

Abstract
Transthyretin (TTR) is a tetrameric transport protein highly conserved through vertebrate evolution and synthesized in the liver, choroid plexus, and retinal pigment epithelium. TTR transports the thyroid hormone thyroxine and the retinol-binding protein (RBP) bound to retinol (vitamin A). Mutations in TTR are associated with inherited transthyretin amyloidosis (ATTRv), a progressive, debilitating disease that is ultimately fatal and is characterized by misfolding of TTR and aggregation as amyloid fibrils, predominantly leading to cardiomyopathy or polyneuropathy depending on the particular TTR mutation. Transthyretin amyloid cardiomyopathy can also occur as an age-related disease caused by misfolding of wild-type TTR. Apart from its transport role, little is known about possible additional physiological functions of TTR. Evidence from animal model systems in which TTR has been disrupted via gene knockout is adding to our cumulative understanding of TTR function. There is growing evidence that TTR may have a role in neuroprotection and promotion of neurite outgrowth in response to injury. Here, we review the literature describing potential roles of TTR in neurobiology and in the pathophysiology of diseases other than ATTR amyloidosis. A greater understanding of these processes may also contribute to further clarification of the pathology of ATTR and the effects of potential therapies for TTR-related conditions.
Keywords: Alzheimer’s disease, Amyloidosis, Neurodegeneration, Neuroprotection, Transthyretin
Key Summary Points
| Transthyretin is a highly conserved protein that transports the thyroid hormone thyroxine and the retinol-binding protein bound to retinol (vitamin A). |
| Mutations in TTR are associated with ATTR amyloidosis, a progressive, debilitating, and ultimately fatal disease. |
| ATTR amyloidosis can also occur as a spontaneous, age-related disease in individuals with non-mutated wild-type TTR. |
| Aside from its function as a transport protein, there is a growing body of evidence for a role for TTR in neuroprotection and promotion of neurite outgrowth in response to injury. |
| The advent of treatments for ATTR amyloidosis based on stabilizing the TTR structure, or reducing expression of TTR, means that a clearer understanding of the role of TTR in neurobiology and pathophysiology is increasingly important. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12988436.
Introduction
Transthyretin (TTR) is a 55 kDa tetrameric transport protein comprising four identical subunits of 127 amino acids. TTR is synthesized in the liver, choroid plexus, and retinal pigment epithelium, before it is secreted into the bloodstream, cerebrospinal fluid (CSF), and eye, respectively [1].
In vivo TTR can dissociate into fragmented and full-length monomers, which aggregate as amyloid fibrils. These fibrils accumulate extracellularly in tissues and organs, including mainly the peripheral nerves and heart [2, 3]. This results in transthyretin (ATTR) amyloidosis, a serious progressive disease that displays substantial heterogeneity, with individual differences in disease susceptibility, clinical expression, and symptom presentation [2, 3].
There are two forms of ATTR amyloidosis: hereditary (ATTRv; v for variant) and wild-type (ATTRwt). These apparently share common substantial physiopathological mechanisms [4]. Mutations in the TTR gene can lead to dominantly inherited ATTR amyloidosis in adult life. This might occur from approximately age 30 onward, but more commonly after 50 years of age, with clinical and geographic differences between early-onset and late-onset forms of the disease [5]. In ATTRwt, the normal protein typically aggregates in the heart, resulting in a progressive pseudohypertrophic, restrictive cardiomyopathy related to aging [3]. Males are more susceptible to ATTRwt, but the reasons behind this gender bias are still unknown.
Transthyretin amyloid cardiomyopathy can also occur in carriers of TTR mutations that are associated with a propensity for amyloid fibril aggregation in cardiac tissue; examples of specific mutations that primarily lead to cardiac disease include Val122Ile [6], Leu111Met [7], Thr60Ala [8], and Ile68Leu [9].
The most common presentation of ATTRv is polyneuropathy (ATTR-PN) [10]. This accounts for the majority of ATTRv cases worldwide, with endemic foci in Portugal, Japan, and Sweden. The predominant genotype is Val30Met. ATTR-PN is characterized by axonal, length-dependent sensorimotor polyneuropathy that progresses upward from the feet and hands, is associated with autonomic dysfunction, and proceeds to death within an average of 10 years [10].
