

Abstract
Purifying extracellular vesicles (EVs) from complex biological fluids is a critical step in analyzing EVs molecularly. Plasma lipoprotein particles (LPPs) are a significant confounding factor as they outnumber EVs >104-fold. Given their overlap in size, LPPs cannot be completely removed using standard size-exclusion chromatography. Density-based separation of LPPs can be applied but is impractical for routine use in clinical research and practice. Here we report a new separation approach, dubbed DMC (dual-mode chromatography), capable of enriching plasma EVs and depleting LPPs. DMC conveniently integrates two orthogonal separation steps in a single column device: i) size exclusion to remove high-density lipoproteins (HDLs) that are smaller than EVs; and ii) cation exchange to clear positively-charged ApoB100-containing LPPs, mostly (very) low-density lipoproteins (V)LDLs, from negatively-charged EVs. The strategy enabled DMC to deplete most LPPs (>97% of HDLs and >99% of (V)LDLs) from human plasma, while retaining EVs (>30% of input). Furthermore, the two-in-one operation is fast (15 min/sample) and equipment-free. With abundant LPPs removed, DMC-prepared samples facilitated EV identification in imaging analyses and improved the accuracy for EV protein analysis.
Keywords: extracellular vesicles, lipoproteins, liquid biopsies
Graphical Abstract
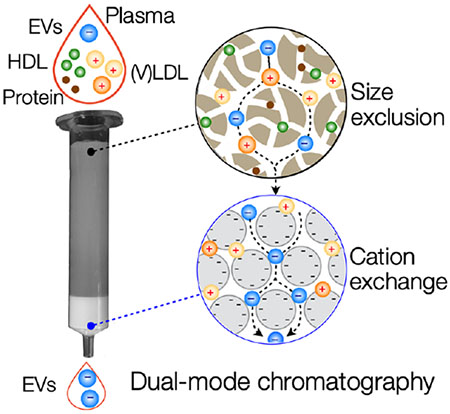
Extracellular vesicles (EVs) are a promising source of disease biomarkers. A new, single-step chromatography approach for EV enrichment, dual-mode chromatography (DMC) was developed. DMC effectively removes most plasma lipoproteins, >60-fold more efficient than size-exclusion chromatography. Consequently, DMC produces an enriched EV population and improves analytical outcomes of EV immunoassays by lowering biological background.
Extracellular vesicles (EVs) are important disease biomarkers;[1] they are ubiquitously present in bodily fluids and carry molecular cargo from their respective parental cells (e.g., transmembrane and intracellular proteins, mRNA, DNA, and microRNA).[2]. EVs thus can serve as a blood-based analytical method to obtain and monitor diseases’ molecular traits,[3] promoting better-informed clinical decisions. Indeed, EVs were found superior to conventional protein markers for tumor detection.[4–6] Key mutations (e.g., EGFRvIII, IDH1R132H, EGFRT790M) have also been detected in EVs.[6,7] Essential to diagnostic EV analysis is the ability to perform a given molecular test in true EV fractions, rather than in contaminated mixtures. In other words, EVs exist in complex heterogeneous matrices (Figure 1a) and retrieving pure vesicle populations is a pivotal first assay step.
Figure 1. Rationale and principle of the dual-mode chromatography.
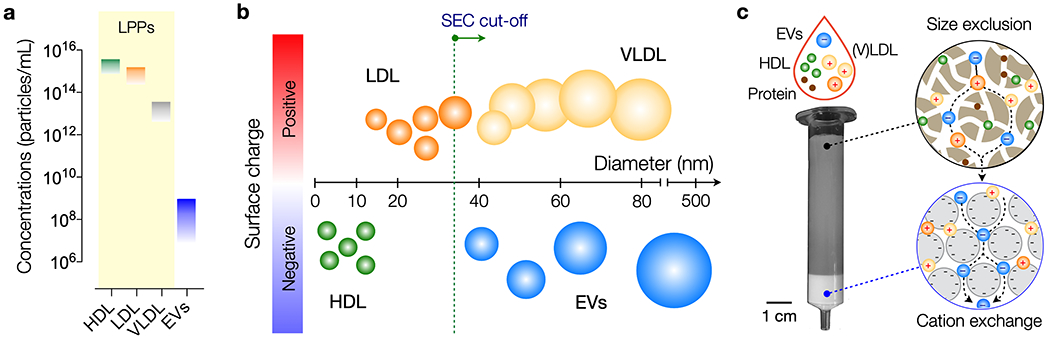
(a) Typical concentrations of lipoprotein particles (LPPs) and extracellular vesicles (EVs) in human plasma. LPP numbers are considerably higher (>104) than EV numbers and also fluctuate during the day. HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein. (b) Schematic representation of the most common particle species found in plasma, discriminated by size and surface charge. Note that EVs overlap in size with (V)LDL, but have opposite surface charge. Green dotted line indicates a commonly used cut-off for size-exclusion chromatography (SEC) that separates EVs from other particles. (c) Design of dual-mode chromatography (DMC) column. The device has two separation layers in tandem: size exclusion (top) and cation exchange (bottom). The top layer is used to filter out small analytes, including soluble proteins and HDL particles. Filtrates then enter the cation exchange layer where positively-charged particles (e.g., LDL, VLDL) are captured. The resulting sample is depleted from LPPs and enriched in EVs.
