

Abstract
Purpose:
The goal of this study was to assess the scale of low-level parental mosaicism in exome sequencing (ES) databases.
Methods:
We analyzed approximately 2000 family trio ES datasets from the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) and Baylor Genetics (BG). Among apparent de novo single nucleotide variants (SNVs) identified in the affected probands, we selected rare unique variants with variant allele fraction (VAF) between 30-70% in the probands and lower than 10% in one of the parents.
Results:
Out of 102 candidate mosaic variants validated using amplicon-based NGS, droplet digital PCR, or blocker displacement amplification, 27 (26.4%) were confirmed to be low- (VAF between 1-10%) or very low- (VAF <1%) level mosaic. Detection precision in parental samples with two or more alternate reads was 63.6% (BHCMG) and 43.6% (BG). In nine investigated individuals, we observed variability of mosaic ratios among blood, saliva, fibroblast, buccal, hair, and urine samples.
Conclusion:
Our computational pipeline enables robust discrimination between true and false positive candidate mosaic variants and efficient detection of low-level mosaicism in ES samples. We confirm that the presence of two or more alternate reads in the parental sample is a reliable predictor of low-level parental somatic mosaicism.
Keywords: exome sequencing, parental somatic mosaicism, rare variants, Mendelian genomics
INTRODUCTION
A growing body of evidence implicates the importance of somatic mosaicism in the etiology of many human genetic disorders, including both cancer and Mendelian conditions1–8. If a pathogenic single nucleotide variant (SNV) or copy-number variant (CNV) occurs during any of the ~ 1016 mitotic post-zygotic cell divisions, the resulting different cell populations can manifest clinically9. If present in the parental germline cells, the variant can be transmitted to the offspring10–14.
Exome sequencing (ES) has been used extensively in both clinical settings and research studies; however, to date, only a few reports have described more in-depth analyses of somatic mosaicism. Recently, Wright et al. analyzed the trio ES data of 4,293 probands mainly with developmental disorders and identified ~3% causative variants exhibiting post-zygotic mosaicism15. We have analyzed a cohort of ~12,000 samples submitted for clinical ES and identified clinically relevant somatic mosaic variants in ~ 1.5% of probands16.
In 2014, we described low-level (<10%) parental somatic mosaicism for CNV deletions detected in four out of 100 unrelated families17, and more recently, we presented accurate methods for detection and validation of mosaic CNVs18,19. Corroboratively, SNV studies in multi-sibling families using genome sequencing revealed that in parental germline, 3.8% of SNVs were mosaic, resulting in 1.3% of variants being shared by siblings20,21. Notably, the level of somatic mosaicism in the parental blood samples has been shown to positively correlate with the overall recurrence risk20–22. In ES data, parental mosaicism was detected in 0.3-0.5% of the analyzed family trios15,16. Most recently, Breuss et al. reported that autism risk in offspring could be assessed through quantification of male sperm mosaicism, further indicating the correlation between the level of mosaicism and disease recurrence risk23.
Here, we have studied ES data of almost two thousand unrelated trios from Baylor Hopkins Center for Mendelian Genomics (BHCMG) at Baylor College of Medicine (BCM) cohort and trios from Baylor Genetics (BG) Laboratories at BCM, respectively. We describe a new approach to identify low- (<10%) and very low- (<1.0%) level somatic mosaicism in the parents and provide a classification tool enabling more accurate assessment of the level of somatic mosaicism in ES samples.
MATERIALS AND METHODS
Ethics Statement
The research studies at BHCMG were approved by the Institutional Review Board (IRB) for Human Subject Research at BCM under the protocols H-29697. All analyzed samples were de-identified. All studied BG samples were de-identified using the IRB waiver protocols H-41191 and H-42680. To study different somatic tissues, written informed consent was obtained from nine participants or their legal guardians. The research was IRB approved at BCM under the protocol H-28088.
Baylor-Hopkins Center for Mendelian Genomics dataset
ES was performed previously on a research basis in 7,790 individuals enrolled in BHCMG at BCM to accelerate the discovery of a variant allele and contributory genetic locus underlying a wide-range of Mendelian conditions (http://bhcmg.org/, accessed in June 2019). To study low-level parental somatic mosaicism, we have selected ES data with the complete BAM (reads were mapped to GRCh37.p13) and VCF files from 823 family trios included in the BHCMG cohort. DNA samples were processed according to the protocols previously described24. In addition, all variants identified by the Mercury pipeline (v3.2)25 were also annotated using Variant Effect Predictor (VEP, v96)26 that incorporates GENECODE release 19 for gene annotations. Average read depth across analyzed samples was ~90x with > 95% having 20x base coverage.
