

Abstract
The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase (LCAD) is expressed at high levels in human alveolar type II (ATII) cells in the lung. A common polymorphism causing an amino acid substitution (K333Q) was previously linked to a loss of LCAD antigen in the lung tissue in sudden infant death syndrome. However, the effects of the polymorphism on LCAD function has not been tested. The present work evaluated recombinant LCAD K333Q. Compared to wild-type LCAD protein, LCAD K333Q exhibited significantly reduced enzymatic activity. Molecular modeling suggested that K333 is within interacting distance of the essential FAD cofactor, and the K333Q protein showed a propensity to lose FAD. Exogenous FAD only partially rescued the activity of LCAD K333Q. LCAD K333Q protein was less stable than wild-type when incubated at physiological temperatures, likely explaining the observation of dramatically reduced LCAD antigen in primary ATII cells isolated from individuals homozygous for K333Q. Despite the effect of K333Q on activity, stability, and antigen levels, the frequency of the polymorphism was not increased among infants and children with lung disease.
Keywords: long-chain acyl-CoA dehydrogenase, respiratory distress, type II alveolar pneumocytes, mitochondria, fatty acid oxidation
Introduction
Long-chain acyl-CoA dehydrogenase (LCAD), encoded by the ACADL gene, is one of four mitochondrial acyl-CoA dehydrogenases (ACADs) that catalyze the first enzymatic step in fatty acid β-oxidation (FAO). The other three ACADs, short-chain acyl-CoA dehydrogenase (SCAD), medium-chain acyl-CoA dehydrogenase (MCAD), and very long-chain acyl-CoA dehydrogenase (VLCAD), are all widely expressed in human tissues that rely upon FAO for energy such as liver, heart, and muscle. Mutations in the genes encoding MCAD and VLCAD are associated with inborn errors of metabolism (1, 2), while most patients with SCAD mutations remain asymptomatic (3). In contrast, LCAD is not widely expressed in human tissues, and no LCAD-deficient patients have been reported (4, 5). We previously observed abundant LCAD expression in human liver and in alveolar type II (ATII) cells, a highly specialized cell type of the lung (6). ATII cells conduct the energy-intensive process of pulmonary surfactant synthesis and secretion, and thus are rich in mitochondria and are highly metabolically active. LCAD knockout mice have altered breathing mechanics, altered surfactant composition, and higher mortality when infected with influenza virus (6–8). LCAD also stands out in the ACAD enzyme family for having a high propensity to leak electrons to oxygen to form H2O2 (9), and also for being the only ACAD linked to multiple cancers (10–13). LCAD expression is dramatically reduced in breast and liver cancers (10, 12), and we observed no detectable LCAD antigen in lung cancer cell lines (6). For liver cancer, re-expression of LCAD can greatly slow proliferation and tumor growth (12).
While there are no known disease-causing mutations in the LCAD gene, there is a common polymorphism (rs2286963) in the coding region that changes the lysine residue at position 333 to a glutamine (LCAD K333Q). The frequency of this allele ranges from a low of 0.05 in some African subpopulations up to a high of 0.35 in Americans of European descent. While the commonality of this variant would suggest that it is benign, it has been associated with increased serum lipids (14, 15). C9-carnitine, most likely derived from the LCAD-specific C9 substrate 2,6-dimethylheptanoic acid (16), was elevated in humans with LCAD K333Q. This would suggest that K333Q may decrease the function of the LCAD enzyme.
Previously, we immunoblotted for LCAD antigen in post-mortem lung tissue collected from six infants who had died unexpectedly of unknown causes (6). Two of the six had no detectable LCAD antigen. Both were homozygous for K333Q, while those with detectable LCAD antigen were not homozygous for this allele. While this was a very small cohort of infants and did not statistically link K333Q to sudden death, the observation raised questions about the functional significance of K333Q. In the present work we introduced K333Q into recombinant human LCAD and biochemically evaluated the variant enzyme. We further genotyped for the polymorphism in several cohorts of children with lung disease. We conclude that while K333Q destabilizes the LCAD protein and reduces its activity, the polymorphism is not enriched among neonates with respiratory distress syndrome (RDS) or children with pneumonia.
Materials & Methods
Recombinant LCAD expression—
The QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) was used to introduce the K333Q variant into a His-tagged human LCAD bacterial expression construct that we previously described (9). The wild-type and K333Q LCAD proteins were isolated from bacterial lysates using fast protein liquid chromatography as previously described (9).
Molecular modeling—
Molecular modeling was done using the structure of the related protein isovaleryl-CoA dehydrogenase (IVD) using the Yasara software (version 13.9.8, Yasara Biosciences, Vienna, Austria) and the publicly available PDB file 1IVH.
