

Abstract
Cells encounter a multitude of external and internal stress-causing agents that can ultimately lead to DNA damage, mutations and disease. A cascade of signaling events counters these challenges to DNA, which is termed as the DNA damage response (DDR). The DDR preserves genome integrity by engaging appropriate repair pathways, while also coordinating cell cycle and/or apoptotic responses. Although many of the protein components in the DDR are identified, how chemical modifications to DNA impact the DDR is poorly understood. This review focuses on our current understanding of DNA methylation in maintaining genome integrity in mammalian cells. DNA methylation is a reversible epigenetic mark, which has been implicated in DNA damage signaling, repair and replication. Sites of DNA methylation can trigger mutations, which are drivers of human diseases including cancer. Indeed, alterations in DNA methylation are associated with increased susceptibility to tumorigenesis but whether this occurs through effects on the DDR, transcriptional responses or both is not entirely clear. Here, we also highlight epigenetic drugs currently in use as therapeutics that target DNA methylation pathways and discuss their effects in the context of the DDR. Finally, we pose unanswered questions regarding the interplay between DNA methylation, transcription and the DDR, positing the potential coordinated efforts of these pathways in genome integrity. While the impact of DNA methylation on gene regulation is widely understood, how this modification contributes to genome instability and mutations, either directly or indirectly, and the potential therapeutic opportunities in targeting DNA methylation pathways in cancer remain active areas of investigation.
Introduction
Genetic information is stored in DNA, which must be protected from mutations and alterations that can disrupt cell homeostasis and promote diseases. The integrity of the genome is constantly exposed to various threats including genotoxic agents that can harm the stability of the genome. A few examples include exposure to ultraviolet radiation (UV) from the sun, ionizing radiation (IR), natural products or manmade drugs used during cancer treatments and intrinsic cellular processes that damage DNA including replication errors, metabolic products and alterations in proteome homeostasis [1–3]. Exposure to these endogenous and exogenous DNA damaging agents can result in mutations leading to DNA base changes (i.e. via deamination); impact replication through formation of non-canonical DNA structures including RNA-DNA hybrids (R-loops) and G-quadruplexes; modulate gene expression through changes in methylation patterns at the promoter or gene body and form dangerous DNA lesions such as DNA double-strand breaks (DSBs), all of which can threaten genome integrity (Figure 1) [2,4,5]. To combat these hazards, cells utilize diverse mechanisms that are collectively termed as DNA damage responses (DDR), which act to sense DNA damage and repair it, while coordinating these activities with cellular processes including cell cycle, replication/transcription, programmed cell death or senescence. The essential nature of these balanced pathways in genome integrity are highlighted by the frequent loss of these processes in cancer, in which genome instability is a hallmark observed broadly across many cancer types [6].