Selection criteria for the search of candidate mosaic variants and quality control
To identify low-level parental somatic mosaic variants, we have performed a two-step filtering (Figure 1). First, we have analyzed the VCF files to select variants for which probands were found to be heterozygous. Thus, we calculated the variant allele fraction (VAF, defined as a proportion of the number of alternate allele reads relative to the total number of reads at the variant position) for each particular variant. In our recent study, we showed that more than 95% of apparent de novo autosomal SNVs and X-linked SNVs in females have VAF range between 36% and 64% by NGS analysis16. Here, to eliminate genotype calls erroneously classified as heterozygous, we have used more strict criteria and removed variants with the VAF below 30% or above 70%. In addition, we have required that variants with VAF between 30-70% in the probands were not simultaneously reported by Atlas2 variant caller (v1.4.3)27 in the parental samples, or if detected in the parents, have VAF below 10%. Second, variants with the total depth of coverage below 20x in any samples from the given trios were excluded from further analyses. Subsequently, for each selected SNV, we have retrieved pileup information from the proband and parental BAM files that enabled obtaining more precise data on read depth and VAF in these samples. To further narrow the list of candidate mosaic events, we have required that all variants have a minor allele frequency (MAF) < 0.01% in GnomAD (v2.1) (unpublished data) and < 0.015% in the BHCMG dataset, and are not located within the repetitive sequences or segmental duplication regions as identified by the genomic superDups track28 as well as pseudogenes (except one unique DNA region within segmental duplication for which we were able to design PCR primers) from the University of California, Santa Cruz browser (https://genome.ucsc.edu/). To remove likely false positive (FP) events (i.e. technical artifacts), we have excluded variants that occur in the top 5% trios with the highest number of mosaic candidates.
Figure 1.
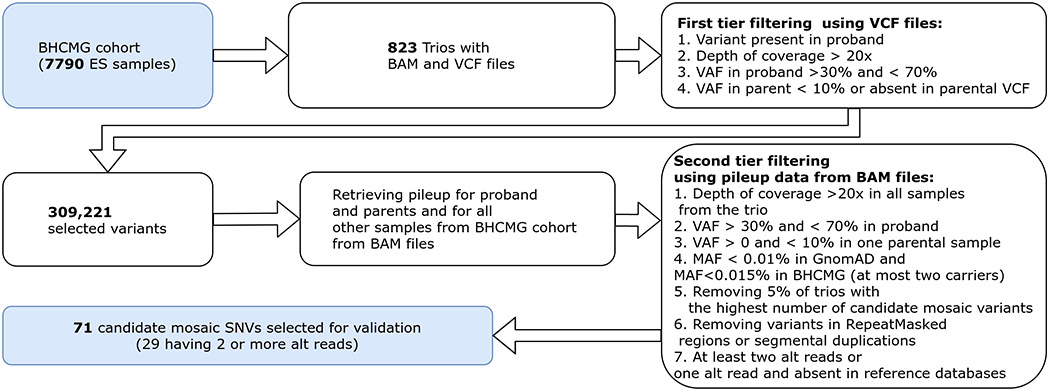
Candidate mosaic variant selection in BHCMG cohort. VCF files from 823 trios from the BHCMG cohort were used to identify variants that are likely heterozygous in probands and have zero or low coverage in one of the parental samples. In the second step, for each selected variant, pileup data from corresponding BAM files was retrieved. This information, along with external annotations (e.g. GnomAD AF), was used to further narrow the list of mosaic candidates.
Baylor Genetics Laboratories dataset
We analyzed family trio ES data from approximately 15,000 patients enrolled in clinical diagnostic studies. Average depth-of-coverage was ~100x with >70% of reads aligned to target, >95% target base covered at >20x, >85% target base covered at >40x. Since ES data in BG have been pre-processed using a different analytical pipeline than BHCMG cohort, we modified the mosaic SNV candidate selection accordingly. We have used three different data subsets, as presented in Supplementary Figure 1. The first subset of parental mosaic variants was derived from the analysis of 3165 apparent de novo heterozygous SNVs in the probands selected previously in the process of clinical analysis. Second subset consists of approximately 1000 trios for which joint VCF files were generated on the Illumina DRAGEN 2 platform. We focused on unique rare variants that occurred in only one family. We also removed any variants that overlapped segmental duplications. Similar to the approach used for the BHCMG cohort, we required a depth of at least 20 reads in each parent, an evidence of heterozygous state in the proband with a VAF of 0.3-0.7 and 0<VAF<0.1 in one parental sample (homozygous reference state in the other parental sample). In the next step, only clinically relevant variants with a read depth ≥50x have been selected, followed by manual analyses of the pileup data of parental samples. Additional 19 samples (third subset) were included after being flagged by the BG directors as suspected somatic mosaic cases during manual analyses of the pileup data.
Exome Sequencing QC
As a quality control measure, each DNA sample undergoing ES in either BHCMG or BG cohorts is analyzed in parallel by a cSNP array (Illumina Human Exome-12v1 array) to ensure correct sample identification and to assess sequencing quality. This approach warrants greater than 99% concordance between both methods29. Concordance ratios above 5% is interpreted as a sample contamination. The sequencing data are further investigated and re-sequenced if needed.