LCAD enzyme activity assays and kinetics—
The acyl-CoA dehydrogenase specific activity of the LCAD proteins was assayed using the electron transferring flavoprotein (ETF) fluorescence reduction assay (17). All acyl-CoA substrates were from Sigma and used at 25 μM concentration, except for the kinetic assay which used C16-CoA at concentrations ranging from 0.5 to 40 μM. In some instances, the recombinant proteins were incubated with 100 μM free FAD at 37°C for 20 minutes and dialyzed to remove unbound FAD prior to activity testing. In these experiments the control proteins were incubated and dialyzed in the same way, but water was added in place of the FAD. For the oxidase activity assay (9), 1 μg of protein was added to 50 mM phosphate buffer pH 7.0 containing HRP (1 U/ml) and Amplex UltraRed (50 μM) at 30°C. Reactions were started by addition of acyl-CoA substrate and fluorescence was monitored for 30 minutes.
LCAD FAD content and stability assays—
To determine FAD content, 1 mg/ml solutions of purified LCAD proteins in 10 mM Tris-HCl pH 8.0 were subjected to absorbance scanning from 200 to 600 nm in a Jasco V-650 spectrophotometer. The FAD peak at 443 nm was divided by the total protein absorbance at 280 nm to provide an indicator of FAD content (18). In the stability assay, the proteins were heated at 37°C for 12 hrs prior to absorbance scanning. Protein unfolding was followed using fluorescence. Aliquots of the LCAD proteins (1 mg/ml in 10 mM Tris-HCl pH 8.0) were incubated in a heating block for 20 minutes at 22, 30, 37, 40, 45, 50, or 60°C and then subjected to tryptophan fluorescence measurements (Ex 335 nm, Em 330 nm) in a Jasco Fp-6300 fluorometer, with the cuvette holder attached to a circulating water bath and pre-warmed to the same temperature as the protein solutions.
Human ATII cells.
Human lungs were obtained from organ donors whose lungs were not suitable for transplantation and donated for medical research through the International Institute for the Advancement of Medicine and National Disease Research Interchange. Donors had no chronic lung diseases and reasonable lung function with a PaO2/FIO2 ratio of >225, X-ray and limited time on a ventilator. ATII cells were isolated as we previously reported (19). The Committee for the Protection of Human Subjects at National Jewish Health and Temple University approved this research.
Immunoblotting of human ATII lysates—
A bank of genomic DNA collected from adult human lungs deemed unsuitable for transplantation, for which ATII cell lysates were also available, was screened for the K333Q polymorphism using a Taqman SNP genotyping assay for LCAD K333Q (Applied Biosystems). Five individuals homozygous for the minor Q allele at LCAD residue 333 and five controls that were homozygous for the major K allele were selected for LCAD immunoblotting. Twenty-five μg of ATII cell lysates were subjected to SDS-PAGE and immunoblotting using a rabbit antibody (1:1000 dilution) raised against the whole rat LCAD protein (gift of Dr. Jerry Vockley). As a loading control, the same blot was probed with anti-actin antibody (Proteintech #HRP60008) at 1:5000 dilution. Image J software was used to quantify the LCAD:actin ratio for each sample.
K333Q frequency in human cohorts—
A Taqman SNP genotyping assay (Applied Biosystems) for LCAD K333Q was used in secondary analyses of genomic DNA samples biobanked during previous studies of five pediatric cohorts: 1) 60 full-term or near full-term Caucasian neonates with diffuse lung disease of undetermined etiology; 2) 52 pre-term Caucasian infants (28–355/7 weeks) with severe respiratory distress syndrome (RDS); 3) 30 full-term Caucasian infants with neonatal respiratory failure and confirmed mutations in the adenosine triphosphate-binding cassette transporter A3 (ABCA3) gene; 4) 474 African-American children presenting to the emergency department or admitted to the hospital with community-acquired pneumonia; and 5) 301 Caucasian children with presenting to the emergency department or admitted to the hospital with community-acquired pneumonia. For the respiratory distress syndrome cohorts (cohorts 1–3), DNA was collected under Institutional Review Board (IRB)-approved studies at Johns Hopkins University. Pneumonia patient DNA (cohorts 4 and 5) was collected under IRB approved studies at Le Bonheur Children’s Hospital, Children’s Memorial Hospital, and Children’s Hospital of Wisconsin. Pneumonia was defined as described previously (20).
Data analysis and statistics—
All statistical analysis, including curve-fitting of enzyme kinetic data and subsequent determination of Vmax and Km, was done using GraphPad Prism 8.0. For the K333Q genotyping assay, Hardy-Weinberg equilibrium was examined in each cohort as a quality control metric (21). Genotyping results showed a modest deviation from Hardy-Weinberg equilibrium (HWE) in the Caucasian pneumonia group (p=0.04), but not other groups. Because the most common cause of HWE departure is genotyping errors, we further validated the genotyping assay by selecting 10% of the DNA samples at random for re-genotyping; 100% concordance was observed. The Caucasian cohort was found to demonstrate loss of heterozygosity, a type of HWE-departure that is ascribed to natural variations in population substructure (22). Association of the K333Q allele with disease was calculated by comparing allele frequency between the disease cohorts and race-matched controls (gnomAD database) using the Fisher exact test with p<0.05 considered statistically significant.
