

SUMMARY
The crosstalk between tumor cells and the adjacent normal epithelium contributes to cancer progression, but its regulators have remained elusive. Here, we show that breast cancer cells maintained in hypoxia release small extracellular vesicles (sEV) that activate mitochondrial dynamics, stimulate mitochondrial movements and promote organelle accumulation at the cortical cytoskeleton in normal mammary epithelial cells. This results in Akt activation, membrane focal adhesion turnover and increased epithelial cell migration. RNA-Seq profiling identified Integrin-Linked Kinase (ILK) as the most upregulated pathway in sEV-treated epithelial cells and genetic or pharmacologic targeting of ILK reversed mitochondrial reprogramming and suppressed sEV-induced cell movements. In a three-dimensional model of mammary gland morphogenesis, sEV treatment induced hallmarks of malignant transformation, with deregulated cell death/cell proliferation, loss of apical-basal polarity and appearance of epithelial-to-mesenchymal transition (EMT) markers. Therefore, sEV released by hypoxic breast cancer cells reprogram mitochondrial dynamics and induce oncogenic changes in a normal mammary epithelium.
Graphical Abstract
eTOC BLURB
The communication between tumor cells and their neighbors is important for disease progression. Here, Bertolini et al show that hypoxic breast cancer cells release small extracellular vesicles that introduce multiple malignant changes in the normal mammary epithelium via reprogramming of mitochondrial functions. This pathway may promote local breast cancer recurrences.
INTRODUCTION
Epithelial tumorigenesis involves an intricate interplay between clones of transformed cells and a complex microenvironment (Gatenby and Gillies, 2008) composed of normal epithelium, immune-inflammatory cells and disparate mesenchymal structures, such as stromal fibroblasts, blood vessels and the extracellular matrix (Quail and Joyce, 2013). How this crosstalk is regulated is largely unknown. However, mechanisms of adaptive evolution (Greaves, 2015), intercellular communication (Tabassum and Polyak, 2015) and metabolic plasticity (Lehuede et al., 2016) select clones with growth advantage and metastatic potential. These pathways may also contribute to the “field effect” of cancerization (Curtius et al., 2018), a poorly understood process that introduces genetic and phenotypic changes in histologically “normal” epithelia, potentially contributing to local and distant disease recurrence (Martincorena et al., 2015).
In this context, extracellular vesicles (EV), including small (40-150 nm) extracellular vesicles (sEV) from either the plasma membrane (ectosomes) or the late endosomal trafficking machinery (exosomes) (Jaiswal and Sedger, 2019), have emerged as pleiotropic mediators of intercellular communication during tumorigenesis (Kalluri and LeBleu, 2020). Modulated by conditions of the microenvironment, such as hypoxia (King et al., 2012; Kucharzewska et al., 2013), and capable of horizontally transferring disparate bioactive molecules and nucleic acids to neighboring cells (Pathan et al., 2019), tumor-derived sEV contribute to multiple oncogenic traits, such as metabolic reprogramming (Fong et al., 2015), drug resistance (Cornell et al., 2019) and various mechanisms of cell migration, invasion and metastatic dissemination (Rodrigues et al., 2019; Wang et al., 2014).
Mechanistically, reprogramming of mitochondrial functions has been recently implicated in multiple aspects of tumorigenesis (Vyas et al., 2016). Fresh evidence has shown that rewiring of oxidative bioenergetics (Anderson et al., 2018), modulation of ROS signaling (Weinberg et al., 2010) and changes in mitochondrial dynamics, a process that controls the size, shape and subcellular position of mitochondria (Eisner et al., 2018), are key drivers of tumor growth, in vivo (Sellers et al., 2015), cancer stemness (Park et al., 2019), and tumor cell invasion and metastasis (Altieri, 2019). What has remained unclear, however, is whether similar mechanisms affect “normal” epithelia in a tumor microenvironment, potentially facilitating the acquisition of transforming traits that enable disease progression (Quail and Joyce, 2013).
In this study, we examined mechanisms of intercellular communication between hypoxic breast cancer cells and the normal mammary epithelium.
RESULTS
Regulation of mammary epithelial cell motility by breast cancer-derived hypoxic sEV.
We began this study by isolating sEV from two HER2−/ER+/PR+ human breast cancer cell lines, MCF7 and T47D under normoxic (5% O2) or hypoxic (1% O2) conditions. sEV produced in hypoxia (sEVHYP) or normoxia (sEVNORM) had comparable shape by cryo-electron microscopy (Figure S1A) and expressed EV-associated markers, Tsg101, CD63 and flotillin 1, by Western blotting (Figure S1B and C). In contrast, cellular markers, calnexin, VDAC, catalase and PRX1 were undetectable (Figure S1B and C). Size exclusion chromatography (SEC) demonstrated that both sEVNORM or sEVHYP produced by MCF7 cells mostly segregated within SEC fractions 7-9 with comparable particle size mean distribution of ~100 nm (Figure S1D and E) (Thery et al., 2018). Only SEC fractions 10-12 contained minor amounts of sEVNORM or sEVHYP particles, whereas other SEC fractions were negative (Figure S1D). Finally, fluorescence (PKH26)-labeled sEVHYP or sEVNORM were efficiently internalized by recipient MCF10A normal mammary epithelial cells starting at 2 h after addition, with comparable uptake by 6 h (Figure S1E and F). Unless otherwise noted, sEVHYP or sEVNORM were used at 3x108 particles per 104 recipient cells for all subsequent experiments. sEV isolated from non-conditioned media were used as control (Ctrl).
Using these conditions, breast cancer-derived sEVHYP increased directional migration of recipient MCF10A cells in a wound closure assay (Figure 1A and B), as well as PET inserts (Figure 1C and D). Treatment with sEVNORM minimally increased cell migration compared to controls (Figure 1A-D). In addition, sEVHYP produced by MCF7 (Figure 1E) or T47D (Figure S2A) cells stimulated 2D motility of recipient MCF10A cells, resulting in greater speed of cell movements and longer distance traveled by individual cells (Figure 1F, Figure S2B). Consistent with these findings, sEVHYP increased chemotaxis of MCF10A cells along a gradient of NIH3T3 cell-derived conditioned medium (Figure 1G), also resulting in increased speed of cell movements and longer distance traveled by individual cells, compared to controls (Figure S2C). In contrast, sEVNORM did not significantly affect 2D (Figure 1E and F, Figure S2A and B) or chemotactic (Figure 1G and Figure S2C) motility of recipient MCF10A cells. A prerequisite of cell movements is heightened focal adhesion (FA) turnover (Roussos et al., 2011). Consistent with this, sEVHYP stimulated FA turnover in recipient MCF10A cells (Figure S2F), characterized by increased formation of new FA and reduction in stable FA, compared to controls (Figure 1H). sEVNORM had no effect on FA turnover (Figure S2F, Figure 1H).
Figure 1. Breast cancer-derived hypoxic sEV (sEVHYP) stimulate motility of normal mammary epithelial cells.

(A) Normal mammary epithelial MCF10A cells treated with small extracellular vesicles (sEV) produced by MCF7 cells under normoxic (eEVNORM) or hypoxic (sEVHYP) conditions and analyzed for directional cell migration in a wound closure assay. Representative images at time (t) 0 h and 12 h. Scale bar, 150 μm.
(B) The conditions are as in (A) and directional motility of sEV-treated MCF10A recipient cells was quantified. Mean±SEM (N=5). Numbers correspond to p values by 2-way Anova with Tukey's multiple comparisons test.
(C and D) MCF10A cells treated with sEVNORM or sEVHYP produced by MCF7 (top) or T47D (bottom) cells were analyzed for migration on PET inserts (C, representative images of DAPI-stained nuclei of migrated cells) and quantified (D). Scale bar, 200 μm. Mean±SEM (N=3). Numbers correspond to p values by 1-way Anova with Tukey's posttest.
(E and F) MCF10A cultures treated with MCF7 cell-derived sEVNORM or sEVHYP were analyzed for cell motility in 2D contour plots (E) and the speed of cell movements (F, left) or total distance traveled (F, right) by individual sEV-treated MCF10A cells was quantified. Each tracing corresponds to an individual cell. The cutoff velocities for slow (black, <1.5 μm/min)- or fast (red, >1.5 μm/min)-moving cells are indicated. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey's posttest.
(G) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP were analyzed for chemotactic cell motility quantified by rose plots. Red arrow, direction of the chemotactic gradient. (N=3).
(H) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP were analyzed for focal adhesion (FA) turnover and the percentage of new, stable or decayed FA was quantified. Mean±SEM (N=3). Numbers correspond to p values by 2-way Anova with Tukey's posttest.
(I and J) MCF10A cells as in (H) were analyzed for mitochondrial motility by time-lapse videomicroscopy (I, representative images) at time (t) 0 and 6 min, and the speed of mitochondrial movements (J, left) and total distance traveled by individual mitochondria (J, right) was quantified. Mean±SD (N=3). Scale bar, 5 μm. Numbers correspond to p values by 1-way Anova with Tukey’s posttest.
We next carried out two sets of experiments to validate the specificity of these findings. First, we found that only SEC fractions 7-9, which contain the majority of breast cancer-derived sEVHYP (Figure S1D), stimulated MCF10A cell motility, increasing the speed of cell movements and the distance traveled by individual cells (Figure S2D). Other SEC fractions from normoxic or hypoxic samples had no effect on 2D cell motility (Figure S2D). Second, we observed that concentrations of sEVHYP as low as 3x105/ml were sufficient to stimulate 2D motility of MCF10A cells, quantitatively indistinguishable from higher concentrations of 3x106-3x108 particles/ml (Figure S2E). Therefore, breast cancer-derived sEV released under conditions of hypoxia potently stimulate membrane dynamics of cell motility in a recipient normal mammary epithelium.
sEVHYP stimulate mitochondrial dynamics in recipient cells.
