

Abstract
Selective inhibition of microRNAs (miRNAs) offers a new avenue for cancer therapeutics. While most of the current anti-miRNA (antimiR) reagents target full length miRNAs, here we investigate novel nanoparticle-delivered short PNA probes containing cationic domains targeting the seed region of the miRNA for effective antimiR therapy. For proof of concept, we tested PNAs targeting miRNA-155 and employed poly(lactic-co-glycolic acid) (PLGA)-based nanoparticle formulation for delivery. A comprehensive evaluation of PLGA nanoparticles (NPs) containing short PNA probes showed significantly superior loading, release profile, and uniform size distribution, compared to conventional non-cationic PNA probes. Confocal microscopy and flow cytometry analyses showed efficient transfection efficiency and uniform distribution of PLGA NPs containing short PNA probes in the cytoplasm. Functional analysis also confirmed efficient miRNA-155 inhibition including an effect on its downstream target proteins. Further, reduced tumor growth was observed after systemic delivery of PLGA nanoparticles containing short PNA probes in vivo in a xenograft mouse model following inhibition of miR-155. There was no evidence of acute or chronic toxicity associated with systemic delivery of PLGA NPs containing short PNA probes in the mice. Overall, in this paper we present a novel antimiR strategy based on PLGA nanoparticle delivered short PNA probes for potential cancer therapy.
Keywords: PNA, PLGA, AntimiR, miR-155, Nanoparticles
Graphical Abstract

INTRODUCTION
The RNA medicine field has made significant progress with the approval of: a) RNAi drugs; Onpattro (patisiran) [1] for treatment of polyneuropathy caused by hereditary ATTR amyloidosis and Givlaari (givosiran) [2] for porphyria, b) RNA-targeting antisense drug Spinraza (nusinersen) for treating spinal muscular atrophy [3]. More recently, the success of Milasen, an antisense drug to treat Batten’s disease is another promising example of RNA-based medicine [4]. Advances have been made in targeting messenger RNA (mRNA) for RNA-based therapies [5]. However, therapies related to microRNA (miRNA) targeting still need to be explored. MiRNAs are short (~22–25 nt) non-coding RNAs that control post-transcriptional gene expression of other RNAs, especially mRNA [6]. The 5’ seed region of miRNAs targets the 3’ untranslated region (UTR) of mRNA by homologous Watson Crick base pairing and hamper its activity by either mRNA degradation or translation inhibition [7]. It has been well established that single miRNAs regulate the network of multiple protein-coding genes and several physiological pathways, including cell survival, differentiation, and proliferation [8]. Therefore, an aberration of miRNA expression (upregulation or downregulation) can result in an uncontrolled cascade of cell signaling activity in the cells that can be oncogenic [9]. miRNAs that are overexpressed in tumors (also known as oncomiRs) play an important role in promoting tumor growth, angiogenesis, and metastasis [9]. Hence, inhibiting oncomiRs selectively using antimiR oligonucleotides have provided a new avenue for cancer therapy [10].
Several chemical modifications have been introduced in antimiR oligonucleotides to increase their binding affinity with cognate miRNA and to improve their enzymatic stability. 2-O-methyl oligonucleotides (e.g. antogomiRs) [11], morpholinos [12], locked nucleic acid (LNA) [13, 14], and peptide nucleic acids (PNAs) [15] represent important classes of synthetic nucleic acids developed to date to silence specific miRNAs via simple rules of Watson Crick (WC) base-pair recognition. Recently, some progress has been made using CRISPR/Cas9 based technology in targeting miRNAs [16].
Unlike other synthetic nucleic acids, PNAs are a unique class in which the phosphodiester backbone is substituted with a neutral-charge achiral N-(2-aminoethyl) glycine backbone. PNAs can bind single-strand DNA/RNA target sequences with high affinity as well as specificity, and are non-susceptible to enzymatic (proteases or nucleases) degradation [17], making PNAs ideal molecules for a myriad of biomedical applications like gene editing [18–23], genomic barcoding for pathogen detection [24], anti-infective agents [25] and miRNA silencing [26–29]. Mechanistically, PNAs bind to target miRNAs by WC base pairing and inhibit their function by sterically blocking miRNA-mRNA interaction [26].
Typically, miRNAs bind mRNAs via a 8mer long stretch of nucleotide sequence called the “seed region” [30]. The 5’ seed region of the miRNAs, are critical for stable Argonaute (AGO) binding and formation of the AGO-miRNA duplex that further activates the RNA induced silencing complex (RISC) [8]. Although the vast majority of reported antisense strategies rely on targeting full-length miRNAs [26, 31], not much progress has been made in using short antimiRs. Prior studies were primarily centered on the use of tiny 8mer LNA probes to target the seed region of miRNAs and regulate gene expression [32, 33]. Further, short PNA probes (9–14 mer) designed to target double-stranded RNA forming a PNA:RNA2 triplex [34–36] and selectively target the mRNA sequence of HOTAIR gene for cancer therapy have been developed [37]. Similarly, we have developed short PNA probes (3mer) to target the double-stranded CUG triplet repeat-containing hairpin RNAs in vitro [38]. Although the aforementioned short oligonucleotide probes can bind to target RNAs, few issues related to specificity, sequence selection, delivery and in vivo validation have not yet been completely resolved. In addition, short PNA-based probes have not been tested exclusively for targeting miRNAs.
Extensive translational applications of PNAs have been stymied by their limited intracellular delivery due to the neutral-charge backbone. A few prior studies noted that poly-arginine conjugated PNAs [39] and guanidinium modified gamma PNAs undergo uptake in mammalian cells [40, 41]. While these strategies show promise for lab-scale setups, important issues such as; 1) cytotoxicity associated with direct exposure to cationic guanidinium domains, 2) endosomal entrapment restricting PNA’s availability to the target site, and 3) challenges of large-scale production including high cost associated with optically pure gamma monomers synthesis [42], are yet to be resolved.
In this report, we developed and optimized a biocompatible nanoparticle delivery system for short PNA probes (also called anti-seed reagents) with superior binding affinity that can selectively bind to the seed region of miRNA and control gene expression both in vitro as well as in vivo. Short seed targeting antimiR PNAs possess numerous advantages over full length antimiRs. During solid-phase synthesis of full-length PNAs, the growing PNA chains fold or aggregate in the neighboring chains, resulting in poor HPLC purification, low yield, and truncated side chain impurities. In contrast, short PNAs can be efficiently prepared and are easily scalable as compared to full-length antimiR PNAs. Also, short PNAs possess superior physicochemical features such as non-aggregation and high hydrophilicity as compared to full length antimiRs. Hence, due to the aforementioned merits, therapeutically active short PNAs can be comparatively easier to translate into clinical applications.
Herein, we investigated short cationic PNA probes with superior efficacy for targeting the miR-155 seed region and inhibiting its activity. miR-155 is up-regulated in many sub-types of lymphoma (including diffuse large B-cell lymphoma [43]) and leukemia, breast, colon and lung cancers among others, and is the functional product of the B-cell integration cluster (Bic) oncogenic RNA [44, 45]. These studies show that miR-155 levels could be useful in both diagnosis as well as prognosis of cancer [46].
Diffuse large B-cell lymphoma (DLBCL) represents ~30–40% of all Non-Hodgkin Lymphoma (NHL) and accounts for >80% of the cases of aggressive lymphoma in the world. miRNA signatures show that miR-155 is significantly elevated in DLBCL [47]. The carcinogenic potential of miR-155 in DLBCL is related to its role in MYC-associated pathways contributing to the transformation of B-cells [48]. It has been demonstrated in vitro that high levels of miR-155 assist DLBCL cells to move from G to S1 phase and inhibit apoptosis [49]. Furthermore miR-155 also regulates the phosphatidylinositol-3 kinase (PI3K)-protein kinase B (AKT) signaling pathway inducing proliferation of DLBCL cells [50]. miR-155 negatively regulates the expression of transcription factors FOXO3A and BACH1, both of which exhibit tumor suppressing activities. FOXO3A belongs to the family of forkhead box O (FOXO) transcription factors which regulate the expression of pro-apoptotic factors by binding to the FOXO recognition elements (FRE) in DNA, hence activating their transcription [51]. The direct transcriptional activation of Bcl-2 family members, including Bim and bNIP3, increases mitochondrial permeability and induces apoptosis [52]. In addition, FRE is also present in the promoter region of pro-apoptotic gene FasL which induces mitochondrial independent apoptosis [53]. The BTB and CNC homology (BACH1) is another transcription factor which negatively regulates the expression of heme-oxygenase-1 (HO-1). HO-1 is implicated in oncogenesis and chemoresistance, and high levels of BACH1 can reduce the transcription of HO-1 gene mitigating its anti-apoptotic activity [54]. Therefore, inhibiting oncogenic miR-155 results in upregulation of transcription factors, FOXO3A and BACH1, which induce apoptosis of DLBCL cells presenting a viable therapeutic approach for treatment of lymphomas.
We developed short cationic PNA probes containing three arginine amino acids, designed to bind the miR-155 seed sequences with high binding affinity to regulate its gene expression. We synthesized a series of short cationic PNA probes and performed comprehensive biophysical characterization to examine their binding affinity as compared to the conventional PNAs. Further, we developed poly(lactic-co-glycolic acid) (PLGA) based nanoparticle (NP) formulations for optimal intracellular delivery of the short cationic PNA probes. PLGA has been shown to provide excellent transfection efficiency with minimal toxicity specifically for PNA delivery, and is among the most effective nano-carriers reported in literature [55, 56]. We encapsulated short cationic antimiR-155 PNAs in PLGA NPs and performed a series of physico-biochemical characterizations to determine optimum morphology, size distribution, surface charge, payload, and nucleic acid release profiles of NPs. Further, in vitro transfection efficiency and therapeutic efficacy of NPs were evaluated in a battery of cell culture studies. Cellular uptake of NPs was assessed by confocal and flow cytometry analyses and therapeutic efficacy was measured by performing RT-PCR and cell viability analysis in the lymphoma cell lines. To validate our results, gene expression as well as protein levels of the transcription regulators BACH1 and FOXO3A, the direct targets of miR-155 [27], were quantified. Additionally, the safety profile of formulated PLGA NPs containing short cationic antimiR-155 PNAs was determined in primary human embryonic kidney 293 (HEK293) cells in vitro as well as in vivo in immunocompetent mice. In a xenograft mouse model, PLGA NPs containing short PNAs were able to successfully target the tumor on systemic administration. The PLGA NPs treated group showed reduced tumor growth and miR-155 levels in comparison to the control group. Furthermore, the levels of direct downstream targets of miR-155, FOXO3A and BACH1, were upregulated in the PLGA NPs treated group resulting in tumor apoptosis. Overall, the results described in this study constitute an exciting and cost-effective strategy of miRNA inhibition at the interface of synthetic nucleic acid chemistry and nanotechnology for potential cancer therapy.
EXPERIMENTAL
Materials
PNAs were synthesized using Boc-protected monomers which were purchased from ASM Chemicals and Research (Germany). TAMRA (Boc-5-carboxytetramethylrhodamine) dye was purchased from VWR (Pennsylvania). Poly(lactic-co-glycolic acid) polymer used for nanoparticle formulation was bought from Lactel Absorbable Polymers (USA). Mini-PEG-3 was obtained from Peptides International (USA). miR-155 target oligonucleotides, both 8 mer and 23 mer were purchased from Midland Certified Reagent (USA). MTS reagent and cell membrane staining dye were bought from Promega (USA) and Biotium (USA), respectively. Lysotracker™ green DND-26 was purchased from Invitrogen (#L7526). Click-iT plus TUNEL assay (#C10617) purchased from Invitrogen was used for detecting apoptosis in tumor samples. Caspase3 antibody (#9662) was purchased from cell signaling technology. Following miR-155 targets were used during studies. miR-155 target (8mer): 5’ TTAATGCT 3’. miR-155 target (23mer): 5’ TTAATGCTAATCGTGATAGGGGT 3’.