Among various EV isolation strategies,[1] size-exclusion chromatography (SEC) is increasingly adopted as a preferable isolation method for clinical samples, considering its low cost, much faster turn-around time compared to ultracentrifugation, and ease-of-operation.[8] SEC separates analytes based on their differential retention time in porous gels. When applied to plasma or serum, SEC produces well-defined vesicle fractions with most soluble proteins removed.[9] The current approaches, however, often fail to differentiate EVs from certain types of lipoprotein particles (LPPs), predominantly (very) low-density lipoproteins (V)LDL, due to size overlap.[10] With LPPs (~1015 particles/mL)[11] significantly outnumbering EVs (~107-109 particles/mL in healthy individuals),[12] SEC-prepared samples are susceptible to artifacts, including overestimation of actual EV counts, steric hindrance in immunoassays, and increased biological noise. This is especially important in the discovery phase of EV protein biomarkers, as the abundance of LPPs can confound the search for less-abundant EV-associated proteins. Density-gradient ultracentrifugation can be incorporated before or after SEC to separate EVs from LPPs but the combined process negates SEC’s practical advantages.
Here, we report a substantially improved chromatography method for rapid EV isolation from plasma samples. We noticed the contrast in surface charge properties between EVs, that are negatively charged,[13] and ApoB100-containing LPPs, which carry an abundance of positive charges.[14] While previous work to discriminate EVs and LPPs predominantly focused on size and/or density, we reasoned that these charge discrepancies could be exploited for better particle separation (Figure 1b). Indeed, ApoB100 lipoproteins are known to interact with glycosaminoglycans on the arterial wall in vivo,[15] which has led to the use of polymers bearing sulphate groups for plasmapheresis[16] or general lipoprotein isolation.[17] We thus explored combining ion exchange in tandem with size-exclusion chromatography to obtain an LPP-depleted EV population. The strategy allowed us to remove the majority of plasma LPPs, >97% of high-density lipoprotein/HDL and >99% of (V)LDL, while maintaining a relatively high EV isolation yield comparable to that of SEC. We also constructed a layered monolithic device, termed a DMC (dual-mode chromatography) column, to perform orthogonal separations through a one-time sample loading. This one-time loading approach is particularly important compared to sequential separation strategies as it minimizes loss of rare EV fractions. DMC operation was simple, fast (15 min per sample) and equipment-free. Furthermore, DMC-prepared samples led to better analytical outcomes in single vesicle imaging and EV protein analyses.
The DMC column consists of a top, size-exclusion layer and a bottom, cation exchange layer (Figure 1c). The top layer separates small analytes (e.g., soluble proteins, protein aggregates, HDL) from larger particles through differential retention time. The bottom layer receives filtrates from the top and captures positively-charged (V)LDL particles. For the top layer, we used a conventional SEC resin (Sepharose CL-4B, exclusion limit ~35 nm). The resin volume was 10 mL, identical to that of gravity-driven SEC columns commonly used for EV isolation.[8,9]
To determine an optimal material for (V)LDL capture, we compared a panel of resins with varying functional groups and physical characteristics. We made cation exchange columns, each packed (2 mL) with a single resin type. Test samples were prepared by filtering human plasma through SEC and collecting the EV-enriched fractions (see Experimental sections), enriching for (V)LDL and EVs. We then processed these fractions with the cation exchange columns, assessing each column’s ability to remove (V)LDL by measuring ApoB100. Fractogel EMD SO3− was found to be most efficient, removing up to 70-fold more ApoB100 than other tested resins (Figure S1a, b), possibly due to its ‘tentacle’ structure which promotes interaction between resin and target.[18] The final removal efficiency of ApoB100 was ~98%. Doubling the resin volume (to 4 mL) resulted in only a minor improvement in efficiency (Figure S1c). The final DMC thus had the following dual layer structure: Sepharose (10 mL, top) and Fractogel (2 mL, bottom).
