

Abstract
Neurogenic orthostatic hypotension (nOH) is among the most debilitating non-motor symptoms of patients with Parkinson disease (PD) and other synucleinopathies. Patients with PD and nOH generate more hospitalizations, make more emergency room visits, create more telephone calls-mails to doctors, and have earlier mortality than those with PD but without nOH. Overall, the health-related cost in patients with PD and OH is 2.5-fold higher compared to patients with PD without OH. Hence, developing effective therapies for nOH should be a research priority. In the last few decades, improved understanding of the pathophysiology of nOH has led to the identification of therapeutic targets and the development and approval of two drugs, midodrine and droxidopa. More effective and safer therapies, however, are still needed, particularly agents that could selectively increase blood pressure only in the standing position because supine hypertension is the main limitation of available drugs. Here we review the design and conduct of nOH clinical trials in patients with PD and other synucleinopathies, summarize the results of the most recently completed and ongoing trials, and discuss challenges, bottlenecks and potential remedies.
Keywords: Dysautonomia, Orthostatic hypotension, Clinical trials, Design, Endpoints
INTRODUCTION
Dysfunction of the autonomic nervous system, causing neurogenic orthostatic hypotension (nOH), is a well-known feature of patients with Parkinson disease (PD) and other synucleinopathies, occurring at all stages of the disease. Moreover, there is now strong evidence indicating that nOH can be one of the earliest prodromal manifestations of the disease, occurring years, and sometimes decades before motor impairment occurs [1, 2].
OH is a sustained fall in blood pressure (BP) on standing. The current definition of OH, based on expert consensus [3], is a fall of at least 20 mmHg in systolic BP or 10 mmHg in diastolic BP within 3 minutes of standing or upright tilt. OH can impair blood supply above the heart, resulting in symptoms of tissue hypoperfusion most notably in the brain including the retina, but also in the neck muscles. Symptoms can be very disabling, reduce quality of life, and increase morbidity, particularly syncope and falls [4, 5] and mortality [6, 7]. In patients with PD or other synucleinopathies [dementia with Lewy bodies, multiple system atrophy (MSA), and pure autonomic failure (PAF)], in which there is abnormal accumulation of α-synuclein (αSyn) in the nervous system, OH is due to reduced norepinephrine release from postganglionic sympathetic nerves, resulting in defective vasoconstriction when in the upright posture [3]. This is referred to as neurogenic OH (nOH) [8, 9].
Patients with PD and nOH generate more hospitalizations and emergency room visits, make more telephone calls-mails to doctors, and have earlier mortality than those with PD but without nOH [10, 11]. Overall, the health-related cost in patients with PD and OH is 2.5-fold higher compared to patients with PD without OH[11]. Hence, developing effective therapies for nOH should be a research priority.
In the few last decades, improved understanding of the pathophysiology of nOH in synucleinopathies has led to the identification of therapeutic targets and the development and approval of effective drug therapies, such as midodrine and droxidopa. Here we review the design and implementation of clinical trials for nOH, summarize the results of the most recently completed and ongoing trials, and discuss challenges, bottlenecks and potential remedies.
PATHOPHYSIOLOGY: THE KEY TO DEVELOPING NOH THERAPIES
Understanding the pathophysiology and neural pathways involved in nOH is key to developing effective treatments. Normally, standing up unloads the baroreceptors and triggers a centrally mediated reflex that results in norepinephrine release from sympathetic post-ganglionic neurons innervating blood vessels. Norepinephrine activates alpha-adrenergic receptors in the vascular walls causing vasoconstriction, which maintains BP upright. In patients with synucleinopathies norepinephrine release in the standing position is blunted so that compensatory vasoconstriction is absent or attenuated, resulting in nOH. Therefore, nOH is best understood as a neurotransmitter disorder. Like dopamine deficiency in the nigrostriatal pathway causes the motor abnormalities, impaired release of norepinephrine from sympathetic postganglionic neurons causes nOH (Figure 1).
Figure 1. Neurogenic orthostatic hypotension as a neurotransmitter disorder in Parkinson disease.
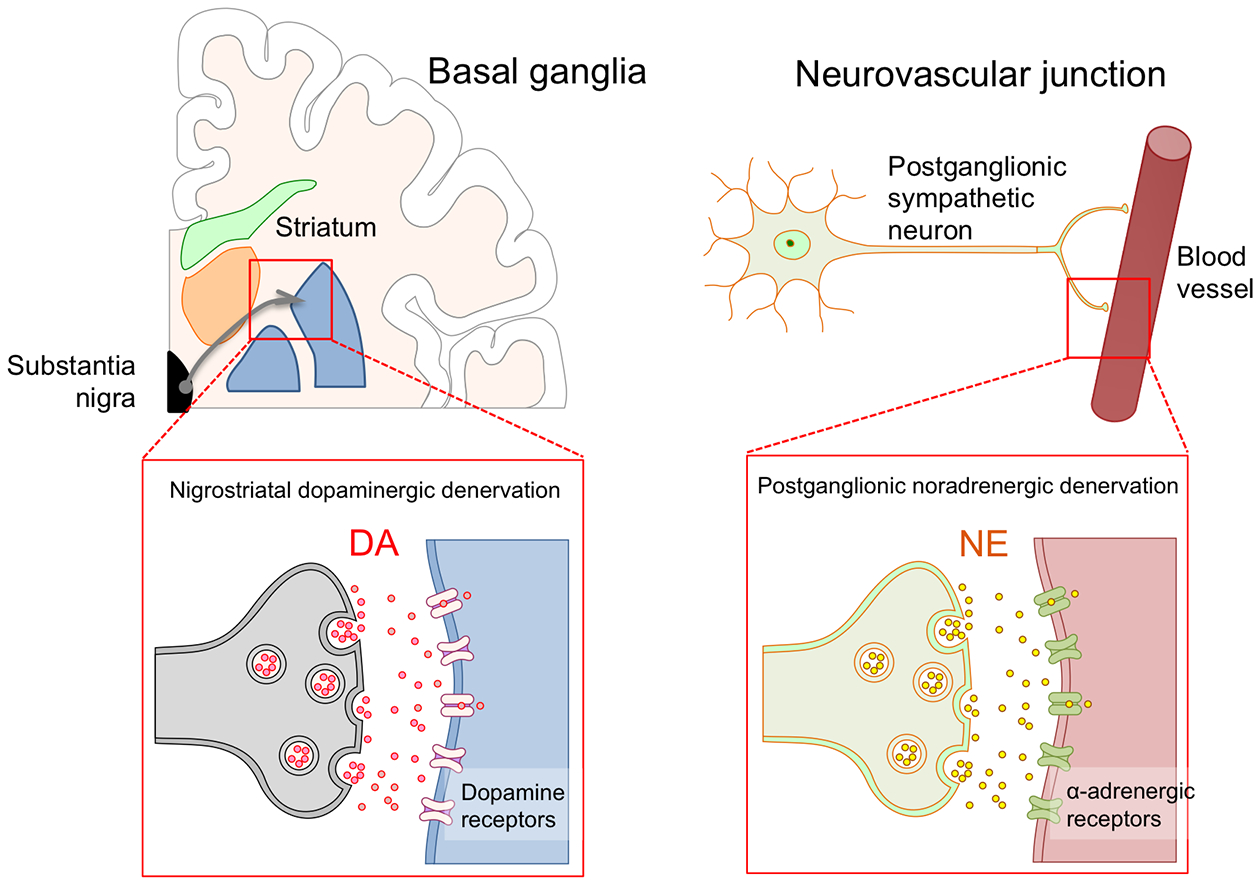
Motor dysfunction in Parkinson disease and other synucleinopathies is mostly due to a dopaminergic deficit in basal ganglia neurons caused by loss of nigrostriatal dopaminergic neurons. Dopamine deficit can be treated with oral administration of the dopamine precursor L-dopa or with dopaminergic agonists. Defective vasoconstriction leading to neurogenic orthostatic hypotension is due to impaired release of norepinephrine from postganglionic sympathetic terminals due to postganglionic sympathetic denervation. Norepinephrine deficit can be treated with oral administration of the norepinephrine precursor droxidopa, or with direct adrenergic agonists like midodrine.
The site of the “autonomic lesion” in the baroreflex pathways responsible for nOH is different in patients with PD and DLB (Lewy body disorders) than in those with MSA. In patients with Lewy body disorders, nOH is predominantly due to degeneration of peripheral post-ganglionic sympathetic neurons. There is robust imaging data showing that post-ganglionic sympathetic neurons innervating the myocardium are functionally affected and neuropathological studies show αSyn deposits and fiber loss [12, 13]. Sympathetic fibers innervating blood vessels are also affected. [13, 14]. Indeed, as expected, plasma norepinephrine levels are lower in patients with PD and nOH than in those without nOH [15].
In contrast, nOH in patients with MSA is caused by degeneration of CNS neurons involved in baroreflex control, [16–20] while only a minority (< 30%) of patients have degeneration of peripheral post-ganglionic sympathetic neurons [12]. Indeed, plasma norepinephrine levels while supine are normal in most patients with MSA,[21] reflecting intact postganglionic sympathetic neurons. These differences between Lewy body disorders and MSA are relevant to explain the different vasopressor responses to norepinephrine reuptake inhibitors, and can be used to develop novel molecules targeting autonomic dysfunction due to CNS (i.e., MSA) versus peripheral (i.e., Lewy body disorders) degeneration.
CHALLENGES IN CLINICAL TRIALS FOR NOH
The natural history of nOH in PD and related disorders is incompletely understood, and the ability to detect clinically meaningful outcomes requires thorough understanding of its occurrence and variability, both of which contribute to difficulties in powering a study. Assembling a large cohort of patients requires a multicenter design sometimes involving several countries. An added difficulty is the “background noise” caused by the wide BP variability characteristic in patients with nOH. Patients report that symptom severity and BP readings vary from day-to-day and fluctuate throughout the day. The morning hours tend to be most difficult, as OH symptoms are aggravated by intravascular volume loss overnight [22]. Meals, particularly carbohydrate-rich, lead to splanchnic vasodilatation and post-prandial hypotension (i.e., fall in BP within 2 hours of eating). Temperature, dehydration and physical activity also induce variations in BP.
Patient-reported outcomes have limitations when studying nOH, and reliance on symptoms alone may not always be an accurate indicator of tissue hypoperfusion. Symptoms of nOH can be nonspecific, and the patients may not identify them as such. For example, fatigue and difficultly concentrating, or immobility sometimes mimicking the levodopa “off” state in PD patients may be due to unrecognized hypotension and without measuring blood pressure standing remains undiagnosed and untreated or mistreated. Conversely, patients may attribute to nOH symptoms due to other causes of lightheadedness/imbalance [23], again resulting in wrong or unnecessary treatments. Of note, nOH is frequently associated with progressive neurodegeneration affecting motor function, worsening of the motor disorder may prevent the patient from standing rather than orthostatic hypotension.
Finally, the response to placebo can at times be quite impressive in some patients with nOH. The reason for this is unclear, but it may be related to a Hawthorne effect, i.e., patients are particularly compliant with non-pharmacological measures to treat nOH (liberalization of salt and water intake, avoiding carbohydrates and alcohol, etc.) because they are participating in a clinical trial and these counter-measures are encouraged. Also, whether there is substantial central or peripheral norepinephrine release in response to placebo in patients with PD (as it is the case with striatal dopamine [24]) is unknown, but definitely worth studying.
