

Summary
Neural stem cells (NSCs) in the dentate gyrus (DG) reside in a specialized local niche that supports their neurogenic proliferation to produce adult-born neurons throughout life. How local niche cells interact at the circuit level to ensure continuous neurogenesis from NSCs remains unknown. Here we report the role of endogenous neuropeptide cholecystokinin (CCK), released from dentate CCK interneurons, in regulating neurogenic niche cells and NSCs. Specifically, stimulating CCK release supports neurogenic proliferation of NSCs through a dominant astrocyte-mediated glutamatergic signaling cascade. In contrast, reducing dentate CCK induces reactive astrocytes, which correlates with decreased neurogenic proliferation of NSCs and upregulation of genes involved in immune processes. Our findings provide novel circuit-based information on how CCK acts on local astrocytes to regulate the key behavior of adult NSCs.
Graphical Abstract

eTOC
Asrican et al. demonstrate that stimulating endogenous CCK neuropeptide release in adult dentate gyrus supports neurogenic proliferation of rNSCs through a dominant astrocyte-mediated glutamatergic signaling cascade. In contrast, reducing dentate CCK induced reactive astrocytes, decreased neurogenic proliferation of rNSCs, and upregulated genes involved in neuroinflammation.
Introduction
Radial-glia like neural stem cells (rNSCs) reside in a specialized niche within the adult dentate gyrus (DG) that consists of highly diverse cells, including excitatory granule cells (GCs) and mossy cells (MCs), distinct inhibitory interneurons, and astrocytes (Bao and Song, 2018; Song et al., 2016). Diverse signaling from these niche cells act on rNSCs to regulate hippocampal neurogenesis. Our previous studies have identified local parvalbumin (PV) interneurons as a critical niche component regulating rNSCs through tonic GABAergic signaling (Song et al., 2012). Furthermore, our recent studies showed that long-distance projection neurons send axonal collaterals to rNSCs or DG interneurons to directly or indirectly regulate rNSCs, respectively (Bao et al., 2017; Yeh et al., 2018). For instance, commissural projections from hilar MCs send glutamatergic axons to rNSCs and dentate PV interneurons to dynamically regulate direct and indirect pathways influencing rNSCs (Yeh et al., 2018). In addition, projections from medial septal GABA neurons innervate dentate PV interneurons to regulate rNSCs (Bao et al., 2017). These studies revealed two fundamental modes of rNSC regulation: a direct mode in which local or long-distance circuits directly act on and regulate rNSCs, and an indirect mode in which long-distance projections act on local niche cells to indirectly regulate rNSCs. Whether and how local niche cells interact within the DG to regulate rNSCs remains undefined.
To address this, we focused on DG cholecystokinin (CCK) interneurons, which co-release GABA and the neuropeptide CCK. For nearly 40 years it has been known that CCK is highly abundant in adult hippocampus (Beinfeld et al., 1981). Within the hippocampus, the DG is highly enriched with CCK2 receptors (CCK2Rs) (Kohler and Chan-Palay, 1988; Kritzer et al., 1988). However, the role of CCK signaling in regulating rNSCs and hippocampal neurogenesis remains unknown, largely due to a combination of technical and conceptual challenges, including 1) lack of selective tools to target dentate CCK cells; 2) complications of multiple releasing components from CCK interneurons (GABA, CCK, and possibly glutamate) (Somogyi et al., 2004); and 3) broad actions of CCK on the neurogenic niche cell types expressing CCK2Rs (Deng and Lei, 2006; Deng et al., 2010; Muller et al., 1997).
In this study, we have addressed each of these challenges and reported that dentate CCK interneurons, upon activation, utilize endogenous CCK to regulate multiple cell types in the neurogenic niche, including MCs, GCs, PV interneurons, and astrocytes. Though all of these niche cells can potentially regulate rNSCs, we found that CCK release induces depolarization of rNSCs and promotes neurogenic proliferation of rNSCs through a dominant astrocyte-mediated glutamatergic signaling cascade. In contrast, reducing dentate CCK induces reactive astrocytes, which correlates with reduced neurogenic proliferation of rNSCs and upregulation of genes involved in immune processes, thus highlighting an anti-inflammatory role of CCK in adult DG.
Results
Activation of dentate CCK interneurons induces robust depolarization of rNSCs through CCK2R signaling
Historically, selective expression of transgenes in CCK cells has been difficult due to off-target labeling of other cell types, including DG GCs and CA3/CA1pyramidal cells (Taniguchi et al., 2011). Previously, intersectional genetics in triple transgenic mice has been employed (Whissell et al., 2019). However, this approach labels all CCK neurons in the brain, prohibiting region-specific manipulation. In our approach we tested several serotypes of Cre-dependent AAVs in CCK-Cre mice to selectively target dentate CCK interneurons. We found that properly titered AAV2 (300 nl at 1.08 × 1011 viral genomes/ml) labeled hilar CCK interneurons without off-target effects (Figure 1A). Moreover, labeling of dentate CCK interneurons by mCherry remained restricted to the hilus throughout the dorsal-ventral axis (Figure S1A). Next we validated the specificity and efficiency of targeting dentate CCK interneurons with an established pro-CCK antibody (Beinfeld, 1985). We found that AAV2 labeled ~70% of pro-CCK+ cells; and nearly 100% of labeled cells were pro-CCK+ (Figure 1B–C). Importantly, almost all cells were GABA+, indicating that CCK interneurons are GABAergic (Figure 1D–E). Activity-dependent neuropeptide release requires a “priming” step in which release of Ca2+ from intracellular stores prepares large dense core vesicle fusion with the cellular membrane (Ludwig et al., 2002). Therefore, we adopted a chemogenetic approach using CNO-activated Designer Receptors Exclusively Activated by Designer Drugs (Gq-DREADDs) to induce Ca2+ mobilization from intracellular stores (Armbruster et al., 2007). We evaluated chemogenetic activation of CCK interneurons by slice electrophysiology. Firing rates of CCK cells labeled with hM3Dq were robustly increased upon bath application of CNO in acute brain slices. (Figure 1F–H). Interestingly, dentate CCK interneurons exhibited distinct responses: some were quiet at baseline, and CNO application depolarized them and increased their firing rate (Figure 1F); while others were relatively active at the baseline, and CNO application increased their firing rate without significant baseline shift (Figure 1G). These results highlight the heterogeneity of dentate CCK interneurons and confirmed the efficacy of chemogenetic activation of dentate CCK interneurons.
Figure 1. Local CCK interneurons depolarize rNSCs through CCK2R-mediated signaling.

(A) AAV2, AAV5, or AAV8 expression of DIO-hM3Dq-mCherry in CCK-Cre mice using equivalent titers. Scale bar 200 μm.
(B) Colocalization of mCherry with ProCCK in hilar CCK interneurons after AAV2 injection in CCK-Cre mice. Scale bars 100 μm, 50 μm.
(C) Specificity (left) and efficiency (right) of DREADD expression in hilar CCK interneurons. Bars indicate mean ± SD for 4 animals.
(D) Colocalization of GABA in hM3D+ CCK interneurons. Arrows highlight colocalized cells. Scale bar 25 μm.
(E) Quantification of GABA colocalization. Bar indicates mean ± SD for 3 animals.
(F-G) Whole-cell recordings in acute slice of an initially silent (F) or initially active (G) hM3Dq+ CCK interneuron upon application of 10 μM CNO. Arrows indicate start of drug application.
(H) Quantification of fold increase in spiking at peak chemogenetic activation. Bars indicate means ± SEM for 4 animals.
(I) hM3Dq-mCherry expression in hilar CCK interneurons near GFP+ rNSCs. Scale bar 100 μm.
(J-K) Sample whole-cell recordings of GFP+ rNSCs showing (J) large depolarization due to chemogenetic activation of CCK interneurons or (K) maintained hyperpolarization in 2 μM YM022.
(L) Quantification of rNSC Vm before and during CNO application in ACSF, or YM022. Bars indicate mean Vm ± SEM. p=0.02(ACSF), 0.09(YM022) by paired t-test for (9,7) cells.
(M) Magnitude of depolarization due to CCK activation. p=0.02 by Wilcoxon-Mann-Whitney test for (9,7) cells from (6,6) animals. Bars indicate mean ± SEM for responding rNSCs (>10 mV; circles). Triangles = non responding rNSCs.
(N) Percent of cells that depolarized by at least 10 mV. See also Figure S1.
To examine the net functional effect of endogenous CCK release on rNSCs at the circuit level, we generated double-transgenic CCK-Cre::Nestin-GFP mice and used Gq DREADDs (Figure 1I) to chemogenetically activate CCK interneurons and record the membrane potential (Vm) of GFP+ rNSCs in acute brain slices. At baseline, rNSCs maintain an average Vm of −78.8 ± 3.2 mV (Figure 1L). Interestingly, chemogenetic activation of CCK interneurons depolarized rNSCs to −23.1 ± 7.2 mV (Figure 1J, L–N), which was prevented by CCK2R antagonist YM022 (Figure 1K, L–N). To confirm that YM022 did not affect dentate CCK interneurons during chemogenetic activation, we performed Ca2+ imaging to record spontaneous Ca2+ events as an activity readout for dentate CCK cells (Figure S1B). During the time window in which we observed CCK2R-dependent depolarization of rNSCs, CNO increased the mean frequency (not area) of Ca2+ events in DG hM3Dq+ CCK cells, and was not affected by YM022 (Figure S1C–D). This confirmed that CNO-induced activation of DG CCK cells was not affected by CCK2R antagonism. Together, these results suggest that activation of CCK interneurons induces rNSC depolarization via neuropeptide signaling, instead of GABA or glutamate transmission.
CCK interneuron activity induced rNSC depolarization is mediated by a CCK2R-dependent glutamatergic relay
We sought to address whether CCK activity induced depolarization of rNSCs represents a direct or indirect effect on rNSCs through other niche components. First, we recorded the Vm of Nestin-GFP+ rNSCs upon bath application of non-sulfated CCK8 peptide, the most abundant form of CCK in the brain and selective agonist for CCK2Rs (Huang et al., 1989). Bath application of CCK8 in the presence of TTX, ionotropic GluR (iGluR) and group I metabotropic GluR (mGluR) antagonists (APV+NBQX+AIDA), and GABAAR antagonist (bicuculline) did not alter the Vm of rNSCs (Figure 2A, D, E). In contrast, CCK8 application without these antagonists induced prominent rNSC depolarization (Figure 2B, D, E), which was absent in YM022 (Figure 2C–E). These data suggest the involvement of intermediates that relay CCK signaling onto rNSCs.
Figure 2. CCK induced rNSC depolarization is mediated by a glutamatergic relay.

(A-C) Sample changes in rNSCs Vm to 300 nM CCK8 peptide in the presence of (A) 100 μM APV, 10 μM NBQX, 100 μM AIDA, 50 μM bicuculline, and 1 μM TTX, (B) in ACSF, or (C) in 1 μM YM022.
(D) Comparison of peptide induced depolarization of rNSCs. p=(0.003,0.003) for (7,7,7) cells in (6, 4, 5) animals by Wilcoxon-Mann-Whitney test. Bars indicate mean ± SEM for rNSCs that depolarized >10 mV from baseline (circles). ANBAT = APV, NBQX, bicuculline, AIDA, TTX.
(E) Percent of rNSCs in (D) that responded >10 mV to CCK8 application.
(F-H) Sample recordings of rNSC Vm to chemogenetic activation of CCK interneurons during (F) GABAAR blockade, (G) combined blockade of GABAARs, iGluRs and mGluRs, and (H) when only GABAARs and iGluRs were blocked or when only GABAAR and mGluRs were blocked. Curve fits to data in red, yellow diamonds indicate delay time to Vm deflection.
(I) Comparison of depolarization magnitude during CNO application under indicated conditions. One-way ANOVA showed an effect by condition F(4,41)=10.18, p=8.1*10−6, and a post hoc Dunnett analysis gave individual p-values vs ACSF of (0.95,0.018,0.002,0.0008) for (9,11,9,10,7) cells from (6,10,8,7,5) animals. Bars indicate mean ± SEM for rNSCs that depolarized >10 mV (circles).
(J) Percent of rNSCs in (I) that depolarized >10 mV.
(K) Times from CNO application to initiation of depolarizing event. Bars indicate mean ± SEM. One-way ANOVA showed no effect of condition; F(3,23)=1.66, p=0.2 for (6,10,5,5) responding cells from (5,9,4,4) animals. See also Figure S2.
To dissect potential intermediates, we recorded rNSCs upon CNO application in the presence of various combinations of GABAR or GluR antagonists. In the presence of bicuculline, rNSCs exhibited a robust depolarization that was not significantly different from ACSF (Figure 2F, I, J), suggesting that interneuron-mediated GABAergic components do not contribute to the rNSC depolarization. Next we added NBQX, APV, and AIDA to block both iGluRs and group I mGluRs. As a result, the rNSC depolarization was completely abolished upon adding CNO (Figure 2G, I, J), suggesting that glutamatergic components are necessary for CCK interneuron mediated rNSC depolarization. Furthermore, we separately examined the contribution of iGluRs or group I mGluRs in mediating CCK interneuron induced rNSC depolarization. We recorded rNSCs in the presence of NBQX + APV or AIDA to isolate the contribution from iGluRs or group I mGluRs, respectively. As a result, we observed significant reductions in the resultant depolarization magnitude in rNSCs (Figure 2H, I). In addition, the percent of responding rNSCs (Figure 2J) upon chemogenetic activation of dentate CCK interneurons was greatly reduced as compared to ACSF or bicuculline. Delay times to depolarization in rNSCs after CNO application varied, with an average of 13.4 ± 3.7 min, and were not significantly different among various treatment conditions (Figure 2K). Furthermore, we directly confirmed the presence of functional iGluRs and group I mGluRs in rNSCs. Specifically, bath application of AMPA, NMDA, or DHPG in the presence of TTX depolarized rNSCs (Figure S2). Together, these data suggest that CCK interneuron activity induced rNSC depolarization requires activation of both iGluRs and group I mGluRs.