We characterized the DMC column performance and compared it to a standard SEC column. We started by assessing EV recovery ratio. EV-only samples (cell-line derived EVs in a buffer) were passed through the columns and EV counts before versus after separation were measured via nanoparticle tracking analysis (NTA; Figure 2a). EV recovery ratios were in the same order of magnitude: 0.34 (DMC) and 0.78 (SEC). DMC’s slightly lower value could be attributed to its longer filtering path. We next measured the capacity for LPP removal and EV enrichment by DMC and SEC columns. We mimicked “clinical samples” by spiking human plasma (0.5 mL) samples with EVs (~1010 /mL) from cancer cell lines (Experimental sections). Representative lipoprotein markers were ApoA1 for HDL and ApoB100 for (V)LDL, while CD63 was chosen as EV marker. We observed a significant reduction in ApoB100 after DMC separation (Figure 2b). When an equal amount (~18 µg) of SEC and DMC sample proteins were analyzed, ApoA1 levels were found to be similar (Figure 2c), which may reflect the same HDL depletion mechanism (size exclusion) in both columns. CD63 level, however, was markedly higher in the DMC filtrate (Figure 2c) due to efficient ApoB100 removal. Figure 2d summarizes the calculated LPP removal efficiency. DMC and SEC columns both effectively cleared HDL particles from plasma, with an efficiency of ~97%. For (V)LDL particles, however, DMC was far superior to SEC: only 0.4% of ApoB100 in the input plasma remained after the DMC filtering, whereas the number was ~25% with the SEC column.
Fig. 2. DMC characterization.
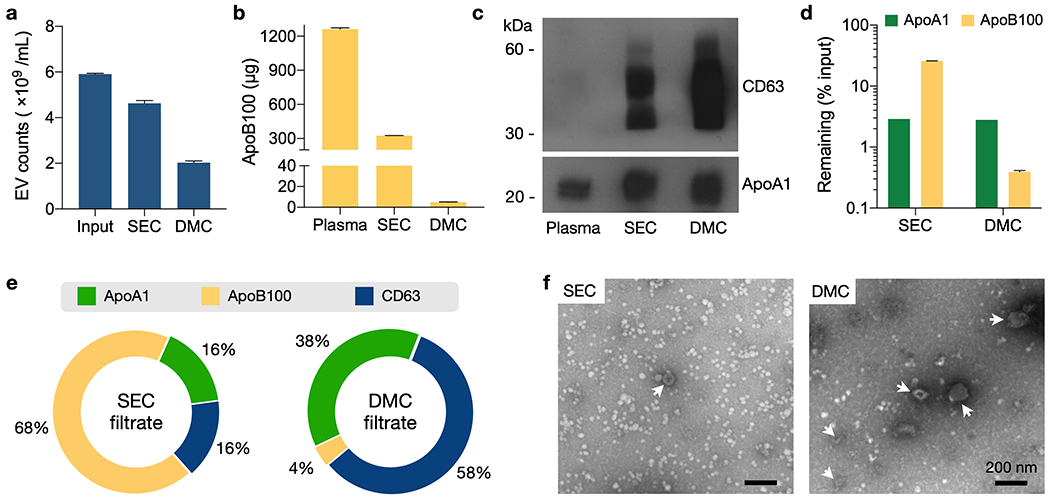
(a) EV recovery was estimated using samples with a known number of cell-line derived EVs in PBS (input). Equal amount of samples were passed through a SEC or DMC column, and particle numbers after preparation were measured via nanoparticle tracking analysis (NTA). Recovery ratios were 78% (SEC) and 34% (DMC). Data from technical triplicates are displayed as mean ± s.d. (b) DMC and SEC columns were used to process mock clinical samples which were human plasma spiked with cell-line derived EVs. Most ApoB1 proteins were removed by DMC. Data from technical duplicates are displayed as mean ± s.d. (c) The same amount of plasma and EV sample proteins from (b) were analyzed. ApoA1 levels were comparable between SEC and DMC. CD63 was significantly enriched in DMC samples. (d) Capacity for LPP removal by DMC and SEC were compared. ApoA1 (HDL) and ApoB100 (VLDL and LDL) contents were measured before and after filtration of human plasma (0.5 mL). Both DMC and SEC showed a similar efficiency (~97%) in ApoA1 depletion. For ApoB100 removal, DMC (efficiency: 0.4%) significantly outperformed SEC (25%). Data from technical duplicates are displayed as mean ± s.d for ApoB100. ApoA1 quantification was based on band intensity from Western blotting. (e) Relative mass-ratios of ApoA1, ApoB100, and CD63 were estimated in SEC and DMC filtrates. (V)LDL was the major vesicle population in the SEC filtrate, whereas EVs were the dominant component in the DMC-prepared sample. (f) Transmission electron micrographs of SEC- and DMC-prepared human plasma. EVs (arrowheads) were negatively stained; LPPs appeared white. Note LPP reduction and EV enrichment in the DMC sample. Close-up images are provided in Figure S2.
Based on these data, we estimated the relative mass ratio of the most significant proteins in SEC- or DMC-prepared samples. Figure 2e show the relative mass fraction of ApoA1, ApoB100, and CD63; the amount of each protein target was estimated from ELISA or Western blotting, and was normalized against the sum of these three proteins. EV protein (CD63) was the dominant fraction (>80%) in the DMC sample, while HDL and (V)LDL were largely removed. The reverse trend was observed in the SEC sample, with LPPs being the major component. Electron microscopy confirmed these observations (Figure 2f and Figure S2). DMC-prepared plasma samples contained more EVs and less LPPs than SEC samples. Indeed, overall protein analysis by SDS-PAGE followed by Coomassie blue staining revealed differences in protein profile between these two samples (Figure S3a). In agreement with lipoprotein particle removal, overall cholesterol levels were found to be ~5 times less in DMC samples (Figure S3b). As a consequence of lipoprotein removal, DMC-prepared samples appear much clearer than SEC samples (Figure S3c).