Consistent with a role of mitochondrial dynamics in cell motility (Caino et al., 2016), sEVHYP increased the subcellular trafficking of mitochondria in recipient MCF10A cells (Figure 1I), resulting in faster speed of organelle movements and greater distance traveled by individual mitochondria (Figure 1J). This was associated with increased accumulation of mitochondria at the cortical cytoskeleton of MCF10A cells in response to T47D (Figure 2A and B)- or MCF7 (Figure S3A and B) cell-derived sEVHYP. sEVNORM had no effect on mitochondrial movements (Figure 1I and J) and did not promote mitochondrial accumulation at the cortical cytoskeleton on MCF10A cells (Figure 2A and B, Figure S3A and B). In control experiments, breast cancer-derived sEVHYP or sEVNORM had no effect on mitochondrial inner membrane potential of MCF10A cells (Figure 2C).
Figure 2. Breast cancer-derived sEVHYP regulate mitochondrial dynamics.

(A and B) MCF10A cells treated with T47D cell-derived sEVNORM or sEVHYP were imaged after 24 h for subcellular mitochondrial distribution by confocal fluorescence microscopy (A, representative images) and the percentage of mitochondria localized to the cortical cytoskeleton was quantified (B). Insets, magnification of indicated areas. Scale bar, 5 μm. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s posttest.
(C) MCF10A cells treated with MCF7- or T47D-cell-derived sEVNORM or sEVHYP were analyzed for changes in mitochondrial inner membrane potential by TMRM labeling and flow cytometry. Mean±SD (N=3).
(D) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP for 8 h were analyzed by Western blotting. p, phosphorylated. (N=3).
(E and F) sEVNORM- or sEVHYP-treated MCF10A cells were analyzed for co-localization of Ser616-phosphorylated Drp1 (pDrp1) and mitochondria by confocal microscopy (E, representative images) and quantified (F). Insets, image magnification of indicated areas. Mean±SD (N=3). Scale bar, 5 μm. Numbers correspond to p values by 1-way Anova with Tukey’s posttest. PCC, Pearson Correlation Coefficient.
(G and H) sEV-treated MCF10A cells were analyzed for changes in mitochondrial volume indicative of organelle fusion (>1.5-fold, positive y-scale) or fission (<1.5-fold, negative y-scale) over 30-sec intervals (G) and the number of mitochondrial fusion and fission events was quantified (H) Each line corresponds to changes in mitochondrial volume in a single cell. Mean±SEM (N=3). Numbers correspond to p values by 2 way-Anova with Sidak's multiple comparisons test. See also Figure S3.
(I and J) sEV-treated MCF10A cells transfected with control non-targeting siRNA (siCtrl) or siRNA directed to MNF1 (siMFN1) or Drp1 (siDrp1) were analyzed by Western blotting (I) and cell migration on PET inserts (J). Mean±SD (N=3).
In addition, sEVHYP treatment of MCF10A cells was associated with activating (Ser616) phosphorylation of dynamin-related protein 1 (Drp1) (Figure 2D), a key effector of mitochondrial fission (Eisner et al., 2018). This was accompanied by increased association of Ser616-phosphorylated Drp1 with mitochondria of recipient cells, whereas sEVNORM had no effect (Figure 2E and F). Co-culture with sEVHYP, but not sEVNORM, also increased the expression of mitofusin-1 (MFN1) and −2 (MFN2), which mediate mitochondrial fusion (Figure 2D). Taken together, real-time quantification of changes in mitochondrial volume by time-lapse videomicroscopy (Figure 2G) demonstrated that sEVHYP increased the rates of both mitochondrial fusion (>1.5-fold mitochondrial volume) and fission (<1.5-fold mitochondrial volume) events in recipient MCF10A cells (Figure 2H). Conversely, sEVNORM had no effect on mitochondrial volume, compared to controls (Figure 2G and H). Mechanistically, silencing of MFN1 or Drp1 by small interfering RNA (siRNA) in recipient MCF10A cells exposed to sEVNORM or sEVHYP (Figure 2I) abolished cell migration stimulated by sEVHYP (Figure 2J). In contrast, MFN1 or Drp1 depletion had no effect on MCF10A cell migration in the presence of sEVNORM indistinguishable from controls (Figure 2J). Taken together, these findings demonstrate that sEVHYP stimulate multiple aspects of mitochondrial dynamics in recipient cells, including organelle motility, subcellular distribution and fusion/fission events.
sEVHYP-ILK reprogramming of normal mammary epithelial cells.
To understand the mechanisms of sEVHYP signaling, we next profiled recipient MCF10A cells by RNA-Seq. Mirroring the observed increase in cell migration, sEVHYP treatment was associated with a global transcriptional response in recipient cells, with activation of multiple pathways of cell movements, chemotaxis, cytoskeletal organization and cell-to-cell contact (Figure 3A). In addition, sEVHYP-treated cells exhibited reduced cell death and increased pro-inflammatory responses (Figure 3A), including NFκB-, IL6- and IL8-associated pathways (Figure 3B). Consistent with these data, sEVHYP treatment induced markers of pro-migratory epithelial-to-mesenchymal transition (EMT) in recipient cells, with increased expression of the EMT transcriptional activator, SNAIL as well as mesenchymal markers, N-cadherin and vimentin and lower levels of β-catenin (Figure 3C). Functionally, this was associated with increased nuclear accumulation of SNAIL compared to controls, whereas sEVNORM had no effect (Figure 3D and Figure S3C). In addition, sEVHYP, but not sEVNORM, increased the phosphorylation of Akt (Ser473 and Thr308) in recipient MCF10A cells, as well as upregulation of MMP9 (Figure 3E), two critical mediators of cell motility and invasion.
Figure 3. sEVHYP-induced reprogramming of normal mammary epithelial cells.

(A) Ingenuity Pathway Analysis of genes significantly modulated (red, activated; blue, inhibited) in MCF10A cells treated with sEVHYP by RNA-Seq (p<0.05).
(B) Bioinformatics analysis of gene pathways regulated by sEVHYP in recipient MCF10A cells by RNA-Seq. The activation Z-score and p values are indicated.
(C) sEVNORM or sEVHYP-treated MCF10A cells were analyzed by Western blotting (N=3).
(D) The conditions are as in (C) and MCF10A cells were analyzed for nuclear accumulation of SNAIL by fluorescence microscopy (representative images, N=3). Scale bar, 25 μm.
(E) sEVNORM or sEVHYP-treated MCF10A cells were analyzed by Western blotting (N=3).
(F) Whole cell extracts (WCE) or the indicated sEV-containing SEC fractions isolated from normoxic (top) or hypoxic (bottom) MCF7 cells were analyzed by Western blotting (N=2).
(G) WCE, unfractionated sEV or CD63-positive (CD63+) or -negative (CD63−) sEV isolated from normoxic (top) or hypoxic (bottom) MCF7 cells were analyzed by Western blotting. An antibody to CD9 was used as a control (N=3).
(H) MCF10A cells treated with total sEV, IgG-negative sEV (sEVIgG−) or CD63-negative sEV (sEVCD63−) isolated from MCF7 or T47D cells were analyzed for 2D motility with quantification of speed of cell movements (top) and total distance traveled by individual cells (bottom). Non-binding-sEV to IgG-coated beads were used as control. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s posttest.
Bioinformatics analysis of RNA-Seq data identified Integrin-Linked Kinase (ILK) as the pathway most activated in sEVHYP-treated MCF10A cells (Figures 3B). Consistent with this, exposure to hypoxia increased the accumulation of ILK in breast cancer-derived sEV (Figure S3D). This effect was specific because ILK levels were not affected in whole cell extracts from hypoxic or normoxic samples (Figure S3D). Next, we mapped the association of ILK with sEV-containing SEC fractions from normoxic or hypoxic MCF7 cells. We found that ILK was prominently enriched in SEC fractions 7-9 from sEVHYP (Figure 3F), containing sEV markers, CD63 and Tsg101, but not ER marker, Calnexin (Figure 3F), and directly mediating MCF10A cell motility (Figure S2D). A pool of ILK was also present in SEC fractions 10-12 from sEVHYP (Figure 3F). In contrast, SEC fractions 7-9 or 10-12 isolated from normoxic MCF7 cells showed little to no ILK (Figure 3F). Using an independent approach, ILK co-localized with CD63-expressing sEV by confocal microscopy in hypoxic, and, to a lesser extent, normoxic breast cancer cells (Figure S3E). As control, siRNA silencing of ILK abolished the co-localization of ILK with CD63-positive sEV in hypoxic or normoxic MCF7 or T47D cells, whereas a control, non-targeting siRNA had no effect (Figure S3E).
To establish the relevance of CD63-ILK association, we next immuno-depleted CD63-reactive material from normoxic or hypoxic sEV. In these experiments, CD63 depletion removed ILK expression from sEVNORM or sEVHYP (Figure 3G). Conversely, ILK was abundantly present in CD63-expressing sEV or unfractionated sEV from hypoxic, and, to a lesser extent, normoxic MCF7 cells (Figure 3G). Similar results were obtained by flow cytofluorometric staining of CD63-positive sEV, where ILK was highly expressed in hypoxic, compared to normoxic samples (Figure S3F). Under these conditions, immuno-depletion of CD63 abolished sEVHYP-induced 2D motility of recipient MCF10A cells, normalizing the speed of cell movements and the total distance traveled by individual cells to levels of control cultures (Figure 3H). Consistent with the data above, depletion of CD63-sEV with CD63-coated beads abolished the effect of sEV on cell motility, whereas incubation of sEV with IgG-coated beads had no effect on sEV-stimulated cell motility (Figure 3H). Finally, co-culture with sEVHYP increased ILK protein levels in recipient MCF10A cells, whereas sEVNORM had no effect on ILK levels in recipient cells (Figure S3G). Consistent with these data, only sEV-containing SEC fractions 7-9 transferred ILK to recipient MCF10A cells, whereas SEC fractions 10-12 had no effect (Figure S3H). Therefore, sEVHYP globally reprogram the transcriptome of recipient mammary epithelial cells accompanied by delivery of ILK in these settings.
Role of ILK in sEVHYP regulation of mitochondrial dynamics.
