

Abstract
Therapeutic cancer vaccines require robust cellular immunity for the efficient killing of tumor cells, and recent advances in neoantigen discovery may provide safe and promising targets for cancer vaccines. However, elicitation of T cells with strong antitumor efficacy requires intricate multistep processes that have been difficult to attain with traditional vaccination approaches. Here, a multifunctional nanovaccine platform has been developed for direct delivery of neoantigens and adjuvants to lymph nodes (LNs) and highly efficient induction of neoantigen-specific T cell responses. A PEGylated reduced graphene oxide nanosheet (RGO-PEG, 20–30 nm in diameter) is a highly modular and biodegradable platform for facile preparation of neoantigen vaccines within 2 h. RGO-PEG exhibits rapid, efficient (15–20% ID/g), and sustained (up to 72 h) accumulation in LNs, achieving >100-fold improvement in LN-targeted delivery, compared with soluble vaccines. Moreover, RGO-PEG induces intracellular reactive oxygen species in dendritic cells, guiding antigen processing and presentation to T cells. Importantly, a single injection of RGO-PEG vaccine elicits potent neoantigen-specific T cell responses lasting up to 30 days and eradicates established MC-38 colon carcinoma. Further combination with anti-PD-1 therapy achieved great therapeutic improvements against B16F10 melanoma. RGO-PEG may serve a powerful delivery platform for personalized cancer vaccination.
Keywords: reduced graphene oxide, nanosheet, cancer vaccine, cancer immunotherapy, positron emission tomography
Graphical Abstract
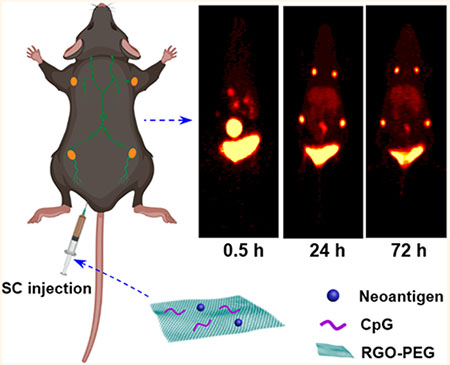
T cell proliferation requires a vaccination strategy that can deliver a high dose of antigen to antigen-presenting cells (APCs) in lymphoid tissues and sustain antigen presentation for at least several days1,2 However, exogenous antigens are rapidly degraded upon phagocytosis by dendritic cells (DCs) due to acidification of endolysosomes and protease activation during the normal DC maturation process, which leads to excessive antigen processing, loss of MHC-I epitopes, and CD4+ T cell-skewed cellular immunity.3–6 In addition, patient-specific neoantigens derived from tumor mutations have recently emerged as promising targets.7–10 Unlike shared tumor antigens, which have been the traditional target of cancer vaccines, neoantigens are absent in healthy tissues, thus potentially offering a more immunogenic and safer alternative for specific elimination of cancer cells.7 Nevertheless, the major hurdles to overcome for neoantigen vaccines include how to reduce the protracted time window currently required from the initial tissue biopsy to the production of patient-specific vaccines capable of eliciting concerted CD4+ and CD8α+ T cell responses.7–11 To address these challenges, various vaccine delivery technologies are currently pursued, including liposomes, polymeric and inorganic nanoparticles (NPs), nanodiscs, albumin “hitchhiking” systems, and scaffolds.12–18 However, it remains to be seen how to design a multifunctional, modular vaccine platform that can be plugged in with patient-specific neoantigens in a convenient manner, while simultaneously supporting antigen transport to lymphoid tissues and antigen presentation on DCs for potent T cell responses.
Here, we report the development of reduced graphene oxide nanosheets (RGO) designed as a multifunctional vaccine platform for neoantigen-based cancer vaccination (Figure 1a). Notably, graphene oxide nanosheets (GO) as well as RGO have been widely studied for drug delivery applications, including photothermal-enhanced immunotherapies and cancer vaccines;19–24 however, most studies to date have utilized GO or RGO that are over 100 nm, and potential concerns of long-term exposure remain, as they are thought to be non-biodegradable. We have developed a PEGylated RGO nanoplatform (RGO-PEG) sized 20 to 30 nm in diameter as a convenient, biodegradable nanoplatform that allows for facile loading of neoantigens and CpG oligodeoxynucleotide (CpG ODN, a Toll-like receptor-9 agonist) within 2 h. Using positron emission tomography (PET imaging) with radioisotope 64Cu, we show that RGO-PEG drastically improves vaccine delivery to lymph nodes (LNs) (>100-fold enhancement compared with soluble vaccines) after subcutaneous vaccination. Furthermore, RGO-PEG triggers intracellular reactive oxygen species (ROS) in DCs, resulting in alkalization of endolysosomes and strong and sustained antigen presentation to T cells. When administered in vivo, a single dose of RGO-PEG vaccination elicits potent and durable neoantigen-specific CD8α+ T cell responses and eliminates established tumors. Overall, RGO-PEG is a promising multifunctional nanoplatform ideally suited for personalized cancer vaccination.
Figure 1.
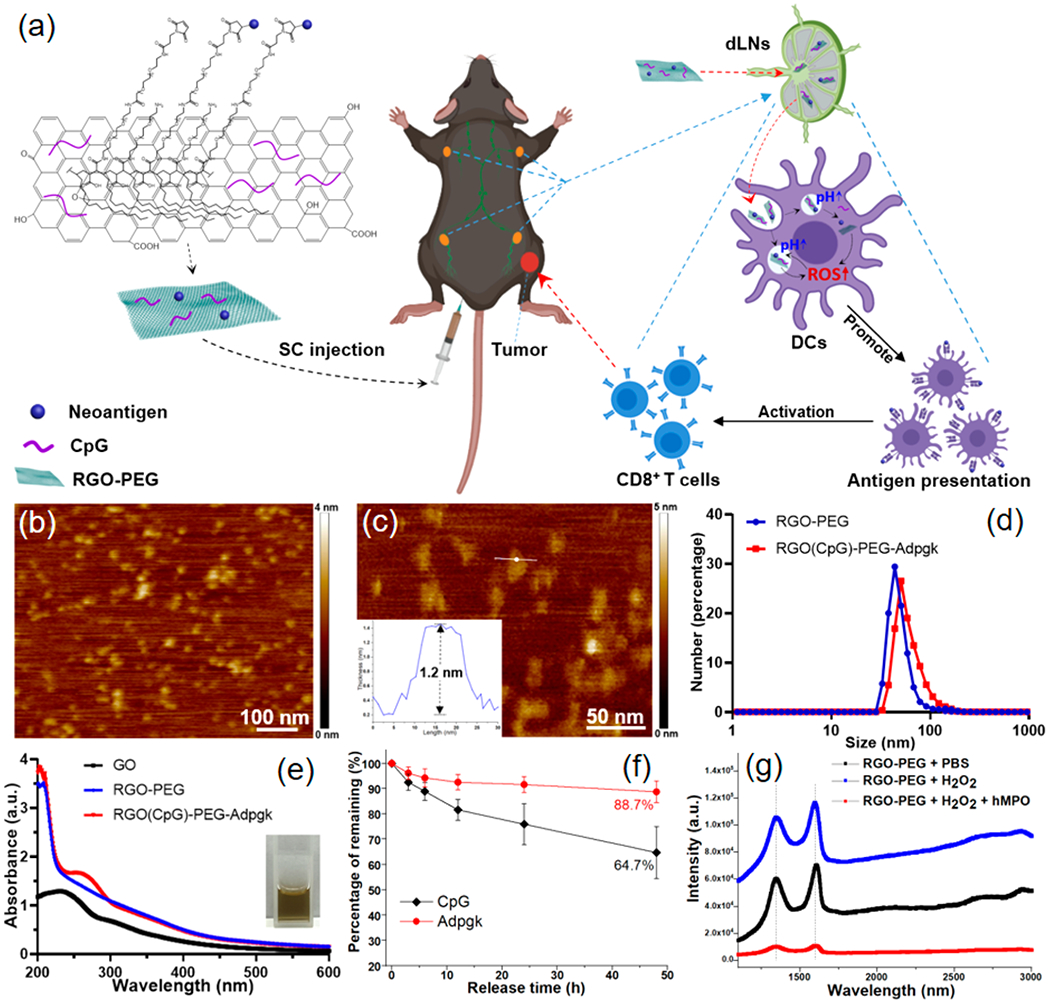
RGO-PEG nanoplatform for cancer vaccination. (a) Schematic illustration of RGO(CpG)-PEG-neoantigen for LN-targeted delivery of antigens and adjuvants. RGO-PEG nanoplatform coloaded with neoantigen peptides and CpG is administered subcutaneously, leading to efficient delivery to APCs in local LNs, generation of intracellular ROS, and elicitation of robust antitumor T cell immunity. (b, c) AFM images and height analysis (inset) of RGO-PEG. (d) Hydrodynamic size analysis of RGO-PEG and RGO(CpG)-PEG-Adpgk. (e) UV–vis absorption spectrum of GO, RGO-PEG, and RGO(CpG)-PEG-Adpgk. (f) Release profile of Adpgk and CpG from RGO(CpG)-PEG-Adpgk kept in PBS at RT. (g) Raman spectra of RGO-PEG, RGO-PEG incubated with H2O2 (50 mM), and RGO-PEG incubated with human myeloperoxidase (hMPO, 1 mg/mL) + H2O2 (50 mM) at 37 °C for 72 h.