PATIENT-REPORTED OUTCOME MEASURES FOR CLINICAL TRIALS OF NOH
The US FDA strongly advocates the use of patient-reported outcomes in drug trials. Outcomes should identify the most clinically important consequences of the disease and be easily measured [25]. The ideal outcome measure should assess how patients feel, function and survive and be responsive to a therapeutic intervention in a defined time period. In this regard, it is important to take into consideration that not all patients with nOH are symptomatic. Typical symptoms of nOH are lightheadedness, dizziness, blurry vision, and, when the fall in BP is pronounced, loss of consciousness and postural tone (syncope). In the most severe cases, patients are unable to leave the supine position as even sitting causes symptoms. In less severe cases symptoms occur only when standing, rarely when sitting. In all cases symptoms always abate when lying down, a defining diagnostic feature of nOH which easily distngiushes it from other disorders. Symptoms of cerebral hypoperfusion emerge when the BP falls below the lower limit of the individual’s cerebral autoregulatory range. In patients with PD, this usually occurs when the mean standing BP is below 75 mmHg, which corresponds to ~90/60 mmHg (systolic/diastolic) at the heart level [26]. Symptoms of nOH abate when prone because gravity passively restores cerebral blood flow. Sitting or physical countermaneuvers can also restore cerebral blood flow if they raise blood pressure just above the lower limit of autoregulatory capacity. The chronic nature of nOH allows remarkable adaptive changes in cerebral autoregulatory mechanisms [27]. Indeed, patients with nOH are frequently able to tolerate wide swings in BPs and often remain conscious at pressures that would otherwise induce syncope in healthy subjects [28]. As mentioned above, symptoms of nOH can be nonspecific, including fatigue and cognitive difficulties [29], and may sometimes mimic a levodopa “off” motor state in PD patients. Conversely, postural lightheadedness mimicking nOH may be caused by abnormal postural reflexes, vestibular deficits or orthostatic tremor [30]. Thus, BP needs to be measured in the standing position to ascertain the cause of the symptoms. Ideally, patient-reported outcomes used in clinical trials of nOH should take these factors into consideration.
There are several patient-reported questionnaires that include items related to cardiovascular autonomic dysfunction including the Composite Symptoms Autonomic Score (CASS) [31], the Scales for Outcomes in Parkinson’s disease-Autonomic (SCOPA-AUT) [32], and the Autonomic Symptom Profile (ASP) [33]]. Other global quality of life questionnaires have been developed for patients with autonomic failure [e.g., Multiple System Atrophy Quality of Life scale (MSA-QoL)[34, 35]]. However, these instruments are not optimized for patients with nOH, as none of these questionnaires specifically quantify the impact of nOH on daily activities.
To overcome this, we developed a novel clinical rating scale specifically for patients with nOH, the Orthostatic Hypotension Questionnaire (OHQ) [36]. The OHQ is a self-reported, validated symptom assessment tool made up of two components: a) the OH symptom assessment (OHSA) scale, with 6 separate items to measure the presence and severity of symptoms; and b) the OH daily activity scale (OHDAS) with 4 individual items to measure the impact of orthostatic symptoms on daily activities. The OHQ uses a scale from 0 (no symptoms/no interference) to 10 (worst possible/complete interference), and asks the patient to rate their symptoms for the previous week. Each of the 10 individual items within the OHQ can be used individually or as a composite summing together the total score of all items (Figure 2).
Figure 2. Orthostatic Hypotension Questionnaire (OHQ).
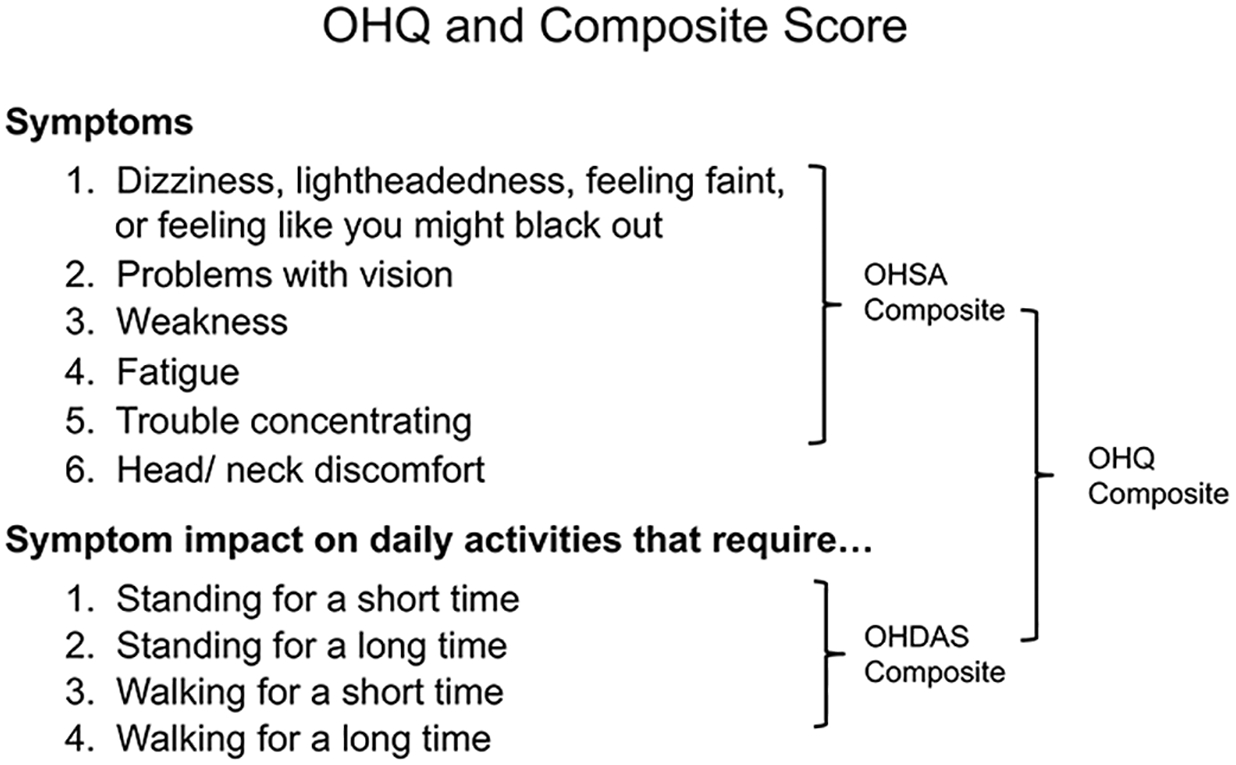
The orthostatic hypotension questionnaire (OHQ) is a validated symptom assessment tool made up of two components: a) the OH symptom assessment (OHSA) scale, with 6 separate items, and b) the OH daily activity scale (OHDAS) with 4 individual items. The OHQ uses a scale from 0 (no symptoms/no interference) to 10 (worst possible/complete interference) and asks the patient to rate their symptoms for the previous week.
The psychometric properties of the OHQ were evaluated using data from a phase-4 multi-center, randomized, placebo-controlled cross-over trial to assess the clinical benefit of the alpha-adrenergic agonist, midodrine, in a group of patients with synucleinopathies and nOH [37]. The OHQ was shown to accurately evaluate the severity of symptoms and the functional impact of nOH, as well as assess the efficacy of treatment [36]. The OHQ was later validated for routine clinical use [38]. Both item 1 of the OHSA scale (symptoms of dizziness/lightheadedness), and the composite overall OHQ score have been used as primary end-points in successful phase 3 clinical trials that led to drug approval.
The OHQ has several strengths. First, it focuses on the full range of symptoms relevant to patients with nOH. Second, the assessment of activities is specific for the most prominent activity impairments imposed by OH: standing or walking for short or long periods of time. Third, the OHQ can accurately measure the symptoms and impact of nOH in a valid and reliable way and, of particular importance for an outcome measure used to assess the impact of treatment interventions, can appropriately detect change over time. Finally, it is brief, making it quick and easy for the patient to complete with minimal burden, a common concern for patient-reported outcomes in clinical trials.
A limitation of the OHQ is that it has a high rate of false positives when used alone, i.e., without accompanying BP readings. Patients complaining of “lightheadedness” due to causes other than OH (e.g., vertigo, hypoglycemia, anxiety and psychosomatic disorders) can score high on the OHQ. Of course, using the OHQ only in populations who have OH confirmed by BP measurements in the supine and standing position theoretically solves this problem. Another minor limitation is that the OHQ asks the patient to rate his symptoms of OH in the last week. Therefore, its properties to accurately measure short-term changes in symptoms (e.g., before and 1-hour after administration of medication) are not known.
The OHQ has been translated to French, Spanish, German and Italian. The OHQ underwent translation and linguistic validation that involved two forward translations, forward translation reconciliation, two back translations and back translation review.
Another patient- and clinician-reported endpoint frequently used as secondary outcome measure in clinical trials of nOH is the Clinical Global Impression (CGI), a tool initially developed for mental health research [39], with two subscales. The CGI-Severity is a 7-point scale scored from 1 (no symptoms) to 7 (severe symptoms), and the CGI-Improvement is a 7-point scale scored from 1 (very much improved) to 7 (very much worse). The patient and the clinician rate both scales independently.
OBJECTIVE OUTCOME MEASURES FOR CLINICAL TRIALS OF NOH
In-office blood pressure readings
The most obvious objective outcome measure in clinical trials of drugs for the symptomatic treatment of nOH is blood pressure. The BP values after 1 or 3 minutes of active standing following drug administration were used in several clinical trials of nOH. How long after drug administration BP should be measured depends on the pharmacodynamics and pharmacokinetics of the tested drug. For instance, in clinical trials of midodrine, the BP taken after 1 minute standing 1-hour post-dose was used as primary endpoint [37], because the prodrug (desglimidodrine) is readily absorbed and converted to active agent, reaching peak plasma levels 1–2 h after oral administration [40].
It is the BP level when standing that is relevant clinically, as it determines cerebral blood flow and symptoms, rather than the BP fall from the supine position. Indeed, the change in BP from the supine to standing position (i.e., ΔBP) is not a useful endpoint, as drugs for nOH typically increase the BP when the subject is supine and standing, therefore, the ΔBP could be similar or even more pronounced when comparing ΔBP before and after drug administration, or ΔBP after active agent vs. placebo.
Other trials have used seated BP 1-hour post-dose, instead of standing BP, as the endpoint [41]. This can be particularly advantageous to enroll patients with very severe nOH, in whom standing for more than a few seconds might be challenging.
It is important that subjects remain blind to the BP results after drug or placebo administration because seeing their BP on the monitor screen may affect their reporting and could potentially unblind the drug allocation.
Finally, some trials of nOH used the “time to symptoms after standing” as a patient-reported endpoint [42]. The primary outcome was the time since the tilt table is positioned in the 60-degree angle until the patient experiences symptoms of syncope or near-syncope and asks to be tilted down. This endpoint has obvious advantages. Because inability to remain in the standing position is one of the main disability of patients with nOH, determining the clinical relevance of the results is straightforward. It might be difficult to establish what a 1- or 2-point reduction in the OHQ, or an increase in 5–10 mmHg in standing blood pressure represent in terms of functional impairment. In contrast, time to symptoms after standing is easier to understand and represents a clearcut measure of functional change: i.e., “the subject was able to remain in the standing position without symptoms for 20 seconds with the placebo, and for 2 minutes with the active agent”.
Ambulatory blood pressure monitoring
In patients with nOH, symptom severity and BP vary from day-to-day and fluctuate throughout the day with activities and meals, due in part to small changes in extracellular fluid volume. BP is often elevated during the night while the patient sleeps supine. Office BP readings do not assess this variability nor the circadian variation of BP [43]. To overcome this limitation, ambulatory BP monitoring (ABPM) can be used to evaluate the response to treatment throughout a typical day [43]. In addition to capturing BP variations while awake, ABPM can assess nocturnal BP. Daytime and particularly nighttime supine hypertension is often worsened by the therapies used to treat nOH, which creates a difficult dilemma [44–47]. Only one clinical trial examined ABPM profiles in patients with nOH on and off droxidopa [48]. The endpoints were mean SBP and DBP during 24-hours, and during the night.
