

Abstract
By acting as a ligand-dependent transcription factor the glucocorticoid receptor (GR) mediates the actions of glucocorticoids and regulates many physiological processes. An impaired regulation of glucocorticoid action has been associated with numerous disorders. Thus, the elucidation of underlying signaling pathways is essential to understand mechanisms of disrupted glucocorticoid function and contribution to diseases. This study found increased GR transcriptional activity upon overexpression of protein phosphatase 1 alpha (PP1α) in HEK-293 cells and decreased expression levels of GR-responsive genes following PP1α knockdown in the endogenous A549 cell model. Mechanistic investigations revealed reduced phosphorylation of GR-Ser211 following PP1α silencing and provided a first indication for an involvement of glycogen synthase kinase 3 (GSK-3). Thus, the present study identified PP1α as a novel post-translational activator of GR signaling, suggesting that disruption of PP1α function could lead to impaired glucocorticoid action and thereby contribute to diseases.
Keywords: Glucocorticoid receptor, protein phosphatase 1 alpha, glycogen synthase kinase 3, phosphorylation, signaling
1. Introduction
Glucocorticoids (GC) are steroid hormones naturally produced and secreted by the adrenal gland in order to maintain body homeostasis. They act highly tissue-specifically and affect almost every organ in the human body by regulating energy homeostasis (carbohydrates, lipids, proteins), bone metabolism, cell growth and differentiation, apoptosis, stress and immune responses, as well as brain function (Ahmad et al., 2019; Blum et al., 2003; Buckingham, 2006; Garabedian et al., 2017; Gross et al., 2008; Quax et al., 2013). GC are essential for life and dysregulation of their signaling is involved in numerous disorders including metabolic and cardiovascular diseases, asthma and chronic obstructive pulmonary disease (COPD), mood and cognitive disorders, immune diseases and cancer. Because of their anti-inflammatory, immunosuppressive and pro-apoptotic effects, synthetic GC are amongst the most widely prescribed drugs for the treatment of inflammation, autoimmune disorders and cancer.
GC exert their effects by binding to the glucocorticoid receptor (GR), a ligand-dependent transcription factor belonging to the nuclear receptor superfamily. The GR consists of an N-terminal transactivation domain (NTD), a DNA binding domain (DBD) and a C-terminal ligand-binding domain (LBD) (Zhou et al., 2005). The NTD contains a transcriptional activation function domain (AF-1) responsible for post-translational modifications and protein-protein interactions. In the absence of ligand, the GR is sequestered in the cytoplasm in a heat-shock protein 90 (HSP-90)-chaperone complex. Ligand binding induces conformational changes that allow the release of the cytosolic GR from the HSP-90-chaperone complex, followed by receptor dimerization and translocation into the nucleus. The hormone-activated GR binds then to specific palindromic DNA sequences in the promoter regions of target genes (glucocorticoid response elements, GREs) and regulates their transcription. Induction or suppression of target gene transcription can occur, depending on the cell type, promoter context and cofactor recruitment. Upon prolonged exposure to GC, the GR is subjected to degradation by the ubiquitin-proteasome pathway, resulting in termination of the transcriptional response (Connell et al., 2001; Galigniana et al., 2004; Kinyamu et al., 2003; Sengupta et al., 2001; Wallace et al., 2001; Wang et al., 2005; Webster et al., 1997).
In addition to ligand binding, cross-talk with other signaling pathways can modulate GR activity by post-translational modifications, including phosphorylation, ubiquitination, and SUMOylation (Faus et al., 2006; Garza et al., 2010; Ismaili et al., 2004; Le Drean et al., 2002; Wallace et al., 2001). GR phosphorylation affects its interaction with co-regulators, subcellular localization, DNA binding and protein stability. Phosphorylation is paramount in receptor regulation and highly cell-, tissue-, species- and promoter-specific and the vast diversity of kinases and phosphatases mediates the high variety of cellular responses to GC (Galliher-Beckley et al., 2009; Zhou et al., 2005). Various kinases, including cyclin-dependent kinases (CDKs), p38 mitogen activated protein kinases (MAPKs), c-Jun N-terminal kinases (JNKs), extracellular signal-regulated kinases (ERKs), protein kinase B (Akt), and glycogen synthase kinase 3 (GSK-3) were shown to modulate GR activity by phosphorylation at several sites, including serine residues 134, 203, 211, 226 and 404 (DeFranco et al., 1991; Galliher-Beckley et al., 2008; Galliher-Beckley et al., 2011; Habib et al., 2017; Itoh et al., 2002; Kino et al., 2007; Krstic et al., 1997; Miller et al., 2005; Rogatsky et al., 1998; Rogatsky et al., 1998; Webster et al., 1997).
GR function is regulated by numerous phosphatases. Recent studies emphasized the importance of protein phosphatases in regulating GR and contributing to the cytokine-induced GC insensitivity seen in patients with severe asthma (Bouazza et al., 2012; Kobayashi et al., 2011; Pazdrak et al., 2016). Protein phosphatase 2A (PP2A) was found to dephosphorylate JNK, resulting in decreased GR-Ser226 phosphorylation and enhanced GR nuclear translocation, while protein phosphatase 5 (PP5) was proposed to negatively regulate GR activity by direct dephosphorylation of GR-Ser211. Besides, PP2A was suggested to form a trimeric complex with GR and the scaffold protein striatin-3 (Petta et al., 2017). In the presence of striatin-3, PP2A might be recruited to facilitate dephosphorylation of GR-Ser211, thereby resulting in diminished GR transcriptional activity.
Protein phosphatase 1 alpha (PP1α) has been shown to control the expression and activity of several steroid receptors, including the mineralocorticoid receptor (MR), the androgen receptor (AR) and the estrogen receptor-α (ER) (Bollig et al., 2007; Liu et al., 2016; Nagarajan et al., 2017). PP1α was found to bind to the LBD of these steroid receptors, thereby stabilizing their expression by dephosphorylation and inactivation of the E3 ubiquitin-ligase mouse double minute 2 (MDM2). In contrast to PP2A and PP5, it remained unclear whether PP1α also regulates GR function. In rat fibroblasts, the subcellular distribution of the GR was influenced by inhibiting activities of PP1 and PP2A using okadaic acid, a potent but unspecific inhibitor of PP1 (IC50 = 15-20 nM) and PP2A (IC50 = 0.1 nM) (Bialojan et al., 1988; Cohen et al., 1989; DeFranco et al., 1991; Somers et al., 1992). The PP1 holoenzyme is a serine/threonine-specific phosphoprotein phosphatase consisting of a catalytic subunit (PP1c) and multiple regulatory subunits. PP1c has four isoforms PP1α, PPβ, and the splice variants PPγ1 and PPγ2, encoded by three different genes PPP1CA, PPP1CB and PPP1CC. These isoforms share a high level of sequence homology and are ubiquitously expressed, except of PPγ2, which is restricted to the testis (Ceulemans et al., 2004; Cohen, 2002; Moorhead et al., 2007).
The present study evaluated the impact of PP1α on GR function by performing a reporter gene assay in HEK-293 cells expressing recombinant human GRα and PP1α. To investigate the effect of PP1α on endogenously expressed GR activity, the transcriptional expression of GC-induced genes was followed after knockdown of PP1α in A549 human lung carcinoma cells. Furthermore, to assess whether PP1α regulates GR through dephosphorylation of MDM2, as shown for the related steroid receptors, or by a direct mechanism similarly to PP2A and PP5, the pathways underlying the PP1α-mediated effect on GR stimulation were assessed in A549 cells after silencing of PP1α.
2. Materials and methods
2.1. Chemicals and reagents
Cortisol (CAS Nr. 50-23-7) was purchased from Steraloids (Newport, RI). Inhibitors of JNK (SP600125, CAS Nr. 129-56-6), p38 MAPK (SB203580, CAS Nr. 152121-47-6), Akt1/2 kinase (CAS Nr. 612847-09-3), MEK1/2 (PD98059, CAS Nr. 167869-21-8), and GSK-3 (CHIR99021, CAS Nr. 252917-06-9), as well as the GR antagonist mifepristone (RU-486) were obtained from Sigma-Aldrich (Buchs SG, Switzerland). Stock solutions (10 mM) were prepared in dimethyl sulfoxide (DMSO, CAS Nr. 67-68-5; AppliChem, Darmstadt, Germany).
2.2. Cell culture and treatments
Human embryonic kidney-293 (HEK-293) cells and A549 human alveolar carcinoma cells were obtained from ATCC (Manassa, VA, USA). Cell culture media were purchased from Sigma-Aldrich. HEK-293 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 4.5 g/L glucose, 100 U/mL penicillin/streptomycin, 2 mM L-glutamine, 10 mM HEPES, pH 7.4, and 10% MEM non-essential amino acid solution. A549 cells were grown in Kaighn's Modification of Ham's F-12 medium (F-12K medium) completed with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin.
Lipofectamine RNAiMax (Thermo Fisher Scientific, Waltham, MA, USA) was used for small interfering RNA (siRNA) delivery. Cells (150’000/6-well) were reverse transfected with 100 pmol siRNA against PP1α and/or 50 pmol siRNA against GSK-3 and 3.75 μL lipofectamine reagent for 64-66 h. The target sequences recognized by the siRNAs are: mock (negative control siRNA with a randomly selected non-targeting sequence) 5′-UGGUUUACAUGUUUUCUGA-3′, PPP1CA (PP1α) 5'-CAAGAGACGCUACAACAUC-3' (both from Microsynth AG, Balgach, Switzerland); GSK3A (L-003009-00-0005) 5'-UCACAAGCUUUAACUGAGA-3', 5'-GAAGGUGACCACAGUCGUA-3', 5'-GAGUUCAAGUUCCCUCAGA-3', 5'-CUGGACCACUGCAAUAUUG-3' and GSK3B (L-003010-00-0005) 5'-GAUCAUUUGGUGUGGUAUA-3', 5'-GCUAGAUCACUGUAACAUA-3', 5'-GUUCCGAAGUUUAGCCUAU-3', 5'-GCACCAGAGUUGAUCUUUG-3' (all from Dharmacon ON-TARGET plus SMART POOL; Dharmacon, Lafayette, CO, USA).