DNA extraction
Initial ES in the BHCMG and BG cohorts was performed on the blood samples in greater than 95% of cases. In the remainder of cases, it was saliva. For validation experiments, peripheral blood DNA was extracted using the Gentra Puregene Blood kit (Qiagen, Germantown, MD, USA). For the selected cases from the BG cohort, at least five hairs with follicles were collected, and DNA was extracted using the QIAamp DNA Investigator Kit (Qiagen). Saliva was collected using the ORAgene Discover OGR-500 kit (DNA Genotek, Ottawa, Canada). Buccal cells were collected using the ORAcollect OC-175 kit (DNA Genotek). Both saliva and buccal cell DNA were extracted using the prepIT-L2P (DNA Genotek). DNA from urine was extracted using the Quick-DNA Urine Kit (Zymo Research, Irvine, CA, USA). All procedures followed the manufacturer’s instructions.
Validation of candidate mosaic variants using molecular methods
To validate a putative parental somatic mosaicism of the selected variants, we have used three different molecular techniques: amplicon-based next generation sequencing (NGS), droplet digital PCR (ddPCR), or blocker displacement amplification (BDA).
Amplicon-based NGS
PCR primers targeting the putative mosaic variants were designed using BatchPrimer3 v1.0 and Primer3 v. 0.4.0 tools. The tested parental samples were amplified by PCR using recombinant Taq DNA Polymerase (ThermoFisher Scientific, Waltham, MA, USA). Each 150 μl reaction contains 1x Taq Buffer with (NH4)2SO4, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.5 μM forward and reverse primer, 3.75 U of Taq polymerase, and 200 ng of DNA. The PCR products were purified by QIAquick PCR Purification Kit (Qiagen) according to the manufacturer’s instructions. Concentration of the purified PCR amplicons was quantified by Qubit dsDNA BR Assay (ThermoFisher Scientific) using the Qubit 4 Fluorometer (ThermoFisher Scientific). The purified amplicons of 300-338 bp were sequenced using the HiSeq 2500 platform (Illumina, San Diego, CA, USA) with 300 bp paired-end (PE) reads at BGI (San Jose, CA, USA) or using the HiSeq X system (Illumina) with PE150 reads at CloudHealth Genomics (Shanghai, China). Integrative Genomics Viewer (IGV, v2.3) software30 was used to analyze the data, as well as in-house developed scripts implemented in the R programming language.
Droplet digital PCR
DNA oligo primers as well as mutation and wild type specific FAM or HEX labeled-probes targeting the potential mosaic variants were designed and purchased from IDT (Coralville, IA, USA). In each 20 μl reaction, 10 μl of ddPCR Supermix for Probes (No dUTP) (Bio-Rad, Hercules, CA, USA), 0.5 μM forward and reverse primer, 4 units of HindIII-HF restriction enzyme (New England Biolabs, Ipswich, MA, USA), and 100 ng of DNA were added. For each family, the proband’s DNA sample was utilized as a positive control and an unrelated wild type DNA from blood sample was used as a negative control. A non-template control was used to confirm no DNA contamination was present in the starting reagents and workflow. The ddPCR reactions were carried out using QX200 AutoDG Droplet Digital PCR System (Bio-Rad) and analyzed with QuantaSoft Analysis Pro software v1.7.4 (Bio-Rad) (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/QuantaSoft-Analysis-Pro-v1.0-Manual.pdf) according to the manufacturer’s protocols. Each parental sample was run in at least three triplicates.
Blocker Displacement Amplification
To determine the VAF in parental DNA, 12 samples were tested using BDA with the probands’ DNA samples as positive controls. BDA principles were previously described in detail by Wu et al.31. qPCR assays were performed with the use of PowerUp SYBR Green Master Mix (ThermoFisher Scientific) with 400 nM of each primer, 4 μM of blocker, and 10 ng of DNA per well. The amplification of GC-rich fragments was carried out with the addition of betaine (Sigma Aldrich, St. Louis, MO, USA) at a final concentration of 1 M. Reactions in the total volume of 10 μl were performed using CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Each reaction was repeated at least twice. The qPCR products from two experiments were purified, Sanger sequenced, and analyzed using the ApE software (v2.0) (https://jorgensen.biology.utah.edu/wayned/ape/ https://openwetware.org/wiki/ApE_-_A_Plasmid_Editor_(software_review) ) 31.
Code availability
The source code of our filtering pipeline is publically available at https://github.com/tgambin/LowLevelMosaicVariantCaller.
RESULTS
BHCMG cohort
Computational analyses
We have obtained 309,221 genotype calls fulfilling the initial inclusion criteria. After removal of the low-quality sequencing samples and variants with MAF >0.01%, we have found 3,156 apparent de novo variants in 768 probands. In the parental samples, 71 candidate SNVs, previously undetected by routine ES algorithms, met all filtering criteria (Figure 1). Their VAFs ranged from 0.17% to 9.0%, with an average of 2.8%. Forty-two mosaic candidates absent in GnomAD database had one alternate read supporting the variant allele, whereas the remaining 29 variants had two or more alternate reads. Among the 71 putative mosaic SNVs, 37 are exonic, including missense (n=23), synonymous (n=13), and nonsense (n=1) variants. In addition, we have also selected variants mapping to the non-coding regions (n=33) or at the splice site (n=1).
