

Abstract
目的
探讨FGFR抑制剂帕那替尼对内源性表达FGFR2-CCDC6融合蛋白的新型胆管癌患者异种移植小鼠模型的抗肿瘤作用。
方法
通过一个晚期肝内胆管细胞癌(iCCA)患者的肺转移瘤组织建立了一个表达了FGFR2-CCDC6融合蛋白异种移植(PDX)小鼠模型。将PDX小鼠模型分为4组(每组10只,帕那替尼组9只可供评估)。对照组(柠檬酸盐缓冲液)、帕那替尼组(20 mg/kg,溶于柠檬酸盐溶液中)、吉西他滨(50 mg/kg,每周腹腔注射)+顺铂(2.5 mg/kg,每周腹腔注射)组、帕那替尼联合吉西他滨+顺铂组(上述等剂量),用免疫组织化学染色对p-FGFR、p-FRS2、p-AKT、p-ERK、CD31、Ki-67进行染色评估其表达。通过裂解半胱天冬酶-3(CC3)染色和TUNEL染色试验评估细胞凋亡。Western blot检测FGFR2、p-FGFR、AKT、p-AKT、ERK、p-ERK、FRS2和p-FRS2的表达。
结果
与对照组相比,帕那替尼组小鼠肿瘤生长体积明显减小(P < 0.0001),Ki-67染色显示肿瘤细胞增殖减少,裂解半胱天冬酶-3染色和TUNEL染色试验显示肿瘤细胞凋亡显著增加。蛋白质印迹和IHC显示,FGFR及其下游信号标记FRS2、AKT和ERK的磷酸化均降低。与对照组相比,吉西他滨+顺铂组显著抑制肿瘤生长(P < 0.0001),且明显比单独使用帕那替尼更有效(P < 0.0001)。但没有降低FGFR或下游信号分子FRS2、AKT和ERK的磷酸化。
结论
FGFR抑制剂帕那替尼在含有FGFR2融合的胆管癌肿瘤中调节FGFR信号传导、抑制细胞增殖和诱导凋亡的能力,可作为FGFR2融合的胆管癌患者的治疗选择方案。
Keywords: 肝内胆管细胞癌, FGFR2, 帕那替尼, 吉西他滨, 顺铂
Abstract
Objective
To investigate the antitumor effect of ponatinib on the growth of cholangiocarcinoma xenograft derived from a clinical patient in a mouse model expressing FGFR2-CCDC6 fusion protein.
Methods
Lung metastatic tumor tissue was collected from a patient with advanced intrahepatic cholangiocarcinoma and implanted subcutaneously a NOD/SCID/ Il2rg-knockout (NSG) mouse. The tumor tissues were harvested and transplanted in nude mice to establish mouse models bearing patient-derived xenograft (PDX) of cholangiocarcinoma expressing FGFR2-CCDC6 fusion protein. The PDX mouse models were divided into 4 groups for treatment with citrate buffer (control group), intragastric administration of 20 mg/kg ponatinib dissolved in citrate buffer (ponatinib group), weekly intraperitoneal injections of 50 mg/kg gemcitabine and 2.5 mg/ kg cisplatin (gemcitabine group), or ponatinib combined with gemcitabine and cisplatin at the same doses (10 mice in each group, and 9 mice were evaluated in ponatinib group). The expressions of p-FGFR, p-FRS2, p-AKT, p-ERK, CD31, and Ki-67 in the xenografts were evaluated with immunohistochemistry, and cell apoptosis was analyzed with cleaved caspase-3 (CC3) staining and TUNEL staining. Western blotting was used to detect the expressions of FGFR2, p-FGFR, AKT, p-AKT, ERK, p-ERK, FRS2 and p-FRS2 in the tumor tissues.
Results
Compared with those in the control group, the mice in ponatinib group showed a significantly reduced tumor volume (P < 0.0001) and suppressed tumor cell proliferation with significantly increased cell apoptosis. Western blotting and immunohistochemistry revealed obviously lowered phosphorylation level of FGFR and its downstream signal markers FRS2, AKT and ERK in the xenografts from ponatinib-treated mice. Gemcitabine treatment combined with cisplatin more effectively inhibited tumor growth than ponatinib alone (P < 0.0001) but did not further decrease the phosphorylation levels of FGFR or its downstream signaling molecules FRS2, AKT and ERK.
Conclusion
Ponatinib can regulate FGFR signaling to inhibit the proliferation and induce apoptosis of tumor cells in mice bearing patient-derived cholangiocarcinoma xenograft with FGFR2 fusion. FGFR inhibitor can serve as a treatment option for patients with cholangiocarcinoma with FGFR2 fusion.