Results
The K333Q polymorphism reduces LCAD enzymatic activity.
Residue K333 is conserved across species and is just downstream of two other lysines (K318 and K322, see Fig 1A) that were previously implicated in regulating LCAD enzymatic activity (18). Because mutations at K318/K322 reduce LCAD activity, we hypothesized that the common polymorphism K333Q would also reduce activity. To test this, we expressed LCAD K333Q as a recombinant protein and purified it to homogeneity (Fig 1B). The wild-type LCAD and LCAD K333Q proteins were then tested for enzymatic activity using a saturating amount (25 μM) of long-chain acyl-CoA substrates ranging from 12 to 18 carbons (C12 to C18). The ACAD activity of LCAD K333Q was significantly lower than wild-type LCAD across all four substrates tested (Fig 1C). We previously showed that LCAD also has low-level activity as an acyl-CoA oxidase, leaking electrons directly to oxygen to produce H2O2 (9). To determine whether the K333Q polymorphism altered the oxidase activity of LCAD, the two recombinant proteins were also compared using an oxidase enzyme activity assay. C18-CoA was used as substrate as this chain length produces the highest H2O2 production from wild-type LCAD (9). As with ACAD activity, the presence of the K333Q substitution significantly reduced oxidase activity (Fig 1D).
Figure 1. LCAD K333Q polymorphism reduces enzymatic activity.
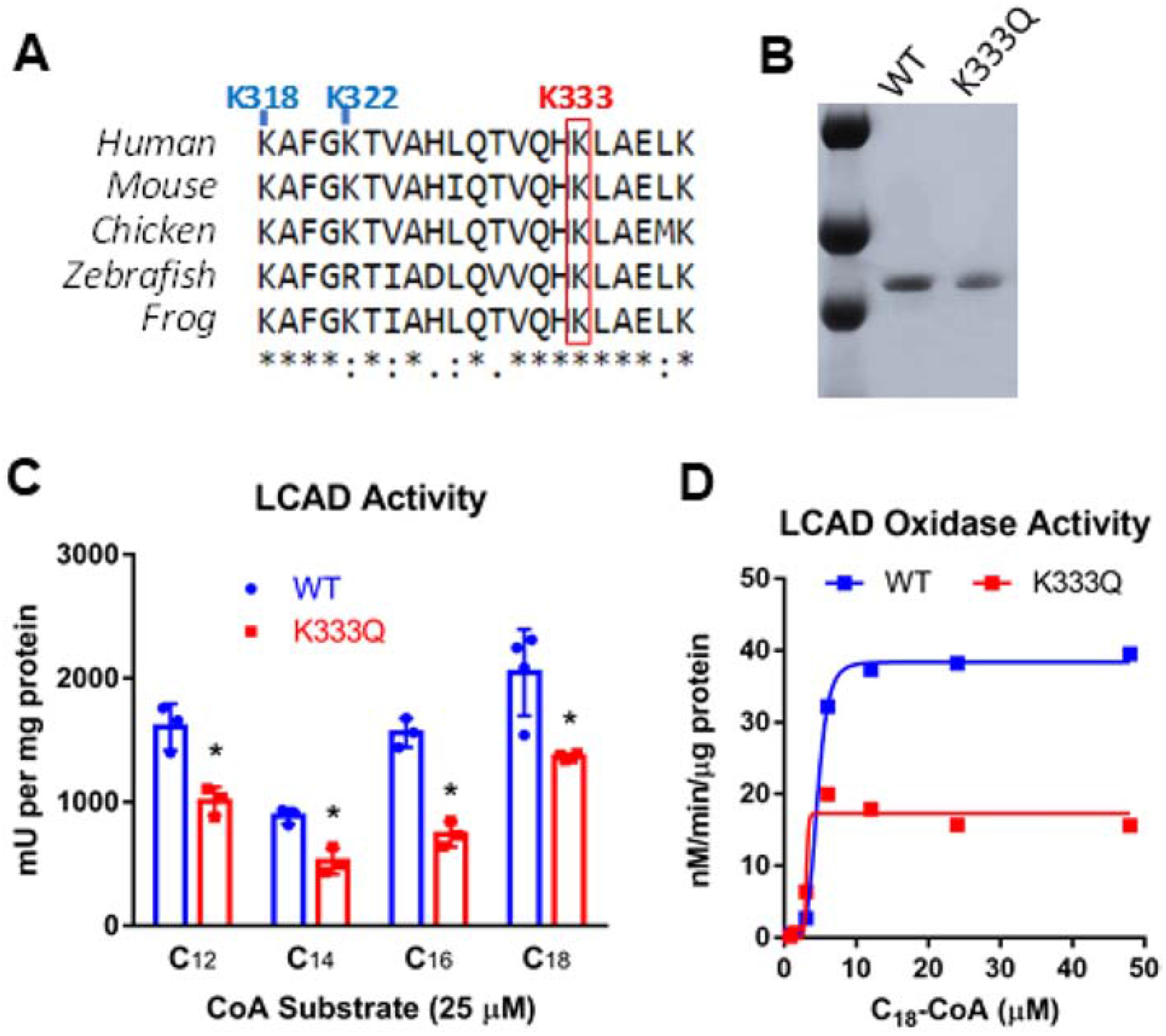
A) An amino acid sequence alignment shows that K333 is conserved across species and is just downstream of two other lysine residues known to impact LCAD function, K318 and K322. B) SDS-PAGE and Coomassie blue staining of 1 μg of protein demonstrate the purity of recombinant LCAD and LCAD K333Q preparations. C) Acyl-CoA dehydrogenase activity of recombinant LCAD (WT) and K3333Q proteins with four different acyl-CoA substrates. Assays were conducted in triplicate. D) Acyl-CoA oxidase activity kinetics of recombinant LCAD (WT) and K3333Q proteins with stearoyl-CoA as substrate (C18). Each data point represents duplicate assays. *P<0.01.
