

Abstract
The purpose of this experiment was to analyze the microbial community diversity in three Daqu samples displaying different characteristics in the same Daqu fermentation chamber. A high throughput sequencing technique was used to detect the microbial abundance and diversity in these Daqu samples. Of the three samples, the microbial diversity in the Black sample (sample B) was significantly higher than in the other two. At the genus level, Saccharopolyspora, Bacillus, Lentibacillus, Staphylococcus, Kroppenstedtia, and Thermoactinomyces were the primary bacterial groups in the sesame-flavored liquor, while Thermomyces, Thermoascus, and Aspergillus represented the main fungal groups. In sample B, the dominant bacteria were Thermoactinomyces, Saccharopolyspora, and Pseudomonas. In the White sample (sample W), Thermoactinomyces was the most abundant, followed by Saccharopolyspora and Lentibacillus. Staphylococcus dominated in the Yellow sample (sample Y), followed by Bacillus and Kroppenstedtia. Regarding the fungi in the three samples, Thermomyces accounted for 93.70% in sample B, and Aspergillus dominated in sample W, while the Thermoascus and Aspergillus content were similar in the sample Y. This study examined the microbial diversity in liquor Daqu with different sesame flavors, providing a foundation for microbial regulation, while investigating the relationship between flavored liquor compounds and microorganisms.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02500-1) contains supplementary material, which is available to authorized users.
Keywords: Sesame flavored liquor, Daqu, Microbial composition, Community diversity, High-throughput sequencing
Introduction
Baijiu is one of the world’s best-selling distilled spirits (Liu et al. 2018). According to flavor types, there are 11 traditional Chinese liquors, which includes a sauce flavor, a strong flavor, and light flavor. Sesame-flavored liquor is an innovative form of traditional liquor in China. In terms of production, it integrates strong (Wang et al. 2008), light, and sauce flavors by combining flavor materials and style characteristics. This is typically representative of Shandong liquor, displaying uniquely “fragrant, soft and mellow” qualities (Wu et al. 2017; He et al. 2019). Daqu plays an essential role in the production of fermented products, such as liquor and vinegar (Tang et al. 2019; Liu et al. 2017). The quality of Daqu is directly related to the high-temperature fermentation process, which can reach 65 °C (Xie et al. 2020).
The quality of the Daqu determines the quality of the liquor and significantly affects the flavor and taste of baijiu (Liu et al. 2017; Huang et al. 2017). Daqu is produced via the complex solid-state fermentation of grains and bran and is prepared by grounding and shaping the grains before treatment (Li et al. 2015), after which they are placed in the Daqu fermentation room at a temperature of 55–70 °C. The fermentation process takes about 50 days, while the incubation lasts one month (Zheng et al. 2014a, b). Because the entire process occurs in the Daqu fermentation room, the microbial system is derived from the air, the raw materials, and the placement environment. The microorganisms and their metabolites are responsible for the unique flavor of the liquor (Huang et al. 2017; Wang et al. 2017a, b). The various flavors of Daqu present different microbial communities (Yao et al. 2015). For example, Yao et al. found that the Bacillus and Lactobacillus numbers in the sauce flavored Daqu liquor were closely related to pyrazine and esters, while Aspergillus was associated with pyrazine, esters, and aromatics (Yao et al. 2015; Jin et al. 2019); Li et al. found that the thermophilic bacterium Laceyella sacchari FBKL4.010 could produce a key enzyme affecting pyrazine metabolism while uncovering its related gene (Li et al. 2019). In addition, Chai et al. revealed that two synthetic pathways of butyrate (butyrate kinase and butyryl-CoA: acetate CoA-transferase), a compound that denotes the key aroma of strong-flavored liquor, were inseparable from microorganisms (Chai et al. 2019).
Although advanced technological innovation has significantly increased the industrial production of liquor Daqu, it is difficult to control this production process without fully understanding its characteristics (Liu et al. 2017). Therefore, a more extensive comprehension regarding the production of Daqu using natural fermentation may prove more conducive to the development of related technology (Fan et al. 2020). For this experiment, three kinds of sesame-flavored liquor Daqu exhibiting different properties are selected from the same storage batch in a single Daqu fermentation chamber. The diversity of the microbial communities of the three types of Daqu are compared and analyzed using high-throughput sequencing technology. This work promotes an understanding of the similarities and differences between the microbial compositions of different sesame-flavored liquor Daqu while providing a scientific basis for microbial control during the fermentation process of sesame-flavored liquor in the future.
Materials and methods
Sample collection
The Daqu samples were collected from the high-temperature (55–65 °C) fermentation room at the Shandong Bandaojing Co. Ltd, Shandong Province, China, after which they were stored at 4 °C. In the Daqu fermentation chamber, the temperatures in the up layer (white Daqu sample, 35–45 °C), middle layer (yellow Daqu sample, 45–55 °C), and low layer (black Daqu sample, 55–65 °C) were different, resulting in a change in the color of the Daqu. Samples were collected from the different temperature layers to obtain the upper white Daqu (sample W), the middle yellow Daqu (sample Y), and the lower black Daqu (sample B). These three samples were collected from different areas of the same Daqu fermentation room (Supplementary Fig.1). Furthermore, the three samples were randomly collected, each with three parallel samples.
DNA extraction
Here, 2.0 g from each Daqu sample was used for the extraction of the total DNA using the Fast DNA® SPIN Kit for Soil (MP Biomedicals, Solon, OH, USA) following the manufacturer’s instructions.