To determine effects of PP1α knockdown on endogenous GC-induced transcripts, A549 cells were cultured in steroid-free medium for 16-18 h prior to incubation with increasing concentrations of cortisol (18.75 nM - 1200 nM) or vehicle (DMSO) for 4 h. To study effects of kinase inhibitors on PP1α-dependent downregulation of GC-induced genes, A549 cells were treated with indicated inhibitors (10 μM, except for GSK-3 inhibitor (5 μM)) in serum-free medium for 16-18 h prior to incubation with cortisol (500 nM) or vehicle (DMSO) for another 4 h. Following treatments, total RNA was isolated and quantitative polymerase chain reaction performed to quantify GILZ (glucocorticoid-induced leucine zipper), IGFBP1 (insulin-like growth factor binding protein 1) and SDPR (serum deprivation-response protein).
Cellular fractionation and phosphorylation of GR was assessed by pre-incubating the cells with steroid-free medium overnight following treatment of cortisol for another 1 h prior to cell lysis. Cellular fractionation experiments in HEK-293 and A549 cells were performed using 50 nM and 500 nM cortisol, respectively. GR phosphorylation in A549 cells was analyzed in the presence of 10 nM and 50 nM cortisol. In all cell treatments, the final concentration of DMSO did not exceed 0.05%.
2.3. GR-dependent reporter gene assay
HEK-293 cells (100,000 cells/well) were seeded in poly-L-lysine coated 24-well plates, incubated for 24 h and co-transfected by calcium phosphate precipitation with the reporter gene TAT3- TATA luciferase (0.375 μg/well), pCMV-Renilla constitutive luciferase transfection control (0.03 μg/well) and the indicated plasmids coding for human GRα, Flag-PP1α and Myc-MDM2 (at a ratio of 1:4:4). Empty vector pcDNA3.1 was supplemented to equalize the total amount of DNA in the transfection. After 4 h, cells were washed with phosphate-buffered saline (PBS) and incubated in DMEM for another 18 h. Cells were then adapted to charcoal-treated DMEM (cDMEM) for 2 h. Cells were exposed 24 h post-transfection to DMSO control, cortisol (0.1 – 100 nM) or mifepristone (RU-486; 1 μM), followed by incubation for another 24 h. Cells were then lysed in passive lysis buffer (Promega, Madison, WI, USA) and fire fly and Renilla luciferase activity were determined according to the Dual-Luciferase® reporter assay kit (Promega, Madison, WI, USA) using a SpectraMax-L luminometer (Molecular Devices, Devon, UK).
2.4. RNA isolation and quantitative polymerase chain reaction (RT-qPCR)
For isolation of total RNA, the RNeasy Mini Kit (QIAGEN, Hilden, Germany) was applied on a QIAcube extraction robot (QIAGEN) according to the manufacturer. The extracted RNA was treated with the RapidOut DNA Removal Kit (Thermo Fisher Scientific) and complementary DNA (cDNA) was subsequently synthesized using the GoScript Reverse Transcription System (Promega) following the manufacturer’s protocol. RT-qPCR comprising 40 cycles of 95°C for 10 s, 60°C for 15 s, followed by a final extension at 72°C for 20 s, and a dissociation curve (72°C to 95°C) was performed in technical triplicate for each sample in the Rotor-Gene Q (QIAGEN) by using KAPA SYBR Fast qPCR Master Mix (2X) Kit (KAPA Biosystems, Woburn, MA, USA). For relative quantification, expression levels were normalized to those of the endogenous control gene cyclophilin A (CYPA) according to the comparative 2 −ΔCt method (Livak et al., 2001). CYPA was chosen as housekeeping gene since its expression did not alter between experimental conditions and complied with the quality criteria for reference genes (Taylor et al., 2010). Table 1 shows sequences of oligonucleotide primers.
Table 1.
Sequences of gene-specific primers used for RT-qPCR.
| Gene name | Forward primers (5’-3’) | Reverse primers (5’-3’) |
|---|---|---|
| CYPA | CATCTGCACTGCCAAGACTGA | TGCAATCCAGCTAGGCATG |
| GILZ | ACAAGATCGAACAGGCCATG | TTGCCAGGGTCTTCAACAG |
| IGFBP1 | CCATGTCACCAACATCAAAAA | TCGTAGAGAGTTTAGCCAAGGC |
| SDPR | AAGAGCGCATGGATAGGCAG | GTTTCACAAACACGCTGGCA |
2.5. Western blot analysis
Upon treatment, cells were washed in PBS and lysed for 30 min on ice in RIPA buffer (Sigma-Aldrich) containing protease inhibitor cocktail (Roche, Rotkreuz, Switzerland). All steps of the lysis process were performed at 4°C, which was sufficient to drastically reduce the activity of phosphatases. An initial experiment showed no effect of phosphatase inhibitor cocktail (data not shown), therefore it was not added to the lysis buffer. After centrifugation (16’000 × g, 15 min, 4°C), the supernatant was analyzed using a Pierce® biocinchonic acid protein assay kit (Thermo Fisher Scientific). Laemmli solubilization buffer (LSB; 60 mM Tris-HCl, 10% glycerol, 2% (w/v) sodium dodecyl sulfate, 0.01% bromophenol blue, pH 6.8) supplemented with 5% β-mercaptoethanol (Promega) was added and samples were boiled for 3 min. Total protein (20 μg) of whole-cell extracts was resolved on 8% or 12% bis-acrylamide gels by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All proteins were transferred to Immun-Blot® polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA, USA), which were subsequently blocked with 5% defatted milk in Tris-buffered saline (20 mM Tris buffer (pH 7.6), 150 mM NaCl) containing 0.1% Tween-20 (TBS-T) for 1 h. Membranes were incubated with the indicated primary antibodies in blocking solution overnight at 4°C. After washing with TBS-T, the membranes were probed with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (A0168) or goat anti-rabbit secondary antibody (A0545; both 1:10’000, from Sigma-Aldrich) for 1 h at room temperature. Visualization and detection of the protein bands was performed on a Fujifilm ImageQuant™ LAS-4000 (GE Healthcare, Glattbrugg, Switzerland) using the Immobilon Western Chemiluminescent HRP substrate kit (Merck, Kenilworth, NJ, USA). Quantitation of at least two independent experiments was done by using NIH ImageJ software and densitometry values were corrected for loading differences by normalization to those of β-actin or CYPA housekeeping control.
The following primary antibodies were used: Anti-GR antibody (1:2’000; sc-1003; validated earlier (Mani et al., 2016)), anti-PP1α antibody (1:5’000; sc-271762), and antβ-actin (1:10’000; sc-47778; all from Santa Cruz Biotechnology, Dallas, TX, USA); anti-pSer134 GR antibody (1:2’000; 85060), anti-pSer211 GR antibody (1:2’000; 4161; verified earlier (Oakley et al., 2017)), anti-pSer226 GR antibody (1:10’000; 97285), and anti-GSK-3 antibody (1:5’000; 5676; all from Cell Signaling Technology, Danvers, MA, USA); anti-pSer203 GR antibody (1:2’000; orb127112; Biorbyt Ltd., Cambridge, United Kingdom); anti-Flag antibody (1:2’000; MA1-91878; Thermo Fisher Scientific), anti-pSer404 GR antibody (1:1’000; described earlier (Galliher-Beckley et al., 2008)), anti-CYPA (1:10’000, ab41684; Abcam Inc, Cambridge, United Kingdom).
2.6. Cellular fractionation
Treated cells were washed with PBS, scraped from culture dishes on ice and centrifuged at 500 × g for 5 min at 4°C. Cell pellets were resuspended in ice-cold lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, pH 7.6) supplemented with protease inhibitor cocktail (Roche) and 1 mM dithiothreitol (DTT, AppliChem). After incubation for 15 min, cells were permeabilized with 0.57% IGEPAL CA-630 (Sigma-Aldrich), vortexed vigorously for 10 s and centrifuged at 8’000 × g for 5 min at 4°C. The supernatant was collected as cytosolic fraction and clarified by centrifugation (16’000 × g, 5 min, 4°C) following protein quantification. After removing the cytosolic fraction, the pellet was washed twice with PBS, centrifuged (800 × g, 5 min, 4°C) and the supernatant discarded. The pellet was resuspended in extraction buffer (20 mM HEPES, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% (v/v) glycerol, pH 7.9) supplemented with protease inhibitor cocktail (Roche) and 1 mM DTT for 20 min at 4°C and 1 ‘400 rotations/min on an orbital shaker (Thermomixer, Vaudaux-Eppendorf, Buchs, Switzerland). The homogenate was centrifuged at 21’000 × g for 5 min at 4°C and the supernatant removed as nuclear fraction, followed by protein quantification. Nuclear and cytoplasmic fractions were electrophoretically separated and blotted to PVDF membranes. Membranes were probed with anti-β-actin or anti-α-tubulin (1:10’000; GTX628802; GeneTex, Irvine, CA, USA) antibodies as cytoplasmic markers and anti-HDAC1 antibody (1:5’000; 5356; Cell Signaling Technology) as nuclear marker.
2.7. Co-immunoprecipitation
For co-immunoprecipitation experiments using Pierce™ NHS-Activated Agarose Slurry (Thermo Scientific), anti-GR antibody (sc-1003, Santa Cruz Biotechnology) was immobilized on beads according to the manufacturer. Cells suspended in lysis buffer (Atanasov et al., 2008) and proteins (1000 μg) were immunoprecipitated with antibody-coupled beads overnight at 4°C. As a control of antigen-antibody binding specificity, lysates were incubated with rabbit IgG antibody (sc-2027, Santa Cruz Biotechnology). After elution of the precipitated protein, Western blot analysis was performed.
Co-immunoprecipitation with Pierce™ Protein A/G Plus Agarose (Thermo Scientific) was performed as described previously (Petrillo et al., 2019). Briefly, 1000 μg of whole-cell extract proteins were incubated with anti-GR antibody (3660, Cell Signaling Technology) or anti-PP1α antibody (sc-271762, Santa Cruz Biotechnology) overnight at 4°C. Rabbit IgG antibody (12-370, Merck, Darmstadt, Germany) or mouse IgG antibody (sc-2025, Santa Cruz Biotechnology) was used as a control of non-antigen specific binding. Beads were added to each sample for another 3 h at 4°C. The beads were washed twice with Co-IP buffer (50 mM Tris buffer, pH 7.6), 150 mM NaCl, 1% NP-40, 10% glycerol, 5 mM MgCl2) containing protease inhibitor cocktail and 1 mM phenylmethanesulfonyl fluoride (PMSF; AppliChem) and twice with PBS. After eluting, the proteins from the beads by adding LSB supplemented with 5% β-mercaptoethanol and heating for 5 min at 99°C, they were subjected to Western blot analysis.