Based on these data, we next asked if sEV-vehiculated ILK participated in mitochondrial reprogramming and cell motility in recipient cells. We found that sEV isolated from hypoxic MCF7 cells transfected with ILK-directed siRNA failed to stimulate mitochondrial trafficking to the cortical cytoskeleton in recipient MF10A cells (Figure 4A and B). Instead, sEVHYP from MCF7 cells transfected with non-targeting siRNA potently stimulated mitochondrial repositioning to the cortical cytoskeleton (Figure 4A and B). In addition, siRNA silencing of HIF1α in sEV-producing cells also suppressed sEVHYP-mediated mitochondrial trafficking (Figure 4A and B). Consistent with these data, treatment with a small molecule ILK inhibitor, Cpd22, which was identified by a cell-based assay to inhibit ILK-mediated signaling (Lee et al., 2011), suppressed sEVHYP-induced mitochondrial trafficking in recipient MCF10A cells (Figure S4A and B). Cpd22 also inhibited Ser616-phosphorylated Drp1 recruitment to mitochondria mediated by sEVHYP (Figure S4C and D). In contrast, Cpd22 did not affect mitochondrial trafficking to the cortical cytoskeleton (Figure S4A and B) or Drp1 association with mitochondria (Figure S4C and D) in the presence of sEVNORM. Consistent with a potential direct effect on mitochondrial dynamics (Onodera et al., 2018), a pool of ILK associated with mitochondria in transfected cells, by confocal microscopy (Figure S4E and F).
Figure 4. ILK regulation of ExoHYP-induced mitochondrial dynamics.

(A and B) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP after transfection with siCtrl, siHIF1α or siILK were analyzed for subcellular mitochondrial localization by confocal fluorescence microscopy (A, representative images) and the percentage of mitochondria accumulated at the cortical cytoskeleton was quantified (B). Insets, image magnification of indicated areas. Scale bar, 5 μm. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(C) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP after transfection with siCtrl or siILK (left) or treated with small molecule ILK inhibitor, Cpd22 were analyzed for FA turnover by time-lapse videomicroscopy and the percentage of new, stable and decayed FA events under the various conditions tested was quantified. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(D) MCF10A cells co-cultured with MCF7 cell-derived sEVNORM or sEVHYP after transfection with siCtrl, siHIF1α or siILK were analyzed for 2D motility and the speed of cell movements (top) or total distance traveled by individual cells (bottom) was quantified. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(E) MCF7 cells transfected with siCtrl or siILK were reconstituted with sEVNORM or sEVHYP and analyzed after 8 h by Western blotting (N=3).
(F and G) MCF7 cells transfected with siCtrl or siILK were treated with sEVNORM or sEVHYP and analyzed for co-localization of Ser616-phosphorylated Drp1 (pDrp1) with mitochondria by confocal microscopy (F, representative images) and quantified (G, N=3). Insets, image magnification of indicated areas. Scale bar, 5 μm. Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test. PCC, Pearson Correlation Coefficient.
(H) The reconstitution conditions are as in (E) and MCF7 cells were analyzed for subcellular accumulation of mitochondria at the cortical cytoskeleton. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(I) Normal primary mammary epithelial cells transfected with ILK or L207W ILK mutant (ILK-Mut) were analyzed for 2D cell motility and the speed of cell movements (top) and total distance traveled by individual cells (bottom) was quantified. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
Using an independent experimental approach, siRNA silencing of ILK, but not control siRNA suppressed mitochondrial dynamics induced by sEVHYP, reducing both fusion and fission events to levels of control cultures (Figure S4G). In contrast, knockdown of ILK had no effect on mitochondrial dynamics in the presence of sEVNORM (Figure S4G). Under these conditions, siRNA knockdown of ILK or treatment with Cpd22 prevented the increased turnover of FA mediated by sEVHYP, lowering the percentage of new FA while increasing the number of stable FA (Figure 4C). In contrast, ILK targeting by siRNA or Cpd22 treatment did not affect FA turnover in sEVNORM-co-cultured cells (Figure 4C). Finally, treatment with Cpd22 (Figure S4H) or HIF1α or ILK silencing (Figure 4D) suppressed the increase in 2D motility stimulated by sEVHYP in MF10A cells, normalizing the speed of cell movements and the total distance traveled by individual cells to levels of control cultures (Figure S4H and Figure 4D). Together, these data demonstrate that sEVHYP stimulation of ILK signaling mediates reprogramming of mitochondrial functions in recipient mammary epithelial cells.
Molecular requirements of ILK-regulation of mitochondrial functions in recipient cells.
To test the specificity of the findings above, we next carried out three sets of complementary experiments. First, we asked if reconstitution with sEVHYP could restore ILK activity in ILK-silenced breast cancer cells. First, co-culture with sEVHYP restored ILK protein expression in siILK-knockdown cells (Figure 4E). Functionally, reconstitution with sEVHYP rescued the recruitment of Ser616-phosphorylated Drp1 to mitochondria (Figure 4F and G) and promoted mitochondrial accumulation at the cortical cytoskeleton, compared to controls (Figure 4H). As a second approach, we reconstituted ILK-silenced MCF7 cells with wild type (WT) ILK or ILK L207W dominant-negative mutant that interferes with the pseudocatalytic site and disrupts downstream signaling leading to stress fiber formation, cell spreading and migration (Vaynberg et al., 2018). Reconstitution of ILK knockdown cells with WT ILK (Figure S5A) restored mitochondrial trafficking to the cortical cytoskeleton (Figure S5B and C) and normalized mitochondrial dynamics reestablishing cycles of organelle fusion and fission events comparably to control transfectants (Figure S5D and E). In contrast, reconstitution with L207W ILK mutant had no effect on mitochondrial trafficking to the cortical cytoskeleton (Figure S5B and C) and did not restore mitochondrial dynamics in transfected cells (Figure S5D and E).
Third, to rule out that this response was selective for MCF10A cells, we transfected primary, normal mammary epithelial cells with WT or L207W ILK mutant. Similar to the data above, WT ILK, but not ILK L207W mutant transfected in primary mammary epithelial cells (Figure S5F) associated with mitochondria, by confocal microscopy (Figure S5G and H). Functionally, WT ILK stimulated subcellular mitochondrial trafficking to the cortical cytoskeleton (Figure S5I and J) and promoted 2D motility of normal mammary epithelial cells, increasing the speed of cell movements and the total distance traveled by individual cells (Figure 4I). Conversely, transfection of L207W ILK mutant did not promote increase mitochondrial trafficking (Figure S5I and J) or 2D cell motility (Figure 4I) in primary mammary epithelial cells. Therefore, mechanisms of pseudocatalytic ILK signaling regulate mitochondrial dynamics and subcellular organelle trafficking.
Requirement of Akt for sEVHYP signaling in normal mammary epithelial cells.
The downstream signaling requirement for sEV-ILK regulation of mammary epithelial cell responses were next investigated. In these experiments, siRNA silencing of ILK or HIF1α in sEV-producing breast cancer cells abolished Akt phosphorylation (Ser473) induced by sEVHYP in recipient MCF10A cells (Figure 5A). Similar results were obtained with a small molecule Akt inhibitor, MK2206, which also suppressed Akt phosphorylation (Ser473) mediated by sEVHYP in MCF10A cells (Figure S6A). MK2206 treatment also moderately reduced total Akt levels in these cells (Figure S6B). In addition, MK2206 inhibited the association of Ser616-phosphorylated Drp1 with mitochondria (Figure 5B and C) and suppressed mitochondrial trafficking to the cortical cytoskeleton in sEVHYP-treated cultures (Figure S6B and C). Consistent with these data, MK2206 blocked mitochondrial motility (Figure 5D) and suppressed mitochondrial dynamics mediated by sEVHYP, decreasing the rate of fusion and fission events to levels of sEVNORM treatment (Figure 5E). Finally, MK2206 blocked the increase in 2D motility mediated by sEVHYP in recipient cells, lowering the speed of cell movements and the total distance traveled by individual cells to levels of sEVNORM-treated cultures (Figure S6D).
Figure 5. Akt regulation of sEVHYP-dependent mitochondrial dynamics.

(A) MCF10A cells treated with sEVNORM or sEVHYP isolated from MCF7 cells transfected with siCtrl, siHIF1α or siILK were analyzed by Western blotting. p, phosphorylated (N=3).
(B and C) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP were incubated with vehicle (Veh) or small molecule Akt inhibitor, MK2206 and co-localization of Ser616-phosphorylated Drp1 (pDrp1) with mitochondria was analyzed by confocal microscopy (B, representative images) and quantified (C). Scale bar, 5 μm. PCC, Pearson Correlation Coefficient. Mean±SEM (N=3) Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(D) MCF10A cells treated with MCF7 cell-derived sEVNORM or sEVHYP were treated with vehicle (Veh) or MK2206 and analyzed for mitochondrial motility by time-lapse videomicroscopy. Each symbol corresponds to an individual mitochondrion. Representative experiment (N=3).
(E) MCF10A cells co-cultured with MCF7 cell-derived sEVNORM or sEVHYP were treated with vehicle (Veh) or MK2206 and analyzed for changes in mitochondrial dynamics by time-lapse videomicroscopy and fusion and fission events were quantified over 30-sec intervals. Mean±SD (N=3). Numbers correspond to p values by 2-way Anova with Tukey's posttest.
(F and G) MCF10A cells treated with sEVNORM or sEVHYP isolated from MCF7 cells transfected with siCtrl, siAkt1 or siAkt2 were analyzed for subcellular mitochondrial localization by confocal microscopy (F, representative images) and the percentage of mitochondria accumulated at the cortical cytoskeleton was quantified (G). Insets, image magnification of indicated areas. Scale bar, 5 μm. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test. ns, not significant.
(H) MCF10A cells transfected with siCtrl or siAkt were reconstituted with wild type (WT) Akt or dominant-negative (DN) Akt cDNA and analyzed by Western blotting. (N=3).
