

Abstract
FLT3-ITD mutations occur in 20–30% of AML patients and are associated with aggressive disease. Patients with relapsed FLT3-mutated disease respond well to 2nd generation FLT3 TKIs but inevitably relapse within a short timeframe. In this setting, until overt relapse occurs, the bone marrow microenvironment facilitates leukemia cell survival despite continued on-target inhibition. We demonstrate that human bone marrow derived conditioned medium (CM) protects FLT3-ITD+ AML cells from the 2nd generation FLT3 TKI quizartinib and activates STAT3 and STAT5 in leukemia cells. Extrinsic activation of STAT5 by CM is the primary mediator of leukemia cell resistance to FLT3 inhibition. Combination treatment with quizartinib and dasatinib abolishes STAT5 activation and significantly reduces the IC50 of quizartinib in FLT3-ITD+ AML cells cultured in CM. We demonstrate that CM protects FLT3-ITD+ AML cells from the inhibitory effects of quizartinib on glycolysis and that this is partially reversed by treating cells with the combination of quizartinib and dasatinib. Using a doxycycline-inducible STAT5 knockdown in the FLT3-ITD+ MOLM-13 cell line, we show that dasatinib-mediated suppression of leukemia cell glycolytic activity is STAT5-independent and provide a preclinical rationale for combination treatment with quizartinib and dasatinib in FLT3-ITD+ AML.
Keywords: acute myeloid leukemia, FLT3, targeted therapy, TKI, signal transducer, activator of transcription
Introduction
FLT3 is a receptor tyrosine kinase (RTK) expressed in CD34+ hematopoietic stem and progenitor cell subsets important in regulation of steady-state hematopoiesis (1, 2). The binding of FLT3 ligand induces FLT3 receptor homodimerization, triggering a mitogenic signaling cascade that results in enhanced proliferation of early hematopoietic precursors (3–5). FLT3 internal tandem duplications (FLT3-ITDs) are observed in up to 30% of newly diagnosed cases of adult acute myeloid leukemia (AML) and confer a poor prognosis (6, 7). ITDs can range in size from 3–400 base pairs and are primarily located in the RTK juxtamembrane (JM) domain. Unlike FLT3, which requires binding of FLT3 ligand to dimerize and initiate downstream signaling, FLT3-ITD results in disruption of the normal autoinhibitory function of the JM domain, resulting in ligand-independent constitutive kinase activation (8). Signaling via FLT3 promotes leukemia cell survival and proliferation via activation of the STAT5, PI3K/AKT and MAPK pathways (9).
Apart from all-trans retinoic acid in acute promyelocytic leukemia, FLT3 tyrosine kinase inhibitors (TKIs) were the first molecularly targeted treatment to demonstrate an improvement in overall survival in AML, prompting the 2017 FDA approval of midostaurin in combination with standard 7+3 chemotherapy for newly diagnosed FLT3-mutated AML (10). Midostaurin, however, is a multikinase inhibitor with poor FLT3 selectivity, and it is unclear whether the positive results from the RATIFY trial are entirely attributable to FLT3 inhibition, especially as improvements in overall survival extended to patients with low FLT3-ITD allelic ratios (10). In contrast to midostaurin monotherapy, which led to zero complete responses (CR) in relapsed/refractory (R/R) FLT3-mutated AML patients (11), a group exhibiting profound FLT3 oncogene addiction (12), the more selective 2nd generation FLT3 TKIs quizartinib and gilteritinib each demonstrated composite CR rates of ~50% as single agents in phase 3 trials in the R/R setting (13, 14). Unfortunately, clinical responses to these newer FLT3 TKIs are not durable, and patients typically relapse within a few months due to emergence of secondary kinase mutations in FLT3, most commonly at D835 (activation loop) or F691 (gatekeeper residue), resulting in reactivation of FLT3-ITD signaling (13–17). As the clinical use of 2nd generation FLT3 inhibitors increases, strategies to circumvent resistance and prolong clinical responses are needed. The observation that peripheral leukemic blasts are more easily cleared from the blood than leukemic blasts residing in the bone marrow (BM) has led to the idea that the BM microenvironment provides a protective niche, allowing leukemia cells to survive until overt mutational resistance is acquired (18, 19).
Constitutively activated tyrosine kinases such as FLT3-ITD in AML and BCR-ABL1 in chronic myeloid leukemia (CML) drive potent STAT5 activation, which is critical for leukemogenesis (9, 20). We and others have previously shown that medium harvested from human HS-5 BM stromal cells protects CML cells from the cytotoxic effects of TKIs via extrinsic activation of pSTAT3Y705, while pSTAT5Y694 levels remain under the control of BCR-ABL1 kinase (21, 22). We hypothesized that extrinsic activation of STAT3 and/or STAT5 may contribute to BM stroma-mediated leukemia cell survival upon 2nd generation FLT3 inhibition in AML.
Materials and Methods
Cell lines
K562, MOLM-13 and MOLM-14 cell lines were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany) and MV411 cells were generously donated by Dr. Ryan O’Connell at the University of Utah. Cell lines were authenticated using the GenePrint 24 kit (Promega, Madison, WI, USA) and the DSMZ Online STR Analysis database at the DNA Sequencing Core at the University of Utah. MOLM-13, MOLM-14 and MV411 cells were genotyped for presence of the published ITD variant by PCR amplification and direct sequencing of genomic DNA. Cells were cultured in regular medium (RM) consisting of RPMI medium supplemented with 20% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA), 2 mM L-glutamine, and 100 U/mL penicillin/streptomycin (RF10), in HS-5 conditioned medium (CM), or in direct contact (DC) with HS-5 cells. HS-5 CM was generated in large batches by culturing HS-5 human BM stromal cells to approximately 80% confluency in RPMI medium supplemented with 10% FBS, L-glutamine and penicillin/streptomycin. Following 24 hours of incubation, HS-5 CM was harvested and filtered (0.22 μm) and final FBS concentration was increased to 20%.
Patient AML samples
Primary samples were obtained from AML patients treated at the University of Utah. Informed consent was obtained from all donors (University of Utah Institutional Review Board #45880). Cells were subjected to Ficoll separation and red blood cell lysis. An AutoMACS Pro Separator (Miltenyi Biotech, Bergisch Gladbach, Germany) was used to purify CD34+ cells. Purity was determined to be >90% by fluorescence activated cell sorting. Prior to assays, primary cells were cultured in RM at 37°C without addition of cytokines. FLT3 mutation status was determined by referencing next-generation sequencing results performed as clinical standard of care by ARUP Laboratories at the University of Utah. FLT3-ITD variant allele frequency in CD34+ primary cells was determined using PCR followed by capillary gel electrophoresis on genomic DNA at ARUP Laboratories.