Activation of dentate CCK interneurons increases GABAergic inputs onto dentate glutamatergic neurons
We sought to identify sources of CCK-induced glutamatergic signaling onto rNSCs. Our recent electron microscopy and immunolabeling work established that bushy processes of rNSCs wrap around MC/GC glutamatergic synapses (Yeh et al., 2018). Therefore, we performed Ca2+ imaging in MCs and GCs to examine the functional impact of CCK interneuron activity on these cells (Figure 3A). Interestingly, we observed a significant decrease in both frequency and area of Ca2+ events in MCs upon CNO-induced chemogenetic activation of CCK interneurons, which was abolished in the presence of CCK2R blocker YM022 (Figure 3B, C, E). These results suggest that CCK interneuron activation leads to decreased MC activity through a CCK2R-mediated mechanism. In contrast, there was no significant change in frequency or area of Ca2+ events in GCs upon CNO in ACSF or YM022 (Figure 3G, H, J). We further quantified the percent of MCs and GCs that increased or decreased Ca2+ activity based on individual brain slices under various pharmacological conditions. Slice-wise analyses showed that 78% of MCs exhibit decreased frequency and 67% of MCs exhibit decreased area of Ca2+ events upon CNO activation of CCK interneurons (Figure 3D, F), and 58% of GCs exhibit decreased Ca2+ frequency (not area) upon the same manipulation (Figure 3I, K). Importantly, the percent of MCs and GCs with decreased Ca2+ signals was significantly reduced in the presence of YM022 (Figure 3D, I, F, K). These data suggest that CCK interneuron activation recruits CCK2R-dependent inhibitory inputs to both MCs and GCs.
Figure 3. CCK interneurons increase GABAergic inputs onto dentate glutamatergic neurons in a CCK2R manner.
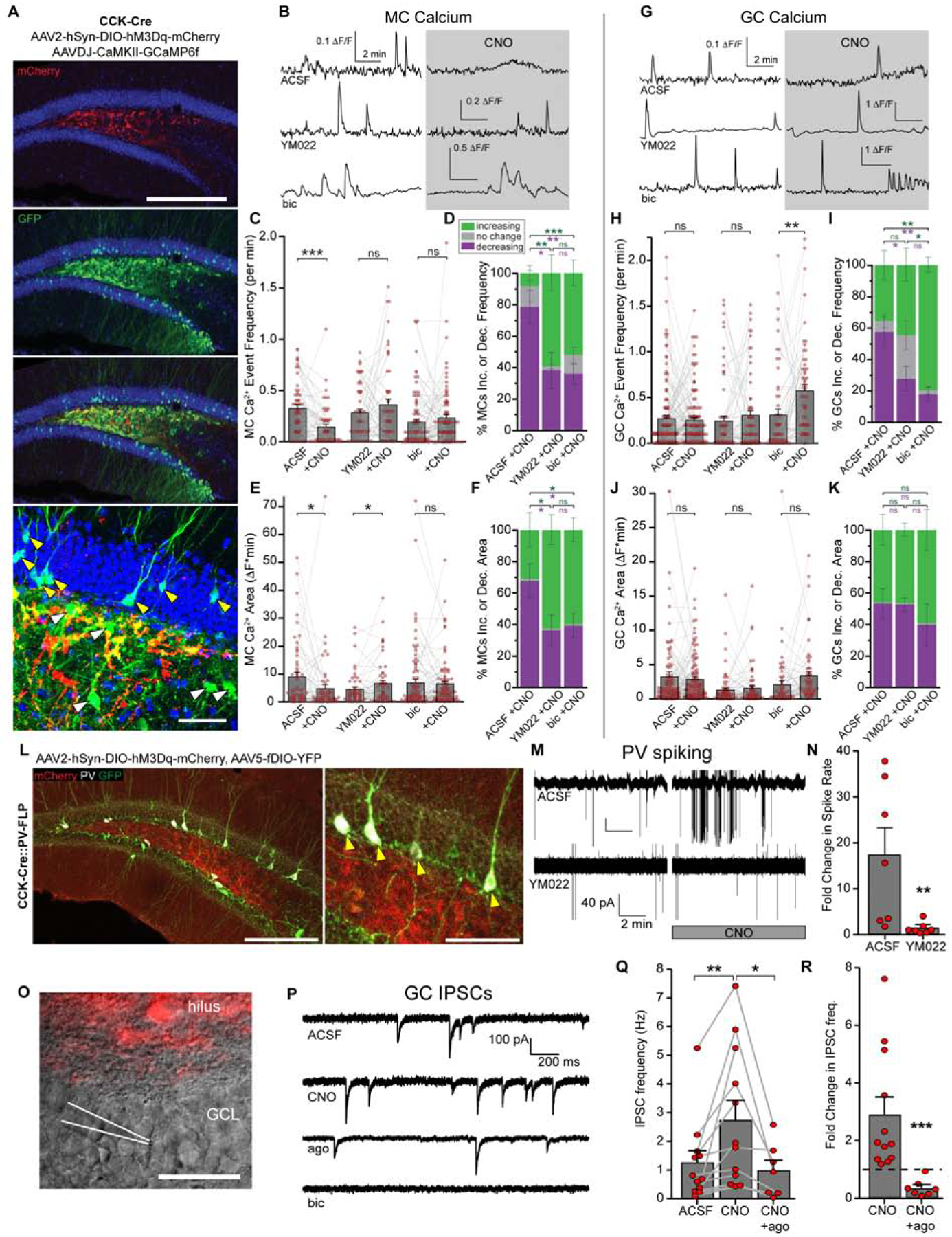
(A) Expression of hM3Dq-mCherry in CCK cells and GCaMP6 in MCs and GCs. Yellow arrows indicate GCaMP6+ GCs, white arrows indicate GCaMP6+ MCs. Scale bars 200 μm, 50 μm.
(B) Ca2+ transients in MCs before and during chemogenetic activation of CCK interneurons in ACSF, 1 μM YM022, or 50 μM bicuculline. Gap indicates a 10 min pause of image acquisition for CNO application.
(C) Frequency of MC Ca2+ events due to chemogenetic activation of CCK cells in ACSF, YM022, or bicuculline. Lines and markers indicate paired measurements in individual MCs. Bars indicate mean ± SEM Ca2+ frequency. p=(1*10−5,0.27,0.30) by paired t-test for (56,52,87) MCs in (5,3,5) animals.
(D) Slice-wise analysis of the percent increasing (green), decreasing (purple), or unchanging (grey) MC Ca2+ frequencies per slice due to CNO application. Stacked bars indicate means ± SEM. p=0.003(ACSF:YM022), 0.60(YM022:bic), 0.0001(ACSF:bic) increasing and 0.02(ACSF:YM022), 0.86(YM022:bic), 0.004(ACSF:bic) decreasing for (10,7,13) slices in (5,3,5) animals by unpaired t-test.
(E) Area under MC Ca2+ traces during baseline and chemogenetic activation of CCK cells as in (C). p=(0.032,0.039,0.70).
(F) Slice-wise analysis of MC Ca2+ area as in (D). p=0.047(ACSF:YM022), 0.81(YM022:bic), 0.047(ACSF:bic).
(G) Sample Ca2+ transients in GCs as in (B).
(H) Frequency data of GC Ca2+ as in (C) for (130,68,58) GCs in (5,3,3) animals. p=(0.71,0.39,0.003).
(I) Slice-wise analysis of GC frequencies (H) as in (D). p=0.52(ACSF:YM022), 0.018(YM022:bic), 0.0013(ACSF:bic) increasing and 0.032(ACSF:YM022), 0.31(YM022:bic), 0.003(ACSF:bic) decreasing for (9,6,6) slices in (5,3,3) animals.
(J) Area data of GC Ca2+ (H) as described in (E). p=(0.52,0.52,0.07).
(K) Slice-wise analysis of GC Ca2+ area (J) as described in (F). p=0.96(ACSF:YM022), 0.39(YM022:bic), 0.43(ACSF:bic).
(L) YFP expression in PV interneurons and mCherry expression in hM3Dq hilar CCK cells. Arrows indicate YFP+ PV+ cells. Scale bars 200 μm, 100 μm.
(M) Cell-attached recordings of PV spiking in ACSF or 1 μM YM022. The gap in the traces indicates a pause of recording during CNO addition.
(N) Quantification of fold-changes in spike rates for (7,7) cells from (5,5) animals. p=0.003 by Wilcoxon-Mann-Whitney test.
(O) Hilar CCK-cell targeted expression of hM3Dq-mCherry and patch pipette for recordings of GCs. Scale bar 50 μm.
(P) Sample inward IPSCs in GCs in ACSF, CNO, CNO + 200 nM ω-agotoxin TK, or CNO + 50 μM bicuculine, using high chloride internal solution.
(Q) Quantification of mean ± SEM IPSC frequencies for (12,7) GCs at baseline, during CNO, and then in CNO + ago. p=(0.007,0.048) by paired t-test in (10,6) animals.
(R) Fold change in GC IPSC frequency for cells in (Q). p=3*10−5 by unpaired t-test. See also Figure S3.
Next we sought to examine the inhibitory inputs to both MCs and GCs upon CCK interneuron activation. First, we repeated the previous Ca2+ experiments in MCs and GCs in the presence of bicuculline to block the GABAAR-mediated component. We found the CCK-induced inhibitory effects onto MCs were completely abolished in the presence of bicuculline (Figure 3C, E), similar to that observed in YM022, suggesting that all GABAergic inputs on MCs induced by CCK interneuron activation are CCK2R-dependent. In contrast, blocking the GABAAR-mediated component induced a significant increase in the frequency (not area) of Ca2+ events in GCs (Figure 3H, J), as well as a further reduction of the percent of GCs with reduced Ca2+ frequency on top of that observed in YM022 (Figure 3I). This suggests that the GABAergic inputs on GCs induced by CCK interneuron activation contain both CCK2R dependent and independent components. It is interesting to note that CCK interneuron activation recruited GABAergic inputs to both MCs and GCs, but only MCs exhibited decreased Ca2+ activity, suggesting that MCs and GCs use differential mechanisms to maintain their activity.
As GCs and MCs are well known to be inhibited by local PV interneurons (Acsady et al., 2000; Espinoza et al., 2018), we sought to determine whether dentate PV interneurons respond to CCK activity by recording PV interneuron excitability upon CCK interneuron activation in CCK-Cre::PV-FLP mice (Figure 3L). We found significantly increased spiking rates in dentate PV interneurons upon CNO application as measured by cell-attached recordings. Importantly, this increase was abolished by YM022 (Figure 3M, N), suggesting that dentate PV interneurons represent CCK2R-dependent inhibitory inputs onto both MCs and GCs. To confirm this, we recorded spontaneous inhibitory postsynaptic currents (sIPSCs) in GCs upon CCK interneuron activation in ω-agotoxin TK, a selective blocker for P/Q type Ca2+ channels unique to PV cells (Hefft and Jonas, 2005; Wilson et al., 2001) (Figure 3O, P). As a result, we observed a 3-fold increase in sIPSCs upon CNO-induced activation of CCK cells (Figure 3Q, R), which was reversed to 36% of the initial CNO-level frequency in ω-agotoxin TK. All inward currents were confirmed GABAergic by addition of bicuculline (Figure 3P). Furthermore, by considering the fractional proportion of sIPSC frequencies that were ω-agotoxin TK sensitive and insensitive upon CNO and at baseline (Figure S3A), we estimated the PV contribution to the CCK-dependent increase in GC sIPSCs to be ~77% (Figure S3B, also see STAR methods). Together, these data confirmed that dentate CCK interneuron activation recruited both CCK2R-dependent PV inputs and other CCK2R-independent GABAergic inputs onto GCs.
Dentate astrocytes respond to CCK activity and astrocyte activation is sufficient to induce rNSC depolarization
Since the activities of local glutamatergic neurons are either inhibited or unchanged during CCK interneuron activation, we sought to identify an alternative source of glutamate-mediated rNSC depolarization. Ca2+ elevation in astrocytes has been widely shown to promote release of gliotransmitters, including glutamate and D-serine (Fiacco and McCarthy, 2018; Hamilton and Attwell, 2010; Savtchouk and Volterra, 2018), which could act on GluRs to depolarize rNSCs. However, it is yet unclear if DG astrocytes respond to CCK, and whether astrocytes act as mediators of the CCK induced changes in rNSCs. To address this, we performed Ca2+ imaging in astrocytes during chemogenetic activation of dentate CCK cells. Confocal imaging confirmed that DG CCK processes reside adjacent to astrocytes (Figure 4A), thus providing morphological support for the interaction between these cells. Functionally, we observed a significant increase in the frequency (not area) of astrocytic Ca2+ events upon chemogenetic activation of CCK cells (Figure 4B–D), which was abolished in YM022 (Figure 4C, D). Since GABA signaling can activate astrocytes (Mederos and Perea, 2019) and our study found that CCK interneurons recruit GABAergic inputs, we recorded astrocyte Ca2+ in the presence of GABAA and GABAB receptor antagonists. As a result, we still observed significant CNO-induced increases in the frequency of Ca2+ events (Figure 4C, D), suggesting that GABA components do not play a major role in CCK-dependent astrocyte activation. Furthermore, bath application of CCK8 peptide in the presence of TTX increased frequency and area of Ca2+ events in astrocytes (Figure 4E, F), suggesting that CCK directly acts on astrocytes without other neuronal intermediates.