(I) MCF10A cells reconstituted as in (H) and treated with MCF7 cell-derived sEVNORM (top) or sEVHYP (bottom) were quantified for mitochondrial accumulation at the cortical cytoskeleton by confocal microscopy. Mean±SD (N=3). Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test. ns, not significant. See also Figures S5 and Figure S6.
As an independent approach, we next silenced by siRNA individual Akt isoforms, Akt1 or Akt2 in recipient cells (Figure S6F). This inhibited Akt phosphorylation in MCF10A cells (Figure S6E) and shut off the accumulation of mitochondria at the cortical cytoskeleton mediated by sEVHYP treatment (Figure 5F and G). In contrast, depletion of Akt1 or Akt2 had no effect on mitochondrial trafficking in the presence of sEVNORM (Figure 5F and G). Finally, we reconstituted MCF10A cells transfected with siCtrl or siAkt with wild type WT Akt or dominant negative (DN) Akt mutant (Figure 5H). In these experiments, reconstitution with WT Akt restored the increase in mitochondrial trafficking to the cortical cytoskeleton mediated by sEVHYP, indistinguishably from control transfectants (Figure 5I). In contrast, transfection of DN Akt did not rescue the defect of mitochondrial motility induced by Akt knockdown, whereas reconstitution with WT or DN Akt had no effect on mitochondrial trafficking in the presence of sEVNORM (Figure 5I). Altogether, these findings demonstrate that an ILK-Akt signaling axis is required for mitochondrial dynamics and downstream regulation of cell motility in mammary epithelial cells.
sEVHYP regulation of mammary gland morphogenesis.
Based on these data, we next examined the effect of sEVHYP in a three-dimensional (3D) model of mammary gland development. In these experiments, addition of sEVHYP increased the surface area of MCF10A 3D acini, in vitro, compared to controls (Figure 6A and B). This was associated with extensive morphological changes in 3D acini, with decrease in multiacinar sphericity (Figure 6A and B) and more extensive filling of the acinar lumen (Figure 6C). Co-culture with sEVNORM had no effect on MCF10A 3D acini in culture, compared to controls (Figure 6A-C). In addition, MCF10A 3D acini treated with a single addition of sEVHYP (Figure 6D and E) or sequentially with sEVHYP additions every 4 d (Figure 6F) exhibited comparable increase in cell proliferation, by Ki67 staining and decreased apoptosis (cleaved caspase-3+ cells). Instead, treatment with sEVNORM did not induce changes in cell proliferation or apoptosis in MCF10A 3D acini after single (Figure 6D and E) or sequential (Figure 6F) addition.
Figure 6. sEVHYP regulation of 3D mammary acini development.

(A) MCF10A cells were seeded in 3D acini culture in the presence of Ctrl, sEVNORM or sEVHYP for 21 d and imaged by brightfield microscopy. Insets, image magnification of indicated areas. Scale bar, 100 μm. (N=3).
(B) The conditions are as in (A) and the surface area (top) and circularity (bottom, 1=perfect circle) of sEV-treated MCF10A acini in 3D culture were quantified. Each dot represents the mean of MCF10A acini in one 10x microscopy field. An average of 12 independent microscopy fields were used for counting. Data are expressed as boxplots (min to max) of at least three independent experiments. Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(C) The conditions are as in (A) and the lumen filling of sEV-treated MCF10A acini in 3D was quantified. Data are expressed as boxplots (min to max) of at least three independent experiments. Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(D) sEV-treated MCF10A acini in 3D as in (A) were analyzed for Ki67 (top) or cleaved caspase 3 (bottom) reactivity after 8 d, by immunohistochemistry. Representative equatorial section images. Scale bar, 10 μm.
(E and F) The conditions are as in (D) and the percentage of Ki67+ nuclei and cleaved caspase 3-expressing cells was quantified for sEV added once at t=0 (E) or sequentially every 4 d (F). MFI, mean fluorescence intensity. Data are expressed as boxplots (min to max) of at least three independent experiments. Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test.
(G) The conditions are as in (D) and sEV-treated MCF10A acini in 3D were analyzed for expression of pAkt (Ser473, top), nuclear accumulation of SNAIL (middle) or levels of vimentin (bottom) after 21 d, by immunohistochemistry. Representative confocal microscopy images are shown; inset, magnification of selected field. Scale bar, 10 μm.
(H) The conditions are as in (G) and the percentage of pAkt+ cells (max projection), SNAIL+ nuclei and expression of vimentin was quantified. Data are expressed as boxplots (min to max) of at least three independent experiments (8 independent acini per condition). MFI, mean fluorescence intensity. Numbers correspond to p values by 1-way Anova with Tukey’s multiple comparisons test, N=3. See also Figures S7.
Consistent with the data above, sEVHYP-treated 3D acini exhibited sustained phosphorylation of Akt (Ser473), persisting up to 21 d after seeding (Figure 6G and H), and appearance of EMT markers, including nuclear accumulation of SNAIL and increased expression of vimentin, compared to controls or sEVNORM-treated acini (Figure 6G and H). Sequential treatment with sEVHYP every 4 d gave similar results, with sustained phosphorylation of Akt (Ser473), nuclear accumulation of SNAIL and increased levels of vimentin, compared to controls (Figure S7A and B).
In addition, sEVHYP treatment disrupted the apical-basal polarity of 3D acini assessed 21 d after seeding, as reflected by deregulated topography of markers of apical polarity (GM130), basolateral polarity (hDlg), cell cortex-cytoskeletal linkages (phosphorylated ezrin, radixin and moesin complex, pERM), as well as disorganized actin filament arrangement (phalloidin) (Figure 7A). 3D acini in control cultures or acini treated with sEVNORM maintained apical-basal polarity and a well-defined arrangement of cell cortex-cytoskeleton and actin filaments (Figure 7A). Sequential addition of sEVHYP every 4 d during a 21-d culture comparably disrupted apical-basal polarity and actin filament organization in 3D acini, whereas sequential treatment with sEVNORM had no effect, compared to controls (Figure 7A, Figure S7C). The above changes in mammary gland morphogenesis, together with deregulated cell proliferation, reduced apoptosis and appearance of EMT markers are all hallmarks of malignant transformation and a potential correlation between ILK expression and breast cancer progression was next investigated. In analysis of publicly available databases, we found that highs levels of ILK strongly correlated with reduced overall survival in patients with triple-negative breast cancer (Figure 7B).
Figure 7. Role of sEVHYP-ILK signaling in mammary gland transformation.

(A) MCF10A acini in 3D culture seeded in the presence of Ctrl, sEVNORM or sEVHYP (added once at t=0) were analyzed after 21 d for expression of apical polarity marker, GM130, basolateral polarity marker, hDlg, plasma membrane marker, pERM or actin cytoskeleton marker, phalloidin by immunohistochemistry. Representative confocal microscopy images (N=3).
(B) Correlation between ILK expression and overall survival in two independent series of triple-negative breast cancer patients. Kaplan-Meier curves for each dataset are indicated. HR, hazard ratio.
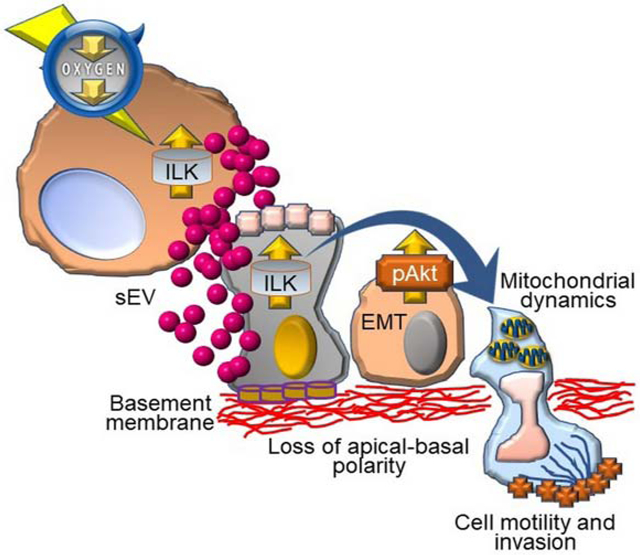
(C) Proposed model for sEVHYP regulation of mitochondrial functions and cell motility in transformation of the normal mammary epithelium via ILK and Akt signaling. See text for details. See also Figures S7.
DISCUSSION
In this study, we have shown that small EV released by hypoxic breast cancer cells, sEVHYP, target mitochondrial dynamics in recipient normal mammary epithelium, activating cycles of fusion and fission, promoting organelle motility and repositioning mitochondria at the cortical cytoskeleton (Figure 7C). In turn, this fuels heightened FA turnover, chemotaxis and mammary cell migration (Figure 7C). Mechanistically, sEVHYP signaling involved transfer of ILK to recipient cells, with downstream activation of Akt and transcriptional upregulation of various cell motility pathways. As a result, co-culture with sEVHYP introduced traits typical of oncogenic transformation in 3D models of mammary gland morphogenesis, with deregulated cell proliferation, reduced apoptosis, appearance of EMT markers and disruption of apical-basal polarity (Figure 7C).
EV are recognized mediators of autocrine signaling for tumor cells, but less is known about their effect(s) on neighboring “normal” cells in the microenvironment (Kalluri and LeBleu, 2020). There is evidence that breast cancer-derived EV, or exosomes reprogram normal epithelial cells and fibroblasts to activate metabolic changes (Fong et al., 2015), mitogenic responses (Antonyak et al., 2011) and oncogenic transformation (Melo et al., 2014), but a paracrine role of sEVHYP on normal mammary epithelial cell motility has not been previously reported. Here, this pathway was unexpectedly centered on sEVHYP activation of mitochondrial dynamics in recipient cells (Eisner et al., 2018), a mechanism not been previously associated with EV signaling, involving upregulation of MFN1 and MFN2, recruitment of Ser616-phosphorylated Drp1 to mitochondria, and increased cycles of organelle fusion and fission.
