

Abstract
The anomeric aminooxy GM3 trisaccharide cancer antigen (Neu5Acα2,3Galβ1,4Glcβ-ONH2) has been chemically synthesized using a linear glycosylation approach. The key step involves a highly α(2,3)-stereoselective sialylation to a galactose acceptor. The Neu5Acα2,3Gal intermediate was functionalized as a donor for a [2 + 1] glycosylation including a glucose acceptor that featured an O-succinimidyl group on the reducing end as an aminooxy precursor. The fully deprotected anomeric aminooxy GM3 trisaccharide was then conjugated to the immunologically relevant zwitterionic polysaccharide PS A1 via an oxime link.
Graphical Abstract

Oxime ligation has emerged as an attractive conjugation method in vaccine formulation for a number of reasons: 1) oxime bonds are hydrolytically stable in physiological conditions,1–3 2) the oxime bond is immunologically “silent” as compared to other conjugation linkages,4–5 3) the condensation of aminooxy functions are fast, quantitative, and only produce water as a byproduct.6–7 Our group has used the oxime ligation strategy to create entirely carbohydrate immunogens capable of generating protective immune responses against tumor associated carbohydrate antigens (TACAs).8–11 These glycoconjugates feature the unique zwitterionic polysaccharide PS A1 which is isolated from the commensal organism Bacteroides fragilis (ATCC 25285/NCTC 9343). The alternating charge character on the repeating unit of PS A1 provides distinct properties that permit binding to the MHC II complex on antigen presenting cells and downstream activation of CD4+ T cells through the MHC II:αβTCR complex pathway.12–13 Formation of these immunogens is materialized by the oxime condensation of a chemically derived aldehyde on PS A1 and with an aminooxy derivative of a chosen TACA (Figure 1). This vaccine platform has been successfully demonstrated with Tn-PS A18, 14 and STn-PS A110 where we observed robust IgG antibody production, cellular immunity, and tumor cell lysis.
Figure 1.

General structure of TACA-PS A1 glycoconjugates and aminooxy derivatives of TACAs.
Progress of these initial developments led us to expand our TACA toolbox to incorporate the ganglioside GM3 (Neu5Acα2,3Galβ1,4Glcβ-Cer). GM3 is overexpressed in carcinomas of the breast and skin and has implications in tumor cell proliferation and metastasis.15–16 GM3 overexpression is also positively correlated with tumor malignancy leading to poor prognosis.16 Due to these characteristics, GM3 has been prioritized within the top 50 cancer antigens for immune intervention.17
The biological relevance of the GM3 antigen implicates necessity for preparative ease and availability for immunological experiments. Indeed, both chemo-enzymatic and chemical methods exist for the production of the GM3 antigen, but they have limitations.18–25 Chemo-enzymatic methods offer excellent regio- and stereo-selective control of glycan production but are typically demonstrated on smaller scales.19, 26 On the other hand, synthetic reports of the GM3 glycan relay poor stereoselectivity forming the challenging α(2→3) glycosidic bond. Up to date, only one synthesis of the aminooxy GM3 glycan has been reported through a sialyltransferase catalyzed glycosylation.19 Herein, we describe our chemical synthetic platform to produce the aminooxy GM3 glycan by using a linear approach that features a high α-selective sialylation in good yield. The final step involves a microwave assisted global deprotection of -OAc/-OBz protecting groups and an O-succinimidyl (OSu) protecting group to achieve the corresponding alcohol and aminooxy functions, respectively. The fully deprotected aminooxy GM3 glycan was then conjugated to PS A1 by oxime ligation to form the GM3-PS A1 glycoconjugate.
The synthetic route to the aminooxy GM3 glycan (1) must feature high α-selectivity in the Neu5Ac2,3Gal bond forming step. To achieve this, we elected to use the α-directing transfused 4O,5N-acetyl oxazolidinone ring system on the sialic acid donor.10, 27–28 This moiety exhibits strain on the cyclic system which destabilizes the formation of the glycosyl oxocarbenium ion intermediate, and instead a β-triflate intermediate is more likely formed. This pushes the mechanism of glycosylation towards more of an SN2-like associative mechanism as opposed to an unselective SN1 dissociative mechanism.29 Additionally, a highly reactive anomeric leaving group, such as a phosphate ester,30–31 was desired based on our previous experiences.10 These two features led us to adopt the sialyl phosphate donor 3 (Scheme 1, Scheme S1). The synthetic strategy can be further broken down through pathway A or pathway B (Scheme 1). Reports that describe pathway A in the synthesis of the GM3 glycan discuss high yields and α-selectivity in the α(2→3) bond forming step, but feature a greater number of overall synthetic steps than pathway B.24 Contemporary reports of GM3 synthesis describe pathway B with the use of lactosyl acceptors, but these reports describe low yields in the α(2→3) bond forming step.25 We must also account for the introduction of -OSu on the reducing end of the protected trisaccharide as the precursor for an aminooxy group.9–10 This requires limited use of benzyl ether protecting groups, as long exposure times of hydrogenation can cause undesired cleavage of the -OSu N-O bond.32 We proceeded to screen various glycosyl acceptors for the α(2→3) bond forming step using sialyl phosphate 3 as the donor (Table 1).
Scheme 1.

Retrosynthetic Analysis of the Aminooxy GM3 Glycan
Table 1.
Optimization of α(2,3)-Glycosylationa







Typical conditions: 1.5 equivalents of donor, 1.0 equivalent of acceptor, 1.5 equivalents of TMSOTf, in dry CH2Cl2 at −78 °C for 1 hour.
Isolated yield.
Determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture.
We commenced with screening lactose acceptors 4 (Table 1, entry 1) and 5 (Table 1, entry 2, Scheme S2). Both acceptors feature a 3’,4’-diol and differ on their reducing ends with 4 containing thiophenol and 5 containing an O-succinimidyl. Both acceptors 4 and 5 provided the β-anomer as the major product with α:β ratios of 1:5 and 1:4 respectively. Poor selectivity is in line with previous reports and is a consequence of the disarming effect of acetyl group protections.25, 33 We then looked toward the 3, 2’, 3’, 4’-unprotected lactoside 6 (Table 1, entry 3) which has been described to provide improved α-formation as compared to 4 and 5 due to the relatively weaker disarming effect of pivaloyl groups.34 Acceptor 6 resulted in an improved α:β ratio at 1:1, although this ratio remained unsatisfactory. Based on these observed limitations of lactose acceptors, we investigated the use of galactose acceptor 735 which featured a 3,4-diol with a 2-O-benzoyl-6-O-benzyl protection strategy (Table 1, entry 4). The resulting α:β ratio was drastically improved to provide the α-anomer in an 8:1 ratio in 89% yield. The greatly improved α:β ratio led us to explore galactose acceptor 831 which contains the conformation restricting 4,6-O-benzylidene acetal (Table 1, entry 5). Galactose acceptor 8 provided excellent α-selectivity with an α:β ratio of 16:1 in 87% yield. It was logical at this point to proceed through pathway A using galactosyl acceptor 8.
With disaccharide 13 in hand, we turned our attention to the D-glucose acceptor 16 as shown in Scheme 2. Here, thio-donor 14 was activated using N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) in the presence of N-hydroxysuccinimide (NHS) to afford compound 15 in 84% yield as a pure β-anomer. Selective ring opening of the 4,6-benzylidene acetal, on compound 15, was performed with triethylsilane (Et3SiH) as the hydride source and boron trifluoride diethyl etherate (BF3·Et2O) as the catalyst. Successful regioselective ring opening to afford 16 was confirmed using HMBC as the C-6 position nicely correlated with the benzylic protons on the benzyl ether.
Scheme 2.

Synthesis of Glc-OSu Acceptor 16
Disaccharide 13 (Table 1, entry 5) was subjected to Zemplén sodium methoxide conditions to remove the oxazolidinone, and the resulting free hydroxyls were acetylated to provide compound 17 as shown in Scheme 3. Interestingly, the C-2 O-benzoylated protection on compound 13 was not removed during the Zemplén method. This is likely due to the steric hinderance of this position, especially within disaccharide systems.21 With disaccharide donor 17 and glucose acceptor 16 in hand, we sought to perform this [2 + 1] glycosylation under typical
Scheme 3.

Attempted 2 + 1 Glycosylation Through Intermediate 13
NIS/TMSOTf activation conditions
TLC analysis of the reaction mixture revealed partial consumption of the donor (Rf = 0.4) accompanied by the presence of a new spot (Rf = 0.2). Investigation of the reaction revealed the new spot to be the glycosyl succinimide product (S6). Indeed, this activation system produces the weakly nucleophilic succinimide as a by-product, which can outcompete weakly nucleophilic or sterically hindered alcohols on glycosyl acceptors. This phenomenon has been described for donor/acceptor pairs with mismatched reactivities.36 Notably, we also discovered partial proton exchange with trimethyl silane on acceptor 16. We attempted the [2 + 1] coupling using different activation systems such as NIS/trifluoromethanesulfonic acid (TfOH), dimethyl disulfidetriflic anhydride (Me2S2-Tf2O), and a trichloroacetamidate (TCA) donor (18). The results led to a glycosyl succinimide by-product, a complex mixture, and the Chapman rearrangement37 respectively (Scheme 3). These experiments offer further evidence of the torsional disarming effect from 4,6-O-acetal bicyclic donors.38
We sought to deviate the synthetic route through the more armed disaccharide donor compound 12 (Table 1, entry 4). As illustrated in Scheme 4, preparation of the disaccharide donor 20 mirrored the preparation of donor 17, with the C-2-O-benzoyl protection remaining intact. Donor 20 was then activated with the NIS/TfOH system in the presence of acceptor 16 and provided the desired, fully protected trisaccharide 2 in 87% yield as the β-anomer. No evidence of the glycosyl succinimide was present.
Scheme 4.

Synthesis of the Aminooxy GM3 Glycan
Deprotection of key intermediate 2 began with the cleavage of the benzyl ethers using hydrogen gas at 1 atm and a suspension of Pearlman’s catalyst. TLC indicated full consumption of starting material after 1 hour of mixing and the reaction was purged with argon gas to remove dissolved hydrogen gas. The reaction was filtered, concentrated, and passed through a short silica column to afford the debenzylated product S7 in 89% yield with no evidence of -OSu N-O cleavage. Deprotection of the Neu5Ac C-1 methyl ester was accomplished using the Krapcho-like demethylation by lithium iodide in pyridine.10 This mixture was refluxed at 110 °C for 3 hours in the absence of light. Pyridine was then removed in vacuo and the crude material was purified by silica flash chromatography to afford compound 21 in 68% yield.
In the final deprotection step, we utilized excess hydrazinehydrate to remove the remaining -OAc/-OBz protecting groups and to remove -OSu to expose the aminooxy functionality. However, based on the results of Zemplén sodium methoxide conditions as described for 12 and 13 we did not expect deprotection of the C-2-O-benzoylatd positions using hydrazine hydrate at room temperature. Additionally, literature precedent suggested long reaction times24 and elevated temperatures25 in hydroxide or methoxide conditions for C-2-O-benzoyl removal, which would then cause an undesirable ring opening of the installed -OSu group and failure of aminooxy deprotection. To overcome this limitation, we turned to microwave assistance in the final deprotection step. Compound 22 was dissolved in ethanol followed by the addition of excess hydrazine hydrate. The mixture was then stirred for 1 hour at room temperature and MS analysis did not detect any formation of product. The reaction was then subjected to microwave irradiation at 200 Watts with constant temperature of 40 °C for 20 minutes. Once again, MS analysis indicated little-to-no deprotection of compound 21. We then increased the temperature to a constant 90 °C for 30 minutes with no change in maximum Watts. Following this procedure, MS analysis of the reaction mixture revealed partial deprotection of 21. Repetitive cycling (flow-chemistry techniques) of the aforementioned conditions led to MS analysis clearly depicting fully deprotected 21. The reaction mixture was then concentrated, resuspended in water, and passed through a P2 BioGel® column using water as the eluent. Fractions identified to contain compound 1 by TLC staining were collected, frozen, and lyophilized. The resulting white solid was characterized and compound 1 was definitively confirmed by NMR and HRMS (See supporting information).