Amplicon library preparation and sequencing
The bacteria were identified via PCR amplification of the v3-v4 region of the 16S rRNA gene, using the 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) primers (Klindworth et al. 2013). An 8 bp barcode was added to the 5′ end of the 806R reverse primer for sample differentiation. The identification of the fungi occurred via PCR amplification in the ITS1 region, and the primers were ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS1R (5′-GCTGCGTTCTTCATCGATGC-3′). An 8 bp barcode was added to the 5′ end of the ITS1F forward primer for sample differentiation. The reaction volume was 50 μL, which comprised 27 μL ddH2O, 2 μL (5 μM) each of the forward and reverse primers, 2.5 μL (10 ng) of template DNA, 5 μL (2.5 mM) of deoxynucleoside triphosphates, 10 μL of 5 × FastPfu buffer, 0.5 μL of bovine serum albumin and 1 μL of TransStart FastPfu Polymerase (TransGen, Beijing, China). Bacterial amplification occurred via predenaturation at 95 °C for 5 min, after which each thermal cycle was denatured at 94 °C for 30 s, annealed at 55 °C for 35 s, and elongated at 72 °C for 30 s. After 30 cycles, the final extension step was performed at 72 °C for 8 min. Furthermore, the predenaturation of the fungi occurred at 95 °C for 5 min, after which each thermal cycle was denatured at 94 °C for 30 s, annealed at 54 °C for 40 s, and elongated at 72 °C for 30 s. After 30 cycles, the final extension step was performed at 72 °C for 8 min. The PCR products were purified using a gel recovery kit (Life Technology, USA), while Qubit3.0 (Life Technology, USA) was used for quantitative analysis. The purified amplicons were pooled in equimolar concentrations, and library-specific sequencing adapters were added via a NEBNext Ultra (NEB#e7370S/ L) assay, following the manufacturer's instructions. Dual index sequencing of the paired-end 250 bp reads was run on an Illumina HiSeq2500 instrument (Illumina, San Diego, CA, USA).
Sequence data processing and statistics
The original data obtained from the machine were filtered using NGS toolkit software (Patel et al. 2012) according to the default parameter values to remove low-quality reads. The coincidence pairs were merged using FLASH software. Split_libraries_fastq.py in the QIIME software (Magoč et al. 2011) was used to separate the data according to the barcode at the 5′ end of the primer to obtain each sequence. The mothur software (Caporaso et al. 2010) was used to edit the barcodes (Schloss et al. 2009) and primers in the tags. The sequences were filtered and denoised to remove chimeras, after which they were classified using the Bayesian method against a database derived from the RDP 16S rRNA reference, db128 (Cole et al. 2014). Non-16S sequences (e.g., unknown, archaea, chloroplast, mitochondria, and cyanobacteria) were removed (Xie et al. 2020).
The edited sequence was divided into OTUs at a distance of 97% using the VSEARCH software (Rognes et al. 2016). The representative OTU sequences were denoted by the most abundant sequences in each OTU. The OTU table was simplified to the minimum sequence number between samples to directly compare the alpha and beta diversity values of each flora. Principal coordinate analysis (PCA) based on the UniFrac distance matrix (estimates the similarity according to the distance between samples), as well as two-way permutational multivariate analysis of variance (PERMANOVA) based on the Bray − Curtis dissimilarity was used to assess the variances in the compositions of the microbial communities using mothur and the R package vegan (version 2.2).
Results
Quality control of the sequencing data
The raw high-throughput sequencing data were submitted to the NCBI database under the BioProject number PRJNA612757. A total of 1.55 M raw sequences was obtained after sequencing, while 1.45 M available reads were acquired after the quality control process. After filtering out the nontarget fragments, a total of approximately 0.43 M bacterial 16S rDNA fragment sequences (ranging from 9387 to 86,575) and 1.02 M fungi ITS DNA fragment sequences (ranging from 71,752 to 188,576) were obtained. Figure 1 shows that regardless of whether it was bacteria or fungi, the rarefaction curve based on the OTU number, approaching the saturation plateau, suggested that the sequencing depth in this research was adequate to represent the microbial diversity in the samples, indicating that the selected sample was representative of different sesame-flavored Daqu.
Fig. 1.
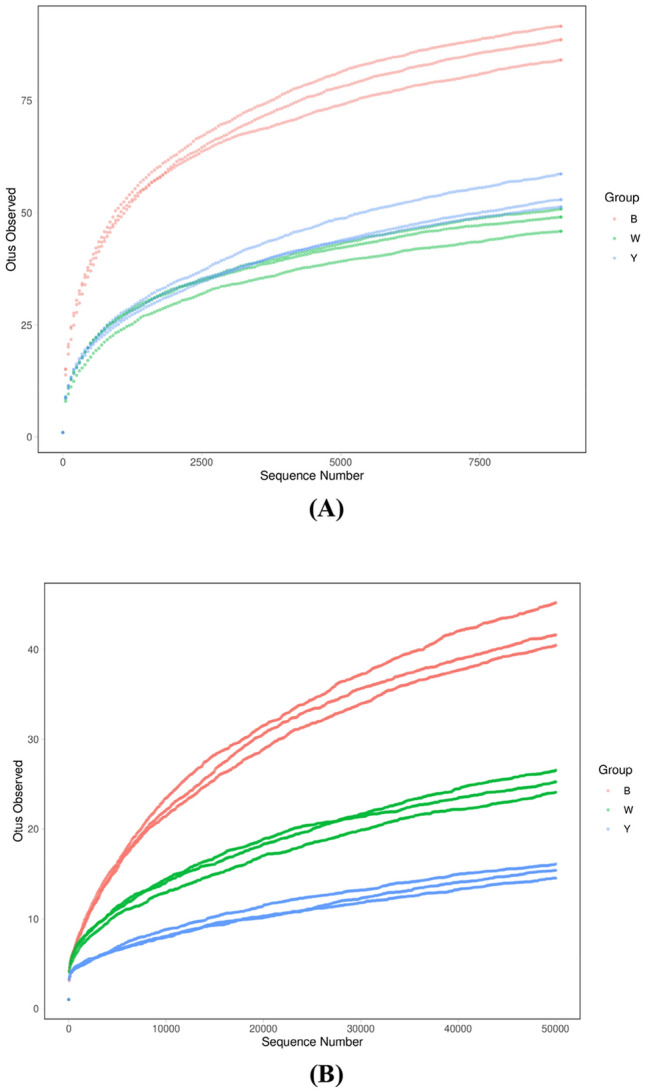
The rarefaction curves. a Bacteria, and b Fungi. B black Daqu sample, W white Daqu sample, Y yellow Daqu sample. Each sample has three parallels based on the number of OTUs. The curves are close to equilibrium, indicating the representativity of the Daqu samples
The microbial diversity in the three Daqu samples
The diversity indices, including the number of OTUs, ACE, Chao, Shannon, and Simpson values, are shown in Table 1. The number of OTUs, ACE, and Chao indices describe the total number of species. The total number of bacterial and fungal species in sample B exceeded that of both sample Y and sample W, while the bacterial level in sample W was lower than in sample Y. However, the fungal levels exhibited the opposite trend. The Shannon and Simpson indices describe species diversity. Unlike the Shannon index, the higher the Simpson index value, the lower the community diversity. Table 1 shows that the bacterial diversity of sample B was the highest, while the fungal diversity was the lowest. The bacterial diversity in samples W and Y was similar, but the fungal diversity in sample W was significantly higher than that of sample Y.