Aberrant mitochondrial dynamics is commonly observed in tumors (Senft and Ronai, 2016), and mechanistically linked to drug resistance (Zhang et al., 2016), cancer stemness (Wu et al., 2019) and metastatic competence (Caino et al., 2015; Jung et al., 2016). As a component of this process, subcellular mitochondrial motility has attracted attention as a prerequisite of heightened cell motility and invasion (Altieri, 2019) for both normal and transformed cell types. In particular, oncogenes, such as Myc (Agarwal et al., 2019), have been recently shown to hijack a cellular machinery for anterograde and retrograde mitochondrial movements originally characterized in neurons (Birsa et al., 2013) to reposition mitochondria at the leading edge of migration. In turn, this is though to fuel FA turnover, membrane lamellipodia dynamics and heightened cell motility and invasion, in vivo (Caino et al., 2016; Cunniff et al., 2016). A similar paradigm emerged here for sEVHYP signaling, where increased mitochondrial dynamics in recipient cells enabled the redistribution of mitochondria to the cortical cytoskeleton to sustain FA turnover, chemotaxis and cell movements.
A key regulator of this response uncovered here was ILK (Qin and Wu, 2012), which was packaged in breast cancer-derived sEVHYP via an HIF1α-dependent manner. There is precedent for a role of HIF1α in regulating the content of EV produced in hypoxia, including incorporation of MMP13 (Shan et al., 2018) or Notch ligand, Jagged-1 (Gonzalez-King et al., 2017) in released EV that affect metastatic dissemination and angiogenesis, respectively. ILK was originally proposed as a kinase regulating integrin-mediated cell-extracellular matrix polarization and cell motility (Hannigan et al., 1996). However, its kinase activity has remained controversial (Qin and Wu, 2012), and recent studies have suggested that ILK acts non-catalytically to regulate stress fiber formation, cell polarization and cell movements (Vaynberg et al., 2018).
Here, a pool of ILK was localized to mitochondria, whereas targeting of ILK with genetic (siRNA silencing), molecular (L207W mutant) (Vaynberg et al., 2018) or pharmacologic (small molecule Cpd22 inhibitor) approaches suppressed mitochondrial trafficking, organelle dynamics and cell motility. Accordingly, ILK has been recently implicated in blocking retrograde mitochondrial movements and regulate cell invasion via inhibition of RhoT1-TRAK2 complex(es) at the organelle outer membrane (Onodera et al., 2018). Whether a similar mechanism is involved here is currently not known. What is known is that ILK contributes to the activation of Akt (Qin and Wu, 2012), a critical mediator of mitochondrial trafficking and associated cell movements (Caino et al., 2015), potentially by targeting it to the plasma membrane via its binding partner Parvin (Fukuda et al., 2003) that directly binds to Akt (Kimura et al., 2010).
A key consequence of sEVHYP-mediated signaling was a general disruption of normal mammary epithelial morphogenesis, with loss of apical-basal polarity in 3D cultures, persistent cell proliferation with concomitantly reduced apoptosis and acquisition of EMT markers, including nuclear accumulation of SNAIL. Although a role of EV in mammary epithelial morphogenesis has not been previously proposed, there is abundant evidence that the phenotype observed here is one of the earliest events in carcinoma initiation (Martin-Belmonte and Perez-Moreno, 2011). Mechanistically, this is thought to involve deregulated gene expression circuitries maintained by cell-cell contacts (Balda and Matter, 2000), disruption of a developmentally timed cell death-cell proliferation balance (Anderson et al., 2010) and impaired control of cell cycle entry (Chandramouly et al., 2007). The molecular details of how sEVHYP drive oncogenic transformation of the normal mammary epithelium remain to be elucidated. It is intriguing that exposure to sEVHYP triggered extensive transcriptional changes in recipient cells, affecting not only intercellular communication, cell movements and cell death pathways but also NFkB signaling, which has been associated with mammary gland tumorigenesis (Liu et al., 2010). There is also evidence that sustained phosphorylation of Akt, which in our system persisted up to 21 d after co-culture with sEVHYP, contributes to Slug upregulation and EMT induction (Carpenter et al., 2015).
In terms of biological context, the oncogenic changes induced by sEVHYP in “normal” mammary epithelium may contribute to a “field effect” of cancerization (Curtius et al., 2018). Although linked to somatic mutations (Martincorena et al., 2015) and epigenetic changes (Ushijima and Hattori, 2012), it seems plausible that sEVHYP reprogramming of mitochondrial dynamics, with disruption of developmental tissue morphogenesis and induction of EMT (Leung and Brugge, 2012), may expand the footprint of mammary epithelial transformation to enable both local and distant disease recurrence(s) (Gatenby and Gillies, 2008). As a key effector of sEV signaling, this model is consistent with an important oncogenic role of ILK (Hannigan et al., 2005), including in mammary tumorigenesis (Hinton et al., 2008) and linked to abbreviated patient survival (this study). On the other hand, therapeutic targeting of mitochondrial reprogramming (Zhang et al., 2016) as well as ILK (Hannigan et al., 2005) may provide viable strategies to disrupt early pro-tumorigenic changes in the microenvironment (this study) as well as mechanisms of metastatic competence (Caino et al., 2016).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Dario Altieri (daltieri@wistar.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The RNA-seq data was submitted to NCBI GEO under accession number GSE154340.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Breast cancer epithelial MCF7 (female) and T47D (female) cells and normal breast epithelial MCF10A (female) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). MCF7 and T47D cells were maintained in culture in RPMI 1640 medium with 10% fetal bovine serum (FBS) plus 1% streptomycin and penicillin. MCF10A cells were grown in DMEM F12 medium with 5% horse serum (HS), 20 ng/ml epidermal growth factor (EGF), 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin and 1% streptomycin and penicillin. Conditioned media used for cell invasion assays was prepared from exponentially growing cultures of NIH 3T3 cells (ADCC) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4.5 g/liter d-glucose, sodium pyruvate, 10 mM HEPES, and 10% FBS for 48 h. All cell lines were maintained at 37°C.
METHOD DETAILS
Antibodies and reagents.
PKH67 was purchased from Sigma, MitoTracker deep red and CellLight™ Mitochondria-RFP, BacMam 2.0 were obtained from ThermoFisher. Growth Factor Reduced Matrigel was obtained from BD Biosciences (BD No. 354230). Antibodies to Ser616-phosphohry lated Drp1, Drp1, MFN1, MFN2, Ser473-phosphorylated AKT, Thr308-phosphorylated Akt, Akt, EGFR, MMP9, ILK, HIF1α, Calnexin, VDAC, Flotillin 1, E-cadherin, Vimentin, SNAIL, Ki67, cleaved-Caspase 3, phosphorylated Ezrin-Radixin-Moesin (pERM) complex were obtained from Cell Signaling. An antibody to hDlg was from Santa Cruz. An antibody to Tsg101 was from ProteinTech. Antibodies to CD63 and CD9 was from Abcam. An antibody to β-actin was from Sigma. The Akt inhibitor MK2206 (1 μM) was purchased from Selleck Chemicals and ILK inhibitor Cpd22 (600 nM) was purchased from Sigma.
Small extracellular vesicles (sEV) isolation.
MCF7 or T47D cells at 70% of confluency were cultured in RPMI medium with EV-depleted FBS for 24 h in normoxic or hypoxic condition (5% or 1% O2, respectively). At the collection of supernatants, cells were counted and cell death was evaluated by trypan blue staining (death cells < 5%). Cells and debris were removed by sequential centrifugation (350 g for 5 min and 1000 g for 10 min) and the supernatants were concentrated using Amicon Ultra centrifugal filter tubes (Merk Millipore). Larger vesicles were eliminated by centrifugation at 10,000 g for 30 minutes at 4°C. Finally, EV were isolated by qEV size exclusion column (SEC – iZON) as described (Lobb et al., 2015). Briefly, 500 μl aliquots of concentrated supernatant were overlaid on qEV columns followed by elution with PBS, 500 μl phases were harvested in Eppendorf tubes and EV were collected in phases 7-8-9 (Witwer et al., 2013). To confirm the presence of sEV in fraction 7-8-9, the first 15 fractions (500 μl each) were collected and analyzed by NTA (nanoparticle tracking analysis). In particular, the fractions werepooled (1 to 3, 4 to 6, 7 to 9, 10 to 12 and 13 to 15) and analyzed using ZetaView NTA (Particle Metrix). Isolated EV were stored at −80°C before analysis or RNA/protein extraction. As control, fresh RPMI medium was processed as culture medium and stored at −80°C. The concentration and size distribution of particles were analyzed using Nanosight NS300 (Malvern, Instruments Ltd Worcestershire). Unless otherwise noted, 3x108 EV were used per every 104 recipient cells. For internalization experiments, sEV were precipitated by ultracentrifugation (100,000 g for 2 h at 4°C), suspended in 400 μl of diluent C, stained with 5 μl PKH67 for 5 min at 22°C and quenched by addition of 100 μl of 20% BSA. Subsequent sEV isolation was carried out using qEV SEC as described above.
Immuno-affinity depletion of CD63.
Five-hundred μl of isolated sEV were incubated overnight at 4°C with CD63-coated microbeads (10606D, Thermo Fisher) and/or Dynabeads protein A (10001D, Thermo Fisher) following the manufacturer’s instructions. Briefly, 100 μl of magnetic beads were washed with PBS – 0.1% BSA and mixed with sEV solution and incubated overnight. Unbound sEV were collected and transferred to a fresh tube, and concentration and size distribution of CD63-depleted sEV were analyzed using the ZetaView NTA. For protein isolation, CD63-depleted sEV were concentrated by ultracentrifugation at 100.000 g at 4°C for 2 h and then lysed with RIPA buffer. For CD63+ vesicles, RIPA buffer was directly added to the magnetic beads, incubated for 2 h at 4°C. For flow cytometry analyses, CD63+ vesicles coated to CD63-microbeads were stained according to the manufacturer’s specifications. Briefly, sEV were permeabilized with Triton 0.5% for 15 min at 22°C and incubated overnight with a primary antibody to ILK and/or IgG isotype control at 4°C on a sample shaker. The bead-bound sEV were washed twice with PBS – 0.1% BSA and incubated with a fluorescent secondary antibody for 1 h at room temperature on a sample shaker. Data were acquired on a BD Biosciences (BD) FACSCelesta, and analysis after acquisition was performed using FlowJo version 9.9.6 (Tree Star Inc).