Keywords: intrahepatic cholangiocarcinoma, FGFR2, ponatinib, gemcitabine, cisplatin
肝内胆管细胞癌(iCCA)是一种致命的胆道癌,治疗效果不佳。最近的一份报告显示,东西方国家的iCCA发病率都有所上升[1]。胆管癌患者的生存率很低,5年生存率不到10%,这是由其典型的晚期表现和有限的治疗手段所决定的[2-3]。尽管外科治疗对于一些特定的患者有潜在的疗效,由于疾病本身的特点,大多数患者都错过了外科手术的最佳时期。此外,目前吉西他滨和顺铂的标准联合化疗并不能使患者达到长期生存的目标[4-5]。因此开发新的抗癌药物极为迫切。成纤维细胞生长因子受体(FGFR)途径具有调节细胞增殖、侵袭和血管生成等功能,这与组织器官发育、成熟和体内平衡密切相关[6]。而异常的FGFR途径则可导致恶性转化并促进肿瘤进展。组成型活性FGFR信号可由功能增益突变、基因扩增和染色体易位等引起。也有研究显示[7],在iCCA和其他癌症中,FGFR2与多个配偶体的基因发生融合。而在使用新一代测序的研究中,在13%~ 50%的iCCA患者中发现了FGFR2融合体[8-13]。13.6%的iCCA患者中发现了包含FGFR2-BIC1和FGFR2-AHCYL1的FGFR2融合体,FGFR2融合几乎只发生在肝内胆管癌亚型中,而几乎没有发生在肝门周围胆管癌或远端胆管癌中[9]。这些观察结果也已通过荧光原位杂交(FISH)分析的研究得到证实,其中在13%(96例中的12例)的iCCA中观察到了FGFR2融合,但在任何肝门周围或远端胆管癌中没有观察到[11]。FGFR2融合阳性胆管癌具有明显的病理特征,包括导管腔内或沿导管壁生长模式,且无干细胞样标记物。含有FGFR融合配体的肿瘤已显示出对FGFR抑制剂明显的敏感性,这提示靶向FGFR2激酶抑制可能会使具有FGFR2融合体特征的胆管癌患者受益[9, 13]。另外一项实验中[10],在用帕唑帕尼治疗的FGFR2-MGEA5融合的iCCA患者和在先用帕唑帕尼后用帕那替尼治疗的FGFR2-TACC3融合阳性iCCA患者中均观察到了这两种药物初步的抗肿瘤活性。基于这些数据和临床前研究,以及为进一步研究FGFR小分子抑制剂在携带FGFR2融合的iCCAs中活性,许多前瞻性的的临床试验正在启动并进行当中[14-16]。因此,发现的FGFR2融合体可能为iCCA的治疗提供一个新的分子分类,并为带有这些融合体的iCCA患者提供一个新的治疗机会。在本研究中,我们建立了一种新的iCCA患者衍生异种移植物(PDX)小鼠模型,命名为LIV31,该模型可内源性表达FGFR2-CCDC6融合蛋白。然而,FGFR2的下游分子作用及通路机制暂未明确,本文旨在通过帕那替尼等药物以揭示其对表达FGFR2融合通路的胆管细胞癌的影响及作用机制,以为肝内胆管细胞癌的靶向治疗提供进一步的证据。
1. 材料和方法
1.1. 试剂和材料
帕那替尼(AP24534)(Ariad Pharmaceutical); 吉西他滨(Sun Pharmaceutical); 顺铂(APP Pharmaceutical); 抗p-FGFR、抗p-AKT、抗AKT、抗p-ERK、抗ERK、抗p-FRS2和抗CC-3(裂解半胱天冬酶-3)一级抗体(Cell Signaling); 抗β肌动蛋白抗体(Sigma); 抗FGFR2抗体(Santa Cruz Bio-technology); 抗Ki-67抗体(Epitomics); 抗CD31抗体(Dako); 抗FRS2与小鼠/兔特异性HRP/ DAB(ABC)IHC检测试剂盒(Abcam Inc); 用于TUNEL染色的原位细胞死亡检测试剂盒(Roche).RNAeasy迷你套件(Qiagen)。
1.2. 建立肿瘤模型
将来自四期iCCA患者的肺转移瘤的新切除组织切碎,与等体积的0.1 mL基质凝胶混合,并皮下注射到6~8周龄NOD/SCID/Il2rg基因敲除(NSG)小鼠的右侧翼。待肿瘤生长到足够体积后将肿瘤切除,切碎并与基质凝胶混合,再并分别注射到各裸鼠的侧翼。
1.3. 荧光原位杂交分析和G带核型分析
通过运用G显带、断裂FISH和染色体涂片等技术,对LIV31细胞和肿瘤组织石蜡切片进行染色体核型分析[13]。
使用从样品中培养的细胞制备载玻片,并用胰蛋白酶和利什曼染色进行G显带分析。从至少两张载玻片中获取20个处分裂中期的细胞,并对其进行分析。分析完成后,在观察到的每一个代表性克隆细胞中制备染色体组型。
通过断裂FISH分析FGFR2的重排基因。使用加州大学圣克鲁斯分校(UCSC)2009年2月人类基因组组件hg19进一步鉴定了侧翼包含FGFR2基因区域的人—细菌人工染色体((BACs)。3’克隆(RP11-878D21、CTD-2542P10、RP11-984I17、CTD-2291K12、CTD-3237E5)通过nick translation(缺刻前移)用绿色光谱标记,5’克隆(CTD-2312O10、RP11-879C17、RP11-454I6、RP11- 135O16)用橙色光谱标记。将带有浅绿色光谱标记的Abbott分子CEP 10(D10Z1)加入到FGFR2裂解分析中,以帮助鉴定10号染色体。将各标记的克隆组合起来,形成双色、断裂探针组。再通过此探针组鉴定10号染色体。
1.4. 药物研究