The K333Q polymorphism alters the LCAD active site.
The lysine residues K318/K322, immediately upstream of K333, are involved in coordinating the FAD in the active site (18). While the crystal structure of LCAD has not been solved, the structure of isovaleryl-CoA dehydrogenase (IVD), a highly homologous ACAD family member, is available. Both LCAD and IVD are homotetrameric enzymes with an FAD cofactor non-covalently bound to each subunit. In IVD the lysine homologous to K333 is K325, which in the IVD structure is seen to lie within interacting distance of the FAD cofactor (Fig 2A). To determine whether the K333Q substitution affects LCAD FAD binding, we first measured the FAD content of the purified recombinant protein. Absorbance scanning of LCAD produces a distinct spectrum with a strong FAD-associated peak at around 443 nm (18), which can be used to quantify the FAD content relative to total protein absorbance at 280 nm. Recombinant wild-type and K333Q LCAD proteins were each purified in three separate purifications and subjected to absorbance scanning to determine the average FAD content. Purified LCAD K333Q protein contained about 35% less FAD than wild-type LCAD (Fig 2B, C).
Figure 2. The K333Q polymorphism alters the LCAD active site.

A) In human IVD, an enzyme closely related to LCAD, the lysine equivalent to K333 is residue K325. In the crystal structure of IVD, K325 is seen to lie within interacting distance of the FAD cofactor and the catalytic base E408 (inset). B,C) Absorbance scanning of recombinant K333Q protein reveals significantly less FAD per unit protein, indicated by the ratio of FAD absorbance (A443) to total protein (A280). Bars in panel C represent three separate protein preparations.
The reduced activity of LCAD K333Q is caused by impaired FAD binding.
The loss of FAD in the LCAD K333Q protein could explain the significantly reduced enzymatic activity we observed compared to wild-type LCAD. Native rat liver LCAD stripped of its FAD has been shown to rebind the cofactor upon incubation with free FAD (23). We therefore incubated both the recombinant wild-type and K333Q LCAD proteins with 100 μM free FAD. After incubation of wild-type LCAD with FAD, the A443/A280 ratio, an indicator of enzyme-bound FAD content, increased from 0.12 to 0.15, and surprisingly, this increase in FAD content was sufficient to nearly double enzymatic activity (Fig 3A). Incubation of LCAD K333Q with FAD only partially rescued the impaired enzyme function. 100 μM free FAD increased the A443/A280 ratio from 0.08 to 0.10, and activity increased by 40% (Fig 3A), but neither the FAD content or the activity reached that of the wild-type enzyme. This lack of enzyme rescue suggests that K333Q alters the active site in such a way as to impair FAD binding.
Figure 3. The reduced activity of LCAD K333Q is caused by impaired FAD binding.