Synthesis of PNA Oligomers
Based on previously reported protocols, solid phase synthesis was used for synthesizing the PNAs using 4-Methylbenzhydrylamine (MBHA) resin and Boc-protected monomers [57]. TAMRA was coupled to the N-terminus or 5’ end of the PNAs and Mini-PEG-3 was used as the linker. In order to cleave the PNAs from the MBHA resin, cleavage cocktail (m-cresol: thioanisole: trifluoromethanesulfonic acid: trifluoroacetic acid, 1:1:2:6) was used and PNA was precipitated using diethyl ether. High performance liquid chromatography (HPLC) was used for purifying the PNAs and molecular weight was confirmed using mass spectroscopy (matrix assisted laser desorption/ionization-time of flight, MALDI). The extinction coefficient of PNAs was calculated via combining the extinction coefficient of each monomer and concentration was measured using UV-Vis spectroscopy.
Gel shift assays
The binding affinity of PNAs with the target was determined as previously described [58]. PNAs were incubated with the target miR-155 (1 μM) at different ratios in buffer mimicking the physiological conditions at pH 7.4 and a temperature of 37°C in the thermal cycler (Bio-Rad) over a period of one hour or 16 hours. The gels were run on polyacrylamide gel at 120V for a duration of 35 minutes. SYBR gold (Invitrogen) was used for visualization of bound and unbound fractions of the target.
Thermal melting analysis
Incubation of PNAs with the target miR-155, both 8 mer and 23 mer, was done in physiological conditions at a 1:1 ratio and a concentration of 4 μM. Samples were subjected to thermal cycling with the first ramp from 95°C to 25°C, second ramp from 25°C to 95°C and absorbance was measured at 260 nm every 30 seconds in a UV-Vis spectrophotometer as reported previously [50, 51].
Nanoparticles formulation
Based on prior protocols, a double emulsion solvent evaporation technique was used for formulation of PLGA nanoparticles [58]. Dichloromethane (DCM) was used to dissolve the polymer and PNAs were used at 2 nanomole/mg of polymer for encapsulation. PNA dissolved in water was added to the DCM containing polymer and ultrasonicated to form the first emulsion (w/o), which was then added to the 1 ml of polyvinyl alcohol (5% w/v, aqueous) followed by ultrasonication to form the double emulsion (w/o/w). The final emulsion was added to 10 ml of 0.3% aqueous polyvinyl alcohol and stirred overnight. Nanoparticles were washed with cold water three times to remove excess PNA or polyvinyl alcohol. The final nanoparticle pellet was resuspended in 5% (w/v, aqueous) trehalose and lyophilized overnight using the benchtop freeze dryer (Labconco). Nanoparticles obtained were weighed and covered with parafilm, followed by storage at −20°C.
Characterization of NPs
Nanoparticle tracking analysis (NTA) was used to measure the hydrodynamic diameter using a NanoSight NS500 (Malvern Panalytical Inc., Westborough, MA, USA) equipped with 640 nm laser. NTA measurements were taken using water as a dispersant at 25°C. For each sample three measurements were recorded for 60 seconds and the standard deviation was calculated from three measurements of each sample. The surface charge of nanoparticles was determined by measuring the zeta potential using an electrophoresis technique (Malvern Panalytical Inc, USA). The measurements were recorded at 25°C and Smoluchowski approximation was selected in the software to obtain the final values.
Loading study
The loading of PNA in nanoparticles was determined by an organic solvent extraction technique according to the previous protocol [58]. Nanoparticles were dispersed in dichloromethane followed by shaking at 37°C for about 3 hours. PNAs released from the dissociated nanoparticles were extracted using equal volume of 1X tris/EDTA buffer by shaking at 37°C for 0.5 hour. The concentration of PNA was determined using Nanodrop One (ThermoFisher) in the buffer layer.
Release study
As described in a prior protocol, the release of PNA from nanoparticles was measured in phosphate buffer at pH 7.4 and 37°C [58]. The release of PNA3 NPs was also studied at acidic pH (6.5, 5.5, and 4.5). Nanoparticles were suspended in phosphate buffer and samples were collected at different time points after centrifugation (15000 rpm, 10 mins). Nanoparticles were resuspended in fresh buffer at each time point. The concentration of PNA in the PBS samples at different time points was determined using Nanodrop One (ThermoFisher).
Scanning Electron Microscopy (SEM) imaging of NPs
Nanoparticles were mounted on the carbon tape and sputter coating was done for 2 minutes. Imaging of nanoparticles was done using FEI NanoSEM 450 (2.0 kV). ImageJ software was used to process the images and obtain the size distribution.
Cell culture
HeLa (ATCC® CCL-2™), SUDHL-5 (ATCC® CRL-2958™), HEK-293 (ATCC® CRL-1573™), A549 (ATCC® CCL-185™) were obtained from ATCC (USA) and U2932 was purchased from Leibniz Institute (DSMZ, Germany). Petri-dishes (10 cm) were used for expanding the HeLa, A549, and HEK-293 cells in presence of eagle’s minimum essential medium (EMEM) which was supplemented with 10% fetal bovine serum (FBS) in the absence of antibiotics. Suspended cell lines including SUDHL-5 and U2932 were cultured in 75 cm2 flasks using RPMI-1640 medium with 10% FBS without any antibiotic.
Confocal microscopy
The uptake of nanoparticles in HeLa and A549 cells was studied according to the prior protocol [29]. HeLa or A549 cells (50,000 to 100,000) were seeded in 12 or 24 well plates containing a glass coverslip of 1 mm thickness. Cells were incubated with nanoparticle suspension for 2, 4, 8, 12, and 24 hours. To stain the cell membrane, HeLa cells were incubated with membrane staining dye in the incubator for 5 mins. Lysosomes were stained by incubating the HeLa cells in 200 nM of lysotracker at 37°C for 40 mins. Cells were washed thoroughly with PBS and fixed in 4% PFA at room temperature. After 10 mins, cells were washed with PBS and permeabilization was done using 0.1% triton at room temperature. Cells were then washed with PBS and the coverslip was mounted on a slide using prolong diamond antifade mounting media with DAPI (Life Technologies). Images were captured using a 60X oil lens on confocal microscope (Nikon A1R) at the same intensity.
Flow cytometry analysis
The uptake of nanoparticles in HeLa cells was quantified as reported previously [29]. HeLa cells (100,000 cells) were seeded in 12 well plates and treated with PNA8 and PNA8 PLGA NPs. After 24 hours, cells were washed and trypsinized (Gibco, Life Technologies). Trypsinized cells were centrifuged followed by washing with PBS (2X). The final cell pellet was then suspended in 4% PFA and analyzed using LSR Fortessa X-20 Cell Analyzer (BD Bioscience). The data was processed in FlowJo analysis software.
RT-PCR studies
The cells (SUDHL-5 or U2932) were treated with nanoparticles and a cell pellet was collected after 48 hours of treatment. The levels of miR-155 were quantified using the previously described protocol [29, 58]. The RNeasy Mini Kit (Qiagen) was used for extraction of total RNA from the cell pellets. The cDNA was synthesized using RT primers for miR-155 (assay ID 467534_mat) and U6 (assay ID: 001973), 100 mM dNTPs, RT buffer, RNase inhibitor in T100 thermal cycler (Bio-Rad) under the conditions specified in the assay. The cDNA of miR-155 and U6 was then amplified using respective primers and universal master mix II, with UNG at conditions specified in the assay and detected using the CFX Connect Real-Time PCR detection system (Bio-Rad). For measuring the levels of FOXO3A and BACH1 mRNA, a high capacity cDNA reverse transcription kit was used and cDNA was amplified under conditions specified in the assay (Hs00818121_m1: FOXO3A, Hs00230917_m1: BACH1). The quantification values obtained were processed via the 2−ΔΔCt method to calculate the fold change in target gene levels.
MTT assay
HEK-293 and HeLa cells were incubated with different doses of nanoparticles in 96 well plate for 24, 48, and 72 hours. After washing with PBS (2X), cells were then incubated in fresh media containing MTS reagent (CellTiter, Promega) for an hour in the incubator (37°C, 5% CO2). After one hour, the absorbance was read at 490nm on iMark plate reader (Bio-Rad). The viability of cells was measured by calculating the fold change in absorbance of treated cells in comparison to the control.
Trypan blue assay
The cells (SUDHL-5 or U2932) were treated with nanoparticles in 96 well plate. After 48 hours, trypan blue dye was used to stain the dead cells and % cell viability was measured using the cell counter (Bio-Rad, USA).
Clonogenic assay
As previously described, HeLa cells were seeded in 24 well plate and treated with nanoparticles for 24 hours [29, 58]. After trypsinization, cells were counted using a cell counter (Bio-Rad, USA). Based on the cell count, equal number of treated as well as untreated cells were then passaged in 6 well plates (n=6 wells/sample). The cells were allowed to expand until the untreated cells showed colonies with more than 50 cells under the inverted microscope. The colonies were fixed using acetic acid: methanol (1:7 v/v) at room temperature and washed with PBS (2X). The colonies were then stained using 0.5% w/v crystal violet and washed under tap water after 2 hours. The colonies were allowed to dry at room temperature before counting.
Western blot analysis
SUDHL-5 cells were collected after 48 hours of treatment with 2.5 nmol PNA equivalent NPs dose. Cells were lysed using 1X RIPA buffer (Cell Signaling Technology) with protease inhibitor cocktail (Thermo Scientific) on ice for 30 min. The whole cell lysates were then clarified to remove cellular debris by centrifugation at 10,000 rpm for 10 min at 4°C. Protein concentration was measured using DC protein assay (#5000112, Bio-Rad). Equal amount of proteins (30 μg) were separated using SDS/PAGE 4–20% MP TGX Stain-Free gels (Bio-Rad) and transferred to PVDF membrane (Bio-Rad). After the transfer of proteins, blots were blocked using 5% milk in 1X tris-buffered saline (TBS) for one hour at RT. BACH1 protein was probed using mouse monoclonal primary antibody (sc-271211, Santa Cruz Biotechnology) at 1:200 dilution. FOXO3A protein was probed by mouse monoclonal antibody (66428–1-Ig, Proteintech) at 1:100 dilution. GAPDH was used as control and was detected using mouse monoclonal GAPDH antibody (sc-47724, Santa Cruz Biotechnology) at 1:2000 dilution. The desired bands were detected using Mouse m-IgGκ BP-HRP (sc-516102, Santa Cruz Biotechnology) secondary antibody (1:3000) and immobilon western chemiluminescent HRP substrate (MilliporeSigma). Intensity of bands was measured using ImageJ 1.52a software (National Institute of Health, Bethesda, MD) and normalized against GAPDH.
Transfection of hsa-miR-155–5p inhibitor
SUDHL-5 cells were transfected with hsa-miR-155–5p inhibitor (#4464084, mirVana® miRNA inhibitors, Thermo Fisher Scientific) at a dose of 50 picomoles using reverse transfection technique. hsa-miR-155–5p inhibitor was incubated with Lipofectamine RNAiMAX Transfection Reagent (#13778100, Invitrogen) at room temperature for 20 minutes in Opti-MEM™ reduced serum medium (#31985062, Gibco). Cell suspension was then added to the medium and incubated at 37°C and 5% CO2 for 48 hours. Cells were collected by centrifugation at 1000 rpm for 4 min at 4°C, followed by RNA extraction and RT-PCR to measure miR-155 levels.
In vivo studies
Female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, strain 005557) were purchased from Jackson labs. Mice were maintained at Beth Israel Deaconess Medical Center (BIDMC) animal facility in accordance with the institutional animal care and use committee (IACUC) rules and guidelines. 1 × 107 U2932 cells were injected subcutaneously on the right flank of 5–6 weeks old mice. When the tumor volume reached about 100–200 mm3 after 9–12 days, mice were divided randomly into four treatment groups (n≥5) and each group was treated with phosphate buffered saline (PBS), PNA3, PNA3 NPs, and PNA5 NPs respectively. NPs were suspended and sonicated in PBS and administered by tail vein injection. Three doses were administered over the course of 16 days. Tumor volume/size was measured with caliper every 48 hours until the tumor volume reached 2000 mm3. We did not observe any toxic effects or weight loss in the experimental group when compared to the PBS-treated group of mice. The tumor, liver and kidney harvested from these mice were stained with H&E. The tumor samples from control and PNA3 NPs treated group were also stained for Ki67, a proliferation marker and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and caspase3 for detecting apoptosis.