Syncope, presyncope and falls
The drop in BP upon standing in patients with nOH results in cerebral hypoperfusion, which, when severe, can cause near fainting or syncope. Because this, in turn, can result in falls, both syncope and falls could be potentially used as an endpoint in clinical trials of nOH.
Using imminent syncope/presyncope as an endpoint is feasible in the in-office setting. Indeed, a phase-4 tilt-table placebo-controlled trial with midodrine used time to symptoms of syncope or presyncope during passive head-up tilt as the main outcome measure [42]. As discussed above, this is a patient-reported outcome that represents a straightforward functional improvement.
Using falls as an endpoint is challenging and has two obvious limitations. First, not all falls in patients with PD are caused by nOH. The underlying cause of falls in PD may be complex and multifactorial. Risk factors include older age, female sex, polypharmacy, fear of falling, depression, alcohol use, visual impairment, muscle weakness, use of an assistive device, OH, and cardiac arrhythmia, among others. PD–specific risk factors include prior falls, worse disease severity, PD medications (higher daily levodopa dosage, dopamine agonist, or anticholinergic use), slow or shuffling gait, freezing of gait, postural instability (balance impairment), flexed posture, axial rigidity, dyskinesia, cognitive impairment, urinary incontinence, and deep brain stimulation. In a study that analyzed the direct causes of falls, sudden falls were most common (31%), followed by freezing and festination (20%), neurologic and sensory disturbances (mostly vertigo; 12%), postural instability (11%), OH (4%), and severe dyskinesia (4%); 6.2% of falls were unclassified [49–51]. Therefore, using falls as an outcome measure in clinical trials for nOH in PD and related disorders would require a complex statistical analysis to take into consideration all the other potential causes of falls.
The second limitation is that the retrospective self-report of the number and severity of falls or syncopal episodes relies on the patient’s recall, which is prone to bias. Patients may remember the most severe syncope or falls resulting in fractures or other injuries, but overlook minor events without consequences. Even when prospectively filling fall/syncope diaries, patients may not remember to write down the events. Moreover, patients with PD significantly underreport adverse events related to medications [52]. This limitation could be potentially overcome with the use of portable devices (e.g., accelerometers or mobile apps) that can prospectively detect and record the number of falls without requiring the patient’s intervention, as will be later discussed. In placebo-controlled trials, it is also necessary to perform a stratified randomization based on the frequency of falls.
Development of biometric monitoring devices for clinical trials of nOH
There is an increasing interest in the development of biometric monitoring devices (BMD) to accurately and objectively reflect the disease process and status of patients during their daily lives to be used as endpoints in clinical trials [53, 54]. In the case of patients with nOH, the most straightforward application of BMD would be that of providing measurements of BP in conjunction with body position (i.e., sitting, standing, flat) at specific times, or even constantly. This would require smaller and lighter devices than the currently available for ABPM. Ideally, they would transmit data wirelessly in real time. Other potential applications of BMD to be used in clinical trials of nOH include measurement of falls and mobility, as well as quality of life surrogates such as socialization. This could potentially be accomplished with biosensors measuring biological responses incorporated to wearables such as watches, clothing, implants, ingestible sensors, or remote biosensors in specific locations (beds, chairs, doors). The use of in-home monitoring data may reduce sample sizes required in clinical trials [55]. However, before BMD can be used in clinical trials a rigorous path of validation must be followed [54].
NOH CLINICAL TRIAL DESIGN
The length and design of a trial depends on the primary objective. Symptoms of nOH are chronic but the trigger is acute and well known (i.e. standing up). This is in contrast to clinical trials of symptoms that may take several weeks/months to change (e.g., depression). To determine the acute efficacy of a drug, a clinical trial of nOH can last from hours to days; to determine durability of the effect, weeks to months are required. For short acting drugs such as alpha-adrenergic agonists, clinical trials for symptomatic nOH may produce the necessary information after one dose. The endpoints can be symptoms and SBP after standing for 1-min, measured 1-hour or longer post-dose depending on the peak plasma concentration of the drug. Long-acting compounds may require several days to achieve a steady state and their effects on the circulation may be more complex (e.g., fludrocortisone, or the investigational agent ampreloxetine).
Trials of nOH may have an initial dose titration stage to determine the safest dose that patients repond to. This was the case in the droxidopa trials, as explained at length in following sections.
The double-blind, randomized placebo-controlled design remains the gold standard for clinical trials of nOH because it eliminates several known sources of bias. Trials can have either a parallel or crossover design, withdrawal design or n-of-1 design (Figure 3).
Figure 3. Designs of clinical trials for neurogenic orthostatic hypotension.
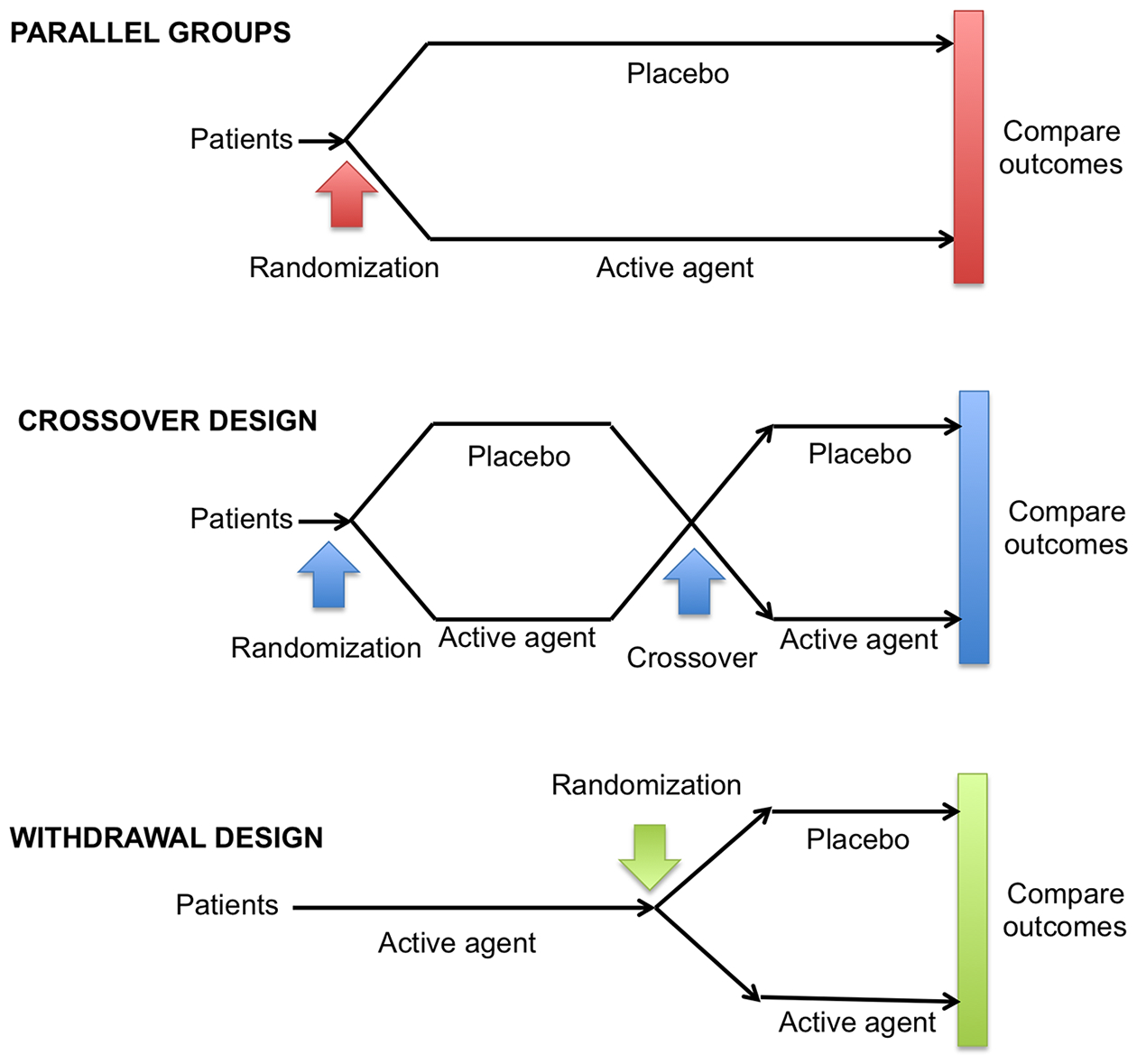
The gold standard design for clinical trials of nOH remains the double-blind randomized parallel-group placebo-controlled trial. This design allows for easier interpretation of the findings and eliminates several known sources of bias. Other designs, however, are used frequently in clinical trials of nOH, including the crossover design and the withdrawal design (see text for details).
Parallel design
In a typical parallel study, each subject receives only one intervention or placebo during the trial. In the simplest parallel design, subjects are randomized to either A (e.g., active agent at a certain dose) or B (e.g., placebo or active agent at a different dose) and remain receiving that compound for the whole duration of the trial. Although this is the most commonly used design in clinical trials, it has several limitations. It typically requires large sample sizes compared to other designs (e.g., cross-over, see below) [56]. Also, in clinical trials of nOH, some subjects may not want to participate in a trial if there is a probability of receiving placebo for a long time. Some subjects may be willing to take the risk if the trial is relatively short (e.g., 1–2 weeks) or they have the possibility of getting the active drug at some point during or after the trial. However, recruitment for long (e.g., 1-month) parallel studies for nOH with a placebo arm may be extremelly challenging.
Cross-over design
In a typical crossover trial, each subject receives more than one intervention or placebo during the different periods of the trial. In the simplest crossover design, a AB:BA design, subjects are randomized to either the AB sequence, where they receive A (e.g., active agent) in the first period, followed by treatment B (e.g., placebo) in the second period, or to the BA sequence, where the treatment order is reversed. Crossover designs are ideal for chronic conditions that remain stable over a period of months, and are unlikely to progress (or improve) significantly over a short period of time. This is frequently, but not always the case with nOH. There is a key consideration in crossover designs, which is the carryover, i.e., the lingering effect of a treatment given in one period into the subsequent period of the clinical trial. To mitigate the carryover effect, a washout period (i.e., a period of time between A and B when the patient receives no study drug) is typically required. This is the simplest and usually favored method to reduce or eliminate carryover effects. If the effect of drug A is reasonably rapid and the outcome is related to the physiological concentration of the drug, then using a washout period is a straightforward strategy. However, other situations might be more complicated if pharmacological effects persist beyond the physical elimination of the drug. Placebo-controlled crossover designs were used in a phase 2 trial of droxidopa [57], phase 3 and phase 4 trials of midodrine [37, 42], and phase 2 trials of atomoxetine [41, 58, 59] and acarbose for postprandial hypotension [60]. This design was recently used in a trial of a servo-controlled inflatable abdominal binder vs. midodrine [61].
Withdrawal design
There are several variations of a withdrawal design. In the one used in clinical trials of nOH, participants receive the active agent (Period 1) for a fixed length of time and responders are then randomized to receive active agent or placebo in a second period (Period 2). Period 1 and Period 2 do not have to be of equal length. Period 1 should be long enough to allow the detection of a change in the selected outcome measure, and Period 2 is chosen to be long enough to eliminate (or washout) any symptomatic effect of the treatment from Period 1 in subjects receiving placebo, so that they return to their baseline. This withdrawal design was used in the NOH302 clinical trial of droxidopa [62]. Period 1 identified responders that were randomized during Period 2. With this design, the exposure of patients to placebo during Period 2 may be shorter than in a randomized placebo-controlled trial. The limitation is that there is no blinding of treatment received during Period 1 and participant retention may become a problem in the placebo group during Period 2, particularly if it is lengthy, as participants will be aware that they may not be receiving the active agent.