Figure 4. Dentate astrocytes respond to CCK activity and are capable of inducing rNSC depolarization.

(A) Expression of hM3Dq-mCherry in CCK cells and GCaMP6-LCK in astrocytes. Arrows highlight hM3Dq+ processes adjacent to GFAP+ astrocyte. Scale bars 100 μm, 20 μm.
(B) Sample astrocyte Ca2+ transients evoked by CCK interneuron activation. The gap in traces indicates a 10 min pause of image acquisition for CNO addition.
(C) Astrocyte Ca2+ frequencies due to activation of CCK cells in ACSF, 1 μM YM022, or 50 μM bicuculline + 1 μM CPG. Lines and markers indicate paired measurements of individual ROIs. Bars indicate mean ± SEM Ca2+ frequency for (282, 278, 219) ROIs in (3, 3, 3) animals. p=(1.3*10−12,0.49,1.8*10−9) by paired t-test.
(D) Area under Ca2+ traces in (C). p=(0.083,0.47,0.50) by paired t-test.
(E) Astrocyte Ca2+ events frequencies to 300 nM CCK8 peptide while in 1 μM TTX for 142 ROIs from 3 animals. p=0.014 by paired t-test.
(F) Area under Ca2+ traces in (E). p=0.03 by paired t-test.
(G) Astrocytic targeting of hM3Dq for chemogenetic stimulation. Arrows highlight colocalized expression in GFAP+ cells. Scale bars 100 μm, 20 μm.
(H) Sample astrocyte Ca2+ transients in hM3D+ astrocytes due to CNO application. The gap in traces indicates a 10 min pause of image acquisition for CNO addition.
(I) Frequencies of astrocyte Ca2+ due to chemogenetic activation of astrocytes for 166 regions from 3 animals. p=0.0006 by paired t-test.
(J) Area under Ca2+ traces in (I). p=0.31 by paired t-test.
(K) Expression of hM3Dq in astrocytes in Nestin-GFP mouse. Scale bar 100 μm.
(L) Recording of a GFP+ mCherry− rNSC during chemogenetic stimulation of astrocytes.
(M) Vm of rNSCs before and during CNO application. Bars indicate mean ± SEM. p=0.006 by paired t-test for 7 cells.
(N) Magnitude of CNO induced depolarization in either ACSF, or in 100 μM APV + 10 μM NBQX + 100 μM AIDA. p=0.038, by unpaired t-test for all (7,7) cells from (4,3) animals. Bars indicate mean ± SEM for rNSCs responding >10 mV (circles). Triangles = non-responders.
(O) Percent of cells that depolarized >10 mV.
We next sought to determine whether direct activation of astrocytes could depolarize rNSCs. We infected DG astrocytes with Gq-DREADDs (Figure 4G) and found that CNO application significantly increased the frequency (not area) of intracellular astrocyte Ca2+ (Figure 4H–J). We then recorded the Vm of rNSCs from Nestin-GFP mice upon astrocyte activation. (Figure 4K). We found that chemogenetic activation of dentate astrocytes was sufficient to induce depolarization of rNSCs (Figure 4L–O). Furthermore, this depolarization was significantly reduced by iGluR and group I mGluR antagonists, and resulted in fewer responding rNSCs. Together, these results indicate that dentate astrocytes are responsive to CCK interneuron activation in a CCK2R-dependent manner, and this activation is sufficient to cause rNSC depolarization presumably via release of gliotransmitters (such as glutamate and D-serine) that act on iGluRs and group I mGluRs on rNSCs.
CCK activity induced rNSC depolarization requires dentate astrocyte intermediates
To determine if dentate astrocytes serve as an intermediate in CCK induced depolarization of rNSCs, we interfered with astrocyte signaling by application of a glial-specific Krebs cycle inhibitor Fluorocitric acid (FC) (Pabst et al., 2016; Swanson and Graham, 1994). Strikingly, chemogenetic activation of dentate CCK cells failed to induce rNSC depolarization when astrocyte signaling was disrupted by FC (Figure 5A, B). Importantly, rNSCs retained their ability to depolarize in response to glutamate (Figure S4A), suggesting that FC does not disrupt the machinery for glutamatergic receptor signaling.
Fig 5. CCK induced rNSC depolarization requires astrocyte intermediates.

(A) Reduced rNSC depolarization when slices are preincubated in 100 μM FC.
(B) Vm of rNSCs due to chemogenetic activation of CCK cells in FC treated slices. Bars indicate mean Vm ± SEM. p=0.02 by paired t-test for 7 cells from 4 animals. Darker blue lines = rNSCs responding >10 mV.
(C) Expression of mCherry and GCaMP6-LCK in GFAP+ astrocytes for pipette targeting and BAPTA filling. Scale bars 100 μm, 20 μm.
(D) Ca2+ traces in hilar astrocytes at baseline and then after BAPTA infusion into a single mCherry+ astrocyte.
(E) Frequencies of nearby astrocyte Ca2+ events due to intracellular BAPTA infusion. Lines and markers indicate paired measurements in individual ROIs. Bars indicate mean ± SEM frequencies for 107 astrocytic ROIs in 3 animals. p=2.3*10−13 by paired t-test.
(F) Area under Ca2+ traces in (E). p=1.9*10−5 by paired t-test.
(G) CCK-Cre::Nestin-GFP animal with (top) BAPTA filling pipette on a mCherry+ astrocyte in the hilus and (bottom) recording configuration from GFP+ rNSC. Scale bar 50 μm.
(H) Vm recording from a GFP+ rNSCs during CCK chemogenetic activation after high BAPTA.
(I) Vm of rNSCs due to CCK activation after astrocytic BAPTA. p=0.16 by paired t-test for 7 cells in 6 animals. Darker purple lines = rNSCs responding >10 mV.
(J) mRFP-p130PH and GCaMP6-LCK expression in DG astrocytes. Arrows indicate colocalization in GFAP+ astrocytes. Scale bars 100 μm, 20 μm.
(K) Basal astrocyte Ca2+ frequencies in control or p130PH injected hemispheres. Bars indicate mean ± SEM frequency. p=0.11 for (275,236) astrocyte ROIs in 3 animals by unpaired t-test.
(L) Area under Ca2+ traces in (K). p=0.009.
(M) Frequencies of DG astrocyte Ca2+ events due to 300 nM CCK8 peptide in p130PH or control hemisphere. Baseline data is identical to (K). p=(0.005,0.11) for (275,236) astrocyte ROIs in 3 animals by paired t-test.
(N) Area under Ca2+ traces in (M). p=(0.3,0.16).
(O) Vm from a GFP+ rNSC from p130PH injected DG upon chemogenetic activation of CCK cells.
(P) Vm of rNSCs due to CCK activation in p130PH injected DGs. p=0.13 by paired t-test for 10 cells in 5 animals. Bars indicate mean Vm ± SEM. Darker red lines = rNSCs responding >10 mV.
(Q) Magnitude of depolarization due to chemogenetic CCK activation in indicted conditions. One-way ANOVA showed a significant effect of condition F(3,29)=9.01,p=0.002, and Dunnett post hoc analysis showed each are significantly reduced from ACSF. p=(0.009,0.009,0.004) for (9,7,7,10) cells from (6,4,6,5) animals. Bars indicate mean ± SEM for rNSCs responding >10 mV (circles). Triangles = non responding rNSCs.
(R) Percent of cells that depolarized >10 mV. See also Figure S4.
As chemogenetic activation of dentate CCK cells increased the Ca2+ transients in astrocytes, we attempted to quench astrocytic Ca2+ signaling through infusion of the Ca2+ chelator BAPTA in astrocytes using a patch pipette (Pabst et al., 2016). Because astrocytes are coupled by gap junctions, BAPTA infusion of a single astrocyte has the ability to buffer Ca2+ in large astrocyte networks (Araque et al., 1998; Pabst et al., 2016). We first confirmed that infusing BAPTA into a single astrocyte was sufficient to interfere with the DG astrocyte network as reflected by significant reductions in both frequency and area of astrocyte Ca2+ events (Figure 5C–F). We then recorded the Vm of rNSCs upon chemogenetic activation of CCK cells after astrocytic Ca2+ depletion (Figure 5G, Figure S4B), and found that CCK activity-induced rNSC depolarizations were largely abolished (Figure 5H, I). Importantly, rNSCs remained responsive to bath application of glutamate after astrocyte BAPTA infusion (Figure S4C).
CCK-evoked Ca2+ increases in astrocytes are independent of extracellular Ca2+ concentrations (Muller et al., 1997) and likely involve PLC induced release of Ca2+ from intracellular stores via IP3 mediated intracellular cascades (Dufresne et al., 2006; Lee et al., 2011). We therefore disrupted dentate astrocyte function by buffering mobile cytosolic IP3 through expression of the pleckstrin homology domain of PLC-like-protein p130 (p130PH) (Xie et al., 2010). We injected AAVs containing p130PH construct along with GCaMP6-LCK, and confirmed co-expression in GFAP+ astrocytes (Figure 5J). We first validated the efficacy of p130PH in disrupting Ca2+ activity of DG astrocytes by bilaterally injecting mice with p130PH + GCaMP6 on one side, and mRFP + GCaMP6 on the other side. As a result, we found a significant reduction in the area of Ca2+ events in dentate astrocytes (not frequency) at the p130PH side as compared to the control side at baseline (Figure 5K, L). Furthermore, CCK8 induced a significant increase in the frequency (not area) of astrocytic Ca2+ events on the control side, with no significant change in either frequency or area of astrocytic Ca2+ events on the p130PH side (Figure 5M, N). Using the ability of p130PH to suppress the activity of astrocytes, we then determined whether dentate astrocytes served as the relay that couples CCK activity to rNSC depolarization. Whole-cell electrophysiological recordings from GFP+ rNSCs displayed a significant reduction in the depolarization magnitude of Vm with fewer responding rNSCs upon chemogenetic activation of CCK neurons when astrocyte IP3 mobilization was disrupted (Figure 5O, P). Quantification of the depolarization magnitude and percent of responding rNSCs was consistently reduced in FC, BAPTA, or p130PH conditions, significantly different from controls (Figure 5Q, R). Together, these data confirmed that dentate astrocytes are the major glutamatergic intermediaries for CCK activity-induced rNSC depolarization.
CCK interneuron stimulation promotes neurogenic proliferation of rNSCs via MAPK/ERK signaling and increases neurogenesis
Vm has been implicated as a functional determinant of proliferation and differentiation in non-neuronal cells, such as embryonic stem cells (Levin, 2014). Quiescent cells remain hyperpolarized, whereas proliferation events are often accompanied by membrane depolarization (Aprea and Calegari, 2012). Transduction of extracellular niche signals activates cell surface receptors that in turn trigger intracellular signaling cascades that control activation and quiescence in adult rNSCs. It has been shown that both NMDARs and group I mGluRs activate MAPK/ERK1/2 pathways via upstream effectors in vitro (Wang et al., 2007). We sought to determine whether pERK signaling facilitates rNSC activation upon CCK interneuron stimulation in vivo. We first examined expression of immediate early gene c-Fos in dentate CCK interneurons 1.5 hours after CNO intraperitoneal injection (Figure 6A). The density of c-Fos+ mCherry+ CCK interneurons in the hM3Dq group was significantly higher compared to controls (Figure 6B, C), thus confirming in vivo activation of CCK interneurons.
Figure 6. CCK interneuron stimulation promotes proliferation of rNSCs via MAPK/ERK signaling and increases neurogenesis.

(A) Experimental paradigm for in vivo stimulation of CCK interneurons and c-Fos analysis.
(B) DG c-Fos expression after CCK chemogenetic activation. Arrows indicate hilar mCherry+ c-Fos+ cells. Scale bars 100 μm, 20 μm.
(C) Density of c-Fos labeled hilar mCherry+ cells. p=0.034 for (3,4) animals by Student’s t-test.
(D) Experimental paradigm for quantification of phospho-ERK, phospho-RB, or EdU uptake in response to chemogenetic stimulation of CCK interneurons.
(E) Phospho-ERK+ rNSCs in the DG 80 minutes after IP CNO. Scale bar 10 μm. Arrows highlight pERK+ Nestin+ adult rNSCs.
(F) Density of pERK+ rNSCs for (4,5) animals. Bars indicate means ± SD. p=0.013 by Student’s t-test.
(G) Phospho-RB+ rNSCs in the DG 5 hours after IP CNO. Scale bar 10 μm. Arrow highlights a pRB+ Nestin+ rNSC.
