

Abstract
Next-generation sequencing (NGS) has been widely adopted for clinical HLA typing and advanced immunogenetics researches. Current methodologies still face challenges in resolving cis-trans ambiguity involving distant variant positions, and the turnaround time is affected by testing volume and batching. Nanopore sequencing may become a promising addition to the existing options for HLA typing. The technology delivered by the MinION sequencer of Oxford Nanopore Technologies (ONT) can record the ionic current changes during the translocation of DNA/RNA strands through transmembrane pores and translate the signals to sequence reads. It features simple and flexible library preparations, long sequencing reads, portable and affordable sequencing devices, and rapid, real-time sequencing. However, the error rate of the sequencing reads is high and remains a hurdle for its broad application. This review article will provide a brief overview of this technology and then focus on the opportunities and challenges of using nanopore sequencing for high-resolution HLA typing and immunogenetics research.
Keywords: Nanopore sequencing, next-generation sequencing, human leukocyte antigen, consensus sequence, sequencing error
Introduction
Next-generation sequencing (NGS) technologies have revolutionized high-resolution HLA typing in recent years [1]. Many clinical laboratories have quickly adopted these technologies for patient and donor HLA typing in the form of commercially available assays [2–4] or bona fide laboratory-developed tests [5, 6]. Two types of platforms enabled this shift in HLA typing methodology: 1) those generating short sequencing reads of hundreds of bases, such as Roche 454 [7], Illumina [3–5], and Ion Torrent [2], and 2) the PacBio platform that generates long reads of thousands or more bases [8, 9]. The outcomes have been excellent shorter turnaround time, decreased ambiguities, increased throughput, and more productive donor searching results [5, 6, 10, 11]. The implementation of these technologies has also electrified the field of immunogenetics research, empowering the recent International HLA & Immunogenetics Workshop and expanding the existing HLA reference database [12–14].
However, innovations in biotechnology never rest. Alternative HLA typing strategies not relying on PCR-based target enrichment, such as the hybrid capture of HLA genes followed by NGS, are being developed to overcome the limitations of PCR dropout and allelic imbalance (e.g., AlloSeq®Tx 17 from CareDx, Brisbane, CA). Novel and robust bioinformatic tools are emerging to infer HLA typing using data from whole-genome sequencing (WGS), whole-exome sequencing, and RNA sequencing [15–23]. Also, there is always the next gadget on the horizon that can potentially make a big difference.
Here, we provide a concise review of nanopore sequencing with a focus on its potential benefit for HLA typing and immunogenetics research. The prototype of nanopore sequencing was pioneered by Branton, Church, Deamer, and colleagues in their academic laboratories in the early 1990s [24]. The first commercial nanopore sequencing device, MinION, was released by Oxford Nanopore Technologies (ONT) in 2014 and has been successfully employed by independent laboratories for diverse sequencing applications since then.
Anatomy of the MinION device and how it works
Nanopore sequencing on the MinION device is fulfilled by transmembrane protein channels with nanoscopic apertures just big enough for single-stranded nucleic acid polymers to pass through [25]. The pore is situated on an electrically resistant membrane that separates two voltage-biased compartments. During the translocation of single-stranded DNA through the pore, changes in ionic currents can be recorded continuously by sensors. The signals are subsequently segmented as discrete events and deciphered computationally into shifting nucleotide sequences occupying the pore. Single-stranded RNA can be sequenced directly as well [26]. Another critical component of the chemistry is a helicase enzyme, or motor protein, that unwinds double-stranded DNA and pulls single-stranded DNA through the pore. This controlled, ratcheting-like process increases the signal-to-noise ratio to allow discrimination of nucleotides at a single-base resolution [25].
The average speed of translocation of single DNA strands through the pore is 450 bases per second [27], which translates into less than 10 seconds to sequence a full-gene amplicon of class I HLA genes (~4 kb). Once a DNA strand is completely translocated, the same pore will be immediately available to sequence the next. A total of 512 active channels, each consists of a group of four nanopores within their individual scaffold, are embedded on a MinION flow cell. High throughput can be achieved by sequencing of DNA molecules that sequentially pass through hundreds of active pores on a flow cell. The array of sequencing channels on the flow cell is mounted on top of a sensory array and an Application Specific Integrated Circuit (ASIC), which is connected to the base of MinION through connector pins. The entire MinION device, including the flow cell and base, weighs about 100 grams and can fit into a regular-sized pocket.
Depending on the library preparation kit used, the workflow from input DNA samples to sequencing data takes minutes to a few hours. For amplicon-based library, PCR amplicons are typically purified and end-repaired and then ligated to a sequencing adapter before sequencing (Fig. 1A, right panel). The Y-shaped adapter is associated with a tether on one strand and a motor protein on the other, which allow the library fragments to be recruited to and ratcheted through the pores. After loading the library onto the flow cell, a laptop or desktop computer is connected to the MinION device via a USB port to power and control the MinION during sequencing. The segmented sequential events (“squiggles”), or the raw ionic current signals, are then basecalled using a neural network algorithm and separated into high-quality (pass) or low-quality (fail) reads for downstream analysis.
Fig. 1.
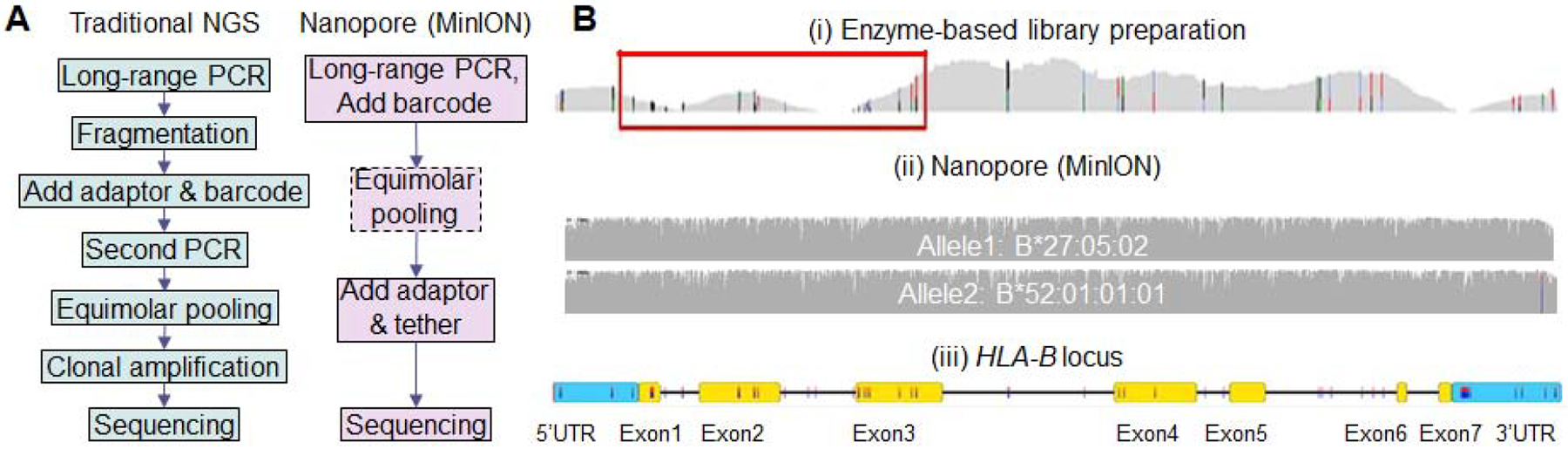
Gaps in HLA typing technologies. (A) Comparison of workflows by traditional NGS (commercial assays on Illumina and Ion-torrent platforms) and nanopore sequencing. Equimolar pooling is optional for MinION if not multiplexing. (B) Selected coverage plots of the HLA-B locus. Library preparation using enzyme-based fragmentation can produce uneven coverage patterns. Key exons may be poorly covered (red boxes) in affected samples leading to error or ambiguity (i). In contrast, nanopore reads cover the entire gene without significant variations (ii). Panels (i) and (iii) are adapted from reference [43]; panel (ii) was generated in the author’s research laboratory.