For biodistribution studies, NPs containing PNA-TAMRA (dye) were administered by tail vein injection when the tumor volume reached about 100–200 mm3. Mice were sacrificed by CO2 inhalation after 4 hours, 48 hours and 72 hours of injection. Tissue samples were collected and prepared for histology. Tissue samples were fixed in OCT embedding media and sectioning was done by the Histology Core of BIDMC (Boston, MA). Tissue sections (5–10 μm of thickness) were stained with DAPI and slides were imaged using a 60x oil lens on a Nikon A1 confocal microscope.
In vivo toxicity analysis was performed with male C57BL/6J (#000664) mice of age 4–6 weeks purchased from Jackson labs. Mice were randomized into four groups (Acute toxicity: Control and PNA3 NP treated, Chronic toxicity: Control and PNA3 NP treated, n=5/group). NPs were systemically administered after resuspending in 100 μl PBS via retro-orbital injection. Control group mice were injected with 100 μl PBS. These mice were sacrificed after 8 hours of treatment for acute toxicity analysis, and 48 hours post-injection for chronic toxicity study. Plasma was collected and separated from blood by centrifugation at 5000 rpm for 10 min at 4°C.
Blood chemistry analyses inlcuded creatinin, blood urea nitrogen (BUN), alkaline phosphatase, lactate dehydrogenase (LDH), alanine aminotransferase (ALT), aspartate aminotransferase (AST) and was performed by Antech diagnostics (Irvine, CA). Organs including spleen, lung, liver, heart and kidney were weighed and fixed in 10% neutral buffered formalin solution (MilliporeSigma) for more than 48 hours. Paraffin embedding of tissues followed by sectioning and staining with H&E for routine histopathology was performed.
Cytokine array
Plasma samples collected from C57BL/6J mice were analyzed to measure pro-inflammatory cytokine levels (IL-10, IL-6, IL-4, IL-2, IL-1β, IFNγ) using the R&D mouse pre-mixed kit (ThermoFisher Scientific). Cytokine array was performed using Luminex 200 at Clinical Research Center, UConn Health Farmington, CT.
RESULTS
Rational design and synthesis of antimiR-PNA oligomers
A series of PNA oligomers were synthesized as shown in Figure 1. PNA1–3 and PNA6–8 are short length PNAs (8mer) designed to bind the seed sequence of miR-155. Seed sequences are the functional region of the miRNA that interacts with the target mRNA, subsequently activating RISC and inducing mRNA degradation. PNA1 consists of regular PNA units. PNA2 and PNA3 consist of regular PNA units conjugated with cationic residues, three lysine and arginine amino acids at N-terminus, respectively. Cationic residues were selected to increase the binding affinity of short PNAs with the target miR-155. In addition to WC base pairing between seed sequence of miR-155 and short PNA nucleobases, cationic residues on the 5’ or N termini of the short PNAs are expected to interact electrostatically with the negatively charged backbone of the non-seed region of miR-155 resulting in increased binding of miRNA-PNA hetero-duplex. We selected only three cationic residues (arginine or lysine) as excessive positive charge often leads to cytotoxicity [40, 41, 59]. Full length PNA5 (with arginine) targeting miR-155 was also tested to compare its efficacy to the short length PNA3. To examine the cellular uptake properties, a series of fluorescent tetramethylrhodamine (TAMRA) dye labeled PNAs (PNA6–11) were synthesized. To decipher the role of arginine, both in NPs formulation and in cellular uptake studies, TAMRA conjugated control PNA6 and PNA10 (containing no arginine residues) were also tested. Scrambled PNAs; PNA4 and its TAMRA conjugated form (PNA9), were also synthesized. Full length TAMRA containing PNA11 (with arginine) was tested to compare its cytoplasmic delivery with short length PNA8. PNA oligomers without cationic residues result in sticky crude PNA that is difficult to purify and characterize. Hence, addition of cationic residues (lysine or arginine) at the C-terminus was performed on all the designed PNAs [60]. All PNA oligomer sequences were synthesized, cleaved and precipitated using Boc-based synthetic protocols [57, 61]. PNAs were purified by RP-HPLC using water/acetonitrile based gradient solvent system (Figure S1, S2, and S3) and characterized by MALDI-TOF analysis (see the supporting information, Table 1).
Figure 1.
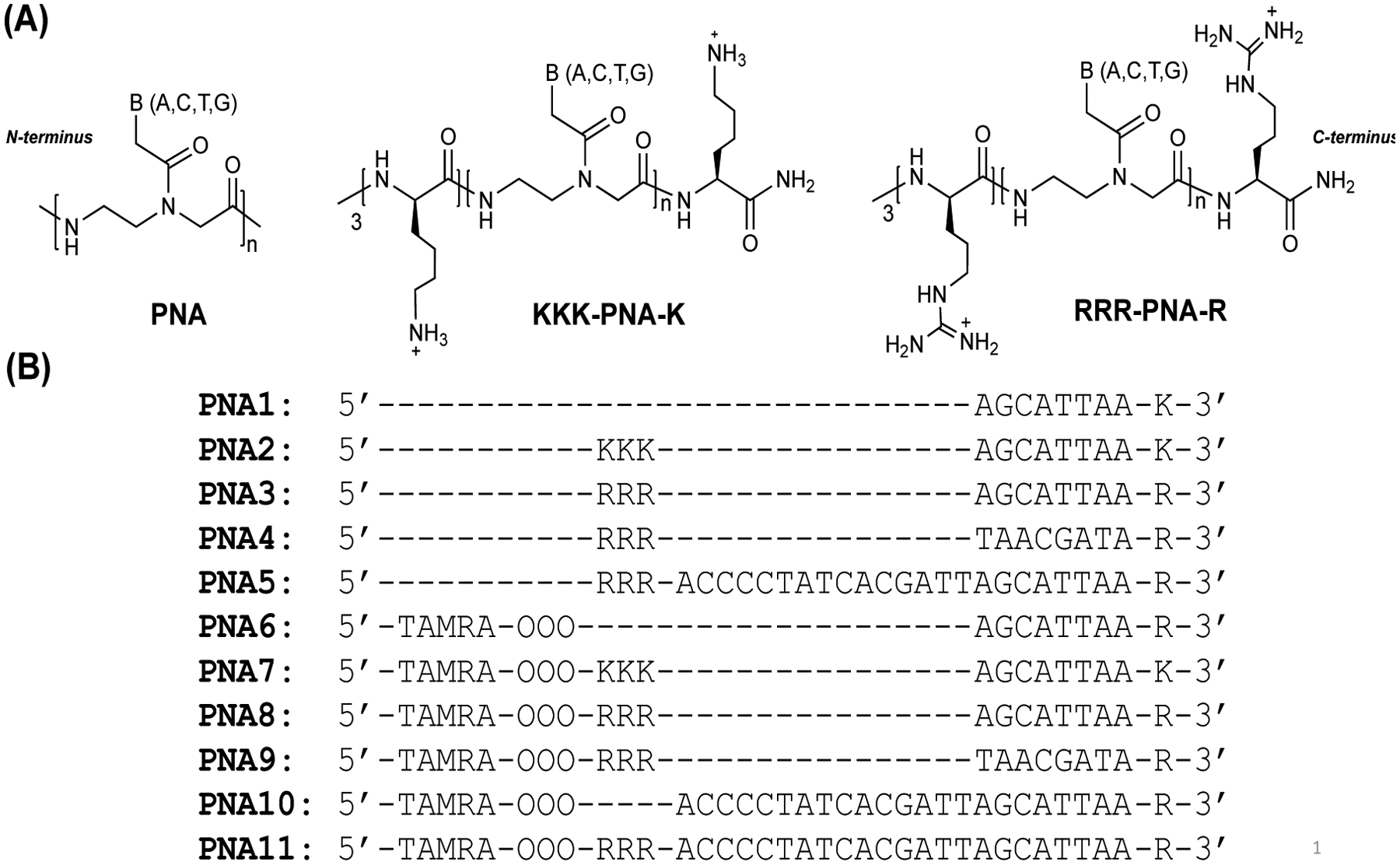
(A) Chemical structure of regular PNA, lysine conjugated PNA (KKK-PNA-K) and arginine conjugated PNA (RRR-PNA-R). (B) The PNA sequences used in this study to bind to miR-155 target are shown. PNA1-PNA3 are designed to bind seed region and PNA5 is designed to bind full length site of miR-155. PNA4 and PNA9 is a scrambled version of PNA3 and PNA8 with the same base composition, respectively. PNAs have either three lysine (K) or arginine (R) residues conjugated to N-terminus and one lysine or arginine (R) appended to C-terminus. PNAs6–11 are conjugated with 5-Carboxytetramethylrhodamine (TAMRA) dye for imaging purpose. OOO represents 8-amino-2,6,10-trioxaoctanoic acid residues (Mini-PEG-3). This is used to form flexible linker connecting the TAMRA and WC binding regions of the PNAs.
Binding affinity of short PNAs with a synthetic miR-155 target
Gel shift-based assays were used to measure the binding affinity of short PNAs. Synthetic miR-155 oligonucleotides (miR-155 target) were incubated with different concentrations of PNA1, PNA2, PNA3, and PNA4 in simulated physiological buffer conditions for 1 hour and 16 hours at 37°C (Figure 2 and S5). Following this, gel shift assays were performed and the bands visualized by SYBR gold staining. As expected, we did not observe any binding with PNA1 even at PNA/miR-155 ratio of 2:1 (Figure 2, lane 8 for PNA1). However, in the case of PNA2 and PNA3, we noted the appearance of a higher molecular weight band (Figure 2, lane 8 for PNA2 and PNA3).
Figure 2.
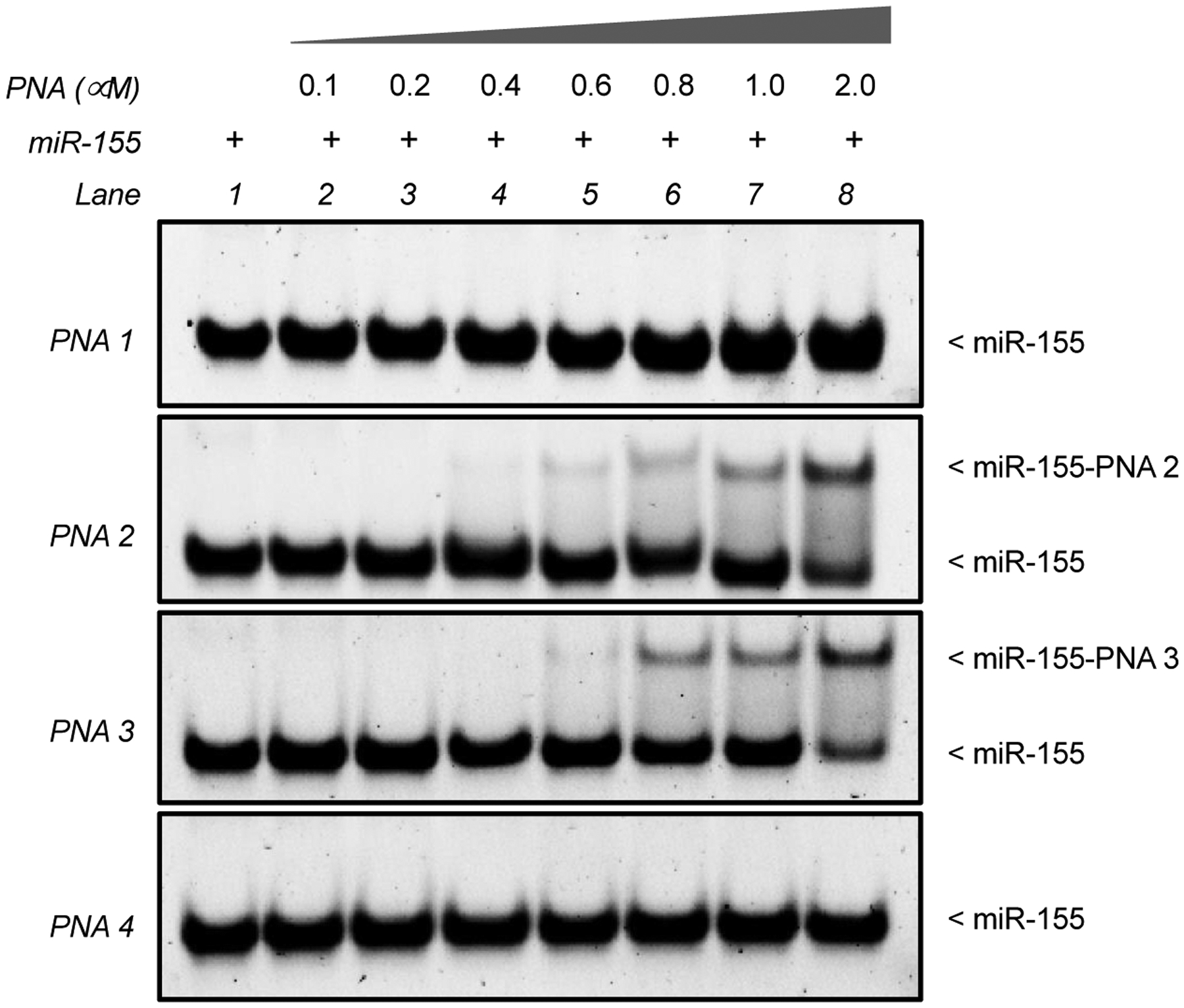
A dose dependent gel-shift assay of miR-155 target (1 μM) and indicated PNAs after 1 hour of incubation. Incubations were performed in simulated physiological buffer conditions (10 mM NaPi, 150 mM KCl and 2 mM MgCl2) for 1 hour at 37°C followed by non-denaturing PAGE separation and SYBR Gold staining.