Table 1.
Species richness estimator and diversity index
| Group | Num of OTUs1 | Ace | Simpson | Shannon | Chao2 | |
|---|---|---|---|---|---|---|
| Bacteria | B | 86.33 ± 5.69a | 98.98 ± 5.74a | 0.14 ± 0.01c | 2.56 ± 0.08a | 94.26 ± 5.33a |
| W | 47.33 ± 1.53b | 79.77 ± 3.91b | 0.38 ± 0.03a | 1.56 ± 0.07c | 68.24 ± 8.55b | |
| Y | 52.33 ± 7.02b | 67.27 ± 12.53b | 0.27 ± 0.01a | 1.78 ± 0.01b | 69.40 ± 16.84b | |
| Fungi | B | 37.33 ± 2.87a | 73.21 ± 28.08a | 0.88 ± 0.01a | 0.34 ± 0.01c | 59.75 ± 20.39a |
| W | 20.67 ± 2.08b | 35.61 ± 8.50a | 0.40 ± 0.01c | 1.1 ± 0.02a | 28.67 ± 5.10b | |
| Y | 13.33 ± 2.52c | 73.55 ± 81.45a | 0.63 ± 0.01b | 0.71 ± 0.02b | 26.33 ± 8.62b |
1 The number of OTUs in each group reflects microbial richness; 2 Nonparametric statistical predictions of the total OTU richness; Values with superscript letters a, b, c, and d are significantly different across columns (p < 0.05)
A Venn diagram was created based on the samples (Fig. 2), which was used to investigate whether exclusively shared OTUs existed. At a 97% similarity level, the number of bacterial OTUs in sample B, sample W, and sample Y was 115, 94, and 110, while the number of fungal OTUs was 70, 42, and 34, respectively. The number of OTUs in sample B was the highest for both bacteria and fungi. Although 73 bacterial OTUs and 25 fungal OTUs were evident in the three samples, each specimen presented its own unique bacterial and fungal OTUs. Similarly, the independent bacterial and fungal OTUs in sample B far exceeded that of the other two samples. The proportion of independent bacterial OTUs in samples B, W, and Y was 12.2, 3.2, and 4.5%, while that of the independent fungal OTUs was 44.3, 9.5, and 5.9%, respectively. Thermoactinomyces, Sphingomonas, Saccharopolyspora, Kroppenstedtia, and Bacillus denoted the primary bacteria in the sesame flavored liquor Daqu, while Thermomyces, Aspergillus, Sagenomella, and Thermoascus signified the dominant fungi. However, the microbial content differed in the individual samples.
Fig. 2.
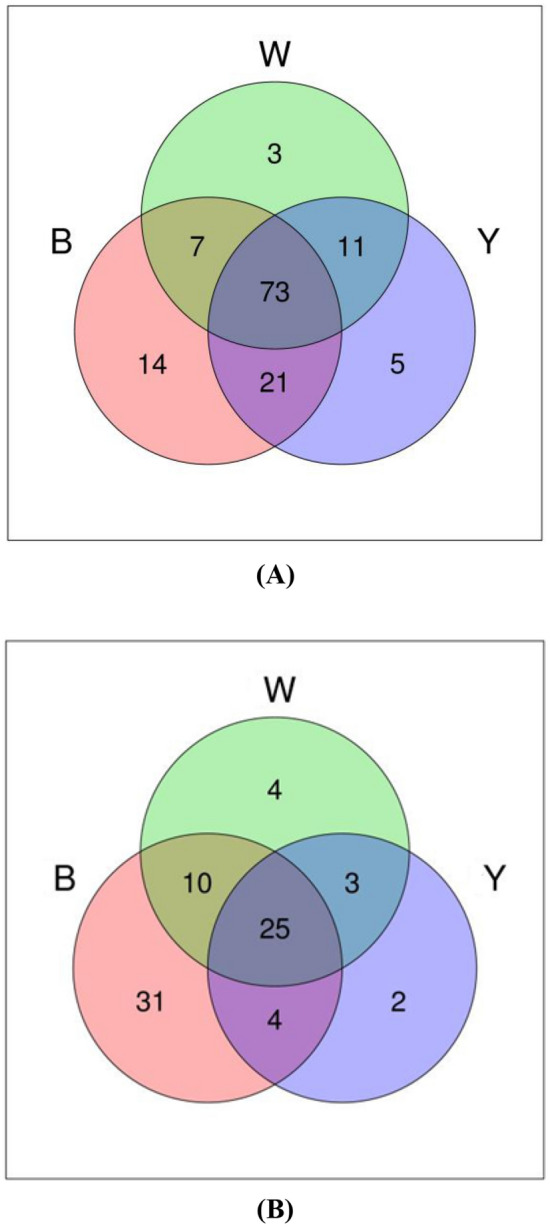
A Venn diagram showing the shared and unique OTUs at 97% identity among the Black Daqu, White Daqu, and Yellow Daqu samples, with OTU abundance < 1% removed
The diversity and composition of the microbial communities in the three Daqu samples
The Mantel test calculation showed a strong correlation (r = 0.9887, p < 0.001) between the Bray − Curtis dissimilarity matrices (based on the bacterial genera level classification and OTUs). Figure 3a shows the distribution comparison rules of the phyla of the three sample bacterial species. Overall, Firmicutes was the most abundant in the three samples, specifically in sample Y (90.70%). Proteobacteria (25.57%) was significantly higher in sample B than in samples W (1.63%) and Y (2.13%). Actinobacteria (23.17%) was substantially higher in sample B than in samples W (14.77%) and Y (7.17%), while Firmicutes were considerably lower in sample B (50.60%). As shown in Fig. 3b, the bacteria in the three samples were different at the genus level. The main bacteria in sample B included Thermoactinomyces (29.47%), Saccharopolyspora (17.60%), Pseudomonas (10.60%), and Kroppenstedtia (7.57%). The bacteria in sample W were mainly Thermoactinomyces (58.63%), Saccharopolyspora (11.33%), and Lentibacillus (10.43%). Sample Y primarily included Thermoactinomyces (7.20%), Staphylococcus (45.20%), Kroppenstedtia (16.87%), and Bacillus (16.63%) (Table 2).