将从NSG小鼠获得的肿瘤组织剪碎并混入0.1 mL基质凝胶,再皮下注射到6~8周的无胸腺雌性裸鼠的右侧。每周记录肿瘤体积(体积=L×S2/2,L和S分别代表肿瘤的最长和最短直径)及体质量。当肿瘤体积达到140~200 mm3时,将动物随机分为4组(每组10只,帕那替尼组,实际9只可供评估),分别给予赋形剂(柠檬酸盐缓冲液,pH2.75)、帕那替尼(20 mg/kg,溶于柠檬酸盐缓冲液中每日管饲)、吉西他滨(50 mg/kg,每周腹腔注射)和顺铂(2.5 mg/kg,每周腹腔注射)或与等剂量的帕那替尼联合吉西他滨和顺铂。根据文献和预实验确定帕那替尼20 mg/kg的剂量,在这种剂量下,小鼠能够耐受这3种药物的组合。当肿瘤达到2000 mm3的大小或在治疗9周后时,处死小鼠并取出肿瘤进行进一步分析。
1.5. IHC和TUNEL(原位末端标记法)试验
从每组中随机选择的3~4只小鼠的肿瘤组织切片进行HE染色,同时针对CD31、Ki-67、p-FGFR、p-FRS2、p-AKT、p-ERK。对CC-3进行TUNEL和免疫组化染色。从福尔马林固定的石蜡包埋组织中切下5 μm组织切片。脱蜡后,切片再水合并用0.3%过氧化氢阻断内源过氧化物酶活性10 min,然后在0.01 mol/L的柠檬酸钠(pH=6)中微波提取抗原5 min。再在用10%山羊血清封闭10 min后,切片置于4 ℃孵育过夜,然后使用上述多克隆抗体针对p-FGFR(1:100)、p-FRS2(1:200)、p-AKT(1:200)、p-ERK(1:400)、MMP2(1:500)、MMP3(1:500)、MMP9(1:500)、CC-3(1:400)、CD31(1:600)和Ki-67(1:400)进行实验。最后用山羊二抗孵育30 min。根据使用说明,使用小鼠/兔特异性HRP/DAB IHC检测试剂盒进行免疫染色。随后,切片用苏木精复染并装在二甲苯中。
TUNEL染色使用原位细胞死亡检测试剂盒进行。每张切片的TUNEL阳性细胞在6个随机高功率场中计数。依次计算微血管密度(MVD)、Ki-67和CC-3阳性细胞百分比IHC分数(强度分数乘以阳性细胞的百分比)。蛋白质染色在光学显微镜下放大400倍的进行评估。染色强度分数通过单盲法由一名病理学家手动评分,评分准则为:0=无染色,1=弱染色,2=中度染色,3=强染色。以此计算5个等级中阳性染色肿瘤细胞的百分比(0~100%)。
1.6. Western blot
在小鼠胆管癌组织的裂解物上进行了FGFR2、p-FGFR、AKT、p-AKT、ERK、p-ERK、FRS2和p-FRS2的免疫印迹。通过变性凝胶电泳分离20至60 μg蛋白质。转移到PVDF膜后,用5%牛血清白蛋白(BSA)封闭膜,并用抗p-FGFR(1:1000)、FGFR2(1:200)、p-FRS2(1:1000)、FRS2(1:1000)、p-ERK(1:1000)、ERK(1:1000)、p-AKT(1:1000)、AKT(1:1000)和β-肌动蛋白(1:5000)的一级抗体孵育过夜。然后用与辣根过氧化物酶(HRP)结合的二级抗体孵育,并使用杂交辣根过氧化物酶检测试剂盒观察信号。测量β-肌动蛋白作为对照。使用Image J软件对信号进行定量。
1.7. RNA提取和RT-PCR验证FGFR2-CCD6融合
使用RNA简易微型试剂盒从LIV31 PDX组织中提取总核糖核酸。使用高容量cDNA逆转录试剂盒进行cDNA合成,以转录2 μg总核糖核酸。使用铂Taq高保真聚合酶进行PCR扩增,孵育条件为在95 ℃,持续5 min,随后是34个循环(95 ℃,持续20 s,53.2 ℃持续20 s,68 ℃持续3 min 45 s)。PCR产物通过桑格测序法使用大染料终止子试剂盒直接测序。扩增产物通过在68 ℃下额外孵育10 min而进一步延伸。然后将PCR产物装载到含有溴化乙锭的1%琼脂糖凝胶上。
FGFR2-CCD6融合引物如下:
前导链:AGGACCGGGGATTGGTACCGTAAC,
后随链:TAATGAATTCTTCTTCCTGCTC
使用重组FGFR2-CCDC6质粒: PentR 5'FGFR2-CCDC 6克隆mp# 1.7作为阳性对照。
1.8. 美国典型培养物保藏中心(ATCC)对LIV31细胞的短串联重复基因分型
使用Promega的PowerPlex 18D试剂盒扩增17个短串联重复序列(STR)基因座以及性别决定基因座——釉原蛋白。使用ABI棱镜3500xl遗传分析仪处理细胞系样品。数据使用GeneMapper ID-X v1.2软件进行分析。对提交的每个样品进行适当的阳性和阴性对照,并进行确认。(ATCC销售订单号:SOJ32770)。
1.9. 统计学方法
数据表示为来自至少3个独立实验的平均扫描电镜。所有的统计测试都是使用GraphPad Prism 8.0软件进行的。使用双尾t检验来比较各组之间的差异。使用曲线的非线性比较来比较肿瘤生长曲线。使用对数秩检验比较存活曲线。P < 0.05即认为差异具有统计学意义。
2. 结果
2.1. 建立和鉴定一种新的肝内胆管癌肺转移异种移植物模型
转移性肺结节取自一名四期肝内胆管细胞癌(iCCA)患者(图 1A、B)。肿物大小为8 cm×7.5 cm× 6.5 cm,病理检查提示为3级胆管腺癌。有两个最大直径分别为2.5 cm和1 cm的相邻卫星结节,并且发现有静脉侵犯。手术切缘未见肿瘤浸润,最小肿瘤游离切缘为2 cm。切除的5个淋巴结亦未见肿瘤侵犯。肿瘤分期为AJCCpT2bN0。切除后2月,选用吉西他滨和顺铂进行辅助化疗,4月后停止化疗。肝脏肿瘤切除9月后,随访影像发现肺转移(图 1B箭头)。随后患者开始服用卡培他滨和奥沙利铂继续治疗,4月以及6月后均发现了肺转移病灶。转移瘤被切除,一部分组织植入NSG小鼠的侧翼。肝肿瘤和转移性肺结节的组织病理学显示典型的腺增生,具有胆管癌丰富的间质反应特征(图 1C、D)。LIV31 iCCA PDX肿瘤随后在裸鼠的侧翼成功传代(图 1F、G)。植入裸鼠体内的LIV31肿瘤体积在大约7~8周内达到了1000 mm3。
1.