(H) Density of pRB+ rNSCs for (5,4) animals. Bars indicate means ± SD. p=0.008 by Student’s t-test.
(I) EdU uptake in rNSCs after 4 days of CNO drinking water. Scale bars 100 μm, 20 μm. Arrow highlights an EdU+ Nestin+ adult rNSC.
(J-M) Proportion (J) and density (K) of proliferating adult neural stem cells in the DG, density of all proliferating cells (L), and total rNSC pool (M). Bars indicate means ± SD. p=(0.03,0.05,0.80,0.005) for (6,5) animals by Student’s t-test.
(N) Experimental paradigm for quantification of neurogenic proliferation in response to in vivo chemogenetic activation of CCK interneurons.
(O) DCX+ adult-born immature neurons after 3 weeks of CNO drinking water. Scale bars 100 μm, 20 μm. Arrows indicate DCX+ EdU+ cells.
(P-Q) Density (P) of DCX+ EdU+ immature neurons and rNSC pool (Q) after in vivo chemogenic stimulation of CCK interneurons. Bars indicate means ± SD; p=(0.047,0.29) for (4,6) animals by Student’s t-test. See also Figure S5.
Next we examined expression of pERK in rNSCs 80 minutes after CNO intraperitoneal injection (Figure 6D, E, Figure S5A), and observed a significant increase in the density of pERK+ rNSCs in hM3Dq mice compared to control mice (Figure 6F). As a mitogen-activated protein kinase (MAPK), ERK regulates cell proliferation through promotion of G1/S cell cycle progression (Chambard et al., 2007; Zhang and Liu, 2002). Specifically, activation of ERK signaling drives hyper-phosphorylation of retinoblastoma protein (RB), releasing RB’s inhibition of E2F transcription factors, which in turn activates genes required for the G1/S transition (Duronio and Xiong, 2013). To address whether hyper-phosphorylated RB (pRB) mediates CCK interneuron activity dependent rNSC activation, we examined pRB expression in rNSCs 5 hours after CNO injection (Figure 6D, G, Figure S5B). As a result, we observed a significant increase in the density of pRB+ rNSCs (Figure 6H), suggesting that CCK interneuron activity promotes G1/S cell cycle progression in rNSCs. Then we examined the incorporation of EdU in rNSCs and newborn progeny (Figure 6D). We assessed EdU incorporation by counting EdU+ Nestin+ rNSCs with radial processes (Figure 6I) after 4 days of CNO drinking water. Stereological analysis showed that chemogenetic activation of local CCK interneurons led to a significant increase in the percent and density of Nestin+ EdU+ rNSCs (Figure 6J, K). As activated rNSCs give rise to proliferating progeny, we examined the density of EdU+ cells, and observed a significant increase of EdU incorporation upon chemogenetic activation of CCK interneurons (Figure 6L) without altering the Nestin+ rNSC pool (Figure 6M). These results suggest that dentate CCK interneuron activation promotes neurogenic proliferation of rNSCs and increases production of proliferating progeny. We also examined the integrity of activated astrocytes, and found that chemogenetic activation of CCK interneurons did not induce changes in astrocyte morphology or GFAP expression in hM3Dq animals (Figure S5C, D).
To determine the long-term effects of dentate CCK interneuron activity in regulating neurogenesis and the rNSC pool, we labeled a starting proliferating population with 5 EdU injections followed by 3-week chronic stimulation of dentate CCK interneurons through CNO drinking water (Figure 6N). Stereological analysis revealed a significant increased density of DCX+ EdU+ immature neurons without exhaustion of the rNSC pool (Figure 6O–Q). Together, these results suggest that CCK interneuron stimulation promotes stable neurogenic proliferation via MAPK/ERK signaling and activation of G1/S cell cycle regulators, resulting in increased production of proliferating progeny and immature neurons without depleting the rNSC pool.
Reduced dentate CCK induces reactive astrocytes, decreased neurogenic proliferation of rNSCs, and upregulation of genes involved in neuroinflammation
To complement the gain-of-function approach for increased CCK, we took a loss-of-function approach to reduce dentate CCK by AAV delivery of shRNA against CCK (shCCK) (Arey et al., 2014). We confirmed efficiency of shCCK by western blot (Figure 7A, B) and immunostaining (Figure 7C) 1 month after viral injection. Based on our finding that increased CCK induces astrocyte gliotransmission, we wondered whether reduced CCK contributes to an adverse state of astrocytes. To address this, we examined GFAP expression, an established marker for astrocyte reactivity (Liddelow and Barres, 2017), in the dentate astrocytes by quantifying the volume of GFAP immunofluorescence in mice injected with shCCK or scrambled control (shScr). Strikingly, GFAP expression in shCCK injected mice was significantly increased as compared to shScr controls (Figure 7D, E), suggesting that reduced CCK induces reactive astrocytes. To examine the functional effects of reduced CCK on rNSCs, we labeled dividing cells through 4 EdU injections 1 month after AAV injection and examined EdU incorporation in Nestin+ rNSCs and EdU+ proliferating progeny. Stereological quantification showed a significant decrease in the percent and density of EdU+ Nestin+ rNSCs (Figure 7F, G) and total EdU+ proliferating progeny (Figure 7H) without altering the Nestin+ rNSC pool (Figure 7I).
Figure 7. Reduced dentate CCK induces reactive astrocytes and decreases rNSCs proliferation potentially via neuroinflammatory-related gene profiles.
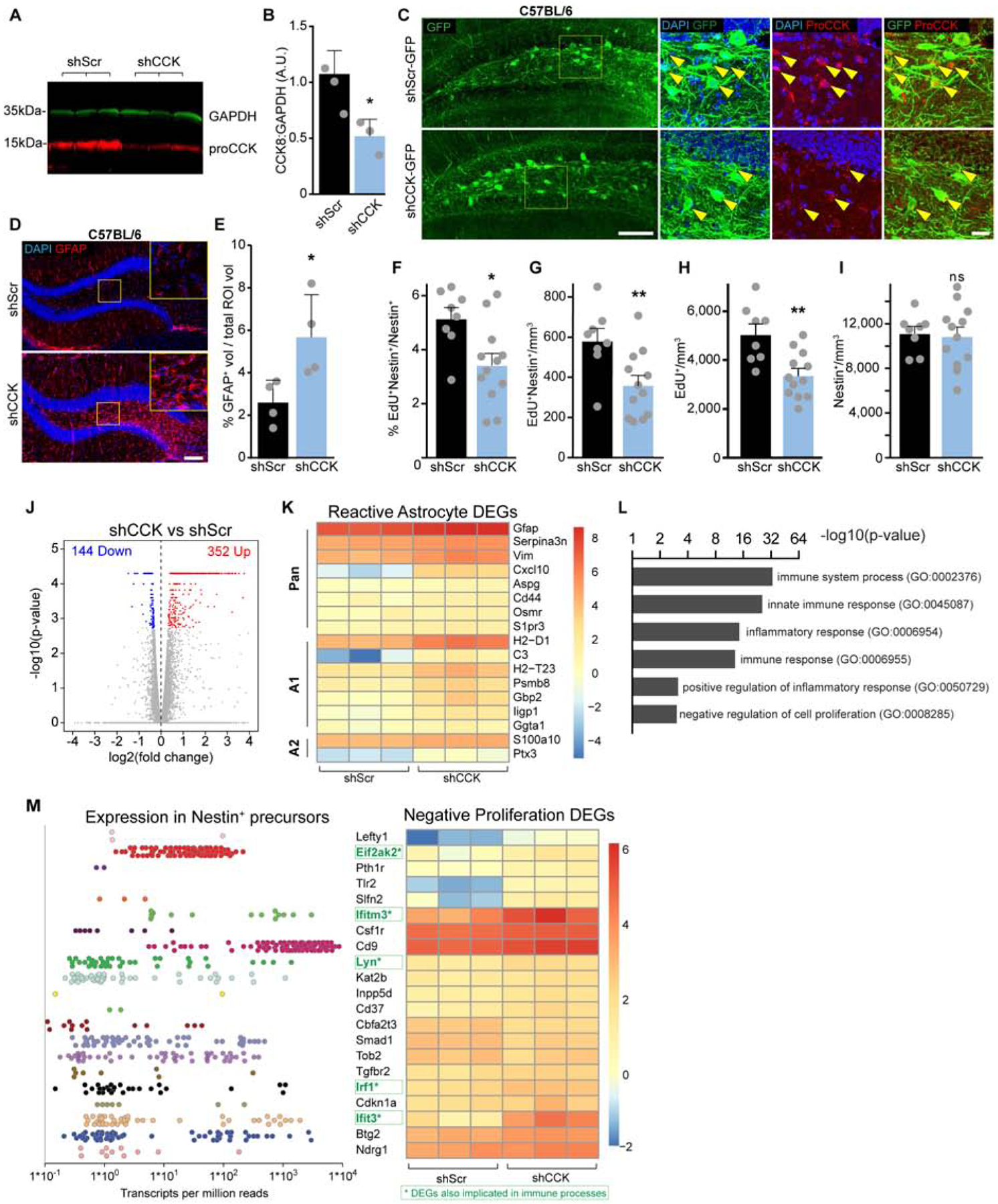
(A-B) Western blot and densitometry of hippocampal lysates 1 month after injection of shCCK or shScr AAVs. p=0.02 from (3,3) animals by Student’s t-test.
(C) Reduced CCK immunofluorescence in the hilus of shCCK treated animals. Arrows indicate targeted GFP+ hilar cells. Scale bars 100 μm, 20 μm.
(D) GFAP reactivity after knockdown of DG CCK. Scale bar 100 μm.
(E) Quantification of %GFAP immunofluorescence per volume for (4,4) animals. p=0.03 by unpaired t-test.
(F-I) Proportion (F) and density (G) of proliferating rNSCs, density (H) of total proliferating cells, and total (I) rNSC pool after shCCK. Bars indicate means ± SD. p=(0.011,0.009,0.002,0.81) for (8,12) animals by Student’s t-test.
(J) Volcano plot of fold changes in gene expression due to shCCK, highlighting upregulated and downregulated DEGs.
(K) Heatmap of 17 DEGs related to reactive astrocyte states from 3 animals in each treatment group. Pan-reactive, type A1, and type A2 reactive astrocyte genes are indicated. Color indicates direction and relative magnitude of FPKMs.
(L) Gene Ontology terms identified from analysis of all 496 DEGs identifying immune processes and negative regulation of proliferation.
(M) Transcript expression in rNSCs (based on Shin et al 2015) and heatmap of 21 DEGs related to negative control of proliferation. Color indicates direction and relative magnitude of FPKMs. Green highlighted genes are additionally implicated in immune processes. See also Figure S6.
To explore mechanisms linking reactive astrocytes and deceased rNSC proliferation upon CCK knockdown, we performed RNA-Seq of DGs microdissected from shCCK or shScr mice (Table S1, Table S2). Our RNA-Seq analysis identified 496 differentially expressed genes (DEGs), including 144 downregulated and 352 upregulated transcripts (Figure 7J, Table S3). We compared the RNA-Seq data with a recently published transcriptional profile of reactive astrocytes induced by neuroinflammation or ischemia (Clarke et al., 2018) and found 17 reactive-astrocyte associated genes were differentially expressed in the DGs of shCCK mice (Figure 7K, Table S4). These DEGs included standard reactive astrocyte genes (PAN: 8/17), such as GFAP and Vimentin, A1-reactive genes associated with neuroinflammation (A1: 7/17), such as C3 and Cxcl10, and A2-reactive genes associated with ischemia (A2: 2/17). These data suggest that reducing CCK induces astrocyte reactivity, especially A1 reactive astrocytes. Additionally, gene ontology and biological pathway analysis revealed many DEGs enriched in biological pathways involved in immune processes and innate inflammatory responses (Figure 7L, Table S5), supporting the involvement of innate immune cells, including astrocytes. Furthermore, of 21 DEGs relating to negative regulation of cell proliferation (Table S6), 19 were also expressed in neural precursor cells based on a published single-cell RNA-Seq profile of Nestin+ cells in adult DG (Shin et al., 2015) (Figure 7M). These data support our observed correlation between reduced CCK and decreased proliferation of rNSCs (Figure 7F–I). Interestingly, several of these highly expressed DEGs in rNSCs, including Eif2ak2, Ifitm3, Lyn, Irf1, and Ifit3, were jointly implicated in both negative proliferation pathways and immune system processes (Figure 7M, Figure S6). Together, these results suggest that reduced CCK induces astrocyte reactivity and increases innate immune responses, which in turn leads to decreased proliferation of rNSCs and newborn progeny.
Discussion
We demonstrated that stimulating CCK release supports neurogenic proliferation of rNSCs through the trophic effects of CCK on dentate astrocytes to promote gliotransmission onto rNSCs (Figure 8A). In contrast, reducing dentate CCK induced reactive astrocyte states, which correlated with decreased neurogenic proliferation of rNSCs and upregulation of genes involved in inflammatory responses (Figure 8B).
Figure 8. Model of dentate CCK interneuron regulation of rNSCs through differential astrocyte states.