Strength and weakness of nanopore sequencing
Nanopore sequencing is the only NGS technology not dependent on DNA synthesis catalyzed by the polymerase. Several attractive features make this technology a promising option for a wide range of clinical and research applications. First, rapid and flexible library preparation protocols can effectively shorten the time from sample to data. Second, nanopore sequencing can produce extremely long reads of thousands to millions of bases [28], which is only limited by the sizes of DNA fragments in the library. For genomics analysis, these long reads are suitable for phasing distant variants and interrogating structural variants and low-complexity regions [28, 29], which have been a weakness for short-read sequencing platforms. Third, the affordable pricing of MinION, currently available at as low as $1000 for two flow cells and a sequencing kit (MinION Starter Pack), allows users to access NGS without significant capital investments. Fourth, MinION is a portable device capable of delivering NGS without the conventional constraint of space and time. It was famously transported to West Africa during the Ebola virus disease epidemic to sequence the virus genome for real-time monitoring of outbreaks [30, 31]. Finally, nanopore sequencing can discriminate not only the regular DNA bases but also uracil in RNA [32] and nucleobases with epigenetic modifications such as methylation [33], and this unique capability is leading genomics research into the uncharted territory of direct sequencing of RNA and genomic DNA molecules.
The above features of nanopore sequencing can potentially address several unmet needs in the field of HLA typing and immunogenetics research, as outlined in the following sections. On the other hand, the error rate of the nanopore sequencing reads, approximately 10–15%, as documented in recent reports [28, 34, 35], remains significantly higher than those of short-read sequencing platforms. The error rate of nanopore reads is influenced by the sequencing chemistry and basecalling algorithm, as reviewed recently in an excellent article by Rang and colleagues [27]. Given the complexity of HLA genes with densely packed single-nucleotide variants (SNVs) and occasional indels, the success of nanopore sequencing in HLA typing will be hinged on continued improvement in read accuracy and the development of robust bioinformatics tools to overcome any persisting errors.
Nanopore sequencing for HLA typing
Several recent publications have reported some early attempts of nanopore sequencing for HLA typing. These studies mostly enriched the target HLA genes by long-range PCR and sequenced the amplicons using a variety of library preparation methods and nanopore sequencing chemistries, all amidst the fast-paced development of the technology itself [15, 36–42]. Both commercial software and academic bioinformatics tools were employed to determine the HLA typing with encouraging results (Table 1).
Table 1.
Recent publications that used nanopore sequencing data for HLA typing
| Reference | Target genes and library preparation | ONT Chemistry | Bioinformatics | Accuracy |
|---|---|---|---|---|
| Ammar et al., 2015 [36] | HLA-A and -B (n=1), genomic DNA Sequencing protocol (SQK-MAP003) | R7.3, 2D reads | BLASR for read alignment, GATK HLACaller package | Discordant for all four alleles |
| Liu et al., 2018 [37, 38] | HLA-A, -B, and -C (n=20), full-gene amplicon, native barcode ligation | R9.4, 2D reads (n=10) and 1D reads (n=10) | BLASR for read mapping, Athlon pipeline for genotyping | Evaluated key exons only; 100% concordant at 2-field resolution |
| Ton et al., 2018 [39] | HLA-B (n=49), key-exon amplicon, PCR barcoding kit (EXP-PBC096) | R9.4, 2D reads | SeqPilot v.4.3.1 (JSI medical systems) + Nanopolish pipeline | 100% concordant at the G group resolution among 13 samples with reference typing results |
| Klasberg et al., 2019 [40, 62, 63] | HLA-A, -B, -C, -DRB1, -DQB1, and -DPB1 (n=96), full-gene amplicon followed by barcoding PCR and ONT adapter ligation | Not reported (conference abstract) | Nanotyper pipeline (DR2S long-read path) | Accuracy of 100% and 92% for HLA-A and -B at 4-field resolution; further adaptions needed for HLA-C and class II genes |
| Dilthey et al., 2019 [15] | HLA-A, -B, -C, -DQA1, -DQB1, and -DRB1 full-gene amplicon sequencing (n=1) and whole-genome sequencing (n=1) | R9.4, 1D reads | HLA-LA program | Average accuracy of 98% at the G group resolution |
| Montgomery et al., 2020 [41] | Class I HLA genes (n=12), cDNA-PCR sequencing kit (SQK-PCS108) | R9.4, 1D reads | Minimap2 for read mapping, Athlon pipeline for genotyping | Accuracy of 94% at the 2-field G group resolution. |
| Matern et al., 2020 [48] | HLA-DRA full-gene amplicon sequencing (n=98) | Flow cell version not reported, 1D2 reads | Nanopore Prospector and GenDx NGSEngine (V2.11) | Identification of 22 novel DRA alleles due to intronic and 3’ UTR variants. |
| De Santis et al., 2020 [42] | HLA-A, -B, -C, -DQA1, - DQB1, -DRB1, -DRB345, - DPA1 and -DPB1 full-gene or partial-gene amplicon sequencing (n=42, SQK-LSK109 kit) | R9.4, 1D reads generated on Flongle flow cells | GenDx NGSEngine (V2.13) | 100% concordance at 2-field resolution. Some manual editing required at high-background or homopolymer regions. |
Streamlined library preparation
The library preparations for sequencing on the Illumina and Ion Torrent platforms typically require enzyme-based fragmentation of PCR amplicons of target HLA genes. The overlapping, short sequencing reads of hundreds of bases are pieced together during the analysis to identify the HLA alleles present in the sample. This “shotgun” approach typically requires a long process of many steps (Fig. 1A, left panel). Bias may also be introduced to regions with high GC content during amplification and library preparation, causing inadequate coverage of key exons (Fig. 1B). This problem may be protocol dependent and has been shown to cause ambiguity or typing errors of several HLA genes with some protocols [43] but not as pronounced with others as reported by the same laboratory [44]. Increasing the data available for analysis by generating more sequencing reads per locus may mitigate such risk but at the cost of efficiency. Alternatively, additional primers have been spiked into the initial PCR reaction to further enrich the key exons in certain assays to compensate for the negative bias during library preparation [45].