Molecular verification of the candidate variants
Out of the 71 mosaic candidates predicted using our computational approach, we have evaluated 48 (68%) variants in the available DNA samples using at least one molecular method, amplicon-based NGS (n=48), BDA (n=12), or ddPCR (n=18) (Supplementary Table 1). We have verified positive somatic mosaicism in 16 (33%) samples (Table 1, Figure 2). The precision [TP/(TP+FP), where TP is the number of true positives and FP is the number of false positives] in the group of variants with two or more alternate reads at the variant position was 63.6% (14 out of 22). Furthermore, when VAF was greater than 5% in the ES data, the prediction of somatic mosaicism was more reliable in that seven out of eight (87.5%) SNVs were confirmed as mosaic events (Supplementary Figure 2). The precision among candidates having a single read supporting the variant allele was 7.7% (two out of 26). To delineate additional predictors of true mosaicism in the group of candidate variants with a single alternate read, for each genomic position of a putative mosaic SNV, we have retrieved the pileup information from the remaining 7,788 ES samples. For each variant, we have calculated the FracSupp value, defined as the fraction of samples having at least one alternate read at the position of the given candidate mosaic event. We have hypothesized that the presence of reads supporting an alternate allele at a given genomic position in the multiple samples from the BHCMG cohort may represent technical artifacts or recurrent sequencing errors rather than the true mosaic variants. Interestingly, we have found that in the group of variants with a single alternate read, the two candidates confirmed as TP mosaic events had significantly lower FracSupp value (Wilcoxon rank sum test, p = 0.046) than the remaining 24 FP events (Supplementary Figure 3). In two subjects, VAFs measured by different methods (including ES) varied significantly between 6.4-19.4% in BAB5936 and between 1.2%-20.5% in WPW160 (Table 1, Figure 2).
Table 1:
Parental low-level mosaicism rates in BHCMG cohort measured using ES, amplicon-based NGS, ddPCR, and BDA.
| Case | Variant (hg19) | ES (Variant/Total reads) | Amplicon-based NGS (Variant/Total reads) | ddPCR | BDA |
|---|---|---|---|---|---|
| 144-25-03 | chr9:g.84528435C>A | 17/338 (5.0%) | 1117/21427 (5.2%) | 4.2% | NT |
| 144-57-03 | chr9:g.404956C>T | 6/103 (5.8%) | 991/19330 (5.1%) | NT | NT |
| BAB3771 | chr2:g.44556121C>T | 6/82 (7.3%) | 245/2715 (9.0%) | 5.6% | 5.2% |
| BAB5936 | chr4:g.22421644A>G | 5/78 (6.4%) | 643/5805 (11.1%) | TF | 19.4% |
| BAB9818 | chr3:g.150661610G>C | 5/143 (3.5%) | 39/692 (5.6%) | NT | NT |
| BAB9852 | chr19:g.50920420C>T | 4/87 (4.6%) | 169/3548 (4.8%) | TF | NT |
| BAB8129 | chr2:g.180835608C>T | 4/57 (7.0%) | 865/16536 (5.2%) | TF | 1.3% |
| BAB8833 | chr2:g.232576565G>C | 1/29 (3.4%) | 260/3281 (7.9%) | TF | NT |
| Fam9-3 | chr3:g.101395501G>A | 11/121 (9.1%) | 214/3072 (7.0%) | 6.6% | NT |
| LP89-036f | chr15:g.65771401A>G | 2/44 (4.5%) | 529/38139 (1.4%) | NT | NT |
| OAVS-PT1F | chr6:g.84632035G>A | 1/49 (2.0%) | 4296/62505 (6.9%) | NT | NT |
| UT0133 | chr4:g.115544174C>T | 2/107 (1.9%) | 25/5001 (0.5%) | 0.3% | 0.3% |
| WPW070 | chr2:g.170129381T>G | 3/81 (3.7%) | 30/1660 (1.8%) | 1.5% | 1.7% |
| WPW160 | chr2:g.89161156A>G | 2/27 (7.4%) | 20/1721 (1.2%) | 15.6% | 20.5% |
| WPW405 | chr16:g.15732966A>G | 8/89 (9.0%) | 286/2508 (11.4%) | TF | NA |
| WPW421 | chr3:g.119367355G>C | 4/68 (5.9%) | 220/2701 (8.1%) | 7.7% | 10% |
NT – not tested, TF – technical failure
Figure 2.