We also noticed a potential pitfall when interpreting NTA data for plasma EV numbers. After SEC or DMC preparation, we found that NTA particle counts were directly proportional to ApoB100 amounts in samples (Figure S4a). This result strongly suggests the majority of counted particles could actually be (V)LDLs rather than EVs. Western blotting supported this observation. When we analyzed SEC and DMC samples with the same NTA particle counts, we saw negligible CD63 and low ApoA1 levels in the SEC samples (Figure S4b). Simply counting particle numbers in SEC filtrates, without any molecular specificity, thus can lead to overestimation of EV numbers, even by as much as an order of magnitude. By extension, this implies that calculating particle/protein ratio for estimating EV purity might be less suitable for plasma-derived samples.[19]
We hypothesized that removing LPP contamination increases the accuracies in EV immunoassays by lowering biological background noise. To test this hypothesis, we processed universal human samples using DMC and SEC columns, and subjected filtered samples to two different analytical modalities, single particle imaging[20] and the integrated magnetoelectrochemical exosome (iMEX) assay.[5] For single EV imaging, we used a lipophilic dye to stain overall lipid particle populations and a fluorophore-conjugated anti-CD63 antibody for EV labeling (Figure 3 and Figure S5). The SEC-only sample had a large lipid particle population but only a small fraction (~4%) of them were CD63-positive (Figure 3a, top). In contrast, the DMC sample was less crowded but more lipid particles (~85%) were CD63-positive; this led to robust EV identification and counting (Figure 3a, bottom). Note that EV-free controls showed no measurable signal in both fluorescent channels after labeling (Figure S6).
Fig. 3. EV assays with DMC and SEC samples.
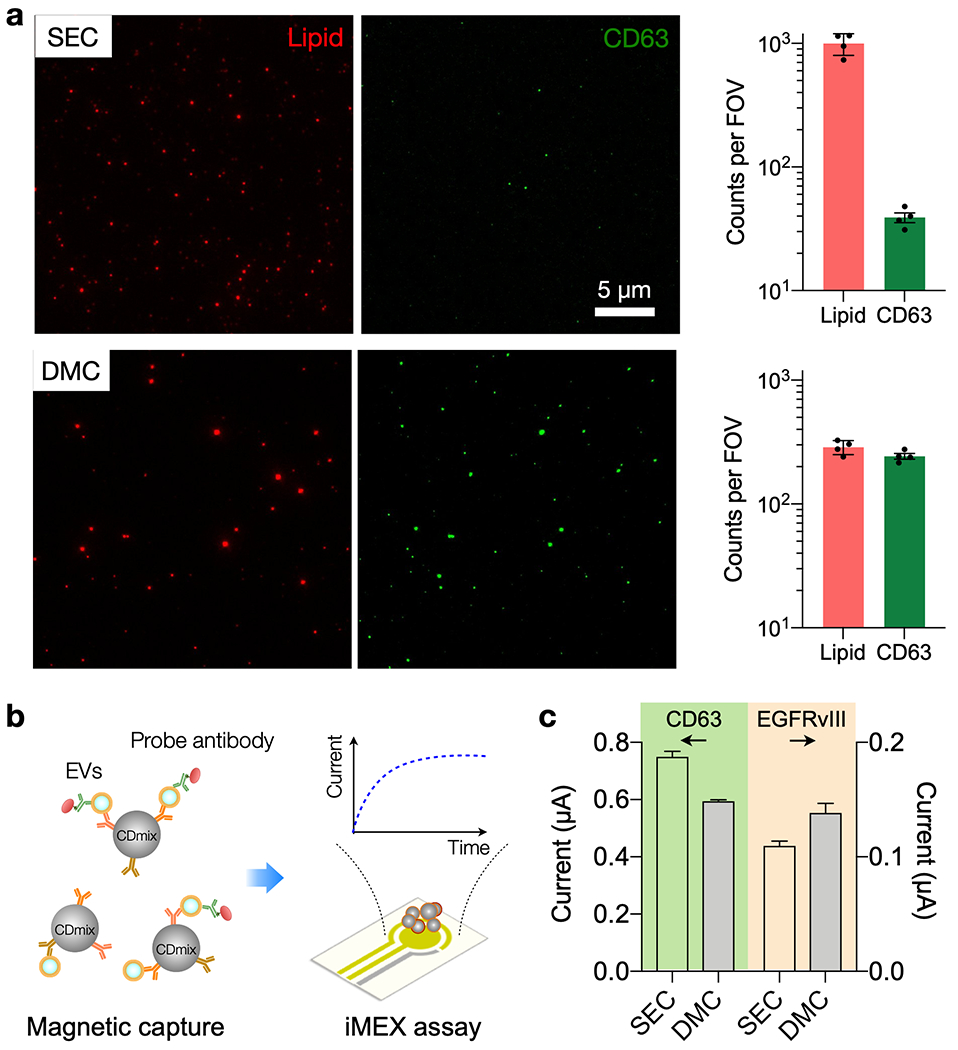
(a) Single particle imaging. SEC and DMC samples were labeled with the general lipid dye (red) and fluorescent CD63 antibodies (green). The SEC sample contained a large number of lipid particles per field-of-view (FOV, 77 × 65 µm2), but few of them were CD63-positive (top row). The DMC sample, in contrast, was enriched with CD63-positive lipid particles (bottom row). The graphs show particle counts (mean ± s.d.) from four FOVs. (b) Integrated magneto-electrochemical exosome (iMEX) EV protein assay. EVs are captured on magnetic beads based on EV-specific surface markers (CD63, CD81, CD9) and further labeled with probe antibodies to detect target protein markers. Probe antibodies, conjugated with oxidizing enzymes, generate electrical currents through electrochemical reaction. (c) Human plasma was spiked with EVs from Gli36 EFGRvIII mutation. Following SEC or DMC preparation, samples were assessed for CD63 and EGFRvIII expression via iMEX. The DMC sample showed a slightly lower CD63 signal than SEC, reflecting lower EV recovery. EGFRvIII signal, however, was higher in the DMC sample, presumably due to reduced interference from biological background. The data are from technical duplicates and displayed as mean ± s.d.