2.8. Statistical analysis
Statistical evaluation was conducted in GraphPad Prism version 5.0. Statistical significance of differences between treatments was calculated using an unpaired two-tailed Student’s t test, one-way ANOVA followed by Tukey’s multiple comparisons test or two-way ANOVA followed by Bonferroni’s multiple comparisons test to adjust the false discovery rate. Values represent mean ± SD and levels of significance are: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
3. Results
3.1. PP1α stimulates GR activity independently of MDM2
Although PP1α was found to enhance the activity of AR and MR through a mechanism involving MDM2-dependent control of receptor protein stability (Liu et al., 2016; Nagarajan et al., 2017), it remained unclear whether PP1α would stimulate GR activity and if so by which mechanism. To investigate the effect of PP1α on GR activity, HEK-293 cells were transiently transfected with human GR, PP1α and a GR-dependent luciferase reporter gene. An excess of PP1α over GR expression plasmid of 4:1 optimally stimulated GR-dependent luciferase reporter activity. Co-transfection with PP1α significantly increased basal and cortisol-dependent GR activity, by a cortisol concentration-dependent manner (Fig. 1A). In PP1α-overexpressing cells, the maximal GR transcriptional activity was observed when cells were treated with 3.7 nM cortisol, resulting in a 7-fold higher GR-mediated luciferase activity compared with cells expressing GR alone. In the absence of PP1α, maximal GR activity was detected in the presence of 30 nM cortisol. PP1α overexpression significantly affected the affinity of GR for cortisol (EC50 GR = 5.9 ± 2.8 nM; EC50 GR+pp1α = 1.4 ± 0.1 nM; P value = 0.01). To exclude non-specific effects of PP1α on the promoter, HEK-293 cells transiently expressing GR and PP1α were treated with the GR antagonist mifepristone (RU-486) in the absence or presence of cortisol (Suppl. Fig. 1A). As mifepristone is known to exhibit also some weak agonistic effects (Jewell et al., 1995; Qi et al., 1990; Zhang et al., 2007), co-expression with PP1α slightly increased the activity of GR upon incubation with mifepristone. However, in the presence of cortisol, the pronounced PP1α-dependent activation was almost completely reversed by co-treatment with mifepristone. Furthermore, in the absence of GR, i.e. in HEK-293 cells transfected with the GR-dependent luciferase reporter gene and PP1α alone, no significant induction of the reporter by PP1α was observed in both the absence and presence of cortisol (Suppl. Fig. 1B). This indicates that the PP1α-dependent stimulation is mediated via GR.
Fig. 1. Effect of PP1α overexpression on GR activity and nuclear translocation.
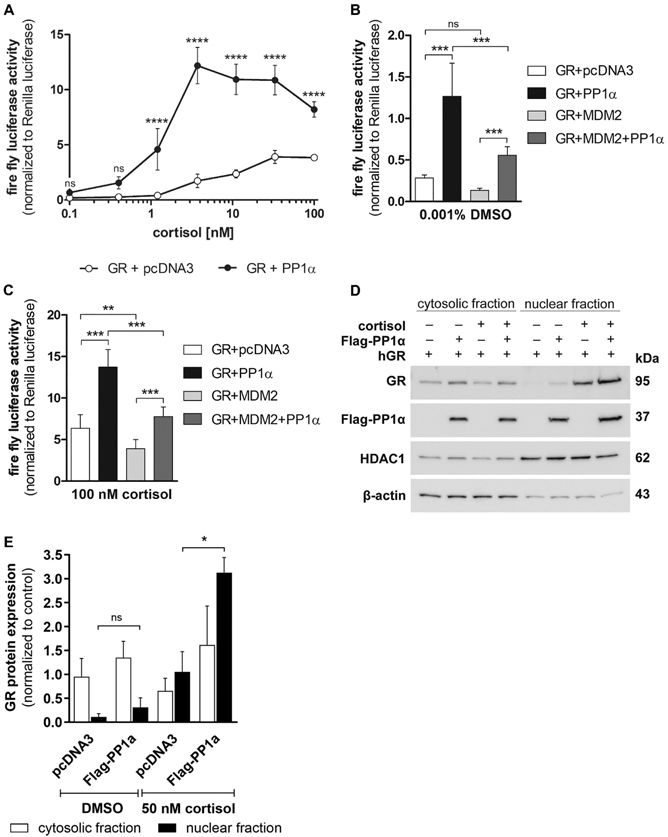
(A, B and C) HEK-293 cells were transiently transfected with plasmids coding for GR with or without PP1α, a luciferase reporter gene and a Renilla luciferase transfection control. Empty vector pcDNA3.1 was supplemented to equalize the total amount of DNA in the transfection. After 24 h of transfection, cells were incubated with vehicle or increasing concentrations of cortisol (A) for another 24 h. The luciferase reporter activity was normalized to the internal Renilla control. (B and C) Same as (A) but with co-transfection of MDM2. Data represent mean ± SD from at least two independent experiments, each performed in triplicate, ****P < 0.0001, ***P < 0.001, **P < 0.01, ns not significant. (D and E) HEK-293 cells were transfected with GR, with and without PP1α for 24 h, incubated overnight in charcoal-treated medium and then exposed to vehicle or 50 nM cortisol for 1 h. Cytosolic and nuclear fractions were analyzed by Western blot using antibodies against GR and the Flag-tag on PP1α. As controls, the fractions were reprobed with ant-β-actin (cytosolic) and anti-HDAC1 (nuclear) antibodies. A representative blot (D) and analysis of band density (E) from two independent experiments are shown. Values are depicted as mean ± SD, *P < 0.05, ns not significant.
Since the effects of PP1α on MR and AR were assumed to be mediated by inhibition of MDM2-dependent receptor degradation (Liu et al., 2016; Nagarajan et al., 2017), the impact of MDM2 on GR activity was assessed in HEK-293 cells expressing the respective recombinant proteins. Although MDM2 overexpression led to decreased GR-mediated reporter gene activity, the PP1α-dependent stimulation of GR transactivation could not be prevented, neither under basal (Fig. 1B) nor under cortisol-stimulated (Fig. 1C) conditions. In the basal state, PP1α enhanced the GR transactivation in the absence of MDM2 4.5-fold and in its presence 4-fold. Similarly, in the presence of 100 nM cortisol PP1α stimulated GR activity about 2-fold.
Furthermore, to test whether PP1α alters the translocation of cytosolic GR into the nucleus, nuclear and cytosolic fractions of HEK-293 cells transfected with GR in the absence or presence of PP1α were analyzed by Western blotting (Fig. 1D and 1E). As expected, cortisol treatment induced nuclear translocation of GR; however, PP1α overexpression significantly enhanced GR translocation by about 3 times when the cells were treated with cortisol (Fig. 1E).
3.2. PP1α knockdown decreases GR-dependent gene transcription in A549 cells
In order to support the stimulatory influence of PP1α on GR activity that was observed in the HEK-293 overexpression model, A549 human alveolar carcinoma cells endogenously expressing PP1α and GR were applied. PP1α was downregulated using siRNA, followed by detection of GR protein by Western blot analysis. PP1α protein was efficiently down regulated (Fig. 2A and 2B), but without significantly affecting GR protein expression (Fig. 2A and 2C). Next, cytosolic and nuclear fractions of A549 cells transfected with PP1α siRNA were analyzed by Western blotting (Fig. 2D and 2E), whereby no significant alterations of cytosolic and nuclear GR protein amounts could be detected. PP1α was found to be mainly located in the nucleus. In contrast, a homogenous distribution of PP1α between the cytoplasm and the nucleus along with an increased GR translocation was observed in HEK-293 cells upon overexpression of PP1α and GR (Fig. 1D and 1E). This difference to the A549 cells might be a result of limited availability of GR and PP1α associated regulatory proteins in HEK-293 cells. Nevertheless, these data suggest that the PP1α-dependent GR activation is not a cause of altered receptor protein stability and unlikely mediated through MDM2.
Fig. 2. Impact of PP1α knockdown on endogenous GR protein expression, translocation and GC-induced transcripts.
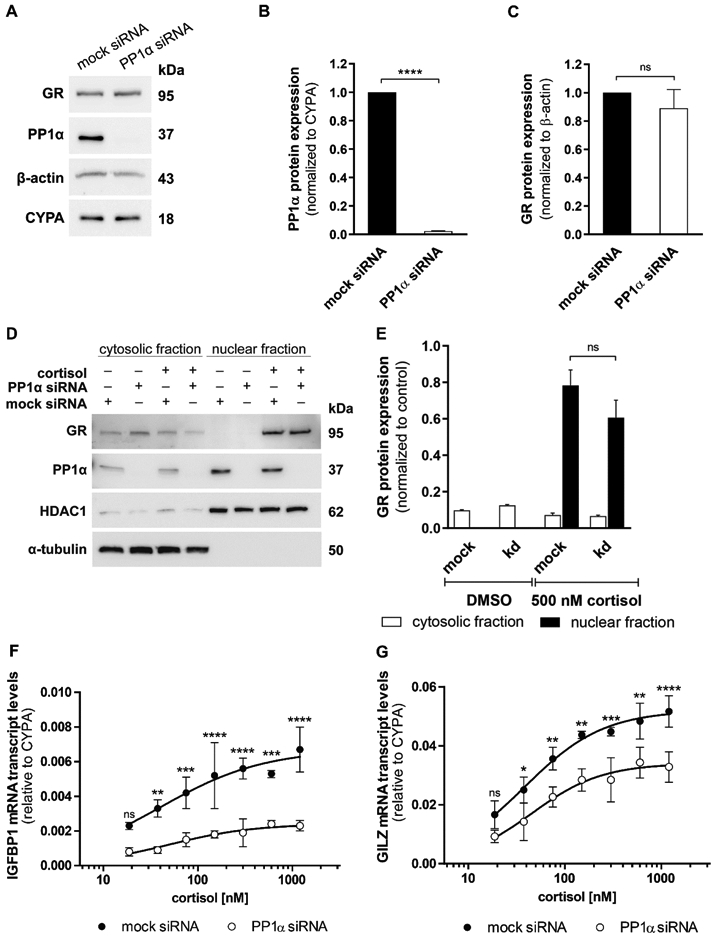
(A, B and C) Western blot analysis for protein expression of GR and PP1α was performed after mock or anti-PP1α siRNA treatment in A549 cells. A representative blot (A) and densitometry analysis of PP1α (B) and GR (C) from three independent experiments are shown. Data are normalized to mock siRNA samples (mean ± SD, ****P < 0.0001, ns not significant). (D and E) A549 cells were transfected with mock or anti-PP1α siRNA for 48 h, incubated overnight in serum-free medium and treated with vehicle or 500 nM cortisol for 1 h. Western blot analysis for cytosolic and nuclear fractions was performed using antibodies against GR and PP1α. As controls, the fractions were reprobed with anti-α-tubulin (cytosolic) and anti-HDAC1 (nuclear) antibodies. A representative blot (D) and analysis of band density (E) from two independent experiments are shown. Values are presented as mean ± SD, ns not significant. (F and G) At 48 h after knockdown of PP1α, A549 cells were cultured in steroid-free medium overnight prior to incubation with vehicle or increasing concentrations of cortisol (18.75 nM - 1200 nM) for 4 h. Cortisol-induced transcription of the GR-responsive genes IGFBP1 (F) and GILZ (G) was measured by RT-qPCR in technical triplicate for each sample. Data are represented as 2−ΔCt mean ± SD from three independent experiments, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns not significant.