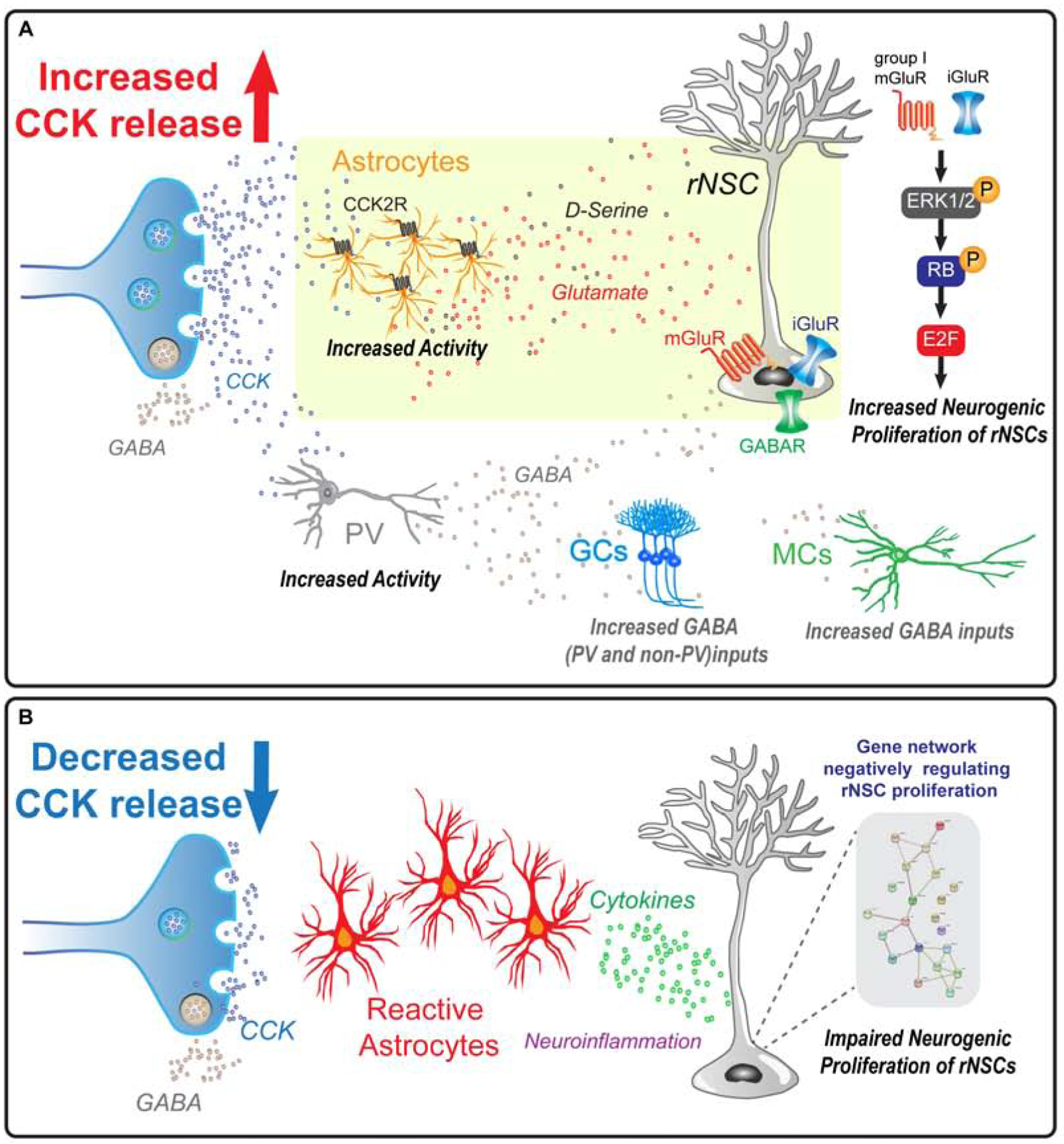
(A) Dentate CCK terminals, via CCK2Rs, induce glutamate release from astrocytes, which activate iGluRs and mGluRs on rNSCs and induce intracellular signaling cascades including phosphorylation of ERK1/2, translocation to the nucleus, hyperphosphorylation of RB, and unbinding of transcription factor E2F. Unbound E2F can drive transcription of genes that promote cell cycle progression. PV interneurons, meanwhile are excited by CCK, and contribute to GABAergic inhibition of local glutamatergic circuitry.
(B) Conversely, reduced CCK levels reflect pathogenic states, and result in immune activation, neuroinflammation, and reactive astrocyte states, leading to inactivation of rNSCs likely through cytokine signaling and gene expression changes that negatively regulate proliferation.
NSCs are capable of incorporating dominant niche signals to determine their actions
Many diverse cell types and signaling pathways within the specialized local niche of the DG can simultaneously act on rNSCs and influence their behavior. This raises a key question: how do rNSCs integrate such diverse signals to ultimately remain quiescent or become activated? We found that activation of dentate CCK interneurons increased the activity of both dentate astrocytes and PV interneurons through CCK2R-mediated mechanisms. Our previous studies implicated inhibitory transmission with rNSC quiescence, since bath application of a GABAAR agonist hyperpolarized rNSCs (Yeh et al., 2018) and optogenetic activation of dentate PV interneurons reduced activation of rNSCs through GABAAR signaling (Song et al., 2012). In contrast, our current study found that CCK interneurons promote rNSC depolarization through an astrocytic intermediary and increased rNSC proliferation via GluR signaling. Simultaneous activation of dentate astrocytes and PV interneurons should therefore exert opposing effects on the Vm and proliferation status of rNSCs. Despite such opposing niche signals, the dominant effects on rNSCs appeared to be rNSC depolarization and increased neurogenic proliferation of rNSCs through GluR-mediated ERK/MAPK signaling cascades. However, in the case of reduced CCK in the DG, we found decreased rNSC proliferation, suggesting that glutamatergic niche signaling was no longer dominant in this context. Indeed, besides increased gliotransmission of glutamate (Madeira et al., 2015; Pirttimaki et al., 2013), reactive astrocytes release cytokines, which appear to be the dominant niche signals that contribute to reduced proliferation of rNSCs (Wu et al., 2012).
Potential role of other niche cells recruited by dentate CCK interneuron stimulation
Besides activating dentate PV interneurons and astrocytes, stimulating CCK interneurons led to increased GABAergic inhibition of both MCs and GCs. Interestingly, the GABAergic inputs to MCs were CCK2R dependent, while GABAergic inputs to GCs were both CCK2R dependent and independent. Importantly, blocking the GABA component upon CCK interneuron stimulation abolished the inhibition of MCs and induced significant increases in GC activity, suggesting that CCK interneuron stimulation exerts an inhibitory tone on dentate excitatory neurons in order to prevent hyperexcitability of the DG. The inhibitory tone in the neurogenic niche is critical for maintaining the NSC pool by preventing DG hyperexcitability (Bao et al., 2017; Sierra et al., 2015). In addition, activation of dentate PV interneurons promoted survival of neuroblasts, and increased adult-born GCs (Song et al., 2013). Consistent with this, we found that chronic activation of local CCK interneurons promoted survival of newborn neurons and preserved the rNSC pool. This suggests that the combination of astrocyte-mediated glutamatergic signaling and inhibitory tone in the DG induced by CCK interneuron activation collectively supports sustainable hippocampal neurogenesis.
Signaling mechanisms underlying CCK activity dependent regulation of local astrocytes
Our study identifies dentate CCK interneurons as a critical niche component that utilizes an endogenous neuropeptide to regulate rNSCs via local astrocytes. Specifically, local astrocyte states are regulated by endogenous levels of dentate CCK: high dentate CCK promotes neurogenic proliferation of rNSCs and production of newborn progeny through an astrocyte-mediated glutamatergic signaling cascade; while low dentate CCK reduces neurogenic proliferation of rNSCs and production of newborn progeny potentially through neuroinflammatory signaling cascades associated with reactive astrocytes. The signaling mechanisms underlying CCK modulation of dentate astrocytes remain to be determined. We show that dentate astrocytes respond to CCK signals with increased Ca2+ events via CCK2Rs, suggesting a direct mechanism mediated by astrocyte CCK2Rs. It remains to be determined whether reduced CCK leads to aberrant Ca2+ signals in reactive astrocytes, which could contribute to proinflammatory cytokine release. Accumulating evidence has suggested that reactive astrocytes exhibit aberrant Ca2+ signals, and the patterns of aberrant Ca2+ depend upon distinct pathological conditions (Shigetomi et al., 2019; Vincent et al., 2010). Future studies will be required to characterize the Ca2+ signals of reactive astrocytes associated with reduced dentate CCK.
Reduced CCK, reactive astrocytes, and NSC regulation
One striking finding from our study is that reduced dentate CCK appears to exert a detrimental effect on local astrocytes by switching them to an A1 reactive state associated with neuroinflammation, suggesting that CCK normally acts as an anti-inflammatory molecule in adult DG. The anti-inflammatory role of CCK has been implicated in the peripheral system (Meng et al., 2002; Miyamoto et al., 2012; Oehlers et al., 2017), but not in the central nervous system nor in the context of adult rNSC regulation. Since CCK knockdown in the DG induced reactive astrocytes and neuroinflammation, this raised a question on the potential mechanism linking reactive astrocytes and deceased rNSC proliferation. Interestingly, our RNA-Seq analysis found that the majority of DEGs involved in both negative proliferation and immune processes were associated with interferon-γ (IFNγ) signaling, including Eif2ak2, Ifitm3, Irf1, and Ifit3 (based on GeneCards), posing the possibility that upregulated IFNγ signaling upon reduced CCK may contribute to decreased rNSC proliferation. Supporting this, recent studies showed increased proliferation of adult precursor cells in the adult olfactory bulbs and DGs of IFNγ knockout mice (Monteiro et al., 2016; Pereira et al., 2015). The classical sources of IFNγ are T cells and natural killer cells in the peripheral system (Monteiro et al., 2017), however, our RNA-Seq data provided strong support for the involvement of resident immune cells, including astrocytes and microglia. Based on our findings that astrocytes are extremely responsive to dentate CCK, we speculate that reactive astrocytes may be the source of IFNγ upon reduced CCK. Future experiments with specific manipulation of IFNγ in astrocytes upon reduced CCK could identify the role of IFNγ in regulating rNSC behavior.
Recent human studies have provided tremendous support for reduced CCK in Alzheimer’s disease (AD) patients. For instance, a recent microarray analysis of human hippocampal tissues showed that physical exercise significantly increased hippocampal CCK transcription, while AD significantly reduced it (Berchtold et al., 2019). Another study showed that higher levels of CCK in cerebrospinal fluid is negatively correlated with AD susceptibility (Plagman et al., 2019). Moreover, emerging evidence has revealed impaired rNSC behavior and hippocampal neurogenesis in AD mouse models (Fu et al., 2019; Mu and Gage, 2011) and human patients (Crews et al., 2010; Gomez-Nicola et al., 2014; Jin et al., 2004; Li et al., 2008; Lovell et al., 2006; Moreno-Jimenez et al., 2019; Perry et al., 2012). Furthermore, human AD patients display aberrant astrocytes with ramified processes and altered GFAP isoforms (Kamphuis et al., 2012; Kamphuis et al., 2014) and AD mice exhibit significant astrogliosis (Oakley et al., 2006). Therefore, AD pathological hallmarks, including plaques and tangles, may induce reduced endogenous CCK and reactive astrocytes as a potential mechanism for impaired rNSCs and hippocampal neurogenesis. Given the critical role of adult-born neurons in regulating cognitive functions, increasing CCK release to promote neurogenic potential of NSCs and reduce inflammatory responses may represent a novel strategy to treat neuropathological conditions associated with aberrant hippocampal neurogenesis and neuroinflammation, such as aging, AD, and epilepsy.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Information and requests should be directed to and will be fulfilled by the lead contact, Juan Song, Ph.D. (juansong@email.unc.edu).
Materials Availability
Viral reagents generated in this study (AAV5-CMV-shCCK-eGFP and AAV5-CMV-shScrambledeGFP) will be made available without restriction by request to the lead contact.