Viability assays
Apoptosis assays were performed with allophycocyanin-conjugated annexin V and 7-aminoactinomycin D (BD Biosciences, San Jose, CA, USA). Cells were analyzed for fluorescence on a Guava easyCyte HT Flow Cytometer (MilliporeSigma, Burlington, MA, USA) or a BD Canto flow cytometer.
Tyrosine kinase inhibitors
Quizartinib, fibroblast growth factor receptor 1 (FGFR1) inhibitor PD1703074, spleen tyrosine kinase (SYK) inhibitor PRT062607, ibrutinib and dasatinib were purchased from Selleckchem (Houston, TX, USA). Ruxolitinib was purchased from Chemietek (Indianapolis, IN, USA).
Plasmids
The pLMP-GFP-shSTAT3 retroviral vector was obtained by subcloning the STAT3-specific sequence (5’-TGCTGTTGACAGTGAGCGAAATGTTCTCTATCAGCACAATTAGTGAAGCCACAGATGTAATTGT GCTGATAGAGAACATTCTGCCTACTGCCTCGGA-3’) beginning at nucleotide 1008 of human STAT3 into the pLMP-miR-122 vector (Thermo Fisher Scientific, Waltham, MA, USA). The pLMP-miR-122 vector lacking the STAT3-specific sequence was used as an empty vector (EV) control. Custom doxycycline-inducible lentiviral shSTAT5 expression constructs (pRSIT17-U6Tet-sh-CMV-TetRep-2A-TagGFP2–2A-Puro) were obtained from Cellecta (Mountain View, CA, USA). Doxycycline-inducible lentiviral shSTAT5 constructs containing tagged RFP were generated from the original plasmid by subcloning the RFP site from pRSIT16-U6Tet-sh-HTS6-CMV-TetRep-2A-TagRFP-2A-Puro (Cellecta).
Immunoblot analysis
Cells were harvested and washed with cold PBS, then lysed in radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology (CST); 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin; with 1 mM PMSF added immediately before use). Equal amounts of protein were separated by SDS-PAGE (Bio-Rad) and transferred to nitrocellulose. Antibodies included: β-actin (cat. #3700S), STAT3 (cat. #9139), pSTAT3Y705 (cat. #9145), pSTAT5Y694 (cat. #9359), pLynY507 (cat. #2731), pSrcY527 (cat. #2105) (all from CST, Danvers, MA, USA), and STAT5 (cat. #610191) (BD). Images were obtained using a Licor Odyssey CLx Infrared Imaging System.
Cell proliferation assays
Cells were seeded in triplicate at 5e3 cells/well in a 96-well plate ± doxycycline (for knockdown) or inhibitors. Cell proliferation was measured at 72 or 96 hours after plating using the CellTiter 96 AQueous One Solution MTS Reagent (Promega, Madison, WI, USA) according to manufacturer’s instructions. Optical density was measured at 490 nm using an Epoch Microplate Spectrophotometer (BioTek Instruments, Winooski, VT, USA).
STAT5 phospho-flow cytometry
Flow cytometry was performed with Alexa Fluor 647 mouse anti-pSTAT5Y694 (clone 47, BD Biosciences). MOLM-13 cells were cultured in CM for 4 hours prior to administration of quizartinib and candidate inhibitors. Inhibitors were identified by literature review of kinases implicated in STAT5 phosphorylation in AML. Cells were harvested following 4 hours of culture with inhibitor, fixed (BD Phosflow Fix Buffer I) and permeabilized (BD Phosflow Perm Buffer III). Cells were analyzed for fluorescence intensity on a BD LSRFortessa cell analyzer.
Metabolic phenotyping
Glycolytic activity of viable cells grown for 24 hours in RM versus CM was measured using the Seahorse XFe96 Analyzer (Agilent, Santa Clara, CA, USA) and Seahorse Glycolysis Stress Test kit per manufacturer’s instructions. Seahorse X96 plates were coated with 20 μL/well of Cell-TAK adhesive at 22.4 μg/mL in 0.1M NaHCO3 prior to addition of cells at a concentration of 1.0–1.5e5 cells/well suspended in XF base medium (DMEM without NaHCO3). Plates were centrifuged at 200 × g for 3 minutes and incubated in a CO2-free incubator prior to analysis. Glycolysis was measured on a Seahorse XFe96 Analyzer under basal conditions and following sequential addition of 10 mM glucose, 1 μM oligomycin and 50 mM 2-deoxy-D-glucose. For STAT5 knockdown experiments, MOLM-13 cells containing doxycycline-inducible shSTAT5 constructs were cultured with doxycycline and inhibitors in CM for 24 hours prior to Seahorse assay.
Statistical analysis
Results are provided as mean ± SEM. Data was analyzed with Student’s t test or 1-way ANOVA using Tukey’s corrections for multiple comparisons testing. IC50 curves and calculations were performed using GraphPad Prism software v8.01.
Results
HS-5 cells protect FLT3-ITD+ AML cells from quizartinib mostly through STAT5Y694 activation
The FLT3-ITD+ AML cell lines, MOLM-13, MOLM-14 and MV411, were cultured with 10 nM quizartinib: (i) in RM, (ii) in HS-5 CM (see Materials and Methods section for details on medium components), or, (iii) in direct contact (DC) with HS-5 stromal cells. In RM, quizartinib induced apoptosis of MOLM-13, MOLM-14 and MV411 cells, with no effect on K562 cells (negative control) (Fig. 1a). HS-5 CM or DC reduced quizartinib-mediated apoptosis in all three FLT3-ITD+ AML lines, with mostly comparable effects between CM and DC. Cell proliferation experiments demonstrated two- to nine-fold increases in the IC50 of quizartinib in FLT3-ITD+ AML cell lines cultured in CM compared to RM (Fig. 1b). Analogous cell proliferation experiments using primary CD34+ cells from FLT3-ITD+ AML patients produced similar results (Fig. 1c). These data indicate that BM stromal cells protect FLT3-ITD+ AML cells from FLT3 inhibition and that this process involves soluble factors.
Figure 1. Culture in HS-5 CM protects FLT3-ITD+ AML cells from quizartinib.