We next used the iMEX assay to measure EV surface protein levels in SEC and DMC samples. We prepared test samples by spiking human plasma (1 mL) with EVs (~1010 /mL) from a human glioblastoma cell line (Gli36 with EGFRvIII overexpression). Aliquots (0.5 mL) were processed with either SEC or DMC. To capture EVs, we used magnetic beads specific to tetraspanins (CD63, CD81, CD9). Bead-EV complexes were then further labeled probing antibodies (CD63 or EGFRvIII) for signal generation (Figure 3b). The iMEX results (Figure 3c) showed that CD63 levels effectively followed the EV recovery ratio of SEC and DMC (Figure 2a), whereas EGFRvIII signal was more pronounced in the DMC sample. This observation may result from differences in target molecule concentrations. CD63 level in plasma would be higher than that of EGFRvIII, which makes CD63 detection less affected by biological background (i.e., LPPs); for the less abundant EGFRvIII, removing interfering LPPs makes the analytical signal more robust.
In summary, we developed a new, single-step chromatography approach for EV enrichment, DMC, and evaluated its performance with plasma, the most commonly used bodily fluid for EV analyses. DMC effectively removed most plasma LPPs: >97% HDL, similar to SEC; and >99% (V)LDL, >60-fold more efficient than SEC. Consequently, DMC produced enriched EV population and improved analytical outcomes of EV immunoassays with lower biological background. We envision further investigations with different biofluids (e.g., CSF, saliva) and analytical modalities (e.g., nucleic acid detection) to broaden DMC’s applicability. Such efforts would further confirm DMC as a powerful EV preparation strategy that can seamlessly replace the current SEC-based EV isolation.
Experimental Section
Plasma.
The sodium-EDTA plasma was acquired from Rockland Immunochemicals, Inc.
Size-exclusion chromatography (SEC) and dual-mode chromatography (DMC).
Sepharose CL-4B (GE Healthcare), Fractogel EMD SO3− (M) (Millipore Sigma), Capto S (GE Healthcare) and SP Sepharose Fast Flow (GE Healthcare) resins were washed three times with PBS buffer. A nylon net with 11 μm pore size (NY1102500, Millipore Sigma) was placed on the bottom of a 10 mL syringe (BD Biosciences). For the SEC column, this was followed by stacking of 10 mL washed Sepharose. For the DMC column, 2 mL of ion exchange resin was stacked first, followed by careful layering of 10 mL Sepharose on top. After adding 0.5 mL plasma sample, individual fractions of 1 mL eluate were collected. EV-containing fractions (first 2 fractions after the void volume, which was 3 mL for SEC and 3.8 mL for DMC) were pooled and concentrated using Amicon Ultra-2 10K filters (Millipore Sigma).
EV preparation for spike-in experiments.