Data and Code Availability
RNA-seq data reported in this paper has been submitted to Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE137052.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Single or double transgenic young adult mice (6–12 weeks, males and females) were used and randomly assigned to experimental groups. C57BL/6J, CCK-IRES-Cre (B6N.Cg-Ccktm1.1(cre)Zjh/J), and PV-Flp (B6.Cg-Pvalbtm4.1(flpo)Hze/J), were obtained from Jackson laboratory; Nestin-GFP mice (Mignone et al., 2004) were obtained from Dr. Grigori Enikolopov at Stony Brook University. Animals were group housed and bred in a dedicated husbandry facility with 12/12 light-dark cycles with food and water ad libitum and received veterinary supervision. Animals subjected to surgical procedures were moved to a satellite housing facility for recovery with the same light-dark cycle. No immune deficiencies or other health problems were observed in these lines, and all animals were experimentally and drug naive before use. Electrophysiology or Ca2+ imaging experiments were typically performed on animals at 2–4 months of age. All animal procedures followed NIH Guide for the Care and Use of Laboratory Animals and were approved by Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
METHOD DETAILS
Stereotaxic Injections
In brief, mice were anesthetized under 1–2% isoflurane in oxygen at 0.6–0.8 LPM flow rate. AAV vectors were injected under stereotaxic control to the DG using the following coordinate: AP −2.0 mm, ML +/− 1.5 mm, DV −2.1 mm from Bregma, with microsyringe (Hamilton, 33GA) and microinjection pump (Hamilton) at a speed of 75 nl/min. The needle then remained in place for 5 min before being withdrawn. Mice were allowed to recover for 2 weeks from the surgery before in vivo or in vitro experiments. For in vivo DREADD experiments, CCK-IRES-Cre mice were injected with 300 nl AAV2-hSyn-DIO-hM3Dq-mCherry or AAV2-hSyn-DIO-mCherry (Addgene/UNC Vector Core) bilaterally into the DG. Several measures were taken to increase the specificity of viral labeling by restricting the spread of the virus to the hilus by: (1) reducing flow rate to 75 nl/min (2) packaging the DREADD in AAV2, which doesn’t spread as far from the injection site as other serotypes do, and (3) diluting the virus to a titer of 1.08 × 1011 viral genomes/ml. For EdU/proliferation quantifications, CNO was dissolved in drinking water at a concentration of 5 mg/200 ml for 4 days. CNO was from NIH as part of the Rapid Access to investigative Drug Program funded by National Institute of Neurological Disorders and Stroke (NINDS). On the last day, animals were given IP injections of EdU (40 mg/kg, Carbosynth) for 4 times at 2-hour intervals. Animals were perfused 2 hours after the last injections of EdU. For quantifications of phospho-ERK and phospho-RB in rNSCs, CNO was administered via a single IP injection (5 mg/kg) and animals were then perfused either 80 minutes (phospho-ERK) or 5 hours (phospho-RB) later. For in vivo shRNA experiments, mice were injected with 1 μl of either AAV5-CMV-shCCK-eGFP or AAV5-CMV-shScrambled-eGFP (AAV vectors from Ralph DiLeone, Yale Univ.; short-hairpin sequences were adapted from published work (Arey et al., 2014) and packaged by the Duke Vector Core). After 4 weeks, animals were given IP injections of EdU and perfused as previously described. For slice recording, viral injections were conducted similarly. Additional AAV vectors including AAVDJ-EF1α-DIO-GCaMP6s (Dr. Stuber, U. Washington), AAV5-EF1a-fDIO-EYFP-WPRE (UNC Vector Core), AAV8-GFAP104-mCherry (UNC Vector Core), AAV2/5-GFAP-GCaMP6-LCK (Addgene), AAV9-Syn-DIO-jGCaMP7f (Addgene), AAV8-GFAP-hM3Dq-mCherry (Dr. McCarthy, Univ of North Carolina at Chapel Hill), and AAV2/5-gfaABC1D-mRFP-p130PH and AAV2/5-gfaABC1D-mRFP (Dr Ding, Univ. Missouri-Columbia) were injected into the DG of adult mice for various experiments. Mice with viral expression outside the region of interest (DG) were excluded from tissue processing and further analysis. Please note, AAVDJ is a hybrid serotype vector created by shuffling 8 different AAV serotypes (Grimm et al., 2008) and is commonly used to achieve good efficiency of expression in vitro and in vivo (used by Stanford Vector Core, for example).
Electrophysiological recordings
Adult mice were used for slice preparation and electrophysiological recording 2–4 weeks after viral injection. No specific replication designs were used, but multiple animals were used as indicated in the figure legends. Sample size estimation was based on previous publications and power analysis. Mice with viral expression outside the region of interest (DG) were excluded from further analysis.
Animals were anesthetized under isoflurane and briefly perfused intracardially with 10 ml of ice-cold NMDG solution (Ting et al., 2014) containing (in mM): 92 NMDG, 30 NaHCO3, 25 glucose, 20 HEPES, 10 MgSO4, 5 sodium ascorbate, 3 sodium pyruvate, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, 0.5 CaCl2 (pH 7.24, 300 mOsm, bubbled with 95% O2 and 5% CO2). The brains were then quickly removed into additional ice-cold NMDG solution for slicing. Transverse slices were cut using a Leica VT1200S vibratome at 280 μm thickness, and warmed to 36°C for 10 min. Slices were transferred to room temperature (22–24°C) HEPES holding solution(Ting et al., 2014) containing (in mM): 92 NaCl, 30 NaHCO3, 25 glucose, 20 HEPES, 5 sodium ascorbate, 3 sodium pyruvate, 2.5 KCl, 2 thiourea, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, (pH 7.24, 300 mOsm, bubbled with 95% O2 and 5% CO2) for 1 – 2 hr. Solutions intended for use with CCK recordings, PV recordings, GC recordings, or for Ca2+ imaging also contained 12 mM N-acetyl-L-cysteine in the NMDG and HEPES solutions to improve glutathione synthesis (Ting et al., 2014).
Whole-cell electrophysiological recordings were obtained at 22–24°C in artificial CSF (ACSF) containing (in mM): 125 NaCl, 26 NaHCO3, 20 glucose, 2.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, (pH 7.24, 300 mOsm, bubbled with 95% O2 and 5% CO2) on a Scientifica SliceScope. GFP+ neural stem cells within the sub-granule zone, or hilar mCherry+ CCK interneurons were visualized by DIC and fluorescence microscopy with a 40X water-dipping objective (LUMPlanFL NA, 0.8; Olympus). rNSC identity of Nestin-GFP+ cells were confirmed by stereotypical type-1 responses to current pulses (non-rectifying, high-resistance). Microelectrodes (3–6 MΩ) were pulled from 1.5 mm diameter borosilicate glass capillaries (WPI) and filled with potassium based internal solution containing (in mM): 130 K-gluconate, 20 HEPES, 4 MgCl2, 4 Na2-ATP, 2 NaCl, 0.5 EGTA, 0.4 Na-GTP (pH 7.24, 310 mOsm).
Cell-attached recordings from YFP+ parvalbumin neurons or visually identified non-fluorescent mature GCs were made by forming ≥ 200 MOhm resistance seals using similar microelectrodes filled with filtered ACSF and no applied holding voltage. Because of the sparsity of GC spiking, CaCl2 and MgCl2 in the ACSF were adjusted to 2.2 and 1.1 mM for those experiments.
Spontaneous IPSCs from GCs were made by whole-cell recordings of non-fluorescent mature granule cells identified by DIC microscopy in the mid to outer GCL using a high-Cl internal solution containing (in mM): 140 KCl, 10 EGTA, 8 HEPES, 5 QX-314, 2 Mg2-ATP (pH 7.3, 310 mOsm).
BAPTA-filling of astrocytes was made in mCherry+ hilar astrocytes using a K-BAPTA internal solution containing (in mM): 70 sucrose, 50 K-gluconate, 20 K4-BAPTA, 10 HEPES, 4 MgCl2, 4 Na2-ATP, 2 NaCl, 0.5 EGTA, 0.4 Na-GTP (pH 7.24, 310 mOsm). Internal solution was permitted to intracellularly perfuse the astrocyte for 20 minutes through a patch pipette in whole-cell configuration.
Slices treated with FC (100 μM) were preincubated for 1 hour in bubbled ACSF containing the drug before recording, and remained in FC-ACSF during patch-recording.
Data were collected using a Multiclamp 700B amplifier and digitized with a DigiData 1440A (Axon Instruments) at 10 kHz using pClamp10 software. The whole-cell patch-clamp configuration was employed in current-clamp mode (with I = 0) to freely monitor membrane potential changes for rNSC recordings and for monitoring CCK neuron action potential firing. Unstable whole-cell recordings (monitored by membrane test pulse) were excluded from analysis.
Pharmacological agents (Tocris) were used at the following final concentrations in the bath as indicated: bicuculline (50 μM), CPG (1 μM), NBQX (10 μM), APV (100 μM), TTX (1 μM), AIDA (100 μM), AMPA (1 mM), NMDA (1 mM), DHPG (200 μM), CCK8 (300 nM), YM022, (1 μM), L-glutamic acid (1 mM). CNO (obtained from NIH) was bath applied at 10 μM. The ω-agotoxin TK (200 nM) was purchased from Alamone Labs. All other chemicals, including DL-Fluorocitric acid (100 μM), were purchased from Sigma.
Ca2+ Imaging
GCaMP was selectively expressed by AAV-mediated expression of GFAP-GCaMP6-LCK, CaMKII-GCamp6s, or DIO-jGCaMP7f. Acute brain slices were prepared and observed as for electrophysiology. A CoolLED pE-100 at 470 nm was used for excitation, and GCaMP fluorescence was acquired through a standard GFP filter cube and captured with an Optimos sCMOS camera (Q-Imaging), using Micro Manager acquisition software. Image sequences were acquired at 100 ms frame rates with 5 focal-plane sections in 10 minute episodes. Actual imaging rate after microscope movements and z-stack completion effectively reduced the time resolution to ~2.8 s. Imaging sessions of mossy cells, granule cells, CCK interneurons, or astrocytes were paused for 10 minutes during pharmacological exchange to allow adequate perfusion of drugs before resuming image acquisition. In experiments with astrocyte BAPTA loading, imaging was paused during the 20 minute infusion, and were resumed after the pipette was withdrawn.
Tissue process and Immunohistochemistry
Mice were anesthetized with a mixture of ketamine and xylazine (100 mg/kg and 10 mg/kg respectively) until no longer responsive to toe pinch, or subjected to isofluorane inhalation at 5% until nonresponsive to toe pinch. Brains were fixed via transcardial perfusion of ice-cold 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) at pH 7.4. Brains were collected and post-fixed in 4% PFA overnight, followed by 30% sucrose in PBS for 2–3 days. Brains were sectioned into 40 μm coronal sections with a microtome and stored in cryoprotectant solution containing 30% sucrose, 30% ethylene glycol in 0.1M phosphate buffer.
To label proliferating cells, sections from mice injected with EdU were first rinsed in Tris-buffered saline (TBS) and then 0.05% Triton X-100 in TBS, followed by permeabilization in 0.5% Triton X-100 in TBS for 30 min. Sections were then incubated in click reaction buffer (0.1 M Tris, 0.5–1mM CuSO4, 30μM 488 Alexa azide (A10266, Invitrogen), and 50–100 mM ascorbic acid) for 1 hour for EdU labelling. Tissue was then rinsed 4–6X in 0.05% Triton X-100 in TBS.
For immunostaining, sections were first rinsed in TBS and then 0.05% Triton X-100 in TBS, followed by incubation in 0.5% Triton X-100 in TBS for 30 min (this step was omitted if tissue had already undergone the click reaction and so was already permeabilized). Sections were then incubated in blocking buffer (5% donkey serum in 0.05% Triton X-100 in TBS) for 30–60 min followed by incubation in primary antibody (primary antibody + 5% donkey serum diluted in 0.05% Triton X-100 in TBS). The following primary antibodies were incubated with tissue at 4C overnight: rat anti-mCherry (1:1000, Invitrogen), rabbit anti-GABA (1:500, Sigma), chicken anti-Nestin (1:250, Aves), rabbit anti-phospho RB (1:500, Cell Signaling), mouse anti-GFAP (1:500, Millipore), goat anti-DCX (1:500, SCBT), rabbit anti-PV (1:500 Swant), and goat anti-GFP (1:500, Rockland). Rabbit anti-proCCK (1:500, a gift from Margery Beinfeld) was incubated for 48 hours at room temperature. Mouse anti-CCK8 (1:500, no. 9303 CURE) and anti phospho-ERK (1:100, Cell Signaling) were incubated for 72 hours at 4C. c-Fos immunostaining was the same as others with the exception that there was 20 minute incubation in 1 mg/ml solution of sodium borahydride in PBS prior to any incubations, and the rabbit anti c-Fos primary (1:1000, Sysy) was incubated for 72 hours at 4C. The sections were then rinsed with 0.05% Triton X-100 in TBS and incubated in secondary antibody (secondary antibody + 5% donkey serum diluted in 0.05% Triton X-100 in TBS) at room temperature (RT) for 2 hours followed by additional rinsing in 0.05% Triton X-100 in TBS. All the brain sections were mounted in an aqueous medium.
Antigen retrieval is required for immunostaining with Nestin, phospho-ERK, phospho-RB, and CCK8. Sections requiring antigen retrieval were first mounted onto charged slides and incubated in a beaker containing 0.1M citrate buffer and kept at between 92 – 95°C for a period of 7 mins under constant stirring. Slides then remained in citrate buffer after being removed from heat source and put on ice to cool down to room temperature. Sections then underwent permeabilization and blocking as previously described.
QUANTIFICATION AND STATISTICAL ANALYSIS
Electrophysiological quantification
Electrophysiology traces were analyzed using a combination of the NeuroMatic package (Rothman and Silver, 2018) and custom built analysis routines, written for Igor Pro (Wavemetrics). Resting Vm was reported for sample traces throughout the figures. Depolarization magnitudes were computed by averaging several minutes of baseline activity, subtracted from the smoothed peak membrane potential during drug application. Responding cells were considered as those that depolarized by ≥ 10 mV, and were color-coded in the figures. Cells that depolarized too suddenly (within less than 2 minutes) were not included for analysis, as it may reflect unexpected loss of whole-cell configuration.
Calculations of delay time were accomplished by fitting the membrane potential data from responding rNSCs to a Sigmoid function and drawing a tangent line at the midpoint of inflection. The intersection of the tangent line with the baseline level marked the initiation time of depolarization. The difference in time between CNO application and the intersection point determined the reported delay time. Cells that were poorly fit by the Sigmoid or had computed delay times beyond 40 minutes were excluded.