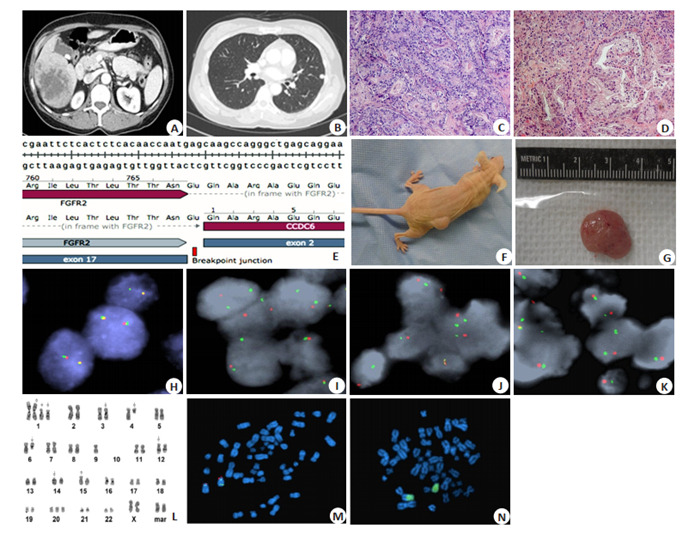
内源性表达FGFR2-CCDC6融合蛋白的异种移植物(LIV31)的构建与验证
Establishment and verification of a xenograft (LIV31) endogenously expressing FGFR2-CCDC6 fusion protein. A: Abdominal contrast-enhanced CT image of the patient before surgery. B: Chest contrast-enhanced CT image after resection (9 months later). C-D: HE staining of primary carcinoma tissue and lung metastatic tumor. E: Sanger sequencing results of RT-PCR products. F: LIV31 grows subcutaneously on the flanks of nude mice. G: Tumor tissue excised from LIV31 xenograft in nude mice. H-K: FGFR2 break-apart FISH results from control tissues, primary liver tumors, lung metastases and xenografts. L: G-banded karyogram of the LIV31 cell line. M: Metaphase state of LIV31 cell line using FGFR2 BAC and D10Z1 probes. N: Metaphase spread of LIV31 cell line with the application of whole chromosome paint 10 probe.
LIV31肿瘤的RNA测序揭示了FGFR2基因的第17外显子和CCDC6基因的第2外显子之间的融合,产生了嵌合的mRNA。这种产物随后通过RT-PCR和桑格测序(图 1E)得到证实,并通过胆管癌组织中与肝脏原发肿瘤、转移性肺结节和作为小鼠皮下肿瘤建立的PDX组织的断裂FISH分析得到验证(图 1H~K)。通过断裂分析和G显带也证明了LIV31中的FGFR2存在融合。LIV31细胞系的G-带状核型为48,XX,+add(1) (p13)x2,idic(1)(p13),del(3)(p13p21),del(4)(q21),del (6)(q13q25),-9,-10,-10,dup(12)(q13q15),add(14) (q32),add(15)(p11.2),+20,+2mar(图 1L)。而应用FGFR2 BAC和D10Z1探针的LIV31细胞系显示出两条衍生染色体10 s,一条丢失了3'FGFR2,另一条重排了FGFR2基因(图 1M)。应用全染色体涂10探针的LIV31在细胞系图像显示,衍生染色体10 s中的一条完全由10号染色体组成,另一条由染色体10与一条未知染色体间的不平衡易位有关(图 1N)。
2.2. 帕那替尼显著抑制LIV31异种移植瘤的生长
有研究表明[19],在一个具有FGFR2过表达的胃癌模型中,用30 mg/kg的帕那替尼治疗小鼠,可导致小鼠皮肤出现脱落,而将帕那替尼的剂量降低到20~25 mg/kg时,小鼠皮肤未出现不良反应。故本实验将帕那替尼的剂量定在20 mg/kg。与用赋形剂治疗的对照小鼠相比,单独以20 mg/kg/d的帕那替尼剂量口服管饲小鼠,显著降低了其肿瘤生长(P < 0.0001)(图 2A、4A)。在治疗的第63天,帕那替尼治疗小鼠的肿瘤体积明显小于用载体治疗的对照组小鼠,平均肿瘤体积分别为619.3± 119.5 mm3和1417±206.3 mm3。存活曲线显示单独使用帕那替尼治疗后存活率显著提高(通过肿瘤体积 < 400 mm的小鼠数量评估,图 2B)。在治疗结束时,用帕那替尼治疗的9只小鼠中有4只(44%)的肿瘤体积小于400 mm,而用载体治疗的10只小鼠中没有一只的肿瘤体积小于400 mm(P < 0.05,图 2B)。各组异种移植物的H & E染色显示出相似的中分化腺癌细胞特征(图 2C)。帕那替尼治疗后,肿瘤总大小显著减小(图 2D)。Ki-67染色显示,与对照组相比,帕那替尼组肿瘤细胞增殖减少(图 2C、E)。CD31染色显示,与对照组相比,帕那替尼治疗LIV31肿瘤的微血管密度没有显著差异(图 2C、E)。裂解半胱天冬酶-3(CC3)染色和TUNEL染色试验显示,与对照组相比,帕那替尼治疗的肿瘤中细胞凋亡显著增加(图 2C、E)。通过蛋白质印迹和IHC可以证明,帕那替尼降低了FGFR及其下游信号标记FRS2、AKT和ERK的磷酸化(图 3A~D)。逆转录聚合酶链反应显示,用帕那替尼治疗后,FGFR-CCDC6融合体的表达持续存在(图 4C)。
2.