With the aminooxy GM3 glycan complete, we pursued oxime ligation with PS A1 (Scheme 5, 22). The repeating unit of PS A1 contains the side chain D-galactofuranose with a 1o and 2o vicinal diol that can be chemo- and regio-selectively oxidized by sodium periodate to afford an aldehyde.8–11 After PS A1 oxidation, the aldehyde moiety was then subjected to four equivalents of 1 and subsequently stirred for 18 hours in a 0.1 M sodium acetate buffer at a pH of 5. The reaction mixture was then filtered and rinsed through a 10 kDa molecular weight cutoff filter. The filtered material was then resuspended in water, frozen, and lyophilized to produce 23 as a white (cotton-like) substance. NMR analysis of this material at 60 °C provided the evidence we needed to note successful conjugation with PS A1. The defining peaks included an oxime proton peak (δ = 7.99–8.00), an additional anomeric proton peak (δ = 5.37), an additional N-acetyl peak (δ = 2.31), and the presence of the Neu5Ac H-3eq (δ = 3.03–3.06) and H-3ax peaks (δ = 2.04–2.08). Based on integration analysis of the Neu5Ac H-3eq and PS A1 AAT methyl peak, we determined the relative ratio of GM3:PS A1 to be 1:2.33, which translates into approximately 51.5 GM3 conjugation sites per PS A1.
Scheme 5.

Synthesis of GM3-PS A1
In conclusion, we have discussed the first fully chemical synthetic method to provide the aminooxy GM3 glycan. This synthesis features highly α-selective chemistries in the formation of the Neu5Ac(2→3)Gal bond. In these experiments we showed the benefit of using the armed galactose acceptors as opposed to the disarmed lactose acceptors, which gave undesirable α:β ratios. This synthesis also features a 3-step deprotection strategy of the fully protected trisaccharide. Notably, the final step utilized microwave assisted chemistry to deprotect the sterically hindered C-2 O-benzoylations in excellent yield, after reiterations. Lastly, we highlighted the utility of the aminooxy functionality by conjugating the GM3 glycan to the immunologically relevant PS A1 to generate the GM3-PS A1 glycoconjugate. The GM3-PS A1 vaccine candidate will be investigated for its immunological characteristics in due time.
EXPERIMENTAL SECTION
General Information
Relevant reagents and solvents were purchased from commercial sources and used without further purification. Molecular sieves (4 Å) were dried overnight in a vacuum oven set to 140 °C, and further dried under high vacuum using a heat gun for 5 minutes and cooled to room temperature under high vacuum. Synthesized compounds were purified using flash chromatography with SiliCycle Inc. 60 Å 230–400 mesh silica gel or size exclusion chromatography with Bio-Gel P-2 (Bio-Rad Laboratories, Inc). Thin layer chromatography (TLC) was performed with SiliCycle Inc. silica gel TLC 250 μm w/h F-254. Proton and carbon NMR spectra were recorded using a Bruker Avance III 600 Ultrafield Cryoprobe spectrometer. CDCl3, CD3OD, and D2O were used as solvents, and the chemical shifts were referenced relative to residual solvent. Low resolution mass spectrometry data were taken on an LCQ Deca ESI-MS machine. High resolution mass spectrometry data were collected using an Orbitrap Fusion Tribrid Mass Spectrometer.
Experimental Procedures
Methyl (phenyl-5-acetamido-7,8,9-tri-O-acetyl-5-N,4-Ocarbonyl-3,5-dideoxy-2-thio-d-glycero-α,β-d-galacto-non-2ulopyranoside)onate (S2)
Compound S128 (5.20 g, 8.91 mmol, 1.0 equiv) was suspended in 75 mL of MeOH with vigorous stirring. To the suspension was added MeSO3H (2.90 mL, 44.6 mmol, 5.0 equiv) at room temperature and the suspension was refluxed for 24 hours. The mixture was then cooled to room temperature and the reaction quenched with excess TEA. The solution was then concentrated under reduced pressure and dried under vacuum. The resulting crude concentrate was then dissolved in mL MeCN and mL H2O followed by the addition of NaHCO3 (3.74 g, 44.6 mmol, 5.0 equiv). The mixture was stirred vigorously to a uniform suspension and cooled to 0 °C. A separate solution of triphosgene (7.93 g, 26.7 mmol, 3.0 equiv) dissolved in mL MeCN was then added to the vigorously stirred suspension via dropping funnel at a slow rate of ~1 drop/5 seconds. The reaction was maintained at 0 °C for 5 hours. EtOAc ( mL) was then added to the mixture and vigorously stirred for 10 minutes. The organic and aqueous layers were separated, and the aqueous layer was extracted with EtOAc ( mL, 2X). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The resulting residue was passed through a short, thick bed of silica eluting with EtOAc:MeOH (9:1). Fractions containing the intermediate were pooled and concentrated under reduced pressure and then set under high vacuum overnight to give the crude intermediate (3.02 g, 7.56 mmol, 85% yield of crude). The crude material was then dissolved in pyridine (35 mL) followed by the addition of DMAP (0.18 g, 1.5 mmol, 0.2 equiv). Gas was exchanged with Ar and the reaction mixture was cooled to 0 °C with vigorous stirring. Ac2O (14.3 mL, 151 mmol, 20 equiv) was then injected into the reaction mixture and the reaction was stirred for 12 hours while the reaction was allowed to slowly warm to room temperature. The reaction was then quenched with MeOH at 0 °C and then concentrated under reduced pressure followed by co-distillation with toluene (3X). The crude material was then dissolved in CH2Cl2 (50 mL) and washed with saturated NaHCO3 (50 mL, 3X). The resulting organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude material was then purified by silica flash chromatography eluting with hexanes:EtOAc (1:1) to give compound S2 as a white solid (4.00 g, 7.05 mmol, 79% yield over 3 steps). 1H NMR (CDCl3, 600 MHz): δ = 1.96 (3H, s), 2.09 (3H, s), 2.15 (3H, s), 2.34 (1H, t, J = 13.0 Hz), 2.53 (3H, s), 2.91 (1H, dd, J = 3.7, 13.1 Hz), 3.63 (3H, s), 3.76 (1H, dd, J = 9.2, 11.3 Hz), 3.90 (1H, dd, J = 8.2, 12.1 Hz), 4.37 (1H, dd, J = 2.5, 12.0 Hz), 4.78 (1H, td, J = 4.3, 15.0 Hz), 4.89 (1H, dd, J = 11.7, 6.6 Hz), 5.00 (1H, td, J = 2.2, 8.2 Hz), 5.56 (1H, t, J = 2.3 Hz), 7.36 (2H, m), 7.43 (1H, m), 7.50 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.7, 20.8, 21.1, 24.7, 36.0, 52.8, 59.6, 62.9, 72.7, 73.9, 75.1, 75.7, 88.4, 128.2, 129.2, 130.2, 136.8, 153.6, 167.8, 169.7, 170.4, 171.2, 172.5. LRMS (ESI) m/z: [M + Na]+ calcd for C25H29NO12SNa 590.1; Found 590.2.
Methyl (5-acetamido-7,8,9-tri-O-acetyl-5-N,4-O-carbonyl2-(dibutylphosphoryl)-3,5-dideoxy-d-glycero-α,β-d-galactonon-2-ulopyranoside)onate (3)
Compound S2 (1.00 g, 1.76 mmol, 1.0 equiv) was dried by co-distillation with toluene (3X) followed by high vacuum for 3 hours. The dry compound was then dissolved in mL of dry CH2Cl2 followed by the addition of 4 Å molecular sieves (4 g) and dibutyl phosphate (1.75 mL, 8.80 mmol, 5.0 equiv). Gas in the reaction vessel was exchanged with Ar and this solution was then stirred for 30 minutes at room temperature, followed by the addition of NIS (1.19 g, 5.28 mmol, 3.0 equiv). Gas was then exchanged with Ar again and the reaction was stirred at room temperature for 12 hours. After 12 hours, the reaction was diluted with CH2Cl2 (30 mL) and quenched with a saturated solution of Na2S2O3 and NaHCO3 (30 mL). The quenched reaction was then filtered, and the collected organic layer was washed with saturated Na2S2O3 and NaHCO3 (30 mL, 2X). The resulting organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated under reduced pressure. The crude compound was then purified via flash chromatography using hexanes:EtOAc (1:1) to give sialyl donor 3 as an anomeric mixture and as a yellow oil (1.05 g, 1.57 mmol, 89% yield). (3α) 1H NMR (CDCl3, 600 MHz): δ = 0.96 (6H, t, J = 7.4 Hz), 1.43 (4H, m), 1.68 (4H, m), 2.06 (3H, s), 2.13 (3H, s), 2.17 (3H, s), 2.52 (3H, s), 2.71 (1H, t, J = 12.7 Hz), 3.03 (1H, dd, J = 4.0, 12.2 Hz), 3.86 (3H, s), 3.89 (1H, t, J = 10.1 Hz), 4.14 (6H, m), 4.43 (1H, dd, J = 2.8, 12.3 Hz), 4.78 (1H, dd, J = 1.4, 9.5 Hz), 5.34 (1H, m), 5.69 (1H, dd, J = 1.4, 7.5 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 13.6, 18.6, 18.7, 20.8, 20.8, 21.0, 24.7, 32.1, 32.1, 32.1, 35.9, 35.9, 53.5, 58.3, 62.5, 68.0, 68.1, 68.1, 68.2, 69.8, 71.5, 74.2, 98.2, 98.2, 153.6, 167.3, 167.3, 169.9, 170.0, 170.7, 171.9. LRMS (ESI) m/z: [M + Na]+ calcd for C27H42NO16PNa 690.2; Found 690.4. (3β) 1H NMR (CDCl3, 600 MHz): δ = 0.96 (6H, dt, J = 4.2, 7.4 Hz), 1.42 (4H, m), 1.68 (4H, m), 2.05 (3H, s), 2.12 (3H, s), 2.15 (3H, s), 2.32 (1H, dt, J = 2.8, 12.7 Hz), 2.52 (3H, s), 2.91 (1H, dd, J = 3.7, 12.8 Hz), 3.79 (1H, dd, J = 9.5, 11.3 Hz), 3.88 (3H, s), 4.13 (5H, m), 4.56 (1H, dd, J = 2.6, 12.2 Hz), 4.60 (1H, td, J = 3.7, 11.4, 12.6 Hz), 4.75 (1H, dd, J = 2.0, 9.5 Hz), 5.28 (1H, m), 5.64 (1H, dd, J = 2.0, 4.0 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 13.6, 18.6, 18.6, 20.8, 21.0, 24.7, 32.1, 32.1, 32.1, 32.2, 36.1, 36.1, 53.5, 58.9, 62.9, 68.4, 68.4, 68.5, 68.6, 71.8, 72.6, 74.0, 76.7, 98.8, 98.9, 153.5, 165.6, 169.8, 170.6, 170.7, 172.2. LRMS (ESI) m/z: [M + Na]+ calcd for C27H42NO16PNa 690.2; Found 690.4.