ES-2 cells and Gli36 cells carrying an EGFRvIII overexpression (Gli36 EGFRvIII) were cultured in DMEM supplemented with 10% fetal bovine serum. Cells were washed 3 times with serum free medium and cultured in DMEM supplemented with 1% EV-depleted FBS (Invitrogen) for 24 hours. Conditioned medium (CM) from ~2 × 108 cells was collected and centrifuged at 300 g (10 min) and 2000 g (20 min). Next, CM was concentrated approximately 300 times using a Centricon Plus-70 centrifugal filter device with a 10K nominal molecular weight limit (Millipore Sigma). Subsequently, a discontinuous OptiPrep density gradient (ODG) was constructed as described previously.[21]. 1 mL of concentrated CM was overlaid onto the top of the gradient, which was then centrifuged for 18 hours at 100,000 g and 4 °C (SW 27.1 Ti rotor, Beckman Coulter). Next, fractions of 1 mL were collected from the top of the gradient, with F9 and 10 (density ~1.1 g mL−1) being pooled and used for subsequent SEC-based separation of EVs from the iodixanol polymer, using Sepharose CL-2B as resin. EV-containing fractions (F4-7) were pooled, concentrated to 100 µL, aliquoted, and stored at −80 °C. In Figure 2b–e, 3a, S3 and S4, the results were obtained using plasma samples spiked with ES2 EVs (~7×109 mL−1); in Figs. 2f and 3c, plasma samples spiked with Gli36 EGFRvIII EVs (~1×1010 mL−1). To demonstrate bona fide EV isolation, presence of CD9, CD63 and CD81 was analyzed using bead-based flow cytometry (Figure S7).[22]
Nanoparticle tracking analysis (NTA).
We used a NanoSight LM10 microscope (Malvern) equipped with a 405 nm laser. Three 30-second videos were recorded of each sample with camera level 15. After each video, the sample was advanced through the chamber to avoid repeated measurement of identical particles in the field of view. Videos recorded for each sample were analyzed with NTA software version 3.2 with detection threshold kept constant at 3. Samples were diluted with PBS buffer until particle concentration was within the linear concentration range of the NTA software (3×108 – 1×109 mL−1).
Western blot and Coomassie blue.
EV samples were lysed in 0.2% SDS and protein concentration measured by Qubit assay (Thermo Fisher). Samples were lysed with non-reducing LDS sample buffer (Invitrogen), boiled for 5 min at 95 °C, and loaded on a 5-12% gradient gel (Invitrogen). Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunostained for 1h with the following antibodies: anti-CD63 (clone H5C6, BD Biosciences, 1:200 dilution) and anti-ApoA1 (clone B-10, Santa Cruz, 1:1000 dilution). HRP-conjugated secondary antibodies were added for 1h, blots were washed, followed by addition of chemiluminescence substrate (WesternBright Sirius, Advansta). Blots were then developed using autoradiographic films. Films were digitized and quantification of signal intensity was performed using ImageJ. For Coomassie blue staining, gel was stained with SimplyBlue SafeStain (Thermo Fisher) for 1 hour at room temperature, followed by destaining overnight in dH20 at 4 °C. Gel was imaged on a Sapphire Biomolecular Imager (Azure Biosystems).
ELISA.
Human Apolipoprotein B Quantikine ELISA Kit (R&D Systems) was used according to manufacturer’s instructions. Standards and samples were assayed in duplicate.
Cholesterol assay.
The MyQubit Amplex® Red Cholesterol Assay (Thermo Fisher) was used according to manufacturer’s instructions.
Electron microscopy.
EV samples (5 µL) were overlaid with Formvar carbon-coated grids and incubated 20 min. Grids were then washed in PBS and fixed for 5 min with 1% glutaraldehyde. Grids were washed in dH2O and incubated on 2% uranylacetate for 5 min. Excess stain was removed by blotting and grids were air-dried. Images were taken using a Tecnai G2 Spirit BioTWIN microscope.
Single EV imaging.
EV-containing SEC and DMC fractions were incubated with CM-DiI (Thermo Fisher Scientific) for 30 min at room temperature. EV-free samples were subjected to the same labeling processes. Dye aggregates were removed by Millex-GV syringe filter (0.22 μm pore size, Millipore Sigma). Filtered EVs were captured on a glass slide. Following 30 min incubation at room temperature, the slide was washed with PBST (PBST buffer containing 0.001% Tween 20). After incubation with fixation buffer (4% formaldehyde) and blocking buffer (Superblock, Thermo Fisher Scientific), EV samples were incubated with anti-human CD63 antibody (Ancell) for 90 min at room temperature. After washing, Alexa Fluor 488-labeled secondary antibody was introduced and incubated for 30 min at room temperature. After final wash steps, fluorescence images were taken with BX-63 upright fluorescent microscope (Olympus) with 40× objective. Acquisition settings (i.e., objective, exposure time, camera setting, illumination) were kept constant for all images.
iMEX protocol.
