations in 102 primary gastric cancers by comparative genomic hybridization: gain of 20q and loss of 18q are associated with tumor progression [J]. Mod Pathol, 2004, 17(11): 1328-37.] [DOI] [PubMed] [Google Scholar]
- 25.Hyman E, Kauraniemi P, Hautaniemi S, et al. Impact of DNA amplification on gene expression patterns in breast cancer. http://www.ncbi.nlm.nih.gov/pubmed/12414653. Cancer Res. 2002;62(21):6240–5. [Hyman E, Kauraniemi P, Hautaniemi S, et al. Impact of DNA amplification on gene expression patterns in breast cancer[J]. Cancer Res, 2002, 62(21): 6240-5.] [PubMed] [Google Scholar]
- 26.Wong MP, Lam WK, Wang E, et al. Primary adenocarcinomas of the lung in nonsmokers show a distinct pattern of allelic imbalance. http://www.ncbi.nlm.nih.gov/pubmed/12154056. Cancer Res. 2002;62(15):4464–8. [Wong MP, Lam WK, Wang E, et al. Primary adenocarcinomas of the lung in nonsmokers show a distinct pattern of allelic imbalance [J]. Cancer Res, 2002, 62(15): 4464-8.] [PubMed] [Google Scholar]
- 27.Tewhey R, Bansal V, Torkamani A, et al. The importance of phase information for human genomics. Nat Rev Genet. 2011;12(3):215–23. doi: 10.1038/nrg2950. [Tewhey R, Bansal V, Torkamani A, et al. The importance of phase information for human genomics[J]. Nat Rev Genet, 2011, 12(3): 215-23.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu MC, Lee S, Cai TX, et al. Rare-variant association testing for sequencing data with the sequence kernel association test. http://www.sciencedirect.com/science/article/pii/S0002929711002229. Am J Hum Genet. 2011;89(1):82–93. doi: 10.1016/j.ajhg.2011.05.029. [Wu MC, Lee S, Cai TX, et al. Rare-variant association testing for sequencing data with the sequence kernel association test[J]. Am J Hum Genet, 2011, 89(1): 82-93.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brinza D, Schultz M, Tesler G, et al. RAPID detection of gene-gene interactions in genome-wide association studies. Bioinformatics. 2010;26(22):2856–62. doi: 10.1093/bioinformatics/btq529. [Brinza D, Schultz M, Tesler G, et al. RAPID detection of gene-gene interactions in genome-wide association studies[J]. Bioinformatics, 2010, 26(22): 2856-62.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaFramboise T, Dewal N, Wilkins K, et al. Allelic selection of amplicons in glioblastoma revealed by combining somatic and germline analysis. PLoS Genet. 2010;6(9):e1001086–93. doi: 10.1371/journal.pgen.1001086. [LaFramboise T, Dewal N, Wilkins K, et al. Allelic selection of amplicons in glioblastoma revealed by combining somatic and germline analysis[J]. PLoS Genet, 2010, 6(9): e1001086-93.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geng Y, Zhao Z, Cui D, et al. An expanded association approach for rare germline variants with copy-number alternation[C] // Bioinformatics and Biomedical Engineering, IWBBIO, 2017: 81-94.
- 32.Wang JY, Zhao ZM, Cao Z, et al. A probabilistic method for identifying rare variants underlying complex traits. BMC Genomics. 2013;14(Suppl 1):S11–20. doi: 10.1186/1471-2164-14-S1-S11. [Wang JY, Zhao ZM, Cao Z, et al. A probabilistic method for identifying rare variants underlying complex traits[J]. BMC Genomics, 2013, 14(Suppl 1): S11-20.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geng Y, Zhao ZM, Zhang XP, et al. An improved burden-test pipeline for identifying associations from rare germline and somatic variants. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4133-4. BMC Genomics. 2017;18(Suppl 7):753–60. doi: 10.1186/s12864-017-4133-4. [Geng Y, Zhao ZM, Zhang XP, et al. An improved burden-test pipeline for identifying associations from rare germline and somatic variants[J]. BMC Genomics, 2017, 18(Suppl 7): 753-60.] [DOI] [PMC free article] [PubMed] [Google Scholar]