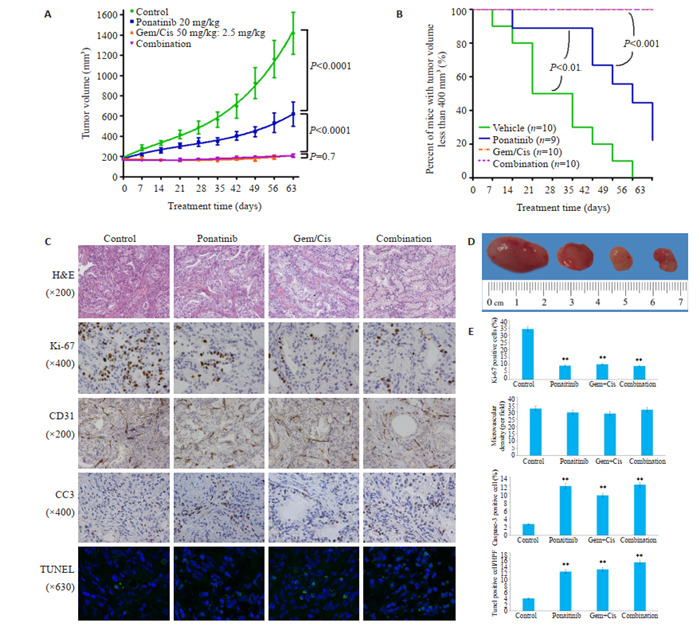
不同药物对裸鼠皮下建立的LIV31异种移植物的作用
Effect of different treatments on growth of subcutaneous LIV31 xenografts in nude mice. A: Tumor growth curves in each group. B: Percentage of mice in each group with tumor volume less than 400mm3. C: HE staining and immunohistochemistry in each group. D: Representative tumors collected from each group. E: Quantification analysis of the results of immunohistochemical staining (Ki-67, CD31, cleaved caspase-3 and TUNEL) in the 4 groups. **P < 0.01 vs the control group.
4.
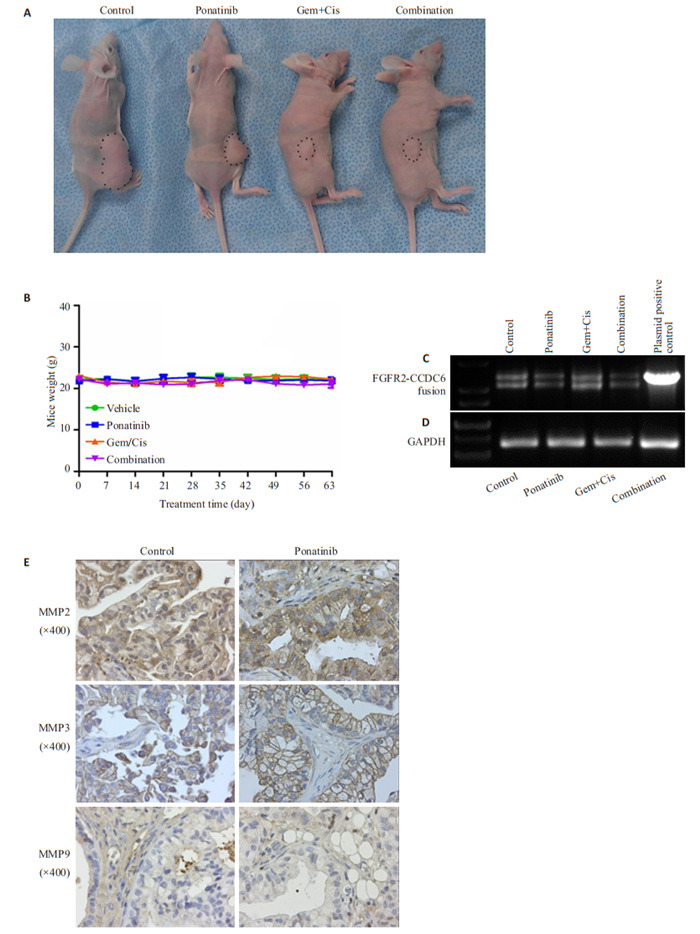
各组小鼠肿瘤及体质量变化,FGFR2-CCDC6融合蛋白表达情况
Changes in tumor volume and body weight of the mice in each group and expression of FGFR2-CCDC6 fusion protein and matrix metalloproteinase. A: A mouse from each group at 63 days after initiation of treatment. B: Changes of body weight of the mice during the treatment. C, D: Expression of FGFR2-CCD6 fusion compared with the control group. E: Immunohistochemical staining.
3.
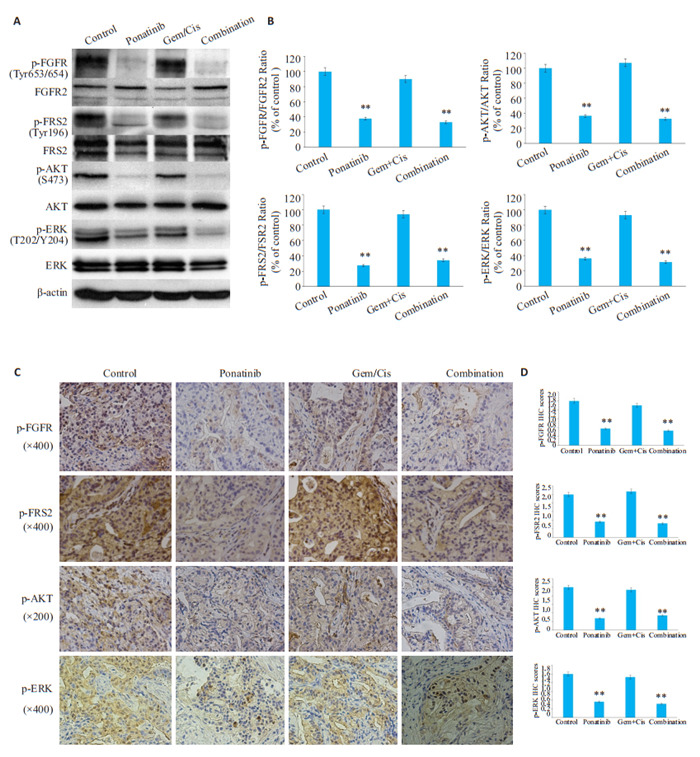
不同药物对LIV31异种移植物FGFR信号通路的影响
Effects of different treatments on FGFR signaling pathway in LIV31 xenograft. A: Western blotting for detecting the downstream markers of FGFR signaling pathway. B: Quantitative analysis of the results of Western blotting. C: Immunohistochemical staining of the tumor tissues in each group. D: Quantitative analysis of the results of immunohistochemical staining. **P < 0.01 vs the control group.