Furthermore, co-immunoprecipitation assays using A549 cell lysates were performed according to two different protocols using two antibodies against different epitopes on GR in order to determine whether PP1α and the GR interact directly. Moreover, pull-down experiments were conducted once with an antibody against GR (Suppl. Fig. 2A and 2B) and once with an antibody against PP1α (Suppl. Fig. 2C and 2D). Nevertheless, there was no evidence for a physical interaction between PP1α and GR, both in the absence (Suppl. Fig. 2A and 2C) and presence (Suppl. Fig. 2B and 2D) of cortisol.
Importantly, the cortisol-induced expression of the endogenous GR-responsive genes IGFBP1 and GILZ was markedly downregulated, in a concentration-dependent manner, in PP1α siRNA transfected cells compared to mock siRNA transfected cells (Fig. 2F and 2G). In addition, using a different PP1α-specific siRNA confirmed the effect of PP1α silencing on the expression of GILZ and IGFBP1 (Suppl. Fig. 3).
3.3. PP1α knockdown results in reduced GR-Ser211 phosphorylation
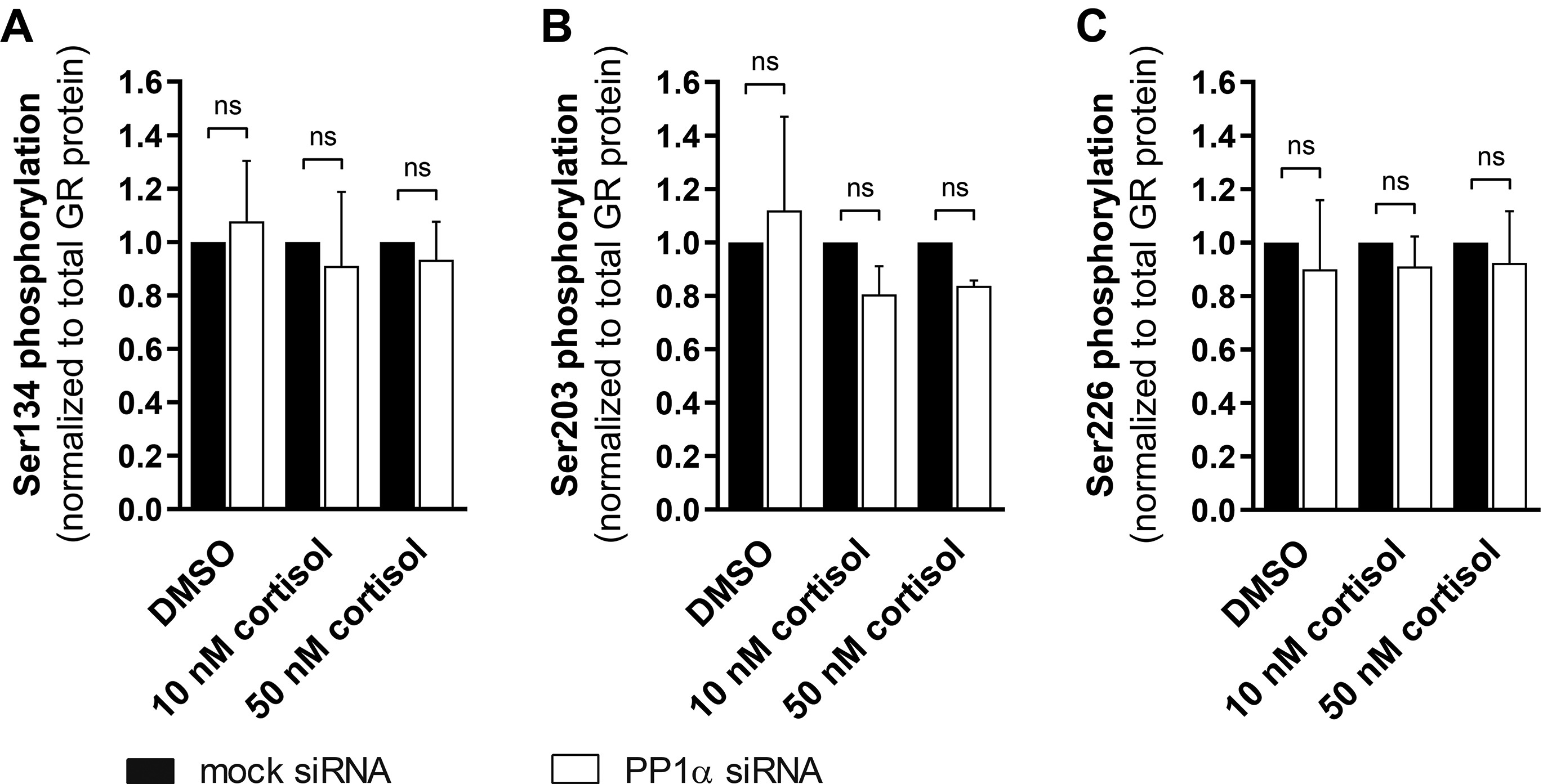
Since PP1α stimulated the activity of the GR but did not seem to influence its stability nor directly interact with the receptor, it was next assessed whether altered phosphorylation at four well-characterized sites on GR, namely serine residues 134, 203, 211 and 226, may affect the transcriptional response to GC. For this purpose, A549 cells were transfected with siRNA against PP1α, treated with 10 nM or 50 nM cortisol for 1 h, prior to cell lysis, and Western blot analysis using phospho-specific and total anti-GR antibodies was performed to determine the serine residues influenced (Fig. 3A). Silencing of PP1α did not affect GR phosphorylation of serine residues 134, 203 and 226 (Suppl. Fig. 4A-C), but reduced the phosphorylation status of Ser211 in a GC-independent manner (Fig. 3B). Influences on the phosphorylation of Ser404 were also tested. The antibody signal for GR-Ser404 was very weak and several other bands were detected, not allowing densitometry analysis; however, there was no evidence supporting altered phosphorylation upon PP1α knockdown (data not shown).
Fig. 3. Effect of PP1α knockdown on GR phosphorylation.
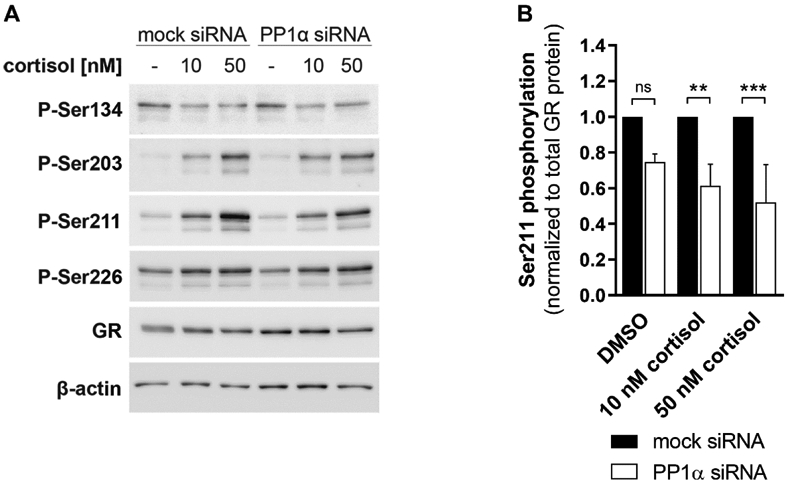
A549 cells were transfected with mock or anti-PP1α siRNA for 48 h, incubated in serum-free medium for 16-18 h, treated with vehicle or cortisol (10 nM and 50 nM cortisol) for 1 h, followed by cell lysis and Western blot analysis. A representative Western blot image (A) and densitometry values (B) are presented. Levels of phosphorylated Ser211 from three independent experiments were normalized to total GR levels and are depicted as values normalized to mock siRNA control (mean ± SD, ***P < 0.001, **P < 0.01, ns not significant).
3.4. Impact of various kinase inhibitors on the PP1α-mediated effect on GR-induced transcripts
Next, in an attempt to start to understand the mechanism underlying the influence of PP1α on GR-Ser211 phosphorylation, A549 cells transfected with mock or anti-PP1α siRNA were exposed to inhibitors of several known kinases. The mRNA expression levels of the two GR-responsive genes IGFBP1 and GILZ were then measured in order to identify the kinase(s) involved in the PP1α-dependent GR stimulation. In both, the basal and cortisol-activated state, a significant decrease in the GILZ and IGFBP1 mRNA expression was observed in cells transfected with siRNA against PP1α compared to mock siRNA treated cells (Fig. 4A-D). This effect of PP1α knockdown was retained when the cells were exposed to inhibitors of p38, MEK1/2, JNK and Akt1/2. In contrast, the PP1α-mediated effect on both genes was reversed by treatment with the GSK-3 inhibitor, i.e. expression levels in mock treated cells were reduced to those of cells transfected with anti-PP1α siRNA, indicating an involvement of GSK-3 in the PP1α-dependent GR stimulation. However, IGFBP1 expression was abrogated in the presence of GSK inhibitor. IGFBP1 carries a thymine-rich insulin response element (TIRE) in its promoter region (Finlay et al., 2004), implying that GSK-3 has to be active for efficient gene transcription. Therefore, IGFBP1 is not appropriate to demonstrate the impact of GSK-3 on the PP1α-mediated GR activation, and another GR-dependent gene, SDPR, was analyzed under both basal and cortisol-stimulated conditions (Fig. 4E and 4F). The results support an involvement of GSK-3 in the PP1α-related GR modulation (see Fig. 6 for an overview of the proposed mechanism). It needs to be noted that, when comparing mock transfected cells incubated with DMSO or cortisol vs inhibitors, p38 and MEK1/2 inhibitors did not exert a pronounced effect on GILZ and IGFBP1 gene expression levels, whereas JNK and Akt1/2 inhibitors markedly increased the expression levels of these genes in both basal and cortisol-stimulated state. This suggests that JNK and Akt1/2 pathways are active in A549 cells and their inhibitors influenced GILZ and IGFBP1 expression independent of PP1α.