In contrast, MinION can sequence long-range amplicons spanning a full HLA gene without the need for fragmentation. Neither does nanopore sequencing require clonal amplification on the Illumina flow cell or ion-sphere particles of the Ion Torrent platform. Therefore, the library preparation for nanopore sequencing can be significantly simplified (Fig. 1A, right panel). Barcodes can be added to the long-range amplicon by two main strategies. First, a four-primer PCR can amplify target genes using HLA-specific inner primers with 5’ adapter sequences, followed by a second amplification using outer primers with overlapping adapter sequences and unique barcode sequences. Second, barcode fragments can be directly ligated to the long-range amplicons. Next, the sequencing adapter with the motor protein and tether can be added to the DNA duplexes by ligation, or through a proprietary rapid attachment reaction within a minute. The hands-on time to complete these procedures is typically 1–2 hours. As the nanopore approach removes the bias intrinsic to enzyme-based fragmentation, uniform coverage can be achieved throughout target genes, including regions with high-GC content (Fig. 1B).
There used to be an option of sequencing both strands of a duplex molecule that are connected by a hairpin linker at one end; the other end was ligated to a Y-shaped sequencing adapter with the motor protein and tether. The template read and the linked complementary read were combined by pairwise alignment to generate the so-called 2D reads. This library preparation option is no longer available. Instead, 1D reads can be generated by adding Y-shaped adapters to both ends of a duplex DNA and sequencing the two strands independently of each other. These reads have a higher error rate than the 2D reads [27], but the 1D approach increases the efficiency of library preparation and sequencing throughput. The feasibility of using 1D reads for accurate HLA typing has been demonstrated in several recent studies [40, 46]. Another method called 1D2 allows sequential sequencing of the template and complementary strands of a duplex molecule without physically linking them with a hairpin [47, 48]. This method also results in improved read accuracy but is not yet compatible with amplicon sequencing.
To improve the accuracy of nanopore sequencing reads, Li and colleagues developed a library preparation method called INC-seq (Intramolecular-ligated Nanopore Consensus Sequencing) [49]. Double-stranded DNA molecules are self-ligated to form circular DNA molecules followed by rolling-circle amplification. The amplicons are sequenced on MinION to generate sequences with repeating units. Consensus sequence based on repeating units of the same origin showed a median accuracy of over 97%, allowing accurate 16S rRNA-based bacterial profiling at the species level. The application of INC-seq to HLA typing has not been reported to our knowledge. INC-seq is analogous to the single-molecule real-time (SMRT) sequencing on the PacBio platform, where double-stranded DNA molecules are converted to single-stranded circular DNA to be sequenced repeatedly around the circle. Multiple sub-reads of the same origin are generated and can be combined to create highly accurate consensus reads for HLA typing [8].
Long-reads for improved haplotype phasing
Although NGS on the short-read sequencing platforms have reduced the cis-trans ambiguity involving variants that are hundreds of bases apart, the ability of short sequencing reads to phase distant variants is limited. In our recent evaluation of a commercial assay on the Ion Torrent S5 instrument for two-field resolution HLA typing [50], we encountered cis-trans ambiguity across different exons in 26 out of 1685 genotypes (1.5%) primarily affecting the HLA-A, -B, -DPB1 and -DQB1 loci. Most of the ambiguous genotypes include one or two rare alleles, which can be reported with NMDP codes, and the limitation may not be unique to the method or sequencer we evaluated. For Illumina sequencing libraries, despite the short read length, the inclusion of longer fragments with pair-end sequencing can improve the phasing of distant variants to some extent [51].
The long reads from nanopore sequencing can readily phase distant variant positions. One example is the genotype DQB1*06:03 and DQB1*06:04 versus the alternative genotype DQB1*06:39 and DQB1*06:41. The two genotypes cannot be distinguished with short reads from Ion Torrent due to the cis-trans ambiguity across exon 2 and exon 3. Sequencing reads must bridge two variant positions that are 2668 bases apart to be able to resolve the ambiguity (Fig. 2). Paired-end reads from the Illumina platform may not be able to resolve these genotypes either due to the lack of library fragments that happen to span these variant positions. We performed nanopore sequencing of full-gene amplicons of DQB1, and sequencing reads with an average length of 6654 bases were generated, which unequivocally support the assignment of DQB1*06:03 and DQB1*06:04 in a sample we tested (Fig. 2).
Fig. 2.

Resolving cis-trans ambiguity with nanopore sequencing long reads. Reads from full-length amplicon sequencing of DQB1 were fully aligned to the genotype DQB1*06:03:01:01 and DQB1*06:04:01:01 with uniform coverage, but not the alternative genotype DQB1*06:39 and DQB1*06:41. The cis-trans ambiguity involves two variant positions in exon 2 and exon 3 that are 2668 bases apart, and Ion Torrent sequencing did not resolve this ambiguity. The fold coverage is display at the upper left corner of each coverage plot.
Affordable, scalable, and portable HLA typing
The advantages in library preparation and variant phasing described above are not unique to nanopore sequencing, as similar progress has been made on the PacBio platform as well. However, PacBio sequencers require significant capital investment and laboratory space, while nanopore sequencing removes such requirement, allowing smaller HLA laboratories with limited resources to access NGS. It will not be surprising if nanopore sequencing starts to penetrate the molecular testing and HLA typing market in developing countries.
The multiplexing capability and scalability of nanopore sequencing also impact the cost of HLA typing on this platform. A MinION device can generate up to 50 Gb of data per run under optimized conditions (https://nanoporetech.com). This data yield can theoretically provide a theoretical 2000x coverage for 11 HLA genes and approximately 50 samples multiplexed in a run, even after cutting the run time by half to 24 hours and excluding low-quality reads (~50%) that cannot be basecalled. Currently, up to 96 samples can be indexed using the PCR Barcoding kits from ONT. The above data output and multiplexing capacity would probably suit the need of most hospital-based HLA laboratories. But in case a higher throughput is desired, the platform can be scaled up with the GridION and PromethION devices that can generate up to 250 Gb and 5.2 Tb per device, respectively.