Until 2011, liver transplant was the only approach for treating ATTRv. This worked by replacing a variant TTR-producing liver with a normal, wild-type TTR-expressing organ. Over 2000 patients with ATTR amyloidosis have received liver transplants, and this has improved life expectancy in well-selected patient populations [11, 12]. Nevertheless, the complexity, costs, and risks associated with liver transplantation have fueled a search for alternative and less intrusive treatments for ATTR amyloidosis.
Validated treatment options for ATTR amyloidosis presently fall into two main categories: (1) TTR tetramer stabilization to prevent cleavage and dissociation into monomers with subsequent amyloid fibril formation; and (2) reduction of TTR protein expression through targeted gene silencing [13–17].
While the medical impact of ATTR amyloidosis is becoming clearer as this under-recognized disease becomes more widely known, knowledge of the role of TTR in healthy individuals remains limited. To facilitate greater understanding of ATTR amyloidosis, particularly with respect to treatments that stabilize/silence TTR, it will be necessary to improve the characterization of the physiological TTR functions. To aid this understanding, this article aims to review the evidence of the role of TTR in normal physiology and in the pathophysiology of disease other than ATTR amyloidosis.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
The Role of TTR in the Transport of Thyroid Hormones and Retinol
TTR was first characterized as a transporter of the thyroid hormone thyroxine and the retinol-binding protein (RBP) bound to retinol (vitamin A) [18]. TTR appeared during the early phase of vertebrate evolution and, overall, its sequence is highly conserved.[19, 20]. The major differences in sequence between TTR in fish and terrestrial vertebrates involve the residues that form the binding site for RBP [21], and it is worth noting that fish TTR does not bind RBP. In spite of these differences, the quaternary structure and overall shape of the native protein is almost identical among different species [22]. Therefore, it appears that the most conserved function is the transport of thyroid hormones and that vitamin A transport came later in the evolution of terrestrial vertebrates.
How Essential is TTR in Living Organisms?
The complete absence of TTR in a human has not been reported. Mouse TTR shares 80% amino acid homology with human TTR, so it may be expected that insights into the physiological function of human TTR could be gained through the generation of TTR knockout (KO) mice. Such mice are viable, appear phenotypically normal, and remain fertile. One notable difference with respect to wild-type (WT) animals is that levels of serum retinol, RBP, and thyroid hormone are significantly reduced, demonstrating that TTR has a role in maintaining normal levels of these metabolites in plasma [23]. In tissue samples from the liver, kidney, cortex, cerebellum, and hippocampus from TTR KO mice, thyroxine levels are normal, confirming that TTR is not crucial for thyroxine delivery to tissues [24]. RBP levels in the liver of TTR KO mice are 60% higher than those of wild-type mice, suggesting that the absence of TTR may reduce secretion of RBP-retinol from the liver [25]. Further phenotypic investigation of TTR knockout mice suggests a broader range of functions, particularly in the physiology of the nervous tissue, as discussed in the following sections.
TTR and Neuroprotection
Due to the impact of TTR in the peripheral nervous system (PNS) in ATTR-PN, a putative physiological function for the protein in nerve biology was investigated. Studies with TTR KO mice demonstrated sensorimotor impairment and a delayed functional recovery following sciatic nerve crush in the absence of TTR. This phenotype was rescued by expression of human TTR in the TTR null background, demonstrating that TTR has an important role in peripheral nerve function and repair [26, 27]. The effect of TTR on nerve regeneration was related to neuritogenic activity of the protein mediated by the megalin receptor, and to an impact on axonal retrograde transport in PNS neurons [27].
TTR was also shown to have a neuroprotective role in the central nervous system (CNS). This was initially investigated by studies demonstrating an impact of TTR on memory. Studies by Sousa et al. showed that 5-month-old TTR KO mice had evidence of memory impairment compared with WT mice, an effect that was not seen in aged animals. Moreover, in aged WT mice, the levels of TTR were reduced compared with young mice [28]. These observations suggest that the absence of TTR accelerates the decline in cognitive performance commonly associated with aging. Supporting this hypothesis, a reduction in TTR expression was demonstrated in aged memory-impaired rats compared with aged memory-unimpaired rats [29]. In this study, experiments with mice showed that TTR KO mice presented age-related memory deficits, a phenotype that was rescued by administration of retinoic acid [29].