RESULTS AND DISCUSSION
Synthesis and Characterization of RGO-PEG Nanoplatform.
We have synthesized an RGO(CpG)-PEG-neoantigen nanoplatform (RGO vaccine) for personalized cancer immunotherapy (Figure 1a). Briefly, RGO was synthesized from GO nanosheets by thermal reduction, followed by surface PEGylation to improve its solubility and stability in physiological conditions.25,26 RGO-PEG was visualized by atomic force microscopy (AFM) and transmission electron microscopy (TEM) (Figures 1b,c and S1a), revealing their size of 20–30 nm with a thickness of ~1.2 nm (Figure 1c, inset). Neoantigen peptides (modified with a cysteine-serine-serine linker) were conjugated to RGO-PEG (with maleimide groups at the end of PEG), followed by simple adsorption of CpG mediated by π–π stacking interactions on the surfaces of RGO. Notably, RGO-PEG can be stored up to 1 year after synthesis, and coloading of neoantigens and CpG is completed in less than 2 h, thus offering a convenient platform for production of patient-specific neoantigen vaccines. After RGO-PEG was coloaded with CpG and Adpgk neoantigen peptide derived from MC-38 colon carcinoma,27 the average hydrodynamic size of RGO-PEG increased from 42.8 ± 14.1 nm to 50.6 ± 19.4 nm (Figure 1d), and the surface zeta potential became more negative (from −10.7 mV to −24.9 mV, Figure S1b). Two distinct absorption peaks at 205 and 260 nm (characteristic absorption peaks for peptide and DNA, respectively) were observed in the UV–vis spectrum of RGO(CpG)-PEG-Adpgk, suggesting the successful loading of Adpgk and CpG (Figure 1e). Liquid chromatography–mass spectrometry (LC-MS) and UV–vis spectrum analysis confirmed a fairly high drug loading capacity of RGO-PEG with simultaneous loading of 15.4 μg Adpgk and 34.1 μg CpG per 100 μg of RGO-PEG (Figure S1c–e) and steady drug release profiles over at least 48 h in vitro (Figure 1f). As a recent study has reported biodegradation of GO by myeloperoxidase (MPO) secreted from human neutrophils,28 we examined whether biodegradation of RGO-PEG was feasible. Raman spectroscopic analyses showed that plain RGO-PEG incubated with or without H2O2 had prominent D and G bands (Figure 1g), whereas RGO-PEG incubated with H2O2 and human MPO for 3 days exhibited markedly decreased intensities of D and G bands, with the ratio of D and G bands increasing from 0.79 to 1.31, indicating hMPO-mediated oxidation and defect formation on the surfaces of RGO-PEG. Overall, these results indicate RGO-PEG may serve as a promising nanocarrier for antigens and adjuvant molecules.
Dendritic Cell Activation and Antigen Cross-Presentation.
We next examined DC activation with RGO-PEG. Murine bone marrow-derived dendritic cells (BMDCs) were incubated with RGO-PEG, CpG, or RGO(CpG)-PEG and analyzed for the expression of costimulatory markers and cytokines. All formulations exhibited high biocompatibility with BMDCs (Figure S2). Whereas blank RGO-PEG induced minimal DC activation, RGO(CpG)-PEG promoted significant up-regulation of CD40, CD80, and CD86 and robust secretion of TNFα and IL-12p70 (Figure 2a,b). Compared with soluble CpG, RGO(CpG)-PEG induced significantly higher levels of costimulatory markers and cytokines. The RGO nanoplatform also significantly improved DC uptake of antigen peptide, compared with free peptide (Figure S2c). Confocal microscopy revealed that RGO-PEG-Cy5 was localized in endolysosomes in BMDCs at the 1 h time point (Figure 2c), but by 24 h, the RGO-PEG-Cy5 signal was primarily localized in the cytosol with minimal overlap with the Lysotracker signal. Importantly, significantly increased endolysosomal pH was detected in BMDCs treated with RGO-PEG, RGO(CpG)-PEG, and lipopolysaccharide (LPS) (used as a positive control), as shown by the decreased LysoSensor fluorescence (Figure 2d and Figure S3a). Interestingly, both RGO-PEG and RGO(CpG)-PEG significantly increased the intracellular levels of ROS in BMDCs and bone marrow derived macrophages (BMDMs) (P < 0.05, Figure 2e, Figure S3b), in line with a recent report showing increased intracellular ROS among macrophages treated with graphene oxide.29 When we added N-acetyl-l-cysteine (NAC, a ROS scavenger) to the culture, NAC significantly decreased intracellular ROS (Figure 2e) and abrogated RGO-PEG- and RGO(CpG)-PEG-mediated alkalization of endolysosomes (Figure 2d and Figure S3a). We also validated these results by using another pH-sensitive dye, SNARF-4F, as well as by showing that the environmental pH, rather than ROS or RGO-PEG themselves, was responsible for changing the LysoSensor signal (Figure S3c,d). Taken together, these results show that RGO-PEG increases intracellular ROS, leading to the increased endolysosomal pH, probably due to ROS-mediated consumption of protons in endolysosomes.30
Figure 2.
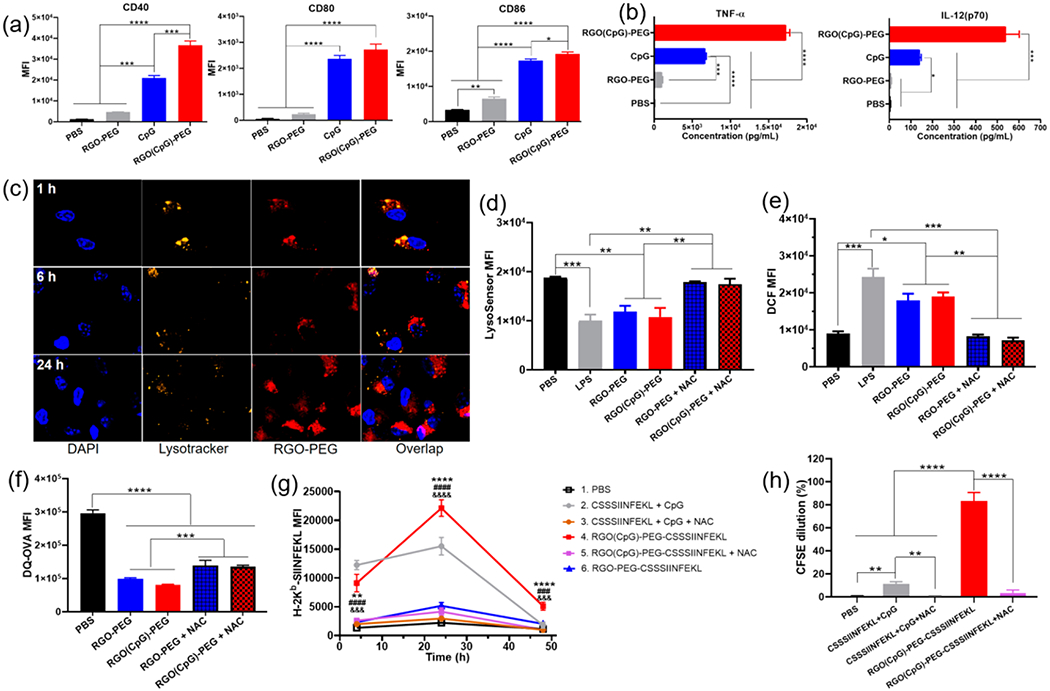
RGO-PEG promotes intracellular ROS, antigen presentation on DCs, and induction of CD8α+ T cells. (a) CD40, CD80, and CD86 expression levels on BMDCs and (b) TNF-α and IL-12(p70) secretion by BMDCs incubated with PBS, RGO-PEG, soluble CpG, or RGO(CpG)-PEG for 8 h. (c) Confocal microscopy images of BMDCs incubated with RGO-PEG-Cy5 (red). BMDCs were stained with Lysotracker dye. (d) LysoSensor fluorescence intensity and (e) intracellular DCF; (f) DQ-OVA fluorescence intensity in BMDCs incubated with the indicated formulations with or without ROS inhibitor NAC (5 mM) for 24 h (d, f) or 4 h (e). (g) SIINFEKL-H-2Kb presentation was quantified on BMDCs after incubation with the indicated formulations for 24 h. *, #, and & indicate statistical differences between group 4 and group 2 (*), 5 (&), or 6 (#). (h) CFSE dilution of OT-I CD8α+ T cells after 48 h of coculture with BMDCs pretreated as in (g). Data represent mean ± SEM from a representative experiment (n = 3) from three independent experiments (a–e) or two independent experiments (f–h). Data were analyzed by one-way ANOVA (a, b, d–f, h) or two-way ANOVA (g) with Tukey’s HSD multiple comparison post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next studied the downstream processes of antigen presentation and cross-priming of CD8α+ T cells. BMDCs cultured with self-quenched DQ-OVA (based on a model antigen ovalbumin) exhibited a high BODIPY fluorescence signal, indicating unquenching of the fluorescence signal due to degradation of OVA (Figure 2f).31 In contrast, BMDCs cultured with DQ-OVA in the presence of RGO-PEG or RGO(CpG)-PEG exhibited significantly decreased BODIPY fluorescence (P < 0.0001), which was somewhat reversed by adding NAC (P < 0.001, Figure 2f). These results show that RGO prevents rapid degradation of antigens in endolysosomes, a prerequisite for efficient presentation of MHC-I-restricted peptides and cross-priming of CD8α+ T cells.5,6 Indeed, BMDCs treated with RGO(CpG)-PEG-CSSSIINFEKL displayed significantly higher levels of SIINFEKL-MHC-I at 24 h (P < 0.0001) and 48 h (P < 0.0001) (Figure 2g), compared with the soluble CSSSIINFEKL peptide + CpG group. In contrast, antigen presentation was compromised by RGO-PEG-CSSSIINFEKL without CpG or by the addition of NAC to RGO(CpG)-PEG-CSSSIINFEKL (P < 0.001, Figure 2g), suggesting crucial roles of both CpG and intracellular ROS in antigen presentation. Furthermore, when CFSE-labeled SIINFEKL-specific OT-I CD8α+ T cells were cocultured with BMDCs pretreated as in Figure 2g, we observed a 7.9-fold greater OT-I CD8α+ T cell proliferation in the RGO(CpG)-PEG-CSSSIINFEKL group (P < 0.0001, Figure 2h and Figure S4), compared with the soluble peptide + CpG group. Notably, NAC added to the coculture abrogated CD8α+ T cell proliferation. Taken all together, RGO(CpG)-PEG induces potent CD8α+ T cell cross-priming in vitro, in part by promoting DC maturation, intracellular ROS generation, endolysosomal alkalization, and antigen presentation.