Spike counting and detection of sIPSCs were performed by semi-automated threshold detection and additional timing/level parameters in NeuroMatic analysis package (Rothman and Silver, 2018) in Igor Pro, and manually reviewed for accuracy. Non-typical events and any recording artifacts were excluded from analysis. Fold change in spiking occasionally was determined as infinite (inf) when zero spikes were detected at baseline. Some recording artifacts were digitally removed for presentation purposes.
Estimation of the fractional contribution of PV interneurons to the increase in sIPSCs seen in GCs under CNO conditions was calculated by the following equation:
Where total IPSC frequency in ACSF (ACSFfreq) was found in bar 1 of Figure 3Q, total IPSC frequency in CNO (CNOfreq) was found in bar 2 of Figure 3Q. The fraction of ago-sensitive (FsenACSF) and ago-resistant (FresACSF) IPSCs in ACSF was determined experimentally from the average IPSC frequencies given in Figure S3A. The fraction of ago-sensitive (FsenCNO) and ago-resistant (FresCNO) IPSCs in CNO was determined experimentally from the average IPSC frequencies given in Figure 3Q. See Figure S3B for additional derivation.
Calcium analysis
Ca2+ fluorescent signals were analyzed using hand-drawn ROIs in NIH ImageJ software and computed using custom procedures written in Igor Pro (Wavemetrics). Sample Ca2+ traces presented in the figures originate from the same cell in both the baseline and experimental conditions. Displayed Ca2+ signals were determined from surround subtracted regions of interest (ROIs) at the best focal plane, and converted into ΔF/F0 values using a running F0 (Jia et al., 2011). Quantification of event frequency was determined using peak detection algorithms in Igor Pro (using a smooth value of 200, and a box detection width of 5 points) and manual thresholding (using the larger of either 0.2 ΔF/F units or 1 SD above the non-signaling portion (defined as the 5th through 95th percentile of the ΔF/F). Area measurements were simply the integrated sum under the ΔF/F trace and were limited to be >= 0. Excessive data points near zero were occasionally slightly adjusted in the figures to fit all the data into the plotting area for display purposes only. Fold changes were calculated per cell as Frequency2/Frequency1, or Area2/Area1. As frequencies can often be zero during Frequency1, infinite fold changes were recalculated as 2 * Frequency2 / minimum_Frequency1 [i.e. 1 event / imaging time period]. Fold changes became zero if there were no events in Frequency2. To eliminate bias from brain sections that might have higher numbers of labeled cells to analyze, stacked bar plots depicting the mean percent of increasing and decreasing responses were made by counting the total number of cells having >1, 1, or <1 fold change in each slice.
Stereological quantification
Coronal sections through the entire hippocampus were collected in a serial order. Tiled images were acquired with an Olympus FLUOVIEW1000/Zeiss, or LSM780/Zeiss, or LSM710 confocal microscopes, using either 40x Oil (Olympus and Zeiss LSM780) or 20X (Zeiss LSM710) objectives, XY-resolution 0.4975 μm/pixel, and Z-resolution 1.0 μm/slice. Images were stitched using either Olympus FluoView or Zeiss ZEN imaging software. All positive cell type marker quantifications were done manually throughout z-stacks using ImageJ with the exception of GFAP+ cells. Due to high density, it is difficult to accurately identify individual GFAP+ cells. Volume of GFAP+ labeling was quantified using Imaris volumetric analysis and normalized to the volume of the hilus. Slide identities were blinded during quantification to avoid bias.
Microdissection of the dentate gyrus
Two months after unilateral injection of 8 week-old, wild-type, C57BL/6J mice with either AAV5-CMV-shScrambled-eGFP or AAV5-CMV-shCCK-eGFP, mice were anesthetized using 5% isoflurane and sacrificed by decapitation. The whole brain was then removed from the skull and immediately placed in cold PBS. Still in cold PBS, the brain was then bisected, and the dentate gyrus (DG) was then dissected out from the injected hemisphere. DGs from each hemisphere were then placed in RNAlater Stabilization Solution (Thermo Fisher).
RNA-Seq library prep and analysis
Total RNA was extracted from fresh DG using TRIzol Reagent (Invitrogen) and Direct-zol RNA Mini Prep kit (Zymo Research) according to the manufacturer’s instructions. Bulk mRNA-seq libraries were generated from triplicated samples per condition by Novogene. An Agilent 2100 BioAnalyzer and a DNA1000 kit (Agilent) were used to quantify amplified cDNA and to control the quality of the libraries. Illumina HiSeq4000 was used to perform 150-cycle pair-end sequencing. Image processing and sequence extraction were performed using the standard Illumina pipeline. Paired-end reads were first aligned to mouse genome assembly and transcriptome annotations (mm9) by Tophat v2.1.1 with default settings. The numbers of mapped reads for each sample can be found in Table S2. FPKM (fragments per kilobase of transcript per million mapped reads) values were calculated by Cufflinks v2.2.1. Pairwise comparison between shCCK and shScrambled conditions were performed to detect differentially expressed genes (DEGs) using Cuffdiff v2.2.1. DEGs are defined as those with q-value less than 0.05. Gene ontology (GO) analyses on biological processes were performed by the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8. To identify significantly enriched DIRECT GO terms, a Benjamini–Hochberg procedure was used to control the false discovery rate (FDR) at 0.05. KEGG pathway enrichment analyses were performed by the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) update 2019 with adjusted p-value no greater than 0.05. Heatmaps and volcano plots were made in R v3.5.1 by pheatmap and ggplot2 packages.
Statistics
Mice from both sexes were used for all experiments, and no other covariates were identified. For stereological analysis, two-tailed unpaired Student’s t-test was used to examine the difference by using Graphpad Prism (Graphpad Software). Test for normality were not performed. For all experimental results, the number of animals used was reported in figure legends. All measurements are from distinct samples and were not repeated measurements. Data throughout are presented as mean ± SD or mean ± SEM as reported in the figure legends.
Statistics for electrophysiology were calculated using two-sample tests in Igor Pro (Wavemetrics), and were checked for randomness and equal variances. Data were presented as means ± SEM. All data points (responding and nonresponding cells) were included for statistical analysis. The p values from t-tests are reported in the figure legends, or by Wilcoxon-Mann-Whitney test if the variance was determined as unequal. In each figure, animals and number of cells recorded are reported. In cases comparing changes in spike rates or IPSC rates, paired t-tests were used. Changes of Vm from baseline levels to depolarization levels also used paired t-tests. All others were unpaired. The number of animals used was reported in figure legends, and slices were changed after each cell recording to eliminate residual effects of prior pharmacology. One-way ANOVA was occasionally employed with Dunnett post-hoc analysis when comparing multiple groups to control.
Statistics for Ca2+ imaging were calculated using two-sample tests in Igor Pro, and were checked for randomness and equal variances. Data were presented as means ± SEM. For frequency and area comparisons, p values were generated by paired t-test and are reported in the figure legends. Unpaired t-tests were used to determine % increasing or decreasing cells across different conditions. The number of ROIs (cells or astrocyte domains) that were defined manually in the image stacks and number of animals used were reported in figure legends.
Statistical outcomes were based on p< 0.05 and displayed throughout figures as: ‘ns’ not significant, ‘*’ p < 0.05, ‘**’ p< 0.01, ‘***’ p < 0.001.
Supplementary Material
Table S1. Gene Expression Table (All Genes). Related to Figure 7.
Table S3. DEGs (Full List). Related to Figure 7.
Table S5. Significantly Enriched GO Terms Analysis. Related to Figure 7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rat anti-mCherry | Thermo Fisher Scientific | Cat# M11217, RRID: AB_2536611 |
| rabbit anti-GABA | Sigma | Cat# A2052, RRID: AB_477652 |
| rabbit anti-cFos | SySy | Cat# 226003, RRID: AB_2231974 |
| chicken anti-nestin | Aves | Cat# NES, RRID: AB_2314882 |
| rabbit anti-phospho RB | Cell Signaling | Cat# 8516, RRID: AB_11178658 |
| rabbit anti-proCCK | Dr. Margery Beinfeld | RRID: AB_2313591 |
| mouse anti-CCK8 | CURE | Cat# 9303, RRID: AB_10013360 |
| rabbit anti-phospho-ERK | Cell Signaling | Cat# 4370, RRID: AB_2315112 |
| rabbit anti-GAPDH | Abcam | Cat# ab9485, RRID: AB_307275 |
| mouse anti-GFAP | Millipore | Cat# MAB360, RRID: AB_11212597 |
| goat anti-DCX | SCBT | Cat# sc-8066, RRID: AB_2088494 |
| goat anti-GFP | Rockland | Cat# 600-101-215, AB_218182 |
| rabbit anti-PV27 | Swant | Cat# Swant PV27, AB_2631173 |
| Bacterial and Virus Strains | ||
| AAV2-hSyn-DIO-mCherry | Addgene | RRID: AddGene_50459 |
| AAV2-hSyn-DIO-hM3Dq-mCherry | Addgene | RRID: AddGene_44361 |
| AAVDJ-CaMKII-GCaMP6s | Garret Stuber (U. Washington) (Otis et al., 2017); Also available from Stanford Vector Core: GVVC-AAV-88 | NA |
| AAV5-CMV-shCCK-eGFP | Backbone plasmid from Ralph DiLeone (Yale)/ shCCK (Arey et al. 2014) Packaged by Duke Vector Core | NA |
| AAV5-CMV-shScrambled-eGFP | Backbone plasmid from Ralph DiLeone (Yale)/ shScrambled (Arey et al. 2014) Packaged by Duke Vector Core | NA |
| AAV5-EF1a-fDIO-EYFP-WPRE | UNC Vector Core | NA |
| AAV8-GFAP104-mCherry | UNC Vector Core | NA |
| AAV2/5-GFAP-GCaMP6-LCK | Addgene | RRID: AddGene_52924 |
| AAV9-Syn-DIO-jGCaMP7f-WPRE | Addgene | RRID: AddGene_104488 |
| AAV8-GFAP-hM3Dq-mCherry | Ken McCarthy (UNC) and UNC Vector Core | NA |
| AAV2/5-gfaABC1D-mRFP-p130PH | Shinghua Ding (University of Missouri-Columbia, MO) (Xie et al. 2010) | NA |
| AAV2/5-gfaABC1D-mRFP | Shinghua Ding (University of Missouri-Columbia, MO) (Xie et al. 2010) | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| bicuculline | Tocris | 0131 |
| NBQX | Tocris | 0373 |
| APV | Tocris | 0105 |
| TTX | Tocris | 1078 |
| AIDA | Tocris | 0904 |
| AMPA | Tocris | 0254 |
| NMDA | Tocris | 0114 |
| DHPG | Tocris | 0805 |
| CCK8ns | Tocris | 1150 |
| YM022 | Tocris | 1408 |
| EdU | Carbosynth | NE08701 |
| DL-Fluorocitric Acid barium salt | Sigma | F9634 |
| BAPTA tetrapotassium salt | Invitrogen | B1204 |
| CNO | NIH | C-929 |
| ω-agotoxin TK | Alamone Labs | STA-530 |
| CPG 52432 | Tocris | 1246 |
| L-glutamic acid | Tocris | 0218 |
| Deposited Data | ||
| Raw and analyzed RNAseq data | This paper | GSE137052 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: CCK-IRES-Cre | Jackson Laboratory | RRID: IMSR_JAX:012706 |
| Mouse: Nestin-GFP | Dr. Grigori Enikolopov at Stony Brook University | RRID: IMSR_JAX:033927 |
| Mouse: Pvalb-T2A-FlpO-D | Jackson Laboratory | RRID: IMSR_JAX:022730 |
| Software and Algorithms | ||
| Imaris | Bitplane | RRID: SCR_007370 |
| FIJI | ImageJ | RRID: SCR_002285 |
| pClamp | Molecular Devices | RRID: SCR_011323 |
| Micro-Manager | Micro-manager.org | RRID: SCR_000415 |
| Igor Pro | Wavemetrics | RRID: SCR_000325 |
| Neuromatic | Thinkrandom.com | RRID: SCR_004186 |
| Olympus FluoView | Olympus | RRID: SCR_014215 |
| Zeiss ZEN | Zeiss | RRID: SCR_013672 |
| Tophat v2.1.1 | (Kim et al., 2013) | RRID: SCR_013035 |
| Discovery (DAVID) v6.8 | (Huang da et al., 2009a, b) | RRID: SCR_001881 |
| GEne SeT AnaLysis Toolkit | (Liao et al., 2019) | RRID: SCR_006786 |
| STRING v11.0 | (Szklarczyk et al., 2019) | RRID: SCR_005223 |
| Cufflinks v2.2.1 | (Trapnell et al., 2013) | RRID: SCR_014597 |
| Prism | Graphpad | RRID:SCR_002798 |
Highlights.
Stimulating dentate CCK release promotes rNSC depolarization and proliferation
CCK modulates local astrocytes to regulate rNSCs via glutamatergic gliotransmission
CCK interneuron stimulation recruits GABAergic inputs onto excitatory neurons
Reduced CCK induces reactive astrocytes and impairs neurogenic potential of rNSCs
Acknowledgements:
We thank members of Song lab for comments and discussions. We also thank Dr. Bryan Roth lab (UNC) for chemogenetic approach, Dr. Ralph DiLeone (Yale Univ.) for AAV knockdown vector, Dr. Garret Stuber (Univ. Washington) for GCaMP virus, Dr. Margery Beinfeld (Tufts Univ.) for pro-CCK antibody, and Dr. Shinghua Ding’s lab for AAV2/5-GFAP-p130PH (University of Missouri-Columbia). This work was supported by grants awarded to: J.S. from American Heart Association, Whitehall Foundation, and NIH (MH111773-01, MH122692-01 AG058160, NS104530); and NARSAD Young Investigator Award to B.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Statement
The authors declare no competing interests.