Fig. 3.

The distribution of the predominant bacteria in the Black Daqu, White Daqu, and Yellow Daqu samples (three biological replicates in each group). The relative abundance in each sample at the phylum level (a) and the genus level (b)
Table 2.
The predominant species of Daqu bacteria in each sample
| OTU ID | B (%) | W (%) | Y (%) |
|---|---|---|---|
| Actinopolysporaceae | 4.33 | 3.33 | 1.50 |
| Saccharopolyspora | 17.60 | 11.33 | 5.50 |
| Bacillus | 2.70 | 1.40 | 16.63 |
| Lentibacillus | 3.10 | 10.43 | 1.87 |
| Staphylococcus | 4.40 | 3.20 | 45.20 |
| Kroppenstedtia | 7.57 | 3.50 | 16.87 |
| Thermoactinomyces | 29.47 | 58.63 | 7.20 |
| Weissella | 0.37 | 5.67 | 0.43 |
| Pseudomonas | 10.60 | 0.23 | 0.83 |
| Stenotrophomonas | 2.93 | 0.03 | 0.20 |
| Lactobacillus | 0.37 | 0.00 | 0.07 |
As shown in Fig. 4a, the fungi in the three samples were uniformly distributed at the phylum level, with Ascomycetes as the most abundant, while some Basidiomycetes and Zygomycetes were also apparent. The Ascomycetes level was 99.8% in sample B, 99.9% in sample W, and nearly 100% in sample Y. Figure 4b shows the variation in the three samples at the fungal genus level. Thermomyces (93.70%) dominated in sample B, while Thermomyces (54.60%), Aspergillus (28.23%), and Sagenomella (13.90%) were the primary fungi in sample W. Furthermore, Thermomyces (78.07%) was the most abundant in sample Y, while the disparity between Thermoascus (11.10%) and Aspergillus (10.17%) was minimal (Table 3).
Fig. 4.

The distribution of the predominant fungi in the Black Daqu, White Daqu, and Yellow Daqu samples (three biological replicates in each group). The relative abundance in each sample at the phylum level (a) and the genus level (b)
Table 3.
The predominant species of Daqu fungi in each sample
| OTU ID | B (%) | W (%) | Y (%) |
|---|---|---|---|
| Thermomyces | 93.70 | 54.60 | 78.07 |
| Thermoascus | 1.37 | 2.67 | 11.10 |
| Aspergillus | 2.37 | 28.23 | 10.17 |
| Sagenomella | 1.60 | 13.90 | 0.60 |
PCA (Principal coordinate analysis) showed significant differences between the microbial communities of the three Daqu samples (Fig. 5). Although the dominant bacterial (top 20) and fungal genera (top 12) were almost identical in each sample, the microbial abundance was significantly different between the three groups. The statistical differences between the microbial populations in the two groups were identified using bidirectional PERMANOVA based on Bray Curtis similarity.
Fig. 5.

The clustering in the Black Daqu sample, White Daqu sample, and Yellow Daqu sample (three biological replicates in each group), obtained via Principal Coordinate Analysis (PCoA) and based on the Thetayc-beta distance. The first two principal coordinates are plotted on the x and y axes, respectively (representing 98.9% of the total variation). The distance between two points represents the similarity between two samples
Discussion
The metabolic activity of the Daqu microbe is essential to the unique flavor of sesame flavored liquor (He et al. 2020), a traditional Chinese drink. High-temperature fermentation is responsible for the main characteristics of sesame-flavored liquor Daqu. During the production process of Daqu, most thermophilic yeasts and molds were eliminated by increasing the temperature, while the microorganisms formed a microbial community dominated by high-temperature bacteria (Yi et al. 2019). This study analyzed the microbial communities of three kinds of sesame-flavored liquor Daqu with different properties. The microbial communities of samples B, W, and Y were similar, as were their physical and chemical indices (the data has not been published yet). The determination of the microbial communities will provide a foundation for the correlation between the brewing of sesame-flavored liquor and the Daqu microorganisms, as well as its physicochemical properties. Furthermore, it will be useful in follow-up research involving the unique flavor compounds and optimization of sesame-flavored liquor.
In 2015, Yao et al. used a culturally based method to separate sesame flavored Daqu samples by employing traditional separation techniques. The dominant bacterial genera that were evident during the drying stage were Bacillus sp. (23.15%), Thermoactinomyces sp. (15.68%) and Lactobacillus sp. (7.47%). (Yao et al. 2015). In 2017, Sha et al. demonstrated that Pantoea, Weissella, Lactobacillus, and Thermoactinomyces were the dominant bacterial genera via an independent culture method using a 16S rDNA clone library (Sha et al. 2017). In 2019, Fan et al. used high-throughput techniques to reveal that the dominant bacteria in sesame-flavored Daqu samples were Bacillus (26.9%), and Thermoactinomyces (12.1%), while the dominant fungi were Thermomucor, Absidia, and Gregarina (Fan et al. 2020). In the same year, based on simulated fermentation experiments, Fan et al. performed a comprehensive analysis of three sesame-flavored Daqu of different grades, evaluating the physical and chemical indices, volatile compounds, microbial community diversities and their correlation. The results differed significantly from those acquired in this study in terms of the diversity and abundance of the microbial communities. Firmicutes were the most abundant bacterial phylum in all the Daqu samples, followed by Proteobacteria. At the genus level, the detection results of the three samples were completely different. The first dominant genera in the three samples were Kroppenstedtia (18.9%), Thermoactinomyces (25.8%), and Weissella (18.0%), respectively. Lactobacillus was the only dominant genus with a relative abundance of more than 1% shared by the three samples. The eukaryotic microorganism communities in all the Daqu samples were similar. Ascomycota and Zygomycota represented the dominant eukaryotic communities. The fungal genera consisted mainly of Absidia, and Thermomucor (Fan et al. 2019). According to a study by Xie et al., Kroppenstedtia, Lactobacillus, Weissella, Lentibacillus, Bacillus, and Saccharopolyspora represented the primary bacterial groups in the high-temperature Daqu of the Chinese sesame-flavored liquor (Xie et al. 2020).