In Vivo PET Imaging and Biodistribution.
We investigated whether the small size of RGO-PEG allows its lymphatic transport to local dLNs.32 Radioisotope 64Cu (t1/2 = 12.7 h) and chelator NOTA33 were conjugated to Adpgk neoantigen peptide or RGO(CpG)-PEG-Adpgk and administrated subcutaneously (SC) in the tail base of C57BL/6 mice. We performed serial PET scans over 72 h and quantified the 64Cu signal in the draining LNs and major organs. In mice administered with 64Cu-NOTA-Adpgk, no obvious free peptide signal was detected in dLNs throughout 72 h (Figure 3a); however, a high 64Cu signal was observed in kidneys and bladder at 0.5 and 3 h postinjection (PI) time points, suggesting rapid renal clearance. In stark contrast, SC administration of 64Cu-NOTA-RGO(CpG)-PEG-Adpgk resulted in bright signal in inguinal LNs as well as in the secondary axillary dLNs, starting 3 h PI and reaching a peak at 48 h PI (19.9% ID/g and 16.8% ID/g, respectively), followed by sustained signal up to 72 h (18.7% ID/g and 16.6% ID/g, respectively. Figure 3a,b). Furthermore, at 72 h PI, we detected a significantly brighter and broader 64Cu signal at the injection site of 64Cu-NOTA-RGO(CpG)-PEG-Adpgk (26.7% ID/g), compared with that of 64Cu-NOTA-Adpgk (5.5% ID/g) (Figure 3a,b). Throughout the study, 64Cu-NOTA-RGO-(CpG)-PEG-Adpgk exhibited <2.0% ID/g total uptake in blood and other major organs. Ex vivo quantification of 64Cu radioactivity at 72 h PI revealed 106-fold and 136-fold higher 64Cu-NOTA-RGO(CpG)-PEG-Adpgk signal in inguinal and axillary LNs (25.5% ID/g and 20.4% ID/g, respectively), compared with 64Cu-NOTA-Adpgk (0.24% ID/g and 0.15% ID/g, respectively) (P < 0.0001, Figure 3c).
Figure 3.
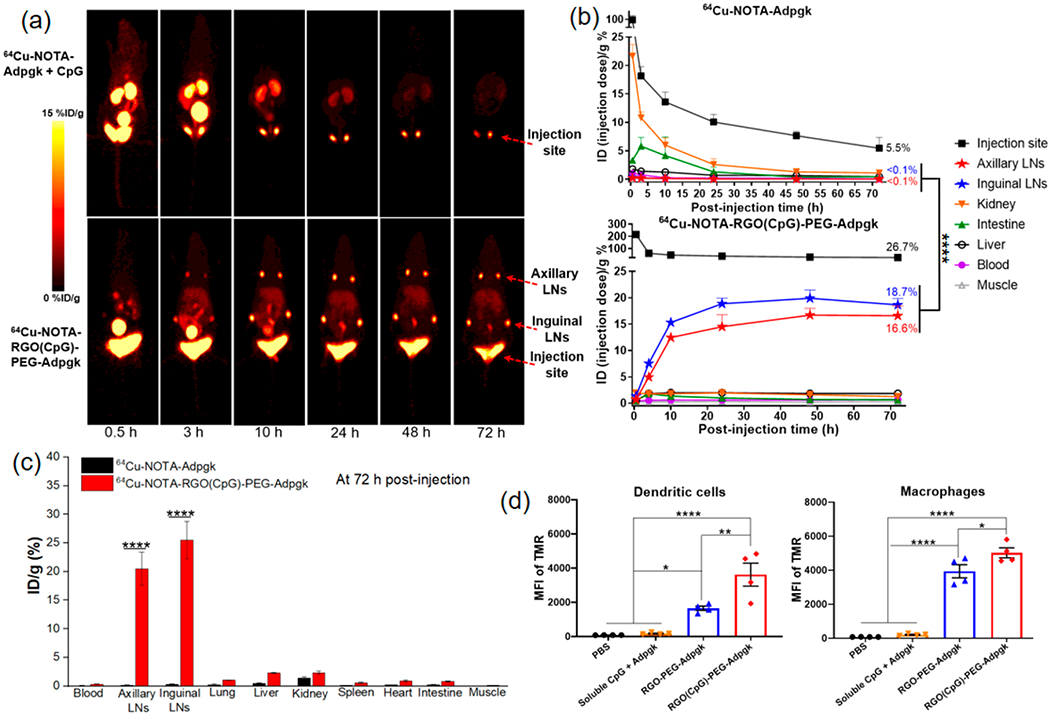
RGO-PEG promotes efficient antigen delivery to APCs in LNs. (a) Serial PET images of C57BL/6 mice after SC administration of 64Cu-NOTA-Adpgk + CpG and 64Cu-NOTA-RGO(CpG)-PEG-Adpgk. Inguinal and axillary LNs and injection site are indicated by red arrowheads. (b) Time–radioactivity curves of injection site, inguinal and axillary LNs, kidney, intestine, liver, blood, and muscle. (c) 64Cu radioactivity in major organs at 72 h postinjection. (d) Cellular uptake of Adpgk and RGO(CpG)-PEG-Adpgk in CD45+CD11c+ DCs and CD45+F4/80+ macrophages in inguinal LNs at 24 h postinjection. Data represent mean ± SEM from a representative experiment (n = 4) from two independent experiments (a–d). Data were analyzed by one-way ANOVA (b, d) or two-way ANOVA (c) with Tukey’s HSD multiple comparison post hoc test. *P < 0.05, **P < 0.01, ****P < 0.0001.
We also labeled Adpgk peptide and RGO(CpG)-PEG-Adpgk with tetramethylrhodamine (TMR) and analyzed their cellular uptake in inguinal LNs via flow cytometric analysis. At 24 h after SC administration, RGO(CpG)-PEG-Adpgk-TMR mediated 21-fold and 25-fold greater antigen uptake among DCs and macrophages, respectively, compared with soluble Adpgk-TMR + CpG (P < 0.0001, Figure 3d). Notably, RGO-PEG-Adpgk-TMR without CpG exhibited reduced antigen delivery to DCs and macrophages. To test whether the small size of RGO (20–30 nm) was crucial for LN-targeted delivery, we prepared graphene oxide with comparable drug loading and surface chemistry but with a larger diameter (200–300 nm, 64Cu-NOTA-GO(CpG)-PEG-Adpgk), which formed a depot at the injection site with minimal (<0.1% ID/g) delivery to dLNs after SC administration (Figure S5). We also investigated whether robust LN delivery of RGO-PEG was mediated by “hitchhiking” onto albumin, as recently reported for amphiphilic conjugates binding to hydrophobic domains in albumin.12 However, our GPC analyses did not reveal any obvious binding or interaction between RGO-PEG and mouse albumin (Figure S6). Taken together, RGO-PEG with a small diameter (20–30 nm) substantially enhances vaccine delivery to APCs in dLNs.
Prophylactic and Therapeutic Studies in MC-38 Tumor Model.