Figure 1. Potential roles of DNA methylation in genome maintenance.
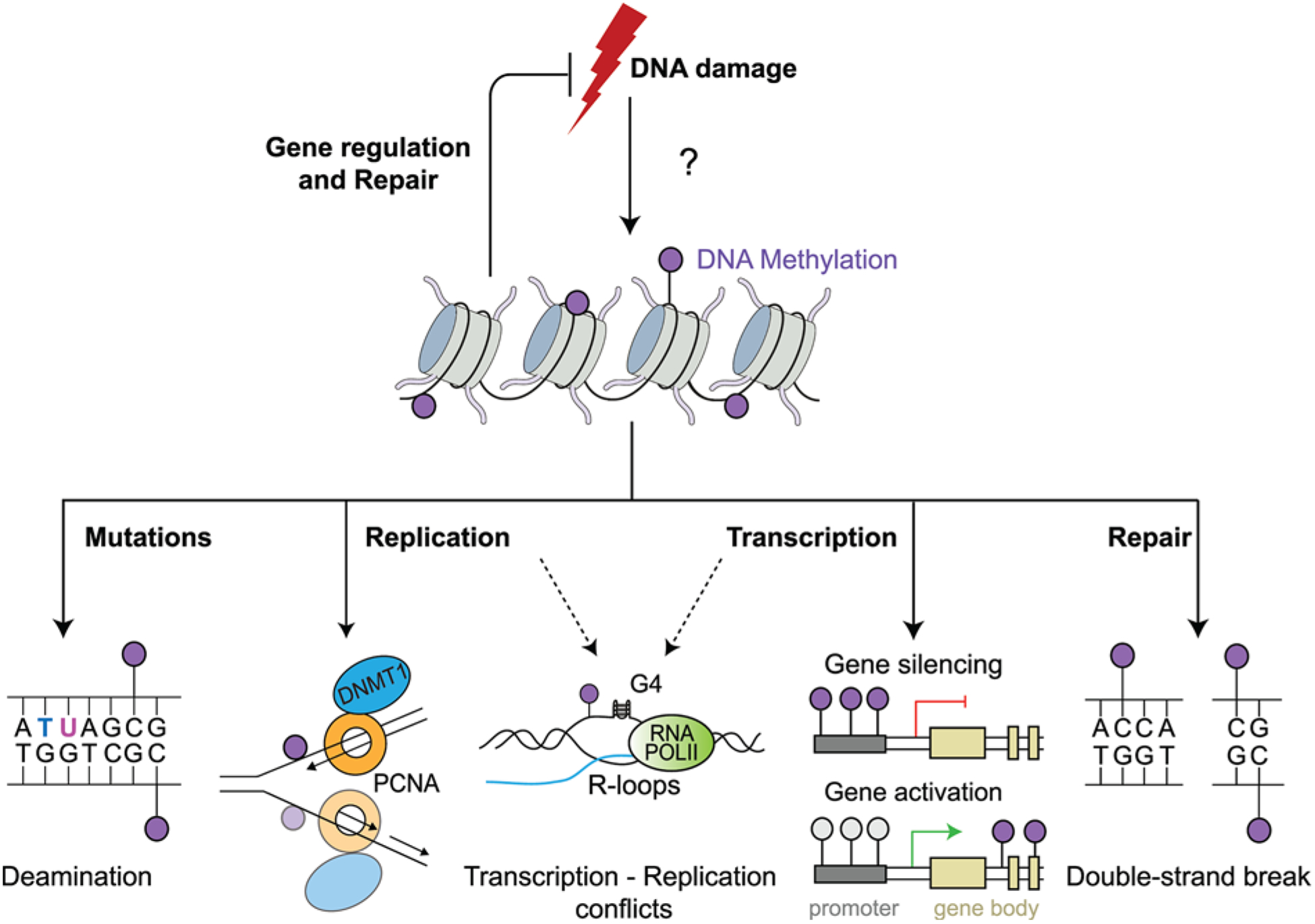
A simplified model depicting the putative roles of DNA methylation in the maintenance of genome integrity. DNA is methylated commonly at CpG sites, transposable elements, sites of tissue-specific gene silencing, X-chromosome inactivation and genome imprinting. DNA can undergo spontaneous deamination causing mutations or encounter roadblocks during replication from secondary structures such as R-loops and G-quadruplexes (G4), which may confer aberrant methylation patterns across the genome affecting gene transcription or impacting DNA DSB repair in response to DNA damage. Collectively, DNA methylation has the potential to affect the DDR as illustrated. Likewise,alterations in these pathways could also alter DNA methylation, which warrants consideration.
DNA methylation is a dynamic epigenetic mark mediated by DNA methyltransferases (DNMTs). While promoter methylation is generally associated with transcriptional gene silencing, gene body hypermethylation correlates with gene activation [7]. Methylation is also essential for X-chromosome inactivation, development and differentiation [8]. Since DNA methylation modifies the potential function and physical properties of the base, changes in methylation could also influence genome integrity and cancer by altering various processes either directly through mutations involving base changes and coding outcomes or more broadly through the DDR and DNA repair.
DNA methylation
Methyltransferases
DNA methylation is catalyzed by a family of DNMTs. DNMTs mediate the transfer of the labile methyl group (-CH3) from S-adenosyl-l-methionine (SAM) cofactor to the C5 position of the cytosine ring forming 5-methylcytosine (5mC) (Figure 2A). Mammalian DNMTs include DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. DNMT1 is a large protein that preferentially binds to hemi-methylated CpG dyads through its functional partner Ubiquitin-like, containing PHD and RING finger domains 1 (UHRF1). This complex functions to maintain DNA methylation during DNA replication [9,10]. De novo DNA methylation activity is catalyzed by DNMT3A and DNMT3B, which are responsible for establishing methylation patterns during development [11]. The other member of the DNMT3 family is DNMT3L, which is catalytically inactive and is required for gene imprinting, and regulation of DNMT3A/B activity [12,13]. Interestingly, DNMT2 has no DNMT activity, although it shares sequence similarities with other DNMT family members, and was subsequently identified as an RNA methyltransferase. DNMT2 has been shown to catalyze the methylation of tRNAAspGUC at position 38 [14] and other tRNAs (reviewed in [15]) to yield 5-methylcytosine (m5C).