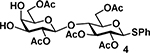
Phenyl (2,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)2,3,6-tri-O-acetyl-1-thio-β-d-glucopyranoside (4)
Acceptor 4 was prepared as previously described.39 Acceptor 4 was purified by silica flash chromatography using 2% MeOH in CH2Cl2 as the eluent to provide 4 as a white solid (0.50 g, 0.78 mmol). 1H NMR (CDCl3, 600 MHz): δ = 2.03 (3H, s), 2.08 (3H, s), 2.09 (3H, s), 2.10 (3H, s), 2.11 (3H, s), 3.59 (1H, dd, J = 3.5, 9.8 Hz), 3.62 (1H, t, J = 6.5 Hz), 3.69 (2H, m), 3.86 (1H, d, J = 3.0 Hz), 4.16 (1H, dd, J = 5.5, 11.8 Hz), 4.22 (1H, dd, J = 6.6, 11.4 Hz), 4.32 (2H, m), 4.51 (1H, dd, J = 1.9, 12.0 Hz), 4.69 (1H, d, J = 10.1 Hz), 4.89 (1H, dd, J = 8.0, 9.8 Hz), 4.93 (1H, t, J = 9.7 Hz), 5.19 (1H, t, J = 8.9 Hz), 7.31 (3H, m), 7.47 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 21.1, 21.1, 21.2, 21.2, 62.7, 68.8, 70.4, 72.6, 73.0, 73.7, 74.1, 76.5, 77.1, 85.9, 101.2, 128.5, 129.2, 133.0, 169.9, 170.8, 170.8, 171.3, 171.7. HRMS (ESI) m/z: [M + Na]+ calcd for C28H36O15SNa 667.1667; Found 667.1769.
Trichloroacetimidatyl (2,6-di-O-acetyl-3,4-O-isopropylidene-β-d-galactopyranosyl)-(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (S4)
Compound S339 (0.67 g, 0.98 mmol, 1.0 equiv) was dissolved in an acetone:H2O (4:1, mL) mixture and then cooled to 0 °C. While stirring the mixture, NBS (0.52 g, 2.9 mmol, 3.0 equiv) was added to the reaction gradually over the duration of 30 minutes. The reaction was then stirred for an additional 30 minutes at room temperature. The solution was then concentrated under reduced pressure until the solution became turbid. The turbid mixture was diluted with CH2Cl2 (20 mL) and then quenched and washed with saturated Na2S2O3 and NaHCO3 (15 mL, 3X). The organic layer was then washed with brine, dried with Na2SO4, filtered, and then concentrated under reduced pressure to give the crude hemiacetal intermediate as a white solid (0.51 g, 0.86 mmol, 88% yield of crude). The crude hemiacetal was then co-distilled with toluene under reduced pressure (3X) and set under high vacuum for 3 hours. The crude hemiacetal was then dissolved in dry CH2Cl2 ( mL) followed by the addition of 4 Å molecular sieves (2 g) and trichloroacetonitrile (345 μL, 3.44 mmol, 4.0 equiv), and then gas was exchanged with Ar. The solution was stirred at room temperature for 30 minutes and was then cooled to 0 °C followed by the addition of DBU (26 μL, 0.17 mmol, 0.2 equiv). The reaction was then stirred for 1 hour at 0 °C. The reaction was filtered and then concentrated under reduced pressure to provide the crude lactosyl donor. The crude material was then purified by silica flash chromatography using toluene:acetone (3:2) as the eluent to provide lactosyl donor S4 as a white solid (0.44 g, 0.60 mmol, 62% yield over 2 steps). 1H NMR (CDCl3, 600 MHz): δ = 1.31 (3H, s), 1.54 (3H, s), 2.01 (3H, s), 2.07 (3H, s), 2.08 (3H, s), 2.09 (3H, s), 2.12 (3H, s), 3.83 (1H, t, J = 9.8 Hz), 3.93 (1H, m), 4.14 (3H, m), 4.19 (1H, dd, J = 4.5, 12.2 Hz), 4.31 (2H, m), 4.41 (1H, d, J = 7.3 Hz), 4.43 (1H, dd, J = 2.0, 12.2 Hz), 4.85 (1H, dd, J = 6.1, 7.3 Hz), 5.06 (1H, dd, J = 3.8, 10.1 Hz), 5.55 (1H, t, J = 9.8 Hz), 6.48 (1H, d, J = 3.8 Hz), 8.64 (1H, s). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.7, 21.0, 21.0, 21.0, 21.1, 26.2, 27.4, 61.9, 63.3, 69.3, 70.1, 71.0, 71.2, 72.9, 73.1, 75.8, 77.0, 90.8, 93.1, 100.8, 111.0, 161.1, 169.4, 169.8, 170.3, 170.5, 170.9. LRMS (ESI) m/z: [M + Na]+ calcd for C27H36Cl3NO16Na 758.1; Found 758.3.
Succinimidyl (2,6-di-O-acetyl-3,4-O-isopropylidene-β-d-galactopyranosyl)-(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (S5)
Lactosyl donor S4 ( mg, 0.11 mmol, 1.0 equiv) and NHS (63 mg, 0.55 mmol, 5.0 equiv) were co-distilled with toluene under reduced pressure (3X) followed by high vacuum overnight. The mixture was then dissolved in 5.0 mL of dry CH2Cl2 and then 4 Å molecular sieves (0.2 g) were added to the solution. Gas was then exchanged with Ar and the solution was stirred at room temperature for 30 minutes. TMSOTf (4.0 μL, 0.022 mmol, 0.2 equiv) was then introduced into the mixture at room temperature and the reaction was stirred for 30 minutes. The reaction was then quenched with TEA, filtered, and then concentrated under reduced pressure. The crude material was then purified by silica flash chromatography using 1% MeOH in CH2Cl2 to provide compound S5 as a white solid (69 mg, 0.10 mmol, 90% yield). 1H NMR (CDCl3, 600 MHz): δ = 1.31 (3H, s), 1.53 (3H, s), 2.08 (3H, s), 2.08 (3H, s), 2.10 (3H, s), 2.12 (3H, s), 2.12 (3H, s), 2.73 (4H, s), 3.74 (1H, m), 3.94 (1H, m), 4.10 (1H, dd, J = 8.5, 10.0 Hz), 4.14 (2H, m), 4.19 (1H, dd, J = 5.8, 12.0 Hz), 4.27 (1H, dd, J = 7.5, 11.7 Hz), 4.33 (1H, dd, J = 4.7, 11.7 Hz), 4.39 (1H, d, J = 7.7 Hz), 4.41 (1H, dd, J = 2.3, 12.0 Hz), 4.86 (1H, dd, J = 6.4, 7.6 Hz), 5.13 (1H, d, J = 6.3 Hz), 5.17 (1H, t, J = 6.8 Hz), 5.20 (1H, t, J = 7.9 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.8, 20.9, 21.0, 21.0, 25.5, 26.3, 27.5, 62.1, 63.3, 70.2, 71.1, 72.5, 72.8, 73.1, 73.2, 75.5, 77.0, 100.7, 102.7, 111.0, 169.4, 169.6, 169.9, 170.3, 170.6, 171.0. LRMS (ESI) m/z: [M + Na]+ calcd for C29H39NO18Na 712.2; Found 712.1.
Succinimidyl (2,6-di-O-acetyl-β-d-galactopyranosyl)(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (5)
Compound S5 (69 mg, 0.10 mmol, 1.0 equiv) was dissolved in 5 mL of an AcOH:H2O (4:1) mixture and was heated at 80 °C for 1 hour with a condenser attached to the reaction flask. The reaction was then concentrated under reduced pressure followed by co-distillation with toluene under reduced pressure (3X). The crude was then purified by silica flash chromatography using 2% MeOH in CH2Cl2 as the eluent to provide lactosyl acceptor 5 as a white solid (55 mg, 0.084 mmol, 84% yield). 1H NMR (CDCl3, 600 MHz): δ = 2.08 (3H, s), 2.11 (3H, s), 2.11 (3H, s), 2.12 (3H, s), 2.13 (3H, s), 2.73 (4H, s), 3.00 (1H, br), 3.28 (1H, br), 3.61 (1H, dd, J = 2.7, 9.4 Hz), 3.64 (1H, t, J = 6.5 Hz), 3.75 (1H, m), 3.84 (1H, br), 4.10 (1H, t, J = 9.4 Hz), 4.21 (2H, m), 4.36 (1H, dd, J = 6.4, 11.5 Hz), 4.40 (1H, d, J = 7.9 Hz), 4.47 (1H, dd, J = 2.3, 12.0 Hz), 4.86 (1H, dd, J = 8.0, 9.6 Hz), 5.13 (1H, d, J = 6.4 Hz), 5.19 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.8, 21.0, 21.0, 21.0, 21.0, 25.5, 62.1, 62.4, 68.4, 70.1, 72.3, 72.7, 72.9, 73.1, 73.9, 75.7, 101.0, 102.7, 169.6, 170.2, 170.4, 170.7, 171.2, 171.8. HRMS (ESI) m/z: [M + Na]+ calcd for C26H35NO18Na 672.1746; Found 672.1750.
Phenyl (6-O-pivaloyl-β-d-galactopyranosyl)-(1→4)-2,6-diO-pivaloyl-1-thio-β-d-glucopyranoside (6)
Acceptor 6 was prepared as previously described.34 Acceptor 6 was purified by silica flash chromatography using toluene:acetone (1:1) as the eluent to provide 6 as a white solid (0.31 g, 0.45 mmol). 1H NMR (CDCl3, 600 MHz): δ = 1.19 (9H, s), 1.21 (9H, s), 1.26 (9H, s), 3.46 (1H, t, J = 9.2 Hz), 3.60 (1H, m), 3.66 (1H, m), 3.77 (4H, m), 3.91 (1H, t, J = 3.3 Hz), 4.16 (2H, m), 4.27 (3H, m), 4.36 (1H, dd, J = 4.1, 11.9 Hz), 4.50 (1H, d, J = 3.8 Hz), 4.69 (1H, d, J = 10.9 Hz), 4.72 (1H, d, J = 10.2 Hz), 4.90 (1H, t, J = 9.7 Hz), 7.28 (3H, m), 7.50 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 27.1, 27.1, 27.2, 38.7, 38.8, 38.9, 63.4, 63.7, 68.6, 70.7, 70.9, 73.2, 73.3, 74.4, 76.8, 81.5, 86.2, 104.2, 127.8, 128.9, 132.2, 133.3, 176.8, 178.8, 179.2. LRMS (ESI) m/z: [M + Na]+ calcd for C33H50O13SNa 709.3; Found 709.2.
Phenyl (6-O-benzyl-2-O-benzoyl-1-thio-β-d-galactopyranoside (7)
Acceptor 7 was prepared as previously described.35 Acceptor 7 was purified by silica flash chromatography using hexanes:EtOAc (1:1) as the eluent to provide 7 as a white solid (0.80 g, 1.7 mmol). 1H NMR (CDCl3, 600 MHz): δ = 2.95 (1H, d, J = 3.6 Hz), 3.21 (1H, d, J = 7.6 Hz), 3.78 (1H, t, J = 4.8 Hz), 3.83 (1H, m), 3.88 (2H, d, J = 5.10 Hz), 4.14 (1H, t, J = 3.5 Hz), 4.64 (2H, br), 4.84 (1H, d, J = 10.0 Hz), 5.23 (1H, t, J = 9.6 Hz), 7.28 (3H, m), 7.37 (5H, m), 7.50 (4H, m), 7.63 (1H, m), 8.11 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 69.7, 69.8, 72.2, 73.8, 74.1, 77.2, 86.0, 127.8, 127.9, 128.0, 128.5, 128.5, 128.9, 129.5, 130.1, 132.6, 132.6, 133.5, 137.7, 167.0. LRMS (ESI) m/z: [M + Na]+ calcd for C26H26O6SNa 489.1; Found 489.0.