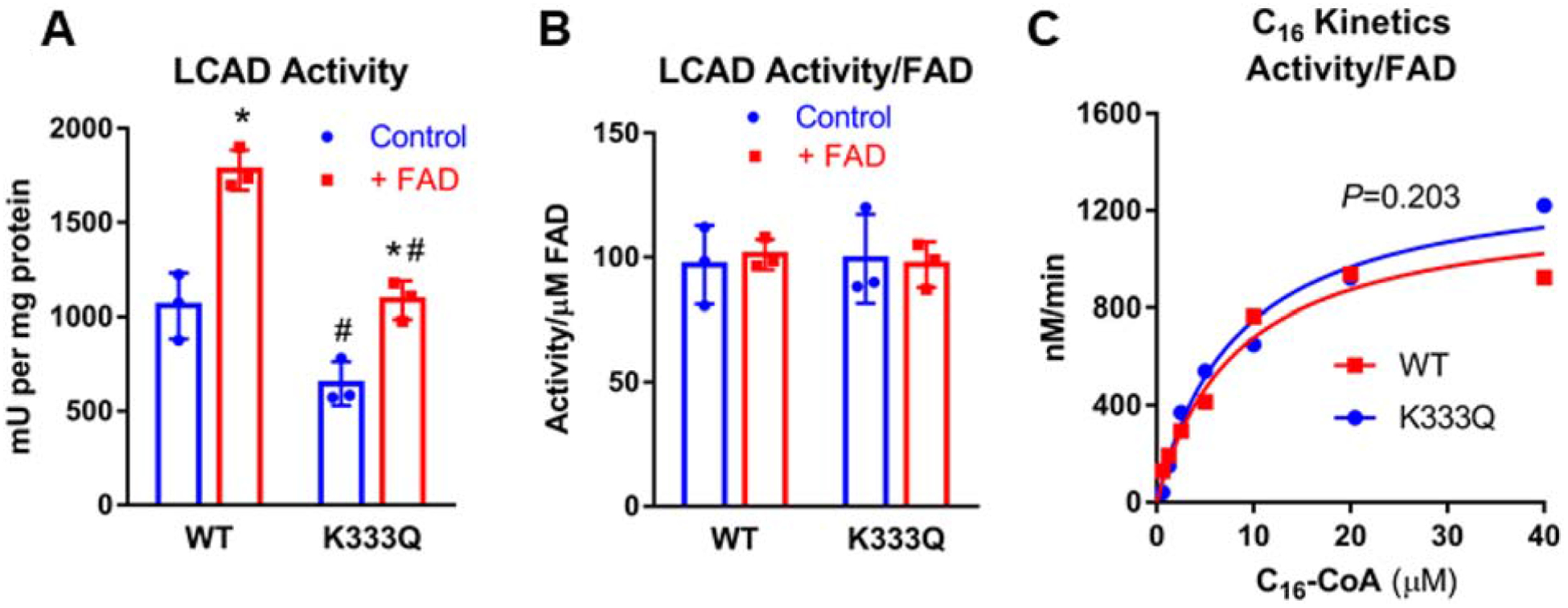
A) Incubation of recombinant LCAD proteins with free FAD increases the ACAD activity of both the WT and K333Q enzymes, but K333Q is only partially rescued. B) Normalizing the activity values from panel A by FAD content rather than protein eliminates the differences in activity, indicating that the loss of activity in K333Q as well as the gain of activity upon FAD incubation are solely a consequence of the amount of FAD bound to the enzymes. C) wild-type LCAD and K333Q are kinetically indistinguishable when activity values are normalized to FAD content rather than protein concentration. Assays were conducted in triplicate for panels A, B and duplicate for panel C.
To determine whether the portion of K333Q protein with bound FAD has normal activity, the enzyme activity values from Fig 3A were recalculated based on bound FAD content rather than protein concentration. When calculated in this manner, both WT and K333Q LCAD enzymes, with and without exogenous FAD incubation, had identical activities (Fig 3B). This suggests that the loss of activity seen in the recombinant K333Q protein were strictly due to impaired binding/retention of the FAD cofactor. This is further supported by enzyme kinetics normalized to bound FAD rather than protein concentration. Wild-type LCAD and K333Q assayed over as range of palmitoyl-CoA concentrations with ETF as the electron acceptor display indistinguishable enzyme kinetics (Fig 3C).
LCAD antigen is significantly reduced in ATII cells from humans homozygous for the K333Q polymorphism.
We previously identified two SIDS cases in which the LCAD antigen was undetectable in tissue lysates, and both were homozygous for K333Q (6). In human lung, LCAD is expressed only in the mitochondria-rich ATII cells (6). To explore the effect of the K333Q polymorphism on LCAD in human primary ATII cells, western blotting was performed on lysates from banked primary human ATII cells that were either homozygous for K333Q (N=5, “Q/Q”) or homozygous for the major K allele (N=5, “K/K”). ATII cells isolated from Q/Q individuals contained 80% less LCAD antigen than those isolated from K/K individuals (Fig 4A, B). We hypothesized that this reduction in antigen may be due to decreased stability, which in turn could be caused by the decreased FAD binding seen in earlier experiments. To test this, we incubated the recombinant LCAD proteins at physiological temperature (37°C) for 12 hr. This incubation resulted in a significant shift in the absorbance spectra of K333Q, but not wild-type LCAD (Fig 4C,D), consistent with protein unfolding. Similarly, the LCAD K333Q protein was seen to unfold at lower temperatures than wild-type LCAD, as observed by monitoring changes tryptophan intrinsic fluorescence (unfolding and exposure of internal tryptophan residues) during a stepwise increase in temperature from 22°C to 60°C (Fig 4E).