Cryo-EM sample preparation and data collection.
A 3-μl aliquot of each sample was applied onto 200-mesh copper grids (Quantifoil R1.2/1.3) that were glow discharged for 60 s. The excess solution was blotted with filter paper for 6 sec using Vitrobot Mark IV (FEI Netherlands) at 4°C and the grids were immediately flash-frozen by rapidly plunging the grid into liquid ethane at - 165°C. CryoEM data for each sample were collected on a Tecnai F20 TEM microscope operated at 200 keV. Images were recorded on Falcon III direct electron detector at a magnification of 25,000x. Each micrograph was generated by averaging individual dose fractionated frames collected at a rate of 40 frames/s for 4 sec exposure. The frames were motion-corrected and summed into a single micrograph. The micrographs collected were in the range of 2.0–4.0 μm under focus.
Plasmid and siRNA transfection.
Small interference RNA (siRNA) experiments were carried out using control, non-targeting small interfering RNA (siRNA) pool (Dharmacon, cat. no. D-001810) or individual siRNA targeting HIFlα (L-004018, Dharmacon), ILK (PDSIRNA2D, Sigma), Aktl (sc-29195, Santa Cruz Biotech) or Akt2 (sc-29197, Santa Cruz Biotech), MFN1 (sc-270320, Santa Cruz Biotech) and Drp1 (sc-43732, Santa Cruz Biotech). For gene silencing experiments, pooled or individual siRNA oligonucleotide sequences were transfected at 10 nM concentrations in the presence of Lipofectamine RNAiMAX in a 1:1 ratio (Invitrogen). Cells were incubated for 48 h, confirmed for target protein knockdown by Western blotting, and processed for subsequent experiments. For rescue experiments, plasmid DNA transfections were performed 48 h after siRNA transfection in MCF10A cells using X-tremeGENE HP DNA transfection reagent (Roche). Plasmids encoding 896 pcDNA3 T7 Akt1 (wild type), 1014 pcDNA3 T7 Aktl K179M T308A S473A (dominant negative) (Addgene) (Ramaswamy et al., 1999) were used. Plasmids encoding WT ILK or loss-of-function L207W ILK mutant were as described (Vaynberg et al., 2018).
Immunofluorescence.
Ten thousand cells were seeded on cover-glasses. MCF7 and T47D cells were transfected with ILK-directed siRNA for 48 h. MCF10A cells were treated with sEV for 24 h, with or without the small molecule Akt inhibitor, MK2206 or ILK inhibitor, Cpd22 and then stained with 300 nM MitoTracker deep red FM dye for 30 min at 37°C. Cells were fixed with 4% paraformaldehyde for 10 min, quenched with 20 mM glycine for green auto-fluorescence for 20 min and then permeabilized with Triton 0.5% for 15 min at 22°C. Samples were saturated with 10% BSA for 1 h at 22°C. Primary antibodies (ILK, SNAIL and Ser616-phosphorylated Drp1, 1:100; CD63, 1:50 in 10% PBS-BSA) were incubated for 18 h at 4 °C, whereas secondary antibodies (1:1000) were incubated for 1 h at 22°C. Nuclei were stained with Hoechst 3342 (Cell Signaling) for 5 min at 22°C and slides were mounted using the Prolong reagent (ThemoFisher Scientific). Confocal images were generated on a Leica TCS SP5 confocal microscope (Leica Microsystems) with a magnification of 40X or 63X. For colocalization experiments, images were acquired with a pixel size <80 nm in xy and a z-stack of 0.12 μm. Images were deconvolved using Hyugens software and co-localization was quantified as Pearson Correlation Index (PCI).
RNA sequencing (RNA-Seq).
Total RNA was extracted from MCF10A cells after 24 h of co-culture with MCF7 cell-derived sEV using Direct-zol RNA MiniPrep kit (Zymo Research). RNA quantity was determined using the Qubit 2.0 Fluorometer (ThermoFisher Scientific) and the quality was validated using the TapeStation RNA ScreenTape (Agilent). One-hundred ng of total RNA was used to prepare a library for Illumina Sequencing using the Quant-Seq 3’mRNA-Seq Library Preparation Kit (Lexogen). Library quantity was determined using qPCR (KAPA Biosystem), and overall library size was determined using the Agilent TapeStation and the DNA High Sensitivity D5000 ScreenTape (Agilent). Equimolar amounts of each sample library were pooled, denatured and Mid-Output, 75 bp paired-end Next Generation Sequencing was carried out on a NextSeq 500 (Illumina). RNA-seq data were aligned using bowtie 2 (Langmead and Salzberg, 2012) against hg19 version of human genome, and RSEM v1.2.12 software (Li and Dewey, 2011) was used to estimate gene-level read counts using Ensemble transcriptome information. Raw gene counts were used in DESeq2 (Love et al., 2014) algorithm to estimate significance of differential expression between group pairs. Expression heatmaps were plotted using mean-centered log2-scaled normalized by DESeq2 expression values. Gene set enrichment analysis was done using QIAGEN’s Ingenuity® Pathway Analysis software (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) on genes that passed nominal p<0.05 using “Disease and Functions” and “Canonical Pathways” options. General cellular functions that passed p<10−5 threshold and pathways that passed p<0.05 threshold were considered significant. Only results with significantly predicted activation state (∣Z-score∣>2) were reported.
Mitochondrial time-lapse videomicroscopy.
To study mitochondrial dynamics two different experimental approaches were utilized. In both cases, MCF10A and/or MCF7 cells (1×104) were seeded on high-optical-quality glass-bottom 35-mm plates (MatTek Corporation) for 24 h. When using MitoTracker, MCF10A cells were co-cultured with sEV and/or treated with small molecule Akt inhibitor, MK2206. For MCF7 cells, cultures were seeded after 48 h of transfection with control non-targeting siRNA (siCtrl) or ILK-directed siRNA (siILK) and then co-cultured with sEV. After 24 h, cells were stained with 300 nM (MCF10A) or 100 nM (MCF7) MitoTracker deep red FM dye for 30 min at 37°C. Alternatively, cells were incubated with 2 μl of CellLight™ Mitochondria-RFP, BacMam 2.0 O.N. before co-culture with sEV and drug treatment.
Using both approaches, cells were imaged on a Leica TCS SP8 X inverted confocal laser scanning microscope using a 63X, 1.40-numerical-aperture (NA) oil objective. Live imaging time-lapse videomicroscopy was performed using a Tokai Hit incubation chamber equilibrated to bidirectional scanning at 8,000 Hz at 37°C with 5% CO2. Images were acquired every 2 sec for a 6-min interval. Individual 12-bit images were acquired using a white-light supercontinuum laser (0.2% at 643 nm) and hybrid detectors at a 4× digital zoom with a pixel size of 70 nm by 70 nm and a step size of 0.260 μm. At least 6 single cells under each condition were collected for analysis. Initial postprocessing of 3D sequences was carried out with Hyugens software to deconvolve the images. Images were imported into ImageJ Fiji and individual mitochondria were automatically tracked using TrackMate plugin (Tinevez et al., 2017) or into Leica LAS X software to study fission and fusion events as described (Wang et al., 2019).
Cortical mitochondria quantification.
MCF10A cells were seeded on coverslip and co-cultured with sEV or transfected with siCtrl or siAkt for 48 h and then co-cultured with sEV or treated with Akt inhibitor, MK2206 or ILK inhibitor, Cpd22. After 24 h, cells were stained with 300 nM MitoTracker deep red FM dye for 30 min at 37°C, washed, and fixed with 4% formaldehyde for 10 min at 22°C. Actin filaments were stained with phalloidin-rhodamine (Molecular Probes) 488 for 1 h at 22°C and nuclei were stained with Hoechst (Molecular Probe) for 5 min at 22°C. Slides were analyzed on a Leica TCS SP5 confocal laser microscope with a 63X oil objective and images were analyzed in ImageJ as described (Agarwal et al., 2019).
Time-lapse videomicroscopy analysis of focal adhesion (FA) dynamics.
MCF10A cells were seeded on high-optical-quality 35 mm glass-bottom plates and transduced with talin-red fluorescent protein (RFP) BacMam virus for 18 h, followed by co-culture with sEV with or without vehicle or ILK inhibitor, Cpd22 for additional 24 h. Time-lapse videomicroscopy was carried out using a Leica TCS SP8 confocal laser scanning microscope system with an HCX PL APO CS 40X, 1.40-NA oil UV objective. Acquisition of live cells using integrated Leica LAS software was performed every 2 min per frame for a total time interval of 30 min. Sequences were imported into ImageJ for further analysis. The initial and final frames were duplicated and assembled as composite images. FA complexes were manually counted and classified (according to the presence in some or all the time frames) into three groups: decaying, newly formed, and stable (merged areas). The analysis was carried out with 10 cells per condition in 3 independent time-lapse experiments using the LASX software package.
ROS and mitochondria depolarization analyses.
To detect total ROS, MCF10A cells were co-cultured with sEV for 24 h, and incubated with 2.5 μM of CellROX Green Reagent (Invitrogen) for 30 min at 37°C, according to the manufacturer’s instructions. After three washes in PBS, pH 7.4, cells were harvested and analyzed on a BD Biosciences FACSCelesta flow cytometer. To evaluate mitochondrial inner membrane depolarization, MCF10A cells co-cultured with sEV for 24 h, were stained with Tetramethylrhodamine (TMRM, Invitrogen, 100 nM) for 30 min at 37°C. Cells were analyzed on a BD Biosciences FACSCelesta flow cytometer and intact cells were gated in the FSC/SSC plot to exclude small debris. The resulting data were plotted on a histogram.
Tumor cell motility and 2D chemotaxis.