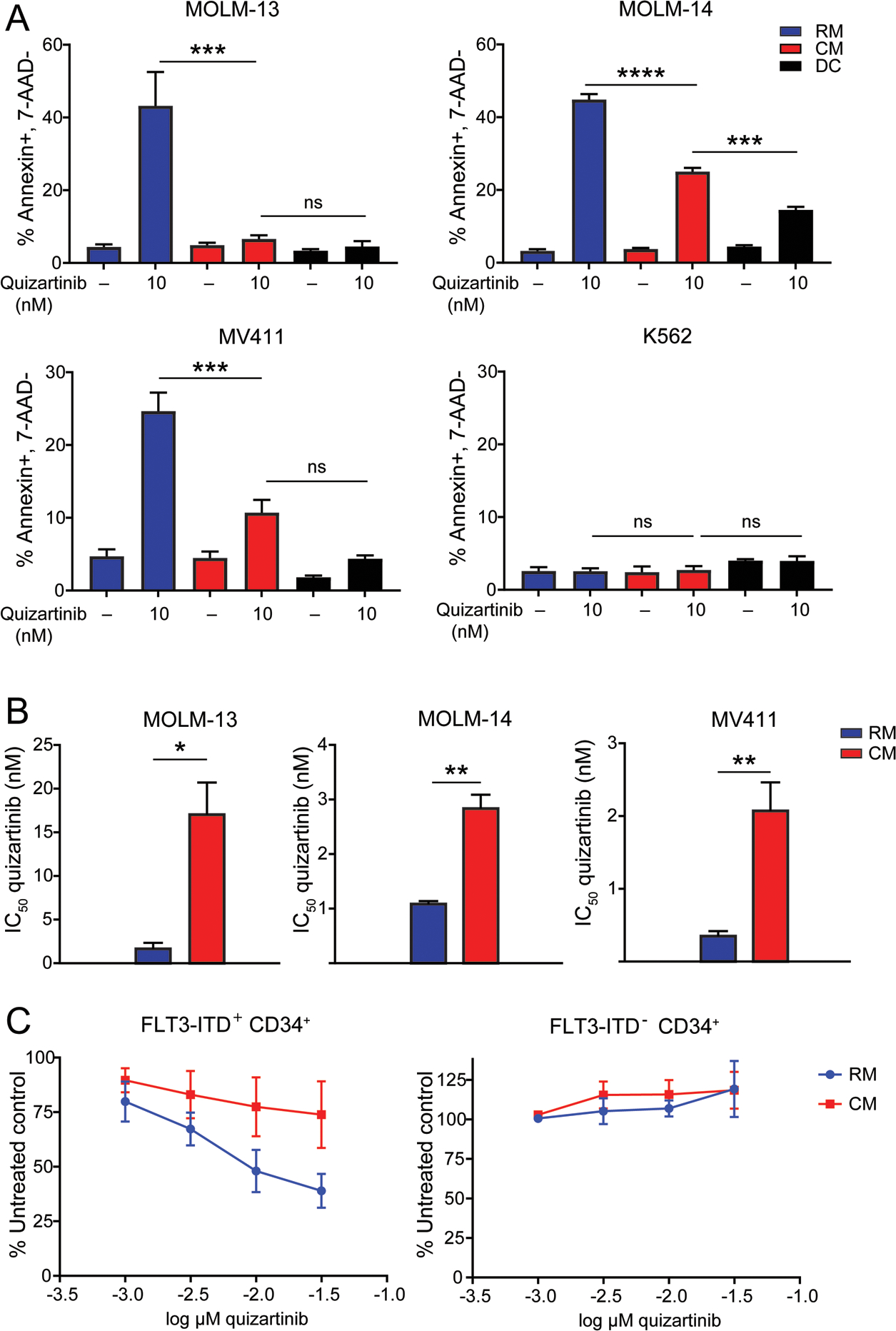
A. Apoptosis at 48 hours following treatment of various FLT3-ITD+ AML cell lines and the K562 cell line (negative control) with quizartinib 10 nM. Treated cells were cultured in medium conditions indicated by the blue, red and black bars. (n=3) B. Bar graphs comparing the IC50 values of quizartinib in FLT3-ITD+ AML cell lines cultured in RM or CM for 72 hours. (n=3) C. Primary CD34+ cells from AML patients with or without FLT3-ITD mutations were cultured in RM or CM and treated with graded concentrations of quizartinib in cell proliferation experiments (n=3)
To assess the role of STAT3 and STAT5 in BM stroma-mediated protection of FLT3-ITD+ AML, K562 (negative control), MOLM-13, MOLM-14, and MV411 cells were grown in RM or HS-5 CM ± 10 nM quizartinib and analyzed by immunoblot (Fig 2a). In FLT3-ITD+ AML cells grown in RM, pSTAT3Y705 expression was consistently low or absent, irrespective of quizartinib concentration; pSTAT5Y694 was readily detected, but abolished with 10 nM quizartinib. In CM, MOLM-13, MOLM-14 and MV411 cells exhibited consistent expression of pSTAT5Y694 that was reduced but not abolished by quizartinib, indicating that STAT5 is activated both by FLT3-ITD and extrinsic factors. Culture of all cell lines in CM resulted in activation of pSTAT3Y705 that was unchanged upon quizartinib treatment. Unlike the FLT3-ITD+ lines, K562 cells demonstrated activation of both STAT3 and STAT5 in RM and CM, neither of which was diminished by quizartinib.
Figure 2. CM activates STAT3 and STAT5 in AML cells and knockdown of STAT5 is associated with impaired cell growth in FLT3-ITD+ AML.