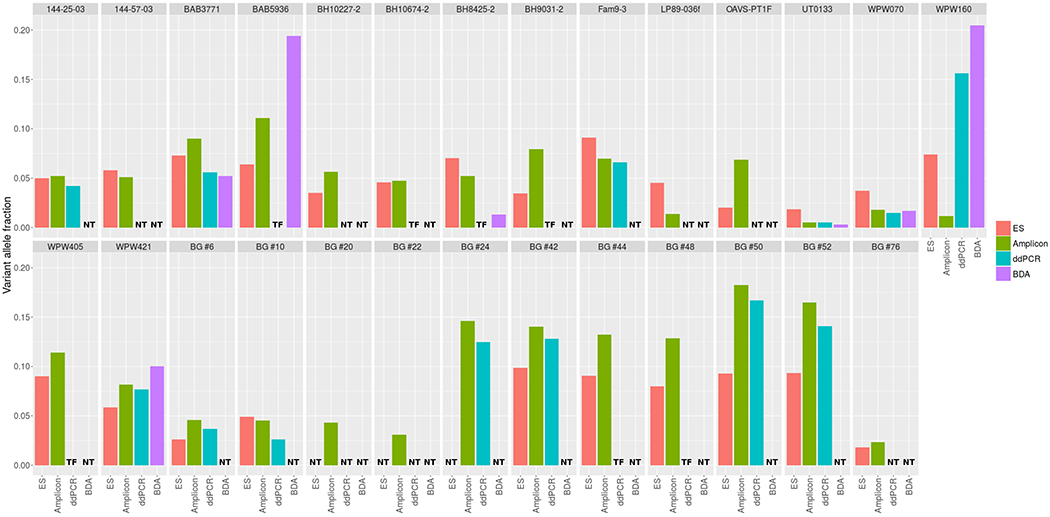
Variant allele fraction (VAF) estimated using four different molecular methods: ES, amplicon-based NGS, BDA, and ddPCR. If there are no results for a particular validation method we indicated that it was either not tested (NT) or validation did not succeed due to technical failure (TF). In most of cases, estimated VAFs were consistent among different experimental methods.
Impact of potential cross-sample contamination
A potential cross-sample contamination is another limiting factor in the detection of mosaicism in ES data that can lead to an increased number of false positives. All ES data used in this study passed quality control (see Materials and Methods); however, to confirm the lack of significant cross-sample contamination and to measure the actual level of contamination more accurately, we have processed the BHCMG samples that underwent orthogonal validation for mosaicism using the GATK CalculateContamination software. We found that on average, each sample yielded contamination of 1%, ranging between 0% and 5% (Supplementary Figure 4) with no significant difference between the cohorts of samples that passed or failed validation. We did not observe any significant contamination (i.e. larger than 5%); however, in 15 samples, we found contamination levels higher than 1% (which was used as expected background noise cutoff in previous work32).
BG cohort
We have analyzed the apparent de novo SNVs detected in the probands. In the parental blood samples, we have selected 46 potentially mosaic exonic SNVs, including missense (n=33), nonsense (n=4), frameshift (n=7), synonymous (n=1), and UTR (n=1) variants. In addition, we have selected eight intronic variants, including six splice site variants. We have examined these variants for somatic mosaicism using amplicon-based NGS (n=54) or ddPCR (n=6). In the 45 samples having pileup data (from 58 labeled as DS1 or DS2 in Supplementary Figure 1), the precision was 17.7% (eight of 45). In the subgroup of variants with two or more alternate reads at the variant position, the precision was 43.6% (seven out of 16), whereas among candidates having a single read supporting the variant allele it was only 3.4% (1 out of 29). In nine studied samples that were flagged by BG directors (DS3) as potential mosaic, three (33.3%) were confirmed as mosaic (Table 2).
Table 2:
Parental low-level mosaicism rates in BG cohort measured using ES and amplicon-based NGS.
| Case | Variant (hg19) | ES (Variant/Total reads) | Amplicon-based NGS (Variant/Total reads) | ddPCR |
|---|---|---|---|---|
| BG #6 | chr2:g.27672430C>T | 3/116 (2.6%) | 459/10034 (4.6%) | 3.7% |
| BG #10 | chr11:g.64402826C>T | 3/61 (4.9%) | 127/2817 (4.5%) | 2.6% |
| BG #20 | chr7:g.129019551C>T | NT | 181/4207 (4.3%) | NT |
| BG #22 | chr9:g.86258554T>G | NT | 101/3284 (3.1%) | NT |
| BG #24 | chrX:g.24007143T>A | NT | 1411/9645 (14.6%) | 12.5% |
| BG #42 | chr19:g.3753762C>T | 8/81 (9.9%) | 681/4858 (14.0%) | 12.8% |
| BG #44 | chr6:g.41554624G>A | 6/66 (9.1%) | 305/2304 (13.2%) | TF |
| BG #48 | chr3:g.182763355T>G | 10/125 (8.0%) | 829/6444 (12.9%) | TF |
| BG #50 | chr14:g.53331537G>A | 13/140 (9.3%) | 1763/9676 (18.2%) | 16.7% |
| BG #52 | chr6:g.28244760A>G | 13/139 (9.4%) | 374/2268 (16.5%) | 14.1% |
| BG #76 | chr14:g.68029158 G>A | 1/55 (1.8%) | 44/1900 (2.3%) | NT |
NT – not tested; TF – technical failure
Distribution of VAFs among different somatic tissues
We had previously detected mosaicism level (calculated as VAF) greater than 10% in the whole blood samples from three parents, M1.1, M3.1, and M8.216. To study somatic mosaicism in other tissues in these individuals, we have assessed their levels using amplicon-based NGS. For parent M1.1, in four tested tissues, the levels of mosaicism were estimated as 27.3%, 23.7%, 29.5%, and 40.2% in whole blood, buccal, fibroblast, and hair samples, respectively. For parent M3.1, 3.3% mosaicism was detected in the buccal sample, 16.7% in the saliva sample, and 17.6% in the blood, whereas no evidence of this variant was found in the hair sample. For parent M8.2, we have identified similar levels of mosaicism in the blood (13.2%), buccal (14.2%), saliva (17.7%), and urine (15.8%) samples, with the exception of low level-mosaicism in the hair (2.5%) (Figure 3). To expand the tissue distribution study, we have also included previously published six probands with somatic mosaicism greater than 10% in their blood samples16. The most outlying VAFs were observed in the hair tissue, where the level of mosaicism was significantly higher in the hair than in the blood in three cases, and significantly lower in five cases. We have also found that in six out of nine cases, VAFs observed in at least one non-blood tissue were higher than VAFs estimated for blood samples (either by ES or amplicon-based NGS) (Figure 3).