Reference
- Acsady L, Katona I, Martinez-Guijarro FJ, Buzsaki G, and Freund TF (2000). Unusual target selectivity of perisomatic inhibitory cells in the hilar region of the rat hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience 20, 6907–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea J, and Calegari F (2012). Bioelectric state and cell cycle control of Mammalian neural stem cells. Stem Cells Int 2012, 816049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, and Haydon PG (1998). Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 18, 6822–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arey RN, Enwright JF 3rd, Spencer SM, Falcon E, Ozburn AR, Ghose S, Tamminga C, and McClung CA (2014). An important role for cholecystokinin, a CLOCK target gene, in the development and treatment of manic-like behaviors. Molecular psychiatry 19, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proceedings of the National Academy of Sciences of the United States of America 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Asrican B, Li W, Gu B, Wen Z, Lim SA, Haniff I, Ramakrishnan C, Deisseroth K, Philpot B, et al. (2017). Long-Range GABAergic Inputs Regulate Neural Stem Cell Quiescence and Control Adult Hippocampal Neurogenesis. Cell Stem Cell 21, 604–617 e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, and Song J (2018). Treating Brain Disorders by Targeting Adult Neural Stem Cells. Trends in molecular medicine 24, 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinfeld MC (1985). Cholecystokinin (CCK) gene-related peptides: distribution and characterization of immunoreactive pro-CCK and an amino-terminal pro-CCK fragment in rat brain. Brain research 344, 351–355. [DOI] [PubMed] [Google Scholar]
- Beinfeld MC, Meyer DK, Eskay RL, Jensen RT, and Brownstein MJ (1981). The distribution of cholecystokinin immunoreactivity in the central nervous system of the rat as determined by radioimmunoassay. Brain research 212, 51–57. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Prieto GA, Phelan M, Gillen DL, Baldi P, Bennett DA, Buchman AS, and Cotman CW (2019). Hippocampal gene expression patterns linked to late-life physical activity oppose age and AD-related transcriptional decline. Neurobiology of aging 78, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J, and Lenormand P (2007). ERK implication in cell cycle regulation. Biochim Biophys Acta 1773, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E, Hansen L, and Masliah E (2010). Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 12252–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, and Lei S (2006). Bidirectional modulation of GABAergic transmission by cholecystokinin in hippocampal dentate gyrus granule cells of juvenile rats. J Physiol 572, 425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Jha A, Ramonet D, Matsui T, Leitges M, Shin HS, Porter JE, Geiger JD, and Lei S (2010). Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 5136–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne M, Seva C, and Fourmy D (2006). Cholecystokinin and gastrin receptors. Physiological reviews 86, 805–847. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, and Xiong Y (2013). Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol 5, a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza C, Guzman SJ, Zhang X, and Jonas P (2018). Parvalbumin(+) interneurons obey unique connectivity rules and establish a powerful lateral-inhibition microcircuit in dentate gyrus. Nature communications 9, 4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, and McCarthy KD (2018). Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu CH, Iascone DM, Petrof I, Hazra A, Zhang X, Pyfer MS, Tosi U, Corbett BF, Cai J, Lee J, et al. (2019). Early Seizure Activity Accelerates Depletion of Hippocampal Neural Stem Cells and Impairs Spatial Discrimination in an Alzheimer’s Disease Model. Cell reports 27, 3741–3751 e3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nicola D, Suzzi S, Vargas-Caballero M, Fransen NL, Al-Malki H, Cebrian-Silla A, Garcia-Verdugo JM, Riecken K, Fehse B, and Perry VH (2014). Temporal dynamics of hippocampal neurogenesis in chronic neurodegeneration. Brain 137, 2312–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, and Kay MA (2008). In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. Journal of virology 82, 5887–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton NB, and Attwell D (2010). Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Hefft S, and Jonas P (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8, 1319–1328. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang SC, Yu DH, Wank SA, Mantey S, Gardner JD, and Jensen RT (1989). Importance of sulfation of gastrin or cholecystokinin (CCK) on affinity for gastrin and CCK receptors. Peptides 10, 785–789. [DOI] [PubMed] [Google Scholar]
- Jia H, Rochefort NL, Chen X, and Konnerth A (2011). In vivo two-photon imaging of sensory-evoked dendritic calcium signals in cortical neurons. Nature protocols 6, 28–35. [DOI] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, and Greenberg DA (2004). Increased hippocampal neurogenesis in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 101, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR, Smith VD, Rangarajan S, et al. (2012). GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PloS one 7, e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi EJ, Moeton M, Freriks M, Mizee MR, and Hol EM (2014). Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiology of aging 35, 492–510. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, and Chan-Palay V (1988). Cholecystokinin-octapeptide (CCK-8) receptors in the hippocampal region: a comparative in vitro autoradiographic study in the rat, monkey and the postmortem human brain. Neurosci Lett 90, 51–56. [DOI] [PubMed] [Google Scholar]
- Kritzer MF, Innis RB, and Goldman-Rakic PS (1988). Regional distribution of cholecystokinin receptors in macaque medial temporal lobe determined by in vitro receptor autoradiography. The Journal of comparative neurology 276, 219–230. [DOI] [PubMed] [Google Scholar]
- Lee SY, Foldy C, Szabadics J, and Soltesz I (2011). Cell-type-specific CCK2 receptor signaling underlies the cholecystokinin-mediated selective excitation of hippocampal parvalbumin-positive fast-spiking basket cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 10993–11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M (2014). Molecular bioelectricity: how endogenous voltage potentials control cell behavior and instruct pattern regulation in vivo. Mol Biol Cell 25, 3835–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Yamamori H, Tatebayashi Y, Shafit-Zagardo B, Tanimukai H, Chen S, Iqbal K, and Grundke-Iqbal I (2008). Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol 67, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic acids research 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, and Barres BA (2017). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Geiger H, Van Zant GE, Lynn BC, and Markesbery WR (2006). Isolation of neural precursor cells from Alzheimer’s disease and aged control postmortem brain. Neurobiology of aging 27, 909–917. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Sabatier N, Bull PM, Landgraf R, Dayanithi G, and Leng G (2002). Intracellular calcium stores regulate activity-dependent neuropeptide release from dendrites. Nature 418, 85–89. [DOI] [PubMed] [Google Scholar]
- Madeira C, Lourenco MV, Vargas-Lopes C, Suemoto CK, Brandao CO, Reis T, Leite RE, Laks J, Jacob-Filho W, Pasqualucci CA, et al. (2015). d-serine levels in Alzheimer’s disease: implications for novel biomarker development. Translational psychiatry 5, e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos S, and Perea G (2019). GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 67, 1842–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng AH, Ling YL, Zhang XP, and Zhang JL (2002). Anti-inflammatory effect of cholecystokinin and its signal transduction mechanism in endotoxic shock rat. World journal of gastroenterology 8, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. The Journal of comparative neurology 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Shikata K, Miyasaka K, Okada S, Sasaki M, Kodera R, Hirota D, Kajitani N, Takatsuka T, Kataoka HU, et al. (2012). Cholecystokinin plays a novel protective role in diabetic kidney through anti-inflammatory actions on macrophage: anti-inflammatory effect of cholecystokinin. Diabetes 61, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro S, Ferreira FM, Pinto V, Roque S, Morais M, de Sa-Calcada D, Mota C, Correia-Neves M, and Cerqueira JJ (2016). Absence of IFNgamma promotes hippocampal plasticity and enhances cognitive performance. Translational psychiatry 6, e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro S, Roque S, Marques F, Correia-Neves M, and Cerqueira JJ (2017). Brain interference: Revisiting the role of IFNgamma in the central nervous system. Progress in neurobiology 156, 149–163. [DOI] [PubMed] [Google Scholar]
- Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, and Llorens-Martin M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nature medicine 25, 554–560. [DOI] [PubMed] [Google Scholar]
- Mu Y, and Gage FH (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Heinemann U, and Berlin K (1997). Cholecystokinin activates CCKB-receptor-mediated Ca-signaling in hippocampal astrocytes. J Neurophysiol 78, 1997–2001. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlers SH, Flores MV, Hall CJ, Wang L, Ko DC, Crosier KE, and Crosier PS (2017). A whole animal chemical screen approach to identify modifiers of intestinal neutrophilic inflammation. The FEBS journal 284, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Namboodiri VM, Matan AM, Voets ES, Mohorn EP, Kosyk O, McHenry JA, Robinson JE, Resendez SL, Rossi MA, et al. (2017). Prefrontal cortex output circuits guide reward seeking through divergent cue encoding. Nature 543, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst M, Braganza O, Dannenberg H, Hu W, Pothmann L, Rosen J, Mody I, van Loo K, Deisseroth K, Becker AJ, et al. (2016). Astrocyte Intermediaries of Septal Cholinergic Modulation in the Hippocampus. Neuron 90, 853–865. [DOI] [PubMed] [Google Scholar]
- Pereira L, Medina R, Baena M, Planas AM, and Pozas E (2015). IFN gamma regulates proliferation and neuronal differentiation by STAT1 in adult SVZ niche. Frontiers in cellular neuroscience 9, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Johnson M, Ekonomou A, Perry RH, Ballard C, and Attems J (2012). Neurogenic abnormalities in Alzheimer’s disease differ between stages of neurogenesis and are partly related to cholinergic pathology. Neurobiol Dis 47, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, and Parri HR (2013). alpha7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Abeta effects in a preclinical Alzheimer’s mouse model. PloS one 8, e81828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagman A, Hoscheidt S, McLimans KE, Klinedinst B, Pappas C, Anantharam V, Kanthasamy A, Willette AA, and Alzheimer’s Disease Neuroimaging I (2019). Cholecystokinin and Alzheimer’s disease: a biomarker of metabolic function, neural integrity, and cognitive performance. Neurobiology of aging 76, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, and Silver RA (2018). NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Front Neuroinform 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchouk I, and Volterra A (2018). Gliotransmission: Beyond Black-and-White. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Saito K, Sano F, and Koizumi S (2019). Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. International journal of molecular sciences 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming GL, et al. (2015). Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell 17, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Martin-Suarez S, Valcarcel-Martin R, Pascual-Brazo J, Aelvoet SA, Abiega O, Deudero JJ, Brewster AL, Bernales I, Anderson AE, et al. (2015). Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 16, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi J, Baude A, Omori Y, Shimizu H, El Mestikawy S, Fukaya M, Shigemoto R, Watanabe M, and Somogyi P (2004). GABAergic basket cells expressing cholecystokinin contain vesicular glutamate transporter type 3 (VGLUT3) in their synaptic terminals in hippocampus and isocortex of the rat. Eur J Neurosci 19, 552–569. [DOI] [PubMed] [Google Scholar]
- Song J, Olsen RH, Sun J, Ming GL, and Song H (2016). Neuronal Circuitry Mechanisms Regulating Adult Mammalian Neurogenesis. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Sun J, Moss J, Wen Z, Sun GJ, Hsu D, Zhong C, Davoudi H, Christian KM, Toni N, et al. (2013). Parvalbumin interneurons mediate neuronal circuitry-neurogenesis coupling in the adult hippocampus. Nat Neurosci 16, 1728–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, et al. (2012). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, and Graham SH (1994). Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain research 664, 94–100. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AJ, Gasperini R, Foa L, and Small DH (2010). Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. Journal of Alzheimer’s disease : JAD 22, 699–714. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Fibuch EE, and Mao L (2007). Regulation of mitogen-activated protein kinases by glutamate receptors. Journal of neurochemistry 100, 1–11. [DOI] [PubMed] [Google Scholar]
- Whissell PD, Bang JY, Khan I, Xie YF, Parfitt GM, Grenon M, Plummer NW, Jensen P, Bonin RP, and Kim JC (2019). Selective Activation of Cholecystokinin-Expressing GABA (CCK-GABA) Neurons Enhances Memory and Cognition. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, and Nicoll RA (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31, 453–462. [DOI] [PubMed] [Google Scholar]
- Wu MD, Hein AM, Moravan MJ, Shaftel SS, Olschowka JA, and O’Banion MK (2012). Adult murine hippocampal neurogenesis is inhibited by sustained IL-1beta and not rescued by voluntary running. Brain, behavior, and immunity 26, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wang T, Sun GY, and Ding S (2010). Specific disruption of astrocytic Ca2+ signaling pathway in vivo by adeno-associated viral transduction. Neuroscience 170, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CY, Asrican B, Moss J, Quintanilla LJ, He T, Mao X, Casse F, Gebara E, Bao H, Lu W, et al. (2018). Mossy Cells Control Adult Neural Stem Cell Quiescence and Maintenance through a Dynamic Balance between Direct and Indirect Pathways. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, and Liu HT (2002). MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12, 9–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene Expression Table (All Genes). Related to Figure 7.
Table S3. DEGs (Full List). Related to Figure 7.
Table S5. Significantly Enriched GO Terms Analysis. Related to Figure 7.
Data Availability Statement
RNA-seq data reported in this paper has been submitted to Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE137052.