Phenyl (4,6-O-benzylidene-2-O-benzoyl-1-thio-β-d-galactopyranoside) (8)
Acceptor 8 was prepared as previously described.31 Acceptor 8 was purified by silica flash chromatography using hexanes:EtOAc (1:1) as the eluent to provide 8 as a white solid (1.70 g, 3.66 mmol). 1H NMR (CDCl3, 600 MHz): δ = 2.65 (1H, d, J = 10.8 Hz), 3.61 (1H, s), 3.91 (1H, td, J = 10.1, 3.6 Hz), 4.06 (1H, d, J = 12.3 Hz), 4.26 (1H, d, J = 3.5 Hz), 4.43 (1H, d, J = 12.3 Hz), 4.84 (1H, d, J = 9.8 Hz), 5.28 (1H, t, J = 9.7 Hz), 5.55 (1H, s), 7.28 (2H, m), 7.34 (1H, m), 7.45 (7H, m), 7.60 (3H, m), 8.09 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 69.2, 70.1, 70.7, 73.0, 75.7, 84.9, 101.5, 126.6, 128.3, 128.3, 128.5, 128.9, 129.5, 129.9, 130.0, 131.2, 133.3, 134.0, 137.5, 166.0. LRMS (ESI) m/z: [M + Na]+ calcd for C26H24O6SNa 487.1; Found 487.1.
General procedure for sialylations in Table 1
Acceptors (1.0 equiv) were co-distilled with toluene under reduced pressure (3X) and then were combined with freshly prepared sialyl phosphate donor 3 (1.5 equiv) followed by high vacuum for 3 hours. The donor acceptor pair were then dissolved in dry CH2Cl2 (volume calculated to make 20 mM solution of acceptor), 4 Å molecular sieves were added (mass = 4 × acceptor mass), gas was exchanged with Ar, and the resulting solution was stirred at room temperature for 1 hour. The reaction mixture was cooled to −78 °C and TMSOTf (1.5 equiv) was added to initiate the reaction. The reaction was stirred at −78 °C for 1 hour and the reaction was quenched with saturated NaHCO3 with vigorous stirring as the reaction slowly warmed to room temperature. The quenched reaction mixture was then diluted with CH2Cl2 (10 mL) and the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was then analyzed by 1H NMR to determine α:β ratios based on the integrals of the Neu5Ac C3 equatorial protons. Crudes were then purified by silica flash chromatography using appropriate solvent mixtures to isolate α:β mixtures of the products to obtain the isolated yield. Isolated and pure α anomers were then characterized.
Phenyl (methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N,4-O-carbonyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-(2,6-di-O-acetyl-β-d-galactopyranosyl)(1→4)-2,3,6-tri-O-acetyl-1-thio-β-d-glucopyranoside (9)
Compound 9 was purified by silica flash chromatography using toluene:acetone (1:1) as the eluent to provide 9 as a white solid (0.29 g, 0.26 mmol, 68% yield). 1H NMR (CDCl3, 600 MHz): δ = 2.01 (3H, s), 2.02 (3H, s), 2.03 (3H, s), 2.07 (6H, s), 2.08 (3H, s), 2.10 (3H, s), 2.12 (1H, m), 2.16 (3H, s), 2.49 (3H, s), 2.70 (1H, d, J = 5.0 Hz), 2.83 (1H, dd, J = 3.5, 12.1 Hz), 3.67 (3H, m), 3.77 (2H, m), 3.81 (4H, m), 4.01 (2H, m), 4.09 (1H, t, J = 3.8 Hz), 4.14 (1H, dd, J = 6.3, 11.91 Hz), 4.40 (1H, dd, J = 2.8, 12.2 Hz), 4.51 (2H, m), 4.59 (1H, dd, J = 1.5, 9.4 Hz), 4.68 (1H, d, J = 10.1 Hz), 4.87 (2H, m), 5.16 (1H, dd, J = 7.9, 10.2 Hz), 5.20 (1H, t, J = 9.2 Hz), 5.36 (1H, dt, J = 2.8, 7.6 Hz), 5.56 (1H, dd, J = 1.6, 7.7 Hz), 7.29 (3H, m), 7.46 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.8, 20.9, 20.9, 21.0, 21.0, 21.0, 21.2, 21.6, 24.8, 35.8, 53.5, 53.6, 59.0, 62.5, 63.0, 63.5, 66.4, 69.3, 69.7, 70.4, 71.9, 72.8, 73.5, 73.9, 74.8, 76.0, 76.1, 85.4, 99.3, 100.9, 128.3, 129.0, 132.0, 133.0, 153.6, 168.4, 169.4, 169.8, 169.9, 170.2, 170.3, 170.4, 170.5, 171.2, 172.3. HRMS (ESI) m/z: [M + Na]+ calcd for C47H59NO27SNa 1124.2887; Found 1124.2898.
Succinimidyl (methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N,4O-carbonyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-(2,6-di-O-acetyl-β-d-galactopyranosyl)(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (10)
Compound 10 was purified by silica flash chromatography using toluene:acetone (1:1) as the eluent to provide 10 as a white solid (0.13 g, 0.12 mmol, 81% yield). 1H NMR (CDCl3, 600 MHz): δ = 2.00 (1H, t, J = 12.8 Hz), 2.07 (3H, s), 2.09 (3H, s), 2.11 (3H, s), 2.11 (3H, s), 2.14 (9H, s), 2.23 (3H, s), 2.50 (3H, s), 2.74 (4H, s), 2.97 (1H, dd, J = 3.2, 12.0 Hz), 3.47 (1H, br), 3.73 (3H, m), 3.81 (3H, s), 3.89 (2H, m), 3.98 (1H, dd, J = 7.1, 12.3 Hz), 4.18 (1H, t, J = 9.3 Hz), 4.23 (1H, dd, J = 6.3, 11.9 Hz), 4.27 (2H, d, J = 6.2 Hz), 4.36 (1H, dd, J = 3.0, 10.0 Hz), 4.42 (1H, dd, J = 2.0, 11.8 Hz), 4.48 (1H, dd, J = 2.4, 12.2 Hz), 4.53 (1H, dd, J = 1.7, 9.4 Hz), 4.64 (1H, d, J = 7.9 Hz), 5.05 (1H, dd, J = 8.1, 9.9 Hz), 5.12 (1H, t, J = 6.6 Hz), 5.17 (1H, t, J = 7.2 Hz), 5.24 (1H, t, J = 8.1 Hz), 5.60 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.8, 20.8, 20.9, 20.9, 20.9, 21.0, 21.2, 24.9, 25.5, 36.8, 53.1, 59.3, 62.1, 63.2, 68.9, 69.8, 69.8, 71.0, 72.2, 72.5, 72.9, 73.7, 74.5, 74.9, 75.9, 76.4, 79.3, 99.8, 101.6, 103.3, 154.5, 166.7, 168.9, 169.5, 170.0, 170.2, 170.7, 170.7, 170.8, 171.0, 171.3, 173.0. HRMS (ESI) m/z: [M + Na]+ calcd for C45H58N2O30Na 1129.2967; Found 1129.2948.
Phenyl (methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N,4-O-carbonyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-(6-O-pivaloyl-β-d-galactopyranosyl)(1→4)-2,6-di-O-pivaloyl-1-thio-β-d-glucopyranoside (11)
Compound 11 was purified by silica flash chromatography using toluene:acetone (1:1) as the eluent to provide 11 as a white solid (0.16 g, 0.14 mmol, 52% yield). 1H NMR (CDCl3, 600 MHz): δ = 1.20 (9H, s), 1.21 (9H, s), 1.26 (9H, s), 2.05 (3H, s), 2.13 (3H, s), 2.16 (3H, s), 2.21 (1H, t, J = 13.0 Hz), 2.49 (1H, d, J = 2.64Hz), 2.52 (3H, s), 3.00 (1H, dd, J = 3. 5, 12.3 Hz), 3.20 (1H, d, J = 1.7 Hz), 3.52 (1H, t, J =9.2 Hz), 3.70 (2H, m), 3.78 (4H, m), 3.84 (3H, s), 4.04 (2H, m), 4.09 (1H, dd, J = 7.9, 12.1 Hz), 4.15 (1H, dd, J = 3.3, 9.6 Hz), 4.23 (1H, dd, J = 8.1, 11.9 Hz), 4.31 (1H, d, J = 1.0 Hz), 4.40 (1H, dd, J = 4.2, 11.9 Hz), 4.47 (1H, dd, J = 2.6, 12.3 Hz), 4.51 (1H, d, J = 7.9 Hz), 4.65 (1H, dd, J = 1.1, 9.5 Hz), 4.71 (1H, d, J = 10.3 Hz), 4.88 (1H, dd, J = 1.5, 12.1 Hz), 4.94 (1H, dd, J = 9.4, 10.1 Hz), 5.55 (1H, td, J = 4.1, 10.7 Hz), 5.70 (1H, dd, J = 1.1, 8.5 Hz), 7.28 (3H, m), 7.51 (2H, m). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.7, 20.9, 21.2, 24.7, 27.1, 27.1, 27.2, 36.6, 38.7, 38.8, 53.5, 58.8, 63.2, 63.3, 63.6, 67.9, 68.8, 68.9, 70.7, 71.2, 72.6, 74.4, 74.7, 76.7, 76.9, 77.2, 82.2, 86.6, 97.9, 104.2, 127.6, 128.9, 131.8, 134.0, 153.3, 168.3, 169.8, 170.4, 170.7, 172.0, 176.7, 177.9, 178.6. HRMS (ESI) m/z: [M + Na]+ calcd for C52H73NO25SNa 1166.4085; Found 1166.4072.
Phenyl (methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N,4-O-carbonyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-2-O-benzoyl-6-O-benzyl-1-thio-β-d-galactopyranoside (12)
Compound 12 was purified by silica flash chromatography using hexanes:EtOAc (1:1) as the eluent to provide 12 as a white solid (1.30 g, 1.41 mmol, 89% yield). 1H NMR (CDCl3, 600 MHz): δ = 1.48 (3H, s), 2.02 (3H, s), 2.07 (1H, t, J = 6.3 Hz), 2.13 (3H, s), 2.44 (3H, s), 2.74 (1H, br), 2.86 (1H, dd, J = 3.4, 12.0 Hz), 3.55 (1H, dd, J = 9.4, 11.2 Hz), 3.73 (3H, s), 3.85 (5H, m), 3.95 (1H, dd, J = 7.4, 12.2 Hz), 4.43 (1H, dd, J = 2.5, 12.3 Hz), 4.49 (1H, dd, J = 1.8, 9.4 Hz), 4.52 (1H, dd, J = 3.1, 9.5 Hz), 4.61 (2H, m), 4.96 (1H, d, J = 10.0 Hz), 5.43 (1H, t, J = 9.7 Hz), 5.49 (1H, dd, J = 1.9, 8.7 Hz), 5.55 (1H, td, J = 2.4, 8.7 Hz), 7.25 (3H, m), 7.35 (5H, m), 7.49 (4H, m), 7.61 (1H, t, J = 7.4 Hz), 8.18 (2H, dd, J = 1.0, 8.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.2, 20.8, 21.3, 24.6, 35.9, 53.3, 58.8, 63.4, 67.5, 68.5, 68.9, 69.1, 71.5, 73.6, 74.9,75.2, 75.5, 76.5, 86.7, 97.5, 127.7, 127.7, 127.8, 128.4, 128.5, 128.8, 130.1, 130.2, 132.6, 132.9, 133.3, 138.1, 153.4, 165.3, 168.5, 170.0, 170.5, 170.8, 171.8. LRMS (ESI) m/z: [M + Na]+ calcd for C45H49NO18SNa 946.3; Found 946.2.