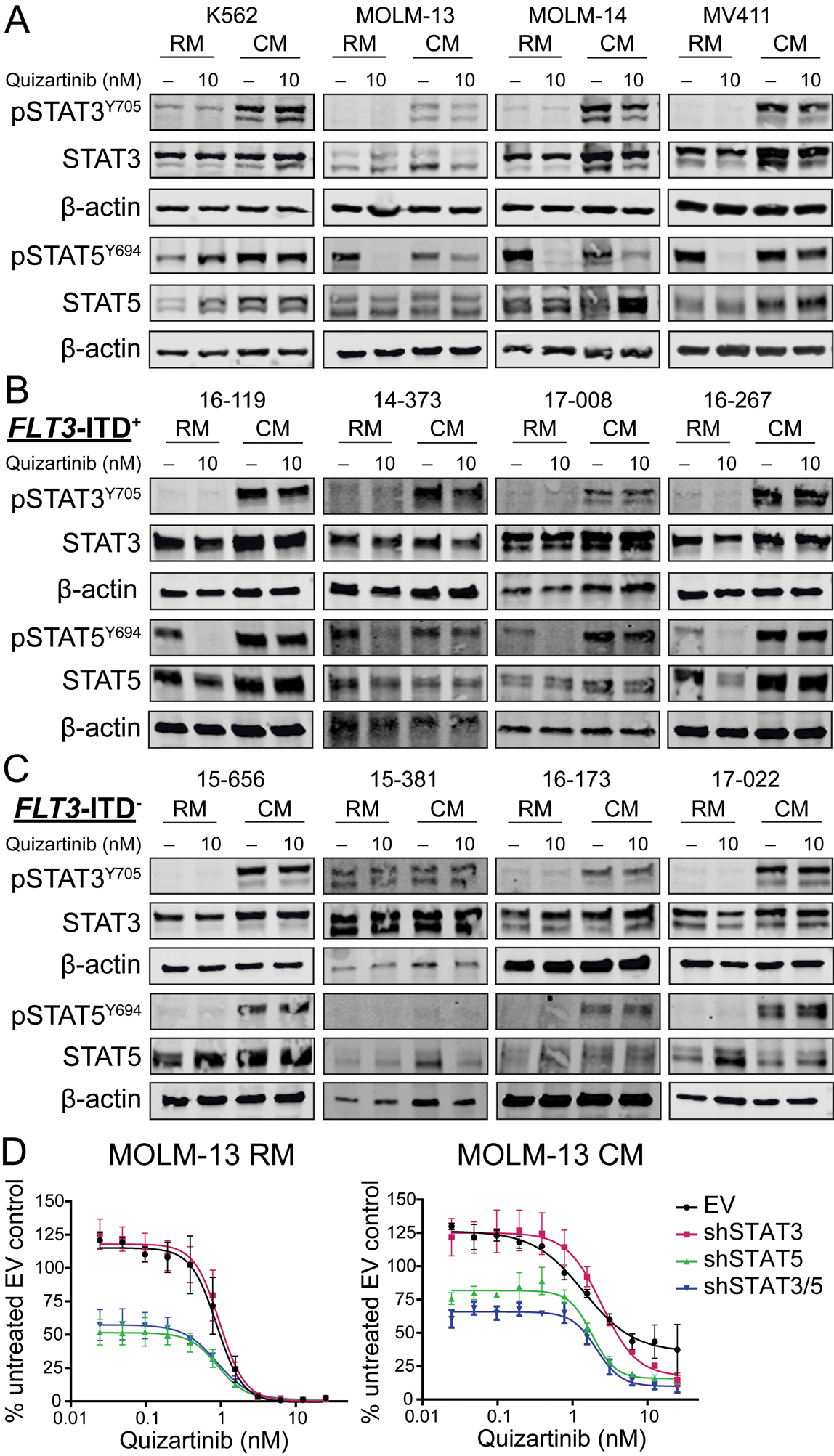
A. Immunoblots demonstrate STAT3 and STAT5 activation in AML cell lines cultured in RM and CM. B and C. Patterns of STAT3 and STAT5 activation in CD34+ cells from patients with FLT3-ITD+ and FLT3-ITD− AML. D. MOLM-13 cells containing an empty vector or shRNA constructs targeting STAT3, STAT5 or both STAT3 and STAT5 were cultured in RM or CM and treated with graded concentrations of quizartinib. Cell proliferation was measured at 72 hours. (n=3)
Next, primary CD34+ cells from four patients with FLT3-ITD+ AML (Supplemental Table 1) were grown in RM and CM ± 10 nM quizartinib and assessed for STAT3 and STAT5 expression by immunoblot (Fig. 2b). The patterns of BM stroma-dependent STAT3 and STAT5 activation observed in primary FLT3-ITD+ AML were similar to those observed in the FLT3-ITD+ cell lines, with prominent quizartinib-independent activation of both STAT3 and STAT5 in CM. Unlike CD34+ cells from the four patients with FLT3-ITD+ AML, primary cells from four AML patients without FLT3-ITD mutations did not express pSTAT5Y694 in RM, while both pSTAT3Y705 and pSTAT5Y694 were expressed in CM (Fig. 2c). One FLT3-ITD− patient sample (15–381) showed activation of STAT3 in RM that was not further increased with CM. As expected quizartinib had no effect on pSTAT3Y705 or pSTAT5Y694 in primary cells from FLT3-ITD− AML patients. Overall, these data suggest CM activates STAT3 and STAT5 in both FLT3-ITD+ and FLT3-ITD− primary AML cells and cell lines in a FLT3 kinase-independent manner.
Extrinsic STAT5 activation drives leukemia cell survival in the setting of FLT3 inhibition
To test whether STAT3 or STAT5 activation in CM is associated with protection from FLT3 inhibition, MOLM-13 cells were infected with short hairpin RNA (shRNA) targeting STAT3, STAT5, or two distinct shRNAs targeting STAT3 and STAT5, respectively. The MOLM-13 cell line was chosen as a model due to the heterozygous presence of the FLT3-ITD mutation (23), recapitulating the genotype most commonly observed in human disease, and because of the concordance between patterns of STAT3 and STAT5 activation in this cell line to those seen with primary FLT3-ITD+ AML samples. Knockdown of STAT3, STAT5 and STAT3/5 was confirmed by immunoblot following 72 hours of culture in RM ± doxycycline (Supplemental Fig. 1). Cells were then cultured in RM or CM ± doxycycline and treated with graded concentrations of quizartinib in cell proliferation assays. In MOLM-13 cells, STAT3 knockdown alone did not significantly affect leukemia cell proliferation in RM or CM, while STAT5 and combined STAT3/5 knockdown significantly impaired cell growth in both medium types (Fig. 2d). In CM, at lower concentrations of quizartinib, combined STAT3/5 knockdown only marginally increased growth inhibition over STAT5 knockdown alone. In total, these results suggest that the majority of protection conferred by the BM microenvironment in FLT3-ITD+ AML results from extrinsic activation of STAT5 and not STAT3.
Dasatinib decreases pSTAT5Y694 in MOLM-13 cells in CM and overcomes BM stroma-mediated resistance in combination with quizartinib
We performed a literature search to identify proximal kinases potentially implicated in STAT5 activation in AML by CM. Candidate kinases included Janus kinase 2 (JAK2), Bruton’s tyrosine kinase (BTK), fibroblast growth factor receptor 1 (FGFR1), spleen tyrosine kinase (SYK) and Src family kinases (SFK) (24–29). Corresponding kinase inhibitors were chosen to interrupt signaling, including: ruxolitinib (JAK2), ibrutinib (BTK), PD1703074 (FGFR1), PRT062607 (SYK) and dasatinib (SFK). Phospho-flow cytometry was used to quantify pSTAT5Y694 in MOLM-13 cells grown for 24 hours in CM in the presence of quizartinib ± inhibitors of candidate kinases. All five inhibitors significantly reduced pSTAT5Y694 when added to quizartinib (Fig. 3a). Increasing the concentration from 0.1 to 1 μM did not significantly decrease the pSTAT5Y694 median fluorescent intensity for any of the inhibitors, and a maximum concentration of 0.1 μM was used for subsequent experiments. To further validate candidate inhibitors for use in combination with quizartinib, we performed cell proliferation experiments in MOLM-13 cells cultured in CM and treated with kinase inhibitors and graded concentrations of quizartinib. The combination of dasatinib and quizartinib proved most effective at decreasing the IC50 of quizartinib in cells grown in CM (Fig. 3b). As expected, dasatinib had no effect on pSTAT5Y694 in MOLM-13 cells grown in RM, irrespective of quizartinib (Fig. 3c). In contrast dasatinib reduced pSTAT5Y694 in the presence of CM and quizartinib (Fig. 3c). SFK activity as assessed by pLynY507 and pSrcY527 was reduced by dasatinib, but not the type of medium, suggesting that SFK activity is re-directed to STAT5 in the presence of CM, but not RM (Fig. 3c and d).