Antibodies for EV capture (mouse monoclonal anti-CD63 (clone H5C6, BD Biosciences), CD9 (clone MM2/57, Millipore Sigma) and CD81 (clone 1.3.3.22, Thermo Fisher)) were coupled to Pierce Protein A magnetic beads (Life Technologies) in a ratio of 10 µg of total antibody per 100 µL of beads by overnight incubation at 4 °C with rotation. Beads were washed three times with 500 μL of PBS/0.001% Tween and resuspended in 100 µL of the same buffer. For the iMEX assay, 100 µL of EV samples were mixed with 10 µL of the immunomagnetic bead solution for 15 min at room temperature. After incubation, magnetic beads were separated from the solution with a permanent magnet and re-suspended in 80 µL of PBS (1% BSA). After 5 seconds of vortexing, the beads were separated and re-suspended in 50 µL of PBS (1% BSA). 10 µL of antibodies of interest (20 µg/mL in PBS) were added to the solution and the mixture was incubated for 15 min at room temperature. The magnetic beads were separated and washed as described before and re-suspended in 50 µL of PBS (1% BSA). 5 µL of streptavidin-conjugated HRP enzymes (1:100 diluted in PBS) were mixed with the beads for 15 min at room temperature. The magnetic beads were separated and washed as described before and re-suspended in 7 µL of PBS. The prepared bead solution and 20 µL of UltraTMB solution (ThermoFisher Scientific) were loaded on top of the screen-printed electrode. After 3 min, chronoamperometry measurement was started with the electrochemical sensing device. The current levels in the range of 50-55 seconds were averaged.
Statistical analysis.
All data were displayed as mean ± s.d. from technical replicates. Samples numbers and relevant statistical tests are indicated in figure legends. P values < 0.05 were considered statistically significant. We used GraphPAD Prism (version 8.0) for analyses. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200025).[23]
Supplementary Material
Acknowledgements
The authors thank Dr. Breakefield (Massachusetts General Hospital) for helpful discussion. This work was supported in part by U.S. NIH Grants P01CA069246 (R.W., H.L.), R01CA229777 (H.L.), 1R01CA204019 (R.W.), U01CA233360 (H.L.), T32CA 79443 (H.-Y.L.), W81XWH1910199 (H.L.), DOD-W81XWH1910194 (H.L.); R00CA201248 (H.I.), R21CA217662 (H.I.), P30AG062421 (H.I); Belgian American Educational Foundation fellowship (J.V.D.); MGH Scholar Fund (H.L.), MGH Fund for Medical Discovery Fellowship (H.-Y.L.); the Institute for Basic Science IBS-R026-D1 (H.L.), South Korea. Huiyan Li thanks a postdoctoral fellowship from the Canadian Institutes of Health Research.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jan Van Deun, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Ala Jo, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Huiyan Li, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Hsing-Ying Lin, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Ralph Weissleder, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA, Department of Systems Biology, Harvard Medical School, Boston, MA 02114, USA.
Hyungsoon Im, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Hakho Lee, Center for Systems Biology and Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA, Center for NanoMedicine, Institute for Basic Science (IBS), Seoul 03722, Republic of Korea, Yonsei-IBS Institute, Yonsei University, Seoul 03722, Republic of Korea.
References
- [1].Shao H, Im H, Castro CM, Breakefield X, Weissleder R, Lee H Chem. Rev 2018, 118, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, Bigner DD. FASEB J. 2009, 23, 1541; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WTJ, Carter BS, Krichevsky AM, Breakefield XO. Nat. Cell Biol 2008, 10, 1470; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Nat. Commun 2011, 2, 180; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Nat. Cell Biol 2007, 9, 654. [DOI] [PubMed] [Google Scholar]
- [3].a) Kaiser J. Science 2018, 359, 259. [DOI] [PubMed] [Google Scholar]; b) Pantel K, Alix-Panabieres C. Cancer Res. 2013, 73, 6384. [DOI] [PubMed] [Google Scholar]