Figure 2. An overview of DNA methylases and demethylases.
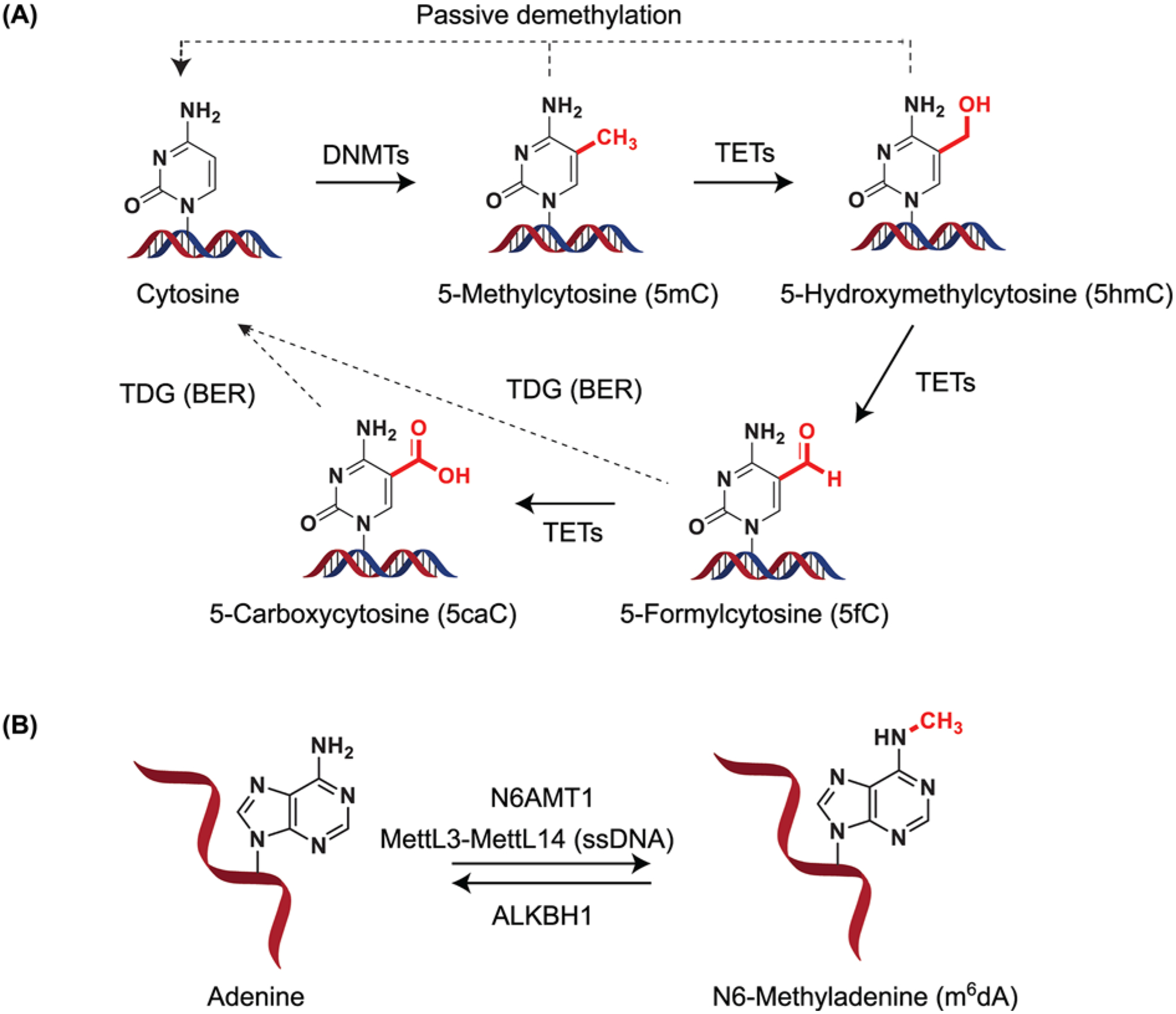
(A) Structural depiction of cytosine methylation to form 5mC. Upon TET activity, 5mC can give rise to 5-hydroxymethylcytosine (5hmC). TETs can iteratively oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC). 5fC and 5caC can be recognized and excised by the action of TDG forming cytosine through BER. (B) DNA methylation in adenine. Adenine can be methylated by N6AMT1 in DNA. The m6A RNA methyltransferase MettL3–MettL14 has been suggested to also methylate ssDNA. Demethylation has been suggested to occur via ALKBH1 activity. DNMT - includes DNMT1, DNMT3A, DNMT3B; TETs - TET1, TET2, TET3. Abbreviations: BER, base excision repair; N6AMT1, N6-adenine-specific DNA methyltransferase 1; TDG, thymine DNA glycosylase.