Phenyl (methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N,4-O-carbonyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-2-O-benzoyl-4,6-O-benzylidene-1-thio-βD-galactopyranoside (13)
Compound 13 was purified by silica flash chromatography using hexanes:EtOAc (1:1) as the eluent to provide 13 as a white solid (0.96 g, 1.0 mmol, 87% yield). 1H NMR (CDCl3, 600 MHz): δ = 1.76 (1H, dd, J = 12.3, 13.5 Hz), 1.82 (3H, s), 2.05 (3H, s), 2.21 (3H, s), 2.46 (3H, s), 2.92 (1H, dd, J = 3.2, 12.1 Hz), 3.48 (3H, s), 3.53 (1H, dd, J = 9.5, 11.2 Hz), 3.73 (1H, br), 3.75 (1H, m), 4.01 (1H, m), 4.12 (1H, d, J = 3.2 Hz), 4.17 (1H, dd, J = 1.4, 12.0 Hz), 4.39 (1H, dd, J = 1.4, 12.1 Hz), 4.47 (2H, m), 4.62 (1H, dd, J = 3.5, 9.7 Hz), 5.00 (1H, d, J = 9.8 Hz), 5.38 (1H, s), 5.44 (1H, t, J = 9.7 Hz), 5.57 (2H, m), 7.25 (2H, m), 7.31 (1H, m), 7.38 (3H, m), 7.43 (2H, m), 7.50 (2H, t, J = 7.8 Hz), 7.61 (3H, m), 8.16 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.7, 21.0, 21.6, 24.8, 37.2, 53.0, 58.9, 63.9, 68.1, 68.3, 69.3, 69.6, 71.6, 73.0, 73.9, 75.0, 75.1, 85.2, 96.9, 101.0, 126.7, 128.1, 128.3, 128.6, 128.8, 129.2, 130.1, 130.5, 131.4, 133.3, 134.1, 137.9, 153.5, 165.0, 168.8, 170.3, 170.6, 171.1, 172.1. LRMS (ESI) m/z: [M + Na]+ calcd for C45H47NO18SNa 944.2; Found 944.1.
Succinimidyl (4,6-O-benzylidene-2,3-di-O-benzoyl-β-d-glucopyranoside (15)
Compound 1440 (1.58 g, 2.78 mmol, 1.0 equiv) and NHS (1.60 g, 13.9 mmol, 5.0 equiv) were co-distilled with toluene under reduced pressure (3X) followed by high vacuum for 3 hours. The mixture was then dissolved in mL of dry CH2Cl2 and 4 Å molecular sieves (5 g) were added to the solution. Gas was then exchanged with Ar and the solution was stirred at room temperature for 30 minutes. The mixture was then cooled to −10 °C using a salted ice bath and then NIS (1.88 g, 8.34 mmol, 3.0 equiv) was quickly added to the reaction vessel. Gas was exchanged with Ar again, and TMSOTf (0.10 mL, 0.56 mmol, 0.2 equiv) was then introduced into the mixture and the reaction was stirred at −10 °C for 30 minutes and then at 0 °C for 1 hour. The reaction was then diluted with CH2Cl2 (10 mL) and quenched with a saturated solution of Na2S2O3 and NaHCO3 (20 mL). The quenched reaction was then filtered, and the resulting organic layer was washed with saturated Na2S2O3 and NaHCO3 (20 mL, 2X). The collected organic layer was then washed with brine, dried over Na2SO4, filtered, and then concentrated under reduced pressure. The crude material was then purified by silica flash chromatography using hexanes:EtOAc (4:1) as the eluent with 10% CH2Cl2 to ensure compound solubility on the column. Compound 15 was isolated as a white solid (1.34 g, 2.34 mmol, 84% yield). 1H NMR (CDCl3, 600 MHz): δ = 2.72 (4H, s), 3.91 (1H, m), 4.02 (1H, t, J = 10.3 Hz), 4.41 (1H, dd, J = 4.9, 10.7 Hz), 4.53 (1H, t, J = 9.3 Hz), 5.49 (1H, d, J = 5.9 Hz), 5.65 (1H, s), 5.76 (2H, m), 7.35 (3H, m), 7.44 (6H, m), 7.56 (2H, m), 8.04 (2H, d, J = 7.1 Hz), 8.10 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 25.4, 66.6, 68.3, 70.9, 72.3, 77.3, 101.5, 103.1, 126.2, 128.3, 128.4, 128.5, 128.9, 129.2, 129.2, 130.0, 130.1, 133.4, 133.6, 136.7, 165.0, 165.5, 170.4. HRMS (ESI) m/z: [M + Na]+ calcd for C31H27NO10Na 596.1527; Found 596.1529.
Succinimidyl (2,3-di-O-benzoyl-6-O-benzyl-β-d-glucopyranoside (16)
Compound 15 (0.80 g, 1.4 mmol, 1.0 equiv) was co-distilled with toluene under reduced pressure (3X), set under high vacuum for 3 hours, and was then dissolved in mL of dry CH2Cl2. Gas was then exchanged with Ar and the solution was cooled to 0 °C followed by the addition of Et3SiH (0.67 mL, 4.2 mmol, 3.0 equiv) into the reaction vessel. This mixture was then stirred for 10 minutes at 0 °C and then BF3 Et2O (0.69 mL, 5.6 mmol, 4.0 equiv) was introduced into the reaction dropwise. The reaction was then stirred for 1.5 hours at 0 °C. The reaction was then quenched with saturated NaHCO3 (20 mL). The aqueous layer was extracted with CH2Cl2 (20 mL, 3X) and the combined organic layers were washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude was then purified by silica flash chromatography using toluene:EtOAc (4:1) as the eluent with 10% CH2Cl2 to ensure compound solubility. Compound 16 was isolated as a white solid (0.65 g, 1.1 mmol, 81% yield). 1H NMR (CDCl3, 600 MHz): δ = 2.67 (4H, m), 3.41 (1H, br), 3.80 (1H, m), 3.85 (1H, m), 3.95 (1H, dd, J = 3.9, 10.6 Hz), 4.06 (1H, t, J = 9.2 Hz), 4.65 (2H, q, J = 11.5 Hz), 5.33 (1H, d, J = 7.8 Hz), 5.5 (1H, t, J = 9.1 Hz), 5.70 (1H, dd, J = 7.9, 9.1 Hz), 7.33 (1H, m), 7.40 (8H, m), 7.55 (2H, m), 8.02 (2H, d, J = 7.1 Hz), 8.08 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 25.4, 69.7, 69.9, 70.6, 73.8, 75.5, 76.6, 103.5, 127.7, 127.9, 128.4, 128.5, 128.5, 128.8, 129.1, 130.0, 130.1, 133.4, 133.7, 137.8, 165.4, 167.2, 170.1. HRMS (ESI) m/z: [M + Na]+ calcd for C31H29NO10Na 598.1684; Found 598.1680.
Phenyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)2-O-benzoyl-4,6-O-benzylidene-1-thio-β-d-galactopyranoside (17)
Compound 13 (0.66 g, 0.72 mmol, 1.0 equiv) was dissolved in MeOH (9 mL) and cooled to 0 °C. Freshly prepared 1.0 M NaOMe in MeOH (1 mL) was then introduced into the reaction mixture to generate a 0.1 M NaOMe solution with the substrate, and was stirred for 15 minutes at 0 °C. The reaction was stirred at room temperature for 1 hour and then quenched with Amberlite® IR120 acidic resin, filtered, and concentrated under reduced pressure to reveal a white solid in quantitative yield. The solid was dried by co-distillation with toluene under reduced pressure (3X) followed by high vacuum for 3 hours. Dry intermediate was then dissolved in pyridine ( mL) followed by the addition of DMAP (18 mg, 0.14 mmol, 0.2 equiv). Gas was exchanged with Ar and then the mixture was cooled to 0 °C. Once cool, Ac2O (1.4 mL, 14 mmol, 20 equiv) was introduced and the reaction was stirred for 12 hours while gradually warming to room temperature. The reaction was then cooled to 0 °C again and quenched with excess MeOH and stirred for 10 minutes. The crude was concentrated under reduced pressure, co-distilled with toluene under reduced pressure (3X) and was reconstituted in CH2Cl2 (20 mL). The crude was then washed with saturated NaHCO3 (20 mL, 3X) and the resulting organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was then purified by silica flash chromatography using toluene:acetone (3:1) as the eluent to reveal 17 as a white solid (0.51 g, 0.55 mmol, 76% yield over 2 steps). 1H NMR (CDCl3, 600 MHz): δ = 1.69 (1H, t, J = 12.7 Hz), 1.82 (3H, s), 1.83 (3H, s), 1.95 (3H, s), 2.07 (3H, s), 2.23 (3H, s), 2.56 (1H, dd, J = 4.5, 12.9 Hz), 3.56 (3H, s), 3.67 (1H, br), 3.90 (2H, m), 4.00 (1H, dd, J = 6.2, 12.2 Hz), 4.05 (1H, d, J = 3.4 Hz), 4.13 (1H, d, J = 11.2 Hz), 4.35 (1H, dd, J = 2.5, 12.4 Hz), 4.39 (1H, d, J = 12.0 Hz), 4.61 (1H, dd, J = 3.4, 9.7 Hz), 4.71 (1H, m), 4.99 (1H, d, J = 9.8 Hz), 5.06 (1H, br), 5.25 (1H, d, J = 9.5 Hz), 5.38 (1H, s), 5.43 (1H, t, J = 9.8 Hz), 5.52 (1H, m), 7.25 (3H, m), 7.38 (3H, m), 7.44 (2H, m), 7.51 (2H, t, J = 7.7 Hz), 7.60 (3H, m), 8.16 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.6, 20.8, 20.9, 21.5, 23.2, 38.2, 49.1, 52.8, 62.6, 66.9, 67.6, 68.1, 68.8, 69.3, 69.6, 72.3, 73.3, 73.6, 85.3, 96.7, 101.0, 126.6, 127.9, 128.1, 128.4, 128.7, 129.1, 130.0, 130.4, 131.6, 133.1, 133.7, 137.8, 164.9, 168.8, 170.1, 170.2, 170.3, 170.8, 170.9. HRMS (ESI) m/z: [M + Na]+ calcd for C46H51NO18SNa 960.2719; Found 960.2715.
Succinimidyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)(2→3)-2-O-benzoyl-4,6-O-benzylidene-1-amino-β-d-galactopyranoside (S6)
Donor 17 ( mg, 0.053 mmol, 1.5 equiv) and acceptor 16 (21 mg, 0.036 mmol, 1.0 equiv) were combined and co-distilled with toluene under reduced pressure (3X) and set under high vacuum for 3 hours. The donor and acceptor pair were dissolved in dry CH2Cl2 (5 mL) followed by the addition of 4 Å molecular sieves (0.2 g) and gas was exchanged with Ar. The mixture was stirred at room temperature for 1 hour before cooling the mixture to 0 °C. NIS (36 mg, 0.16 mmol, 4.5 equiv) was then introduced into the reaction mixture and gas was exchanged with Ar again followed by the addition of TMSOTf (2.0 μL, 0.011 mmol, 0.3 equiv). The reaction was then stirred at 0 °C for 1.5 hours. The reaction was then diluted with CH2Cl2 (5 mL) and quenched with a saturated solution of Na2S2O3 and NaHCO3 (10 mL). The quenched reaction was then filtered, and the resulting organic layer was washed with saturated Na2S2O3 and NaHCO3 (10 mL, 2X). The resulting organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude solid was then purified by silica flash chromatography using toluene:acetone (3:1) as the eluent to reveal compound S6, and not desired compound. This procedure was repeated using TfOH as the acid catalyst and resulted in the same product. 1H NMR (CDCl3, 600 MHz): δ = 1.68 (1H, t, J = 12.8 Hz), 1.84 (3H, s), 1.93 (3H, s), 1.97 (3H, s), 2.09 (3H, s), 2.24 (3H, s), 2.53 (1H, br), 2.56 (1H, dd, J = 4.4, 12.9 Hz), 2.65 (3H, m), 3.49 (3H, s), 3.72 (1H, s), 3.97 (2H, m), 4.04 (1H, dd, J = 6.2, 12.4 Hz), 4.15 (2H, m), 4.24 (1H, dd, J = 1.4, 12.3 Hz), 4.34 (1H, dd, J = 2.6, 12.4 Hz), 4.65 (1H, dd, J = 3.3, 9.9 Hz), 4.70 (1H, m), 5.06 (1H, br), 5.28 (1H, d, J = 10.2 Hz), 5.37 (1H, s), 5.50 (1H, td, J = 3.0, 12.2 Hz), 5.53 (1H, d, J = 9.2 Hz), 6.36 (1H, t, J = 9.6 Hz), 7.34 (1H, m), 7.39 (2H, t, J = 7.3 Hz), 7.47 (2H, t, J = 7.8 Hz), 7.58 (1H, t, J = 7.4 Hz), 7.66 (2H, d, J = 7.0 Hz), 8.02 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.7, 20.8, 20.9, 21.5, 23.2, 27.9, 38.6, 49.3, 52.8, 62.6, 66.8, 67.0, 67.6, 68.2, 68.7, 68.7, 72.5, 72.7, 72.9, 78.7, 96.7, 101.7, 127.1, 128.3, 128.6, 129.2, 129.6, 129.9, 133.4, 137.9, 165.4, 168.8, 170.1, 170.3, 170.8, 171.0, 174.8, 176.2. HRMS (ESI) m/z: [M + Na]+ calcd for C44H50N2O20Na 949.2849; Found 949.2838.
Trichloroacetimidatyl (methyl 5-acetamido-4,7,8,9-tetra-Oacetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-2-O-benzoyl-4,6-O-benzylidene-β-d-galactopyranoside (18)
Compound 17 (0.41 g, 0.44 mmol, 1.0 equiv) was dissolved in an acetone:H2O (4:1) mixture ( mL) and then cooled to 0 °C. While stirring, NBS (94 mg, 0.53 mmol, 1.2 equiv) was added gradually to the reaction and the mixture was stirred for 30 minutes. After 30 minutes of mixing, NBS (78 mg, 0.44 mmol, 1.0 equiv) was added and the mixture was stirred at 0 °C for another 1 hour. The reaction was then concentrated under reduced pressure until the solution became turbid. The turbid mixture was diluted with CH2Cl2 (20 mL) and then quenched and washed with saturated Na2S2O3 and NaHCO3 (15 mL, 3X). The organic layer was then washed with brine, dried with Na2SO4, filtered, and then concentrated under reduced pressure. The crude hemiacetal was passed through a short bed of silica using toluene:acetone (2:1) as the eluent to give the crude hemiacetal intermediate as a white solid (0.31 g, 0.36 mmol, 82% crude). The crude hemiacetal was then co-distilled with toluene under reduced pressure (3X) and set under high vacuum for 3 hours. The crude hemiacetal was then dissolved in dry CH2Cl2 (5 mL) followed by the addition of 4 Å molecular sieves (1 g), trichloroacetonitrile (144 μL, 1.44 mmol, 4.0 equiv), and the gas was exchanged with Ar. The solution was stirred at room temperature for 30 minutes. The mixture was then cooled to 0 °C followed by the addition of DBU ( μL, 0.072 mmol, 0.2 equiv) and the reaction was then stirred for 1.5 hours at 0 °C. The reaction was filtered and then concentrated under reduced pressure to provide the crude donor. The crude material was then purified by silica flash chromatography using toluene:acetone (3:1) as the eluent to provide donor 18 as a white solid (0.32 g, 0.32 mmol, 73% yield over 2 steps). 1H NMR (CDCl3, 600 MHz): δ = 1.79 (1H, t, J = 12.7 Hz), 1.90 (3H, s), 1.96 (3H, s), 2.06 (3H, s), 2.16 (3H, s), 2.21 (3H, s), 2.59 (1H, dd, J = 4.4, 12.9 Hz), 3.52 (3H, s), 4.08 (3H, m), 4.14 (1H, br), 4.21 (1H, d, J = 12.5 Hz), 4.26 (1H, dd, J = 2.7, 12.5 Hz), 4.32 (1H, d, J = 12.6 Hz), 4.42 (1H, d, J = 3.1 Hz), 4.78 (1H, m), 5.11 (1H, d, J = 9.4 Hz), 5.18 (1H, dd, J = 3.3, 10.7 Hz), 5.37 (1H, dd, J = 1.5, 9.8 Hz), 5.47 (1H, s), 5.49 (1H, m), 5.70 (1H, dd, J = 3.3, 10.7 Hz), 6.79 (1H, d, J = 3.3 Hz), 7.37 (3H, m), 7.47 (2H, t, J = 7.8 Hz), 7.54 (2H, d, J = 6.7 Hz), 7.59 (1H, t, J = 7.4 Hz), 8.07 (2H, d, J = 7.1 Hz), 8.49 (1H, s). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.8, 20.9, 21.4, 23.2, 38.7, 49.5, 52.9, 62.5, 65.2, 67.0, 67.4, 68.0, 68.6, 69.0, 72.7, 73.5, 91.2, 95.1, 96.6, 100.6, 126.3, 128.2, 128.3, 128.5, 129.0, 129.1, 129.6, 129.8, 133.4, 137.7, 160.4, 165.5, 169.0, 169.8, 170.0, 170.3, 170.7, 171.0. HRMS (ESI) m/z: [M + Na]+ calcd for C42H47Cl3N2O19Na 1011.2; Found 1011.1.
Phenyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)4-O-acetyl-2-O-benzoyl-6-O-benzyl-1-thio-β-d-galactopyranoside (20)
Compound 12 (1.3 g, 1.4 mmol, 1.0 equiv) was dissolved in MeOH (18 mL) and cooled to 0 °C. Freshly prepared 1.0 M NaOMe in MeOH (2.0 mL) was then introduced to the reaction mixture to generate a 0.1 M NaOMe solution with the substrate, and was stirred for 15 minutes at 0 °C. The reaction was then stirred at room temperature for 1 hour and quenched with Amberlite® IR120 acidic resin, filtered, and concentrated under reduced pressure to reveal a white solid in quantitative yield. The solid was dried by co-distillation with toluene under reduced pressure (3X) followed by high vacuum for 3 hours. Dry intermediate was then dissolved in pyridine ( mL) followed by the addition of DMAP (35 mg, 0.28 mmol, 0.2 equiv). Gas was exchanged with Ar and the mixture was cooled to 0 °C. Once cool, Ac2O (3.3 mL, 35 mmol, 25 equiv) was introduced and the reaction was stirred for 12 hours while gradually warming to room temperature. The reaction was then cooled to 0 °C again and quenched with excess MeOH and stirred for 10 minutes. The crude was concentrated under reduced pressure, co-distilled with toluene under reduced pressure (3X) and was reconstituted in CH2Cl2 (20 mL). The crude was then washed with saturated NaHCO3 (20 mL, X 3) and the resulting organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was then purified by silica flash chromatography using toluene:acetone (3:1) as the eluent to reveal 20 as a white solid (1.02 g, 1.04 mmol, 74% yield over 2 steps). 1H NMR (CDCl3, 600 MHz): δ = 1.43 (3H, s), 1.74 (1H, t, J = 12.4 Hz), 1.80 (3H, s), 1.99 (3H, s), 2.02 (3H, s), 2.08 (3H, s), 2.17 (3H, s), 2.56 (1H, dd, J = 4.6, 12.6 Hz), 3.51 (1H, dd, J = 6.1, 10.0 Hz), 3.61 (2H, m), 3.82 (1H, m), 3.86 (3H, s), 3.90 (1H, dd, J = 5.7, 12.5 Hz), 3.98 (1H, t, J = 6.2 Hz), 4.27 (1H, dd, J = 2.4, 12.5 Hz), 4.48 (1H, d, J = 11.6 Hz), 4.56 (1H, d, J = 11.6 Hz), 4.76 (1H, dd, J = 3.2, 9.6 Hz), 4.85 (1H, m), 4.96 (1H, d, J = 10.3 Hz), 5.10 (2H, m), 5.19 (1H, dd, J = 2.8, 9.5 Hz), 5.32 (1H, t, J = 9.8 Hz), 5.57 (1H, m), 7.25 (3H, m), 7.35 (5H, m), 7.50 (4H, m), 7.59 (1H, t, J = 7.4 Hz), 8.19 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.2, 20.8, 20.8, 21.5, 23.2, 37.4, 48.8, 53.2, 62.3, 66.5, 67.4, 68.4, 68.5, 69.3, 69.4, 71.8, 72.4, 73.5, 75.8, 86.5, 96.8, 127.7, 127.7, 127.7, 128.3, 128.4, 128.8, 130.2, 130.4, 132.3, 133.0, 133.2, 138.0, 165.3, 168.0, 170.1, 170.3, 170.3, 170.5, 170.7, 170.9. HRMS (ESI) m/z: [M + Na]+ calcd for C48H55NO19SNa 1004.2981; Found 1004.2967.
Succinimidyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)(2→3)-(4-O-acetyl-2-O-benzoyl-6-O-benzyl-β-d-galactopyranosyl)-(1→4)-2,3-di-O-benzoyl-6-O-benzyl-β-d-glucopyranoside (2)
Donor 20 (0.36 g, 0.37 mmol, 1.5 equiv) and acceptor 16 (0.14 g, 0.25 mmol, 1.0 equiv) were combined and co-distilled with toluene under reduced pressure (3X) and set under high vacuum for 3 hours. The donor acceptor pair were dissolved in dry CH2Cl2 (5 mL) followed by the addition of 4 Å molecular sieves (1 g) and gas was exchanged with Ar. The mixture was stirred at room temperature for 1 hour before cooling the mixture to 0 °C. NIS (0.25 g, 1.1 mmol, 4.5 equiv) was then introduced into the reaction mixture and gas was exchanged with Ar again followed by the addition of TfOH (6 μL, 0.07 mmol, 0.3 equiv). The reaction was then stirred at 0 °C for 1.5 hours and the reaction was diluted with CH2Cl2 (15 mL) and then quenched with saturated Na2S2O3 and NaHCO3 (20 mL). The organic layer was separated, filtered, and washed with saturated Na2S2O3 and NaHCO3 (20 mL, 2X). The resulting organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude solid was then purified by silica flash chromatography using toluene:acetone (3:1) as the eluent to afford compound 2 as a white solid (0.32 g, 0.22 mmol, 87% yield). Only β anomer was observed due to anchimeric assistance. 1H NMR (CDCl3, 600 MHz): δ = 1.41 (3H, s), 1.71 (1H, t, J = 12.4 Hz), 1.78 (3H, s), 1.94 (3H, s), 1.97 (3H, s), 2.09 (3H, s), 2.13 (3H, s), 2.56 (5H, m), 2.80 (1H, t, J = 9.2 Hz), 2.91 (1H, dd, J = 4.8, 9.5 Hz), 3.46 (2H, m), 3.55 (1H, dd, J = 2.8, 10.7 Hz), 3.65 (1H, d, J = 9.2 Hz), 3.71 (1H, t, J = 7.6 Hz), 3.78 (3H, s), 3.81 (1H, q, J = 10.5 Hz), 3.96 (1H, dd, J = 6.1, 12.5 Hz), 4.02 (1H, d, J = 11.7 Hz), 4.10 (1H, d, J = 11.8 Hz), 4.24 (2H, br), 4.34 (1H, t, J = 9.3 Hz), 4.38 (1H, dd, J = 2.28, 12.42 Hz), 4.58 (1H, dd, J = 3.2, 10.1 Hz), 4.83 (1H, td, J = 4.6, 11.9 Hz), 4.94 (1H, d, J = 3.0 Hz), 4.98 (2H, t, J = 8.9 Hz), 5.19 (2H, dd, J = 7.6, 10.0 Hz), 5.24 (1H, d, J = 7.4 Hz), 5.56 (1H, t, J = 7.6 Hz), 5.67 (2H, t, J = 8.6 Hz), 7.18 (2H, t, J = 7.3 Hz), 7.27 (3H, m), 7.32 (7H, m), 7.39 (2H, t, J = 7.8 Hz), 7.50 (4H, m), 7.57 (1H, t, J = 7.3 Hz), 8.02 (4H, t, J = 6.7 Hz), 8.26 (2H, d, J = 7.2 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.3, 20.7, 20.8, 20.9, 21.5, 23.1, 25.3, 37.2, 48.7, 53.2, 62.6, 66.4, 66.6, 67.2, 67.3, 68.7, 69.5, 70.4, 71.0, 71.7, 71.7, 72.8, 72.9, 74.0, 75.0, 75.7, 96.9, 101.3, 102.9, 127.4, 127.4, 127.4, 127.5, 128.2, 128.3, 128.3, 128.3, 128.6, 129.1, 129.2, 129.8, 129.9, 130.0, 130.1, 130.5, 133.0, 133.2, 138.0, 138.4, 164.9, 165.0, 165.2, 167.9, 170.0, 170.0, 170.2, 170.3, 170.4, 170.8, 170.8. HRMS (ESI) m/z: [M + Na]+ calcd for C73H78N2O29Na 1469.4582; Found 1469.4562.
Succinimidyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-(2→3)-(4-O-acetyl-2-O-benzoyl-β-d-galactopyranosyl)(1→4)-2,3-di-O-benzoyl-β-d-glucopyranoside (S7)
Compound 2 (0.32 g, 0.22 mmol, 1.0 equiv) was dissolved in a mixture of MeOH:EtOAc (1:3, mL) followed by the addition of palladium hydroxide on carbon (55 mg, 0.25 g/mmol substrate) at room temperature. The reaction was then stirred for 1 hour under an atmosphere of H2 gas (balloon, 1 atm). After 1 hour the reaction was diluted with 10 mL of the same MeOH:EtOAc mixture followed by bubbling Ar gas into the reaction solution. The reaction was then filtered over a bed a Celite® followed by a short bed of silica using 1% MeOH in CH2Cl2 as the eluent. Compound S7 was then concentrated under reduced pressure to a white solid (0.25 g, 0.20 mmol, 89% yield). 1H NMR (CDCl3, 600 MHz): δ = 1.46 (3H, s), 1.74 (1H, t, J = 12.5 Hz), 1.80 (3H, s), 1.98 (3H, s), 2.02 (3H, s), 2.13 (3H, s), 2.18 (3H, s), 2.52 (1H, dd, J = 4.5, 12.6 Hz), 2.68 (4H, s), 2.73 (1H, dd, J = 6.8, 12.0 Hz), 2.97 (1H, dd, J = 6.4, 12.0 Hz), 3.50 (1H, t, J = 6.7 Hz), 3.56 (1H, td, J = 3.1, 9.3 Hz), 3.69 (1H, dd, J = 2.7, 10.7 Hz), 3.76 (5H, m), 3.83 (1H, q, J = 10.4 Hz), 3.90 (1H, dd, J = 7.9, 12.2 Hz), 4.37 (1H, t, J = 8.8 Hz), 4.46 (1H, dd, J = 2.5, 12.2 Hz), 4.55 (1H, dd, J = 3.4, 10.1 Hz), 4.69 (1H, d, J = 3.2 Hz), 4.73 (1H, td, J = 6.1, 14.9 Hz), 4.91 (1H, d, J = 7.9 Hz), 4.96 (1H, d, J = 10.3 Hz), 5.13 (1H, dd, J = 2.6, 9.6 Hz), 5.22 (2H, m), 5.62 (2H, m), 5.70 (1H, td, J = 3.9, 12.1 Hz), 7.41 (4H, q, J = 8.2 Hz), 7.53 (4H, m), 7.60 (1H, t, J = 7.3 Hz), 8.02 (4H, d, J = 8.3 Hz), 8.26 (2H, d, J = 7.1 Hz). 13C{1H} NMR (CDCl3, 150 MHz): δ = 20.3, 20.6, 20.8, 20.8, 21.4, 23.1, 25.3, 37.4, 48.7, 53.3, 59.9, 60.5, 63.3, 67.0, 67.1, 68.1, 69.2, 70.1, 71.1, 71.1, 71.9, 73.0, 74.5, 76.3, 96.8, 100.9, 104.1, 128.2, 128.3, 128.4, 128.5, 129.1, 129.1, 129.8, 129.9, 130.0, 130.4, 133.2, 133.2, 133.3, 164.9, 165.2, 165.2, 168.1, 170.2, 170.3, 170.4, 170.6, 170.8, 171.7, 171.9. HRMS (ESI) m/z: [M + Na]+ calcd for C59H66N2O29Na 1289.3643; Found 1289.3627.
Succinimidyl (5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonic acid)(2→3)-(4-O-acetyl-2-O-benzoyl-β-d-galactopyranosyl)(1→4)-2,3-di-O-benzoyl-β-d-glucopyranoside (21)
Compound S7 (0.25 g, 0.20 mmol, 1.0 equiv) was dissolved in mL pyridine followed by the addition of LiI (0.54 g, 4.0 mmol, 20 equiv) in the dark. This solution was then refluxed in the dark for 3 hours, after which pyridine was removed under reduced pressure followed by co-distillation with toluene under reduced pressure (3X). This crude was then purified by silica flash chromatography using 5% MeOH in CH2Cl2 as the eluent to afford compound 21 as a white solid (0.18 g, 0.14 mmol, 68% yield). Note: The white solid pertains to the protonated form of the compound. A yellow oily material may result as the lithiated species. This species has identical NMR characterization as the protonated form but exhibits the lithiated form in MS analysis i.e. [M➔Li + Na]+. 1H NMR (CD3OD, 600 MHz): δ = 1.58 (1H, br), 1.70 (3H, br), 1.78 (3H, s), 1.92 (3H, s), 2.04 (3H, s), 2.08 (3H, s), 2.10 (3H, s), 2.51 (1H, br), 2.66 (4H, s), 3.52 (1H, br, J = 9.6 Hz), 3.62 (2H, m), 3.69 (4H, m), 3.90 (1H, br), 3.94 (1H, dd, J = 5.6, 11.0 Hz), 4.04 (1H, dd, J = 6.2, 12.3 Hz), 4.21 (1H, br), 4.32 (1H, t, J = 9.4 Hz), 4.39 (1H, dd, J = 2.3, 12.4 Hz), 4.46 (1H, br, J = 9.1 Hz), 4.94 (1H, d, J = 8.00 Hz), 5.23 (2H, br), 5.33 (1H, d, J = 8.00 Hz), 5.44 (1H, t, J = 8.6 Hz), 5.54 (1H, br), 5.66 (1H, t, J = 9.2 Hz), 7.43 (4H, m), 7.56 (4H, m), 7.65 (1H, t, J = 7.4 Hz), 7.99 (4H, m), 8.17 (2H, d, J = 7.5 Hz). 13C{1H} NMR (CD3OD, 150 MHz): δ = 19.4, 19.5, 20.1, 21.2, 25.0, 48.2, 49.1, 59.7, 62.4, 62.8, 67.5, 68.1, 70.7, 71.0, 71.2, 72.3, 73.0, 73.5, 74.5, 76.1, 101.0, 103.3, 128.0, 128.1, 128.4, 129.3, 129.5, 129.5, 129.5, 129.7, 129.8, 130.0, 133.0, 133.0, 165.5, 165.8, 170.3, 170.4, 170.5, 170.9, 170.9, 171.3, 171.9, 172.0. HRMS (ESI) m/z: [M + Na]+ calcd for C58H63N2O29LiNa 1281.3569; Found 1281.3561. LRMS (ESI): [M]- calcd for C58H64N2O29 1252.4; Found 1252.1.
Aminooxy (5-acetamido-3,5-dideoxy-d-glycero-α-d-galactonon-2-ulopyranosylonic acid)-(2→3)-(β-d-galactopyranosyl)(1→4)-β-d-glucopyranoside (1)
Compound 21 ( mg, 0.016 mmol, 1.0 equiv) was dissolved in EtOH (1 mL) in a microwave reaction tube. Hydrazine hydrate ( μL, 0.96 mmol, 60 equiv) was then introduced into the vessel which was then capped and stirred at room temperature for 1 hour. Compound 21 was then subjected to microwave conditions (200 W, 90 °C) for 1 hour total in two 30 minute repetitive cycles. MS analysis revealed a single peak at 687.6 [M]−. This peak is the acetone adduct of the aminooxy GM3 which spontaneously forms with residual acetone in the injector of the MS instrument. The reaction mixture was then concentrated under reduced pressure and then passed through a P2 biogel column using H2O as the eluent. Fractions were found to contain final compound by staining silica gel plates. These fractions were frozen and the lyophilized to reveal compound 1 as a white solid (8.0 mg, 0.012 mmol, 77% yield). 1H NMR (D2O, 600 MHz): δ = 1.70 (1H, t, J = 12.1 Hz), 1.94 (3H, s), 2.66 (1H, dd, J =4.6, 12.4 Hz), 3.26 (1H, m), 3.57 (12H, m), 3.77 (4H, m), 3.86 (1H, d, J = 3.1 Hz), 3.92 (1H, dd, J = 2.1, 12.2 Hz), 4.02 (1H, dd, J = 3.2, 9.9 Hz), 4.43 (1H, d, J = 7.9 Hz), 4.51 (1H, d, J = 8.3 Hz). 13C{1H} NMR (D2O, 150 MHz): δ = 22.0, 39.6, 51.6, 59.9, 61.0, 62.5, 67.4, 68.1, 68.3, 69.3, 71.3, 71.7, 72.8, 74.3, 74.7, 75.1, 75.4, 78.0, 99.8, 102.6, 104.8, 173.9, 175.0. HRMS (ESI) m/z: [M + Na]+ calcd for C23H40N2O19Na 671.2117; Found 671.2106.
GM3-PS A1 (23)
Polysaccharide PS A1 (22) was isolated and purified as previously described.8, 41 PS A1 (1.0 mg, 9.1 X 10−9 mol) was dissolved in NaOAc buffer (1.0 mL, 0.1 M, pH = 5). To this solution was added 55 μL of 10 mM NaIO4 solution (5.5 × 10−7 mol). The reaction was allowed to shake gently for 90 min in the dark at room temperature. Excess NaIO4 was quenched by adding ethylene glycol and the mixture was continually shaken for another 20 min in the dark. The oxidized PS A1 was purified with a centrifugal filter (Vivaspin, MWCO = 30 kDa). PS A1 was redissolved in NaOAc buffer (1.0 mL, 0.1 M, pH = 5). Subsequently, 2.9 mg (4.4 × 10−6 mol) of β-aminooxy GM3 (1) was added and the reaction was gently shaken for 18 hours in the dark at room temperature. Unreacted β-aminooxy GM3 (1) was removed using a 30 kDa centrifugal filter and purified GM3-PS A1 (23) conjugate was obtained as a lyophilized white foam (1.0–1.1 mg).