In additional studies assessing behavior in TTR KO mice, it was shown that the absence of TTR was associated with a reduction in depressive-like behavior and an increase in exploratory activity. These behavioral effects of the absence of TTR were thought to be mediated by modulation of the noradrenergic system [30]. Other studies assessed behavioral phenotypes in both TTR KO and RBP KO mice, which showed that both genotypes were associated with mild behavioral phenotypes. Also, both TTR KO and RBP KO mice presented neurodegeneration in the CA3 region of the hippocampus and an impairment in adult neurogenesis. However, these phenotypes were more aggravated in RBP KO mice, suggesting that the behavioral effects of TTR silencing are discrete and unlikely to be related to its function as an RBP carrier [31].
TTR has also been associated with a role in neuroprotection after brain injury caused by ischemia, although this was only seen under conditions in which the heat shock response was compromised [32]. Using models in which brain ischemia was surgically induced by permanent middle cerebral artery occlusion (pMCAO), it was shown that TTR KO mice heterozygous for heat shock factor 1 (and therefore with a compromised heat shock response) had a significant increase in cortical infarction, cerebral edema, and microglial/leukocyte response. Moreover, in WT animals, TTR was localized throughout the infarct area and was shown to be derived from the CSF [32].
Additional studies using the same models demonstrated that the larger infarcts seen in TTR KO mice subjected to pMCAO were related to a downregulation of TTR and megalin in neurons [33]. In these studies, the neuritogenic effect of TTR on CNS neurons was also assessed. Experiments with hippocampal neurons from TTR KO and WT mice incubated with exogenous recombinant TTR showed that addition of TTR significantly boosted neurite outgrowth in both TTR KO and WT neurons, under physiological conditions and under conditions of excitotoxic insults [33]. Interestingly, neuritogenic effects in CNS neurons were also mediated by megalin, which places the receptor as a major player in the TTR neuroprotective function.
Evidence on the role of TTR in neuroprotection is not limited to in vitro and animal model studies, as clinical studies have also provided important insights. For example, the relationship between TTR concentration and stroke severity was determined using the modified Rankin Scale in a study of 585 young patients with cerebral infarction [34]. Multivariate logistic regression modeling showed that TTR was an independent predictor of positive clinical outcome, suggesting that high serum concentration of TTR may be a prognostic indicator for the outcome of cerebral infarction [34]. Additionally, a large study of 68,602 participants from two prospective studies analyzed the association between TTR stability and vascular disease risk and life expectancy over a mean 32-year follow-up duration [35]. Subjects were genotyped for the presence of stabilizing genetic variants of TTR Arg104His and Thr119Met, which were then related to plasma TTR levels, risk of vascular disease, thyroid function, and life expectancy. Results showed an association between genetic stabilization of TTR and a decrease in the risk of cerebrovascular disease, and an increase in life expectancy [35].
Summarizing, the neuroprotective role of TTR is reflected physiologically by its neuritogenic activity, which is observed in both PNS and CNS neurons, and by a function in preservation of memory during aging. In pathological conditions, TTR has a protective role after nerve lesion and brain injury such as ischemia.
TTR and Alzheimer’s Disease
As discussed, TTR appears to have biological functions that are most likely independent of its role as a carrier for thyroid hormones and retinol. In addition to the neuroprotective activity discussed in the previous section, it has been suggested that TTR may modulate neurodegeneration in Parkinson’s disease [36]. However, compelling suggestions for a neuroprotective role for TTR are also derived from different studies related to susceptibility and protection from Alzheimer’s disease (AD). AD is characterized by the accumulation of amyloid-β (Aβ) plaques in the brain, leading to progressive loss of neurologic function [37]. The initial evidence of a role of TTR in AD came from the observation that when Aβ was added to the CSF of patients and controls, it was sequestered by TTR, which is the most abundant Aβ-binding protein in the CSF [38].
Transgenic mice engineered to overexpress a mutant form of human amyloid precursor protein (APP) presenting the Swedish mutation (APPSw) are used as an animal model of AD. In one study using this model, gene expression profiles in mouse cerebellum and hippocampus showed that, relative to age-matched controls, TTR expression was selectively increased in the hippocampus, a brain region with high levels of Aβ, suggesting the TTR upregulation in hippocampal neurons might represent a kind of “protective” response to the increased levels of Aβ [39]. Further studies showed that infusion of an anti-TTR antibody into the hippocampus of APPSw transgenic mice led to increased Aβ deposition, tau phosphorylation, neuronal loss, and apoptosis reminiscent of the neuropathology of AD [40]. Moreover, TTR was shown to be upregulated in hippocampal neurons in patients with AD relative to age-matched controls without dementia [1].