Fig. 4. Effect of various kinase inhibitors on the PP1α-dependent effect on GR-induced transcripts.

A549 cells were transfected with mock or anti-PP1α siRNA for 48 h, incubated overnight with indicated inhibitors (10 μM; except for GSK-3 inhibitor, 5 μM) in serum-free medium prior to further incubation with vehicle (A, C and E) or 500 nM cortisol (B, D and F) for another 4 h. Expression levels of GR-responsive genes IGFBP1 (A and B), GILZ (C and D) and SDPR (E and F) were analyzed by RT-qPCR. Values are relative to those of the endogenous control gene using the comparative 2−ΔCt method, followed by normalization to mock siRNA samples. Data are presented as mean ± SD from three independent experiments, each performed in technical triplicate. **P < 0.01, *P < 0.05, ns not significant.
Fig. 6. Schematic representation of the modulation of GR activity by PP1α and GSK-3.

(A) Upon entering the cell, cortisol binds to GR and leads to receptor dimerization. For gaining full activity, GR is phosphorylated at Ser211 to activate the transcription of target genes such as IGFBP1, GILZ and SDPR. Phosphorylation of Ser211 is mediated, at least in part, by a mechanism involving active GSK-3. PP1α dephosphorylates and activates GSK-3. (B) Knockdown of PP1α prevents GSK-3 activation, and inhibition as well as knockdown of GSK-3 diminishes GR-Ser211 phosphorylation, its activation and stimulation of target gene transcription.
3.5. Inhibition or knockdown of GSK-3 abrogates the PP1α-dependent effect on GR-Ser211 phosphorylation
To assess whether pharmacological inhibition of GSK-3 can diminish the PP1α-dependent effect on GR-Ser211 phosphorylation, A549 cells were treated with GSK-3 inhibitor, followed by Western blot analysis. Upon cortisol stimulation in mock siRNA transfected cells, GSK-3 inhibition significantly decreased GR-Ser211 phosphorylation, reducing the phosphorylation level to that observed in the absence of PP1α, i.e. in anti-PP1α siRNA treated cells (Fig. 5A and 5B), suggesting that GSK-3 mediates the PP1α-dependent activation of GR. To further support a role for GSK-3 and exclude an off-target effect of the GSK-3 kinase inhibitor as well as to examine the involved GSK-3 isoform, isoform-specific anti-GSK-3 siRNAs were applied under cortisol-stimulated conditions (Fig. 5C and 5D). Whereas silencing of either GSK-3α or GSK-3β both only partially reversed the effect of PP1α knockdown on Ser211 phosphorylation, the combination of both siRNAs fully prevented it. These results imply that GSK-3 inhibition could prevent the effect of PP1α on Ser211 phosphorylation (see Fig. 6 for an overview of the proposed mechanism).
Fig. 5. Effect of GSK-3 on the PP1α-mediated impact on GR-Ser211 phosphorylation.
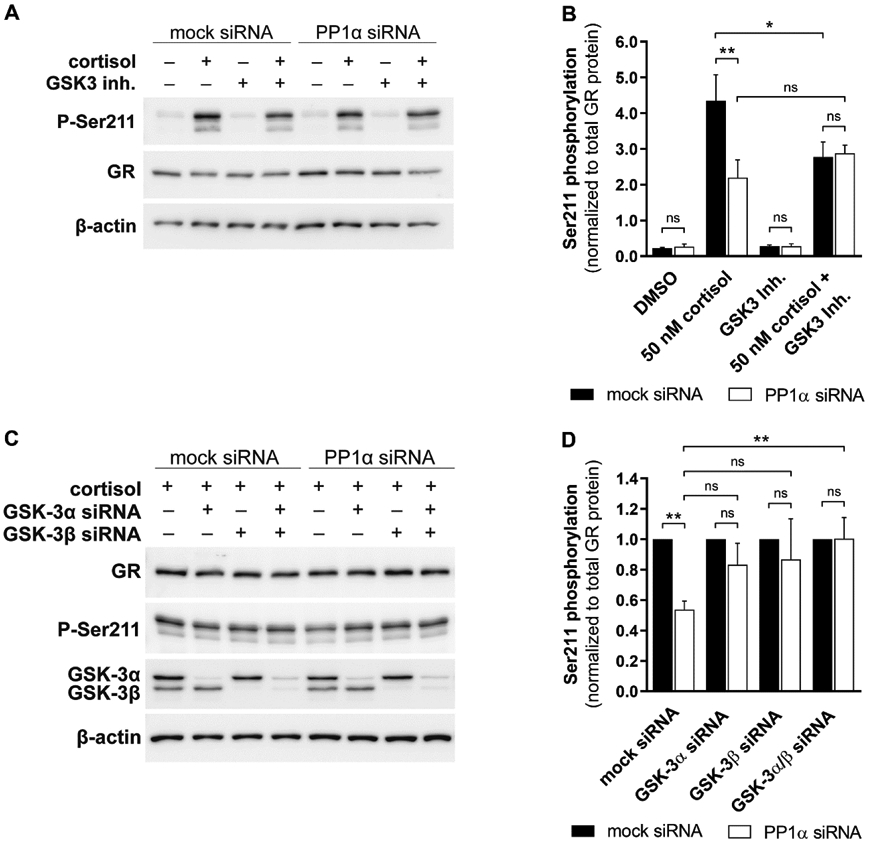
(A and B) After treatment with mock or anti-PP1α siRNA, A549 cells were incubated overnight with 5 μM GSK-3 inhibitor in serum-free medium prior to further incubation with vehicle or 50 nM cortisol for another 1 h. Western blot analysis for expression of total GR protein and Ser211 phosphorylation was performed. A representative blot (A) and densitometry values of Ser211 phosphorylation (B) from two independent experiments normalized to total GR are shown. (C and D) A549 cells were co-transfected with siRNA against PP1α and/or GSK-3α and/or GSK-3β for 48 h, incubated overnight in serum-free medium prior to treatment with 50 nM cortisol for another 1 h. Western blotting for expression of total GR protein and Ser211 phosphorylation was conducted. A representative blot (C) and densitometry analysis of Ser211 phosphorylation (D) from three independent experiments normalized to total GR is presented as values normalized to mock siRNA control. Data represent mean ± SD, **P < 0.01, *P < 0.05, ns not significant.
4. Discussion
The cell- and tissue-specific sensitivity towards GC plays a pivotal role in the fine-tuned regulation of physiological functions as well as for the response to pharmacological doses in the treatment of pathological conditions. Phosphorylation at multiple sites belongs to the most important mechanisms allowing a rapid modulation of the sensitivity of the GR towards GC. The basal and ligand-stimulated phosphorylation of the GR is regulated by various kinases and phosphatases, and an impaired phosphorylation can result in hypersensitivity or resistance to GC, thereby contributing to diseases (Bouazza et al., 2012; DeFranco et al., 1991; Galliher-Beckley et al., 2008; Galliher-Beckley et al., 2011; Habib et al., 2017; Itoh et al., 2002; Kino et al., 2007; Kobayashi et al., 2011; Krstic et al., 1997; Miller et al., 2005; Pazdrak et al., 2016; Rogatsky et al., 1998; Rogatsky et al., 1998; Webster et al., 1997).
PP1α is a ubiquitously expressed protein phosphatase that regulates a vast variety of cellular processes by dephosphorylation of serine/threonine-phosphorylated protein substrates, including cell cycle progression, protein synthesis, mRNA splicing and transcription, calcium signaling and carbohydrate metabolism (Ceulemans et al., 2004; Cohen, 2002). Despite its multiple functions, the impact of PP1α on the modulation of GR activity has not yet been investigated. The current study assessed whether PP1α can stimulate GR function and tested two different hypotheses: First, that PP1α regulates GR activity through suppression of MDM2 activity by dephosphorylating it at Ser166, thereby reducing the MDM2-mediated ubiquitination of GR and the subsequent proteasomal degradation of the receptor, as shown for the MR and AR (Liu et al., 2016; Nagarajan et al., 2017); and second, that PP1α directly dephosphorylates the GR at a particular site to relieve functional repression as demonstrated for PP2A and PP5 (Bouazza et al., 2012; Kobayashi et al., 2011; Pazdrak et al., 2016).
The results revealed a stimulating effect of PP1α on GR activity in HEK-293 cells overexpressing recombinant proteins and a GR-dependent luciferase reporter (Fig. 1A), as well as in A549 cells expressing endogenous GR and PP1α levels by PP1α silencing and assessment of the expression of the GC-target genes IGFBP1 and GILZ (Fig. 2F and 2G). In addition, PP1α overexpression seems to enhance the affinity of GR to cortisol (Fig. 1A). In the absence of ligand, only few receptor molecules exist in the active conformation. The GR might adopt a low affinity antagonist state wherein helix 12 occupies a position covering the coactivator binding site and in which the steroid binding pocket remains closed (Frego et al., 2006; Hu et al., 2011; Kauppi et al., 2003; Quax et al., 2013). Upon ligand binding, the number of receptors in the active conformation increases, indicating that the position of helix 12 enables interaction with coactivator proteins and consists of an open ligand binding pocket. Co-expression with PP1α might further augment the number of receptors in the active conformation, indirectly by promoting phosphorylation of Ser211, leading to increased ligand binding and a shift to higher affinity.
MDM2 has been shown to control the expression of several steroid receptors including the GR (Sengupta et al., 2001). In line with previous observations, co-transfection with MDM2 in HEK-293 cells was found to decrease GR transcriptional activity, both in the basal and cortisol-activated state. However, MDM2 overexpression could not prevent the PP1α-mediated GR activation, with similar relative increases of GR activity in the absence or presence of MDM2. This indicates that PP1α stimulates GR activity by an MDM2-independent mechanism. Moreover, knockdown of PP1α in A549 cells revealed reduced GR-dependent gene expression but did not result in decreased GR protein expression levels, further supporting that PP1α-dependent GR stimulation is independent of MDM2 modulated receptor stability and differs from the mechanism observed for MR and AR (Liu et al., 2016; Nagarajan et al., 2017).