As RGO-PEG efficiently activates DCs and CD8α+ T cells in vitro and promotes LN draining in vivo, we next assessed its potency to elicit CD8α+ T cells in vivo. C57BL/6 mice were vaccinated SC once on day 0 with (1) PBS, (2) RGO(CpG)-PEG, (3) RGO-PEG-Adpgk, (4) soluble CpG + Adpgk, or (5) RGO(CpG)-PEG-Adpgk, followed by flow cytometric analysis of peripheral blood mononuclear cells (PBMCs) over the next 30 days (Figure 4a). On day 8, mice vaccinated with soluble CpG + Adpgk had an average of 7.8% peripheral Adpgk-tetramer+ CD8α+ T cells among PBMCs, which quickly decreased to 3.6% by day 15 and 1.8% by day 30 (Figures 4b and S7). In contrast, mice vaccinated with RGO(CpG)-PEG-Adpgk had significantly higher frequencies of Adpgk-specific T cells on day 8 (P < 0.05), day 15 (P < 0.01), and day 22 (P < 0.01, Figures 4b and S7) Even on day 30 postvaccination, the RGO(CpG)-PEG-Adpgk group had 4.2% Adpgk-specific CD8α+ T cells, compared with 1.8% for the soluble Adpgk + CpG group (P < 0.05, Figures 4b and S7) To assess the functionality of CD8α+ T cells, we challenged these prevaccinated mice with 4 × 105 MC-38 tumor cells on day 40. The RGO(CpG)-PEG-Adpgk group significantly slowed the average tumor growth (P < 0.01, Figure 4c) and prolonged animal survival, with 80% of mice surviving over 60 days after tumor inoculation, compared with 20% survival rate in the soluble CpG + Adpgk group (P < 0.05, Figure 4d).
Figure 4.
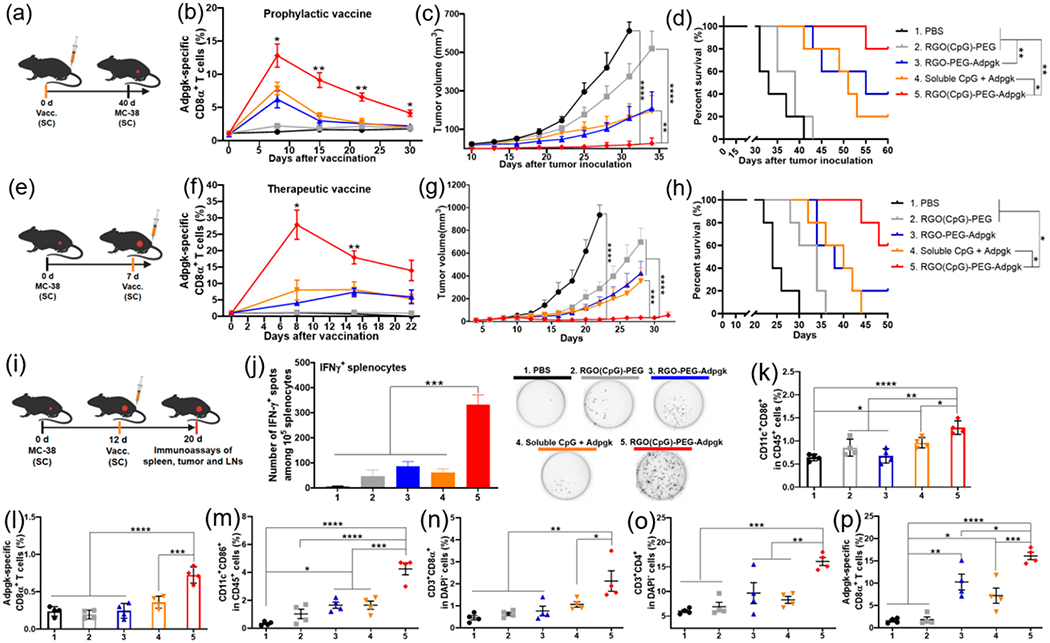
RGO-PEG exerts potent antitumor efficacy against MC-38 colon carcinoma. (a–c) Prophylactic vaccine study. (a) Study design. C57BL/6 mice were vaccinated SC at the tail base with (1) PBS control; (2) RGO(CpG)-PEG; (3) RGO-PEG-Adpgk; (4) soluble CpG + Adpgk; and (5) RGO(CpG)-PEG-Adpgk, followed by MC-38 tumor cell challenge SC in the flank on day 40. Adpgk peptide and CpG doses were both 15 μg in all treatment groups. (b) Adpgk-specific CD8α+ T cells among PBMCs. (c) Tumor growth was measured over time and (d) overall survival curve. (e–h) Therapeutic vaccine study. (e) Study design. MC-38 tumor-bearing mice were vaccinated on day 7 as in (a). (f) Adpgk-specific CD8α+ T cells among PBMCs. (g) Tumor growth was measured over time and (h) overall survival curve. (i–p) MC-38 tumor-bearing mice were treated as in (i), and (j) IFN-γ ELISPOT assay was performed on splenocytes ex vivo restimulated with Adpgk peptide on day 20. In parallel, inguinal LNs (k, l) and tumor tissues (m–p) were analyzed for activated CD45+CD11c+CD86+ DCs (k, m), Adpgk-specific CD8α+ T cells (l, p), CD3+CD8α+ T cells (n), and CD3+CD4+ T cells (o) by flow cytometric analyses. Data represent mean ± SEM from a representative experiment (n = 5, a–h) or (n = 4, i–p) from three independent experiments (a–h) or two independent experiments (i–p). Data were analyzed by one-way ANOVA (j–p) or two-way ANOVA (b, c, f, g) with Tukey’s HSD multiple comparison post hoc test or log-rank (Mantel-Cox) test (d, h). * in (b) and (f) indicates statistical difference between groups 4 and 5. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We also performed a therapeutic vaccine study in MC-38 tumor-bearing mice. Mice were inoculated with MC-38 tumor cells on day 0 and vaccinated SC on day 7 (Figure 4e). As in the prophylactic vaccine study, a single round of RGO(CpG)-PEG-Adpgk vaccination produced a robust, long-lasting CD8α+ T cell response, with 3.5-fold, 2.4-fold, and 2.8-fold higher frequencies of Adpgk-specific CD8α+ T cells among PBMCs on day 8 (P < 0.05), day 15 (P < 0.01), and day 22, respectively (Figures 4f and S8), compared with the soluble Adpgk + CpG group. Accordingly, a single round of RGO(CpG)-PEG-Adpgk vaccination exerted robust antitumor efficacy, with significantly reduced tumor growth (P < 0.001, Figure 4g) and prolonged animal survival (P < 0.05, Figure 4h) in a dose-dependent manner (Figure S9), compared with the soluble Adpgk + CpG group. In addition, antigen and adjuvant molecules were both required for strong antitumor efficacy, as RGO(CpG)-PEG and RGO-PEG-Adpgk vaccines generated significantly dampened antitumor CD8α+ T cell response in both settings of prophylactic and therapeutic vaccination (Figure 4a–h).
Analysis of Systemic and Local Immune Responses.
Having observed potent systemic antitumor efficacy of RGO(CpG)-PEG, we vaccinated MC-38 tumor-bearing mice and analyzed systemic and local immune responses on day 20 (Figure 4i). RGO(CpG)-PEG-Adpgk elicited significantly elevated neoantigen-specific IFN-γ+ T cell response among splenocytes (6.5-fold increase, P < 0.001, Figure 4j), compared with soluble CpG + Adpgk. In addition, RGO(CpG)-PEG-Adpgk induced higher frequencies of activated DCs (CD45+CD11c+CD86+) and Adpgk-specific CD8α+ T cells in inguinal dLNs (P < 0.05 and P < 0.001, respectively, Figure 4k,l), compared with soluble CpG + Adpgk. Furthermore, RGO(CpG)-PEG-Adpgk vaccination induced robust immune activation within the tumor microenvironment, as shown by significantly increased frequencies of activated DCs (2.6-fold increase, P < 0.001), tumor-infiltrating CD3+CD8α+ T cells (2.1-fold increase, P < 0.05), and CD3+CD4+ T cells (2.0-fold increase, P < 0.01), compared with the soluble CpG + Adpgk group (Figure 4m–o). We also observed robust tumor-infiltrating Adpgk-specific CD8α+ T cell response for the RGO(CpG)-PEG-Adpgk group (Figure 4p). Throughout our studies, we did not observe any obvious signs of systemic toxicity (Figure S10a), body weight loss (Figure S11a), or tissue damage of major organs (Figure S10b).
Therapeutic Study in B16F10 Tumor Model with Combination Therapy of Anti-PD-1.