In the other direction, nanopore sequencing can also be scaled down with Flongle, which is a smaller flow cell with 126 channels (vs. 512 for MinION) used in conjunction with a reusable adapter. Almost proportional to the decrease in the number of channels for sequencing, Flongle costs about a quarter of the price of a MinION flow cell. For under $100 per flow cell, up to 2 Gb of data can be generated on Flongle per run. When sequencing amplicons, we regularly obtain a range of 0.2 −1.6 Gb of data per run depending on how much library is loaded onto the flow cells. Even more futuristic is the SmidgION in the pipeline that is probably the smallest sequencing device that can be plugged into a smartphone for sequencing. With these smaller, inexpensive flow cells, it will be affordable to perform NGS for HLA typing on a small batch of samples, or on a single sample without batching at all. De Santis and colleagues successfully implemented single sample typing of 11 HLA loci using Flongle flow cells [42]. This unprecedented level of flexibility will benefit laboratories with low testing volumes as well as larger laboratories that need an option for urgent high-resolution HLA typing.
Rapid, real-time HLA typing
Nanopore sequencing does not require a fixed sequencing time to complete a predetermined number of cycles as on the Illumina or Ion Torrent platforms. Instead, sequencing reads can be generated and basecalled in real time followed by downstream data analysis. These features make it feasible to perform rapid HLA typing by sequencing for as much time as needed to generate sufficient read coverage for the intended application. We have observed that thousands of long reads covering the full-length class I genes can be generated within half an hour of sequencing on a MinION R9.4 flow cell, and accurate HLA typing can be determined using these data (Fig. 3). Considering the entire HLA typing process including DNA extraction (~1 hour), target enrichment by PCR (~3 hours), ONT library preparation (~ 2 hours), nanopore sequencing (~ 0.5–1.5 hours), and data analysis (~0.5 hours), it becomes realistic to develop an assay on the ONT platform to complete a high-resolution HLA typing by NGS within a few hours [42]. That being said, the sequencing speed may vary significantly between different runs (Fig. 3, compare left and right panels), and process standardization will be necessary to achieve a predictable data output.
Fig. 3.
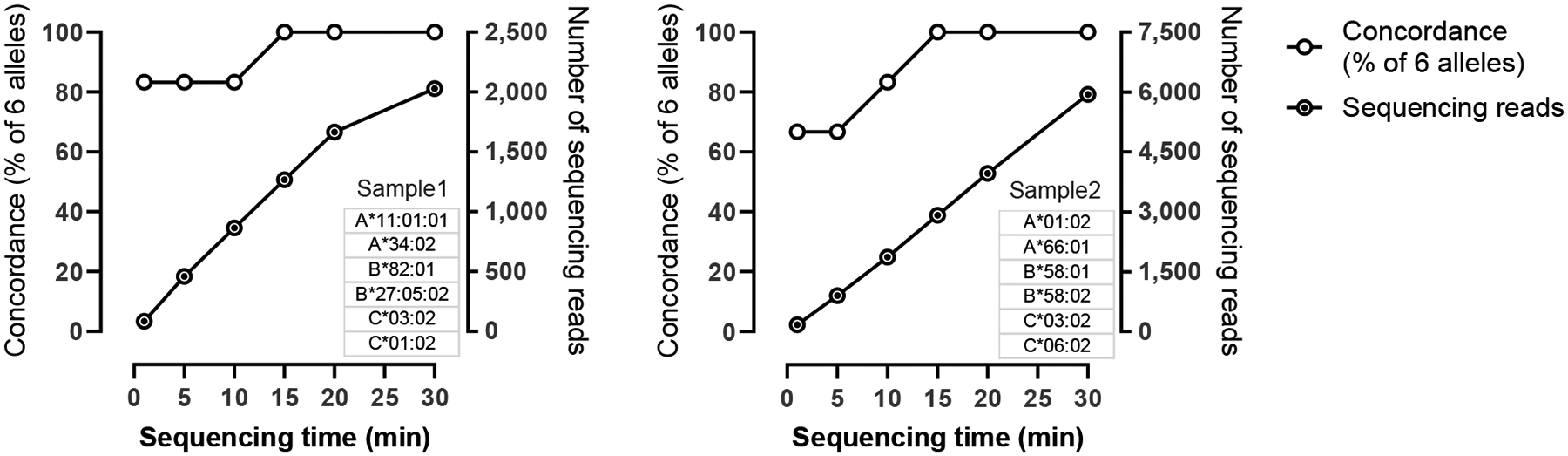
Rapid sequencing of class I HLA genes on MinION. 1D libraries of full-gene amplicons of three class I HLA genes from Samples 1 (left panel) and 2 (right panel) were sequenced on two independent runs using R9.4 MinION flow cells. The number of high-quality sequencing reads (right axis) and the accuracy of typing results based on key exons (left axis) are plotted over time. The consensus sequences fully matched those of the reference alleles (exons 2 and 3) in 15 minutes for both samples. At 1, 5, and 10 minutes, the total numbers of mismatches between the consensus sequences and reference sequences are 1, 1, and 1 for sample 1, and 4, 4, and 1 for sample 2.
Furthermore, the number of on-target sequencing reads per sample and per locus in a multiplexed sequencing assay may vary substantially [50] depending on the PCR reagent and precision of library pooling process. It is critical to ensure balanced representation of all target genes and samples to maximize the multiplexing capability for NGS-based HLA typing. While optimization of the primer mix and process standardization are critical, balanced sequencing of target amplicons can potentially be achieved on the ONT platform through the real-time selective sequencing developed by Loose and colleagues [52]. The process was implemented through an open-source “Read Until” software that matches the ionic current tracing (the segmented sequential events or “squiggle” data) to reference sequences in squiggle space and in real-time. If correct matches were identified for the first 250 events, the amplicon is regarded as from the target region and will be selectively sequenced until a prespecified goal has been reached (e.g., certain coverage depth). Amplicons from the off-target regions were rejected from corresponding pores by a reversal of voltage across these pores. Amplicons from target regions that had been sufficiently covered were also rejected. The approach effectively prioritized the sequencing of multiple target regions and normalized their coverages, which will be beneficial in the setting of amplicon-based typing of multiple HLA genes. Although the method appears to be computationally demanding, it has the potential to be optimized for broader applications.
Bioinformatics
Using long but noisy nanopore sequencing reads for HLA typing requires bioinformatic solutions distinct from those designed for shorter but more accurate reads from the Illumina and Ion Torrent platforms. An excellent and comprehensive review of the bioinformatics for HLA typing using NGS data was published recently [53]. This section will focus on the bioinformatic approaches to HLA typing using nanopore reads.