N-of-1 design
Whereas conventional studies attempt to determine the overall treatment effect on a group of patients, N-of-1 trials are designed to determine which treatment is more effective for a particular individual [63]. They are double-blind randomized trials in which a single patient (i.e., n-of-1) goes through a series of pairs of treatments, one active and one placebo (or alternative treatment) per pair, with the order determined by random allocation [63, 64]. Classic trials generally have a single crossover, but N-of-1 trials frequently have multiple crossovers.
Only drugs with rapid onset and termination of effect, without the need for lengthy treatment, and washout periods between interventions can be studied with N-of-1 trials.
N-of-1 trial design can provide the strongest evidence for individual treatment decisions and have been listed as level 1 evidence in the Oxford University Centre for Evidence-Based Medicine. Moreover, data from multiple N-of-1 trials can be aggregated and with correct statistical tools applied, analyzed to generate population-level data about drug response, while capturing more information about intra- and interindividual heterogeneity than most RCT designs [65, 66].
Enrichment strategies
Enrichment strategies for drug development studies identify a study population most likely to demonstrate the candidate drug’s effects and provide adequate evidence. If the drug is ineffective, such strategies will not falsely suggest effectiveness. There are three enrichment strategies in clinical trials of nOH: 1) Practical enrichment, i.e., decreasing the heterogeneity of the population to reduce variability; 2) Prognostic enrichment, i.e., identifying high risk subjects more likely to experience the outcomes that are being measured; and 3) Predictive enrichment, i.e., selecting subjects that respond to the candidate treatment.
Practical enrichment strategies include prospectively identifying likely compliant subjects who will not drop out of the study, excluding subjects who are too unstable with respect to the signs and symptoms of the disease (e.g., subjects with a mild, occasional, unsustained fall in BP), identifying subjects unlikely to survive long enough to be evaluated (e.g., subjects with MSA and nOH with a disease duration longer than 4 years), and excluding subjects using medications or devices with the same effect as the candidate drug (e.g., using high pressure compression stockings in clinical trials of pharmacological agents against nOH).
Prognostic enrichment strategies include identifying and enrolling patients at high risk of developing the study endpoint (e.g., subjects with an OHQ score of at least 3 points, and who are, therefore, susceptible to have a measurable and clinically significant improvement in the OHQ score after receiving the study drug).
Predictive enrichment strategies include identifying and enrolling patients likely to respond to the study drug. This strategy was used all phase 3 clinical trials with droxidopa [48, 62, 67–70]. The trials had an initial dose optimization period with a forced upward titration from 100 to 600 mg three times daily. Dose titration lasted a maximum of 14 days, with patients receiving escalating dosages in 100 mg three times daily-increments until a) becoming asymptomatic (with a score of 0 in the item 1 of the OHQ [36]), b) developing a sustained supine hypertension >180/110 mmHg, c) reaching the maximum dose of 600 mg three/times day; or d) complaining of intolerable side effects.
Responders were defined as patients who had at least a 1-point improvement in item 1 of the OHQ (dizziness/lightheadedness) in conjunction with a 10 mmHg or more increase in standing systolic BP. Only responders were allowed to continue in the double-blind placebo-controlled phase of the trials.
Telemedicine visits
When clinical trials of nOH use patient-reported outcomes as primary endpoints (e.g., OHQ), it is conceivable that the patient might not need to come to the clinical center for a study visit. Similarly, if BP is used as a secondary outcome measure, the patient can easily measure his/her BP in the supine and standing positions at home, with devices that allow the patient to remain blind to the BP results. These circumstances make telemedicine study visits feasible in clinical trials of nOH. Telemedicine has been proven feasible in clinical trials of other disorders and may overcome geographic barriers to clinical trial participation.
Video conferencing tools in clinical trials must protect data privacy with all audio/video communication securely encrypted and transmitted, such that even the video conferencing company does not have access to identifiable health information. There is the possibility of recording and storing the videoconferences for data quality assurance and auditing. Also, BP readings should be reliable and reproducible. This can be achieved by giving all patients the same BP machine, and by ensuring that all the BP readings required throughout the trial are performed at the same time of the day (e.g., 30 minutes before lunch to avoid postprandial BP effects).
RECRUITMENT STRATEGIES FOR PATIENTS WITH NOH
Advancing drug development for rare diseases such as nOH requires cooperation and collaboration among different stakeholders. Timely and expeditious identification and recruitment of research participants is crucial for success. Specialized treatment centers provide the traditional sites for recruiting patients. Increasingly, patient themselves and patient advocacy groups are becoming well-informed sources for patient recruitment. The development of patient registries with contact information, diagnosis, and demographic information, facilitates identification of potential participants and is a key element to support a clinical research program. Accelerating enrolment through collaboration with patient groups may substantially shorten drug development time. Web-based technologies, including social media, offer a potentially powerful approach to recruiting as well [71].
Many clinical trials are performed internationally to enroll an adequate number of participants. Therefore, special considerations should be made to address the complexities of conducting clinical trials across languages and cultures, and the need for validation of patient-reported outcome measures in different languages.
CLINICAL TRIALS FOR NOH
This section reviews clinical trials for nOH of non-pharmacologic and pharmacologic therapies, with an emphasis on double-blind, placebo-controlled trials. We describe each trial population, outcome measures, main results, and potential biases.
Non-pharmacologic measures
Non-pharmacologic measures are the first step in the management of patients with nOH. That water drinking significantly and rapidly increases BP in patients with nOH was first studied in an open label fashion in 28 patients with MSA and 19 with PAF, as well as in 19 healthy controls. After 30 minutes of drinking 500 ml of tap water, seated SBP/DBP increased 33±5/16±3 mmHg in patients with MSA and 37±7/14±3 mmHg in patients with PAF (P<0.0001 vs. baseline in both disorders). The increase in SBP was sustained for > 60 minutes [72, 73]. Symptoms of nOH were not assessed.
The first attempt to systematically study non-pharmacological measures in patients with PD and nOH was a 3-week open-label trial of 17 patients [74]. The trial design and endpoints were not well defined. The primary outcome measure appeared to be the ΔSBP and ΔDBP after active standing (unclear if after 1 or 3 minutes). The orthostatic domain of the COMPASS scale [31] was used as patient-reported outcome. Patients were then instructed to follow 12 non-pharmacological measures (including but not limited to drinking 1250 ml of water daily, taking 10–20 g of salt daily, elevating the head of the bed 10–15 cm, using thigh-high 30 mmHg compression stockings, having frequent small meals, and avoiding alcohol) that were explained to the patient, printed and given to the patient. Three weeks later, the ΔBP was measured again and the COMPASS was re-administered. There were no significant changes in ΔBP or in the COMPASS score after the 3week open label treatment period. The lack of significant differences is not surprising for a number of reasons. Patients with OH often perceive non-pharmacological measures as inconvenient. Liberalizing water intake increases urine output and, therefore, urinary frequency. Compression garments must be tight to be effective and, consequently, are challenging to put on. Sleeping with the head of the bed raised 30–45 degrees is only effectively accomplished with an electric bed or mattress, rather than just extra pillows. Therefore, even though the reported compliance of the non-pharmacological measures was ~70%, it is likely that patients applied them half-heartedly if no one was monitoring them. Overcoming this limitation can be challenging, but remote biometric monitoring devices could be potentially helpful. Second, as discussed before, the COMPASS is not specifically validated to measure symptoms of OH, so its use is challenging. It must be said, however, that the OHQ had not been developed yet at the time this trial was conducted.
The second trial studying non-pharmacological measures enrolled 15 patients with PD and OH who were randomized to either use a compressive abdominal binder (20 mmHg of abdominal pressure) or a sham abdominal binder (3 mmHg of abdominal pressure) during a head-up tilt procedure [75]. The primary endpoint was the mean BP after 3 minutes of head-up tilt. Use of the compression abdominal binder elicited an increase of 7.7 mmHg compared to a decrease of −2.7 mmHg of the sham abdominal binder. This was followed by a 4 week open label phase where all patients used the compression abdominal binder, which resulted in a reduction of orthostatic symptoms denoted by a reduction of the OHQ of 2.2 points compared to baseline (P=0.003)[75].
The third and most recent trial used a much more sophisticated, servo-assisted splanchnic compression device (40 mmHg of abdominal pressure), which actives only upon standing up [61]. Twenty-three patients with nOH (3 of which had PD) were randomized in a crossover manner to receive either a single oral dose of placebo, midodrine 2.5 to 10 mg, placebo combined with abdominal binder (40 mmHg), or midodrine 2.5 to 10 mg combined with abdominal binder (40 mmHg). The primary outcome was the change from baseline in orthostatic tolerance, defined as the AUC of upright SBP calculated by the trapezoidal rule (ΔAUCSBP: upright SBP multiplied by standing time). This is a composite score that integrates both the standing time and the upright SBP [59]. On standing, inflation of the abdominal binder and midodrine produced a similar significant ΔAUCSBP (i.e., improved orthostatic tolerance) compared to placebo (195±35 and 197±41 vs. 19±38 mmHg x min on the placebo day; P=0.019 and P=0.010, respectively). The same number of patients in the binder and midodrine groups (n=14) were able to stand for the full 10 minutes of the post-treatment orthostatic test, whereas only 10 did in the placebo group. In clinical practice, the effectiveness of abdominal binders in patients with nOH is limited by their difficulty of use and low compliance.
Fludrocortisone
Fludrocortisone (9α-fluorocortisol) is a synthetic mineralocorticoid that increases renal sodium and water re-absorption, thus expanding intravascular volume and increasing BP in all positions. Despite limited studies, and not being FDA-approved for this indication, fludrocortisone is frequently used for the treatment of nOH.
There are no placebo-controlled clinical studies evaluating the efficacy of fludrocortisone in patients with nOH. Two crossover trials compared fludrocortisone to another active agent in patients with PD and nOH. In the first one, 13 patients with PD and nOH were randomized to receive either fludrocortisone 0.1 mg/day or domperidone 30 mg/day for 3 weeks, and, after a 1 week washout period, were crossed-over to receive the other medication for another 3 weeks [74]. The ΔSBP after 3 minutes, ΔDBP after 3 minutes, and the maximum reduction in BP within 5 minutes as well as the COMPASS and CGI were measured as endpoints. There were no statistical differences in any of the BP measurements after taking the medications, even though there were significant improvements in the COMPASS and CGI scores. The second, more recent study was an multicenter, double blind, randomized trial [76]. Nine patients were randomized to receive either fludrocortisone 0.2 mg/day or pyridostigmine 60 mg/day for 14 days, and then crossed over to the other agent, with a 21-day washout period in between. For unclear reasons, the primary outcome measure was the ΔDBP within 3 minutes of active standing, and not the ΔSBP. Symptomatic burden was measured with the OHSA. When on fludrocortisone, patients had a statistically significant less severe pronounced ΔDBP within 3 minutes of active standing compared to baseline (post-hoc comparisons P=0.0016), but not when compared to pyridostigmine. There were no differences in ΔSBP (P=0.26) or in the OHSA score either [76].
Midodrine
Midodrine is an oral α1-adrenoceptor agonist that induces vasoconstriction and increases BP [37, 42, 77, 78]. Midodrine, along with droxidopa, are the only two FDA-approved drugs for the treatment of symptomatic nOH. Midodrine raises BP in the standing, sitting, and supine positions, and its pressor effect is noticeable ~30–45 minutes after consumption, reaching a maximum after ~1 hour, and persists for a total of 2–3 hours. Treatment should begin with a 2.5 or 5 mg dose, which can then be increased up to 10 mg taken up to 3 times a day. Supine hypertension is common; hence patients should not take midodrine within 3–4 hours before bedtime. Other side effects owing to activation of α1-adrenergic receptors are piloerection (“goosebumps”), itching of the scalp, and urinary retention. Midodrine has no effect on heart rate, as it does not activate β-adrenoreceptors and, given its poor diffusion across the blood-brain barrier, it has no CNS adverse effects [79].