The traditional Chinese Maotai-flavored liquor Daqu is fermented at high temperatures of up to 60–70 °C (Jin et al. 2017). Based on the previous research, the dominant bacteria in Maotai Daqu were Thermoactinomyces, Saccharopolyspora, Acinetobacter, Pseudomonas, Bacillus, Lactobacillus, and Weissella, while molds were represented by Aspergillus, Monascus, Thermoascus, and Thermomyces, and the yeasts were Saccharomyces and Pichia (Wang et al. 2008; Jin et al. 2017; Zheng et al 2014a, b; Tang et al. 2017; Li et al. 2014). During the fermentation process, the local temperature of sample B was the same as that of the high-temperature Daqu, and the dominant bacteria were Thermoactinomyces Saccharopolyspora, Pseudomonas, Kroppenstedtia, Staphylococcus, Actinopolysporaceae, Lentibacillus, Stenotrophomonas, Bacillus, and Lactobacillus. The most abundant strains in sample B were similar to high-temperature Daqu and included Thermoactinomyces, Saccharopolyspora, Pseudomonas, Bacillus, Lactobacillus, and Weissella. The Luzhou-flavored liquor Daqu was fermented at a medium temperature of 50–60 °C (Jin et al. 2017). According to the previous study, the dominant bacteria in the Luzhou-flavored liquor Daqu include Weissella, Lactobacillus, Staphylococcus, Pediococcus, Bacillus, Kroppenstedtia, Thermoactinomyces, Lactococcus, and Enterobacter. The dominant fungi were Pichia, Saccharomycopsis, Aspergillus, Rhizopus, Lichtheimia, Thermomyces, Thermoascus, Absidia, and Geotrichum (Yang et al. 2017,2018; Du et al. 2019; Gou et al. 2015). The fermentation temperature of sample Y was 45–55 °C, while the dominant bacteria included Staphylococcus, Kroppenstedtia, Bacillus, Thermoactinomyces, Saccharopolyspora, Lentibacillus, Actinopolysporaceae, Pseudomonas, Weissella, Stenotrophomonas, and Lactobacillus. The same strains were found in sample Y and the medium temperature Daqu, and included Staphylococcus, Kroppenstedtia, Bacillus, Thermoactinomyces, Saccharopolyspora, Weissella, and Lactobacillus. The light-flavored Daqu was fermented at a low temperature of 40–50 °C (Jin et al. 2017), while the primary bacteria were represented by Pantoea, Klebsiella, Lactobacillus, Bacillus, Kroppenstedtia, Staphylococcus, Weissella, Acinetobacter, and Lentibacillus, and the primary fungi were Saccharomycopsis, Pichia, and Aspergillus (Wang et al. 2017a, b; Fan et al. 2018; Zhang et al. 2014). The fermentation temperature of sample W was 35–45 °C, and the dominant bacteria were Thermoactinomyces, Saccharopolyspora, Lentibacillus, Weissella, Kroppenstedtia, Actinopolysporaceae, Staphylococcus Bacillus, Pseudomonas, and Stenotrophomonas. The bacterial strains in sample W and the low-temperature Daqu were the same, and were represented by Bacillus, Kroppenstedtia, Staphylococcus, Weissella, and Lentibacillus.
High-throughput sequencing revealed the presence of primarily three types of bacteria in the three sesame-flavored liquor Daqu samples, namely Firmicutes, Actinobacteria, and Proteobacteria, of which Firmicutes was the dominant bacteria. According to the previous research reports, as well as the results of this study, Thermomyces, Thermoascus, Aspergillus, and Sagenomella were the dominant fungi in sesame-flavored Daqu. Furthermore, Actinopolysporaceae, Saccharopolyspora, Bacillus, Lentibacillus, Thermoactinomyces, Kroppenstedtia, and Aspergillus were abundant in all three types of Daqu. Significant differences were evident in the content of four bacterial genera, namely Weissella, Stenotrophomonas, Pseudomonas, and Lactobacillus, as well as three fungal genera, namely Thermomyces, Thermoascus, Sagenomella. The compositions and concentrations of the volatile compounds, such as esters, alcohols, and acids, substantially influenced the unique flavor and quality of the liquor (Liu et al. 2020). Minimal studies exist regarding the unique fried sesame flavor substances of sesame-flavored liquor. Hu et al. found that pyrazines and other nitrogen-containing heterocyclic compounds and sulfur compounds (notably methionol) are important contributors to the aroma of sesame flavor baijiu (Hu et al. 2017). Several studies have shown that the combination of pyrazines (trimethylpyrazine and tetramethylpyrazine), furans, and phenolic compounds denote the primary flavor substances in sesame-flavored liquor (Qi et al. 2009; Sun et al. 2018). Various common flavor compounds were present in the sesame-flavored liquor samples, such as ethyl hexanoate, ethyl pentanoate, ethyl butanoate, ethyl furoate, ethyl 4-methylpentanoate, 2-furfurylthiol, 3-methyl-1-butanol, methional, hexanoic acid, butanoic acid, 3-methylbutanal, 2-acetyl furan, and trimethyl pyrazine (Liu et al. 2018; Wu et al. 2014).