Figure 3. The combination of dasatinib and quizartinib decreases STAT5 activation in CM and overcomes stroma-based resistance to FLT3 TKI.
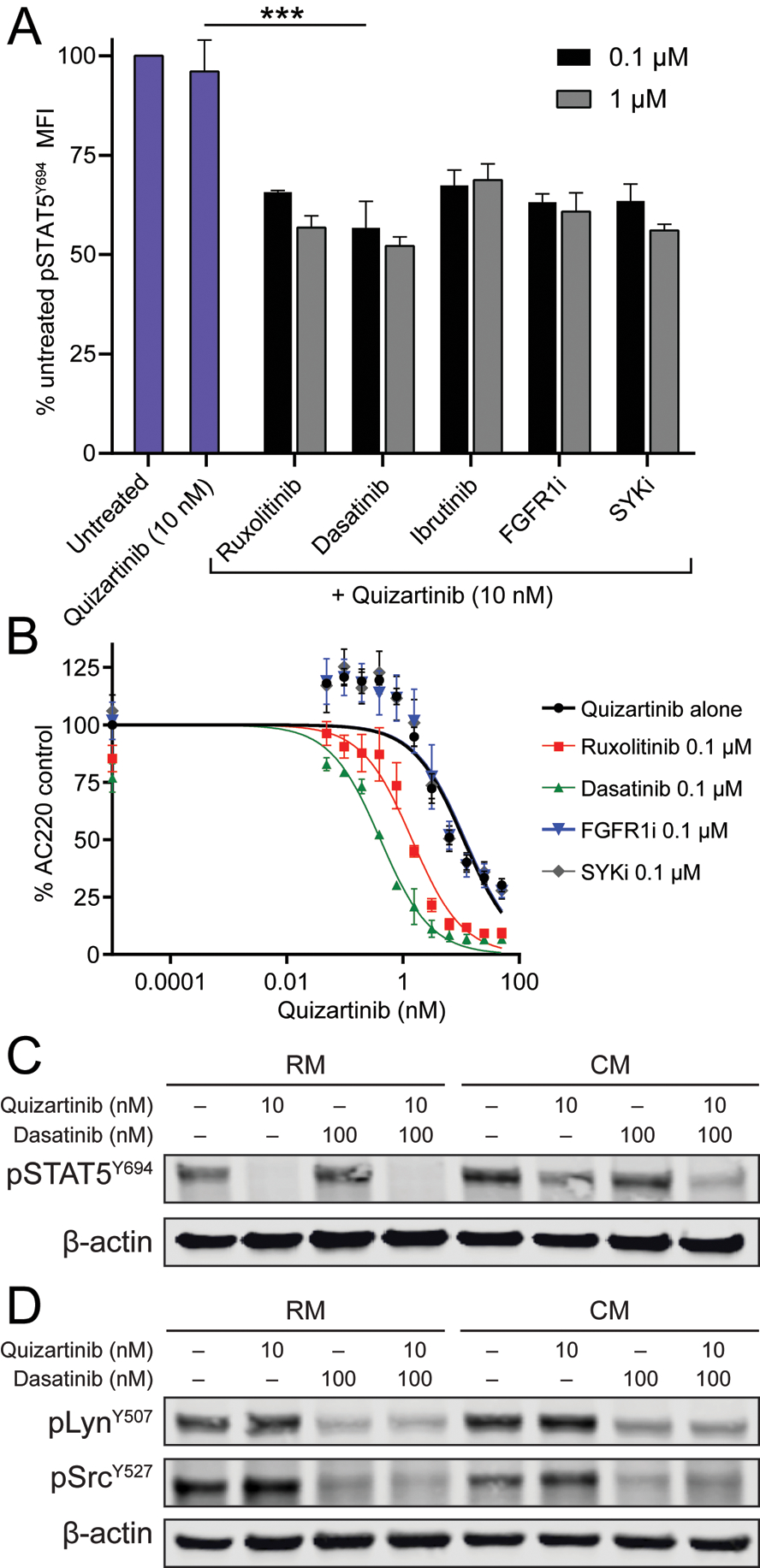
A. MOLM-13 cells cultured in CM were treated with quizartinib 10 nM alone or in combination with fixed doses (0.1 μM) of various inhibitors. Following 4 hours of culture, cells were harvested and STAT5 phosphorylation was assessed by flow cytometry. (n=3) B. Cell proliferation assay in MOLM-13 cells cultured in CM and treated with graded doses of quizartinib alone or in combination with fixed doses (0.1 μM) of inhibitors for 72 hours. (n=3) C and D. STAT5, Lyn and Src activation in MOLM-13 cells following culture in RM or CM and treatment with quizartinib ± dasatinib for 4 hours.
Treatment with dasatinib and quizartinib differentially inhibits glycolysis in FLT3-ITD+ AML cells grown in HS-5 CM in a STAT5-independent manner
It has been reported that FLT3-ITD+ AML cells harbor a highly glycolytic phenotype that can be partially suppressed by FLT3 TKIs (30). We hypothesized that culturing FLT3-ITD+ cells in CM may protect against TKI-mediated suppression of glycolysis. As expected glycolysis and glycolytic capacity were greatly reduced in the presence of quizartinib cultured in RM. However glycolysis and glycolytic capacity were partially restored in the presence of CM (Fig. 4a). We next tested whether addition of dasatinib would abrogate the CM-mediated increase in glucose utilization. Dasatinib alone reduced glycolysis and glycolytic capacity irrespective of medium type. Notably, the combination of quizartinib and dasatinib decreased the glycolytic activity of cells grown in CM to that observed in cells grown in RM and treated with quizartinib alone (Fig. 4a). The relative contribution of dasatinib to the reduction in glycolytic activity observed with combination treatment versus quizartinib alone was larger in cells grown in CM than RM, suggesting that soluble factors contained in CM activate signaling pathways inhibited by dasatinib.
Figure 4. The combination of dasatinib and quizartinib suppresses stroma-enhanced glycolysis in a STAT5-independent manner in FLT3-ITD+ AML.