Figure 4. LCAD antigen is significantly reduced in human ATII cells homozygous for K333Q.
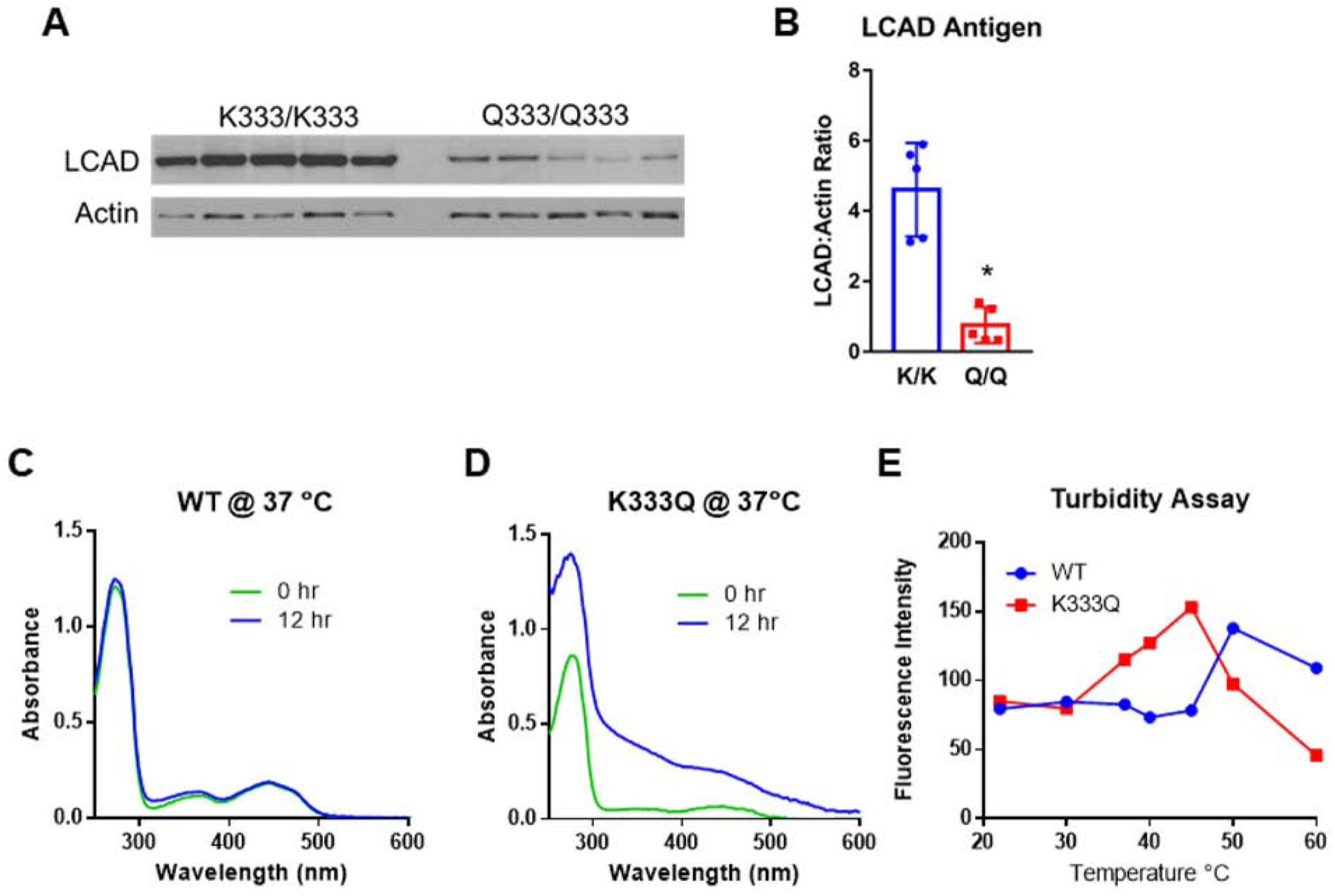
A) Western blot showing reduced LCAD antigen in primary ATII cell lysates from five people homozygous for the Q allele at residue 333, compared to those homozygous for the major K allele. B) Densitometric analysis of the blot in panel A. C,D) Absorbance scans of recombinant proteins following incubation at physiological temperature (37°C) for 12 hr suggest unfolding has occurred in the K333Q enzyme. E) Fluorescence-based assay for thermal unfolding shows that the K333Q protein unfolds at lower temperatures compared to the wild-type protein. *P<0.01. Scans in panels C, D were done twice with similar results, and the data points in panel E represent duplicate assays.
LCAD K333Q is not associated with respiratory disease in children.