2.3. 帕那替尼与吉西他滨和顺铂联合用药没有协同作用
顺铂(2.5 mg/ (kg·周)腹腔注射)联合吉西他滨(50 mg/ (kg·周)腹腔注射)治疗的标准方案显著抑制肿瘤生长(P < 0.0001),且明显比单独使用帕那替尼更有效(P < 0.0001,图 2A、B)。然而,帕那替尼与吉西他滨和顺铂的联合并不比吉西他滨和顺铂的联合更有效(P=0.7)。帕那替尼在体内对胆管癌有抗癌作用,但与吉西他滨和顺铂联合给药时没有附加或协同作用。单独用帕那替尼,单用吉西他滨和顺铂,及联合帕那替尼与吉西他滨和顺铂治疗的小鼠的体质量与用赋形剂治疗的小鼠的体重没有差异(图 4B)。Ki-67染色显示吉西他滨和顺铂治疗后肿瘤细胞增殖减少(图 2C、E)。当用吉西他滨和顺铂治疗时,胆管癌肿瘤的微血管密度没有降低。CC3染色和TUNEL分析显示,与对照组相比,吉西他滨和顺铂联合治疗的肿瘤具有显著的凋亡活性(图 2C、E)。用吉西他滨和顺铂治疗没有降低FGFR或下游信号分子FRS2、AKT和ERK的磷酸化(图 3A~D)。用吉西他滨和顺铂治疗后,通过非定量RT-PCR评估,FGFR-CCDC6融合体的表达持续存在(图 4C)。
3. 讨论
FGFR融合物已被确定为多种癌症的一种新的、药物化的共基因靶点,包括膀胱癌(FGFR3)[18]、胶质母细胞瘤(FGFR1和FGFR3)[17]、乳腺癌(FGFR2)[8]和最近的CCA(FGFR2)。胆管癌和其他癌症中发现了FGFR2与多个配偶体的基因融合,这些配偶体提供促进寡聚化的二聚化结构域,导致FGFR激酶和多种下游信号通路的组成型激活,包括RAS/MASK和PI3K/AKT通路[7, 19]。因此,FGFR2的小分子抑制剂可作为该患者群体的合理疗法。在当前的研究中,我们建立并验证了一种新的iCCA PDX模型,LIV31,其内源性表达FGFR2-CCD6融合蛋白。这种融合基因及表达产物随后在RT-PCR、桑格测序、G显带等一系列方法中得到了验证。研究人员在一名乳腺癌患者中发现了FGFR2与CCDC6的融合基因[8]。包括FGFR-CCDC6融合在内的FGFR融合蛋白的过表达激活细胞外信号调节激酶信号传导,并在体外诱导细胞增殖[8],这说明我们的发现并不是偶然的。FGFR抑制剂帕那替尼在FGFR活性失调的多种人类肿瘤中显示了有效的抗肿瘤活性[17, 20-22]。值得注意的是,与它们的亲本细胞相比,经工程改造过表达FGFR1-4的细胞对帕那替尼显示出更高的敏感性[20, 22]。对于携带FGFR2融合蛋白的晚期iCCA患者,帕那替尼可诱导肿瘤缩小,这包括一例之前对帕唑替尼有反应并随后进展的患者[10]。帕唑替尼优先靶向血管内皮生长因子受体和血小板衍生生长因子受体,但对FGFR也有适度的疗效。因此,我们在这种FGFR2-CCD6驱动的iCCA小鼠模型中检测了帕那替尼的抗肿瘤作用。在20 mg/kg时,帕那替尼显著抑制LIV31胆管癌模型的肿瘤生长,与赋形剂组相比,帕那替尼组同期肿瘤体积明显减小,且存活曲线显示肿瘤体积小于400 mm3的小鼠的百分比明显提高。此外,免疫组化和蛋白质印迹显示帕那替尼抑制FGFR信号的激活,导致LIV31胆管癌肿瘤中细胞增殖的抑制和凋亡的诱导。该模型中帕那替尼的效力类似于先前在FGFR扩增或突变肿瘤和BCR ABL驱动肿瘤模型中观察到的效力[17, 23]。这说明了FGFR2抑制剂帕那替尼对iCCA肿瘤的抑制作用。吉西他滨和顺铂是晚期胆管癌的标准化疗组合,仅达到26.1%的有效率,并将总生存期中位数从8.1月延长至11.7月[5]。因此,研究FGFR抑制剂是否能增强吉西他滨和顺铂在含FGFR2融合的胆管癌中的抗癌作用是非常重要的。在本次实验中,我们将单独使用FGFR2抑制剂帕那替尼、单用吉西他滨和顺铂,以及FGFR2抑制剂帕那替尼联合吉西他滨和顺铂的标准化疗组合做了对比,结果发现,在给定给药剂量下,单用标准化疗方案明显比单独使用帕那替尼更有效,而二者联用时,帕那替尼并没有增强这种联合治疗的效果。帕那替尼与吉西他滨和顺铂缺乏协同作用可能是因为我们给予了相对高剂量的吉西他滨和顺铂;或者,可能是由于最近证实的许多酪氨酸激酶抑制剂阻止吉西他滨在癌细胞积累的作用[24],这使组合应用的效果更差。由于吉西他滨(50 mg/kg)和顺铂(2.5 mg/kg)的组合几乎完全抑制肿瘤生长,因此很难通过与帕那替尼的组合达到额外的效果。因此,下一步可能需要研究FGFR抑制剂和低剂量吉西他滨和顺铂联合治疗的体内效果,包括在使用FGFR抑制剂之前使用吉西他滨序贯给药的研究。信号通路方面,单用帕那替尼对FGFR及其下游信号标记FRS2、AKT和ERK的磷酸化均有明显的抑制作用,而相比之下,标准化疗方案则未见有明显的通路抑制表现,这正是靶向治疗的优势所在,从另一方面而言,FGFR融合基因及其表达产物有可能作为FGFR抑制剂有效性的预测性生物标志物。基质金属蛋白酶可促进肿瘤细胞的增殖和转移,在FGFs和FGFR的某些复合物中被发现与信号转导有关[25-28]。因此我们比较了应用FGFR2抑制剂时MMP在通路中的影响,实验结果显示,在用FGFR2抑制剂治疗后,MMP2、MMP3和MMP9的表达没有显著变化,表明这些基质金属蛋白酶不是FGFR2抑制剂帕那替尼抑制肿瘤发生作用的直接介质。目前一线联合化疗虽然有所进展,但是治疗选择仍然有限,因为没有标准的二线治疗方案。针对EGFR、VEGF和MEK单独或与化疗联合的靶向治疗已在胆管癌进行实验[29]。然而,现有的临床试验数据并不支持它们在胆管癌中的应用。帕唑帕尼,是一种针对VEGFR、PDGFR和FGFR的多靶点TKI药物。最近其已被报道可诱导在一线联合化疗后进展的FGFR2-TACC3融合的晚期胆管癌患者肿瘤的缩小[10]。因此,FGFR抑制剂可用作治疗FGFR遗传改变的胆管癌的二线疗法,特别是存在FGFR2融合的病例。
总之,我们报道了一种新的胆管癌PDX模型,该模型表达FGFR 2-CCD6融合蛋白,在肯定一线化疗的作用基础上证明了非特异性多激酶抑制剂帕那替尼对该肿瘤的临床前抗肿瘤活性。在这个PDX模型上,帕那替尼似乎与标准化疗吉西他滨和顺铂没有相加或协同作用。但与对照组相比之下,单独应用帕那替尼对PDX模型的抑制作用还是显而易见的。
Biographies
吴天宇,硕士,E-mail: 13211163652@163.com
蒋晓青,主治医师,硕士,E-mail: wyjxq.521@163.com
Funding Statement
广东省自然科学基金(2018A030313659)
Contributor Information
吴 天宇 (Tianyu WU), Email: 13211163652@163.com.
蒋 晓青 (Xiaoqing JIANG), Email: wyjxq.521@163.com.
王 宇 (Yu WANG), Email: wyjxq163@163.com.
References
- 1.Bertuccio P. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol. 2019;71(1):104–14. doi: 10.1016/j.jhep.2019.03.013. [Bertuccio P. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma[J]. J Hepatol, 2019, 71(1): 104-14.] [DOI] [PubMed] [Google Scholar]
- 2.Rizvi S, Gores GJ. Emerging molecular therapeutic targets for cholangiocarcinoma. J Hepatol. 2017;67(3):632–44. doi: 10.1016/j.jhep.2017.03.026. [Rizvi S, Gores GJ. Emerging molecular therapeutic targets for cholangiocarcinoma[J]. J Hepatol, 2017, 67(3): 632-44.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–79. doi: 10.1016/S0140-6736(13)61903-0. [Razumilava N, Gores GJ. Cholangiocarcinoma[J]. Lancet, 2014, 383 (9935): 2168-79.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morizane C, Okusaka T, Mizusawa J, et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase Ⅲ clinical trial. Ann Oncol. 2019;30(12):1950–8. doi: 10.1093/annonc/mdz402. [Morizane C, Okusaka T, Mizusawa J, et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase Ⅲ clinical trial[J]. Ann Oncol, 2019, 30(12): 1950-8.] [DOI] [PubMed] [Google Scholar]