Figure 3.

Distribution of VAFs among six different tissues: blood, saliva, buccal, skin fibroblast, hair, and urine. Analyses were performed for nine individuals, including three unaffected parents and six affected probands. In the case of blood tissue, VAF was estimated based on both ES (labeled as “Blood_ES”) and amplicon-based NGS data (labeled as “Blood”). In six out of eight cases, there was at least one tissue for which VAF was estimated to be higher than VAF in blood.
DISCUSSION
While recent advances in NGS techniques enable the detection of mosaic variants more precisely than Sanger sequencing, the identification of low- and very low-level somatic mosaicism in ES data remains challenging. Variants with VAFs lower than 10% are typically not detected using standard ES variant calling pipelines. To overcome these limitations, we have developed a more sensitive computational screening tool and have verified its robustness in the family trio ES dataset using three independent experimental molecular methods.
The performance of NGS methods depends primarily on a read depth at that given base pair. Theoretically, these methods could detect mosaic variants with a single alternate read (VAF = 1/N, where N is the total read coverage at the variant position). However, based on the experimental data, it has been shown that it is possible to detect mosaic fraction only if it is greater than the sequencing error rate generated at various steps of NGS, including library preparation, PCR amplification, and sequencing15,33. The error rate of routine ES ranges between ~0.1% and 1.0% and cannot be significantly reduced even using the ultra-deep sequencing in amplicon-based NGS33,34. Recent studies have shown that joint analyses of library-level replicates can reduce the false positive signals and facilitate a robust identification of mosaic variants with higher sensitivity and specificity35.
To remove variants that were erroneously called as heterozygous in the probands, we have used conservative filtering criteria (based on the fixed VAF thresholds, i.e. 30%<VAF<70%). In case of detection of parental mosaicism, the additional rationale of using this filter is that highly skewed VAF observed in the proband may indicate the existence of technical biases in a given locus, which increases the chance that a candidate mosaic event in the parental sample is not real. Although this approach helped us to reduce the number of false positives, it may also result in under-detection of variants in regions with DP< 50x, in which the VAF of true heterozygous events may fall outside the 30-70% range. Therefore, in other applications, such as de novo variant calling, one should consider using less stringent filters for the heterozygous state, e.g. p-value based on the binomial distribution of VAF that is dependent on DP and allows higher variability of VAF in poorly covered regions.
It is challenging to distinguish whether the reported value by GATK CalculateContamination, that was greater than 1% in 15 samples, was caused by the real cross-sample contamination or is due to the increased number of technical artifacts. The reason for this is that the background noise level depends on multiple factors such as DNA polymerase, sequencing and alignment errors, index hopping, or incomplete trimming of the adapters36, and it may vary between sequencing experiments. Interestingly, other investigators32 who detected signs of contamination in a significant fraction of their analyzed cohort were able to identify a source of contamination only in 17% percent of samples with the reported contamination > 1%. The abovementioned issues further underline the importance of using orthogonal molecular validation methods to confirm low-level somatic mosaicism in parental samples, and to remove most of the potential technical and biological biases.
Using our computational pipeline in the ES dataset, we were able to identify and orthogonally validate 27 somatic mosaic variants with low- and very low-level somatic mosaic VAFs in the parents from two cohorts. Our approach enabled detection of mosaic variants with VAF >5% with high precision (>85%), whereas identification of variants with lower VAFs turned out to be more challenging, with a precision of ~ 28%. Our data confirms that the presence of a single alternate read in ES dataset is usually an insufficient predictor of somatic mosaicism and more likely denotes a false positive event15. Our results also indicate that the improvement of precision in the group of candidates with a single alternate read is possible by using additional predictors for filtering, such as the FracSupp value (i.e. the fraction of samples from the BHCMG cohort having at least one alternate read at the position analyzed) (Supplementary Figure 3).