DNA demethylation
The mechanism of DNA demethylation remained a puzzle until the characterization of Ten-Eleven Translocation proteins (TET) in 2009 [16,17]. TET proteins (TET1, TET2 and TET3) are Fe(II) and α-ketoglutarate-dependent dioxygenases. They utilize molecular oxygen and α-ketoglutarate as co-substrates to generate oxidized 5mC derivatives along with succinate and carbon dioxide (CO2) as co-products. Using recombinant TET proteins, it was confirmed that they not only oxidize 5mC to 5-hydroxymethylcytosine (5hmC), but also mediate the subsequent oxidation of 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) [18,19] (Figure 2A). 5fC and 5caC are recognized and excised by thymine DNA glycosylase (TDG), which generates apyrimidinic (AP) sites [20] that are corrected by Base Excision Repair (BER). Recently, it was shown that GADD45A and GADD45B form a complex with TDG and TET2, which promotes DNA demethylation in vitro [21]. Activity-induced cytosine deaminases (AIDs) and apolipoprotein B mRNA editing complex (APOBEC) mediates an alternate path to successive oxidation of 5mC and 5hmC [22]. In this pathway, the modified nucleotide is also repaired by BER. The above processes are termed as active DNA demethylation, since it involves the removal of a methyl group by enzyme-initiated reactions. Passive DNA demethylation can also occur when the methyl group of 5mC is lost due to inhibition of DNMT1 followed by successive rounds of DNA replication, which dilute out the methylated DNA [23,24].
DNA methylation readers
Methylated DNA is recognized by methyl-CpG-binding domain (MBD) proteins, UHRF proteins, zinc-finger-containing proteins, and the more recently identified basic leucine-zipper (bZIP) and homeodomain-containing transcription factors (TFs) (Figure 3) [25–28]. MBD-containing proteins encompass MeCP2, MBD1, MBD2, MBD3 and MBD4. The MBD domain of MeCP2 is highly specific to methylated CpG sites, which are adjacent to an A/T-rich sequence [29]. MBD1 has been shown to interact with MBD1-containing chromatin-associated factor (MCAF-1) and histone methyltransferases like Suv39H and SETDB1. Furthermore, MBD1 has been classified as an oncogene or tumor suppressor depending on tumor type [30]. MBD2 is a subunit of the Mi2-NuRD complex that facilitates repression of genes upon its recruitment to methylated promoters [31]. Moreover, MBD2 has been shown to play a key role in the maintenance and spread of DNA methylation [32]. MBD3 and MBD4 are unusual regarding their DNA binding activities, as MBD3 cannot bind DNA directly due to a mutation in the MBD domain and is often found working in concert with MBD2 to enhance its recognition [33,34], while MBD4 binds to DNA but preferentially recognizes a guanine mismatched with thymine, uracil or 5-fluorouracil [35]. MeCP2 is also involved in the recruitment of DNMT1 to hemi-methylated DNA [36].
Figure 3. Readers of DNA methylation.