- 5.Valle J, Harpreet W, Daniel HP, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–81. doi: 10.1056/NEJMoa0908721. [Valle J, Harpreet W, Daniel HP, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer[J]. N Engl J Med, 2010, 362(14): 1273-81.] [DOI] [PubMed] [Google Scholar]
- 6.Lamarca A, Barriuso J, McNamara MG, et al. Molecular targeted therapies: Ready for "prime time" in biliary tract cancer. J Hepatol. 2020;73(1):170–85. doi: 10.1016/j.jhep.2020.03.007. [Lamarca A, Barriuso J, McNamara MG, et al. Molecular targeted therapies: Ready for "prime time" in biliary tract cancer[J]. J Hepatol, 2020, 73(1): 170-85.] [DOI] [PubMed] [Google Scholar]
- 7.Parker BC, Engels M, Annala M, et al. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol. 2014;232(1):4–15. doi: 10.1002/path.4297. [Parker BC, Engels M, Annala M, et al. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours[J]. J Pathol, 2014, 232(1): 4-15.] [DOI] [PubMed] [Google Scholar]
- 8.Wu YM, Su FY, Shanker KS, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47. doi: 10.1158/2159-8290.CD-13-0050. [Wu YM, Su FY, Shanker KS et al. Identification of targetable FGFR gene fusions in diverse cancers[J]. Cancer Discov, 2013, 3(6): 636-47.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arai Y, Yasushi T, Fumie H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427–34. doi: 10.1002/hep.26890. [Arai Y, Yasushi T, Fumie H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma[J]. Hepatology, 2014, 59(4): 1427-34.] [DOI] [PubMed] [Google Scholar]
- 10.Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10(2):e1004135. doi: 10.1371/journal.pgen.1004135. [Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma[J]. PLoS Genet, 2014, 10(2): e1004135.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham RP, Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45(8):1630–8. doi: 10.1016/j.humpath.2014.03.014. [Graham RP, Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma[J]. Hum Pathol, 2014, 45(8): 1630-8.] [DOI] [PubMed] [Google Scholar]
- 12.Ross JS, Kai W, Laurie G, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19(3):235–42. doi: 10.1634/theoncologist.2013-0352. [Ross JS, Kai W, Laurie G, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing[J]. Oncologist, 2014, 19(3): 235-42.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. 2015;6:6087. doi: 10.1038/ncomms7087. [Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma[J]. Nat Commun, 2015, 6: 6087.] [DOI] [PubMed] [Google Scholar]
- 14.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–84. doi: 10.1016/S1470-2045(20)30109-1. [Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study[J]. Lancet Oncol, 2020, 21 (5): 671-84.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahleda R, Italiano A, Hierro C, et al. Multicenter phase i study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res. 2019;25(16):4888–97. doi: 10.1158/1078-0432.CCR-18-3334. [Bahleda R, Italiano A, Hierro C, et al. Multicenter phase i study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors[J]. Clin Cancer Res, 2019, 25(16): 4888-97.] [DOI] [PubMed] [Google Scholar]
- 16.Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–63. doi: 10.1158/2159-8290.CD-16-1000. [Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma[J]. Cancer Discov, 2017, 7(3): 252-63.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):p. 1231–5. doi: 10.1126/science.1220834. [Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science, 2012. 337(6099): p. 1231-5.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morales-Barrera R, Suárez C, González M, et al. The future of bladder cancer therapy: Optimizing the inhibition of the fibroblast growth factor receptor. Cancer Treat Rev. 2020;86:102000. doi: 10.1016/j.ctrv.2020.102000. [Morales-Barrera R, Suárez C, González M, et al. The future of bladder cancer therapy: Optimizing the inhibition of the fibroblast growth factor receptor[J]. Cancer Treat Rev, 2020, 86: 102000.] [DOI] [PubMed] [Google Scholar]