We measured the bound and unbound fraction for all PNAs (Figure S4) and noticed that PNA-miR-155 complex was most efficiently formed for PNA2 and PNA3. These results were consistent with our expectations, since PNA2 and PNA3 are predicted to form a more thermodynamically stable complex due to stronger ionic interaction of cationic domains with the negatively charged backbone of miR-155 in addition to Watson-Crick base pairing. Furthermore, we noted that PNA3 was ~50% bound at 2 uM, while <40% of PNA2 was in the bound form, indicating superior binding affinity of PNA3 than PNA2 towards the target. The most plausible explanation of the finding mentioned above is that PNA3 possesses a high net positive charge due to the presence of a guanidinium group leading to its superior binding affinity as compared to lysine containing PNA2. As expected, we did not notice any binding for scrambled PNA4. Similarly, in comparison to 1 hour, we noticed that ~90% of miR-155 target was bound to the PNA2 and PNA3 after 16 hours of incubation (Figure S5). One plausible explanation of this observation could be the difference in kinetics of binding of short antimiR probes with the miR-155 target.
Thermal melting analysis
To gain insight into the binding affinity, UV-thermal melting experiments were performed. To decipher the interaction of cationic domains of PNAs with overhanging negatively charged non-seed region of miR-155, complementary full length (23mer) and short length (8mer) miR-155 targets were used. Samples containing stoichiometric amount of PNA and miR-155 (4 uM of each) were prepared in simulated physiological buffer and annealed. Their UV absorption at 260nm was recorded as a function of temperature. Our thermal melting results with full length (23mer) miR-155 target indicated significant increase in stabilization for PNA2-miRNA-155 and PNA3-miRNA-155 duplexes with thermal denaturation temperature (Tm); ~54°C and ~61°C respectively as compared to regular PNA1-miR-155 complex (Tm = ~40°C) (Figure S6). However, we did not notice an increase in Tm for short miR-155 target (8mer) duplexes. The higher Tm was observed for PNA3 in comparison to PNA2 in PNA-miR-155 complex and can be attributed to the presence of arginine domains in PNA3 which have more positive charge in comparison to lysine amino acid residues in PNA2 [62, 63]. These results suggest that the increase in the binding affinity of PNA3 with full length target (23mer) is due to complementary WC base pairing and electrostatic interactions of the cationic domain with the negatively charged phosphate groups in the non-seed region. No melting transition was noticed for scrambled PNA4-miR-155 duplex (Tm = <30°C).
Size and surface charge determination of PNA loaded PLGA NPs
For cellular delivery, PNA oligomers were encapsulated into PLGA NPs. Prior studies have established that PLGA NPs (containing 50:50 ratio of poly-lactic acid and poly-glycolic acid) successfully deliver PNAs both ex vivo as well as in vivo [31, 64]. PLGA NPs were formulated using an established protocol of double emulsion solvent evaporation technique [64]. Next, we determined the size distribution and morphology of formulated PLGA NPs using SEM. Formulated NPs demonstrated uniform size (~145nm) and morphology (Figure 3A and Figure S7).
Figure 3.
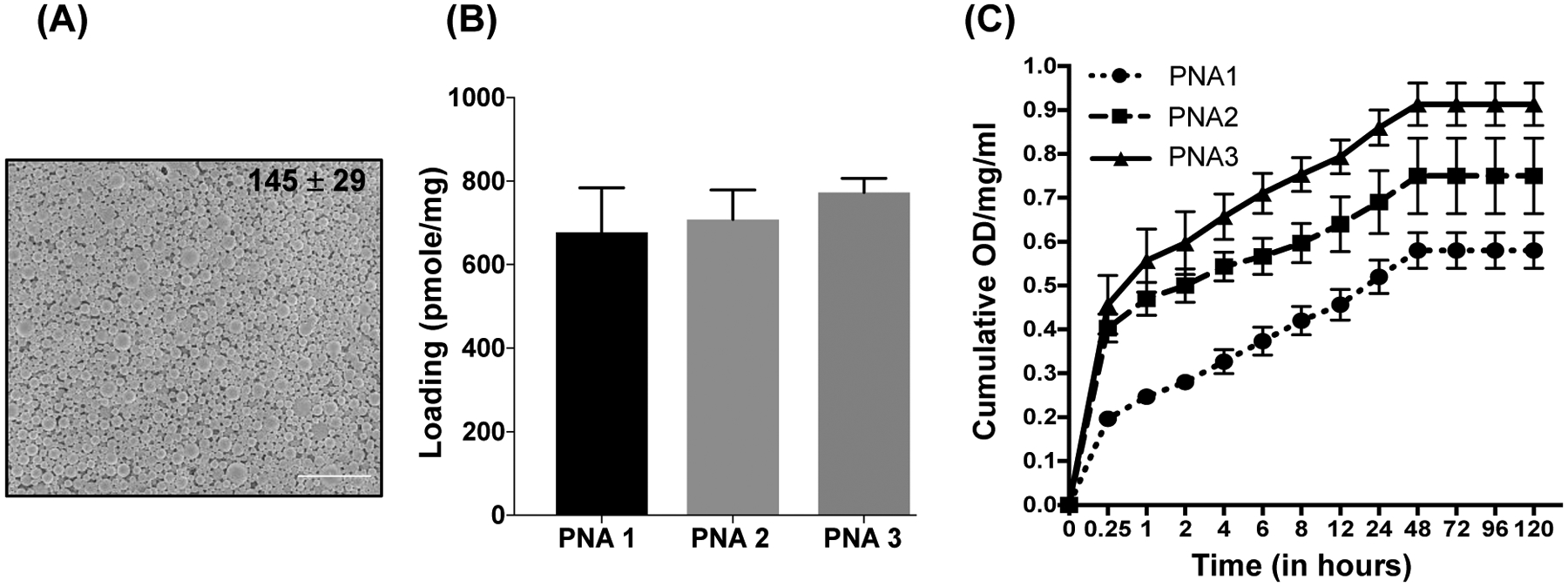
(A) Representative SEM image of PLGA NPs containing PNA3. The image was captured at 10,000X magnification. The scale bar is 4 μm. The average particle diameter (nm) and standard deviation are given for NPs. (B) PNAs loading analysis results. (C) Cumulative release profile data of PNA1, PNA2 and PNA3 from PLGA NPs at a given time point (shown on the X axis). N=3, data are shown as mean ± standard error mean (SEM).
We also measured the hydrodynamic diameter via nanoparticle tracking analysis (NTA). Formulated PLGA NPs showed hydrodynamic diameter between 130–235nm (Table 1). Further, surface charge on the PLGA NPs was determined by measuring the zeta potential. All PLGA NPs contained a negative zeta potential (Table 1) of −18 to −26 mV.
Table 1:
Characterization of PLGA NPs containing PNAs for mode diameter (Nanoparticle Tracking Analysis) and surface charge (zeta potential).
| PLGA polymer (50:50) | Zeta potential (mV) ± SD | Mode Size (nm) ± SD |
|---|---|---|
| Blank NPs | −27.7 ± 2.4 | 250 ± 09 |
| PNA1 | −18.3 ± 1.0 | 207 ± 11 |
| PNA2 | −22.3 ± 1.1 | 173 ± 23 |
| PNA3 | −19.6 ± 0.5 | 211 ± 04 |
| PNA4 | −20.2 ± 1.6 | 196 ± 06 |
| PNA5 | −16.7 ± 0.5 | 229 ± 35 |
| PNA6 | −25.7 ± 1.6 | 235 ± 20 |
| PNA7 | −20.0 ± 2.9 | 129 ± 40 |
| PNA8 | −25.1 ± 1.6 | 165 ± 02 |
| PNA9 | −19.4 ± 1.2 | 209 ± 12 |
| PNA10 | −23.7 ± 4.0 | 208 ± 02 |
| PNA11 | −18.7 ± 4.9 | 213 ± 14 |
Loading and release profile analysis of PNA from NPs
The loading of PNAs in PLGA NPs was measured using an organic solvent extraction method and absorbance was determined at 260nm. All the NPs exhibited uniform loading between 680–810 pmole/mg of PLGA (Figure 3B). In addition, we noticed higher loading for PNA3 (810 pmole/mg) as compared to PNA1 (640 pmole/mg) and PNA2 (690 pmole/mg). This higher loading of PNA3 likely reflected the ionic interactions of its arginine residues with negatively charged PLGA polymer. Release kinetics of PNAs from NPs was determined at physiological temperature by incubating the PLGA NPs in PBS for a defined period of time and the absorbance of released nucleic acids was measured at 260nm. All the NPs showed a slow release of PNAs (Figure 3C and S8A) within 48 hours. We noted increased PNA2 and PNA3 release possibly due to the higher loading into PLGA NPs and faster hydration of cationic PNAs in comparison to regular PNA1 which is relatively hydrophobic.
To determine the effect of pH on the release of PNA3 from NPs, we performed absorbance-based release profile studies in multiple acidic pH conditions (6.5, 5.5, and 4.5) (Figure S8B). We noticed a burst release at the initial time points in the acidic pH followed by a slow-release until 48 hours, which could be due to the acid-mediated degradation of PLGA polymer [65]. Overall, our comprehensive NP characterization established that PLGA NPs containing PNA3 are superior in loading as well as release in comparison to the rest of the formulations. Next, we confirmed the integrity of PNAs1–4 after in vitro release from PLGA NPs (Figure 4 and S9) by gel shift binding studies with the target miR-155. As expected, the released antimiR PNA2 (Figure S9C) and PNA3 (Figure 4B) were bound to miR-155 target. We did not observe binding with control PNA1 (Figure S9B) and scrambled PNA4 (Figure 4B) released from PLGA NPs, as expected. Together these findings imply that PLGA NPs containing PNA3 show superior binding as well as physico-biochemical properties. Hence, cell culture based functional assays and in vivo studies were centered on PLGA NPs containing PNA3 and its TAMRA derivative (PNA8).
Figure 4.