To further investigate the therapeutic efficacy of the RGO cancer vaccine, we employed a highly aggressive and nonimmunogenic B16F10 melanoma model. C57BL/6 mice were inoculated with 2 × 105 B16F10 tumor cells in the SC flank, and when tumors were >30 mm3 on day 7, mice received a single vaccine treatment at the SC tail base with M27 and M30 neoantigens (B16F10-derived MHC-I and MHC-II-restricted neo-epitopes, respectively34) (Figure 5a). RGO(CpG)-PEG-(M27+M30) vaccine (termed RGO vaccine) effectively inhibited the tumor growth (Figure 5b), although there was no statistical difference with the soluble CpG + M27 + M30 (termed soluble vaccine) group. To improve antitumor efficacy, we examined whether strong T cell responses primed by RGO vaccination can synergize with anti-PD-1 ICB therapy against B16F10 tumors, which is resistant to anti-PD-1 monotherapy.35 C57BL/6 inoculated with 2 × 105 B16F10 tumor cells on day 0 were vaccinated on days 7 and 14, and a subset of animals also received intraperitoneal (IP) administration of anti-PD-1 antibody (Figure 5c). The soluble vaccine alone had a minimal impact on tumor growth (Figure 5d), whereas the RGO vaccine alone group exerted a modest antitumor effect similar to the soluble vaccine + αPD-1 group (Figure 5d). RGO vaccine + αPD-1 markedly delayed the B16F10 tumor growth, and much smaller tumors were observed on day 22 (~250 mm3) compared with the soluble vaccine + αPD-1 group (~900 mm3, P < 0.001) and the RGO vaccine only group (~750 mm3, P < 0.05). Furthermore, RGO vaccine + αPD-1 significantly prolonged animal survival compared with all other groups (P < 0.01, Figure 5e), with no obvious body weight loss observed (Figure 5f). An ELISPOT assay performed on day 18 with PBMCs indicated that the RGO vaccine + αPD-1 group elicited strong neoantigen-specific T cell responses, with 9.2-fold higher M27-specific (P < 0.001) and 5.1-fold higher M30-specific IFN-γ+ T cell counts (P < 0.01) compared with the soluble vaccine + αPD-1 group (Figure 5g), and similar results were also found with splenocytes (Figure 5h). RGO vaccine + αPD-1 therapy also significantly increased the intratumoral frequencies of CD3+CD8α+ T cells and activated DCs (CD45+CD11c+CD86+) compared with the soluble vaccine + αPD-1 group (P < 0.001 and P < 0.05, respectively, Figure 5i,j). Taken together, these results show that RGO vaccine combined αPD-1 treatment elicited robust, systemic neoantigen-specific T cell responses with potent antitumor efficacy.
Figure 5.
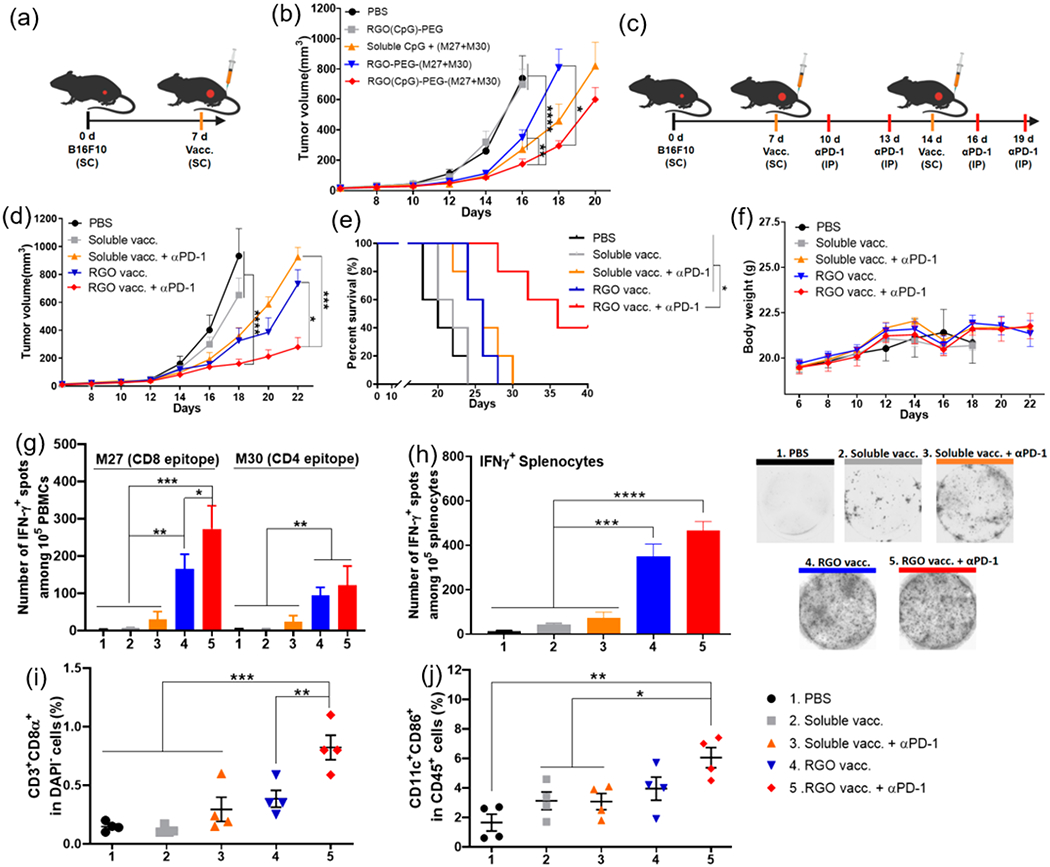
RGO-PEG for combination immunotherapy against B16F10 melanoma. (a, b) C57BL/6 mice were inoculated with B16F10 cells in the SC flank on day 0 and vaccinated on day 7 in the SC tail base with (1) PBS control; (2) RGO(CpG)-PEG; (3) RGO-PEG-(M27+M30); (4) soluble CpG + (M27+M30); or (5) RGO(CpG)-PEG-(M27+M30). Each dose of M27, M30, and CpG was 15 μg in all treatment groups. (b) Average B16F10 tumor growth. (c–j) B16F10 tumor-bearing C57BL/6 mice were vaccinated on days 7 and 14 with the indicated groups with or without 100 μg of anti-PD-1 IgG treatment on days 10, 13, 16, and 19. (d) Average tumor growth; (e) survival curves; and (f) body weight. IFN-γ ELISPOT assay was performed on day 20 with (g) PBMCs or (h) splenocytes ex vivo restimulated with M27 and M30 peptides. On day 20, tumor-infiltrating CD8α+ T cells (i) and activated CD45+CD11c+CD86+ DCs (j) were quantified by flow cytometric analyses. Data represent mean ± SEM from a representative experiment (n = 5, a–f) or (n = 4, g–j) from three independent experiments (a–f) or two independent experiments (g–j). Data were analyzed by one-way ANOVA (g–j) or two-way ANOVA (b, d) with Tukey’s HSD multiple comparison post hoc test or log-rank (Mantel–Cox) test (e). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In this work, we have demonstrated a simple and effective strategy for neoantigen-based cancer vaccination. We have shown that RGO-PEG with sub-50-nm size can be readily loaded with antigen peptides and adjuvants within 2 h (Figure 1). We have employed PET imaging to track and quantify the in vivo biodistribution of radiolabeled neoantigen peptides and RGO(CpG)-PEG-peptides. SC injection of free naked peptides resulted in rapid systemic dissemination, and 72 h PI, there was minimal signal in dLNs or at the injection site (Figure 3). In stark contrast, the RGO nanoplatform achieved >100-fold enhancement in antigen delivery to dLNs with high specificity (<2.0% ID/g RGO-PEG detected in all other major organs). In addition to the small size of RGO-PEG, we believe their surface PEGylation and flexible two-dimensional nanosheets also contribute to their excellent lymphatic delivery.36,37 Even 72 h PI, there was still a large amount of RGO-PEG remaining at the injection site (Figure 3a), suggesting that RGO-PEG vaccine could mediate sustained vaccine delivery to dLNs past the initial 72 h.
RGO as well as GO have been shown to induce ROS, and prior studies have explored this phenomenon for antibacterial and anticancer applications.38,39 In this work, we have exploited this ROS-inducing property of the RGO-PEG nanoplatform to avert acidification of endosomes in DCs and promote antigen presentation. During the DC maturation process, acidification of endosomes activates proteases, leading to rapid and sometimes overt degradation of captured antigens and loss of MHC-I epitopes.3–6 To counter these processes, previous studies have explored pH buffering agents or membrane-destabilizing polymers formulated into NP vaccines.40,41 Here, we show that intracellular ROS generated by RGO-PEG was the key factor for endolysosomal alkalization during phagocytosis by DCs, leading to improved and prolonged antigen presentation to CD8α+ T cells (Figure 2c–g). Furthermore, whereas some carbon nanomaterials (e.g., carbon nanotubes) were reported to be immunosuppressive,42 RGO(CpG)-PEG induced robust secretion of pro-inflammatory cytokines and up-regulation of costimulatory markers on DCs, leading to strong elicitation of CD8α+ T cells (Figure 2a,b,h). Thus, in addition to the LN-targeted delivery aspect, RGO-PEG also has immune-stimulatory properties tailored for potent induction of cellular immunity. Indeed, a single dose of RGO-PEG vaccination in na’ive mice elicited strong and long-lasting neoantigen-specific T cell responses that protected animals against inoculation with MC-38 tumor cells even 50 days PI (Figure 4a–d). Similarly, in MC-38 tumor-bearing mice, a single SC injection of RGO-PEG vaccine achieved ~28% neoantigen-specific CD8α+ T cells in the systemic circulation, accompanied by high frequencies of tumor-infiltrating CD4+ and CD8α+ T cells in the local tumor microenvironment, leading to the eradication of established MC-38 tumors and extension of the animal survival (Figure 4e–p).