Early studies showed that midodrine (25–40 mg/day) significantly increased the average mean BP in the standing position and improved orthostatic tolerance after 7 days of treatment in patients with nOH compared to baseline (i.e., no treatment), but only in a subgroup of subjects [66, 80]. The first large randomized placebo-controlled trial of midodrine for nOH enrolled 97 subjects, 22 of which had PD, 20 had PAF and 18 had MSA [77]. The design of the trial included a 1-week single-blind placebo period. After this period, patients were randomly assigned to one of four treatment groups: placebo, midodrine 2.5 mg, midodrine 5 mg, or midodrine 10 mg, administered orally three times/day for 3 or 4 weeks. The primary outcomes were standing SBP (unclear how long after standing) and symptoms of OH measured dichotomously (improvement yes/no) compared to the patient’s baseline, assessed 1 hour post-dose. Midodrine significantly (p<0.00l) increased standing SBP by 22 mmHg, compared to only 3 mmHg in the placebo group. Symptoms of dizziness/lightheadedness, syncope, depression and standing time also improved significantly in patients taking midodrine compared to placebo [77]. The largest phase 3 trial of midodrine for nOH enrolled 171 subjects in a randomized double-blind placebo-controlled multicenter study [78]. The trial began with a 1 week single-blind placebo period, followed by a 3 week double-blind (midodrine 10 mg three times/day or placebo) period, and a 2 week single-blind placebo period. The primary endpoints were standing BP (not defined after how long standing) and symptoms of lightheadedness using a visual analog scale (0–10). It was unclear how long after the medication dose the outcomes were measured. The secondary endpoint was a symptom composite score of quality of life. Overall, midodrine induced significant increases in standing BP in all study visits, and a significant reduction in symptoms of lightheadedness, but only in the final visits. The most frequently reported adverse events were piloerection, pruritus, and urinary retention [78].
Another double-blind, randomized, dose-response, placebo-controlled, crossover trial of midodrine enrolled 25 patients with nOH. In this trial, subjects were studied on 6 days. On day 1 and day 6 they received no medication. They were then randomized to receive, on successive days, placebo, midodrine 2.5 mg, midodrine 5 mg, or midodrine 10 mg. The end point was the 1 hour post-dose BP after 1 minute of standing. Midodrine resulted in significantly increased standing SBP, peaking at 1 hour post-dose. The global improvement of symptoms score also improved significantly with midodrine [37].
The results of these trials lead to accelerated approval of midodrine by the FDA for the treatment of OH in 1996. However, the FDA considered that the increase in standing BP is only a surrogate marker of effectiveness rather than proof, so they required post-marketing studies to confirm that midodrine provides a clinical benefit for patients with nOH. To that end, a phase 4 tilt-table, double-blind, placebo-controlled, randomized, crossover, multicenter study enrolled 19 patients [42]. Following an open-label 4-week period when they received their usual midodrine dose (2.5 mg/day – 15 mg/day), midodrine was stopped for 1 day. The next day, patients were randomized to receive either midodrine or placebo and undergo a tilt-table test. The following day, patients were crossed-over to the other study drug. The primary endpoint was time to syncope or near-syncope using a 45 minute head-up position with a tilt-table at 1 hour post-dose of midodrine. The time to symptoms in patients receiving midodrine was significantly longer than when receiving placebo (27 minutes vs. 18 minutes, p=0.0013), thus confirming the symptomatic benefit of midodrine in patients with nOH (Table 1).
Table 1.
Placebo-controlled clinical studies with midodrine in patients with neurogenic OH
| Study | Subjects (N) | Arms | Design | Primary endpoints | Results |
|---|---|---|---|---|---|
| Kaufmann et al. 1988 [66] | 7 | Fludrocortisone 0.1 mg + placebo Fludrocortisone 0.1 mg + midodrine |
N-of-1-based trial. 1-week with no therapy as baseline followed by 1-week of midodrine open label at increasing dosages followed by double-blind cross-over of 1-week placebo+fludrocortisone 0.1 mg and 1-week of midodrine+fludrocortisone 0.1 mg (or viceversa). | Standing mean BP 2-h post-dose | Midodrine produced a statistically significant increase in upright blood pressure in 3 patients but not in 4. |
| Jankovic et al. 1993 [77] | 97 | Placebo three times/day Midodrine 2.5 mg three times/day Midodrine 5 mg three times/day Midodrine 10 mg three times/day |
3-week four-arm double-blind randomized placebo-controlled study | Standing SBP (unclear after how long) 1-hour post-dose Symptom improvement (yes/no) 1-hour post-dose |
Subjects on midodrine had significantly higher standing SBP and lower burden of symptoms (p<0.001) than those on placebo. |
| Low et al. 1997 [78] | 193 | Placebo three/times a day Midodrine 10 mg three times/day |
3-week double-blind randomized placebo-controlled study | Standing SBP (unclear after how long) 1-hour post-dose Lightheadedness symptoms (using a 0–10 scale) 1-hour post-dose |
Subjects on midodrine had significantly higher standing SBP and lower burden of symptoms (p<0.001) than those on placebo. |
| Wright et al. 1998 [37] | 25 | Placebo Midodrine 2.5 mg Midodrine 5 mg Midodrine 10 mg |
Double-blind, randomized, dose-response, placebo-controlled, crossover study | Standing SBP after 1-min standing within 3-hours post-dose. | Midodrine increased SBP in a dose-dependent manner, peaking at 1-h post dose |
| Smith et al. 2016 [42] | 19 | Placebo Midodrine (2.5 mg/day- 15 mg/day) |
Double-blind, placebo-controlled, randomized, crossover, tilt-table study | Time to symptoms upon head-up tilt 1-hour post-dose | The time to symptoms in patients receiving midodrine was significantly longer than when receiving placebo (27-mins vs. 18-min p=0.0013). |
Droxidopa
On February 18th 2014, the U.S. FDA approved droxidopa (Northera®), an orally active synthetic precursor of norepinephrine, for the treatment of symptomatic neurogenic orthostatic hypotension (nOH). Droxidopa (L-threo-3,4-dihydroxyphenyl-serine, L-DOPS) is an orally active synthetic amino acid that is converted to norepinephrine in the body [81]. Droxidopa is decarboxylated to norepinephrine by the enzyme aromatic amino-acid decarboxylase (AAAD), the same enzyme the converts L-dopa to dopamine. Extensive clinical experience shows that droxidopa is safe and well tolerated [82–90]. Peak plasma concentrations of droxidopa are reached ~3 hours after oral administration. The dosage used in clinical trials was 100–600 mg three times/day, although clinical experience indicates that the dosage should be tailored to each patient’s needs considering the patient’s daily activities, with higher dosages before he/she is going to be physically active [81, 83, 88].
Phase 2 studies
Table 2 shows a summary of Phase 2 studies with droxidopa for nOH. Some initial studies with droxidopa in patients with nOH were conducted with a racemic mixture of the D- and L-stereoisomers, and showed conflicting results [91]. A subsequent, better-powered double-blind, randomized, placebo-controlled study showed that a single dose of droxidopa (up to 2,000 mg) effectively increased BP and improved symptoms in 19 patients with nOH due to MSA and PAF [57].
Table 2.
Phase 2 clinical studies with L-DOPS and DL-DOPS in patients with neurogenic OH
| Study | Subjects (N) | Active agent | Design | Results |
|---|---|---|---|---|
| Hoeldtke et al, 1984 [97] | 6 (2 PAF and 4 autonomic diabetic neuropathy) | DL-DOPS | Open label crossover study using a single dose of DL-DOPS (600 or 800 mg) and single dose of placebo in separate days. | No change in supine or upright BP after DL-DOPS. |
| Kaufmann et al, 1991 [98] | 6 (2 PAF; 4 MSA) | L-DOPS | Open label dose titration study followed by 2 days of open label administration (700–1000 mg/day) | L-DOPS increased supine BP in MSA and PAF, and standing BP only in MSA. |
| Freeman et al, 1999 [99] | 10 (6 MSA; 4 PAF) | DL-DOPS | Randomized, double-blind, placebo-controlled, crossover study using a single dose of 1000 mg of DL-DOPS and single dose of placebo | DL-DOPS significantly increased supine and upright BP. |
| Mathias et al, 2001 [100] | 32 (26 MSA; 6 PAF) | L-DOPS | Open label incremental study (4 weeks) followed by a 6-week maintenance study (100–300 mg twice/day) | L-DOPS significantly reduced the fall in systolic BP upon standing and the symptoms of OH. |
| Kaufmann et al, 2003 [57] | 19 (11 MSA; 8 PAF) | L-DOPS | Single blind dose titration study followed by a 3-day double blind, placebo-controlled, crossover trial. | L-DOPS significantly increased supine and standing BP for several hours and improved symptoms of OH. After L-DOPS, BP increases were associated with increases in plasma NE levels. |
Phase 3 studies
Because of the variable pressor response in patients with nOH, phase 3 trials had a dose optimization period that lasted a maximum of 14 days with a forced upward titration from 100 to 600 mg three times daily. As explained aboved, dose titration was stopped when patients became asymptomatic [36]), b) developed sustained supine hypertension >180/110 mmHg or intolerable side effects. Responders were defined as patients that had at least a 1-point improvement in item 1 of the OHQ (dizziness/lightheadedness) in conjunction with a 10 mmHg or more increase in standing systolic BP.
Study NOH301 was a double-blind, randomized, placebo-controlled, parallel-group study conducted at 94 US, Canadian and European sites. The trial enrolled 162 patients with nOH due to PD, MSA, PAF or non-diabetic autonomic neuropathy [67]. Following an open-label dose titration phase (starting at 100mg and escalating to 600 mg three times/day), “responders” (162 patients, 62% of enrolled) proceeded to a washout period and were then randomized to receive placebo or droxidopa for 1 week (double-blind). Differences between groups were compared on day 7. Compared with placebo, patients randomized to droxidopa had more improvement in overall OHQ scores (p<0.003) and standing systolic BP that was 7 mmHg higher (p<0.001). Differences between groups were modest, partly because the placebo group failed to worsen back to their original state, suggesting a possible carry over effect of droxidopa, requiring a longer washout period to disappear.
Study NOH302 was a double-blind, placebo-controlled, randomized withdrawal trial including 181 patients with nOH due to PD, MSA, PAF or non-diabetic autonomic neuropathy [62]. Study 302 began with an open label titration and a 1 week treatment phase. After completing the open-label treatment phase, responders were randomized to withdraw to placebo or continue taking droxidopa. Differences between groups were assessed 2 weeks after randomization. The mean dose of droxidopa was 386±178 mg (three times/day), slightly lower than in study 301 (p>0.33). After withdrawal, there were no significant differences in symptoms of dizziness/lightheadedness (OHQ item 1), the primary outcome measure, between droxidopa and placebo patients. This was because placebo patients experienced continuing relief of the dizziness/lightheadedness score and standing SBP at the end of the study compared with baseline. This again raised the possibility of a carryover effect of droxidopa. Substantial carryover effects have been observed for levodopa, which, like droxidopa, undergoes decarboxylation to become a neurotransmitter. Alternatively, patients may have improved simply by their participation in the study, e.g., by better adherence to non-pharmacological treatment recommendations [62]. Post-hoc analysis of the overall composite score for OH symptoms and activities (OHQ composite), however, did show a significant improvement with droxidopa (p=0.026). Standing systolic BP was not different between the two treatment groups. Patients’ self-ratings of clinical global impression (CGI) of symptom severity showed a significant difference favoring droxidopa (p=0.008).