In this study, the microbial diversity in the three Daqu varieties was distinctly different, affecting the liquor flavor obtained as a result. Weissella and Lactobacillus are both typical lactic acid bacteria (LAB) found in liquor Daqu. They can produce lactic acid to provide a substrate for the esterification reaction of yeast, while the ethyl lactate produced by esterification can improve the flavor of the liquor (He et al. 2019; Liu et al. 2020). The Weissella was positively correlated with pentanoic acid and 2-methoxy-5-methylphenol (Liu et al. 2019). The ethyl carbamate (EC; C2H5OCONH2) that naturally exists in fermented foods and beverages, displays carcinogenic and mutagenic effects. The synergistic effect of LAB and yeast can effectively control the level of EC precursors during solid fermentation (Du et al. 2017). Some scholars believe that this synergistic relationship between LAB and yeast during liquor fermentation has not been fully explored. The interaction between LAB with yeast can effectively promote the development of the liquor industry while contributing to the modernization of traditional fermentation technology (Liu et al. 2020). In addition, the synthesis of exopolysaccharides and oligosaccharides by LAB with compounds containing free amino groups during the Maillard reaction can reduce ketones, aldehydes, and heterocyclic compounds, which denote the primary sources of liquor flavor (Xie et al. 2020).
Bacillus is a heat-resistant microorganism commonly found in mature Daqu. A variety of hydrolases, including amylase protease lipase, cellulase, glucanase, are secreted to facilitate the hydrolysis of macromolecules and the production of flavor compounds during the brewing process (He et al. 2019; Yao et al. 2015; Wang et al. 2017a, b; Zhao et al. 2019). One study showed that inoculating Bacillus licheniformis into Daqu significantly improved the amylase action while decreasing the activity of glucose amylase, and lipase (Wang et al. 2017a, b). Furthermore, ethyl octanoate, ethyl hexanoate, hexyl hexanoate, hexanol, and hexanoic acid were positively correlated with Bacillus (He et al. 2020). Lentibacillus and Bacillus are closely related genetically (Xie et al. 2020). As the metaproteomic data show, some Lentibacillus secrete a variety of proteases with a strong ability to metabolize amino acids (Zhang et al. 2018). Changes in the microbial communities, such as in Kroppenstedtia during liquor fermentation lead to an increase in acetic acid, lactic acid, malic acid, and ethyl acetate, while decreasing the ethyl lactate levels (Wang et al. 2017a, b). Thermoactinomyces can also produce lipase and phosphatase, while Saccharopolyspora plays a vital role in compound volatility (Jin et al. 2019). Although Kroppenstedtia and Saccharopolyspora represent the main sesame-flavored Daqu microbes, not much research exists regarding their microbial diversity and biological functions, necessitating further investigation.
The saccharification, liquefaction, and ester production in liquor are related to fungi (Fan et al. 2020). Aspergillus denotes an important filamentous fungus during liquor fermentation, able to produce a variety of enzymes for the formation of starch saccharification, protein hydrolysis, and flavonoids, thus affecting the flavor of the liquor (Chen et al. 2014; Machida et al. 2008). A study involving Maotai-flavored liquor showed that Aspergillus was positively correlated not only with pyrazines, but also with esters and some aromatic compounds (Jin et al. 2019). Thermoascus and Thermomyces produce heat-resistant enzymes that degrade carbohydrates efficiently (McClendon et al. 2012).
In this study, the diversity in the microbial communities of three different sesame-flavored Daqu samples were characterized using high-throughput sequencing technology. This technique facilitated a better understanding of the differences between the microbial communities in Daqu while allowing the detection of various microbial species that was not previously possible using the culture method. Saccharopolyspora, Bacillus, Lentibacillus, Staphylococcus, Kroppenstedtia, and Thermoactinomyces were all dominant in the three Daqu samples, but the abundance values varied significantly for each strain. Thermomyces represented the primary fungi in the three types of Daqu, while a more significant difference was evident for the Aspergillus and Thermoascus content. In general, the microbial diversity and abundance in sample B were significantly higher than in samples W and Y. Daqu is central to liquor brewing, providing raw materials, enzymes, and microorganisms. The study of the microbial communities in different Daqu provides a foundation for examining the relationship between flavor compounds and microorganisms in liquor. At the same time, it provides a scientific basis for the microbial regulation of sesame-flavored Daqu and the industrialization of the winemaking process.
Conclusion
This study reveals the presence of 134 bacterial Daqu OTUs in the evaluated samples, which include the phyla Firmicutes, Proteobacteria, and Actinobacteria. Thermoactinomyces, Saccharopolyspora, Pseudomonas, Kroppenstedtia, Lactobacillus, Weissella, Bacillus, and Lentibacillus represent the central bacterial communities in Chinese sesame-flavored liquor Daqu. Furthermore, 79 fungal OTUs are present in the samples, primarily representing the phylum Ascomycota, while Thermomyces, Thermoascus, Aspergillus, and Sagenomella denote the dominating fungi in the sesame-flavored Daqu. The three samples that are assessed in this study are collected in the same culture conditions. Moreover, the analysis of the three types of Daqu exhibiting different microbial characteristics is central to this experiment.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Miss Ling Xu at Shandong Bandaojing Ltd Co (Shandong Province, China) for assistance with Daqu materials sampling.
Author contributions
XW, RJ, WC and XG designed and participated in all experimental procedures, performed data analysis, and drafted the manuscript. ML and FY participated in the Daqu samples collection and preparation. YY and YL supervised the study and critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Solid-state Fermentation Resource Utilization Key Laboratory of Sichuan Province (No. 2020GTY001), the Open Project Program of the Key Laboratory of Brewing Molecular Engineering of China Light Industry (No. BME-202001) and the Fundamental Research Funds for the Central Universities (No. FRF-TP-18-012A1; FRF-BR-18-009B).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Contributor Information
Yinzhuo Yan, Email: y_y_z@163.com.
Yang Liu, Email: liuyang@ustb.edu.cn, Email: ly81150@163.com.