The first attempt to type HLA-A and -B genes through amplicon sequencing on an earlier version of MinION (R7.3 flow cell) was unsuccessful, with four out of four alleles assigned incorrectly [36]. The result could be explained by the high error rates of earlier nanopore reads and the lack of customized bioinformatic tools at the time. The GATK HLACaller, initially designed for short reads from the 454 platform [54], was used to assign HLA alleles in this study, and the algorithm turned out to be incompatible with error-prone nanopore reads. With the continued improvement in sequencing chemistry and basecalling method, the read error rate of 1D reads from the R9.4/R9.5 flow cells has been hovering around 85–90%, using the recurrent neural network algorithm for basecalling [27]. Subsequent efforts to perform HLA typing using these noisy reads have explored three main strategies as outlined below: 1) consensus allele matching, 2) graph alignment-based allele assignment, and 3) allele-specific read clustering and hierarchical scoring.
Consensus sequence can effectively correct random errors in individual nanopore reads given sufficient coverage. High-quality consensus sequences have been generated using nanopore reads alone for homogenous haploid samples using tools such as Canu [55], Freebayes [56], Nanocorrect [57], and Racon [58]. For example, Loman and colleagues developed Nanocorrect to assemble the E. coli K-12 MG1655 genome de novo and Nanopolish to refine the assembly using the squiggle (signal-level) data. The approach achieved a 99.5% nucleotide identity at a ~29x theoretical coverage [57], demonstrating a potential pathway to overcoming the read-level noise. For consense-based HLA typing by amplicon sequencing on MinION, we developed the Athlon pipeline [37] to first identify one (homozygous) or two (heterozygous) candidate alleles at class I HLA loci through two processes: 1) read mapping to a collection of known reference sequences in the IMGT/HLA database, and 2) comparison of total read depth at the antigen and allele levels. Next, consensus sequences were generated using Freebayes [56] for reads realigned to each candidate allele. Finally, the consensus sequences were compared with the IMGT/HLA database, and the best-matched alleles were selected for the final assignment. This proof-of-concept study considered exons 2 and 3 that encode the antigen-recognitition domains (ARD). At this resolution, Athlon achieved a 100% accuracy using either 2D reads or 1D reads from the R9.4 flow cells. Despite the encouraging results, a relatively high coverage (≥ 1000 1D reads per locus) was required as shown in the downsampling analysis. In addition to the limitation in typing resolution at 2-field level (only considering key exons by the pipeline), Athlon may be susceptible to allele dropout depending on the allele balance in the library and could not handle class II HLA typing at the time of publication. The consensus accuracy of nanopore sequencing reads currently lags behind PacBio (>99.999% at 30x coverage depth) [59]. Even with an identity approaching 99.9% between the consensus sequence and ground truth with nanopore reads [55], about 4 erroneous bases may be present in the consensus for a typical class I allele (~4,000 bases in full length), which will complicate the final allele assignment. This effect may be inconspicuous when Athlon restricted the analysis to key exons with less than 600 bases in total length. These residual errors may be difficult to eliminate completely if they represent systematic errors intrinsic to the nanopore sequencing chemistry or basecalling method.
Instead of mapping sequencing reads to a collection of reference sequences, the graph alignment strategy identifies linear alignments between sequencing reads and a population reference graph (PRG) that combines known reference sequences into a generative model for variations within target genes [60, 61]. For the inference of the final HLA type, all alignments are scored, and the most likely pair of underlying alleles at G group resolution are reported. The strategy was initially implemented as HLA*PRG for whole-genome sequencing (WGS) data from the Illumina platform and achieved an accuracy of 99.4% for 158 alleles analyzed [61]. One caveat of HLA*PRG was its high computational demands. An improved implementation, HLA*LA [15], allows optimization of linear alignments projected on the PRG through a stepwise process, including alignment inspection, polishing, and extension. HLA*LA supports the analysis of more diverse types of NGS data. With exome and low-coverage WGS data from the Illumina platform, HLA*LA demonstrated equal or superior accuracies when compared with other variation-aware alignment approaches, including HLA*PRG [61], Kourami [16], and xHLA [17]. Importantly, HLA*LA is the only graph alignment-based program that has been shown to successfully analyze noisy long reads from PacBio and nanopore platforms, with accuracies ranging from 95% to 100% for targeted sequencing data. If validated with additional nanopore sequencing data, the graph alignment approach may offer an excellent option for HLA typing at the G-group resolution.
Klasberg and colleagues recently reported the nanotyper pipeline for amplicon-based HLA typing, which features read clustering and hierarchical scoring [40, 62, 63]. With this method, nanopore reads were first mapped to a generic reference of the target gene and then clustered into allele-specific read sets. The clustering was implemented based on polymorphic positions that are phase-informative and unlikely to be sequencing artifacts. Next, reads in each allele-specific set were used to create a multiple sequence alignment, and the final genotype was determined by hierarchical scoring that prioritized key exons followed by non-key exons and then non-coding sequences. The authors determined 4-field HLA typings in 94 samples, which were benchmarked against genotypes generated by Illumina and PacBio sequencing [40]. The concordance rates were 99.4–100% for class I genes and 95% for HLA-DQB1, and 60% of these results were unambiguous. Two primary sources of ambiguity were identified, one being the incomplete coverage of the 3’-UTR and the other due to the failure to differentiate homopolymer tracks of different lengths. As the method continues to mature, especially for all relevant class II loci, nanopore sequencing may become a serious contender in ultra-high-resolution HLA typing.
In addition to the above academic bioinformatic tools, some commercial software such as SeqPilot (JSI medical systems GmbH, Germany) and NGSengine (GenDx, Utrecht, Netherland) have also been used to analyze nanopore sequencing data for HLA typing with promising results [39, 42, 64]. Nevertheless, homopolymer errors will continue to be the major challenge in this application as the translocation of extended homopolymer tracks (> 5-mers) through a nanopore does not cause changes in the ionic current signal; the number of bases within a homopolymer region cannot be reliably inferred due to the nonuniform speed of translocation [27]. One compromise is to exclude these difficult regions from decision making during allele assignment, especially when they are located in introns, and accept the increased ambiguity [48, 65]. The relatively high error rate and homopolymer issue of nanopore reads also makes it challenging to ascertain the presence of novel alleles not in the existing database. The hybrid consensus approach combining data from nanopore sequencing and other methods may be useful in this context to achieve excellent phasing and accuracy at the same time [14, 48]. To ultimately root out the problem, innovation in the nanopore design and basecalling method will be necessary. The latest R10.3 nanopore has a longer channel with dual recognition sites that are separate from each other to enhance the resolution of homopolymer tracks [66]. The full validation and integration of this new development are likely to boost the performance of nanopore sequencing and qualify the technology for clinical HLA typing in the near future.