Furthermore, proximity ligation assays suggested an interaction between PP1α and MR, and immunoprecipitation analysis indicated that PP1α interacts with the LBD of the AR (Liu et al., 2016; Nagarajan et al., 2017). In this study, co-immunoprecipitation assays failed to detect a direct interaction between GR and PP1α, further favoring a different mechanism compared to MR and AR. Nevertheless, methods to detect weak and transient interactions such as proximity ligation assays should be performed in follow-on experiments.
To achieve its transcriptional activity, the GR has to translocate from the cytoplasm into the nucleus. Analysis of nuclear and cytosolic fractions of HEK-293 cells overexpressing recombinant GR and PP1α revealed a slight but significantly enhanced cortisol-induced GR translocation from the cytoplasm to the nucleus in the presence of PP1α (Fig. 1D and 1E). In contrast, the amount of GR in the nuclear fraction was not affected by PP1α silencing in A549 cells expressing endogenous receptor levels (Fig. 2D and 2E). This difference in the nuclear import of the GR between the two cell lines might derive from the presence or absence of receptor-associated co-regulators. Furthermore, it should be noted that while the overexpressed PP1α in HEK-293 cells shows an equal distribution between cytoplasm and nucleus, endogenous PP1α in A549 cells is mainly located in the nucleus. This suggests that subcellular localization less likely is a major factor contributing to the PP1α-dependent GR activation.
The site-specific phosphorylation of GR regulates its transcriptional activity (Galliher-Beckley et al., 2009; Zhou et al., 2005). Earlier studies reported that phosphorylation of GR-Ser211 enhances the activity of the receptor, whereas phosphorylation of serine residues 134, 203, 226 and 404 is associated with reduced GR function (Chen et al., 2008; Galliher-Beckley et al., 2008; Galliher-Beckley et al., 2011; Habib et al., 2017; Kino et al., 2007; Krstic et al., 1997; Miller et al., 2005; Miller et al., 2007; Rogatsky et al., 1998; Takabe et al., 2008; Wang et al., 2002). Thus, this study explored whether PP1α-dependent GR stimulation was due to an altered phosphorylation of GR at specific serine residues known to be critical for its activity. The data obtained demonstrated that the level of GR-Ser211 phosphorylation significantly decreased after PP1α silencing (Fig. 3A and 3B) whereas all other phosphorylation sites tested were unaffected (Suppl. Fig. 4). Since phosphorylation of GR-Ser211 is essential for receptor activity, a reduced phosphorylation of this specific residue implicates a decreased transcriptional activity, which was supported by the diminished expression of GC-inducible genes in A549 cells upon PP1α knockdown (Fig. 2F and 2G). The involvement of GR-Ser211 phosphorylation supports the assumption that altered subcellular trafficking is a mechanism less likely contributing to the PP1α-dependent GR activation. Earlier findings have shown that mutation of murine GR at Ser220, the phosphorylation site corresponding to human Ser211, did not affect hormone-dependent nuclear translocation (Webster et al., 1997). The fact that PP1α altered the phosphorylation state of GR-Ser211, and bearing in mind that PP1α is a protein phosphatase, indicates an indirect effect of PP1α on GR, involving another protein.
The present study addressed whether PP1α might dephosphorylate a specific kinase, which in turn is activated, thereby phosphorylating GR at Ser211. Treatment of A549 cells with different inhibitors of known kinases indicated an involvement of GSK-3 in the PP1α-dependent regulation of GC-responsive genes (Fig. 4). Interestingly, inhibitors of JNK and Akt1/2 markedly enhanced mRNA expression levels of GILZ and IGFBP1. JNK was postulated to promote the nuclear export of GR accompanied by termination of GR-dependent transcription through phosphorylation of GR-Ser226 (Itoh et al., 2002), whereas Akt1 was found to delay GR nuclear translocation and reduce GR transcriptional activity by phosphorylation of GR at Ser134 (Habib et al., 2017). Consequently, inhibition of these kinases may result in a decrease in the inhibitory phosphorylation of GR-Ser226 and GR-Ser134, respectively, ultimately leading to increased GR-dependent transcription, as observed in Fig. 4A-D.
Inhibition of GSK-3 prevented the PP1α-mediated difference in the expression levels of GILZ and SDPR as well as completely abolished the effects on IGFBP1. IGFBP1 contains a thymine-rich insulin response element (TIRE) in its promoter region which requires GSK-3 to be active for gene transcription (Finlay et al., 2004), providing an explanation for the repression of IGFBP1 gene expression upon treatment of cells with GSK-3 inhibitor. To assess the role of GSK-3 in the PP1α-dependent effect on GR function, Western blot analysis was performed to determine GR-Ser211 phosphorylation levels. Treatment of A549 cells with GSK-3 inhibitor (Fig. 5A and 5B) and siRNA against GSK-3α/β (Fig. 5C and 5D) resulted in a diminished GR-Ser211 phosphorylation and an abrogated PP1α-mediated effect. Specific knockdown of either GSK-3 a or GSK-3 β partially abolished the PP1α-dependent effect, suggesting that the isoforms might compensate for each other and both are involved in the stimulatory effects of PP1α on GR activity.
GSK-3 is a serine/threonine protein kinase encoded by two genes GSK3A and GSK3B. The closely related isoforms α and β are highly homologous in their catalytic domains but differ significantly in the N- and C-terminal regions (Woodgett, 1990; Woodgett, 1991), suggesting that they could be differentially regulated. However, both proteins are similarly modified at the N-terminal region: phosphorylation of Ser21 on GSK-3α and Ser9 on GSK-3β (Sutherland et al., 1993; Sutherland et al., 1994). PP1α is a known activator of GSK-3β by dephosphorylation of Ser9 (Morfini et al., 2004; Szatmari et al., 2005), and was shown to be engaged in a positive feedback loop with its inhibitor-2 and GSK-3β. GSK-3β can phosphorylate inhibitor-2 of PP1α at Thr72, resulting in PP1α activation, which in turn dephosphorylates and further stimulates GSK-3β (Szatmari et al., 2005; Zhang et al., 2003). It is also assumed that PP1α can dephosphorylate the inhibitory Ser21 on GSK-3α (Spokoini et al., 2010; Zhang et al., 2003). GSK-3 was originally identified as a protein capable of regulating glycogen synthase in order to inhibit glycogen synthesis (Embi et al., 1980) and since then has been described to phosphorylate a wide variety of substrates that are involved in many cellular processes, including glucose metabolism, cell differentiation and apoptosis (Ali et al., 2001; Forde et al., 2007; Soutar et al., 2010). In addition, GSK-3 was found to interact with the GR. In the absence of a ligand, GSK-3α is supposed to be sequestered to the GR and dissociates from it upon exposure to GC (Spokoini et al., 2010). So far, most studies focused on the β-isoform of GSK-3. Inhibition of GSK-3β was reported to suppress GR reporter gene activity and reduce mRNA expression levels of GILZ, implying that GSK-3β has a positive role in GR stimulation (Rubio-Patiño et al., 2012). However, GSK-3β was also proposed to phosphorylate GR-Ser404 and thereby decreasing its function (Galliher-Beckley et al., 2008). Further studies are required to clarify the role of GSK-3 in GR regulation, considering cell type-specific differences. Furthermore, most of the GSK-3 substrates need to be primed by pre-phosphorylation at a specific serine-proline site prior to recognition by GSK-3 (Harwood, 2001). Up to now, such a priming site for GSK-3 within the GR has not been described. Signaling pathways are complex involving a number of components that can cross-talk with other signal transduction pathways. Further research needs to address whether GSK-3 is directly responsible for the PP1α-dependent phosphorylation of GR-Ser211 or whether the observed effect is mediated indirectly through another coregulator. Additionally, it cannot be excluded that GSK-3 dephosphorylation by PP1α is indirect and mediated by a PP1α regulatory protein.
Moreover, the physiological relevance of PP1α-mediated GR signaling needs to be examined. PP1α has been reported to be involved in the progression of several disease states, including memory loss, type II diabetes and cancer (Figueiredo et al., 2014; Ladha et al., 2010; Shastry et al., 2016). PP1α is assumed to suppress learning and memory and acts as a potential mediator of cognitive decline during ageing (Genoux et al., 2002). Excessive GSK-3 activity has been associated with neurodegenerative and psychiatric disorders, and GSK-3 inhibitors as well as GR antagonists are discussed as new potential treatments of Alzheimer’s disease (Canet et al., 2019; Martinez et al., 2008; Muyllaert et al., 2008). In addition, aberrant GC action and excessive activity of GSK-3 have been linked with obesity and type II diabetes (Henriksen et al., 2006; Rose et al., 2010), and regulatory subunits of PP1 have been associated with insulin resistance (Ragolia et al., 1998; Xia et al., 1998). Finally, chronic GC treatment can lead to reduced sensitivity or even resistance in specific cell-types or tissues and underlying mechanisms have been mainly attributed to the impaired GR signaling pathway (Barnes, 2010; Yang et al., 2012). Due to the ability of PP1α to enhance GR activity, one could speculate that cells with impaired PP1α function may have an altered response to GC. Further elucidation of post-translational modification of GR is essential to understand mechanisms of aberrant GC action potentially leading to GC insensitivity.
In this study, PP1α was identified as novel regulator of GR, enhancing its activity but not affecting the expression of the receptor, both under basal and ligand-dependent conditions. Furthermore, the results provided a first insight into the molecular mechanism, proposing GR-Ser211 to be modulated through involvement of GSK-3 (Fig. 6). Thus, PP1α constitutes a new component of the GR signaling pathway, suggesting that impaired activity of PP1α in specific situations could alter GC action and contribute to diseases.
Supplementary Material
Suppl. Fig. 1. Effect of PP1α on GR-dependent luciferase reporter gene activity. (A) To assess whether the PP1α-dependent activation of the reporter gene is mediated by GR, HEK-293 cells were transiently transfected with plasmids coding for a luciferase reporter gene, a Renilla luciferase transfection control, GR, and with or without PP1α. Empty vector pcDNA3.1 was used to adjust the total amount of DNA in the transfection. At 24 h post-transfection, cells were treated with vehicle or 3.7 nM cortisol with or without 1 μM mifepristone (RU-486), followed by incubation for another 24 h. The luciferase reporter activity was normalized to the internal Renilla control. (B) Same as (A) but without transfection of GR and without RU-486 incubations. Values represent mean ± SD from three independent experiments, each performed in triplicate, ***P < 0.001, ns not significant.