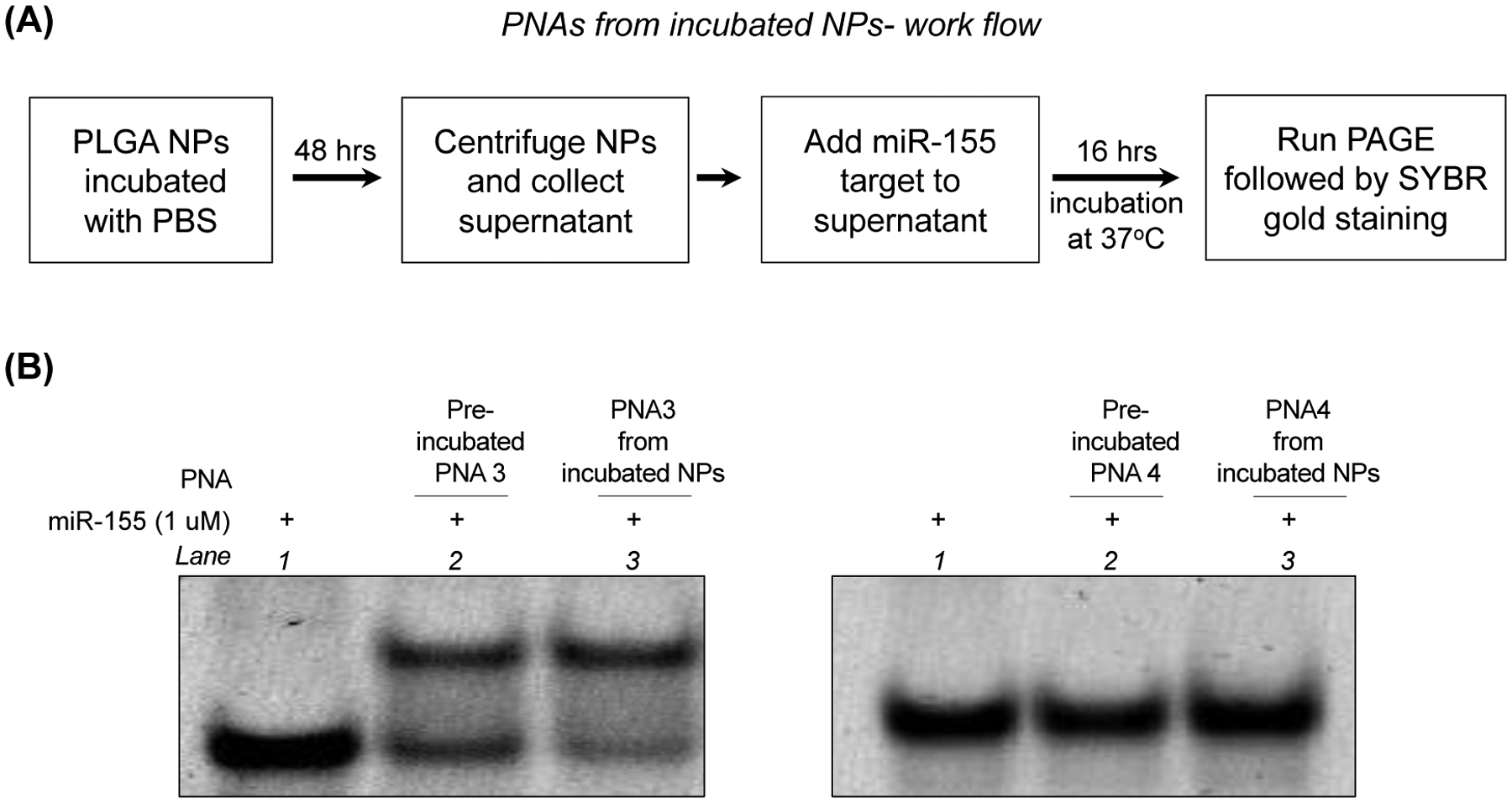
(A) Schematic showing the workflow to evaluate the release and in vitro binding affinity of PNAs with target miR-155 (B) PAGE gel-shift assay following incubation of miR-155 target with PNA3 (perfect match), PNA4 (scramble) and PNA3, PNA4 released from PLGA NPs in simulated physiological salt conditions at 1:1 ratio. Gels were stained using SYBR gold.
Cellular uptake studies
Prior to testing a miR-155 knockdown strategy, we studied the cellular uptake of PLGA NPs containing PNA8 in the cultured cells. HeLa cells cultured in logarithmic phase were incubated separately with control PNA8 and PLGA NPs containing PNA8. To compare transfection efficiency, equimolar quantities of PNA8 were used based on their loading in PLGA NPs. After incubation with HeLa cells for 24 hrs, nuclear (DAPI) and membrane staining (CellBrite) were performed, and cells were imaged by confocal microscopy. Uniform cellular uptake of PLGA NPs containing PNA8 was noticed, as indicated by TAMRA fluorescence intensity, while no uptake was detected for PNA8 alone at the indicated concentration (Figure 5). In order to confirm the distribution of PNA8 NPs, we also evaluated the uptake at multiple time points in HeLa and A549 cell lines (Figure S10). We observed high cellular uptake of PNA8 NPs in both the cell lines starting from 2 hours of incubation. Further, confocal imaging (Figure 6A) and flow cytometry results (Figure 6B) confirmed the dose dependent cytosolic delivery of PLGA NPs containing PNA8 in HeLa cells.
Figure 5.
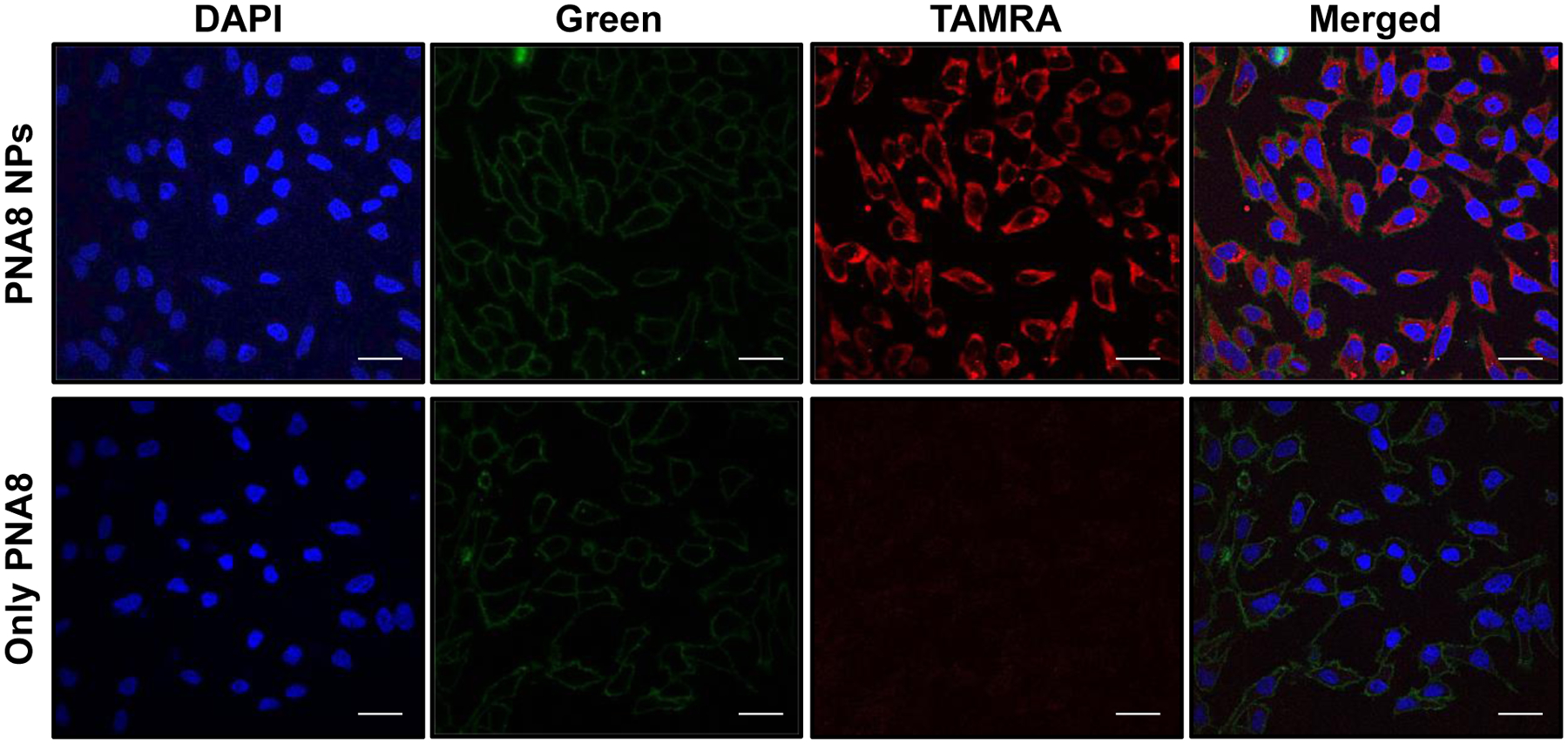
Cellular uptake studies. Confocal images of HeLa cells after 24 hours of incubation with PLGA NPs containing PNA8. Further, DAPI was used for staining the nucleus and cell membrane was stained using MemBrite™ dye. A representative image from at least three fields is shown. Please note the concentration of only PNA8 is the same as that encapsulated in PLGA NPs (1.4 nmole of PNA equivalent dose) for comparative analysis. Blue: DAPI (nucleus), Green: cell membrane, Red: TAMRA PNA. The images were equally enhanced for clarity using ImageJ. The scale bar is 30 μm.
Figure 6.

(A) Dose dependent confocal images of HeLa cells after 24 hours of incubation with PNA8 encapsulated in PLGA NPs and only PNA8. DAPI was used for nuclear staining. Please note the concentration of only PNA8 is provided based on loading of PNAs in PLGA nanoparticles. Red: PNA oligomers (TAMRA), Blue: DAPI (Nucleus). The scale bar is 10 μm. (B) Histogram analysis of dose dependent cellular uptake of PLGA NPs containing PNA8 in HeLa cells.
Prior studies have shown that poly-arginine PNAs as well as guanidinium modified gamma PNAs show puncta of fluorescent signal indicating co-localization of PNAs that could be due to endosomal entrapment or non-specific interaction with other organelles [39, 59]. Here, we noted that PNA8 delivered by PLGA NPs was distributed uniformly in the cytosol with relatively few puncta of TAMRA present after 24 hrs of incubation. Further, we also examined the cytosolic delivery of PLGA NPs containing other set of short PNAs; PNA6, PNA7, and PNA9, respectively (Figure S11). We did not notice uniform distribution of TAMRA for PNA6, instead, puncta of TAMRA signal were noted. In contrast, PLGA NPs containing PNA7 and PNA9 showed significant cytosolic distribution consistent with NPs containing PNA8 results.
Additionally, to compare the cellular uptake of short vs full length arginine PNAs, we performed uptake studies with PLGA NPs containing full length PNA10 (without arginine on 5’ termini) and PNA11 (with arginine residues), respectively. We observed a similar puncta pattern of TAMRA signal for PNA10 as that of PNA6 (Figure S11). We further stained the lysosomes in HeLa cells incubated with PLGA NPs containing PNA8, PNA10, and PNA11 to determine the localization (Figure S12). We observed that the majority of PNA10 was accumulated in the lysosomes, however PNA8 and PNA11 (containing arginine residues) showed both lysosomal accumulation and cytoplasmic distribution. All these findings reflect that irrespective of their length, arginine residues significantly improve the overall cytosolic distribution of PNAs delivered by PLGA NPs in the cells.
Quantification of antimiR activity in cell culture
To investigate whether short PNAs can inhibit miR-155 expression in cultured cells, SUDHL-5 cells, a lymphoma cell line, were treated with PLGA NPs containing PNA1, PNA2, PNA3, and scrambled PNA4 respectively. The PLGA NPs dose was adjusted so that each group was treated with equimolar concentration of PNA. After 48 hours of treatment, miR-155 levels were quantified by RT-PCR.
The RT-PCR results showed a ~50% and ~51% reduced miR-155 expression in the cells treated with PLGA NPs containing PNA1 and PNA2, respectively. Whereas PLGA NPs containing PNA3 treated cells showed a ~72% reduction in miR-155 expression (Figure 7A). These results substantiate our aforementioned findings indicating that PNA3 possess superior efficacy as compared to PNA1 and PNA2 with a potency comparable to commercially available antimiR-155 (mirVana® miRNA inhibitors). In contrast, as expected, we did not notice any decrease in miR-155 expression in the SUDHL-5 cells treated with PLGA NPs containing scrambled PNA4.
Figure 7.

(A) miR-155 levels in SUDHL-5 cells after treatment with PLGA NPs containing antimiR-155 PNAs at the equivalent dose of 2.5 nmol relative to normalized average control U6 (n=3, *p<0.05). AntimiR-155 (mirVana miR-155 inhibitor) transfection was used as a positive control. (B) Dose dependent effect on SUDHL-5 cells treated with NPs containing indicated antimiR PNAs. Cell viability measured using trypan blue dye (n=3, data represented as mean ± standard error mean (SEM)). For statistical analysis student t-test was used.