References
- Adam M, Westphal A, Hallmann J, Heuer H. Specific microbial attachment to root knot nematodes in suppressive soil. Appl Environ Microbiol. 2014;80(9):2679–2686. doi: 10.1128/AEM.03905-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J] Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai LJ, Lu ZM, Zhang XJ, Ma J, Xu PX, Qian W, Xiao C, Wang ST, Shen CH, Shi JS, Zheng-Hong X. Zooming in on butyrate-producing clostridial consortia in the fermented grains of baijiu via gene sequence-guided microbial isolation. Front Microbiol. 2019;10:1397. doi: 10.3389/fmicb.2019.01397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wu Q, Xu Y. Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int J Food Microbiol. 2014;179:80–84. doi: 10.1016/j.ijfoodmicro.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis[J] Nucleic Acids Res. 2014;42(D1):D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Song ZW, Xu Y. ethyl carbamate formation regulated by lactic acid bacteria and nonconventional yeasts in solid-state fermentation of Chinese Moutai-flavor liquor. J Agric Food Chem. 2017;66(1):387–392. doi: 10.1021/acs.jafc.7b05034. [DOI] [PubMed] [Google Scholar]
- Du H, Wang XS, Zhang YH, Xu Y. Exploring the impacts of raw materials and environments on the microbiota in Chinese Daqu starter. Int J Food Microbiol. 2019;297:32–40. doi: 10.1016/j.ijfoodmicro.2019.02.020. [DOI] [PubMed] [Google Scholar]
- Fan GS, Sun BG, Fu ZL, Xia YQ, Huang MQ, Xu CY, Li XT. Analysis of physical indices, volatile flavor components, and microbial community of a light-flavor Daqu. J Am Soc Brew Chem. 2018;76(3):209–218. [Google Scholar]
- Fan GS, Fu ZL, Teng C, Wu QH, Liu PX, Yang R, Karim M, Li XT. Comprehensive analysis of different grades of roasted-sesame-like flavored Daqu. Int J Food Prop. 2019;22(1):1205–1222. [Google Scholar]
- Fan GS, Du YH, Fu ZL, Chen M, Wang Z, Liu PX, Li XT (2020) Characterisation of physicochemical properties, flavour components and microbial community in Chinese Guojing roasted sesame-like flavour Daqu. J Inst Brewing 126(1)
- Gou M, Wang HZ, Yuan HW, Zhang WX, Tang YQ, Kida KJ. Characterization of the microbial community in three types of fermentation starters used for Chinese liquor production. J Inst Brewing. 2015;121(4):620–627. [Google Scholar]
- He GQ, Huang J, Zhou RQ, Wu CD, Jin Y. Effect of fortified Daqu on the microbial community and flavor in Chinese Strong-flavor liquor brewing process. Front Microbiol. 2019;10:56. doi: 10.3389/fmicb.2019.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Huang J, Wu C, Jin Y, Zhou R. Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res Int. 2020;129:108851. doi: 10.1016/j.foodres.2019.108851. [DOI] [PubMed] [Google Scholar]
- Hu YL, Dun YH, Li SN, Fu B, Xiong XM, Nan P, Liang YX, Zhao SM. Changes in microbial community during fermentation of high temperature Daqu used in the production of Chinese ‘Baiyunbian’ liquor. J Inst Brew. 2017;123:594–599. [Google Scholar]
- Huang YH, Yi ZL, Jin YL, Huang MJ, He KZ, Liu DY, Luo HB, Zhao D, He H, Fang Y, Zhao H (2017) Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese Nong-flavor liquor starter. Front Microbiol 8 [DOI] [PMC free article] [PubMed]
- Jin GY, Zhu Y, Xu Y. Mystery behind Chinese liquor fermentation. Trends Food Sci Technol. 2017;63:18–28. [Google Scholar]
- Jin Y, Li DY, Ai M, Tang QX, Huang J, Ding XF, Wu CD, Zhou RQ. Correlation between volatile profiles and microbial communities: a metabonomic approach to study Jiang-flavor liquor Daqu. Food Res Int. 2019;121:422–432. doi: 10.1016/j.foodres.2019.03.021. [DOI] [PubMed] [Google Scholar]
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lian B, Ding YH, Nie CC, Zhang QM. Bacterial diversity in the central black component of Maotai Daqu and its flavor analysis. Ann Microbiol. 2014;64(4):1659–1669. [Google Scholar]
- Li P, Liang HB, Lin WT, Feng F, Luo LX. Microbiota dynamics associated with environmental conditions and potential roles of cellulolytic communities in traditional Chinese cereal starter solid-state fermentation. Appl Environ Microb. 2015;81:5144–5156. doi: 10.1128/AEM.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DN, Huang W, Wang CX, Qiu SY (2019) The complete genome sequence of the thermophilic bacterium Laceyella sacchari FBKL4.010 reveals the basis for tetramethylpyrazine biosynthesis in Moutai‐flavor Daqu. MicrobiologyOpen 8(12):n/a-n/a [DOI] [PMC free article] [PubMed]
- Liu H, Sun B. Effect of fermentation processing on the flavor of Baijiu. J Agric Food Chem. 2018;66:5425–5432. doi: 10.1021/acs.jafc.8b00692. [DOI] [PubMed] [Google Scholar]
- Liu PL, Miao LH. Multiple batches of fermentation promote the formation of functional microbiota in Chinese miscellaneous-flavor baijiu fermentation. Front Microbiol. 2020;11:75. doi: 10.3389/fmicb.2020.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Chen JY, Fan Y, Huang XN, Han BZ. Biochemical characterisation and dominance of different hydrolases in different types of Daqu—a Chinese industrial fermentation starter. J Sci Food Agric. 2017;98(1):113–121. doi: 10.1002/jsfa.8445. [DOI] [PubMed] [Google Scholar]