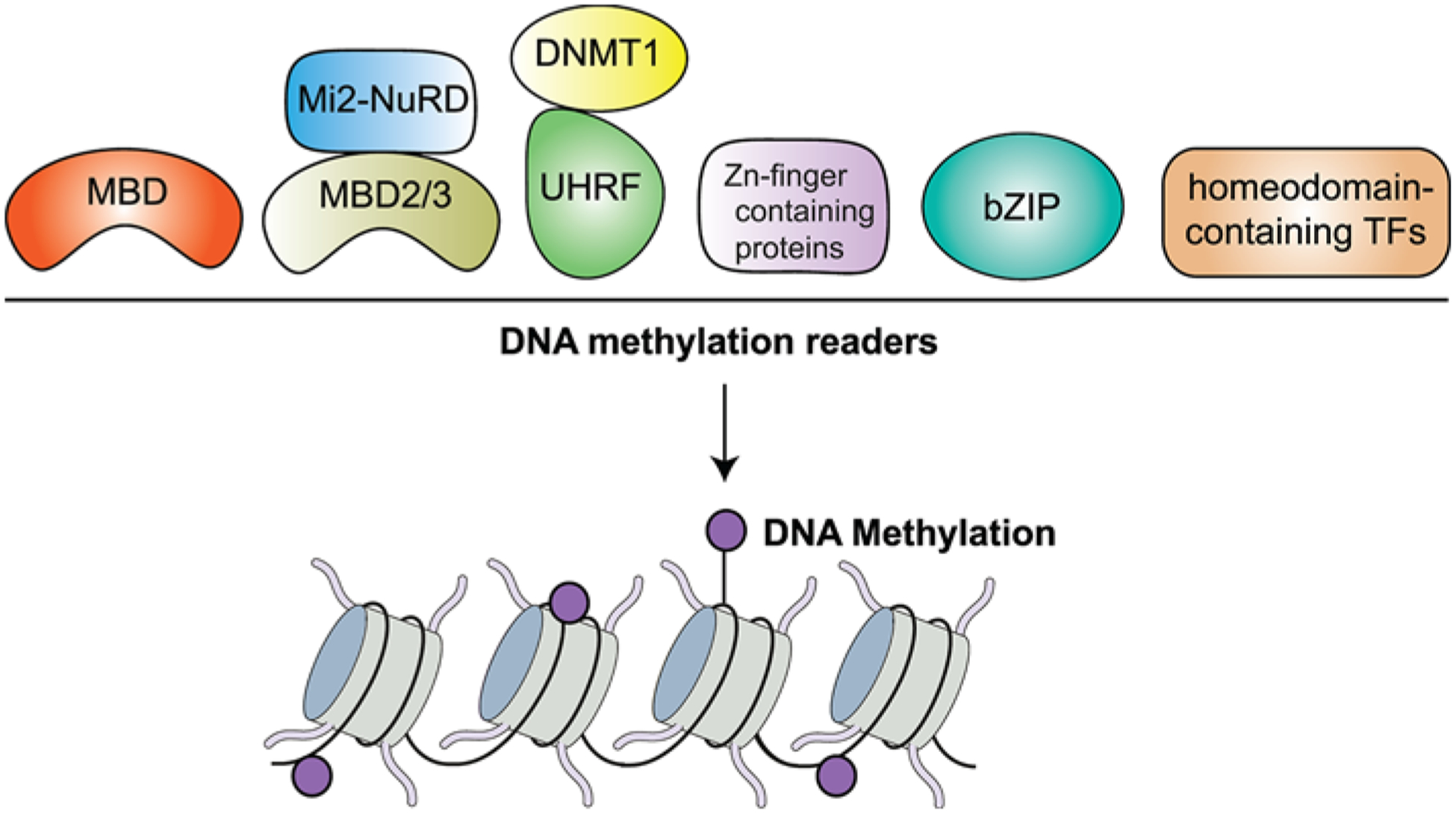
DNA methylation is recognized by several proteins which include: MBD, UHRF, Zn finger domain proteins including ZBTB33 (Kaiso), ZBTB4, ZBTB38, CTCF, KLF4, WT1, EGR1, ZFP57, basic leucine zipper-containing TFs (bZIP) and homeodomain-containing TFs.