To test whether miR-155 inhibition could also affect its downstream targets in SUDHL-5 cells, we measured the levels of FOXO3A and BACH1, predicted targets of miR-155, by RT-PCR analysis as well as western blotting. miR-155 upregulation decreases FOXO3A and BACH1 levels [27]. Using RT-PCR, we found ~40% increase in BACH1 and ~20% increase in FOXO3A gene expression after treatment with PLGA NPs containing PNA3 (Figure S13A). We then performed western blot analysis, and consistent with our gene expression results, we noticed about ~30% increase in BACH1 and ~21% increase in FOXO3A protein levels (Figure S13B). We also noticed that the miR-155 inhibition by PLGA NPs containing PNA3 leads to reduced SUDHL-5 viability in a dose dependent manner as compared to cells treated with PLGA NPs containing scrambled PNA4 (Figure 7B). Next, we evaluated the miR-155 inhibition activity of PLGA NPs containing short length PNA3 as compared to PLGA NPs containing full length PNA5 in treated SUDHL-5 cells.
The cells treated with an equimolar concentration of PNAs in PLGA NPs resulted in ~75% decrease in miR-155 level with PNA5 and ~60% decrease in cells treated with short PNA3 (Figure S14A). We also assessed the levels of miR-155 downstream targets (FOXO3A and BACH1) in the cells treated with PNA5 and PNA3 containing NPs (Figure S14B). Collectively, our results indicated that short cationic PNA3 is relatively comparable in targeting miR-155 vis-à-vis to full-length PNAs.
Similarly, in addition to PNA equivalent dose effect as mentioned above, we also determined miR-155 expression at equivalent doses of PLGA NPs containing antimiR-155 PNAs instead of PNA equivalent dose. Consistent with our prior findings, RT-PCR results showed an ~80% decrease in miR-155 levels in the cells treated with PNA3-containing PLGA NPs (Figure S15A). Consistent with our miR-155 gene expression results, we also observed a 2-fold increase in FOXO3A mRNA levels and 3-fold increase in BACH1 mRNA levels in SUDHL-5 cells treated with PLGA NPs containing PNA3 (Figure S15B).
Further, we confirmed our cell viability results by clonogenic assay. A clonogenic assay was performed on adherent HeLa cells instead of non-adherent SUDHL-5 cells. The cells were treated with PNA3 and PNA4 NPs. After 7 days, colonies were stained with crystal violet and counted for further analysis [66]. We noted that PNA3 NPs treated HeLa cells showed about ~50% reduction in colonies in comparison to the control group and cells treated with PNA4 NPs (Figure S16).
Further, an MTT assay was performed on embryonic kidney cells (HEK293 cells) which are commonly used for cytotoxicity analysis [67, 68], after 24 hrs, 48 hrs, and 72 hrs of treatment with PLGA NPs containing PNA3 at different doses to assess the in vitro safety. As expected PNA3 NPs treated HEK293 cells showed no signs of cytotoxicity at stated dose or time point (Figure S17A). Whereas, in HeLa cells that show overexpression of miR-155, we noted a dose-dependent decrease in cell viability (Figure S17B).
Systemic administration of PLGA NPs cause tumor growth inhibition
In prior work, it has been established that U2932 (another lymphoma cell line) cells reproducibly generate ectopic xenograft mouse models and easily induce tumors in mice [69]. Henceforth, we chose U2932 cells for establishing xenograft tumor mice model for in vivo studies. We analyzed miR-155 expression levels in U2932 cells and noticed >4-fold upregulation of miR-155 in U2932 as compared to SUDHL-5 cells (Figure S18A). Further, prior to in vivo studies, we observed that U2932 cells treated with PLGA NPs containing PNA3 results in ~50% reduction in miR-155 levels (Figure S18B). In addition, we also noticed that PLGA NPs containing PNA3 caused considerable decrease in cell viability of U2932 cell lines in dose dependent manner (Figure S18C).
First, to assess the distribution in vivo, PLGA NPs containing TAMRA PNA were administered systemically and localization of fluorescent TAMRA signal was assessed after 4 hrs, 48 hrs, and 72 hrs in tumor (Figure 8A), liver, kidney, and spleen (Figure S19) by confocal microscopy. We confirmed significant distribution of PNA-TAMRA within the sectioned tumor cells in comparison to other organs after 4 hrs, 48 hrs, and 72 hrs of systemic treatment. Furthermore, the maximum distribution of TAMRA was noticed in the tumor after 48 hrs of systemic treatment.
Figure 8.

(A) Short TAMRA PNA (red) nanoparticles (NPs) within cryosectioned tumor cells, 4 hours, 48 hours, and 72 hours after systemic delivery. Red indicates TAMRA and Blue (DAPI) indicates nuclei, respectively. A representative image from four fields and from two different sections is shown. Scale bar represents 30 μm. (B) Tumor growth fold change in response to systemically administered PLGA NPs containing antimiR-155 PNA3, PNA3 NPs, and PNA5 NPs. N ≥ 5 for each group, data is shown as mean ± SEM. Student t-test was used relative to control group for statistical analysis. (C) Relative miR-155 gene expression level in RNA isolated from U2932 tumor cells from xenograft mice after systemic treatment with indicated nanoparticles. N =4 and data is shown as mean ± SEM, Student t test was used for statistical analysis, *p<0.05. (D) Gene expression level of downstream targets of miR-155; Foxo3A and Bach1 in U2932 tumor cells derived from xenograft tumor. (E) Histopathological studies including H&E, caspase3 and TUNEL (apoptosis markers) on tumors treated with PLGA NPs containing PNA3 and control group. Representative section from each group are shown (n= 6). The images were enhanced equally with ImageJ for clarity. Scale bar represents 200 μm for H&E staining, 100 μm for caspase and 30 μm for TUNNEL staining. In TUNEL staining, blue (DAPI) indicates nucleus and green indicates TUNEL staining.
We also examined the impact on tumor growth upon systemic treatment with only PNA3, PLGA NPs containing short PNA3 and NPs containing full length PNA5 in comparison to control vehicle treated group. For tumor growth delay assay, 2.4mg/kg of PNA3 either alone or encapsulated in PLGA NPs were administered systemically. Tumor growth was assessed 3 times per week (Figure S20). We did not observe any significant change in weights of the mice (Figure S21) during the study. However, mice treated with PNA3 and PNA5 containing PLGA NPs showed a decrease in tumor growth as compared to only PNA3 or the control group (Figure 8B). Further, PNA5 NPs showed similar reduction in tumor growth as that of PNA3 NPs. By day 16, control vehicle treated tumors showed an average increase of ~15-fold in the tumor volume in comparison to PNA3 NPs and PNA5 NPs which showed only ~6-fold and ~10-fold increase at the same time point.
Further, we assessed the miR-155 expression level in RNA isolated from the in vivo treated U2932 tumor cells. As shown in Figure 8C, the PLGA containing short PNA3 treated tumors showed a ~35% decrease in the miR-155 levels as compared to control group. We also examined the gene expression level of miR-155 targets; BACH1 and FOXO3A and noticed that short PNA3 treated U2932 tumors showed increased expression of BACH1 and FOXO3A, respectively (Figure 8D). We also examined tumor histopathology to correlate microscopic findings with the macroscopic response. In addition to the smaller overall size of the tumors treated with PNA3, the histopathology (H&E) of treated tumors showed extensive replacement of tumor with adipose tissue and connective tissue in the PLGA NPs containing PNA3 treated tumors (Figure 8E), whereas control vehicle treated tumors showed confluent tumor without evidence of significant intra-tumoral adipose or connective tissue. We anticipate that the histologic changes observed in the treated tumors may be due to decrease in miR-155 levels in tumor cells. In order to determine proliferation in tumors treated with PNA3 NPs, we performed Ki67 staining and noticed reduced staining of Ki67 antigen in PNA3 NPs treated tumors indicating low proliferation in comparison to the control group (Figure S22). Further, TUNEL assay and caspase3 staining (Figure 8E) confirmed significantly higher apoptosis in PNA3 NPs treated tumors in comparison to the control group.
Systemic administration of PLGA NPs containing short PNAs is not toxic
Next, we tested for an acute and chronic toxicity profile in immunocompetent mice after systemic delivery of PLGA NPs containing PNA3. Groups of mice were euthanized at two different time intervals; 8 hrs (acute) and 48 hrs (chronic) after systemic delivery to determine the acute and chronic toxicity, respectively (Figure S23). Overall, after administration of PLGA NPs containing short PNA3, no gross acute and chronic toxicity was noted in treated mice, including skin reactions, or behavioral changes. Furthermore, an evaluation of blood chemistries did not reveal any signs of renal or hepatic damage (Figure S24). We did not notice significant weight change of the mice (Figure S25) and other major organs for PLGA NPs treated and control group for acute as well as chronic toxicity studies (Figure S26). We also performed histological analysis in liver and kidney to corroborate our findings. We did not observe any sign of toxicity in histology of liver and kidney as compared to control group (Figure S27). We also evaluated the immunogenicity of PLGA NPs containing PNA3 by cytokine array analysis. We tested six different serum cytokine levels in the mice treated with PLGA NPs in comparison to control groups for acute as well as chronic toxicity studies (Figure S28). These results are consistent with the lack of an immune response. Together these results do not indicate any normal tissue toxicity with PLGA NPs containing short PNA3 treatment.
Further, we also performed histological analysis on liver and kidney of NSG mice (Figure S29) used in efficacy studies where multiple doses of PNA3 NPs were administered to check for any toxic response. We did not notice any change in the histology of organs from PNA3 NPs treated mice in comparison to the control group.
DISCUSSION
Advances in the understanding of synthetic nucleic acid analogue chemistry in conjunction with optimization of their delivery strategies have certainly provided a new horizon for the translation of antisense oligonucleotides into the clinic. In particular, miRNA targeted drug candidates, Cobomarsen (targeting miR-155) [70] and RG-012 (targeting miR-21) [71] are in clinical trials for treatment of Cutaneous T cell Lymphoma and Alport syndrome respectively. Most of the present antimiR strategies are centered on targeting full length miRNAs (23mer) and subsequently inhibiting their interaction with the target mRNA. Here, we successfully demonstrated that targeting full length miRNAs by antimiRs is not essential for efficacy. To date, the majority of research in the miRNA field has established the importance of the seed region in miRNA-mRNA interaction. Hence, we tested a novel PNA design; short PNAs containing three arginine amino acids on 5’ end (PNA3) that can target the seed sequence of miRNA-155 with enhanced binding affinity and inhibit its gene expression. Gel shift analysis demonstrated that both lysine as well as arginine containing short PNAs bind to miR-155 at low concentration as compared to non-cationic short PNA. Further, thermal denaturation results indicated that arginine containing short cationic PNAs possess superior binding affinity due to their Watson-Crick recognition between complementary sequences (seed region) and enhanced stabilization of the miRNA-PNA hetero-duplex via electrostatic interaction between negatively charged miRNA backbone (non-seed region) and the arginine containing positive domain. Though prior studies comprising poly-arginine conjugated PNAs (18mer) have shown promise to some extent [39], however, for our study, we chose only three arginine amino acids to minimize cytotoxicity induced by excess positive charge.
Though PNAs have gained considerable attention as therapeutic agents, its intracellular delivery remains a challenge for their broader clinical applications. Prior strategies to increase PNAs traversing across the biological cell membrane include; a) conjugation to poly-arginine tails, and b) gamma modification of PNA backbone with guanidinium based transduction domains (also called guanidinium PNAs). However, these strategies possess a few unresolved challenges. The guanidinium PNA-based strategy demands high cost as it requires multistep synthesis for production of adequate amounts of guanidinium PNA monomers. As stated earlier, cytotoxicity associated with amphipathic polyarginine PNAs can limit its in vivo applications. In addition, for significant antisense effects, poly-arginine and guanidinium containing oligomers require longer incubation time and higher concentration of oligomers (due to endosomal entrapment). As an alternate, we have demonstrated that the PLGA NPs provide an effective and safe strategy to deliver short cationic PNAs. Additionally, supported by comprehensive characterizations, PLGA NPs containing short cationic PNAs exhibit uniform morphology as well as size distribution. Further, we noticed sustained release of short cationic PNA3 in PLGA NPs.