{kind=link}
Suppl. Fig. 2. Co-immunoprecipitation of endogenous GR and PP1α. A549 cells endogenously expressing GR and PP1α were treated with (B and D) or without (A and C) 500 nM cortisol for 1 h. Cell lysates were then immunoprecipitated using an anti-GR antibody (A and B) or an anti-PP1α antibody (C and D) and immunoblots were probed with both, anti-GR and anti-PP1α antibodies
{kind=link}
Suppl. Fig. 3. Effect of PP1α silencing on endogenous GR protein expression and GC induced transcripts using an additional PP1α-specific siRNA. (A-D) In order to confirm the specificity of the siRNA used against PP1α, A549 cells were transfected with mock siRNA, anti-PP1α siRNA or an alternative PP1α-specific siRNA (PP1α siRNA#, Dharmacon; 5'-GAACGACCGUGGCGUCUCU-3') for 48 h. (A) densitometry analysis of PP1α and (B) of GR from two independent Western blot experiments. Cortisol-induced transcription of GR905 responsive genes IGFBP1 (C) and GILZ (D) was measured by RT-qPCR after overnight incubation in serum-free medium and treatment with 500 nM cortisol for another 4 h. Expression levels from two independent experiments in technical triplicate for each sample were standardized to those of the endogenous control gene using the comparative 2−ΔCt method. Data were normalized to mock siRNA samples (mean ± SD, ***P < 0.001, **P < 0.01, *P < 0.05, ns not significant).
{kind=link}
Suppl. Fig. 4. Effect of PP1α knockdown on phosphorylation of GR-Ser134, Ser203 and Ser226. A549 cells were transfected with mock or anti-PP1α siRNA for 48 h, incubated in serum-free medium for 16-18 h and treated with vehicle or cortisol (10 nM and 50 nM cortisol) for another 1 h, followed by Western blot analysis using phospho-specific anti-GR-Ser134, anti-GR-Ser203 and anti-GR-Ser226 antibodies. The levels of phosphorylated Ser134 (A), Ser203 (B) and Ser226 (C) from three independent experiments were normalized to total GR levels and are depicted as values normalized to mock siRNA control (mean ± SD, ns not significant).
{kind=link}
Highlights.
PP1α is a novel post-translational activator of GR signaling.
PP1α modulates GR-Ser211 phosphorylation levels.
The PP1 α-dependent GR activation potentially involves GSK-3.
Acknowledgements
This work was supported by the Swiss Centre for Applied Human Toxicology (SCAHT).
Footnotes
Conflicts of Interest
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad M, Hachemi Y, Paxian K, Mengele F, Koenen M and Tuckermann J, 2019. A Jack of All Trades: Impact of Glucocorticoids on Cellular Cross-Talk in Osteoimmunology. Front Immunol. 10, 2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Hoeflich KP and Woodgett JR, 2001. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 101, 2527–40. [DOI] [PubMed] [Google Scholar]
- Atanasov AG, Nashev LG, Gelman L, Legeza B, Sack R, Portmann R and Odermatt A, 2008. Direct protein-protein interaction of 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the endoplasmic reticulum lumen. Biochim Biophys Acta. 1783, 1536–43. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, 2010. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 120, 76–85. [DOI] [PubMed] [Google Scholar]
- Bialojan C and Takai A, 1988. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 256, 283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A and Maser E, 2003. Enzymology and molecular biology of glucocorticoid metabolism in humans. Prog Nucleic Acid Res Mol Biol. 75, 173–216. [DOI] [PubMed] [Google Scholar]
- Bollig A, Xu L, Thakur A, Wu J, Kuo TH and Liao JD, 2007. Regulation of intracellular calcium release and PP1alpha in a mechanism for 4-hydroxytamoxifen-induced cytotoxicity. Mol Cell Biochem. 305, 45–54. [DOI] [PubMed] [Google Scholar]
- Bouazza B, Krytska K, Debba-Pavard M, Amrani Y, Honkanen RE, Tran J and Tliba O, 2012. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: role of phosphatases. Am J Respir Cell Mol Biol. 47, 464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham JC, 2006. Glucocorticoids: exemplars of multi-tasking. Br J Pharmacol. 147 Suppl 1, S258–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet G, Chevallier N, Perrier V, Desrumaux C and Givalois L, 2019. Targeting Glucocorticoid Receptors: A New Avenue for Alzheimer’s Disease Therapy, in: Singh S and Joshi N (Eds.), Pathology, Prevention and Therapeutics of Neurodegenerative Disease. Springer Singapore, Singapore, pp. 173–183. [Google Scholar]
- Ceulemans H and Bollen M, 2004. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 84, 1–39. [DOI] [PubMed] [Google Scholar]
- Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK and Garabedian MJ, 2008. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 22, 1754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Klumpp S and Schelling DL, 1989. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 250, 596–600. [DOI] [PubMed] [Google Scholar]
- Cohen PT, 2002. Protein phosphatase 1--targeted in many directions. J Cell Sci. 115, 241–56. [DOI] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J and Patterson C, 2001. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 3, 93–6. [DOI] [PubMed] [Google Scholar]
- DeFranco DB, Qi M, Borror KC, Garabedian MJ and Brautigan DL, 1991. Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol Endocrinol. 5, 1215–28. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB and Cohen P, 1980. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 107, 519–27. [PubMed] [Google Scholar]
- Faus H and Haendler B, 2006. Post-translational modifications of steroid receptors. Biomed Pharmacother. 60, 520–8. [DOI] [PubMed] [Google Scholar]
- Figueiredo J, da Cruz ESOA and Fardilha M, 2014. Protein phosphatase 1 and its complexes in carcinogenesis. Curr Cancer Drug Targets. 14, 2–29. [DOI] [PubMed] [Google Scholar]
- Finlay D, Patel S, Dickson LM, Shpiro N, Marquez R, Rhodes CJ and Sutherland C, 2004. Glycogen synthase kinase-3 regulates IGFBP-1 gene transcription through the thymine-rich insulin response element. BMC molecular biology. 5, 15–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde JE and Dale TC, 2007. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 64, 1930–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frego L and Davidson W, 2006. Conformational changes of the glucocorticoid receptor ligand binding domain induced by ligand and cofactor binding, and the location of cofactor binding sites determined by hydrogen/deuterium exchange mass spectrometry. Protein Sci. 15, 722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galigniana MD, Harrell JM, Housley PR, Patterson C, Fisher SK and Pratt WB, 2004. Retrograde transport of the glucocorticoid receptor in neurites requires dynamic assembly of complexes with the protein chaperone hsp90 and is linked to the CHIP component of the machinery for proteasomal degradation. Brain Res Mol Brain Res. 123, 27–36. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG, Collins JB and Cidlowski JA, 2008. Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 28, 7309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliher-Beckley AJ and Cidlowski JA, 2009. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 61, 979–86. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG and Cidlowski JA, 2011. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 31, 4663–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garabedian MJ, Harris CA and Jeanneteau F, 2017. Glucocorticoid receptor action in metabolic and neuronal function. F1000Res. 6, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza AMS, Khan SH and Kumar R, 2010. Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Molecular and cellular biology. 30, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D and Mansuy IM, 2002. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 418, 970–5. [DOI] [PubMed] [Google Scholar]
- Gross KL and Cidlowski JA, 2008. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 19, 331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib T, Sadoun A, Nader N, Suzuki S, Liu W, Jithesh PV and Kino T, 2017. AKT1 has dual actions on the glucocorticoid receptor by cooperating with 14-3-3. Mol Cell Endocrinol. 439, 431–443. [DOI] [PubMed] [Google Scholar]
- Harwood AJ, 2001. Regulation of GSK-3: a cellular multiprocessor. Cell. 105, 821–4. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ and Dokken BB, 2006. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets. 7, 1435–41. [DOI] [PubMed] [Google Scholar]
- Hu X, Du S, Tunca C, Braden T, Long KR, Lee J, Webb EG, Dietz JD, Hummert S, Rouw S, Hegde SG, Webber RK and Obukowicz MG, 2011. The antagonists but not partial agonists of glucocorticoid receptor ligands show substantial side effect dissociation. Endocrinology. 152, 3123–34. [DOI] [PubMed] [Google Scholar]
- Ismaili N and Garabedian MJ, 2004. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 1024, 86–101. [DOI] [PubMed] [Google Scholar]
- Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H and Imai K, 2002. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 16, 2382–92. [DOI] [PubMed] [Google Scholar]
- Jewell CM, Webster JC, Burnstein KL, Sar M, Bodwell JE and Cidlowski JA, 1995. Immunocytochemical analysis of hormone mediated nuclear translocation of wild type and mutant glucocorticoid receptors. J Steroid Biochem Mol Biol. 55, 135–46. [DOI] [PubMed] [Google Scholar]
- Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J and Carlquist M, 2003. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 278, 22748–54. [DOI] [PubMed] [Google Scholar]
- Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, Kawasaki E, Pant HC and Chrousos GP, 2007. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 21, 1552–68. [DOI] [PubMed] [Google Scholar]
- Kinyamu HK and Archer TK, 2003. Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in mdm2 protein expression. Mol Cell Biol. 23, 5867–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Mercado N, Barnes PJ and Ito K, 2011. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PloS one. 6, e27627–e27627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic MD, Rogatsky I, Yamamoto KR and Garabedian MJ, 1997. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 17, 3947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladha J, Donakonda S, Agrawal S, Thota B, Srividya MR, Sridevi S, Arivazhagan A, Thennarasu K, Balasubramaniam A, Chandramouli BA, Hegde AS, Kondaiah P, Somasundaram K, Santosh V and Rao SM, 2010. Glioblastoma-specific protein interaction network identifies PP1A and CSK21 as connecting molecules between cell cycle-associated genes. Cancer Res. 70, 6437–47. [DOI] [PubMed] [Google Scholar]
- Le Drean Y, Mincheneau N, Le Goff P and Michel D, 2002. Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology. 143, 3482–9. [DOI] [PubMed] [Google Scholar]
- Liu X, Han W, Gulla S, Simon NI, Gao Y, Cai C, Yang H, Zhang X, Liu J, Balk SP and Chen S, 2016. Protein phosphatase 1 suppresses androgen receptor ubiquitylation and degradation. Oncotarget. 7, 1754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25, 402–8. [DOI] [PubMed] [Google Scholar]