Successful neoantigen-based vaccines will likely require both CD4 and CD8α+ T cell responses, as recently reported.6,34 We have thus examined whether our RGO-PEG platform can elicit both CD4+ and CD8α+ T cell responses. In a murine model of B16F10 melanoma, a single RGO-PEG vaccination delivering both MHC-I and MHC-II B16F10 neoantigens was able to slow down the tumor growth (Figure 5a,b); however, due to the highly aggressive and immunosuppressive nature of B16F10 melanoma, all mice eventually succumbed to tumor growth. To overcome this limitation, we sought to amplify antitumor effects of T cells with two rounds of RGO-PEG vaccination combined with anti-PD-1 therapy. With this adjusted dosing regimen, the RGO vaccine + anti-PD-1 group induced strong CD4 and CD8α+ T cell responses and activated intratumoral DCs, resulting in markedly improved antitumor efficacy against established B16F10 tumor and animal survival (Figure 5d–j). These results highlight the potency of RGO-PEG for combination cancer immunotherapy. While it is beyond the scope of this study to investigate the long-term accumulation and elimination of RGO-PEG, our in vitro studies have indicated MPO-mediated degradation of RGO-PEG, and we have not observed any signs of systemic toxicity or long-term tissue damage in mice treated with RGO-PEG.
CONCLUSION
In summary, RGO-PEG is a highly versatile vaccine platform that can be adapted to various personalized neoantigen peptides and efficiently deliver them to LNs with high specificity. Furthermore, the multifunctional, ROS-inducing RGO-PEG nanovaccine system is capable of supporting highly modular and facile production of personalized neoantigen vaccines, guiding antigen processing and presentation by DCs to T cells, and promoting robust T cell responses after just a single round of vaccination. RGO-PEG is a promising vaccine delivery platform for personalized cancer immunotherapy.
MATERIAL AND METHODS
Reagents and Materials.
All reagents used in this work were analytical or higher grade. Graphite, C18PMH (poly(maleic anhydride-alt-1-octadecene)), hydrazine monohydrate, myeloperoxidase, N-acetyl-l-cysteine, and Chelex 100 resin were purchased from Sigma-Aldrich. MAL-PEG5k-NHS, DSPE-PEG2k-NH2, and Boc-NH-PEG3k-NH2 were purchased from Creative PEGworks (Winston Salem, NC, USA). S-2-(4-Isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA) was purchased from Macrocyclics, Inc. (Dallas, TX, USA). H2DCFDA, Lysotracker, LysoSensor DND 189, and DQ-OVA were purchased from Thermo Fisher Scientific. Antigen peptides, including Adpgk (CSSASMT-NMELM), M27 (LCPGNKYEM), and M30 (CSSVDWEN-VSPELNSTDQ), were synthesized by RS Synthesis (Louisville, KY, USA). Murine class B CpG ODN 1826 was purchased from Integrated DNA Technologies (Coralville, IA, USA). Anti-mouse PD-1 IgG (clone: RMP1–14) was purchased from BioXcell. All other chemicals and reagents were purchased from Thermo Fisher Scientific.
Characterization.
TEM images were obtained with a JEOL 1400-PLUS, 120 kV field emission. AFM images were obtained with a Veeco Dimension Icon atomic force microscope. Hydrodynamic size and zeta potential measurements were performed on a Nano-ZS90 Zetasizer (Malvern Instruments Ltd.). The UV–vis spectrum was recorded on a Biotek Synergy microplate reader. Raman spectroscopy was obtained with a Renishaw inVia microscope (532 nm).
Synthesis of RGO(CpG)-PEG-antigen.
GO was synthesized from graphite by a modified Hummers method.43 RGO synthesis and PEGylation processes were performed as in previous reports25,26 with some modification. In a typical process, 0.2 g of GO was dissolved in 40 mL of ethylene glycol/diethylene glycol solution (v/v = 1:20). One gram of sodium acrylate and 0.01% v/v of hydrazine monohydrate were added into the solution and then reacted in a Teflon-lined stainless-steel autoclave reactor at 200 °C for 8 h. The resulting RGO was then washed with ethanol and water several times, and C18PMH (poly(maleic anhydride-alt-1-octadecene))-PEG-NH2 was then added and sonicated for 1 h to suspend the solution.26 Centrifugation was then performed at 14000g to remove any aggregates. The supernatant was purified with a 100 kDa MWCO Millipore centrifuge filter at 15000g. To conjugate neoantigen peptides (or NOTA chelator for PET imaging), the second PEGylation of MAL-PEG5k-NHS was performed. A 0.4 mg amount of peptides was added in a 0.5 mg RGO-PEG-MAL suspension and stirred for 1 h at RT. After conjugation, RGO-PEG-Adpgk was washed three times with PBS (using an Amicon Ultracel centrifugal filter 10 kDa) to remove unconjugated or weakly adsorbed peptide. Lastly, 0.2 mg of CpG was added into an RGO-PEG-Adpgk suspension, stirred for 0.5 h, and washed three times with PBS (using Amicon Ultracel centrifugal filter 50 kDa) to remove free CpG. The Adpgk loading capacity was quantified by LC-MS. Briefly, 100 mM glutathione (GSH) was added to RGO-PEG-Adpgk to release Adpgk peptide from RGO-PEG, followed by washing three times in DI water. The amount of Adpgk peptide in the washing solution was measured by LC-MS (Figure S1d). The CpG loading capacity was determined by direct measurement of the UV–vis absorption increase at 265 nm between RGO-PEG-Adpgk and RGO(CpG)-PEG-Adpgk (Figure S1e).
BMDC Activation, Cytokine Secretion, Antigen Cross-Presentation, and CFSE Dilution Assay.
BMDCs were prepared as described in a previous report.44 Cell viability was measure by the WST-1 assay (Sigma-Aldrich). Immature BMDCs were plated at 3 × 106 cells per well in six-well plates overnight. After incubation with PBS, RGO-PEG (4 μg/mL), soluble CpG (0.5 μg/mL), and RGO(CpG)-PEG (2 μg/mL, the same CpG concentration with the soluble group) for 8 h, TNF-α and IL-12(p70) secretion in the medium by BMDCs was measured. The medium was then changed and incubated for another 16 h. Afterward, BMDCs were harvested and stained with fluorophore-labeled antibodies against CD40, CD80, and CD86. The expression levels of CD40, CD80, and CD86 were measured by flow cytometry. Lysosome and intracellular uptake of Cy5-labeled RGO-PEG were tracked at different incubation time points (1, 6, and 24 h) by staining BMDCs with Lysotracker and visualizing with confocal microscopy. BMDCs or BMDMs were pretreated with LPS (0.4 μg/mL), RGO-PEG (4 μg/mL), RGO-(CpG)-PEG (4 μg/mL, CpG: 0.5 μg/mL), and RGO-PEG or RGO(CpG)-PEG with NAC, a ROS inhibitor (5 mM), for 24 h; then BMDCs or BMDMs were incubated with DCFDA (5 μM) for 0.5 h at 37 °C. After that, the ROS generation (DCF fluorescence intensity) was measured by flow cytometry (excitation/emission at 485 nm/535 nm). BMDCs were pretreated with different vaccine groups (same setting as the DCF fluorescence measurement assay indicated above) for 4 h; then BMDCs were incubated with LysoSensor for 0.5 h at 37 °C. The LysoSensor fluorescence intensity was measured by flow cytometry (excitation/emission at 443 nm/505 nm). For the DQ-OVA assay, BMDCs were pretreated with RGO-PEG (4 μg/mL), RGO(CpG)-PEG (4 μg/mL, CpG: 0.5 μg/mL), and RGO-PEG or RGO(CpG)-PEG with NAC (5 mM) for 24 h. After extensive washing with PBS, BMDCs were incubated with 10 μ/mL DQ-OVA for 0.5 h at 37 °C. Then the fluorescence intensity of DQ-OVA was monitored by flow cytometry. For the SIINFEKL-MHC-I presentation study, BMDCs were treated with PBS, soluble CSSSIINFEKL (2 μg/mL) + CpG (0.5 μg/mL), CSSSIINFEKL + CpG with NAC (5 mM), RGO-PEG-CSSSIINFEKL (same peptide concentration), RGO(CpG)-PEG-CSSSIINFEKL (same peptide and CpG concentration), and RGO(CpG)-PEG-CSSSIINFEKL with NAC (5 mM) for 24 h. Then the cells were washed by PBS and stained with H-2Kb-SIINFEKL-PE for 0.5 h at 37 °C, the cells were washed, and SIINFEKL-MHC-I presentation was analyzed by flow cytometry. In vitro cross-priming of OT-I CD8α+ T cells was measured by the CFSE dilution assay. Briefly, 5 × 104 BMDCs were pretreated with different vaccines for 24 h (the same setting as the SIINFEKL presentation assay shown above). Naive OT-I CD8α+ T cells were isolated from OT-I mouse spleens with a magnetic CD8α+ T cell-negative selection kit, prelabeled with CFSE, and added to the BMDC culture at 5 × 104 per well. After co-incubation with BMDCs for 48 h, proliferation of OT-I CD8α+ T cells was determined by flow cytometry.