Study NOH306 was originally designed to evaluate the clinical efficacy of droxidopa over an 8-week double-blind period [70]. In a preplanned interim efficacy analysis, the initial 51 subjects (study NOH306A) [69] showed no significant difference across groups in the change in OHQ composite score, the trial primary endpoint. Exploratory analyses suggested efficacy for the dizziness/lightheadedness score, and led to a change in the trial’s primary efficacy measure while data for subsequent subjects remained blinded. The subsequent 171 enrolled patients formed study NOH306B, multi-center, double-blind, randomized, placebo-controlled, parallel-group study restricted to patients with symptomatic nOH associated with PD, conducted exclusively in the US [70]. Subjects were randomized initially and underwent up to 2 weeks of double-blind titration with either droxidopa or placebo. Patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared with placebo after 1 week of treatment (p = 0.018) [70]. At that time, patients taking droxidopa also had significantly higher systolic BPs in the standing position compared with placebo (p=0.032).
The pooled results of NOH306A and NOH306b (composite NOH306) showed that patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared with placebo after 1 week of treatment (p < 0.01) [70] (Table 3).
Table 3.
Phase 3 clinical studies with droxidopa (L-DOPS) in patients with neurogenic OH
| Study | Subjects (N) | Design | Enrichment design | L-DOPS dosage | Length | Titration | Primary efficacy endpoints | Results |
|---|---|---|---|---|---|---|---|---|
| NOH301 [67] | 162 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, multinational, double blind, randomized controlled trial parallel group induction design | Yes | 100–600 mg three times daily | Initial open label titration, 1-week washout, 7-day randomized placebo controlled phase | Open label – before randomization | OHQ composite score at 1 week | Compared with placebo, patients on droxidopa had a significant improvement in overall OHQ scores (p<0.003); and standing BP that was 7 mmHg higher (p<0.001) |
| NOH302 [62] | 101 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, double blind, randomized controlled trial with withdrawal design | Yes | 100–600 mg three times daily | Initial open label titration, 7-day open label treatment, 2-week of double blind placebo controlled phase | Open label – before randomization | OHQ Item 1 (dizziness / lightheadedness) at 2 weeks | No differences in the OHQ item 1. Significant improvement of the overall OHQ composite score on droxidopa than placebo (p=0.013). Standing systolic BP was not different between the two groups. Patient self-reported symptom severity showed a significant improvement with droxidopa (p=0.008). |
| NOH306A [69] NOH306B [101] |
51 PD (306A) 171 PD (306 B) |
Multicenter, double blind, randomized controlled trial parallel group induction design | No | 100–600 mg three times daily | 2-week double-blind titration; 8 weeks double blind maintenance treatment | Double blind – after randomization | OHQ composite score at 8 weeks (306A) OHQ Item 1 at 1 week (306B) |
For the initial 51 subjects (Study 306A), the primary efficacy measure (OHQ composite score) did not show significant change versus placebo at week 8. For the subsequent 171 subjects (Study 306B) patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared to placebo after 1 week (p = 0.018); and higher systolic BP standing (p=0.032). |
| NOH303 (Unpublished) | 75 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, open-label extension to 301 and 302 studies | No | 100–600 mg three times daily | 3-month open label treatment; 2 week placebo-controlled withdrawal phase; after the controlled phase of the trial some patients continued on open-label droxidopa for up to 2 years | No | OHQ composite score | No significant differences between treatment arms. |
| NOH304 (Unpublished) | 255 (PD, PAF, NDAN, DBHD) | Long term, open-label safety extension to 301, 302 and 306 studies | No | 100–600 mg three times daily | Up to 1–2 years | No | Safety measures | N/A |
| NOH305 [48] | 18 (PD, PAF, NDAN, DBHD) | Multicenter, open-label extension to 301 and 304 studies | No | 100–600 mg three times daily | An off drug 24-hour ambulatory BP assessment was compared to a 4–6 week on-drug 24-hour ambulatory BP assessment | No | Mean 24-hour systolic and diastolic BP | Overall mean 24-hour SBP and DBP were higher compared to off drug (137/81 mm Hg vs. 129/76 mmHg; p=0.017/0.002). Mean daytime SBP was significantly higher with droxidopa (8.4 ± 3.1 mmHg; p=0.014). Nocturnal BP was not significantly higher on droxidopa versus off treatment (p= 0.122). |
Long-term clinical studies
Study NOH303 was a long-term continuation of studies NIH301 and NIH302 and featured a withdrawal design. A total of 75 patients with nOH received droxidopa for a further 3 months and entered into a 2 week randomized withdrawal phase. This study did not show any statistically significant difference between the treatment arms. Potential carryover of droxidopa may have influenced the results, as patients on placebo did not return to their baseline symptom score or blood pressure standing within the two-week withdrawal phase. Moreover, this was an exploratory study not powered to reach significance.
In Study NOH306 overall, differences in change in OHQ item 1 scores from baseline to maintenance weeks 2, 4, and 8 showed numerical trends favoring droxidopa that approached statistical significance (p=0.077 at week 8) [70].
Eventually, these double-blind clinical trials led to the FDA approval of droxidopa in the U.S. The trials showed that patients with symptomatic nOH receiving droxidopa had both symptomatic improvement and higher blood pressure when standing than those on placebo. Integrated analysis and meta-analysis have been published [92, 93]. In summary, 226 patients received droxidopa and 236 received placebo. Symptoms of nOH were measured with the OHQ [36]. Those who received droxidopa improved in virtually all OHQ symptom scores compared to those receiving placebo. Droxidopa also increased upright systolic blood pressure significantly (+11.5 ± 20.5 mmHg vs. placebo +4.8 ± 21.0 mmHg; p < 0.001).
Pyridostigmine
Pyridostigmine, a cholinesterase inhibitor, enhances cholinergic neurotransmission in sympathetic and parasympathetic autonomic ganglia. The evidence for using pyridostigmine in patients with nOH is limited. One double-blind, randomized, cross-over study enrolled 58 patients with nOH [94] who received 4 different treatments (placebo, pyridostigmine 60 mg, pyridostigmine 60 mg + midodrine 2.5 mg, pyridostigmine 60 mg + midodrine 5 mg). For unknown reasons, they did not include a group of midodrine alone. The primary endpoint was the ΔDBP at 1 hour after drug administration. Patients receiving pyridostigmine alone had a ΔDBP of −27.6 mmHg, compared to −34 mmHg receiving placebo (p=0.04). The combination of pyridostigmine and midodrine 5 mg was more effective than pyridostigmine alone (ΔDBP of 27.2 mmHg vs. to 34 mmHg with placebo, p=0.002).
A recent randomized, open-label clinical trial enrolled 87 patients with nOH who were randomized to receive 1 of 3 treatments: midodrine 2.5 mg only, pyridostigmine 30 mg only, or midodrine 2.5 mg + pyridostigmine 30 mg [95]. The primary endpoint was the ΔBP within 3 minutes of standing at 3 months after treatment. Secondary endpoints were improvement of the ΔBP within 3 minutes of standing at 1 month, and the OHQ. The authors concluded that the ΔSBP and ΔDBP were less severe after treatment regardless of the treatment. This trial was not blinded and although the authors report that only patients with nOH were included, no tests confirmed this and most of the patients had diabetes or “idiopathic OH”, so the diagnoses were quite heterogeneous.
Norepinephrine transporter (NET) blockade
Atomoxetine and similar medications increase norepinephrine concentration in the sympathetic neurovascular junction by selectively blocking the norepinephrine transporter (NET). Atomoxetine which is currently FDA-approved for the treatment of attention deficit and hyperactivity disorder (ADHD) has been studied in three randomized, placebo-controlled trials of patients with nOH. In the first one, 17 patients with nOH were administered a single dose of either placebo, yohimbine 5.4 mg or atomoxetine 18 mg, and the combination yohimbine and atomoxetine in a single-blind, crossover study [59]. The primary outcome was seated BP and standing BP 1 hour post-drug. Neither yohimbine nor atomoxetine significantly increased seated systolic blood pressure or orthostatic tolerance compared with placebo. The combination, however, significantly increased seated SBP (p<0.001) in a synergistic manner. The second trial studied 21 patients with nOH who were administered a single dose of atomoxetine 18 mg or placebo in a randomized, single-blind, cross-over fashion [58]. The primary outcome was SBP every 5 minutes for 1 hour post-drug administration. Atomoxetine increased significantly seated and standing SBP in patients with MSA (i.e., central autonomic failure) compared to placebo (p=0.016). However, in patients with PD or PAF (i.e., peripheral autonomic failure), there were not differences in seated or standing BP compared to placebo (p=0.546), suggesting that atomoxetine might be only effective to raise BP in patients with intact post-ganglionic sympathetic nerves (i.e., MSA).
The third, and most recent trial, was a randomized, single-blind, crossover trial with 65 patients with nOH evaluating the effectiveness of a single dose of atomoxetine 18 mg vs. midodrine 5–10 mg [41]. The primary endpoint was the standing SBP after 1 minute, 1 hour post-drug. Secondary endpoints included post-treatment seated SBP and DBP, standing DBP and HR, and OHQ and OHQ item 1 scores. Atomoxetine significantly increased standing SBP by 20 mmHg and DBP by 11 mmHg compared to placebo (p<0.001 for both). Likewise, midodrine increased standing SBP by 12 mmHg and DBP by 7 mmHg compared to placebo (p<0.001 for both). Atomoxetine, however, improved standing SBP to a greater extent than midodrine did (mean difference=7.5 mmHg; 95% CI, 0.6 to 14.5; p=0.03).
An FDA-sponsored phase 2, multi-center double-blind placebo-controlled crossover clinical trial to study the efficacy of atomoxetine for nOH (n=40) is currently underway (ClinicalTrials.gov: NCT02796209). Thus, atomoxetine appears a very promising alternative for the treatment of nOH. However, because atomoxetine has a short biological effect (~3–4 hours), longer active NET blockers are required. In this regard, an investigational long-acting NET blocker and serotonin reuptake inhibitor (ampreloxetine, TD-9855) recently showed promising preliminary results in a small phase-2, single-blind placebo-controlled trial with an open label extension phase. A large multicenter placebo-controlled phase 3 trial is ongoing (ClinicalTrials.gov: NCT03750552).
CONCLUSIONS
Despite significant challenges, there have been important accomplishments in the field of nOH in the last decade, including the development and validation of a disease-specific symptom scale (i.e., OHQ), the approval of droxidopa by the FDA, and several ongoing clinical trials of new antihypotensive agents. Careful consideration of study design, endpoint selection, and patient recruitment and retention strategies are key when planning a clinical trial for nOH. Future priorities are to improve our understanding of nOH (e.g., by developing an animal model of the disease), to define the natural history of nOH in patients with synucleinopathies, to improve collaboration with patient-advocacy groups, and to determine what causes individuals to respond differently to the same medication. In this regard, it is well described that natural human genetic variations are linked to differences to anti-hypertensive drugs, such as β-blockers, angiotensin receptor blockers (ARBs), and angiotensin converting enzyme (ACE) inhibitors (i.e., pharmacogenomics)[96]. Thus, it is likely that potential genetic variations in drug targets of nOH may alter therapeutic efficacy and safety of drugs. Identification of these genetic variations would result in improved enrichment strategies in clinical trials and, ultimately, enhanced personalized medicine.
ACKNOWLEDGEMENTS
Funding support by the Familial Dysautonomia Foundation and the NINDS (U54NS065736).