Our cell culture results illustrate that PNA8 loaded PLGA NPs undergo substantial uptake with uniform distribution in the cytosol of HeLa cells. However, prior studies reported that PLGA nanoparticles undergo cellular uptake at a moderate level via endocytosis [72–75] and further, surface coating or the ligand coating of PLGA nanoparticles will enhance its cytosolic delivery. Hence to establish that arginine PNA containing PLGA NPs shows efficient uptake, we also performed time dependent cellular uptake studies in A549 cell lines. Consistent with our prior results in the HeLa cells, we noted efficient uptake in A549 cells too. Hence, arginine amino acids in short PNAs might play some role in the uptake of PLGA nanoparticles, though, we did not notice an increase in the zeta potential.
Prior studies showed that PLGA NPs containing full length antimiR without cationic domain or polyarginine PNAs undergo significant localization in the endosomes [76]. On the contrary, we noticed that PNA8 containing PLGA NPs demonstrate greater transfection efficiency and undergo uniform distribution in the cytosol. Moreover, prior methods described that higher concentration of poly-arginine PNAs or guanidinium PNAs are required for cellular uptake. Here, we observed that PNA8 alone at small concentrations do not show significant transfection in HeLa cells. We also examined that arginine containing short PNA8 NPs show uniform cellular distribution as compared to non-arginine containing PNA6 NPs or PNA10 NPs. Two plausible explanations for the aforementioned observations are; a) short cationic PNA NPs could undergo substantial solvent penetration that results in faster polymer degradation as compared to full length antimiR PNA containing NPs, resulting in uniform intracellular distribution, and b) arginine residues in short PNAs also assist in endosomal escape due to their interaction with negatively charged endosomal membranes. Additional studies are required to analyze the comprehensive uptake mechanism of short PNAs containing a few stretches of arginine domains, which could help develop more optimal delivery strategies not only restricted to PNA but also for other classes of synthetic nucleic acid mimics.
Further, our gene expression results indicate that arginine containing short cationic PNAs result in increased miR-155 knockdown as compared to lysine containing short PNAs. One possible explanation of higher binding affinity of the arginine versus lysine containing group is that arginine is more positively charged as compared to lysine [77]. Hence arginine containing PNAs can undergo extensive electrostatic interaction with the phosphate clusters of the RNA backbone leading to enhanced binding and subsequently superior antimiR efficacy.
In this study we made two advancements. First, we confirmed that short PNAs containing arginine domains can efficiently target the seed region of miR-155 and inhibit its expression. Second, in conjunction with nanotechnology, we determined that PLGA NPs can be used to encapsulate short cationic PNAs with superior physico-biochemical properties for gene therapy applications. The proposed design offers potential benefits in terms of low cost, better efficacy, minimal cytotoxicity and is clinically translatable. However, there are two possible toxicities, 1) the accumulation of short anti-miRs in the unwanted tissues or organs, and 2) genomic toxicity caused by non-selective binding to complementary off-target sites based on WC recognition, to be evaluated carefully for the success of anti-seed-based miRNA targeting technology. Non-selective accumulation of short PNAs in healthy tissues can be minimized due to promising delivery strategies since PLGA NPs target the tumor by the enhanced permeability and retention (EPR) effect [78]. On the other hand, there is a probability that short PNAs can elicit off-target effects at a genomic scale due to non-selective binding. However, transcriptional and proteomic profiling based prior studies on tiny LNA suggested that anti-miRs targeting the seed region have minimal off-target effects and do not significantly affect other mRNAs gene expression [32, 33, 79, 80]. Hence altogether, we believe that short cationic PNAs with an optimized PLGA based delivery system will further improve the anti-seed based anti-miR technology with minimal adverse reactions.
Finally, supported by comprehensive end point analysis, we provide evidence that short anti-seed cationic PNAs delivered by PLGA NPs are safe and non-toxic after systemic administration in the immunocompetent mice. These indications of safety are in line with previous work that has established PNAs and PLGA NPs with favorable side effect profiles [18, 26, 78]. We also established that PLGA NPs containing short PNAs inhibit miR-155 expression after systemic delivery. Importantly, the PLGA NPs containing PNA3 showed tumor growth inhibition as compared to only PNA3 and controls. Further, PNA3 NPs showed similar or superior efficacy in reducing the tumor growth in comparison to full length PNA5 NPs. In prior studies, full length antimiR-155 PNAs in a lymphoma mouse model showed considerable anti-tumor activity [26, 27]. In one case, antimiR-155 PNAs were encapsulated and cell penetrating peptide coated PLGA NPs were used to increase tumor accumulation and targeting. Whereas in another case, PNAs were delivered to tumor microenvironment by conjugation with tumor selective pH sensitive peptide known as pH low insertion peptide (pHLIP) [27, 79, 80].
In contrast, our present results establish that short cationic PNAs can effectively inhibit an oncogenic miRNA by simple intravenous infusion of regular PLGA NPs without any ligand coating or chemical modification. Though we noticed optimal effects in tumor growth inhibition, the efficacy of our system could be improved through a number of methods. First, next generation gamma modified PNAs (γPNAs) can be used to further increase the therapeutic efficacy of short PNAs. γPNAs are locked into right-handed helical structure due to stereogenic center at the gamma backbone position. γPNAs possess high binding affinity and improved water solubility as compared to regular PNAs [19, 81]. Prior studies demonstrated that γPNAs can be used as effective antisense [59] and gene editing agents [19]. In addition, the enhanced activity of γPNAs can be further augmented by next generation pHLIP and ligand coated delivery systems [82].
Together, these results establish that short anti-seed PNAs can be used as effective therapeutic modality for targeting miRNAs. The development of nanoparticles delivered PNA based probes may eventually lead to novel treatment regimens for cancer with both high specificity as well as low toxicity.
CONCLUSIONS
Overall, we envision that our study will advance miRNA targeted reagent design and delivery for multiple gene therapy-based applications. miRNA is a “druggable” target and in addition to various types of cancer, miRNA plays an important role in a myriad of diseases; ischemia, myocardial infarcts, stroke, tissue trauma, infection, just to name a few. In future, we can foresee that short cationic antimiRs in combination with NP based delivery will present a novel and effective therapeutic strategy for miRNA silencing.
Supplementary Material
Highlights.
Short cationic anti-seed PNAs can efficiently inhibit the action of target oncomiRs.
PLGA NPs containing short arginine-PNAs exhibit superior transfection efficiency.
Short anti-seed PNA loaded PLGA NPs prevents tumor growth in a xenograft mice model of lymphoma.
Proposed anti-seed PNA strategy is cost effective and clinically translatable.
Acknowledgment
We thank Dr. Anisha Gupta (Wesleyan University) for discussions and suggestions. We also acknowledge BIDMC Preclinical Murine Pharmacogenetics Core for their assistance with in vivo studies.
Funding Sources
This work was supported by University of Connecticut startup funds, St. Baldrick Foundation Jack’s Pack - We Still Have His Back, a St. Baldrick’s Hero Fund, UConn START PPOC award provided to R.B, V Foundation award and NIH R35CA232105 to F.J.S, and NIH R01 (1R01CA241194-01A1) grant to R.B and F.J.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Hoy SM, Patisiran: First Global Approval, Drugs, 78 (2018) 1625–1631. [DOI] [PubMed] [Google Scholar]
- [2].Scott LJ, Givosiran: First Approval, Drugs, 80 (2020) 335–339. [DOI] [PubMed] [Google Scholar]
- [3].Corey DR, Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy, Nat Neurosci, 20 (2017) 497–499. [DOI] [PubMed] [Google Scholar]
- [4].Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, Soucy A, Vaze J, Belur NR, Fredriksen K, Stojkovska I, Tsytsykova A, Armant M, DiDonato RL, Choi J, Cornelissen L, Pereira LM, Augustine EF, Genetti CA, Dies K, Barton B, Williams L, Goodlett BD, Riley BL, Pasternak A, Berry ER, Pflock KA, Chu S, Reed C, Tyndall K, Agrawal PB, Beggs AH, Grant PE, Urion DK, Snyder RO, Waisbren SE, Poduri A, Park PJ, Patterson A, Biffi A, Mazzulli JR, Bodamer O, Berde CB, Yu TW, Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease, N Engl J Med, 381 (2019) 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, Bahal R, Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development, J Clin Med, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mishra PJ, Merlino G, MicroRNA reexpression as differentiation therapy in cancer, J Clin Invest, 119 (2009) 2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Slack FJ, Weidhaas JB, MicroRNA in cancer prognosis, N Engl J Med, 359 (2008) 2720–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bartel DP, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell, 116 (2004) 281–297. [DOI] [PubMed] [Google Scholar]
- [9].Slack FJ, MicroRNAs regulate expression of oncogenes, Clin Chem, 59 (2013) 325–326. [DOI] [PubMed] [Google Scholar]
- [10].Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S, Inhibition of microRNA function by antimiR oligonucleotides, Silence, 3 (2012) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Velu CS, Grimes HL, Utilizing antagomiR (antisense microRNA) to knock down microRNA in murine bone marrow cells, Methods Mol Biol, 928 (2012) 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang G, Jin LQ, Hu J, Rodemer W, Selzer ME, Antisense Morpholino Oligonucleotides Reduce Neurofilament Synthesis and Inhibit Axon Regeneration in Lamprey Reticulospinal Neurons, PLoS One, 10 (2015) e0137670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Braasch DA, Corey DR, Cellular delivery of locked nucleic acids (LNAs), Curr Protoc Nucleic Acid Chem, Chapter 4 (2002) Unit 4 13. [DOI] [PubMed] [Google Scholar]
- [14].Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, Sacco A, Liu Y, Ngo H, Quang P, Azab AK, Azab F, Maiso P, Reagan M, Brown JR, Thai TH, Kauppinen S, Ghobrial IM, LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas, Blood, 120 (2012) 1678–1686. [DOI] [PubMed] [Google Scholar]
- [15].Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE, PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules, Nature, 365 (1993) 566–568. [DOI] [PubMed] [Google Scholar]
- [16].Chang H, Yi B, Ma R, Zhang X, Zhao H, Xi Y, CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo, Sci Rep, 6 (2016) 22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, Nielsen PE, Stability of peptide nucleic acids in human serum and cellular extracts, Biochem Pharmacol, 48 (1994) 1310–1313. [DOI] [PubMed] [Google Scholar]
- [18].Bahal R, Ali McNeer N, Quijano E, Liu Y, Sulkowski P, Turchick A, Lu YC, Bhunia DC, Manna A, Greiner DL, Brehm MA, Cheng CJ, Lopez-Giraldez F, Ricciardi A, Beloor J, Krause DS, Kumar P, Gallagher PG, Braddock DT, Mark Saltzman W, Ly DH, Glazer PM, In vivo correction of anaemia in beta-thalassemic mice by gammaPNA-mediated gene editing with nanoparticle delivery, Nat Commun, 7 (2016) 13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bahal R, Quijano E, McNeer NA, Liu Y, Bhunia DC, Lopez-Giraldez F, Fields RJ, Saltzman WM, Ly DH, Glazer PM, Single-stranded gammaPNAs for in vivo site-specific genome editing via Watson-Crick recognition, Curr Gene Ther, 14 (2014) 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Malik S, Asmara B, Moscato Z, Mukker JK, Bahal R, Advances in Nanoparticle-based Delivery of Next Generation Peptide Nucleic Acids, Curr Pharm Des, 24 (2018) 5164–5174. [DOI] [PubMed] [Google Scholar]