Buxbaum's group has elegantly shown that overexpression of human TTR in the APP23 AD transgenic mouse model results in a milder pathological phenotype [41]. High TTR expression in these mice reduces the cognitive deterioration and deficit in spatial learning; cortical and hippocampal deposits are also reduced by 60–75%. Overall, these results suggest that TTR expression is linked to protection against neuronal death induced by Aβ deposition [40].
The above observations have raised the question as to the mechanism by which TTR may impart protection against AD pathology. According to competitive binding assays and transmission electron microscopy studies, it has been suggested that TTR is capable of inhibiting and disrupting Aβ fibril formation, abolishing its neurotoxicity [42]. In another study, the impact of TTR on protection against Aβ toxicity was proposed to be related to proteolytic cleavage of Aβ operated by TTR, as it was shown in vitro that a proteolytically active form of TTR, but not an inactive form, was able to reduce Aβ fibril formation, degrade neuronal-secreted Aβ, and reduce Aβ-induced toxicity in hippocampal neurons [43].
More recently, the impact of TTR as a neuroprotective agent in AD has been proposed to be related to its tetrameric stability, as drug-induced stabilization of TTR was able to increase Aβ protein uptake in cell-based assays [44]. It has also been shown that the TTR tetramer is a stronger inhibitor of β fibril formation than the TTR monomer [45, 46].
Discussion and Conclusions
Although the physiological role of TTR is yet to be fully defined, there is a growing body of evidence suggesting that it might have additional important functions besides the most commonly acknowledged transporter role (Fig. 1). Evidence from studies with both animal models and human samples suggests important activity of TTR in the preservation and regulation of memory function and behavior. An additional role in protection against neurodegeneration in AD models has been well documented, as well as neuroprotection in response to ischemic injury and nerve regeneration and promotion of neurite outgrowth in both PNS and CNS neurons.
Fig. 1.
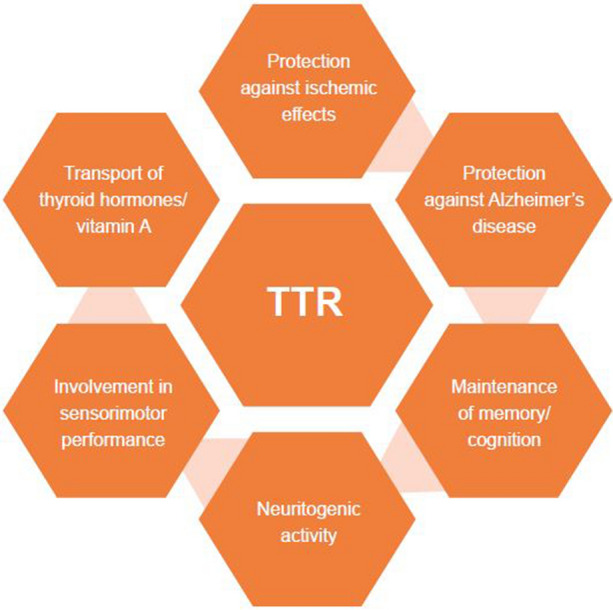
Known biological functions of TTR from mouse models and human studies. TTR transthyretin
With therapies that target TTR now available, it is increasingly relevant to investigate the full biological role of TTR in order to anticipate the potential consequences of partial or total reduction of TTR function. TTR stability has been correlated with neuroprotection. Whether long-term treatments that reduce TTR expression may lead to loss of this neuroprotective function requires further investigation.
Despite the apparent importance of TTR in neuroprotection, animal studies have shown that TTR KO mice are viable, although they exhibit alterations in behavior and memory. Whether partial or total reduction of TTR function in humans will be associated with deleterious effects is, therefore, still not clear.
Additional long-term studies of the effects of TTR therapies that stabilize or reduce TTR may lead to a clearer understanding of the normal biological role of TTR and further contribute to defining the most appropriate therapeutic options for different patients with diseases related to TTR.