In Vivo PET Imaging and Biodistribution Study.
Radioisotope 64Cu was purchased from the University of Wisconsin, Madison.33 64CuCl2 (~200 MBq) was diluted in 0.5 mL of 0.1 M sodium acetate buffer (pH 5.0) and mixed with 0.4 mg of NOTA-RGO(CpG)-PEG-Adpgk or NOTA-Adpgk. The reaction was conducted at 37 °C for 1 h with constant shaking. Then 80 μL of 0.1 M EDTA (ethyl-enediaminetetraacetic acid) was added and incubated for 15 min to remove free 64Cu. The resulting 64Cu-NOTA-RGO(CpG)-PEG-Adpgk or 64Cu-NOTA-Adpgk were purified by a 100 or 2 kDa MWCO Millipore centrifuge filter (15000g for 10 min) and washed three times with PBS to remove free 64Cu. PET scans of C57BL/6 mice were performed using a microPET Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.) at the indicated time points after SC injection of 25–30 MBq of 64Cu-NOTA-RGO(CpG)-PEG Adpgk or 64Cu-NOTA-Adpgk. Detailed data acquisition and region-of-interest (ROI) analysis of PET imaging have been reported previously.33 Quantitative PET data for LNs and major organs were presented as percentage injected dose per gram of tissue (% ID/g). To corroborate the ROI values of PET imaging, we harvested LNs and major organs/tissues at 72 h postinjection, weighed them, and measured radioactivity by a gamma counter.
In Vivo Immunization and Cancer Immunotherapy Study.
All animal experiments were in accordance with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan, Ann Arbor. Female C57BL/6 mice 6–8 weeks of age (Jackson Laboratories) were SC inoculated in the flank with MC-38 or B16F10 cells. For the prophylactic study, female C57BL/6 mice were first immunized with the indicated vaccines on day 0 and then inoculated with MC-38 tumors cells on day 40. For the therapeutic study, when tumor volumes reached ~30–40 mm3 on day 7, mice were immunized SC in the tail base with the indicated vaccine formulations, followed by IP administration of anti-PD-1 IgG as indicated. The CpG dose was 15 μg/injection/mouse in all treatment groups in all animal studies unless otherwise noted. The Adpgk peptide dose was 15 μg/injection/mouse in all treatment groups in the MC-38 tumor studies. The doses of M27 peptide, M30 peptide, and anti-PD-1 were 15, 15, and 100 μg/injection/mouse, respectively, in all treatment groups in the B16F10 tumor studies. Tumor volume was measured every other day and expressed as tumor volume = length × width2 × 0.5. Animals were euthanized when tumors reached 15 mm in any dimension.
Immunological Assays.
For the analysis of neoantigen-specific CD8α+ T cells among PBMCs, submandibular bleeding was performed on the indicated days. After removing red blood cells with ACK lysis buffer, a tetramer staining assay was performed using peptide-MHC tetramer tagged with PE (H-2Db-restricted ASMTN-MELM (Adpgk), from NIH Tetramer Core Facility) as described previously.13 For the analysis of T cells and DCs in LNs and tumor, tissues were cut into small pieces and passed through a 70 μm cell strainer. Then the cell suspensions were washed with FACS buffer (1% bovine serum albumin in PBS) and stained with the following reagents: CD3-FITC, CD4-BV605, CD8α-APC, and Adpgk-tetramer-PE for T cells; CD45-APC, CD11c-FITC, and CD86-PE for DCs. Cells were also stained with DAPI and analyzed by a CytoFLEX cytometer (Beckman Coulter, Indianapolis, IN, USA). In all flow cytometry analyses, only live and intact cells were analyzed. An ELISOPOT (enzyme-linked immunospot) assay was performed with splenocytes or PBMCs. For histopathological analysis, the major organs were harvested 20 days after vaccination and processed for H&E staining.
Statistical Analysis.
Sample sizes were chosen based on preliminary data from pilot experiments. For animal studies, the mice were randomized to match similar primary tumor volume, and all procedures were repeated at least twice in a nonblinded fashion. The results are expressed as mean ± SEM. Statistical analysis was performed with one- or two-way ANOVA, followed by Tukey’s posthoc analyses for multiple comparison tests with Prism 8.0 software (GraphPad Software). Analyses of survival differences were performed using Kaplan–Meier survival analyses with Log-rank Mantel–Cox. Statistical significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH (R01AI127070, R01EB022563, R01CA210273, R01DK125087, U01CA210152). J.J.M. is supported by DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369) and NSF CAREER Award (1553831). K.S.P. acknowledges financial support from the UM TEAM Training Program (DE007057 from NIDCR). We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for the provision of MHC-I tetramers. Opinions interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. Some figures were created by the authors with BioRender.com.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c05062.
Data set of TEM and surface zeta potential of RGO-PEG; in vitro cytotoxicity of RGO vaccine; confocal microscopy images of LysoSensor after RGO-PEG treatment; CFSE dilution study; PET imaging of GO-PEG; GPC of RGO-PEG with albumin; flow cytometry scatter plots in Figure 4; and biosafety profile of RGO vaccine (PDF)
The authors declare no competing financial interest.
Contributor Information
Cheng Xu, Department of Pharmaceutical Sciences and Biointerfaces Institute, University of Michigan, Ann Arbor, Michigan 48109, United States.
Hao Hong, Medical School of Nanjing University, Nanjing, Jiangsu 210093, China.
Yonghyun Lee, Department of Pharmaceutical Sciences and Biointerfaces Institute, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Pharmacy, College of Pharmacy and Pharmacy of Graduate School, Ewha Womans University, Seoul 03760, South Korea.
Kyung Soo Park, Biointerfaces Institute and Department of Biomedical Engineering, University of Michigan, Ann Arbor, Michigan 48109, United States.
Mingjiao Sun, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Tianrui Wang, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Marisa E. Aikins, Department of Pharmaceutical Sciences and Biointerfaces Institute, University of Michigan, Ann Arbor, Michigan 48109, United States
Yao Xu, Department of Pharmaceutical Sciences and Biointerfaces Institute, University of Michigan, Ann Arbor, Michigan 48109, United States.
James J. Moon, Department of Pharmaceutical Sciences, Biointerfaces Institute, and Department of Biomedical Engineering, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Henrickson SE; Mempel TR; Mazo IB; Liu B; Artyomov MN; Zheng H; Peixoto A; Flynn MP; Senman B; Junt T T Cell Sensing of Antigen Dose Governs Interactive Behavior with Dendritic Cells and Sets a Threshold for T Cell Activation. Nat. Immunol 2008, 9, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Johansen P; Storni T; Rettig L; Qiu Z; Der-Sarkissian A; Smith KA; Manolova V; Lang KS; Senti G; Müllhaupt B Antigen Kinetics Determines Immune Reactivity. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Delamarre L; Pack M; Chang H; Mellman I; Trombetta ES Differential Lysosomal Proteolysis in Antigen-Presenting Cells Determines Antigen Fate. Science 2005, 307, 1630–1634. [DOI] [PubMed] [Google Scholar]
- (4).Accapezzato D; Visco V; Francavilla V; Molette C; Donato T; Paroli M; Mondelli MU; Doria M; Torrisi MR; Barnaba V Chloroquine Enhances Human CD8+ T Cell Responses against Soluble Antigens In Vivo. J. Exp. Med 2005, 202, 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shen L; Rock KL Priming of T Cells by Exogenous Antigen Cross-Presented on Mhc Class I Molecules. Curr. Opin. Immunol 2006, 18, 85–91. [DOI] [PubMed] [Google Scholar]
- (6).Joffre OP; Segura E; Savina A; Amigorena S Cross-Presentation by Dendritic Cells. Nat. Rev. Immunol 2012, 12, 557–569. [DOI] [PubMed] [Google Scholar]
- (7).Schumacher TN; Schreiber RD Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [DOI] [PubMed] [Google Scholar]
- (8).Ott PA; Hu Z; Keskin DB; Shukla SA; Sun J; Bozym DJ; Zhang W; Luoma A; Giobbie-Hurder A; Peter L An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sahin U; Derhovanessian E; Miller M; Kloke B-P; Simon P; Löwer M; Bukur V; Tadmor AD; Luxemburger U; Schrörs B Personalized Rna Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [DOI] [PubMed] [Google Scholar]