- [21].Malik S, Oyaghire S, Bahal R, Applications of PNA-laden nanoparticles for hematological disorders, Cell Mol Life Sci, 76 (2019) 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, Quijano E, Polikoff L, Kong Y, Bahal R, Geibel JP, Glazer PM, Saltzman WM, Egan ME, Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium, Nat Commun, 6 (2015) 6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ricciardi AS, Bahal R, Farrelly JS, Quijano E, Bianchi AH, Luks VL, Putman R, Lopez-Giraldez F, Coskun S, Song E, Liu Y, Hsieh WC, Ly DH, Stitelman DH, Glazer PM, Saltzman WM, In utero nanoparticle delivery for site-specific genome editing, Nat Commun, 9 (2018) 2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Singer A, Rapireddy S, Ly DH, Meller A, Electronic barcoding of a viral gene at the single-molecule level, Nano Lett, 12 (2012) 1722–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Montagner G, Bezzerri V, Cabrini G, Fabbri E, Borgatti M, Lampronti I, Finotti A, Nielsen PE, Gambari R, An antisense peptide nucleic acid against Pseudomonas aeruginosa inhibiting bacterial-induced inflammatory responses in the cystic fibrosis IB3–1 cellular model system, Int J Biol Macromol, 99 (2017) 492–498. [DOI] [PubMed] [Google Scholar]
- [26].Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, Slack FJ, Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma, Proc Natl Acad Sci U S A, 109 (2012) E1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM, Saltzman WM, Slack FJ, MicroRNA silencing for cancer therapy targeted to the tumour microenvironment, Nature, 518 (2015) 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gupta A, Quijano E, Liu Y, Bahal R, Scanlon SE, Song E, Hsieh WC, Braddock DE, Ly DH, Saltzman WM, Glazer PM, Anti-tumor Activity of miniPEG-gamma-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy, Mol Ther Nucleic Acids, 9 (2017) 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tahmasbi Rad A, Malik S, Yang L, Oberoi-Khanuja TK, Nieh MP, Bahal R, A universal discoidal nanoplatform for the intracellular delivery of PNAs, Nanoscale, 11 (2019) 12517–12529. [DOI] [PubMed] [Google Scholar]
- [30].Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP, Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs, Nat Struct Mol Biol, 18 (2011) 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Quijano E, Bahal R, Ricciardi A, Saltzman WM, Glazer PM, Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities, Yale J Biol Med, 90 (2017) 583–598. [PMC free article] [PubMed] [Google Scholar]
- [32].Murphy BL, Obad S, Bihannic L, Ayrault O, Zindy F, Kauppinen S, Roussel MF, Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression, Cancer Res, 73 (2013) 7068–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S, Silencing of microRNA families by seed-targeting tiny LNAs, Nat Genet, 43 (2011) 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li M, Zengeya T, Rozners E, Short peptide nucleic acids bind strongly to homopurine tract of double helical RNA at pH 5.5, J Am Chem Soc, 132 (2010) 8676–8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ong AAL, Toh DK, Krishna MS, Patil KM, Okamura K, Chen G, Incorporating 2-Thiouracil into Short Double-Stranded RNA-Binding Peptide Nucleic Acids for Enhanced Recognition of A-U Pairs and for Targeting a MicroRNA Hairpin Precursor, Biochemistry, 58 (2019) 3444–3453. [DOI] [PubMed] [Google Scholar]
- [36].Patil KM, Toh DK, Yuan Z, Meng Z, Shu Z, Zhang H, Ong AAL, Krishna MS, Lu L, Lu Y, Chen G, Incorporating uracil and 5-halouracils into short peptide nucleic acids for enhanced recognition of A-U pairs in dsRNAs, Nucleic Acids Res, 46 (2018) 7506–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ozes AR, Wang Y, Zong X, Fang F, Pilrose J, Nephew KP, Therapeutic targeting using tumor specific peptides inhibits long non-coding RNA HOTAIR activity in ovarian and breast cancer, Sci Rep, 7 (2017) 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bahal R, Manna A, Hsieh WC, Thadke SA, Sureshkumar G, Ly DH, RNA-Templated Concatenation of Triplet Nucleic-Acid Probe, Chembiochem, 19 (2018) 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fabbri E, Manicardi A, Tedeschi T, Sforza S, Bianchi N, Brognara E, Finotti A, Breveglieri G, Borgatti M, Corradini R, Marchelli R, Gambari R, Modulation of the biological activity of microRNA-210 with peptide nucleic acids (PNAs), ChemMedChem, 6 (2011) 2192–2202. [DOI] [PubMed] [Google Scholar]
- [40].Dragulescu-Andrasi A, Rapireddy S, He G, Bhattacharya B, Hyldig-Nielsen JJ, Zon G, Ly DH, Cell-permeable peptide nucleic acid designed to bind to the 5’-untranslated region of E-cadherin transcript induces potent and sequence-specific antisense effects, J Am Chem Soc, 128 (2006) 16104–16112. [DOI] [PubMed] [Google Scholar]
- [41].Thomas SM, Sahu B, Rapireddy S, Bahal R, Wheeler SE, Procopio EM, Kim J, Joyce SC, Contrucci S, Wang Y, Chiosea SI, Lathrop KL, Watkins S, Grandis JR, Armitage BA, Ly DH, Antitumor effects of EGFR antisense guanidine-based peptide nucleic acids in cancer models, ACS Chem Biol, 8 (2013) 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saarbach J, Sabale PM, Winssinger N, Peptide nucleic acid (PNA) and its applications in chemical biology, diagnostics, and therapeutics, Curr Opin Chem Biol, 52 (2019) 112–124. [DOI] [PubMed] [Google Scholar]
- [43].Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A, BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas, J Pathol, 207 (2005) 243–249. [DOI] [PubMed] [Google Scholar]
- [44].Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM, MicroRNA gene expression deregulation in human breast cancer, Cancer Res, 65 (2005) 7065–7070. [DOI] [PubMed] [Google Scholar]
- [45].Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM, A microRNA expression signature of human solid tumors defines cancer gene targets, Proc Natl Acad Sci U S A, 103 (2006) 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC, MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma, JAMA, 299 (2008) 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, Tibshirani R, Lossos IS, Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas, Blood, 113 (2009) 3754–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A, High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma, Genes Chromosomes Cancer, 39 (2004) 167–169. [DOI] [PubMed] [Google Scholar]
- [49].Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR, Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus, J Virol, 84 (2010) 11670–11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang X, Shen Y, Liu M, Bi C, Jiang C, Iqbal J, McKeithan TW, Chan WC, Ding SJ, Fu K, Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma, Am J Pathol, 181 (2012) 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fu Z, Tindall DJ, FOXOs, cancer and regulation of apoptosis, Oncogene, 27 (2008) 2312–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ, Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR L1, Curr Biol, 10 (2000) 1201–1204. [DOI] [PubMed] [Google Scholar]
- [53].Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME, Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor, Cell, 96 (1999) 857–868. [DOI] [PubMed] [Google Scholar]
- [54].Miyazaki T, Kirino Y, Takeno M, Samukawa S, Hama M, Tanaka M, Yamaji S, Ueda A, Tomita N, Fujita H, Ishigatsubo Y, Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1, Cancer Sci, 101 (2010) 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ando S, Putnam D, Pack DW, Langer R, PLGA microspheres containing plasmid DNA: preservation of supercoiled DNA via cryopreparation and carbohydrate stabilization, J Pharm Sci, 88 (1999) 126–130. [DOI] [PubMed] [Google Scholar]
- [56].Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V, PLGA-based nanoparticles: an overview of biomedical applications, J Control Release, 161 (2012) 505–522. [DOI] [PubMed] [Google Scholar]
- [57].Christensen L, Fitzpatrick R, Gildea B, Petersen KH, Hansen HF, Koch T, Egholm M, Buchardt O, Nielsen PE, Coull J, Berg RH, Solid-phase synthesis of peptide nucleic acids, J Pept Sci, 1 (1995) 175–183. [DOI] [PubMed] [Google Scholar]
- [58].Malik S, Bahal R, Investigation of PLGA nanoparticles in conjunction with nuclear localization sequence for enhanced delivery of antimiR phosphorothioates in cancer cells in vitro, J Nanobiotechnology, 17 (2019) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Delgado E, Bahal R, Yang J, Lee JM, Ly DH, Monga SP , beta-Catenin knockdown in liver tumor cells by a cell permeable gamma guanidine-based peptide nucleic acid, Curr Cancer Drug Targets, 13 (2013) 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, Ly DH, Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing gamma-peptide nucleic acids with superior hybridization properties and water solubility, J Org Chem, 76 (2011) 5614–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Manna A, Rapireddy S, Bahal R, Ly DH, MiniPEG-gammaPNA, Methods Mol Biol, 1050 (2014) 1–12. [DOI] [PubMed] [Google Scholar]
- [62].Borders CL Jr., Broadwater JA, Bekeny PA, Salmon JE, Lee AS, Eldridge AM, Pett VB, A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens, Protein Sci, 3 (1994) 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sokalingam S, Raghunathan G, Soundrarajan N, Lee SG, A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein, PLoS One, 7 (2012) e40410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM, Nanotechnology for delivery of peptide nucleic acids (PNAs), J Control Release, 240 (2016) 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Holy CE, Dang SM, Davies JE, Shoichet MS, In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam, Biomaterials, 20 (1999) 1177–1185. [DOI] [PubMed] [Google Scholar]
- [66].Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C, Clonogenic assay of cells in vitro, Nat Protoc, 1 (2006) 2315–2319. [DOI] [PubMed] [Google Scholar]
- [67].Verdugo M, Ogra Y, Quiroz W, Mechanisms underlying the toxic effects of antimony species in human embryonic kidney cells (HEK-293) and their comparison with arsenic species, J Toxicol Sci, 41 (2016) 783–792. [DOI] [PubMed] [Google Scholar]
- [68].Zhao J, Qi X, Dai Q, He X, Dweep H, Guo M, Luo Y, Gretz N, Luo H, Huang K, Xu W, Toxicity study of ochratoxin A using HEK293 and HepG2 cell lines based on microRNA profiling, Hum Exp Toxicol, 36 (2017) 8–22. [DOI] [PubMed] [Google Scholar]
- [69].Mahadevan D, Morales C, Cooke LS, Manziello A, Mount DW, Persky DO, Fisher RI, Miller TP, Qi W, Alisertib added to rituximab and vincristine is synthetic lethal and potentially curative in mice with aggressive DLBCL co-overexpressing MYC and BCL2, PLoS One, 9 (2014) e95184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, Jackson AL, Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma, Br J Haematol, 183 (2018) 428–444. [DOI] [PubMed] [Google Scholar]
- [71].Guo J, Song W, Boulanger J, Xu EY, Wang F, Zhang Y, He Q, Wang S, Yang L, Pryce C, Phillips L, MacKenna D, Leberer E, Ibraghimov-Beskrovnaya O, Ding J, Liu S, Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome, Hum Gene Ther, 30 (2019) 865–881. [DOI] [PubMed] [Google Scholar]
- [72].Jin C, Bai L, Wu H, Song W, Guo G, Dou K, Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells, Pharm Res, 26 (2009) 1776–1784. [DOI] [PubMed] [Google Scholar]
- [73].Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng JX, Kohane DS, Yeo Y, Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited, Mol Pharm, 6 (2009) 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Date AA, Shibata A, Goede M, Sanford B, La Bruzzo K, Belshan M, Destache CJ, Development and evaluation of a thermosensitive vaginal gel containing raltegravir+efavirenz loaded nanoparticles for HIV prophylaxis, Antiviral Res, 96 (2012) 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Risnayanti C, Jang YS, Lee J, Ahn HJ, PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer, Sci Rep, 8 (2018) 7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bahal R, McNeer NA, Ly DH, Saltzman WM, Glazer PM, Nanoparticle for delivery of antisense gammaPNA oligomers targeting CCR5, Artif DNA PNA XNA, 4 (2013) 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Li L, Vorobyov I, Allen TW, The different interactions of lysine and arginine side chains with lipid membranes, J Phys Chem B, 117 (2013) 11906–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Cheng CJ, Saltzman WM, Polymer nanoparticle-mediated delivery of microRNA inhibition and alternative splicing, Mol Pharm, 9 (2012) 1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li N, Yin L, Thevenin D, Yamada Y, Limmon G, Chen J, Chow VT, Engelman DM, Engelward BP, Peptide targeting and imaging of damaged lung tissue in influenza-infected mice, Future Microbiol, 8 (2013) 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM, Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane, Proc Natl Acad Sci U S A, 105 (2008) 15340–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Yeh JI, Shivachev B, Rapireddy S, Crawford MJ, Gil RR, Du S, Madrid M, Ly DH, Crystal structure of chiral gammaPNA with complementary DNA strand: insights into the stability and specificity of recognition and conformational preorganization, J Am Chem Soc, 132 (2010) 10717–10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wahane A, Waghmode A, Kapphahn A, Dhuri K, Gupta A, Bahal R, Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy, Molecules, 25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.