GM3-PS A1 NMR Analysis
NMR analysis was performed using a Bruker Avance III 600 MHz spectrometer and data processed with Bruker TopSpin 4.0.3. GM3-PS A1 1H spectra were obtained with a probe temperature of 60 °C using the zg30 pulse program (D1 = 3.0 sec, AQ = 2.7 sec, NS = 128). Integrations of the key “diagnostic” peaks were obtained using an automated baseline correction. These integrations suggested a 1:2.33 ratio between GM3:PSA1 repeating unit. On average, PS A1 has at least 120 repeating units12 which translates to approximately 51.5 GM3 conjugation sites per PS A1 molecule.
Supplementary Material
ACKNOWLEDGMENT
Support for this work was provided by the National Institutes of Health (NIH NCI R01 CA156661) & (NIH NIGMS U01 GM125271). We acknowledge Dr. Yong-Wah Kim in the Department of Chemistry and Biochemistry at the University of Toledo for providing assistance with the NMR instrument. We also acknowledge Dr. Dragan Isailovic and David Baliu-Rodriguez for their assistance with high resolution mass spectrometry.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
NMR spectra for new or appropriate compounds (.pdf)
REFERENCES
- (1).Johnson RW; Stieglitz J The Velocity of Hydrolysis of Stereoisomeric Hydrazones and Oximes. J. Am. Chem. Soc 1934, 56, 1904–1908. [Google Scholar]
- (2).Kalia J; Raines RT Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem. Int. Ed 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hossain F; Nishat S; Ghosh S; Boga S; Hymel GT; Andreana PR Synthesis of glycoimmunogen Tn-Thr-PS A1 via hydrazone bond and stability optimization of PS A1 monosaccharide mimics under vaccine development conditions. J. Carbohydr. Chem 2020, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Buskas T; Li Y; Boons G-J The Immunogenicity of the Tumor-Associated Antigen Lewisy May Be Suppressed by a Bifunctional Cross-Linker Required for Coupling to a Carrier Protein. Chem. - Eur. J 2004, 10, 3517–3524. [DOI] [PubMed] [Google Scholar]
- (5).Yin Z; Chowdhury S; McKay C; Baniel C; Wright WS; Bentley P; Kaczanowska K; Gildersleeve JC; Finn MG; BenMohamed L; Huang X Significant Impact of Immunogen Design on the Diversity of Antibodies Generated by CarbohydrateBased Anticancer Vaccine. ACS Chem. Biol 2015, 10, 2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jencks WP The Reaction of Hydroxylamine with Activated Acyl Groups. I. Formation of O-Acylhydroxylamine1. J. Am. Chem. Soc 1958, 80, 4581–4584. [Google Scholar]
- (7).Jencks WP The Reaction of Hydroxylamine with Activated Acyl Groups. II. Mechanism of the Reaction. J. Am. Chem. Soc 1958, 80, 4585–4588. [DOI] [PubMed] [Google Scholar]
- (8).De Silva RA; Wang Q; Chidley T; Appulage DK; Andreana PR Immunological Response from an Entirely Carbohydrate Antigen: Design of Synthetic Vaccines Based on Tn−PS A1 Conjugates. J. Am. Chem. Soc 2009, 131, 9622–9623. [DOI] [PubMed] [Google Scholar]
- (9).Bourgault JP; Trabbic KR; Shi M; Andreana PR Synthesis of the tumor associative alpha-aminooxy disaccharide of the TF antigen and its conjugation to a polysaccharide immune stimulant. Org. Biomol. Chem 2014, 12, 1699–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shi M; Kleski KA; Trabbic KR; Bourgault J-P; Andreana PR Sialyl-Tn Polysaccharide A1 as an Entirely Carbohydrate Immunogen: Synthesis and Immunological Evaluation. J. Am. Chem. Soc 2016, 138, 14264–14272. [DOI] [PubMed] [Google Scholar]
- (11).Kleski KA; Trabbic KR; Shi M; Bourgault J-P; Andreana PR Enhanced Immune Response Against the Thomsen-Friedenreich Tumor Antigen Using a Bivalent Entirely Carbohydrate Conjugate. Molecules 2020, 25, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kalka-Moll WM; Tzianabos AO; Bryant PW; Niemeyer M; Ploegh HL; Kasper DL Zwitterionic Polysaccharides Stimulate T Cells by MHC Class II-Dependent Interactions. J. Immunol 2002, 169, 6149–6153. [DOI] [PubMed] [Google Scholar]
- (13).Cobb BA; Wang Q; Tzianabos AO; Kasper DL Polysaccharide Processing and Presentation by the MHCII Pathway. Cell 2004, 117, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).De Silva RA; Appulage DK; Pietraszkiewicz H; Bobbitt KR; Media J; Shaw J; Valeriote FA; Andreana PR The entirely carbohydrate immunogen Tn-PS A1 induces a cancer cell selective immune response and cytokine IL-17. Cancer Immunol., Immunother 2012, 61, 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Xia L; Schrump DS; Gildersleeve JC Whole-Cell Cancer Vaccines Induce Large Antibody Responses to Carbohydrates and Glycoproteins. Cell Chem. Biol 2016, 23, 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng C; Terreni M; Sollogoub M; Zhang Y Ganglioside GM3 and Its Role in Cancer. Curr. Med. Chem 2019, 26, 2933–2947. [DOI] [PubMed] [Google Scholar]
- (17).Cheever MA; Allison JP; Ferris AS; Finn OJ; Hastings BM; Hecht TT; Mellman I; Prindiville SA; Viner JL; Weiner LM; Matrisian LM The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin. Cancer. Res 2009, 15, 5323–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pan Y; Chefalo P; Nagy N; Harding C; Guo Z Synthesis and Immunological Properties of N-Modified GM3 Antigens as Therapeutic Cancer Vaccines. J. Med. Chem 2005, 48, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hudak JE; Yu HH; Bertozzi CR Protein Glycoengineering Enabled by the Versatile Synthesis of Aminooxy Glycans and the Genetically Encoded Aldehyde Tag. J. Am. Chem. Soc 2011, 133, 16127–16135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu KKC; Danishefsky SJ A striking example of the interfacing of glycal chemistry with enzymatically mediated sialylation: a concise synthesis of ganglioside GM3. J. Am. Chem. Soc 1993, 115, 4933–4934. [Google Scholar]
- (21).Sakamoto H; Nakamura S; Tsuda T; Hashimoto S Chemoselective glycosidation strategy based on glycosyl donors and acceptors carrying phosphorus-containing leaving groups: a convergent synthesis of ganglioside GM3. Tetrahedron Lett. 2000, 41, 7691–7695. [Google Scholar]
- (22).Meo CD; Demchenko AV; Boons G-J A Stereoselective Approach for the Synthesis of α-Sialosides. J. Org. Chem 2001, 66, 5490–5497. [DOI] [PubMed] [Google Scholar]
- (23).Lee KJ; Mao S; Sun C; Gao C; Blixt O; Arrues S; Hom LG; Kaufmann GF; Hoffman TZ; Coyle AR; Paulson J; Felding-Habermann B; Janda KD Phage-Display Selection of a Human Single-Chain Fv Antibody Highly Specific for Melanoma and Breast Cancer Cells Using a Chemoenzymatically Synthesized GM3−Carbohydrate Antigen. J. Am. Chem. Soc. 2002, 124, 12439–12446. [DOI] [PubMed] [Google Scholar]
- (24).Liu Y; Ruan X; Li X; Li Y Efficient Synthesis of a Sialic Acid α(2→3)Galactose Building Block and Its Application to the Synthesis of Ganglioside GM3. J. Org. Chem 2008, 73, 4287–4290. [DOI] [PubMed] [Google Scholar]
- (25).Zheng XJ; Yang F; Zheng M; Huo CX; Zhang Y; Ye XS Improvement of the immune efficacy of carbohydrate vaccines by chemical modification on the GM3 antigen. Org. Biomol. Chem 2015, 13, 6399–6406. [DOI] [PubMed] [Google Scholar]
- (26).Sugiarto G; Lau K; Qu J; Li Y; Lim S; Mu S; Ames JB; Fisher AJ; Chen X A Sialyltransferase Mutant with Decreased Donor Hydrolysis and Reduced Sialidase Activities for Directly Sialylating Lewisx. ACS Chem. Biol 2012, 7, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tanaka H; Nishiura Y; Takahashi T Stereoselective Synthesis of Oligo-α-(2,8)-Sialic Acids. J. Am. Chem. Soc 2006, 128, 7124–7125. [DOI] [PubMed] [Google Scholar]
- (28).Crich D; Li W O-Sialylation with N-Acetyl-5-N,4-O-Carbonyl-Protected Thiosialoside Donors in Dichloromethane: Facile and Selective Cleavage of the Oxazolidinone Ring. J. Org. Chem 2007, 72, 2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kancharla PK; Navuluri C; Crich D Dissecting the influence of oxazolidinones and cyclic carbonates in sialic acid chemistry. Angew. Chem 2012, 51, 11105–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Martin TJ; Brescello R; Toepfer A; Schmidt RR Synthesis of phosphites and phosphates of neuraminic acid and their glycosyl donor properties — convenient synthesis of GM3. Glycoconjugate J. 1993, 10, 16–25. [DOI] [PubMed] [Google Scholar]
- (31).Hsu CH; Chu KC; Lin YS; Han JL; Peng YS; Ren CT; Wu CY; Wong CH Highly alpha-selective sialyl phosphate donors for efficient preparation of natural sialosides. Chem. - Eur. J 2010, 16, 1754–1760. [DOI] [PubMed] [Google Scholar]
- (32).Toşa MI; Podea PV; Paizs C; Irimie FD Chemoenzymatic synthesis of (R)- and (S)-1-heteroarylethanols. Tetrahedron: Asymmetry 2008, 19, 2068–2071. [Google Scholar]
- (33).Fraser-Reid B; Wu Z; Udodong UE; Ottosson H Armed/disarmed effects in glycosyl donors: rationalization and sidetracking. J. Org. Chem 1990, 55, 6068–6070. [Google Scholar]
- (34).Zheng M; Ye X-S Synthesis of N-modified ganglioside GM3 derivatives. Tetrahedron 2012, 68, 1475–1482. [Google Scholar]
- (35).Chuang H-Y; Ren C-T; Chao C-A; Wu C-Y; Shivatare SS; Cheng T-JR; Wu C-Y; Wong C-H Synthesis and Vaccine Evaluation of the Tumor-Associated Carbohydrate Antigen RM2 from Prostate Cancer. J. Am. Chem. Soc 2013, 135, 11140–11150. [DOI] [PubMed] [Google Scholar]
- (36).Zhang Z; Ollmann IR; Ye X-S; Wischnat R; Baasov T; Wong C-H Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc. 1999, 121, 734–753. [Google Scholar]
- (37).Christensen HM; Oscarson S; Jensen HH Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res 2015, 408, 51–95. [DOI] [PubMed] [Google Scholar]
- (38).Moumé-Pymbock M; Furukawa T; Mondal S; Crich D Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans–gauche Conformation of C5–C6 Bonds. J. Am. Chem. Soc 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chan J; Lewis AR; Indurugalla D; Schur M; Wakarchuk W; Bennet AJ Transition state analysis of Vibrio cholerae sialidase-catalyzed hydrolyses of natural substrate analogues. J. Am. Chem. Soc 2012, 134, 3748–3757. [DOI] [PubMed] [Google Scholar]
- (40).Bera S; Linhardt RJ Design and Synthesis of Unnatural Heparosan and Chondroitin Building Blocks. J. Org. Chem 2011, 76, 3181–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Baumann H; Tzianabos AO; Brisson JR; Kasper DL; Jennings HJ Structural elucidation of two capsular polysaccharides from one strain of Bacteroides fragilis using highresolution NMR spectroscopy. Biochemistry 1992, 31, 4081–4089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.