- [4].a) Shao H, Chung J, Lee K, Balaj L, Min C, Carter BS, Hochberg FH, Breakefield XO, Lee H, Weissleder R. Nat. Commun 2015, 6, 6999; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang KS, Im H, Hong S, Pergolini I, Del Castillo AF, Wang R, Clardy S, Huang CH, Pille C, Ferrone S, Yang R, Castro CM, Lee H, Del Castillo CF, Weissleder R. Sci. Transl. Med 2017, 9, eaal3226; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yoshioka Y, Kosaka N, Konishi Y, Ohta H, Okamoto H, Sonoda H, Nonaka R, Yamamoto H, Ishii H, Mori M. Nat. Commun 2014, 5, 3591; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang P, Zhou X, He M, Shang Y, Tetlow AL, Godwin AK, Zeng Y. Nat. Biomed. Eng 2019, 3, 438; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Liu C, Zhao J, Tian F, Cai L, Zhang W, Feng Q, Chang J, Wan F, Yang Y, Dai B, Cong Y, Ding B, Sun J, Tan W. Nat. Biomed. Eng 2019, 3, 183; [DOI] [PubMed] [Google Scholar]; f) Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, Lee H. Nat. Biotechnol 2014, 32, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jeong S, Park J, Pathania D, Castro CM, Weissleder R, Lee H. ACS Nano 2016, 10, 1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shao H, Chung J, Balaj L, Charest A, Bigner DD, Carter BS, Hochberg FH, Breakefield XO, Weissleder R, Lee H. Nat. Med 2012, 18, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Figueroa JM, Skog J, Akers J, Li H, Komotar R, Jensen R, Ringel F, Yang I, Kalkanis S, Thompson R, LoGuidice L, Berghoff E, Parsa A, Liau L, Curry W, Cahill D, Bettegowda C, Lang FF, Chiocca EA, Henson J, Kim R, Breakefield X, Chen C, Messer K, Hochberg F, Carter BS. Neuro. Oncol 2017, 19, 1494; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Castellanos-Rizaldos E, Grimm DG, Tadigotla V, Hurley J, Healy J, Neal PL, Sher M, Venkatesan R, Karlovich C, Raponi M, Krug A, Noerholm M, Tannous J, Tannous BA, Raez LE, Skog JK. Clin. Cancer Res 2018, 24, 2944. [DOI] [PubMed] [Google Scholar]
- [8].Lane RE, Korbie D, Trau M, Hill MM. Proteomics 2019, 19, e1800156. [DOI] [PubMed] [Google Scholar]
- [9].Böing AN, van der Pol E, Grootemaat AE, Coumans FA, Sturk A, Nieuwland R. J. Extracell. Vesicles 2014, 3, 23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Karimi N, Cvjetkovic A, Jang SC, Crescitelli R, Hosseinpour Feizi MA, Nieuwland R, Lötvall J, Lässer C. Cell. Mol. Life Sci 2018, 75, 2873; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sódar BW, Kittel Á, Pálóczi K, Vukman KV, Osteikoetxea X, Szabó-Taylor K, Németh A, Sperlágh B, Baranyai T, Giricz Z, Wiener Z, Turiák L, Drahos L, Pállinger É, Vékey K, Ferdinandy P, Falus A, Buzás EI. Sci. Rep 2016, 6, 24316; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Takov K, Yellon DM, Davidson SM. J. Extracell. Vesicles 2019, 8, 1560809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Caulfield MP, Li S, Lee G, Blanche PJ, Salameh WA, Benner WH, Reitz RE, Krauss RM. Clin. Chem 2008, 54, 1307. [DOI] [PubMed] [Google Scholar]
- [12].Coumans FAW, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, Emanueli C, Gasecka A, Hendrix A, Hill AF, Lacroix R, Lee Y, van Leeuwen TG, Mackman N, Mäger I, Nolan JP, van der Pol E, Pegtel DM, Sahoo S, Siljander PRM, Sturk G, de Wever O, Nieuwland R. Circ. Res 2017, 120, 1632. [DOI] [PubMed] [Google Scholar]
- [13].Brownlee Z, Lynn KD, Thorpe PE, Schroit AJ. J. Immunol. Methods 2014, 407, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Olsson U, Camejo G, Olofsson SO, Bondjers G. Biochim. Biophys. Acta 1991, 1097, 37. [DOI] [PubMed] [Google Scholar]
- [15].Lipponen K, Stege PW, Cilpa G, Samuelsson J, Fornstedt T, Riekkola ML. Anal. Chem 2011, 83, 6040. [DOI] [PubMed] [Google Scholar]
- [16].Yokoyama S, Hayashi R, Kikkawa T, Tani N, Takada S, Hatanaka K, Yamamoto A. Arteriosclerosis 1984, 4, 276. [DOI] [PubMed] [Google Scholar]
- [17].Liangsupree T, Multia E, Metso J, Jauhiainen M, Forssén P, Fornstedt T, Öörni K, Podgornik A, Riekkola ML. Sci. Rep 2019, 9, 11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Connell-Crowley L, Nguyen T, Bach J, Chinniah S, Bashiri H, Gillespie R, Moscariello J, Hinckley P, Dehghani H, Vunnum S, G. Vedantham. Biotechnol. Bioeng 2012, 109, 157. [DOI] [PubMed] [Google Scholar]
- [19].Webber J, Clayton A. J. Extracell. Vesicles 2013, 2, 19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Lee K, Fraser K, Ghaddar B, Yang K, Kim E, Balaj L, Chiocca EA, Breakefield XO, Lee H, Weissleder R. ACS Nano 2018, 12, 494; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fraser K, Jo A, Giedt J, Vinegoni C, Yang KS, Peruzzi P, Chiocca EA, Breakefield XO, Lee H, Weissleder R. Neuro. Oncol 2019, 21, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, Bracke M, De Wever O, Hendrix A. J. Extracell. Vesicles 2014, 3, 24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Osteikoetxea X, Sódar B, Németh A, Szabó-Taylor K, Pálóczi K, Vukman KV, Tamási V, Balogh A, Kittel Á, Pállinger É, Buzás EI. Org. Biomol. Chem 2015, 13, 9775. [DOI] [PubMed] [Google Scholar]
- [23].EV-TRACK Consortium, Nat. Methods 2017, 14, 228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.