Other immunogenetics applications beyond HLA typing
Nanopore sequencing offers a lot more opportunities for immunogenetics research than just HLA genotyping. One natural application that takes advantage of the nanopore read length is to generate extended haplotypes of the major histocompatibility complex (MHC) region. In the recent effort to assemble a reference genome for the human GM12878 Utah/Ceph cell line by nanopore sequencing, a protocol was developed to generate ultra-long reads with an N50 of greater than 100kb, meaning that reads with a length of 100kb or greater sum to at least half of the total bases generated [28]. These ultra-long 1D reads made it possible to assemble the 4Mb MHC region into a single contig with haplotype phasing. Due to the error rate and limited depth of nanopore reads, the assembly and phasing were assisted by Illumina reads to call the heterozygous variants. Nevertheless, this accomplishment will lead to future work to better understand the recombination events in the MHC region and to elucidate functional gene and disease associations.
Nanopore sequencing can also discriminate nucleotide with chemical modifications such as cytosine methylation, among others [33, 67]. The direct detection of epigenetic marks on native DNA can overcome some of the limitations of existing methods, including bisulfite sequencing for the profiling of DNA methylation. Selective sequencing of genomic DNA with epigenetic modifications has also become a reality with Cas9-guided adapter ligation to target regions of interest, revealing haplotype-phased methylation patterns [68]. These advances offer abundant opportunities for immunogenetics research. It has been shown that allelic expression of HLA may impact the outcome after hematopoietic stem cell transplantation, as elevated expressions of mismatched host antigens (HLA-DPB1 or -C) have been associated with an increased risk of graft-versus-host disease [69, 70]. Nanopore sequencing may allow direct coupling of HLA genotyping and the allelic epigenetic modification patterns to shed light on the regulatory mechanism of HLA allelic expressions. Similarly, cDNA sequencing or direct RNA sequencing [26, 32] on nanopore platforms can generate reads for full-length transcripts, enabling simultaneous genotyping and transcripts quantification; chemical modification of RNA can also be directly detected [71]. Weimer and colleagues recently performed whole-transcriptome sequencing on MinION to determine the class I HLA typing and gene transcription in donor lymphocytes. Variable gene expression was observed among individual donors, which correlated with the median channel shift on flow crossmatch using sera with a single donor-specific antibody to the donor cells [41]. Finally, except for a small number of classical and non-classical HLA genes [72–75], different splicing isoforms of HLA genes and their physiological relevance have not been extensively examined. Nanopore sequencing has been successfully used to determine the transcriptional variations, including allele-specific isoforms, of complex genes in various cell types [26, 76, 77]; it may prove a useful tool to study the exon connectivity in HLA gene transcripts and its functional impact.
Conclusion and future directions
Nanopore sequencing is a unique technology that sequences DNA/RNA strands through chemistry independent of nucleic acid synthesis. It has the potential to deliver rapid, portable, and inexpensive high-resolution HLA typing free of cis-trans ambiguity. The library preparation process is simple and flexible, which remains a fertile ground for innovation. The lengths of nanopore reads are only limited by the lengths of library fragments, which can span the entirety of HLA genes and transcripts. The technology also has the capability of directly detecting epigenetic modifications, which is a remarkable breakthrough and may lead to novel discoveries in immunogenetics research.
The biggest hurdle for the broad application of nanopore sequencing in clinical molecular testing and HLA typing is the high error rate of the sequencing reads. The cumulative sample size of HLA typing by nanopore sequencing remains small in the literature and is dwarfed by the number of samples sequenced by other NGS platforms [4, 6]. Moreover, the excellent performance of existing NGS platforms are raising our expectations for full-gene characterization at a nearly perfect accuracy. Nanopore sequencing must meet the high standards shaped by existing NGS platforms to qualify for clinical HLA typing. To meet this goal, novel bioinformatic tools and validated commercial software packages are needed to generate accurate typing results from the noisy nanopore reads. And, together with the continued improvement in the nanopore design and basecalling method, nanopore sequencing may fulfill the promise of a robust and versatile platform for HLA typing and immunogenetics research in the near future.
Conflict of interest:
This work was supported by NIH NIAID award number R41 AI142919–01. The author declare no other conflict of interest.
Abbreviations
- ONT
Oxford Nanopore Technologies
- HLA
Human Leukocyte Antigen
- NGS
Next-generation sequencing
- SNV
Single-nucleotide variants
- PCR
Polymerase chain reaction
- INC-Seq
Intramolecular-ligated Nanopore Consensus Sequencing
- SMRT
Single-molecule real-time
- PRG
Population reference graph
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Erlich HA: HLA typing using next generation sequencing: An overview. Hum Immunol 2015;76:887. [DOI] [PubMed] [Google Scholar]
- [2].Barone JC, Saito K, Beutner K, Campo M, Dong W, Goswami CP, et al. : HLA-genotyping of clinical specimens using Ion Torrent-based NGS. Hum Immunol 2015;76:903. [DOI] [PubMed] [Google Scholar]
- [3].Weimer ET, Montgomery M, Petraroia R, Crawford J, Schmitz JL: Performance Characteristics and Validation of Next-Generation Sequencing for Human Leucocyte Antigen Typing. J Mol Diagn 2016;18:668. [DOI] [PubMed] [Google Scholar]
- [4].Gandhi MJ, Ferriola D, Lind C, Duke JL, Huynh A, Papazoglou A, et al. : Assessing a single targeted next generation sequencing for human leukocyte antigen typing protocol for interoperability, as performed by users with variable experience. Hum Immunol 2017;78:642. [DOI] [PubMed] [Google Scholar]
- [5].Lange V, Bohme I, Hofmann J, Lang K, Sauter J, Schone B, et al. : Cost-efficient high-throughput HLA typing by MiSeq amplicon sequencing. BMC Genomics 2014;15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schofl G, Lang K, Quenzel P, Bohme I, Sauter J, Hofmann JA, et al. : 2.7 million samples genotyped for HLA by next generation sequencing: lessons learned. BMC Genomics 2017;18:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gabriel C, Danzer M, Hackl C, Kopal G, Hufnagl P, Hofer K, et al. : Rapid high-throughput human leukocyte antigen typing by massively parallel pyrosequencing for high-resolution allele identification. Hum Immunol 2009;70:960. [DOI] [PubMed] [Google Scholar]