REFERENCES
- 1.Kaufmann H, Norcliffe-Kaufmann L, Palma JA et al. Natural history of pure autonomic failure: A United States prospective cohort. Ann Neurol 2017; 81: 287–297. doi: 10.1002/ana.24877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg D, Postuma RB, Adler CH et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015; 30: 1600–1611. doi: 10.1002/mds.26431 [DOI] [PubMed] [Google Scholar]
- 3.Freeman R, Wieling W, Axelrod FB et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res 2011; 21: 69–72. doi: 10.1007/s10286-011-0119-5 [DOI] [PubMed] [Google Scholar]
- 4.Ooi WL, Hossain M, Lipsitz LA. The association between orthostatic hypotension and recurrent falls in nursing home residents. The American journal of medicine 2000; 108: 106–111 [DOI] [PubMed] [Google Scholar]
- 5.Jonsson PV, Lipsitz LA, Kelley M et al. Hypotensive responses to common daily activities in institutionalized elderly. A potential risk for recurrent falls. Archives of internal medicine 1990; 150: 1518–1524 [PubMed] [Google Scholar]
- 6.Freeman R Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med 2008; 358: 615–624. doi: 10.1056/NEJMcp074189 [DOI] [PubMed] [Google Scholar]
- 7.Masaki KH, Schatz IJ, Burchfiel CM et al. Orthostatic hypotension predicts mortality in elderly men: the Honolulu Heart Program. Circulation 1998; 98: 2290–2295 [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003; 23: 351–363. doi: 10.1055/s-2004-817719 [DOI] [PubMed] [Google Scholar]
- 9.Kaufmann H, Norcliffe-Kaufmann L, Palma JA et al. The Natural History of Pure Autonomic Failure: a U.S. Prospective Cohort. Ann Neurol 2017. doi: 10.1002/ana.24877. doi: 10.1002/ana.24877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstein DS, Holmes C, Sharabi Y et al. Survival in synucleinopathies: A prospective cohort study. Neurology 2015; 85: 1554–1561. doi: 10.1212/WNL.0000000000002086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merola A, Sawyer RP, Artusi CA et al. Orthostatic hypotension in Parkinson disease: Impact on health care utilization. Parkinsonism Relat Disord 2017. doi: 10.1016/j.parkreldis.2017.11.344. doi: 10.1016/j.parkreldis.2017.11.344 [DOI] [PubMed] [Google Scholar]
- 12.Kaufmann H, Goldstein DS. Autonomic dysfunction in Parkinson disease. Handb Clin Neurol 2013; 117: 259–278. doi: 10.1016/B978-0-444-53491-0.00021-3 [DOI] [PubMed] [Google Scholar]
- 13.Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol Dis 2012; 46: 572–580. doi: 10.1016/j.nbd.2011.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palma JA, Kaufmann H. Epidemiology, Diagnosis, and Management of Neurogenic Orthostatic Hypotension. Mov Disord Clin Pract 2017; 4: 298–308. doi: 10.1002/mdc3.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein DS, Holmes CS, Dendi R et al. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology 2002; 58: 1247–1255. doi: 10.1212/wnl.58.8.1247 [DOI] [PubMed] [Google Scholar]
- 16.Nagayama H, Yamazaki M, Ueda M et al. Low myocardial MIBG uptake in multiple system atrophy with incidental Lewy body pathology: an autopsy case report. Mov Disord 2008; 23: 1055–1057. doi: 10.1002/mds.22031 [DOI] [PubMed] [Google Scholar]
- 17.Nagayama H, Ueda M, Yamazaki M et al. Abnormal cardiac [(123)I]-meta-iodobenzylguanidine uptake in multiple system atrophy. Mov Disord 2010; 25: 1744–1747. doi: 10.1002/mds.23338 [DOI] [PubMed] [Google Scholar]
- 18.Goldstein DS. Dysautonomia in Parkinson disease. Compr Physiol 2014; 4: 805–826. doi: 10.1002/cphy.c130026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orimo S, Oka T, Miura H et al. Sympathetic cardiac denervation in Parkinson’s disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002; 73: 776–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braune S The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res 2001; 11: 351–355 [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann H, Oribe E, Miller M et al. Hypotension-induced vasopressin release distinguishes between pure autonomic failure and multiple system atrophy with autonomic failure. Neurology 1992; 42: 590–593 [DOI] [PubMed] [Google Scholar]
- 22.Arnold AC, Biaggioni I. Management approaches to hypertension in autonomic failure. Curr Opin Nephrol Hypertens 2012; 21: 481–485. doi: 10.1097/MNH.0b013e328356c52f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L et al. Orthostatic Hypotension in Parkinson Disease: How Much You Fall or How Low You Go? Mov Disord 2015. doi: 10.1002/mds.26079. doi: 10.1002/mds.26079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Fuente-Fernandez R, Ruth TJ, Sossi V et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science 2001; 293: 1164–1166. doi: 10.1126/science.1060937 [DOI] [PubMed] [Google Scholar]
- 25.Administration FaD. The Voice of the Patient: A Series of Reports from FDA’s Patient-Focused Drug Development initiative. In; 2017
- 26.Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L et al. Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Mov Disord 2015; 30: 639–645. doi: 10.1002/mds.26079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuente Mora C, Palma JA, Kaufmann H et al. Cerebral autoregulation and symptoms of orthostatic hypotension in familial dysautonomia. J Cereb Blood Flow Metab 2016. doi: 10.1177/0271678X16667524. doi: 10.1177/0271678X16667524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clin Auton Res 2001; 11: 363–367 [DOI] [PubMed] [Google Scholar]
- 29.Centi J, Freeman R, Gibbons CH et al. Effects of orthostatic hypotension on cognition in Parkinson disease. Neurology 2017; 88: 17–24. doi: 10.1212/WNL.0000000000003452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palma JA, Norcliffe-Kaufmann L, Kaufmann H. An orthostatic hypotension mimic: The inebriation-like syndrome in Parkinson disease. Mov Disord 2016; 31: 598–600. doi: 10.1002/mds.26516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sletten DM, Suarez GA, Low PA et al. COMPASS 31: a refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc 2012; 87: 1196–1201. doi: 10.1016/j.mayocp.2012.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visser M, Marinus J, Stiggelbout AM et al. Assessment of autonomic dysfunction in Parkinson’s disease: the SCOPA-AUT. Mov Disord 2004; 19: 1306–1312. doi: 10.1002/mds.20153 [DOI] [PubMed] [Google Scholar]
- 33.Robinson-Papp J, Sharma SK, George MC et al. Assessment of autonomic symptoms in a medically complex, urban patient population. Clin Auton Res 2017; 27: 25–29. doi: 10.1007/s10286-016-0384-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrag A, Selai C, Mathias C et al. Measuring health-related quality of life in MSA: the MSA-QoL. Mov Disord 2007; 22: 2332–2338. doi: 10.1002/mds.21649 [DOI] [PubMed] [Google Scholar]
- 35.Schrag A, Geser F, Stampfer-Kountchev M et al. Health-related quality of life in multiple system atrophy. Mov Disord 2006; 21: 809–815. doi: 10.1002/mds.20808 [DOI] [PubMed] [Google Scholar]
- 36.Kaufmann H, Malamut R, Norcliffe-Kaufmann L et al. The Orthostatic Hypotension Questionnaire (OHQ): validation of a novel symptom assessment scale. Clin Auton Res 2012; 22: 79–90. doi: 10.1007/s10286-011-0146-2 [DOI] [PubMed] [Google Scholar]
- 37.Wright RA, Kaufmann HC, Perera R et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology 1998; 51: 120–124 [DOI] [PubMed] [Google Scholar]
- 38.Frith J, Newton JL. Validation of a questionnaire for orthostatic hypotension for routine clinical use. Geriatr Gerontol Int 2016; 16: 785–790. doi: 10.1111/ggi.12553 [DOI] [PubMed] [Google Scholar]
- 39.Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont) 2007; 4: 28–37 [PMC free article] [PubMed] [Google Scholar]
- 40.Zachariah PK, Bloedow DC, Moyer TP et al. Pharmacodynamics of midodrine, an antihypotensive agent. Clin Pharmacol Ther 1986; 39: 586–591. doi: 10.1038/clpt.1986.101 [DOI] [PubMed] [Google Scholar]
- 41.Ramirez CE, Okamoto LE, Arnold AC et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension 2014; 64: 1235–1240. doi: 10.1161/HYPERTENSIONAHA.114.04225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith W, Wan H, Much D et al. Clinical benefit of midodrine hydrochloride in symptomatic orthostatic hypotension: a phase 4, double-blind, placebo-controlled, randomized, tilt-table study. Clin Auton Res 2016; 26: 269–277. doi: 10.1007/s10286-016-0363-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Norcliffe-Kaufmann L, Kaufmann H. Is ambulatory blood pressure monitoring useful in patients with chronic autonomic failure? Clin Auton Res 2014; 24: 189–192. doi: 10.1007/s10286-014-0229-y [DOI] [PubMed] [Google Scholar]
- 44.Umehara T, Matsuno H, Toyoda C et al. Clinical characteristics of supine hypertension in de novo Parkinson disease. Clin Auton Res 2016; 26: 15–21. doi: 10.1007/s10286-015-0324-8 [DOI] [PubMed] [Google Scholar]
- 45.Jordan J, Biaggioni I. Diagnosis and treatment of supine hypertension in autonomic failure patients with orthostatic hypotension. J Clin Hypertens (Greenwich) 2002; 4: 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldstein DS, Pechnik S, Holmes C et al. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension 2003; 42: 136–142. doi: 10.1161/01.HYP.0000081216.11623.C3 [DOI] [PubMed] [Google Scholar]
- 47.Fanciulli A, Gobel G, Ndayisaba JP et al. Supine hypertension in Parkinson’s disease and multiple system atrophy. Clin Auton Res 2016; 26: 97–105. doi: 10.1007/s10286-015-0336-4 [DOI] [PubMed] [Google Scholar]
- 48.Kaufmann H, Norcliffe-Kaufmann L, Hewitt LA et al. Effects of the novel norepinephrine prodrug, droxidopa, on ambulatory blood pressure in patients with neurogenic orthostatic hypotension. Journal of the American Society of Hypertension : JASH 2016; 10: 819–826. doi: 10.1016/j.jash.2016.07.009 [DOI] [PubMed] [Google Scholar]
- 49.Pickering RM, Grimbergen YA, Rigney U et al. A meta-analysis of six prospective studies of falling in Parkinson’s disease. Mov Disord 2007; 22: 1892–1900. doi: 10.1002/mds.21598 [DOI] [PubMed] [Google Scholar]
- 50.Rascol O, Perez-Lloret S, Damier P et al. Falls in ambulatory non-demented patients with Parkinson’s disease. J Neural Transm (Vienna) 2015; 122: 1447–1455. doi: 10.1007/s00702-015-1396-2 [DOI] [PubMed] [Google Scholar]
- 51.van der Marck MA, Klok MP, Okun MS et al. Consensus-based clinical practice recommendations for the examination and management of falls in patients with Parkinson’s disease. Parkinsonism Relat Disord 2014; 20: 360–369. doi: 10.1016/j.parkreldis.2013.10.030 [DOI] [PubMed] [Google Scholar]
- 52.Perez-Lloret S, Rey MV, Fabre N et al. Do Parkinson’s disease patients disclose their adverse events spontaneously? Eur J Clin Pharmacol 2012; 68: 857–865. doi: 10.1007/s00228-011-1198-x [DOI] [PubMed] [Google Scholar]