- 19.Borad MJ, Gores GJ, Roberts LR. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr Opin Gastroenterol. 2015;31(3):264–8. doi: 10.1097/MOG.0000000000000171. [Borad MJ, Gores GJ, Roberts LR. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma[J]. Curr Opin Gastroenterol, 2015, 31(3): 264-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren M, Qin H, Ren R, et al. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia. 2013;27(1):32–40. doi: 10.1038/leu.2012.188. [Ren M, Qin H, Ren R, et al. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities[J]. Leukemia, 2013, 27(1): 32-40.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wynes MW, Hinz TK, Gao DX, et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin Cancer Res. 2014;20(12):3299–309. doi: 10.1158/1078-0432.CCR-13-3060. [Wynes MW, Hinz TK, Gao DX, et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies[J]. Clin Cancer Res, 2014, 20 (12): 3299-309.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chase A, Bryant C, Score J, et al. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica. 2013;98(1):103–6. doi: 10.3324/haematol.2012.066407. [Chase A, Bryant C, Score J, et al. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome[J]. Haematologica, 2013, 98(1): 103-6.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Hare T, William CS, Zhu XT, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–12. doi: 10.1016/j.ccr.2009.09.028. [O'Hare T, William CS, Zhu XT, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance[J]. Cancer Cell, 2009, 16(5): 401-12.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Damaraju VL, Kuzma M, Mowles D, et al. Interactions of multitargeted kinase inhibitors and nucleoside drugs: Achilles heel of combination therapy? http://europepmc.org/abstract/med/25519698. Mol Cancer Ther. 2015;14(1):236–45. doi: 10.1158/1535-7163.MCT-14-0337. [Damaraju VL, Kuzma M, Mowles D, et al. Interactions of multitargeted kinase inhibitors and nucleoside drugs: Achilles heel of combination therapy[J]? Mol Cancer Ther, 2015, 14(1): 236-45.] [DOI] [PubMed] [Google Scholar]
- 25.Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008;27(1):57–66. doi: 10.1007/s10555-007-9105-8. [Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2[J]. Cancer Metastasis Rev, 2008, 27(1): 57-66.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elewa MA, Gayyar MM, Schaalan MF, et al. Hepatoprotective and anti-tumor effects of targeting MMP-9 in hepatocellular carcinoma and its relation to vascular invasion markers. Clin Exp Metastasis. 2015;32(5):479–93. doi: 10.1007/s10585-015-9721-6. [Elewa MA, Gayyar MM, Schaalan MF, et al. Hepatoprotective and anti-tumor effects of targeting MMP-9 in hepatocellular carcinoma and its relation to vascular invasion markers[J]. Clin Exp Metastasis, 2015, 32(5): 479-93.] [DOI] [PubMed] [Google Scholar]
- 27.Ardi VC, Steen PE, Opdenakker G, et al. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J Biol Chem. 2009;284(38):25854–66. doi: 10.1074/jbc.M109.033472. [Ardi VC, Steen PE, Opdenakker G, et al. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway[J]. J Biol Chem, 2009, 284(38): 25854-66.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pintucci G, Yu PJ, Sharony R, et al. Induction of stromelysin-1 (MMP-3) by fibroblast growth factor-2 (FGF-2) in FGF-2-/-microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK-1/2) J Cell Biochem. 2003;90(5):1015–25. doi: 10.1002/jcb.10721. [Pintucci G, Yu PJ, Sharony R, et al. Induction of stromelysin-1 (MMP-3) by fibroblast growth factor-2 (FGF-2) in FGF-2-/-microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK-1/2)[J]. J Cell Biochem, 2003, 90(5): 1015-25.] [DOI] [PubMed] [Google Scholar]
- 29.Onesti CE, Romiti A, Roberto M, et al. Recent advances for the treatment of pancreatic and biliary tract cancer after first-line treatment failure. Expert Rev Anticancer Ther. 2015;15(10):1183–98. doi: 10.1586/14737140.2015.1081816. [Onesti CE, Romiti A, Roberto M, et al. Recent advances for the treatment of pancreatic and biliary tract cancer after first-line treatment failure[J]. Expert Rev Anticancer Ther, 2015, 15(10): 1183-98.] [DOI] [PubMed] [Google Scholar]