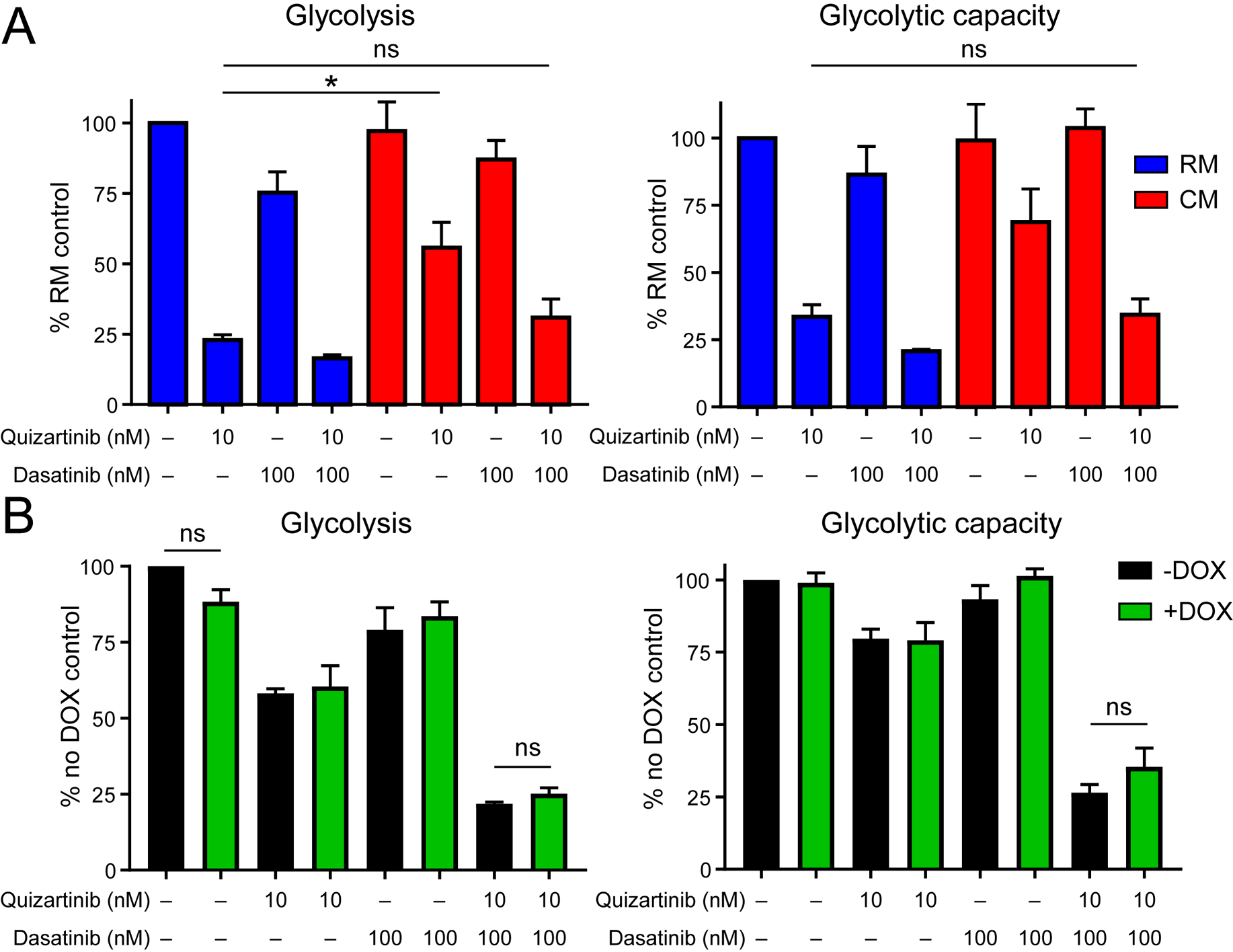
A. Glycolysis and glycolytic capacity were measured in MOLM-13 cells cultured in RM or CM and treated with quizartinib ± dasatinib for 24 hours. (n=3) B. Glycolysis and glycolytic capacity were measured in MOLM-13 cells expressing a doxycycline-inducible shRNA against STAT5. Cells were cultured in CM ± doxycycline (DOX) and treated with quizartinib and/or dasatinib for 24 hours. (n=3)
We hypothesized that dasatinib may preferentially decrease glycolytic activity in FLT3-ITD+ cells cultured in CM through inhibition of FLT3-independent STAT5 activation. To test this, we measured glycolytic activity in MOLM-13 cells transduced with a lentiviral doxycycline-inducible shSTAT5 construct. These cells were cultured for 24 hours in CM ± doxycycline (knockdown confirmed; Supplemental Fig. 2) and treated with quizartinib and/or dasatinib. Results from these experiments indicate that knockdown of STAT5 does not affect glycolysis in AML cells, suggesting that the effect of dasatinib on inhibition of glycolysis is STAT5-independent (Fig. 4b). In addition, these results indicate that STAT5 knockdown alone cannot recapitulate the inhibitory effect of quizartinib on glycolytic activity.
Discussion
After decades of minimal progress, the therapeutic armamentarium available to treat FLT3-ITD+ AML has been enriched by midostaurin in the frontline setting and gilteritinib and quizartinib in R/R disease, both of which have been reported to improve survival (10, 13, 14). Second-generation FLT3 TKIs demonstrate improved on-target inhibition compared to midostaurin and have proven more efficacious as salvage therapies for patients with relapsed FLT3-ITD+ disease, who tend to demonstrate higher mutant allele burdens than at diagnosis (16, 17). Gilteritinib is currently approved in the R/R setting in Europe and the United States, while quizartinib is approved only in Japan, with the FDA citing concerns over clinical benefit and equipoise following review of QUANTUM-R trial results. With several clinical studies ongoing (31), it remains possible that quizartinib may be FDA-approved for FLT3-ITD+ AML in combination with other agents in the not-distant future. As the use of 2nd generation FLT3 TKIs becomes standard, combination therapies to improve the duration and depth of response to FLT3 inhibition will be required to achieve long-term survival in transplant-ineligible patients. In this study, we show that extrinsic cues from BM stromal cells promote leukemia cell survival in the setting of selective FLT3 inhibition and that blocking such signals with dasatinib can overcome extrinsic resistance and markedly improve AML cell killing.