The real frequency of mosaicism can be biased by technical limitations. For example, too high or too low GC content, predicted probe dimerization, or the presence of runs of consecutive nucleotides at the SNV site can substantially affect nucleotide discrimination, precluding testing of some variants using ddPCR. Insufficient amount of DNA was the main limiting factor for variant validation detection using BDA, and ddPCR (Supplementary Table 1). Thus, studies using larger dataset are needed to confirm the utility of our approach.
As somatic mosaic variants may occur at different developmental stages, their distribution may vary substantially among different somatic tissues. However, larger-scale studies of the distribution of mosaicism in different tissues representing the three primary germ layers have not been performed systematically. Growing evidence implicates that whole blood, which is typically tested in the clinical diagnostics setting, may not be the optimal tissue to search for somatic mosaicism37. A pool of whole blood cells may grow at a relatively faster rate and lead to clonal expansion, especially in older subjects38. Therefore, mosaic variations in the blood are more likely to be under- or overrepresented, particularly if the variant influences cell survival or growth. We and others have observed that VAFs in non-blood tissues were usually higher than those in blood samples, suggesting that tissues other than blood (e.g. those exhibiting different VAFs), may serve as more optimal tissue to test somatic mosaicism. Our correlation analyses showed that VAFs identified in hair follicles are the least correlated with VAFs assessed in other somatic tissues (Supplementary Figure 5). However, given that in some cases not all six types of parental or proband tissue were available for screening, the real inter-tissue distribution of mosaic variants may be unrecognized. Further studies in larger cohorts are needed to estimate the mosaic ratios across different tissues.
In most cases, the levels of parental somatic mosaicism measured using three orthogonal molecular experimental methods were comparable, whereas only in a few samples the levels varied significantly. The highest consistency of mosaic fraction was observed between BDA and ddPCR results, confirming our previous observations that these methods can be alternatively used for the accurate quantitation of low-level mosaicism. BDA and ddPCR are both more sensitive than NGS-based approaches. BDA was proven to reliably detect variants with VAF as low as 0.1%31,39. We were able to validate very low-level somatic mosaicism in sample UT0133 with VAF assessed as 0.3% using BDA, 0.3% using ddPCR, and 0.5% using amplicon-based NGS. In the BG samples where the VAFs calculated based on the PCR amplicon NGS data were less than 1.0%, we have elected not to interpret them as real events as they were not verified by any other orthogonal molecular method (Supplemental Table 1).
In conclusion, we describe a customized computational pipeline that enables robust and accurate identification of low- and very low-level parental somatic mosaic variants in ES data that are not detected using standard NGS data processing methods. We show that the number of alternate reads in the parental sample positively correlates with the likelihood of confirming the parental mosaicism in the validation studies. Knowing that a suspected de novo variant may actually be present in a mosaic state in one of the parents is critical in providing an accurate chance of recurrence risk.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to our colleagues who provided their expertise that greatly assisted this research work. We thank Dr. Davut Pehlivan for helpful discussion. This study is supported by the US National Institute of Health (NIH): Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) grant R01HD087292 to Dr. Stankiewicz, National Human Genome Research Institute (NHGRI)/National Heart Lung and Blood Institute (NHLBI) grant UM1HG006542 to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG), and NHGRI grant HG008986 to Dr. Posey.
Footnotes
DISCLOSURE
The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from clinical exome sequencing offered by the Baylor Genetics Laboratories. Authors who are faculty members in the Department of Molecular and Human Genetics at Baylor College of Medicine are identified as such in the affiliation section. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. DYZ and LRW have a patent pending on Blocker Displacement Amplification. DYZ, NGX, and LRW are consultants of NuProbe Global. DYZ consults for Avenge Bio. DYZ owns equity of NuProbe Global and Torus Biosystems. The remaining authors declare that they have no competing interests.