Because LCAD knockout mice have altered pulmonary surfactant composition and breathing mechanics (6), we hypothesized that homozygosity for K333Q may be a risk factor for human respiratory diseases. Specifically, we hypothesized that the presence of the K333Q variant might modify the impact of other factors already linked to respiratory disease, such as prematurity, infection, or inborn errors of surfactant metabolism. To test this, we genotyped DNA from the following cohorts of children with respiratory disease and compared the LCAD allele frequencies to those established in the gnomAD database: 1) infants with respiratory distress due to ABCA3 deficiency, an inborn error of surfactant metabolism (N=30); 2) full-term infants with diffuse lung disease of unknown etiology (N=60); 3) pre-term infants with severe respiratory distress of unknown etiology (N=52); and 4) children with pneumonia (N=775). Due to the much higher prevalence of the minor Q allele among Caucasians compared to African Americans (0.35 versus 0.14), the samples were analyzed separately by race. The frequency of the minor Q allele did not significantly differ from that seen in the control population (European or African gnomAD databases) for any of the cohorts (Table 1). In the pneumonia cohorts we also compared the frequency of the Q allele between those who required mechanical ventilation and those who did not. The frequency of K333Q also did not differ between these groups of pneumonia patients (not shown), suggesting no link between K333Q and the severity of response to lung infection.
Table 1:
Frequency of LCAD K333Q (rs2286963) in patients with pediatric lung disease.
| Cohort | N | Q Allele Frequency |
|---|---|---|
| gnomAD database (European) | 133,460 | 0.35 |
| gnomAD database (African) | 15,294 | 0.14 |
| ABCA3-deficient infants with respiratory failure (Caucasian) | 30 | 0.38 |
| Full-term infants with diffuse lung disease (Caucasian) | 60 | 0.36 |
| Pre-term infants (>28 weeks) with severe RDS (Caucasian) | 52 | 0.39 |
| Children hospitalized with pneumonia (Caucasian) | 301 | 0.33 |
| Children hospitalized with pneumonia (African American) | 474 | 0.17 |
Discussion
Our biochemical evaluation of the recombinant LCAD K333Q protein indicates reduced stability and a propensity for losing the essential FAD cofactor, leading to reduced enzymatic activity. This finding likely explains previous reports linking K333Q to increased serum acylcarnitines (a consequence of reduced activity) and loss of LCAD antigen in lung tissue from SIDS cases (consequence of reduced protein stability). We extended this last observation by examining LCAD antigen levels in primary adult ATII cells, which are the specific cell type that expresses LCAD in human lung. Homozygosity for K333Q was associated with 80% less LCAD antigen. Together, our data suggest that humans homozygous for K333Q would be expected to have considerably reduced LCAD function in vivo.
ATII cells are responsible for synthesis of lung surfactant and several lung diseases have been associated with surfactant deficiency. Neonatal respiratory distress syndrome (RDS) is characterized by defective pulmonary surfactant production or function (24). This can be due to prematurity or to specific mutations in genes critical for surfactant synthesis, secretion, or function, such as surfactant protein-B (SP-B), surfactant protein-C (SP-C), and ABCA3 (24–26). However, RDS has a strong genetic component that is not explained by variants in these genes (27, 28). RDS is more common in white infants, and white ethnicity is considered a risk factor for mortality from RDS (29). Because LCAD K333Q is strikingly more common in those of European descent, and because LCAD knockout mice have altered surfactant function and breathing mechanics (6, 8), we hypothesized that K333Q may associate with RDS. However, this hypothesis was not supported by the data, which indicated no difference in the frequency of K333Q in either pre-term or term infants with RDS (Table 1). However, it must be taken into consideration that our RDS cohort sizes were small and weak associations may have been missed. In contrast, for the pneumonia cohorts we calculate a minimal detectable odds ratio of 1.35 with 90% power. This is sufficient statistical power to detect even modest associations, and we therefore conclude that LCAD K333Q is not associated with hospitalization from pediatric pneumonia. Future studies with larger cohorts of respiratory distress infants, and patients with adult-onset lung disease, are needed to definitively evaluate whether the K333Q polymorphism confers greater risk for lung diseases.