We demonstrate that in both cell lines and primary samples, HS-5 CM consistently protects FLT3-ITD+ AML cells from FLT3 inhibition. Extrinsic activation of STAT5 by CM appears to be the primary mediator of leukemia cell survival and resistance to FLT3 inhibition, in agreement with previous reports in AML (19, 32, 33), but in contrast to BCR-ABL1 inhibition in CML, in which selective STAT3 activation by CM promotes TKI resistance (34, 35). The reason for this discrepancy is unknown, as both FLT3-ITD and BCR-ABL1 drive constitutive STAT5 activation. One plausible explanation is differential expression of cytokine receptors. Medium supplemented with exogenous granulocyte-macrophage colony stimulating factor (GM-CSF) or interleukin-3 (IL-3) has been shown to be sufficient to rescue FLT3-ITD+ AML cells from FLT3 inhibition with crenolanib via activation of the JAK2/STAT5 axis (32), while elevated BM interleukin-6 (IL-6) concentrations have been noted in CML patients and persistent STAT3 activation by IL-6 has been demonstrated to be JAK1-dependent in CML cell lines and patient samples (22, 36, 37). The reported decrease in extrinsic STAT5 phosphorylation seen with the combination of ruxolitinib and quizartinib (32) was similar to that observed with dasatinib and quizartinib in our study, suggesting some degree of activation of the JAK2/STAT5 signal transduction pathway by CM. Indeed, inhibitors of multiple kinases potentially upstream of STAT5 reduced pSTAT5Y694 in FLT3-ITD expressing AML cells, suggesting that full extrinsic activation of STAT5 derives from the activation of multiple upstream pathways, and implying that different upstream kinases may target separate STAT5 pools. Dasatinib-mediated inhibition of multiple RTKs activated by various soluble factors in CM, such as stem cell factor (SCF), platelet derived growth factor (PDGF) and FGF, may explain why combining quizartinib and dasatinib has the strongest effect on pSTAT5Y694 in our study (22, 38, 39). Additionally dasatinib may target other signaling pathways relevant to protective microenvironment effects, such as the Gas6/Axl axis that has been implicated as an effective stroma-based escape route from FLT3 inhibition in FLT3-ITD+ AML (33, 40). Gas6, the ligand for the RTK Axl, is secreted by BM stromal cells, including HS-5 cells (41). Axl bears structural resemblance to PDGFR and FGFR, and can heterodimerize with other RTKs, resulting in downstream SFK and PI3K/Akt/mTOR pathway activation (42–46). While bosutinib, another 2nd generation BCR-ABL1 TKI, has been shown to inhibit both Axl and SFKs (42), there is no evidence that Axl is a direct target of dasatinib (47, 48). This suggests that dasatinib impacts the Gas/Axl pathway primarily via distal SFK inhibition, which may also explain the observation that dasatinib abolishes Gas6/Axl mediated SFK activation in renal cell carcinoma (49). In the same study, dasatinib was also observed to inhibit SFK-dependent activation of MET, highlighting the importance of SFKs as critical intermediaries for cross-talk between divergent signal transduction pathways.
SFKs (in particular Src) are well-known to potentiate the Warburg effect and tumor cell dependence on glycolysis via phosphorylation of hexokinases HK1 and HK2 and pyruvate dehydrogenase (50–52). Similar to our data on FLT3-mutated AML, which is known to be highly dependent on glycolysis (30), inhibition of SFKs results in a decrease of glycolytic activity across multiple cancer types (50–54). Axl activation has been reported to stabilize insulin receptor substrate 1 (IRS-1) through phosphorylation of tensin2 (TNS2), resulting in enhanced expression of GLUT4 and pyruvate dehydrogenase kinase 1 (PDK1), enzymes known to be essential for glucose uptake and aerobic glycolysis (55). Thus it is conceivable that dasatinib blocks distal effector pathways of the Gas6/Axl/TNS2 signaling axis via SFK inhibition to inhibit CM-induced glycolysis. It is important to note that while upregulation of Axl occurs in response to extrinsic STAT5 activation, this is mediated through secretion of stromal cytokines other than Gas6, in keeping with the observation from our study that dasatinib-related effects on glycolytic inhibition are STAT5 independent (33).
Taken together our findings suggest that dasatinib overcomes stroma-based resistance to FLT3 inhibition not only through inhibition of extrinsic STAT5 activation but also through inhibition of glycolytic activity, ultimately re-sensitizing FLT3-ITD+ leukemia cells to FLT3 inhibition by quizartinib. While the BM niche has been observed to influence leukemia cell metabolism through hypoxia-responsive elements and mitochondrial transfer, this is the first report demonstrating that soluble factors secreted by the BM microenvironment directly enhance glycolytic activity in FLT3-ITD+ leukemia cells and mitigate previously described FLT3 TKI effects on glycolysis (30, 56, 57). These data provide a rationale for the combined use of quizartinib and dasatinib in FLT3-ITD mutated AML and support further investigation into the mechanisms underlying the efficacy of this combination. Chemotherapy-free regimens consisting of multiple targeted agents, such as ibrutinib and venetoclax in chronic lymphocytic leukemia, and asciminib and dasatinib in BCR-ABL1+ leukemias, have proven feasible in clinical trials and are likely to dominate the clinical landscape in due time (58–60). Toxicities shared by both quizartinib and dasatinib, including QTc prolongation and myelosuppression, are of potential clinical concern with combination therapy, but could be addressed with dose reduction of one or both drugs. Future short-term studies should investigate whether combining selective SFK inhibitors with FLT3 TKIs or treatment with gilteritinib alone (dual activity against FLT3/Axl) can recapitulate the metabolic effects observed here with dasatinib and quizartinib.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute (NCI) at the National Institutes of health (NIH) through grant R21CA20593601 (M.W.D.), the V Foundation for Cancer Research Translational Research Grant T2017-008 (M.W.D.), NIH grant R01CA178397 (M.W.D. and T.O), and the American Society of Hematology Research Training Award for Fellows (A.B.P). D.Y is supported by the International Award from the Lady Tata Memorial Trust. A.M.E is supported by NIH grant 1K22CA216008. We thank the Metabolic Phenotyping Core Facility at the University of Utah for assistance with Seahorse experiments. This work was funded in part by the University of Utah Flow Cytometry Core Facility and the NCI through award 5P30CA042014-24 awarded to the Huntsman Cancer Institute and the National Center for Research Resources of the NIH award 1S10RR026802-01.
Footnotes
Competing Interests
Michael W. Deininger reports research funding from and is a paid advisory board member and/or consultant for the following companies: Pfizer Inc, TRM Blueprint, Fusion Pharma, Takeda, Ascentage Pharma, Humana, Adelphi, Medscape, Novartis, Incyte and Sangamo. Inc.