- [8].Mayor NP, Robinson J, McWhinnie AJ, Ranade S, Eng K, Midwinter W, et al. : HLA Typing for the Next Generation. PLoS One 2015;10:e0127153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Turner TR, Hayhurst JD, Hayward DR, Bultitude WP, Barker DJ, Robinson J, et al. : Single molecule real-time DNA sequencing of HLA genes at ultra-high resolution from 126 International HLA and Immunogenetics Workshop cell lines. Hla 2018;91:88. [DOI] [PubMed] [Google Scholar]
- [10].Allen ES, Yang B, Garrett J, Ball ED, Maiers M, Morris GP: Improved accuracy of clinical HLA genotyping by next-generation DNA sequencing affects unrelated donor search results for hematopoietic stem cell transplantation. Hum Immunol 2018;79:848. [DOI] [PubMed] [Google Scholar]
- [11].Huang Y, Dinh A, Heron S, Gasiewski A, Kneib C, Mehler H, et al. : Assessing the utilization of high-resolution 2-field HLA typing in solid organ transplantation. Am J Transplant 2019;19:1955. [DOI] [PubMed] [Google Scholar]
- [12].Creary LE, Guerra SG, Chong W, Brown CJ, Turner TR, Robinson J, et al. : Next-generation HLA typing of 382 International Histocompatibility Working Group reference B-lymphoblastoid cell lines: Report from the 17th International HLA and Immunogenetics Workshop. Hum Immunol 2019;80:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Osoegawa K, Vayntrub TA, Wenda S, De Santis D, Barsakis K, Ivanova M, et al. : Quality control project of NGS HLA genotyping for the 17th International HLA and Immunogenetics Workshop. Hum Immunol 2019;80:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Albrecht V, Zweiniger C, Surendranath V, Lang K, Schofl G, Dahl A, et al. : Dual redundant sequencing strategy: Full-length gene characterisation of 1056 novel and confirmatory HLA alleles. Hla 2017;90:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dilthey AT, Mentzer AJ, Carapito R, Cutland C, Cereb N, Madhi SA, et al. : HLA*LA-HLA typing from linearly projected graph alignments. Bioinformatics 2019;35:4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee H, Kingsford C: Kourami: graph-guided assembly for novel human leukocyte antigen allele discovery. Genome Biol 2018;19:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xie C, Yeo ZX, Wong M, Piper J, Long T, Kirkness EF, et al. : Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc Natl Acad Sci U S A 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O: OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 2014;30:3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, et al. : ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res 2013;41:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu C, Yang X: Using Exome and Amplicon-Based Sequencing Data for High-Resolution HLA Typing with ATHLATES. Methods Mol Biol 2018;1802:203. [DOI] [PubMed] [Google Scholar]
- [21].Bai Y, Ni M, Cooper B, Wei Y, Fury W: Inference of high resolution HLA types using genome-wide RNA or DNA sequencing reads. BMC Genomics 2014;15:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ka S, Lee S, Hong J, Cho Y, Sung J, Kim HN, et al. : HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinformatics 2017;18:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boegel S, Bukur T, Castle JC, Sahin U: In Silico Typing of Classical and Non-classical HLA Alleles from Standard RNA-Seq Reads. Methods Mol Biol 2018;1802:177. [DOI] [PubMed] [Google Scholar]
- [24].Deamer D, Akeson M, Branton D: Three decades of nanopore sequencing. Nat Biotechnol 2016;34:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jain M, Olsen HE, Paten B, Akeson M: The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 2016;17:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Workman RE, Tang AD, Tang PS, Jain M, Tyson JR, Razaghi R, et al. : Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat Methods 2019;16:1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rang FJ, Kloosterman WP, de Ridder J: From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol 2018;19:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. : Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M: Improved data analysis for the MinION nanopore sequencer. Nat Methods 2015;12:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hoenen T, Groseth A, Rosenke K, Fischer RJ, Hoenen A, Judson SD, et al. : Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerg Infect Dis 2016;22:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. : Real-time, portable genome sequencing for Ebola surveillance. Nature 2016;530:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, et al. : Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 2018. [DOI] [PubMed] [Google Scholar]
- [33].Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W: Detecting DNA cytosine methylation using nanopore sequencing. Nat Meth 2017;advance online publication. [DOI] [PubMed] [Google Scholar]
- [34].Jain M, Tyson JR, Loose M, Ip CLC, Eccles DA, O’Grady J, et al. : MinION Analysis and Reference Consortium: Phase 2 data release and analysis of R9.0 chemistry. F1000Res 2017;6:760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cretu Stancu M, van Roosmalen MJ, Renkens I, Nieboer M, Middelkamp S, de Ligt J, et al. : Mapping And Phasing Of Structural Variation In Patient Genomes Using Nanopore Sequencing. bioRxiv 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ammar R, Paton TA, Torti D, Shlien A, Bader GD: Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res 2015;4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu C, Xiao F, Hoisington-Lopez J, Lang K, Quenzel P, Duffy B, et al. : Accurate Typing of Human Leukocyte Antigen Class I Genes by Oxford Nanopore Sequencing. J Mol Diagn 2018;20:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu C, Berry R: Rapid High-Resolution Typing of Class I HLA Genes by Nanopore Sequencing. Methods Mol Biol 2020;2120:93. [DOI] [PubMed] [Google Scholar]
- [39].Ton KNT, Cree SL, Gronert-Sum SJ, Merriman TR, Stamp LK, Kennedy MA: Multiplexed Nanopore Sequencing of HLA-B Locus in Maori and Pacific Island Samples. Front Genet 2018;9:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Klasberg S, Putke K, Surendranath V, Schmidt A, Lange V, Schöfl G: P084 Typing in the third generation: A HLA typing approach for nanopore sequencing data. Human Immunology 2019;80:116. [Google Scholar]
- [41].Montgomery MC, Liu C, Petraroia R, Weimer ET: Using Nanopore Whole-Transcriptome Sequencing for Human Leukocyte Antigen Genotyping and Correlating Donor Human Leukocyte Antigen Expression with Flow Cytometric Crossmatch Results. J Mol Diagn 2020;22:101. [DOI] [PubMed] [Google Scholar]
- [42].De Santis D, Truong L, Martinez P, D’Orsogna L: Rapid High Resolution HLA genotyping by MinION Oxford Nanopore Sequencing for Deceased Donor Organ Allocation. HLA 2020. [DOI] [PubMed] [Google Scholar]