Acknowledgments
This work was supported by R01 DK090242 (E.S.G.), R01 HL118171 (B.K.) and the Catalyst Award from the American Lung Association (K.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Yamada K, Taketani T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet 2019;64:73–85. [DOI] [PubMed] [Google Scholar]
- 2.Anderson DR, Viau K, Botto LD, Pasquali M, Longo N. Clinical and biochemical outcomes of patients with medium-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2020;129:13–19. [DOI] [PubMed] [Google Scholar]
- 3.Nochi Z, Olsen RKJ, Gregersen N. Short-chain acyl-CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms. J Inherit Metab Dis 2017;40:641–655. [DOI] [PubMed] [Google Scholar]
- 4.Chegary M, Brinke H, Ruiter JP, Wijburg FA, Stoll MS, Minkler PE, van Weeghel M, et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta 2009;1791:806–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher AC, Mohsen AW, Vockley J, Tarnopolsky MA. Low expression of long-chain acyl-CoA dehydrogenase in human skeletal muscle. Mol Genet Metab 2010;100:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goetzman ES, Alcorn JF, Bharathi SS, Uppala R, McHugh KJ, Kosmider B, Chen R, et al. Long-chain acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J Biol Chem 2014;289:10668–10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shinde A, Luo J, Bharathi SS, Shi H, Beck ME, McHugh KJ, Alcorn JF, et al. Increased mortality from influenza infection in long-chain acyl-CoA dehydrogenase knockout mice. Biochem Biophys Res Commun 2018;497:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otsubo C, Bharathi S, Uppala R, Ilkayeva OR, Wang D, McHugh K, Zou Y, et al. Long-chain Acylcarnitines Reduce Lung Function by Inhibiting Pulmonary Surfactant. J Biol Chem 2015;290:23897–23904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Bharathi SS, Beck ME, Goetzman ES. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol 2019;26:101253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Heng J, Yan J, Guo X, Tang L, Chen M, Peng L, et al. Integrated analysis of gene expression and methylation profiles of 48 candidate genes in breast cancer patients. Breast Cancer Res Treat 2016;160:371–383. [DOI] [PubMed] [Google Scholar]
- 11.Xie BX, Zhang H, Wang J, Pang B, Wu RQ, Qian XL, Yu L, et al. Analysis of differentially expressed genes in LNCaP prostate cancer progression model. J Androl 2011;32:170–182. [DOI] [PubMed] [Google Scholar]
- 12.Huang, Li T, Li X, Zhang L, Sun L, He X, Zhong X, et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep 2014;8:1930–1942. [DOI] [PubMed] [Google Scholar]
- 13.Yu DL, Li HW, Wang Y, Li CQ, You D, Jiang L, Song YP, et al. Acyl-CoA dehydrogenase long chain expression is associated with esophageal squamous cell carcinoma progression and poor prognosis. Onco Targets Ther 2018;11:7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong MG, Karlsson R, Magnusson PK, Lewis MR, Isaacs W, Zheng LS, Xu J, et al. A genome-wide assessment of variability in human serum metabolism. Hum Mutat 2013;34:515–524. [DOI] [PubMed] [Google Scholar]
- 15.Illig T, Gieger C, Zhai G, Romisch-Margl W, Wang-Sattler R, Prehn C, Altmaier E, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet 2010;42:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wanders RJ, Denis S, Ruiter JP, L IJ, Dacremont G. 2,6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation. Biochim Biophys Acta 1998;1393:35–40. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Mohsen AW, Kochersperger C, Solo K, Schmidt AV, Vockley J, Goetzman ES. An acyl-CoA dehydrogenase microplate activity assay using recombinant porcine electron transfer flavoprotein. Anal Biochem 2019;581:113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharathi SS, Zhang Y, Mohsen AW, Uppala R, Balasubramani M, Schreiber E, Uechi G, et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J Biol Chem 2013;288:33837–33847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bahmed K, Messier EM, Zhou W, Tuder RM, Freed CR, Chu HW, Kelsen SG, et al. DJ-1 Modulates Nuclear Erythroid 2-Related Factor-2-Mediated Protection in Human Primary Alveolar Type II Cells in Smokers. Am J Respir Cell Mol Biol 2016;55:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahmer MK, O’Cain P, Patwari PP, Simpson P, Li SH, Halligan N, Quasney MW. The influence of genetic variation in surfactant protein B on severe lung injury in African American children. Crit Care Med 2011;39:1138–1144. [DOI] [PubMed] [Google Scholar]
- 21.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet 2006;7:781–791. [DOI] [PubMed] [Google Scholar]
- 22.Chen B, Cole JW, Grond-Ginsbach C. Departure from Hardy Weinberg Equilibrium and Genotyping Error. Front Genet 2017;8:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda Y, Okamura-Ikeda K, Tanaka K. Purification and characterization of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. Isolation of the holo- and apoenzymes and conversion of the apoenzyme to the holoenzyme. J Biol Chem 1985;260:1311–1325. [PubMed] [Google Scholar]
- 24.Gower WA, Nogee LM. Surfactant dysfunction. Paediatr Respir Rev 2011;12:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, An P, et al. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics 2012;130:e1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garmany TH, Wambach JA, Heins HB, Watkins-Torry JM, Wegner DJ, Bennet K, An P, et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res 2008;63:645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo HS. Genetic risk factors associated with respiratory distress syndrome. Korean J Pediatr 2014;57:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levit O, Jiang Y, Bizzarro MJ, Hussain N, Buhimschi CS, Gruen JR, Zhang H, et al. The genetic susceptibility to respiratory distress syndrome. Pediatr Res 2009;66:693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anadkat JS, Kuzniewicz MW, Chaudhari BP, Cole FS, Hamvas A. Increased risk for respiratory distress among white, male, late preterm and term infants. J Perinatol 2012;32:780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]