References
- 1.Small D, Levenstein M, Kim E, Carow C, Amin S, Rockwell P, et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci U S A. 1994;91(2):459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beaudin AE, Boyer SW, Forsberg EC. Flk2/Flt3 promotes both myeloid and lymphoid development by expanding non-self-renewing multipotent hematopoietic progenitor cells. Exp Hematol. 2014;42(3):218–29 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dehmel U, Quentmeier H, Drexler HG. Effects of FLT3 ligand on human leukemia cells. II. Agonistic and antagonistic effects of other cytokines. Leukemia. 1996;10(2):271–8. [PubMed] [Google Scholar]
- 4.Hannum C, Culpepper J, Campbell D, Mcclanahan T, Zurawski S, Bazan JF, et al. Ligand for Flt3 Flk2 Receptor Tyrosine Kinase Regulates Growth of Hematopoietic Stem-Cells and Is Encoded by Variant Rnas. Nature. 1994;368(6472):643–8. [DOI] [PubMed] [Google Scholar]
- 5.Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia. 1996;10(4):588–99. [PubMed] [Google Scholar]
- 6.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93(9):3074–80. [PubMed] [Google Scholar]
- 8.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21(16):2555–63. [DOI] [PubMed] [Google Scholar]
- 9.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110(1):370–4. [DOI] [PubMed] [Google Scholar]
- 10.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Short NJ, Kantarjian H, Ravandi F, Daver N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther Adv Hematol. 2019;10:2040620719827310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–97. [DOI] [PubMed] [Google Scholar]
- 14.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med. 2019;381(18):1728–40. [DOI] [PubMed] [Google Scholar]
- 15.Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, Garcia-Manero G, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer. 2014;120(14):2142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115(7):1425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020. [DOI] [PubMed] [Google Scholar]
- 18.Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96(1):62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weisberg E, Liu Q, Nelson E, Kung AL, Christie AL, Bronson R, et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia. 2012;26(10):2233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger A, Hoelbl-Kovacic A, Bourgeais J, Hoefling L, Warsch W, Grundschober E, et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia. 2014;28(3):629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eiring AM, Kraft IL, Page BD, O’Hare T, Gunning PT, Deininger MW. STAT3 as a mediator of BCR-ABL1-independent resistance in chronic myeloid leukemia. Leukemia supplements. 2014;3(Suppl 1):S5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Traer E, MacKenzie R, Snead J, Agarwal A, Eiring AM, O’Hare T, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26(5):1140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17(1):120–4. [DOI] [PubMed] [Google Scholar]
- 24.Wu H, Hu C, Wang A, Weisberg EL, Wang W, Chen C, et al. Ibrutinib selectively targets FLT3-ITD in mutant FLT3-positive AML. Leukemia. 2016;30(3):754–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Traer E, Martinez J, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, et al. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016;76(22):6471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weisberg E, Liu Q, Zhang X, Nelson E, Sattler M, Liu F, et al. Selective Akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS One. 2013;8(2):e56473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weisberg EL, Puissant A, Stone R, Sattler M, Buhrlage SJ, Yang J, et al. Characterization of midostaurin as a dual inhibitor of FLT3 and SYK and potentiation of FLT3 inhibition against FLT3-ITD-driven leukemia harboring activated SYK kinase. Oncotarget. 2017;8(32):52026–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook AM, Li L, Ho Y, Lin A, Li L, Stein A, et al. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123(18):2826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang Y, Zhong L, Lin M, Zhou X, Jing H, Ying M, et al. MEK/ERK dependent activation of STAT1 mediates dasatinib-induced differentiation of acute myeloid leukemia. PLoS One. 2013;8(6):e66915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 2018;131(15):1639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fletcher L, Joshi S, Traer E. Profile of Quizartinib for the Treatment of Adult Patients with Relapsed/Refractory FLT3-ITD-Positive Acute Myeloid Leukemia: Evidence to Date. Cancer Management and Research. 2020;2020(12):151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sung PJ, Sugita M, Koblish H, Perl AE, Carroll M. Hematopoietic cytokines mediate resistance to targeted therapy in FLT3-ITD acute myeloid leukemia. Blood Adv. 2019;3(7):1061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dumas PY, Naudin C, Martin-Lanneree S, Izac B, Casetti L, Mansier O, et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica. 2019;104(10):2017–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7(10):3169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eiring AM, Page BDG, Kraft IL, Mason CC, Vellore NA, Resetca D, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2015;29(3):586–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuepper MK, Butow M, Herrmann O, Ziemons J, Chatain N, Maurer A, et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia. 2019;33(8):1964–77. [DOI] [PubMed] [Google Scholar]
- 37.Pricola KL, Kuhn NZ, Haleem-Smith H, Song Y, Tuan RS. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J Cell Biochem. 2009;108(3):577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manshouri T, Estrov Z, Quintas-Cardama A, Burger J, Zhang Y, Livun A, et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011;71(11):3831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordon PM, Dias S, Williams DA. Cytokines secreted by bone marrow stromal cells protect c-KIT mutant AML cells from c-KIT inhibitor-induced apoptosis. Leukemia. 2014;28(11):2257–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furukawa M, Ohkawara H, Ogawa K, Ikeda K, Ueda K, Shichishima-Nakamura A, et al. Autocrine and Paracrine Interactions between Multiple Myeloma Cells and Bone Marrow Stromal Cells by Growth Arrest-specific Gene 6 Cross-talk with Interleukin-6. J Biol Chem. 2017;292(10):4280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh AK, Secreto C, Boysen J, Sassoon T, Shanafelt TD, Mukhopadhyay D, et al. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011;117(6):1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vouri M, Croucher DR, Kennedy SP, An Q, Pilkington GJ, Hafizi S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5(10):e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goruppi S, Ruaro E, Varnum B, Schneider C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997;17(8):4442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116(4):415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51. [DOI] [PubMed] [Google Scholar]
- 48.Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, et al. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18(2):110–22. [DOI] [PubMed] [Google Scholar]
- 49.Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin Y, Cai Q, Shenoy AK, Lim S, Zhang Y, Charles S, et al. Src drives the Warburg effect and therapy resistance by inactivating pyruvate dehydrogenase through tyrosine-289 phosphorylation. Oncotarget. 2016;7(18):25113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Wang S, Jiang B, Huang L, Ji Z, Li X, et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat Commun. 2017;8:13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeNicola GM, Cantley LC. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol Cell. 2015;60(4):514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin L, Chun J, Pan C, Alesi GN, Li D, Magliocca KR, et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene. 2017;36(27):3797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee M, Cui X, Li Z, Yu H, Cai L, Jia X, et al. Na/K-ATPase Y260 Phosphorylation-mediated Src Regulation in Control of Aerobic Glycolysis and Tumor Growth. Sci Rep. 2018;8(1):12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng LC, Chen YL, Cheng AN, Lee AY, Cho CY, Huang JS, et al. AXL phosphorylates and up-regulates TNS2 and its implications in IRS-1-associated metabolism in cancer cells. J Biomed Sci. 2018;25(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. 2016;128(2):253–64. [DOI] [PubMed] [Google Scholar]
- 58.Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. New Engl J Med. 2019;380(22):2095–103. [DOI] [PubMed] [Google Scholar]
- 59.Luskin M, Murakami MA, Stevenson KE, Wadleigh M, McMasters M, Winter P, et al. A Phase I Study of Asciminib (ABL001) in Combination with Dasatinib and Prednisone for Untreated BCR-ABL1-Positive ALL in Older Adults. Blood. 2019;134(Supplement_1):3879-. [Google Scholar]
- 60.DeAngelo DJ, Mauro MJ, Kim D-W, Cortes J, Réa D, Hughes TP, et al. Combination of Asciminib+Nilotinib or Asciminib+Dasatinib in Previously Treated Chronic Myeloid Leukemia (CML) Patients: Phase 1 Study Results. Clinical Lymphoma, Myeloma and Leukemia. 2019;19:S290–S1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.