REFERENCES
- 1.Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14(5):307–320. doi: 10.1038/nrg3424 [DOI] [PubMed] [Google Scholar]
- 2.Boone PM, Bacino CA, Shaw CA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31(12):1326–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartnik M, Derwińska K, Gos M, et al. Early-onset seizures due to mosaic exonic deletions of CDKL5 in a male and two females. Genet Med Off J Am Coll Med Genet. 2011;13(5):447–452. doi: 10.1097/GIM.0b013e31820605f5 [DOI] [PubMed] [Google Scholar]
- 4.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341(6141):1237758. doi: 10.1126/science.1237758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ansari M, Poke G, Ferry Q, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51(10):659–668. doi: 10.1136/jmedgenet-2014-102573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stosser MB, Lindy AS, Butler E, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med Off J Am Coll Med Genet. 2018;20(4):403–410. doi: 10.1038/gim.2017.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krupp DR, Barnard RA, Duffourd Y, et al. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am J Hum Genet. 2017;101(3):369–390. doi: 10.1016/j.ajhg.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim ET, Uddin M, De Rubeis S, et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci. 2017;20(9):1217–1224. doi: 10.1038/nn.4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science. 2013;341(6144):358–359. doi: 10.1126/science.1239503 [DOI] [PubMed] [Google Scholar]
- 10.Acuna-Hidalgo R, Bo T, Kwint MP, et al. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am J Hum Genet. 2015;97(1):67–74. doi: 10.1016/j.ajhg.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genet Med Off J Am Coll Med Genet. 2016;18(7):746–749. doi: 10.1038/gim.2015.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet TIG. 2015;31(7):382–392. doi: 10.1016/j.tig.2015.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldmann JM, Veltman JA, Gilissen C. De Novo Mutations Reflect Development and Aging of the Human Germline. Trends Genet TIG. 2019;35(11):828–839. doi: 10.1016/j.tig.2019.08.005 [DOI] [PubMed] [Google Scholar]
- 14.Møller RS, Liebmann N, Larsen LHG, et al. Parental mosaicism in epilepsies due to alleged de novo variants. Epilepsia. 2019;60(6):e63–e66. doi: 10.1111/epi.15187 [DOI] [PubMed] [Google Scholar]
- 15.Wright CF, Prigmore E, Rajan D, et al. Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat Commun. 2019;10(1):2985. doi: 10.1038/s41467-019-11059-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Y, Tokita MJ, Chen ES, et al. A clinical survey of mosaic single nucleotide variants in disease-causing genes detected by exome sequencing. Genome Med. 2019;11. doi: 10.1186/s13073-019-0658-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95(2):173–182. doi: 10.1016/j.ajhg.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Karolak JA, Grochowski CM, et al. Parental somatic mosaicism for CNV deletions - A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics. Published online May 6, 2020. doi: 10.1016/j.ygeno.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Grochowski CM, Bi W, Lupski JR, Stankiewicz P. Quantitative Assessment of Parental Somatic Mosaicism for Copy-Number Variant (CNV) Deletions. Curr Protoc Hum Genet. 2020;106(1):e99. doi: 10.1002/cphg.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahbari R, Wuster A, Lindsay SJ, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48(2):126–133. doi: 10.1038/ng.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jónsson H, Sulem P, Arnadottir GA, et al. Multiple transmissions of de novo mutations in families. Nat Genet. 2018;50(12):1674–1680. doi: 10.1038/s41588-018-0259-9 [DOI] [PubMed] [Google Scholar]
- 22.Campbell IM, Stewart JR, James RA, et al. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet. 2014;95(4):345–359. doi: 10.1016/j.ajhg.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breuss MW, Antaki D, George RD, et al. Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat Med. 2020;26(1):143–150. doi: 10.1038/s41591-019-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambin T, Jhangiani SN, Below JE, et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. 2015;7(54). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 2014;15(1):30. doi: 10.1186/1471-2105-15-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13(1):8. doi: 10.1186/1471-2105-13-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey JA, Gu Z, Clark RA, et al. Recent Segmental Duplications in the Human Genome. Science. 2002;297(5583):1003–1007. doi: 10.1126/science.1072047 [DOI] [PubMed] [Google Scholar]
- 29.Zastrow DB, Zornio PA, Dries A, et al. Exome sequencing identifies de novo pathogenic variants in FBN1 and TRPS1 in a patient with a complex connective tissue phenotype. Cold Spring Harb Mol Case Stud. 2017;3(1):a001388. doi: 10.1101/mcs.a001388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu LR, Chen SX, Wu Y, Patel AA, Zhang DY. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat Biomed Eng. 2017;1:714–723. doi: 10.1038/s41551-017-0126-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiévet A, Bernard V, Tenreiro H, et al. ART-DeCo: easy tool for detection and characterization of cross-contamination of DNA samples in diagnostic next-generation sequencing analysis. Eur J Hum Genet EJHG. 2019;27(5):792–800. doi: 10.1038/s41431-018-0317-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma X, Shao Y, Tian L, et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019;20(1):50. doi: 10.1186/s13059-019-1659-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19(5):269–285. doi: 10.1038/nrg.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kim D, Lim JS, et al. The use of technical replication for detection of low-level somatic mutations in next-generation sequencing. Nat Commun 2019;10(1):1–11. doi: 10.1038/s41467-019-09026-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costello M, Fleharty M, Abreu J, et al. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics. 2018;19(1):332. doi: 10.1186/s12864-018-4703-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang X, Liu A, Xu X, et al. Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci Rep 2017;7. doi: 10.1038/s41598-017-15814-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shlush LI. Age-related clonal hematopoiesis. Blood. 2018;131(5):496–504. doi: 10.1182/blood-2017-07-746453 [DOI] [PubMed] [Google Scholar]
- 39.Karolak JA, Liu Q, Xie NG, et al. Highly Sensitive Blocker Displacement Amplification and Droplet Digital PCR Reveal Low-Level Parental FOXF1 Somatic Mosaicism in Families with Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins. J Mol Diagn JMD. 2020;22(4):447–456. doi: 10.1016/j.jmoldx.2019.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.