- 53.Espay AJ, Bonato P, Nahab FB et al. Technology in Parkinson’s disease: Challenges and opportunities. Mov Disord 2016; 31: 1272–1282. doi: 10.1002/mds.26642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arneric SP, Cedarbaum JM, Khozin S et al. Biometric monitoring devices for assessing end points in clinical trials: developing an ecosystem. Nat Rev Drug Discov 2017; 16: 736. doi: 10.1038/nrd.2017.153 [DOI] [PubMed] [Google Scholar]
- 55.Dodge HH, Zhu J, Mattek NC et al. Use of High-Frequency In-Home Monitoring Data May Reduce Sample Sizes Needed in Clinical Trials. PLoS One 2015; 10: e0138095. doi: 10.1371/journal.pone.0138095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans SR. Clinical trial structures. J Exp Stroke Transl Med 2010; 3: 8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaufmann H, Saadia D, Voustianiouk A et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation 2003; 108: 724–728. doi: 10.1161/01.CIR.0000083721.49847.D7 [DOI] [PubMed] [Google Scholar]
- 58.Shibao C, Raj SR, Gamboa A et al. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension 2007; 50: 47–53. doi:HYPERTENSIONAHA.107.089961 [DOI] [PubMed] [Google Scholar]
- 59.Okamoto LE, Shibao C, Gamboa A et al. Synergistic effect of norepinephrine transporter blockade and alpha-2 antagonism on blood pressure in autonomic failure. Hypertension 2012; 59: 650–656. doi: 10.1161/HYPERTENSIONAHA.111.184812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shibao C, Gamboa A, Diedrich A et al. Acarbose, an alpha-glucosidase inhibitor, attenuates postprandial hypotension in autonomic failure. Hypertension 2007; 50: 54–61. doi: 10.1161/HYPERTENSIONAHA.107.091355 [DOI] [PubMed] [Google Scholar]
- 61.Okamoto LE, Diedrich A, Baudenbacher FJ et al. Efficacy of Servo-Controlled Splanchnic Venous Compression in the Treatment of Orthostatic Hypotension: A Randomized Comparison With Midodrine. Hypertension 2016; 68: 418–426. doi: 10.1161/HYPERTENSIONAHA.116.07199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biaggioni I, Freeman R, Mathias CJ et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension 2015; 65: 101–107. doi: 10.1161/HYPERTENSIONAHA.114.04035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guyatt G, Sackett D, Taylor DW et al. Determining optimal therapy--randomized trials in individual patients. N Engl J Med 1986; 314: 889–892. doi: 10.1056/NEJM198604033141406 [DOI] [PubMed] [Google Scholar]
- 64.Guyatt GH, Heyting A, Jaeschke R et al. N of 1 randomized trials for investigating new drugs. Control Clin Trials 1990; 11: 88–100 [DOI] [PubMed] [Google Scholar]
- 65.Offord C N-of-1 Trials Take on Challenges in Health Care. In. The Scientist; 2019 [Google Scholar]
- 66.Kaufmann H, Brannan T, Krakoff L et al. Treatment of orthostatic hypotension due to autonomic failure with a peripheral alpha-adrenergic agonist (midodrine). Neurology 1988; 38: 951–956 [DOI] [PubMed] [Google Scholar]
- 67.Kaufmann H, Freeman R, Biaggioni I et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology 2014; 83: 328–335. doi: 10.1212/WNL.0000000000000615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hauser RA, Heritier S, Rowse GJ et al. Droxidopa and Reduced Falls in a Trial of Parkinson Disease Patients With Neurogenic Orthostatic Hypotension. Clinical neuropharmacology 2016; 39: 220–226. doi: 10.1097/WNF.0000000000000168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hauser RA, Hewitt LA, Isaacson S. Droxidopa in patients with neurogenic orthostatic hypotension associated with Parkinson’s disease (NOH306A). Journal of Parkinson’s disease 2014; 4: 57–65. doi: 10.3233/JPD-130259 [DOI] [PubMed] [Google Scholar]
- 70.Hauser RA, Isaacson S, Lisk JP et al. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson’s disease (nOH306B). Mov Disord 2015; 30: 646–654. doi: 10.1002/mds.26086 [DOI] [PubMed] [Google Scholar]
- 71.Close S, Smaldone A, Fennoy I et al. Using information technology and social networking for recruitment of research participants: experience from an exploratory study of pediatric Klinefelter syndrome. J Med Internet Res 2013; 15: e48. doi: 10.2196/jmir.2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jordan J, Shannon JR, Black BK et al. The pressor response to water drinking in humans : a sympathetic reflex? Circulation 2000; 101: 504–509 [DOI] [PubMed] [Google Scholar]
- 73.May M, Jordan J. The osmopressor response to water drinking. Am J Physiol Regul Integr Comp Physiol 2011; 300: R40–46. doi: 10.1152/ajpregu.00544.2010 [DOI] [PubMed] [Google Scholar]
- 74.Schoffer KL, Henderson RD, O’Maley K et al. Nonpharmacological treatment, fludrocortisone, and domperidone for orthostatic hypotension in Parkinson’s disease. Mov Disord 2007; 22: 1543–1549. doi: 10.1002/mds.21428 [DOI] [PubMed] [Google Scholar]
- 75.Fanciulli A, Goebel G, Metzler B et al. Elastic Abdominal Binders Attenuate Orthostatic Hypotension in Parkinson’s Disease. Mov Disord Clin Prac 2016; 3: 156–160. doi: 10.1002/mdc3.12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schreglmann SR, Buchele F, Sommerauer M et al. Pyridostigmine bromide versus fludrocortisone in the treatment of orthostatic hypotension in Parkinson’s disease - a randomized controlled trial. Eur J Neurol 2017; 24: 545–551. doi: 10.1111/ene.13260 [DOI] [PubMed] [Google Scholar]
- 77.Jankovic J, Gilden JL, Hiner BC et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. The American journal of medicine 1993; 95: 38–48 [DOI] [PubMed] [Google Scholar]
- 78.Low PA, Gilden JL, Freeman R et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277: 1046–1051 [PubMed] [Google Scholar]
- 79.McTavish D, Goa KL. Midodrine. A review of its pharmacological properties and therapeutic use in orthostatic hypotension and secondary hypotensive disorders. Drugs 1989; 38: 757–777 [DOI] [PubMed] [Google Scholar]
- 80.Schirger A, Sheps SG, Thomas JE et al. Midodrine. A new agent in the management of idiopathic orthostatic hypotension and Shy-Drager syndrome. Mayo Clin Proc 1981; 56: 429–433 [PubMed] [Google Scholar]
- 81.Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Droxidopa in neurogenic orthostatic hypotension. Expert Rev Cardiovasc Ther 2015; 13: 875–891. doi: 10.1586/14779072.2015.1057504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaufmann H Droxidopa for symptomatic neurogenic orthostatic hypotension: what can we learn? Clin Auton Res 2017; 27: 1–3. doi: 10.1007/s10286-017-0426-6 [DOI] [PubMed] [Google Scholar]
- 83.Gupta F, Karabin B, Mehdirad A. Titrating droxidopa to maximize symptomatic benefit in a patient with Parkinson disease and neurogenic orthostatic hypotension. Clin Auton Res 2017; 27: 15–16. doi: 10.1007/s10286-017-0430-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vernino S, Claassen D. Polypharmacy: droxidopa to treat neurogenic orthostatic hypotension in a patient with Parkinson disease and type 2 diabetes mellitus. Clin Auton Res 2017; 27: 33–34. doi: 10.1007/s10286-017-0435-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kremens D, Lew M, Claassen D et al. Adding droxidopa to fludrocortisone or midodrine in a patient with neurogenic orthostatic hypotension and Parkinson disease. Clin Auton Res 2017; 27: 29–31. doi: 10.1007/s10286-017-0434-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mehdirad A, Karabin B, Gupta F. Managing neurogenic orthostatic hypotension with droxidopa in a patient with Parkinson disease, atrial fibrillation, and hypertension. Clin Auton Res 2017; 27: 25–27. doi: 10.1007/s10286-017-0433-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Claassen D, Lew M. Initiating droxidopa for neurogenic orthostatic hypotension in a patient with Parkinson disease. Clin Auton Res 2017; 27: 13–14. doi: 10.1007/s10286-017-0429-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goodman BP, Claassen D, Mehdirad A. Adjusting droxidopa for neurogenic orthostatic hypotension in a patient with Parkinson disease. Clin Auton Res 2017; 27: 17–19. doi: 10.1007/s10286-017-0431-9 [DOI] [PubMed] [Google Scholar]
- 89.Goodman BP, Gupta F. Defining successful treatment of neurogenic orthostatic hypotension with droxidopa in a patient with multiple system atrophy. Clin Auton Res 2017; 27: 21–23. doi: 10.1007/s10286-017-0432-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gupta F, Kremens D, Vernino S et al. Managing neurogenic orthostatic hypotension in a patient presenting with pure autonomic failure who later developed Parkinson disease. Clin Auton Res 2017; 27: 9–11. doi: 10.1007/s10286-017-0428-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bartholini J, Constantinidis J, Puig M et al. The stereoisomers of 3,4-dihydroxyphenylserine as precursors of norepinephrine. J Pharmacol Exp Ther 1975; 193: 523–532 [PubMed] [Google Scholar]
- 92.Biaggioni I, Arthur Hewitt L, Rowse GJ et al. Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol 2017; 17: 90. doi: 10.1186/s12883-017-0867-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elgebaly A, Abdelazeim B, Mattar O et al. Meta-analysis of the safety and efficacy of droxidopa for neurogenic orthostatic hypotension. Clin Auton Res 2016; 26: 171–180. doi: 10.1007/s10286-016-0349-7 [DOI] [PubMed] [Google Scholar]
- 94.Singer W, Sandroni P, Opfer-Gehrking TL et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63: 513–518. doi: 10.1001/archneur.63.4.noc50340 [DOI] [PubMed] [Google Scholar]
- 95.Byun JI, Moon J, Kim DY et al. Efficacy of single or combined midodrine and pyridostigmine in orthostatic hypotension. Neurology 2017; 89: 1078–1086. doi: 10.1212/WNL.0000000000004340 [DOI] [PubMed] [Google Scholar]
- 96.Hauser AS, Chavali S, Masuho I et al. Pharmacogenomics of GPCR Drug Targets. Cell 2017. doi: 10.1016/j.cell.2017.11.033. doi: 10.1016/j.cell.2017.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoeldtke RD, Cilmi KM, Mattis-Graves K. DL-Threo-3,4-dihydroxyphenylserine does not exert a pressor effect in orthostatic hypotension. Clin Pharmacol Ther 1984; 36: 302–306 [DOI] [PubMed] [Google Scholar]
- 98.Kaufmann H, Oribe E, Yahr MD. Differential effect of L-threo-3,4-dihydroxyphenylserine in pure autonomic failure and multiple system atrophy with autonomic failure. Journal of neural transmission Parkinson’s disease and dementia section 1991; 3: 143–148 [DOI] [PubMed] [Google Scholar]
- 99.Freeman R, Landsberg L, Young J. The treatment of neurogenic orthostatic hypotension with 3,4-DL-threo-dihydroxyphenylserine: a randomized, placebo-controlled, crossover trial. Neurology 1999; 53: 2151–2157 [DOI] [PubMed] [Google Scholar]
- 100.Mathias CJ, Senard JM, Braune S et al. L-threo-dihydroxyphenylserine (L-threo-DOPS; droxidopa) in the management of neurogenic orthostatic hypotension: a multi-national, multi-center, dose-ranging study in multiple system atrophy and pure autonomic failure. Clin Auton Res 2001; 11: 235–242 [DOI] [PubMed] [Google Scholar]
- 101.Hauser RA, Isaacson S, Lisk JP et al. Droxidopa for the Short-Term Treatment of Symptomatic Neurogenic Orthostatic Hypotension in Parkinson’s Disease (nOH306B). Mov Disord 2014. doi: 10.1002/mds.26086. doi: 10.1002/mds.26086 [DOI] [PubMed] [Google Scholar]