- Mani O, Nashev LG, Livelo C, Baker ME and Odermatt A, 2016. Role of Pro-637 and Gln-642 in human glucocorticoid receptors and Ser-843 and Leu-848 in mineralocorticoid receptors in their differential responses to cortisol and aldosterone. J Steroid Biochem Mol Biol. 159, 31–40. [DOI] [PubMed] [Google Scholar]
- Martinez A and Perez DI, 2008. GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer's disease? J Alzheimers Dis. 15, 181–91. [DOI] [PubMed] [Google Scholar]
- Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R and Thompson EB, 2005. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 19, 1569–83. [DOI] [PubMed] [Google Scholar]
- Miller AL, Garza AS, Johnson BH and Thompson EB, 2007. Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell Int. 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead GBG, Trinkle-Mulcahy L and Ulke-Lemee A, 2007. Emerging roles of nuclear protein phosphatases. Nature Reviews Molecular Cell Biology. 8, 234. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U and Brady ST, 2004. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. Embo j. 23, 2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyllaert D, Kremer A, Jaworski T, Borghgraef P, Devijver H, Croes S, Dewachter I and Van Leuven F, 2008. Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 7 Suppl 1, 57–66. [DOI] [PubMed] [Google Scholar]
- Nagarajan S, Vohra T, Loffing J and Faresse N, 2017. Protein Phosphatase 1alpha enhances renal aldosterone signaling via mineralocorticoid receptor stabilization. Mol Cell Endocrinol. 450, 74–82. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Busillo JM and Cidlowski JA, 2017. Cross-talk between the glucocorticoid receptor and MyoD family inhibitor domain-containing protein provides a new mechanism for generating tissue-specific responses to glucocorticoids. J Biol Chem. 292, 5825–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazdrak K, Straub C, Maroto R, Stafford S, White WI, Calhoun WJ and Kurosky A, 2016. Cytokine-Induced Glucocorticoid Resistance from Eosinophil Activation: Protein Phosphatase 5 Modulation of Glucocorticoid Receptor Phosphorylation and Signaling. J Immunol. 197, 3782–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo MG, Oakley RH and Cidlowski JA, 2019. beta-Arrestin-1 inhibits glucocorticoid receptor turnover and alters glucocorticoid signaling. J Biol Chem. 294, 11225–11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I, Bougarne N, Vandewalle J, Dejager L, Vandevyver S, Ballegeer M, Desmet S, Thommis J, De Cauwer L, Lievens S, Libert C, Tavernier J and De Bosscher K, 2017. Glucocorticoid Receptor-mediated transactivation is hampered by Striatin-3, a novel interaction partner of the receptor. Sci Rep. 7, 8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Stasenko LJ and DeFranco DB, 1990. Recycling and desensitization of glucocorticoid receptors in v-mos transformed cells depend on the ability of nuclear receptors to modulate gene expression. Mol Endocrinol. 4, 455–64. [DOI] [PubMed] [Google Scholar]
- Quax RA, Manenschijn L, Koper JW, Hazes JM, Lamberts SW, van Rossum EF and Feelders RA, 2013. Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol. 9, 670–86. [DOI] [PubMed] [Google Scholar]
- Ragolia L and Begum N, 1998. Protein phosphatase-1 and insulin action. Molecular and Cellular Biochemistry. 182, 49–58. [PubMed] [Google Scholar]
- Rogatsky I, Logan SK and Garabedian MJ, 1998. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci U S A. 95, 2050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Waase CL and Garabedian MJ, 1998. Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase-3 (GSK-3). Species-specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK-3 phosphorylation. J Biol Chem. 273, 14315–21. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Vegiopoulos A and Herzig S, 2010. Role of glucocorticoids and the glucocorticoid receptor in metabolism: insights from genetic manipulations. J Steroid Biochem Mol Biol. 122, 10–20. [DOI] [PubMed] [Google Scholar]
- Rubio-Patiño C, Palmeri CM, Pérez-Perarnau A, Cosialls AM, Moncunill-Massaguer C, González-Gironès DM, Pons-Hernández L, López JM, Ventura F, Gil J, Pons G and Iglesias-Serret D, 2012. Glycogen synthase kinas-3β is involved in ligand-dependent activation of transcription and cellular localization of the glucocorticoid receptor. Molecular endocrinology (Baltimore, Md.). 26, 1508–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S and Wasylyk B, 2001. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. 15, 2367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry AH, Thota B, Srividya MR, Arivazhagan A and Santosh V, 2016. Nuclear Protein Phosphatase 1 alpha (PP1A) Expression is Associated with Poor Prognosis in p53 Expressing Glioblastomas. Pathol Oncol Res. 22, 287–92. [DOI] [PubMed] [Google Scholar]
- Somers JP and DeFranco DB, 1992. Effects of okadaic acid, a protein phosphatase inhibitor, on glucocorticoid receptor-mediated enhancement. Mol Endocrinol. 6, 26–34. [DOI] [PubMed] [Google Scholar]
- Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, Snider WD, Gordon-Weeks PR and Sutherland C, 2010. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem. 115, 974–83. [DOI] [PubMed] [Google Scholar]
- Spokoini R, Kfir-Erenfeld S, Yefenof E and Sionov RV, 2010. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol Endocrinol. 24, 1136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA and Cohen P, 1993. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 296 ( Pt 1), 15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C and Cohen P, 1994. The alpha-isoform of glycogen synthase kinase-3 from rabbit skeletal muscle is inactivated by p70 S6 kinase or MAP kinase-activated protein kinase-1 in vitro. FEBS Lett. 338, 37–42. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T and Hetman M, 2005. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 280, 37526–35. [DOI] [PubMed] [Google Scholar]
- Takabe S, Mochizuki K and Goda T, 2008. De-phosphorylation of GR at Ser203 in nuclei associates with GR nuclear translocation and GLUT5 gene expression in Caco-2 cells. Arch Biochem Biophys. 475, 1–6. [DOI] [PubMed] [Google Scholar]
- Taylor S, Wakem M, Dijkman G, Alsarraj M and Nguyen M, 2010. A practical approach to RT-qPCR—Publishing data that conform to the MIQE guidelines. Methods. 50, S1–S5. [DOI] [PubMed] [Google Scholar]
- Wallace AD and Cidlowski JA, 2001. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 276, 42714–21. [DOI] [PubMed] [Google Scholar]
- Wang X and DeFranco DB, 2005. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. 19, 1474–82. [DOI] [PubMed] [Google Scholar]
- Wang Z, Frederick J and Garabedian MJ, 2002. Deciphering the phosphorylation "code" of the glucocorticoid receptor in vivo. J Biol Chem. 277, 26573–80. [DOI] [PubMed] [Google Scholar]
- Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M and Cidlowski JA, 1997. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. 272, 9287–93. [DOI] [PubMed] [Google Scholar]
- Woodgett JR, 1990. Molecular cloning and expression of glycogen synthase kinase-3/factor A. Embo j. 9, 2431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, 1991. cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol. 200, 564–77. [DOI] [PubMed] [Google Scholar]
- Xia J, Scherer SW, Cohen PT, Majer M, Xi T, Norman RA, Knowler WC, Bogardus C and Prochazka M, 1998. A common variant in PPP1R3 associated with insulin resistance and type 2 diabetes. Diabetes. 47, 1519–24. [DOI] [PubMed] [Google Scholar]
- Yang N, Ray DW and Matthews LC, 2012. Current concepts in glucocorticoid resistance. Steroids. 77, 1041–9. [DOI] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N and Klein PS, 2003. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 278, 33067–77. [DOI] [PubMed] [Google Scholar]
- Zhang S, Jonklaas J and Danielsen M, 2007. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids. 72, 600–608. [DOI] [PubMed] [Google Scholar]
- Zhou J and Cidlowski JA, 2005. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 70, 407–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 1. Effect of PP1α on GR-dependent luciferase reporter gene activity. (A) To assess whether the PP1α-dependent activation of the reporter gene is mediated by GR, HEK-293 cells were transiently transfected with plasmids coding for a luciferase reporter gene, a Renilla luciferase transfection control, GR, and with or without PP1α. Empty vector pcDNA3.1 was used to adjust the total amount of DNA in the transfection. At 24 h post-transfection, cells were treated with vehicle or 3.7 nM cortisol with or without 1 μM mifepristone (RU-486), followed by incubation for another 24 h. The luciferase reporter activity was normalized to the internal Renilla control. (B) Same as (A) but without transfection of GR and without RU-486 incubations. Values represent mean ± SD from three independent experiments, each performed in triplicate, ***P < 0.001, ns not significant.
Suppl. Fig. 2. Co-immunoprecipitation of endogenous GR and PP1α. A549 cells endogenously expressing GR and PP1α were treated with (B and D) or without (A and C) 500 nM cortisol for 1 h. Cell lysates were then immunoprecipitated using an anti-GR antibody (A and B) or an anti-PP1α antibody (C and D) and immunoblots were probed with both, anti-GR and anti-PP1α antibodies
Suppl. Fig. 3. Effect of PP1α silencing on endogenous GR protein expression and GC induced transcripts using an additional PP1α-specific siRNA. (A-D) In order to confirm the specificity of the siRNA used against PP1α, A549 cells were transfected with mock siRNA, anti-PP1α siRNA or an alternative PP1α-specific siRNA (PP1α siRNA#, Dharmacon; 5'-GAACGACCGUGGCGUCUCU-3') for 48 h. (A) densitometry analysis of PP1α and (B) of GR from two independent Western blot experiments. Cortisol-induced transcription of GR905 responsive genes IGFBP1 (C) and GILZ (D) was measured by RT-qPCR after overnight incubation in serum-free medium and treatment with 500 nM cortisol for another 4 h. Expression levels from two independent experiments in technical triplicate for each sample were standardized to those of the endogenous control gene using the comparative 2−ΔCt method. Data were normalized to mock siRNA samples (mean ± SD, ***P < 0.001, **P < 0.01, *P < 0.05, ns not significant).
Suppl. Fig. 4. Effect of PP1α knockdown on phosphorylation of GR-Ser134, Ser203 and Ser226. A549 cells were transfected with mock or anti-PP1α siRNA for 48 h, incubated in serum-free medium for 16-18 h and treated with vehicle or cortisol (10 nM and 50 nM cortisol) for another 1 h, followed by Western blot analysis using phospho-specific anti-GR-Ser134, anti-GR-Ser203 and anti-GR-Ser226 antibodies. The levels of phosphorylated Ser134 (A), Ser203 (B) and Ser226 (C) from three independent experiments were normalized to total GR levels and are depicted as values normalized to mock siRNA control (mean ± SD, ns not significant).