- Liu C, Feng S, Wu Q, Huang H, Chen Z, Li S, Xu Y. Raw material regulates flavor formation via driving microbiota in chinese liquor fermentation. Front Microbiol. 2019;10:1520. doi: 10.3389/fmicb.2019.01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida M, Yamada O, Gomi K. Genomics of Aspergillus oryzae: learning from the history of Koji mold and exploration of its future. DNA Res. 2008;15(4):173–183. doi: 10.1093/dnares/dsn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies[J] Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon SD, Batth T, Petzold CJ, Adams PD, Simmons BA, Singer SW. Thermoascus aurantiacus is a promising source of enzymes for biomass deconstruction under thermophilic conditions. Biotechnol Biofuels. 2012;5(1):54. doi: 10.1186/1754-6834-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data[J] PLoS ONE. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YM. Cognition and investigation on the manufacturing of sesame flavour liquor. Modern Applied Science. 2009;3:92–95. [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. Peer J. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities[J] Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha S, Chen S, Qian M, Wang CC, Xu Y. Characterization of the typical potent odorants in Chinese roasted sesame-like flavor type liquor by headspace solid phase microextraction-aroma extract dilution analysis, with special emphasis on sulfur-containing odorants. J Agric Food Chem. 2017;65:123–131. doi: 10.1021/acs.jafc.6b04242. [DOI] [PubMed] [Google Scholar]
- Sun J, Li Q, Luo S, Zhang J, Huang M, Chen F, Zheng F, Sun X, Li H. Characterization of key aroma compounds in Meilanchun sesame flavour style baijiu by application of aroma extract dilution analysis, quantitative measurements, aroma recombination, and omission/addition experiments. RSC Adv. 2018;8:23757–23767. doi: 10.1039/c8ra02727g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Tang XX, Tang M, Zhang XM, Xu XR, Yi Y. Analysis of the bacterial communities in two liquors of Soy Sauce Aroma as revealed by high-throughput sequencing of the 16S rRNA V4 hypervariable region. Biomed Res Int. 2017;2017:1–9. doi: 10.1155/2017/6271358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Liang H, Song J, Lin W, Luo L. Comparison of microbial community and metabolites in spontaneous fermentation of two types Daqu starter for traditional Chinese vinegar production. J Biosci Bioeng. 2019;128(3):307–315. doi: 10.1016/j.jbiosc.2019.03.011. [DOI] [PubMed] [Google Scholar]
- Wang CL, Shi DJ, Gong GL. Microorganisms in Daqu: a starter culture of Chinese Maotai-flavor liquor. World J Microbiol Biotechnol. 2008;24:2183–2190. [Google Scholar]
- Wang P, Wu Q, Jiang XJ, Wang ZQ, Tang JL, Xu Y. Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int J Food Microbiol. 2017;250:59–67. doi: 10.1016/j.ijfoodmicro.2017.03.010. [DOI] [PubMed] [Google Scholar]
- Wang XS, Du H, Zhang Y, XuY (2017) Environmental microbiota drives microbial succession and metabolic profiles during Chinese Liquor Fermentation. Appl Environ Microbiol 84(4) [DOI] [PMC free article] [PubMed]
- Wu Q, Ling J, Xu Y. Starter culture selection for making Chinese sesame-flavored liquor based on microbial metabolic activity in mixed-culture fermentation. Appl Environ Microbiol. 2014;80(14):4450–4459. doi: 10.1128/AEM.00905-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JH, Huo JY, Huang MQ, Zhao MM, Luo XL, Sun BG. Structural characterization of a tetrapeptide from sesame flavor-type Baijiu and its preventive effects against AAPH-induced oxidative stress in HepG2 Cells. J Agric Food Chem. 2017;65(48):10495–10504. doi: 10.1021/acs.jafc.7b04815. [DOI] [PubMed] [Google Scholar]
- Xie MW, Lv FX, Ma GX, Asim F, Li HH, Du Y, Liu Y. High throughput sequencing of the bacterial composition and dynamic succession in Daqu for Chinese sesame flavour liquor. J Inst Brewing. 2020;126:98–104. [Google Scholar]
- Yang JG, Dou X, Han PJ, Bai FY, Zhou J, Zhang SY, Hui Q, Ma YY. Microbial diversity in Daqu during production of luzhou flavored liquor. J Am Soc Brew Chem. 2017;75(2):136–144. [Google Scholar]
- Yang JG, Dou X, Ma YY. Diversity and dynamic succession of microorganisms during Daqu preparation for Luzhou-flavour liquor using second-generation sequencing technology. J Inst Brewing. 2018;124(4):498–507. [Google Scholar]
- Yao S, Liu Y, Li H, Ge YY, Zhang MJ, Xin CH, Xu L, Cheng C. Bacterial communities during the process of high-temperature Daqu production of roasted sesame-like flavor liquor. J Inst Brew. 2015;121:440–448. [Google Scholar]
- Yi ZL, Jin YL, Xiao Y, Chen LC, Tan L, Du AP, He KZ, Liu DY, Luo HB, Fang Y, Zhao H. Unraveling the contribution of high temperature stage to Jiang-flavor Daqu, a liquor starter for production of Chinese Jiang-flavor baijiu, with special reference to metatranscriptomics. Front Microbiol. 2019;10:472. doi: 10.3389/fmicb.2019.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhao J, Du X. Barcoded pyrosequencing analysis of the bacterial community of Daqu for light-flavour Chinese liquor. Lett Appl Microbiol. 2014;58(6):549–555. doi: 10.1111/lam.12225. [DOI] [PubMed] [Google Scholar]
- Zhang LL, Li LJ, Pan XG, Shi ZL, Feng XH, GongB LJ, Wang LS. Enhanced growth and activities of the dominant functional microbiota of chicken manure composts in the presence of maize straw. Front Microbiol. 2018;9:1131. doi: 10.3389/fmicb.2018.01131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Liu Y, Shu L, He Y. Study on metabolites of Bacillus producing soy sauce-like aroma in Jiang-flavor Chinese spirits. Food Sci Nutr. 2019;8(1):97–103. doi: 10.1002/fsn3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XW, Yan Z, Nout MJR, Smid EJ, Zwietering MH, Boekhout T, Han JS, Han BZ. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. Int J Food Microbiol. 2014;182–183:57–62. doi: 10.1016/j.ijfoodmicro.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Zheng XW, Yan Z, Robert Nout MJ, Boekhout T, Han BZ, Zwietering MH, Smid EJ. Characterization of the microbial community in different types of Daqu samples as revealed by 16S rRNA and 26S rRNA gene clone libraries. World J Microbiol Biotechnol. 2014;31(1):199–208. doi: 10.1007/s11274-014-1776-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.