Acknowledgements
Funding
The development of this article, including the Rapid Service Fees, was funded by Pfizer.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by Paul Hassan PhD CMPP, of Engage Scientific Solutions, and was funded by Pfizer.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Teresa Coelho, Marcia Almeida Liz, and Laura Obici have participated as investigators in Pfizer-sponsored clinical trials of the TTR stabilizer, tafamidis. Marcia Almeida Liz is an FCT Investigator (IF/00902/2015). Maria Isabel Fernandez-Arias and Pablo Mallaina are employees of Pfizer and hold stock and/or stock options. Vittorio Bellotti has nothing to declare.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Li X, Buxbaum JN. Transthyretin and the brain re-visited: is neuronal synthesis of transthyretin protective in Alzheimer's disease? Mol Neurodegener. 2011;6:79. doi: 10.1186/1750-1326-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 3.Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 4.Marcoux J, Mangione PP, Porcari R, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7:1337–1349. doi: 10.15252/emmm.201505357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59:1771–1776. doi: 10.1001/archneur.59.11.1771. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22:171–174. doi: 10.3109/13506129.2015.1051219. [DOI] [PubMed] [Google Scholar]
- 7.Svendsen IH, Steensgaard-Hansen F, Nordvag BY. A clinical, echocardiographic and genetic characterization of a Danish kindred with familial amyloid transthyretin methionine 111 linked cardiomyopathy. Eur Heart J. 1998;19:782–789. doi: 10.1053/euhj.1997.0841. [DOI] [PubMed] [Google Scholar]
- 8.Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33:1120–1127. doi: 10.1093/eurheartj/ehr383. [DOI] [PubMed] [Google Scholar]
- 9.Almeida MR, Hesse A, Steinmetz A, et al. Transthyretin Leu 68 in a form of cardiac amyloidosis. Basic Res Cardiol. 1991;86:567–571. doi: 10.1007/BF02190707. [DOI] [PubMed] [Google Scholar]
- 10.Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–1097. doi: 10.1016/S1474-4422(11)70246-0. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015;21:282–292. doi: 10.1002/lt.24058. [DOI] [PubMed] [Google Scholar]
- 12.Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–1854. doi: 10.1097/TP.0000000000000574. [DOI] [PubMed] [Google Scholar]
- 13.Adams D, Gonzalez-Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 14.Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22–31. doi: 10.1056/NEJMoa1716793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–2814. doi: 10.1007/s00415-013-7051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coelho T, Maia LF, da Silva MA, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 18.Raz A, Goodman DS. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J Biol Chem. 1969;244:3230–3237. [PubMed] [Google Scholar]
- 19.Power DM, Elias NP, Richardson SJ, Mendes J, Soares CM, Santos CR. Evolution of the thyroid hormone-binding protein, transthyretin. Gen Compr Endocrinol. 2000;119:241–255. doi: 10.1006/gcen.2000.7520. [DOI] [PubMed] [Google Scholar]
- 20.Zanotti G, Folli C, Cendron L, et al. Structural and mutational analyses of protein-protein interactions between transthyretin and retinol-binding protein. FEBS J. 2008;275:5841–5854. doi: 10.1111/j.1742-4658.2008.06705.x. [DOI] [PubMed] [Google Scholar]
- 21.Santos CR, Anjos L, Power DM. Transthyretin in fish: state of the art. Clin Chem Lab Med. 2002;40:1244–1249. doi: 10.1515/CCLM.2002.215. [DOI] [PubMed] [Google Scholar]
- 22.Eneqvist T, Lundberg E, Karlsson A, et al. High resolution crystal structures of piscine transthyretin reveal different binding modes for triiodothyronine and thyroxine. J Biol Chem. 2004;279:26411–26416. doi: 10.1074/jbc.M313553200. [DOI] [PubMed] [Google Scholar]
- 23.Episkopou V, Maeda S, Nishiguchi S, et al. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci USA. 1993;90:2375–2379. doi: 10.1073/pnas.90.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palha JA, Hays MT, de Escobar MG, Episkopou V, Gottesman ME, Saraiva MJ. Transthyretin is not essential for thyroxine to reach the brain and other tissues in transthyretin-null mice. Am J Physiol. 1997;272:E485–E493. doi: 10.1152/ajpendo.1997.272.3.E485. [DOI] [PubMed] [Google Scholar]