For 2D motility studies, MCF10A cells (1x104) were seeded in 4-well Ph+ chambers (Ibidi) in complete medium. For 2D chemotaxis, MCF10A cells (2×104) were placed in 8-μm chemotaxis slides (Ibidi). After 5 h from sEV addition, time-lapse videomicroscopy was performed over a 10-h interval, as described (Agarwal et al., 2019). Stacks were imported into ImageJ Fiji software for analysis, and 30 cells per each condition tested were tracked using the Manual Tracking plugin for ImageJ Fiji in 3 independent experiments. Tracking data were exported into Chemotaxis and Migration Tool v. 2.0 (Ibidi) for graphing and calculation of means of speed and accumulated distance of movement. For analysis of directional cell migration using a wound closure assay, MCF10A cells were seeded on 24 MW and co-cultured with sEV for 24 h. “Wounds” in the cell monolayer were made with a 20-μl pipette tip and cell debris was washed. Time-lapse imaging of migrating cells was performed using a TE300 inverted microscope (Nikon) equipped with an incubator set at 37°C with 5% CO2 and 95% relative humidity. Each image was acquired using a 10× objective of the same fields at 1-h time intervals for a total of 12 h.
Cell migration.
Cell migration and invasion experiments were performed essentially as described (Agarwal et al., 2019), using growth factor-reduced Matrigel-coated 8-μm PET (cell invasion) and PET (cell migration) Transwell chambers (Corning). MCF10A cells were seeded on Transwell filters (105 cells/well) in medium containing 0.1% bovine serum albumin (BSA) with or without sEV. Conditioned medium from NIH 3T3 fibroblasts was placed in the lower chamber as a chemoattractant. Cells were allowed to invade or migrate for 24 h, non-invading cells were removed whereas invaded cells were fixed in methanol. Membranes were mounted in medium containing DAPI (Vector Laboratories) and analyzed by fluorescence microscopy. Ten fields at 10X magnification were collected and the number of invading/migrating cells were counted using ImageJ.
3D acini formation and immunofluorescence.
Three-dimensional (3D) culture of MCF10A cells was performed as described (Debnath et al., 2003). Each well of a μ-Slide 8 Well chamber (Ibidi) was coated with 40 μl of Growth Factor-Reduced Matrigel (BD Biosciences). MCF10A cells (5000/well) were suspended in Assay Medium (DMEM/F12, 2% horse serum, 0.5 μg/ml Hydrocortisone, 100 ng/ml Cholera Toxin, 10 μg/ml Insulin and Pen/Strep) plus 2% Matrigel and 5 ng/ml EGF with or without sEV and seeded on coated Matrigel. Cultures were supplemented every 4 d with assay medium containing 2% Matrigel and 5 ng/ml EGF. sEV were added once at t=0 or, alternatively, every 4 d. Brightfield images were collected at d 7, 14 or 21 using TE300 inverted microscope (Nikon) equipped with an incubator set at 37°C with 5% CO2 and 95% relative humidity with a 10X objective.
At d 21, MCF10A 3D acini were fixed in 4% formalin for 20 min at 22°C and processed for immunofluorescence (Debnath et al., 2003). Briefly, cultures were permeabilized with 0.5% PBS-Triton for 20 min at 4°C, rinsed three times with PBS/glycine, incubated with 200 μl of IF buffer (130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaNs, 0.1% bovine serum albumin, 0.2% Triton X-100, 0.05% Tween-20) plus 10% goat serum for 1 h at 22°C (primary block). For secondary block, 10% goat serum and 20 μg/ml goat anti-mouse F(ab’)2 fragment (Jackson ImmunoResearch) were added to the IF buffer and cells were incubated for additional 40 min. Finally, primary antibodies were diluted in secondary block solution (GM130 and hDlg, 1:50; pERM, Ki67, cleaved caspase 3, pAKT (Ser473), vimentin and SNAIL, 1:100) and incubated for 18 h at 22°C. Isotype-matched secondary antibodies and phalloidin were diluted 1:200 in IF buffer plus 10% goat serum and further incubated for 1 h at 22°C. Nuclei were counterstained with Hoechst 1:500 in PBS for 15 min at 22°C. Images were captured using a Leica TCS SP8 confocal laser scanning microscopy system with an HCX PL APO CS 63×, 1.40-NA oil UV objective, zoom 2X and a pinhole of 0.7 μm.
Bioinformatics analysis of ILK expression in breast cancer patients.
For these experiments, Kaplan Meier curves for ILK were generated (Gyorffy et al., 2010) using two different databases (Liu_2014 (n=126) and Tang_2018 (n=118)). Both cohorts contained patients with triple-negative breast cancer (HER2-, ER-, PGR-). For the survival analysis generated from the Liu_2014 database, the p value was 0.0367 and FDR > 50%. For the survival analysis generated from the Tang_2018 database, the p value was 0.0068 and FDR 50%.
QUANTIFICATION AND STATISTICAL ANALYSIS.
Statistical analysis was performed using GraphPad Prism 8 software (La Jolla, CA). All biological experiments were performed at least three times (all single experiments have a technical duplicate). To compare means for two groups, a Mann Whitney t-test with Tukey’s pos-test was performed. For three or more group comparison, a one-way Anova test was used. To determine how a response is affected by two factors, a two-way Anova test was used.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Ser616-phosphohrylated Drp1 | Cell Signaling | Cat #4494 |
| Mouse monoclonal anti-Drp1 | Cell Signaling | Cat #14647 |
| Rabbit monoclonal anti-MFN1 | Cell Signaling | Cat #14739 |
| Rabbit monoclonal anti-MFN2 | Cell Signaling | Cat #9482 |
| Rabbit monoclonal anti-Ser473-phosphorylated AKT | Cell Signaling | Cat #9271 |
| Rabbit monoclonal anti-Thr308-phosphorylated Akt | Cell Signaling | Cat #4056 |
| Rabbit monoclonal anti-AKT | Cell Signaling | Cat #9272 |
| Rabbit monoclonal anti-EGFR | Cell Signaling | Cat #4267 |
| Rabbit monoclonal anti-MMP9 | Cell Signaling | Cat # 13667 |
| Rabbit monoclonal anti-ILK | Cell Signaling | Cat # 3862 |
| Rabbit monoclonal anti-HIF1α | Cell Signaling | Cat # 36169 |
| Rabbit monoclonal anti-Calnexin | Cell Signaling | Cat # 2679 |
| Rabbit monoclonal anti-VDAC | Cell Signaling | Cat # 4661 |
| Rabbit monoclonal anti-Flottilin1 | Cell Signaling | Cat # 18634 |
| Rabbit monoclonal anti-E-cadherin | Cell Signaling | Cat # 3195 |
| Rabbit monoclonal anti-N-cadherin | Cell Signaling | Cat # 13116 |
| Rabbit monoclonal anti-SNAIL | Cell Signaling | Cat # 3879 |
| Rabbit monoclonal anti-Cleaved-Caspase 3 | Cell Signaling | Cat # 9661 |
| Mouse monoclonal anti-Ki67 | Cell Signaling | Cat # 9449 |
| Rabbit monoclonal anti-pERM | Cell Signaling | Cat # 3726 |
| Rabbit monoclonal anti-Vimentin | Cell Signaling | Cat # 5741 |
| Mouse monoclonal anti-hDlg (SAP97) | Santa Cruz | Cat # sc-9961 |
| Rabbit monoclonal anti-Tsg101 | Protein Tech | Cat # 14497-1-AP |
| Mouse monoclonal anti-CD63 | Abcam | Cat # ab59479 |
| Rabbit monoclonal anti-CD9 | Abcam | Cat # ab223052 |
| Mouse monoclonal anti-β-actin | Sigma | Cat # A5441 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human Epidermal growth factor | Peprotech | Cat # AF-100-15 |
| Hydrocortisone | Sigma | Cat # H0888 |
| Cholera toxin | Sigma | Cat # C8052 |
| Growth Factor Reduced Matrigel | BD Biosciences | Cat # 354230 |
| Talin-red fluorescent protein (RFP) BacMam | ThermoFisher | Cat # C10612 |
| PKH67 Green Fluorescent Cell Linker | Sigma | Cat # PKH67GL-1KT |
| MitoTracker deep red | ThermoFisher | Cat # M22426 |
| CellLight™ Mitochondria-RFP | ThermoFisher | Cat # C10601 |
| CellROX Green Reagent | ThermoFisher | Cat # C10444 |
| TMRM | ThermoFisher | Cat #M20036 |
| RNAiMAX | ThermoFisher | Cat #13778100 |
| X-tremeGENE HP | Sigma | Cat #366244001 |
| MK-2206 AKT inhibitor | Selleck Chemicals | Cat S1078; CAS: 1032350-13-2 |
| Cpd22 ILK inhibitor | Sigma | Cat #407331–5MG |
| Critical Commercial Assays | ||
| qEV size exclusion column | IZON | qEVoriginal / 70nm |
| Deposited Data | ||
| RNA-seq data | NCBI GEO | Accession number GSE154340 |
| Experimental Models: Cell Lines | ||
| MCF7 | ATCC | ATCC® HTB-22 |
| T47D | ATCC | ATCC® HTB-133™ |
| MCF10A | ATCC | TCC® CRL-10317™ |
| Oligonucleotides | ||
| siRNA targeting HIF1α | Dharmacon | Cat # L-004018 |
| siRNA targeting ILK | Sigma | Cat # PDSIRNA2D |
| siRNA targeting AKT1 | Santa Cruz Biotech | Cat # sc-29195 |
| siRNA targeting AKT2 | Santa Cruz Biotech | Cat # sc-29197 |
| siRNA targeting MFN1 | Santa Cruz Biotech | Cat # sc-270320 |
| siRNA targeting DRP1 | Santa Cruz Biotech | Cat # sc-43732 |
| Recombinant DNA | ||
| 896 pcDNA3 T7 Akt1 | Ramaswamy et al., 1999 | Addgene Cat #9003 |
| 1014 pcDNA3 T7 Akt1 K179M T308A S473A | Ramaswamy et al., 1999 | Addgene Cat #9031 |
| ILK WT | Vaynberg et al., 2018 | N/A |
| L207W ILK mutant | Vaynberg et al., 2018 | N/A |
| Software and Algorithms | ||
| ImageJ | Commercial | https://imagej.nih.gov/ij |
| Prism 8 | Commercial | https://www.graphpad.com/scientificsoftware/prism |
| TrackMate plugin | Commercial | Tinevez et al., 2017 |
| Chemotaxis and Migration Tool v. 2.0 | Ibidi | https://ibidi.com/chemotaxis-analysis/171-chemotaxis-and-migration-tool.html |
| Bowtie 2 | (Langmead and Salzberg, 2012) | N/A |
| RSEM v1.2.12 software | Li and Dewey, 2011 | N/A |
| DESeq2 algorithm | Love et al., 2014 | N/A |
| QIAGEN's Ingenuity® Pathway Analysis software | IPA®, QIAGEN | www.qiagen.com/ingenuity |
HIGHLIGHTS.