- [43].Lan JH, Yin Y, Reed EF, Moua K, Thomas K, Zhang Q: Impact of three Illumina library construction methods on GC bias and HLA genotype calling. Hum Immunol 2015;76:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yin Y, Lan JH, Nguyen D, Valenzuela N, Takemura P, Bolon YT, et al. : Application of High-Throughput Next-Generation Sequencing for HLA Typing on Buccal Extracted DNA: Results from over 10,000 Donor Recruitment Samples. PLoS One 2016;11:e0165810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cargou M, Ralazamahaleo M, Blouin L, Top I, Elsermans V, Andreani M, et al. : Evaluation of the AllType kit for HLA typing using the Ion Torrent S5 XL platform. Hla 2019. [DOI] [PubMed] [Google Scholar]
- [46].Liu C, Xiao F, Hoisington-Lopez J, Lang K, Quenzel P, Duffy B, et al. : Accurate typing of Human Leukocyte Antigen class I genes by Oxford nanopore sequencing. J Mol Diagnostics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cornelis S, Gansemans Y, Vander Plaetsen AS, Weymaere J, Willems S, Deforce D, et al. : Forensic tri-allelic SNP genotyping using nanopore sequencing. Forensic Sci Int Genet 2019;38:204. [DOI] [PubMed] [Google Scholar]
- [48].Matern BM, Olieslagers TI, Voorter CEM, Groeneweg M, Tilanus MGJ: Insights into the polymorphism in HLA-DRA and its evolutionary relationship with HLA haplotypes. HLA 2020;95:117. [DOI] [PubMed] [Google Scholar]
- [49].Li C, Chng KR, Boey EJ, Ng AH, Wilm A, Nagarajan N: INC-Seq: accurate single molecule reads using nanopore sequencing. Gigascience 2016;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu C, Duffy BF, Weimer ET, Montgomery MC, Jennemann J-E, Hill R, et al. : Performance of a multiplexed amplicon-based next-generation sequencing assay for HLA typing. PLoS ONE 2020;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Duke JL, Lind C, Mackiewicz K, Ferriola D, Papazoglou A, Derbeneva O, et al. : Towards allele-level human leucocyte antigens genotyping - assessing two next-generation sequencing platforms: Ion Torrent Personal Genome Machine and Illumina MiSeq. Int J Immunogenet 2015;42:346. [DOI] [PubMed] [Google Scholar]
- [52].Loose M, Malla S, Stout M: Real-time selective sequencing using nanopore technology. Nat Meth 2016;13:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Klasberg S, Surendranath V, Lange V, Schofl G: Bioinformatics Strategies, Challenges, and Opportunities for Next Generation Sequencing-Based HLA Genotyping. Transfus Med Hemother 2019;46:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Erlich RL, Jia X, Anderson S, Banks E, Gao X, Carrington M, et al. : Next-generation sequencing for HLA typing of class I loci. BMC Genomics 2011;12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM: Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Garrison E, Marth G: Haplotype-based variant detection from short-read sequencing. arXiv 2012. [Google Scholar]
- [57].Loman NJ, Quick J, Simpson JT: A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 2015;12:733. [DOI] [PubMed] [Google Scholar]
- [58].Vaser R, Sovic I, Nagarajan N, Sikic M: Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017;27:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nakano K, Shiroma A, Shimoji M, Tamotsu H, Ashimine N, Ohki S, et al. : Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum Cell 2017;30:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dilthey A, Cox C, Iqbal Z, Nelson MR, McVean G: Improved genome inference in the MHC using a population reference graph. Nat Genet 2015;47:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dilthey AT, Gourraud PA, Mentzer AJ, Cereb N, Iqbal Z, McVean G: High-Accuracy HLA Type Inference from Whole-Genome Sequencing Data Using Population Reference Graphs. PLoS Comput Biol 2016;12:e1005151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Klasberg S, Putke K, Surendranath V, Schmidt A, Lange V, Schofl G: Nanotyper: an HLA genotyping algorithm for nanoporesequencing data (https://dkms-lab.de/dateien/EFI2019_P192_nanotyper.pdf). 2019.
- [63].Schofl G, Klasberg S, Surendranath V, Putke K, Schmidt A, Lange V: Typing in the third generation: Class I and Class II HLA genotyping performance using nanopore long-read data (https://dkms-lab.de/dateien/EFI2019_P184_ThirdGenTyping.pdf). 2019.
- [64].van Deutekom HW, Kooter R, Geerligs J, Ruzius F-P, Meulenberg P, Surendranath V, et al. : P177 NGS typing results using oxford nanopore sequencing: Can minion data be reliably used for HLA typing? Human Immunology 2017;78:190.27888067 [Google Scholar]
- [65].van Deutekom HWM, Mulder W, Rozemuller EH: Accuracy of NGS HLA typing data influenced by STR. Hum Immunol 2019;80:461. [DOI] [PubMed] [Google Scholar]
- [66].https://nanoporetech.com/about-us/news/r103-newest-nanopore-high-accuracy-nanopore-sequencing-now-available-store. 2020.
- [67].Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, et al. : Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 2017;14:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, et al. : Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Petersdorf EW, Gooley TA, Malkki M, Bacigalupo AP, Cesbron A, Du Toit E, et al. : HLA-C expression levels define permissible mismatches in hematopoietic cell transplantation. Blood 2014;124:3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Petersdorf EW, Malkki M, O’HUigin C, Carrington M, Gooley T, Haagenson MD, et al. : High HLA-DP Expression and Graft-versus-Host Disease. N Engl J Med 2015;373:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liu H, Begik O, Lucas MC, Ramirez JM, Mason CE, Wiener D, et al. : Accurate detection of m(6)A RNA modifications in native RNA sequences. Nat Commun 2019;10:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hoarau JJ, Cesari M, Caillens H, Cadet F, Pabion M: HLA DQA1 genes generate multiple transcripts by alternative splicing and polyadenylation of the 3’ untranslated region. Tissue Antigens 2004;63:58. [DOI] [PubMed] [Google Scholar]
- [73].Zhang XH, Lian XD, Dai ZX, Zheng HY, Chen X, Zheng YT: alpha3-Deletion Isoform of HLA-A11 Modulates Cytotoxicity of NK Cells: Correlations with HIV-1 Infection of Cells. J Immunol 2017;199:2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hiby SE, King A, Sharkey A, Loke YW: Molecular studies of trophoblast HLA-G: polymorphism, isoforms, imprinting and expression in preimplantation embryo. Tissue Antigens 1999;53:1. [DOI] [PubMed] [Google Scholar]
- [75].Yao YQ, Barlow DH, Sargent IL: Differential expression of alternatively spliced transcripts of HLA-G in human preimplantation embryos and inner cell masses. J Immunol 2005;175:8379. [DOI] [PubMed] [Google Scholar]
- [76].Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, et al. : Nanopore Long-Read RNAseq Reveals Widespread Transcriptional Variation Among the Surface Receptors of Individual B cells. bioRxiv 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Clark MB, Wrzesinski T, Garcia AB, Hall NAL, Kleinman JE, Hyde T, et al. : Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol Psychiatry 2020;25:37. [DOI] [PMC free article] [PubMed] [Google Scholar]