- 25.Wolf G. Retinol transport and metabolism in transthyretin-"knockout" mice. Nutr Rev. 1995;53:98–99. doi: 10.1111/j.1753-4887.1995.tb01528.x. [DOI] [PubMed] [Google Scholar]
- 26.Fleming CE, Saraiva MJ, Sousa MM. Transthyretin enhances nerve regeneration. J Neurochem. 2007;103:831–839. doi: 10.1111/j.1471-4159.2007.04828.x. [DOI] [PubMed] [Google Scholar]
- 27.Fleming CE, Mar FM, Franquinho F, Saraiva MJ, Sousa MM. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J Neurosci. 2009;29:3220–3232. doi: 10.1523/JNEUROSCI.6012-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sousa JC, Marques F, Dias-Ferreira E, Cerqueira JJ, Sousa N, Palha JA. Transthyretin influences spatial reference memory. Neurobiol Learn Mem. 2007;88:381–385. doi: 10.1016/j.nlm.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Brouillette J, Quirion R. Transthyretin: a key gene involved in the maintenance of memory capacities during aging. Neurobiol Aging. 2008;29:1721–1732. doi: 10.1016/j.neurobiolaging.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Sousa JC, Grandela C, Fernandez-Ruiz J, et al. Transthyretin is involved in depression-like behaviour and exploratory activity. J Neurochem. 2004;88:1052–1058. doi: 10.1046/j.1471-4159.2003.02309.x. [DOI] [PubMed] [Google Scholar]
- 31.Buxbaum JN, Roberts AJ, Adame A, Masliah E. Silencing of murine transthyretin and retinol binding protein genes has distinct and shared behavioral and neuropathologic effects. Neuroscience. 2014;275:352–364. doi: 10.1016/j.neuroscience.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 32.Santos SD, Lambertsen KL, Clausen BH, et al. CSF transthyretin neuroprotection in a mouse model of brain ischemia. J Neurochem. 2010;115:1434–1444. doi: 10.1111/j.1471-4159.2010.07047.x. [DOI] [PubMed] [Google Scholar]
- 33.Gomes JR, Nogueira RS, Vieira M, et al. Transthyretin provides trophic support via megalin by promoting neurite outgrowth and neuroprotection in cerebral ischemia. Cell Death Differ. 2016;23:1749–1764. doi: 10.1038/cdd.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao C, Zhang B, Zhang W, Pu S, Yin J, Gao Q. Serum prealbumin (transthyretin) predict good outcome in young patients with cerebral infarction. Clin Exp Med. 2011;11:49–54. doi: 10.1007/s10238-010-0103-8. [DOI] [PubMed] [Google Scholar]
- 35.Hornstrup LS, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Genetic stabilization of transthyretin, cerebrovascular disease, and life expectancy. Arterioscler Thromb Vasc Biol. 2013;33:1441–1447. doi: 10.1161/ATVBAHA.113.301273. [DOI] [PubMed] [Google Scholar]
- 36.Maetzler W, Tian Y, Baur SM, et al. Serum and cerebrospinal fluid levels of transthyretin in Lewy body disorders with and without dementia. PLoS ONE. 2012;7:e48042. doi: 10.1371/journal.pone.0048042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller-Thomas MM, Sipe AL, Benzinger TL, McConathy J, Connolly S, Schwetye KE. Multimodality review of amyloid-related diseases of the central nervous system. Radiographics. 2016;36:1147–1163. doi: 10.1148/rg.2016150172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwarzman AL, Gregori L, Vitek MP, et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buxbaum JN, Ye Z, Reixach N, et al. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of A-beta toxicity. Proc Natl Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa R, Goncalves A, Saraiva MJ, Cardoso I. Transthyretin binding to A-Beta peptide–impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 43.Silva CS, Eira J, Ribeiro CA, et al. Transthyretin neuroprotection in Alzheimer's disease is dependent on proteolysis. Neurobiol Aging. 2017;59:10–14. doi: 10.1016/j.neurobiolaging.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Alemi M, Silva SC, Santana I, Cardoso I. Transthyretin stability is critical in assisting beta amyloid clearance- relevance of transthyretin stabilization in Alzheimer's disease. CNS Neurosci Ther. 2017;23:605–619. doi: 10.1111/cns.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Zhang X, Ladiwala AR, et al. Mechanisms of transthyretin inhibition of beta-amyloid aggregation in vitro. J Neurosci. 2013;33:19423–19433. doi: 10.1523/JNEUROSCI.2561-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cascella R, Conti S, Mannini B, et al. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim Biophys Acta. 2013;1832:2302–2314. doi: 10.1016/j.bbadis.2013.09.011. [DOI] [PubMed] [Google Scholar]