Hypoxic breast cancer cells release signaling small extracellular vesicles (sEV)
sEV activate mitochondrial dynamics in recipient normal mammary epithelium
Mitochondrial reprogramming stimulates ILK-Akt-dependent epithelial cell migration
sEV disrupt normal mammary gland morphogenesis with hallmarks of malignancy
ACKNOWLEDGMENTS
We thank James Hayden and Frederick Keeney for assistance with time-lapse videomicroscopy in the Imaging Core Facility as well as the Genomics and Flow Cytometry Core Facilities at The Wistar Institute. This work was supported by National Institutes of Health (NIH) grants P01 CA140043 (D.C.A. and L.R.L.), R35 CA220446 (D.C.A.), R01 HL58758 (J.Q.), 2 PO1 HL073311 (J.Q. and E.F.P.) and R50 CA211199 (A.V.K.), by the Italian Minister of Health (GR2011-02351626) (V.V.). Support for Core Facilities utilized in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Footnotes
DECLARATION OF INTEREST
The authors declare no competing financial interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agarwal E, Altman BJ, HoSeo J, Bertolini I, Ghosh JC, Kaur A, Kossenkov AV, Languino LR, Gabrilovich DI, Speicher DW, et al. (2019). Myc Regulation of a Mitochondrial Trafficking Network Mediates Tumor Cell Invasion and Metastasis. Mol Cell Biol 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC (2019). Mitochondrial dynamics and metastasis. Cell Mol Life Sci 76, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LR, Sutherland RL, and Butt AJ (2010). BAG-1 overexpression attenuates luminal apoptosis in MCF-10A mammary epithelial cells through enhanced RAF-1 activation. Oncogene 29, 527–538. [DOI] [PubMed] [Google Scholar]
- Anderson RG, Ghiraldeli LP, and Pardee TS (2018). Mitochondria in cancer metabolism, an organelle whose time has come? Biochim Biophys Acta Rev Cancer 1870, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, Holowka DA, and Cerione RA (2011). Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A 108, 4852–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda MS, and Matter K (2000). The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 19, 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Higgs N, Lopez-Domenech G, and Kittler JT (2013). Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem Soc Trans 41, 1525–1531. [DOI] [PubMed] [Google Scholar]
- Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, et al. (2015). PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci U S A 112, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, Hayden JE, Vaira V, Morotti A, Ferrero S, et al. (2016). A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun 7, 13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter RL, Paw I, Dewhirst MW, and Lo HW (2015). Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 34, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, Abad PC, Knowles DW, and Lelievre SA (2007). The control of tissue architecture over nuclear organization is crucial for epithelial cell fate. J Cell Sci 120, 1596–1606. [DOI] [PubMed] [Google Scholar]
- Cornell L, Wander SA, Visal T, Wagle N, and Shapiro GI (2019). MicroRNA-Mediated Suppression of the TGF-beta Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep 26, 2667–2680 e2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunniff B, McKenzie AJ, Heintz NH, and Howe AK (2016). AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27, 2662–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtius K, Wright NA, and Graham TA (2018). An evolutionary perspective on field cancerization. Nat Rev Cancer 18, 19–32. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Eisner V, Picard M, and Hajnoczky G (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O'Connor ST, Li S, Chin AR, et al. (2015). Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Guo L, Shi X, and Wu C (2003). CH-ILKBP regulates cell survival by facilitating the membrane translocation of protein kinase B/Akt. J Cell Biol 160, 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, and Gillies RJ (2008). A microenvironmental model of carcinogenesis. Nat Rev Cancer 8, 56–61. [DOI] [PubMed] [Google Scholar]
- Gonzalez-King H, Garcia NA, Ontoria-Oviedo I, Ciria M, Montero JA, and Sepulveda P (2017). Hypoxia Inducible Factor-1alpha Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem Cells 35, 1747–1759. [DOI] [PubMed] [Google Scholar]
- Greaves M (2015). Evolutionary determinants of cancer. Cancer Discov 5, 806–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, and Szallasi Z (2010). An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123, 725–731. [DOI] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, and Dedhar S (2005). Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer 5, 51–63. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, and Dedhar S (1996). Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379, 91–96. [DOI] [PubMed] [Google Scholar]
- Hinton CV, Avraham S, and Avraham HK (2008). Contributions of integrin-linked kinase to breast cancer metastasis and tumourigenesis. J Cell Mol Med 12, 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R, and Sedger LM (2019). Intercellular Vesicular Transfer by Exosomes, Microparticles and Oncosomes - Implications for Cancer Biology and Treatments. Front Oncol 9, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JU, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, and Sitcheran R (2016). NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr Biol 26, 3288–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and LeBleu VS (2020). The biology, function, and biomedical applications of exosomes. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Murakami T, Kizaka-Kondoh S, Itoh M, Yamamoto K, Hojo Y, Takano M, Kario K, Shimada K, and Kobayashi E (2010). Functional molecular imaging of ILK-mediated Akt/PKB signaling cascades and the associated role of beta-parvin. J Cell Sci 123, 747–755. [DOI] [PubMed] [Google Scholar]
- King HW, Michael MZ, and Gleadle JM (2012). Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 12, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, and Belting M (2013). Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A 110, 7312–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SL, Hsu EC, Chou CC, Chuang HC, Bai LY, Kulp SK, and Chen CS (2011). Identification and characterization of a novel integrin-linked kinase inhibitor. J Med Chem 54, 6364–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehuede C, Dupuy F, Rabinovitch R, Jones RG, and Siegel PM (2016). Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res 76, 5201–5208. [DOI] [PubMed] [Google Scholar]
- Leung CT, and Brugge JS (2012). Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature 482, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, Ojeifo J, Jiao X, Yeow WS, Katiyar S, et al. (2010). The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res 70, 10464–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb RJ, Becker M, Wen SW, Wong CS, Wiegmans AP, Leimgruber A, and Moller A (2015). Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles 4, 27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, and Perez-Moreno M (2011). Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 12, 23–38. [DOI] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Sugimoto H, O'Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, et al. (2014). Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y, Nam JM, Horikawa M, Shirato H, and Sabe H (2018). Arf6-driven cell invasion is intrinsically linked to TRAK1-mediated mitochondrial anterograde trafficking to avoid oxidative catastrophe. Nat Commun 9, 2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HK, Hong JH, Oh YT, Kim SS, Yin J, Lee AJ, Chae YC, Kim JH, Park SH, Park CK, et al. (2019). Interplay between TRAP1 and Sirtuin-3 Modulates Mitochondrial Respiration and Oxidative Stress to Maintain Stemness of Glioma Stem Cells. Cancer Res 79, 1369–1382. [DOI] [PubMed] [Google Scholar]
- Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, Hendrix A, and Mathivanan S (2019). Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res 47, D516–D519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, and Wu C (2012). ILK: a pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr Opin Cell Biol 24, 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, and Sellers WR (1999). Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 96, 2110–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues G, Hoshino A, Kenific CM, Matei IR, Steiner L, Freitas D, Kim HS, Oxley PR, Scandariato I, Casanova-Salas I, et al. (2019). Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos ET, Condeelis JS, and Patsialou A (2011). Chemotaxis in cancer. Nat Rev Cancer 11, 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. (2015). Pyruvate carboxylase is critical for nonsmall-cell lung cancer proliferation. J Clin Invest 125, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft D, and Ronai ZA (2016). Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol 39, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, You B, Shi S, Shi W, Zhang Z, Zhang Q, Gu M, Chen J, Bao L, Liu D, et al. (2018). Hypoxia-Induced Matrix Metalloproteinase-13 Expression in Exosomes from Nasopharyngeal Carcinoma Enhances Metastases. Cell Death Dis 9, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum DP, and Polyak K (2015). Tumorigenesis: it takes a village. Nat Rev Cancer 15, 473–483. [DOI] [PubMed] [Google Scholar]
- Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Ushijima T, and Hattori N (2012). Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res 18, 923–929. [DOI] [PubMed] [Google Scholar]
- Vaynberg J, Fukuda K, Lu F, Bialkowska K, Chen Y, Plow EF, and Qin J (2018). Non-catalytic signaling by pseudokinase ILK for regulating cell adhesion. Nat Commun 9, 4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, and Haigis MC (2016). Mitochondria and Cancer. Cell 166, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, Chaturvedi P, Green JJ, and Semenza GL (2014). Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci U S A 111, E3234–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Agarwal E, Bertolini I, Ghosh JC, Seo JH, and Altieri DC (2019). IDH2 reprograms mitochondrial dynamics in cancer through a HIF-1alpha-regulated pseudohypoxic state. FASEB J, fj201901366R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, Nolte-’t Hoen EN, Piper MG, Sivaraman S, Skog J, et al. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MJ, Chen YS, Kim MR, Chang CC, Gampala S, Zhang Y, Wang Y, Chang CY, Yang JY, and Chang CJ (2019). Epithelial-Mesenchymal Transition Directs Stem Cell Polarity via Regulation of Mitofusin. Cell Metab 29, 993–1002 e1006. [DOI] [PubMed] [Google Scholar]
- Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, et al. (2016). Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 126, 1834–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data was submitted to NCBI GEO under accession number GSE154340.