- (10).Keskin DB; Anandappa AJ; Sun J; Tirosh I; Mathewson ND; Li S; Oliveira G; Giobbie-Hurder A; Felt K; Gjini E Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Alspach E; Lussier DM; Miceli AP; Kizhvatov I; DuPage M; Luoma AM; Meng W; Lichti CF; Esaulova E; Vomund AN Mhc-Ii Neoantigens Shape Tumour Immunity and Response to Immunotherapy. Nature 2019, 574, 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu H; Moynihan KD; Zheng Y; Szeto GL; Li AV; Huang B; Van Egeren DS; Park C; Irvine DJ Structure-Based Programming of Lymph-Node Targeting in Molecular Vaccines. Nature 2014, 507, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Luo M; Wang H; Wang Z; Cai H; Lu Z; Li Y; Du M; Huang G; Wang C; Chen X A Sting-Activating Nanovaccine for Cancer Immunotherapy. Nat. Nanotechnol 2017, 12, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhu G; Lynn GM; Jacobson O; Chen K; Liu Y; Zhang H; Ma Y; Zhang F; Tian R; Ni Q Albumin/Vaccine Nanocomplexes That Assemble In Vivo for Combination Cancer Immunotherapy. Nat. Commun 2017, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li AW; Sobral MC; Badrinath S; Choi Y; Graveline A; Stafford AG; Weaver JC; Dellacherie MO; Shih T-Y; Ali OA A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat. Mater 2018, 17, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Scheetz L; Park KS; Li Q; Lowenstein PR; Castro MG; Schwendeman A; Moon JJ Engineering Patient-Specific Cancer Immunotherapies. Nat. Biomed. Eng 2019, 3, 768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lynn GM; Sedlik C; Baharom F; Zhu Y; Ramirez-Valdez RA; Coble VL; Tobin K; Nichols SR; Itzkowitz Y; Zaidi N Peptide–Tlr-7/8a Conjugate Vaccines Chemically Programmed for Nanoparticle Self-Assembly Enhance Cd8 T-Cell Immunity to Tumor Antigens. Nat. Biotechnol 2020, 38, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu J; Cui L; Losic D Graphene and Graphene Oxide as New Nanocarriers for Drug Delivery Applications. Acta Biomater. 2013, 9, 9243–9257. [DOI] [PubMed] [Google Scholar]
- (20).Kim H; Lee D; Kim J; Kim T.-i.; Kim WJ Photothermally Triggered Cytosolic Drug Delivery Via Endosome Disruption Using a Functionalized Reduced Graphene Oxide. ACS Nano 2013, 7, 6735–6746. [DOI] [PubMed] [Google Scholar]
- (21).Miao W; Shim G; Kang CM; Lee S; Choe YS; Choi H-G; Oh Y-K Cholesteryl Hyaluronic Acid-Coated, Reduced Graphene Oxide Nanosheets for Anti-Cancer Drug Delivery. Biomaterials 2013, 34, 9638–9647. [DOI] [PubMed] [Google Scholar]
- (22).Tao Y; Ju E; Ren J; Qu X Immunostimulatory Oligonucleotides-Loaded Cationic Graphene Oxide with Photothermally Enhanced Immunogenicity for Photothermal/Immune Cancer Therapy. Biomaterials 2014, 35, 9963–9971. [DOI] [PubMed] [Google Scholar]
- (23).Li H; Fierens K; Zhang Z; Vanparijs N; Schuijs MJ; Van Steendam K; Feiner Gracia N. l.; R. De Rycke; De Beer T; De Beuckelaer A Spontaneous Protein Adsorption on Graphene Oxide Nanosheets Allowing Efficient Intracellular Vaccine Protein Delivery. ACS Appl. Mater. Interfaces 2016, 8, 1147–1155. [DOI] [PubMed] [Google Scholar]
- (24).Wang X; Cao F; Yan M; Liu Y; Zhu X; Sun H; Ma G Alum-Functionalized Graphene Oxide Nanocomplexes for Effective Anticancer Vaccination. Acta Biomater. 2019, 83, 390–399. [DOI] [PubMed] [Google Scholar]
- (25).Robinson JT; Tabakman SM; Liang Y; Wang H; Sanchez Casalongue H; Vinh D; Dai H Ultrasmall Reduced Graphene Oxide with High Near-Infrared Absorbance for Photothermal Therapy. J. Am. Chem. Soc 2011, 133, 6825–6831. [DOI] [PubMed] [Google Scholar]
- (26).Yang K; Hu L; Ma X; Ye S; Cheng L; Shi X; Li C; Li Y; Liu Z Multimodal Imaging Guided Photothermal Therapy Using Functionalized Graphene Nanosheets Anchored with Magnetic Nanoparticles. Adv. Mater 2012, 24, 1868–1872. [DOI] [PubMed] [Google Scholar]
- (27).Yadav M; Jhunjhunwala S; Phung QT; Lupardus P; Tanguay J; Bumbaca S; Franci C; Cheung TK; Fritsche J; Weinschenk T Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [DOI] [PubMed] [Google Scholar]
- (28).Mukherjee SP; Gliga AR; Lazzaretto B; Brandner B; Fielden M; Vogt C; Newman L; Rodrigues AF; Shao W; Fournier PM Graphene Oxide Is Degraded by Neutrophils and the Degradation Products Are Non-Genotoxic. Nanoscale 2018, 10, 1180–1188. [DOI] [PubMed] [Google Scholar]
- (29).Ma J; Liu R; Wang X; Liu Q; Chen Y; Valle RP; Zuo YY; Xia T; Liu S Crucial Role of Lateral Size for Graphene Oxide in Activating Macrophages and Stimulating Pro-Inflammatory Responses in Cells and Animals. ACS Nano 2015, 9, 10498–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nathan C; Cunningham-Bussel A Beyond Oxidative Stress: An Immunologist’s Guide to Reactive Oxygen Species. Nat. Rev. Immunol 2013, 13, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gao J; Ochyl LJ; Yang E; Moon JJ Cationic Liposomes Promote Antigen Cross-Presentation in Dendritic Cells by Alkalizing the Lysosomal Ph and Limiting the Degradation of Antigens. Int. J. Nanomed 2017, 12, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Reddy ST; Van Der Vlies AJ; Simeoni E; Angeli V; Randolph GJ; O’Neil CP; Lee LK; Swartz MA; Hubbell JA Exploiting Lymphatic Transport and Complement Activation in Nanoparticle Vaccines. Nat. Biotechnol 2007, 25, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (33).Hong H; Yang K; Zhang Y; Engle JW; Feng L; Yang Y; Nayak TR; Goel S; Bean J; Theuer CP In Vivo Targeting and Imaging of Tumor Vasculature with Radiolabeled, Antibody-Conjugated Nanographene. ACS Nano 2012, 6, 2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kreiter S; Vormehr M; Van de Roemer N; Diken M; Löwer M; Diekmann J; Boegel S; Schrörs B; Vascotto F; Castle JC Mutant Mhc Class Ii Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Curran MA; Montalvo W; Yagita H; Allison JP Pd-1 and Ctla-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells within B16 Melanoma Tumors. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kaminskas LM; Kota J; McLeod VM; Kelly BD; Karellas P; Porter CJ Pegylation of Polylysine Dendrimers Improves Absorption and Lymphatic Targeting Following Sc Administration in Rats. J. Controlled Release 2009, 140, 108–116. [DOI] [PubMed] [Google Scholar]
- (37).Son S; Nam J; Zenkov I; Ochyl LJ; Xu Y; Scheetz L; Shi J; Farokhzad OC; Moon JJ Sugar-Nanocapsules Imprinted with Microbial Molecular Patterns for Mrna Vaccination. Nano Lett. 2020, 20, 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gurunathan S; Han JW; Dayem AA; Eppakayala V; Kim J-H Oxidative Stress-Mediated Antibacterial Activity of Graphene Oxide and Reduced Graphene Oxide in Pseudomonas Aeruginosa. Int. J. Nanomed 2012, 7, 5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Arya N; Arora A; Vasu K; Sood AK; Katti DS Combination of Single Walled Carbon Nanotubes/Graphene Oxide with Paclitaxel: A Reactive Oxygen Species Mediated Synergism for Treatment of Lung Cancer. Nanoscale 2013, 5, 2818–2829. [DOI] [PubMed] [Google Scholar]
- (40).Song C; Noh Y-W; Lim YT Polymer Nanoparticles for Cross-Presentation of Exogenous Antigens and Enhanced Cytotoxic T-Lymphocyte Immune Response. Int. J. Nanomed 2016, 11 , 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Qiu F; Becker KW; Knight FC; Baljon JJ; Sevimli S; Shae D; Gilchuk P; Joyce S; Wilson JT Poly(propylacrylic Acid)-Peptide Nanoplexes as a Platform for Enhancing the Immunogenicity of Neoantigen Cancer Vaccines. Biomaterials 2018, 182, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Laverny G; Casset A; Purohit A; Schaeffer E; Spiegelhalter C; de Blay F; Pons F Immunomodulatory Properties of Multi-Walled Carbon Nanotubes in Peripheral Blood Mononuclear Cells from Healthy Subjects and Allergic Patients. Toxicol. Lett 2013, 217, 91–101. [DOI] [PubMed] [Google Scholar]
- (43).Xu C; Yang D; Mei L; Li Q; Zhu H; Wang T Targeting Chemophotothermal Therapy of Hepatoma by Gold Nanorods/Graphene Oxide Core/Shell Nanocomposites. ACS Appl. Mater. Interfaces 2013, 5, 12911–12920. [DOI] [PubMed] [Google Scholar]
- (44).Lutz MB; Kukutsch N; Ogilvie AL; Rößner S; Koch F; Romani N; Schuler G An Advanced Culture Method for Generating Large Quantities of Highly Pure Dendritic Cells from Mouse Bone Marrow. J. Immunol. Methods 1999, 223, 77–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.