UHRF consists of two proteins that include UHRF1 and UHRF2, which bind 5mC and 5hmC via a SET- and RING-associated DNA-binding domain, respectively [10,37,38]. UHRF proteins aid in maintaining DNA methylation by targeting DNMT1 specifically to hemi-methylated DNA during DNA replication [10]. The zinc-finger domain proteins composed of ZBTB33 (Kaiso), ZBTB4 and ZBTB38 bind to methylated DNA and repress transcription in a DNA methylation-dependent manner [39–41]. Other members of this family include ZFP57, KLF4, WT1, EGR1 and CTCF [25,30]. Aside from ZFP57, all these proteins have shown marginal selectivity toward mCpG (~1.5–1.8 fold) over CpG in vitro, and in cells [25]. In the case of CTCF, binding to methylated DNA has been shown to depend on the position of the CTCF motif recognition [42]. Cytosine methylation status at position 12 of the core consensus motif increased CTCF binding, but was severely reduced when position 2 was methylated [43,44]. Furthermore, CTCF has been shown to maintain genome stability through DSB repair by homologous recombination (HR) [45,46]. These studies revealed that the DNA binding domain of CTCF was required for interacting with DNA lesions. This suggests the potential for DNA methylation to affect CTCF binding and its associated repair process. For example, DSBs are repaired by two main pathways, non-homologous end joining (NHEJ) and HR. Given that CTCF binding is reduced by DNA methylation and CTCF promotes HR specifically, this suggests that breaks within hypo-methylated DNA would be preferentially repaired by HR. DNA methylation changes occurring de novo at break sites could also impact the dynamics of repair. CTCF may also influence the three-dimensional organization of the genome that is also known to be involved in genome maintenance [45]. Structural analysis has indicated that transcription factors containing bZIP and homeodomain bind methylated CpG sequences in vitro [27,28]. However, in vivo characterization of these transcription factors with DNA methylation is required and their potential involvement in genome maintenance remains untested.
DNA methylation of adenine
Although cytosine methylation has been extensively studied, adenine methylation in eukaryotes has only recently been identified using deep sequencing technologies. Adenines in the mammalian genome can undergo a similar SAM-dependent methylation on the exocyclic amino group (-NH2) at the sixth position of the purine ring to form N6-methyldeoxyadenosine (6mA or m6dA) that is mediated by a new family of DNMTs (Figure 2B). A recent study using overexpression and silencing experiments reported that N6-adenine-specific DNMT 1 (N6AMT1) is able to catalyze DNA m6dA methylation (calculated to be ~0.051% of all adenines in the human genome) [47]. Demethylation of this mark is mediated by an alkylated DNA repair protein B (ALKB) homolog, ALKBH1 in humans [47] (Figure 2B). This study also indicated that decreased levels of genomic m6dA levels correlated with tumorigenesis and indicated poor prognosis for cancer patients. Other studies have reported a role of human MettL3–MettL14 (which is more commonly known for m6A deposition in RNA), in adenine methylation on ssDNA, and mtDNA [48,49] (Figure 2B). In mitochondria, hypoxia increased m6dA levels, suggesting a putative connection between adenine methylation and mitochondrial DNA damage [49]. Besides m6dA, human N6-hydroxymethyladenine (hm6dA) has also been described in human genomic DNA [50]. Oxidation of m6dA to hm6dA is catalyzed by human ALKBH1 and its levels increased in lung carcinoma tissues. Thus, the observation of methylated adenines in the human genome, although infrequent, warrants further consideration for its potential involvement in biological processes including the DDR and human diseases such as cancer.
DNA methylation and mutations
In the human genome, 5mC is frequently found in CpG dinucleotides, with these sites being hotspots for mutations including in tumor suppressor genes. For example, CpG island (CGI) mutations within the coding region of p53, a gene involved in genome stabilization and cell cycle/apoptotic responses, contribute to ~25% of its inactivating mutations in cancer [51]. Mutations within CpG sites can occur as a consequence of exposure to agents that are alkylating, oxidizing and hydrolytic. Hydrolytic deamination of cytosine results in the formation of uracil in DNA, which is readily recognized and repaired by uracil DNA glycosylase (UDG) (Figure 4A). However, in the case of 5mC, deamination forms thymine, a naturally occurring DNA base that makes it significantly more difficult to repair by TDG. Deamination of 5mC results in increased C→T transitions, which are among the largest class of mutations found in human cancers (Figure 4B) [52]. Transition mutations also disrupt DNA methylation patterns, potentially causing aberrant transcription. Both C→T transitions and alterations in DNA methylation can contribute to carcinogenesis. Moreover, the presence of 5mC in mammalian DNA enhances the formation of pyrimidine dimers (CC→TT transitions) upon exposure to UV light from sun, which promotes skin cancer [53]. This is due to the higher energy absorption of 5mC compared with cytosine.
Figure 4. DNA methylation and mutations.
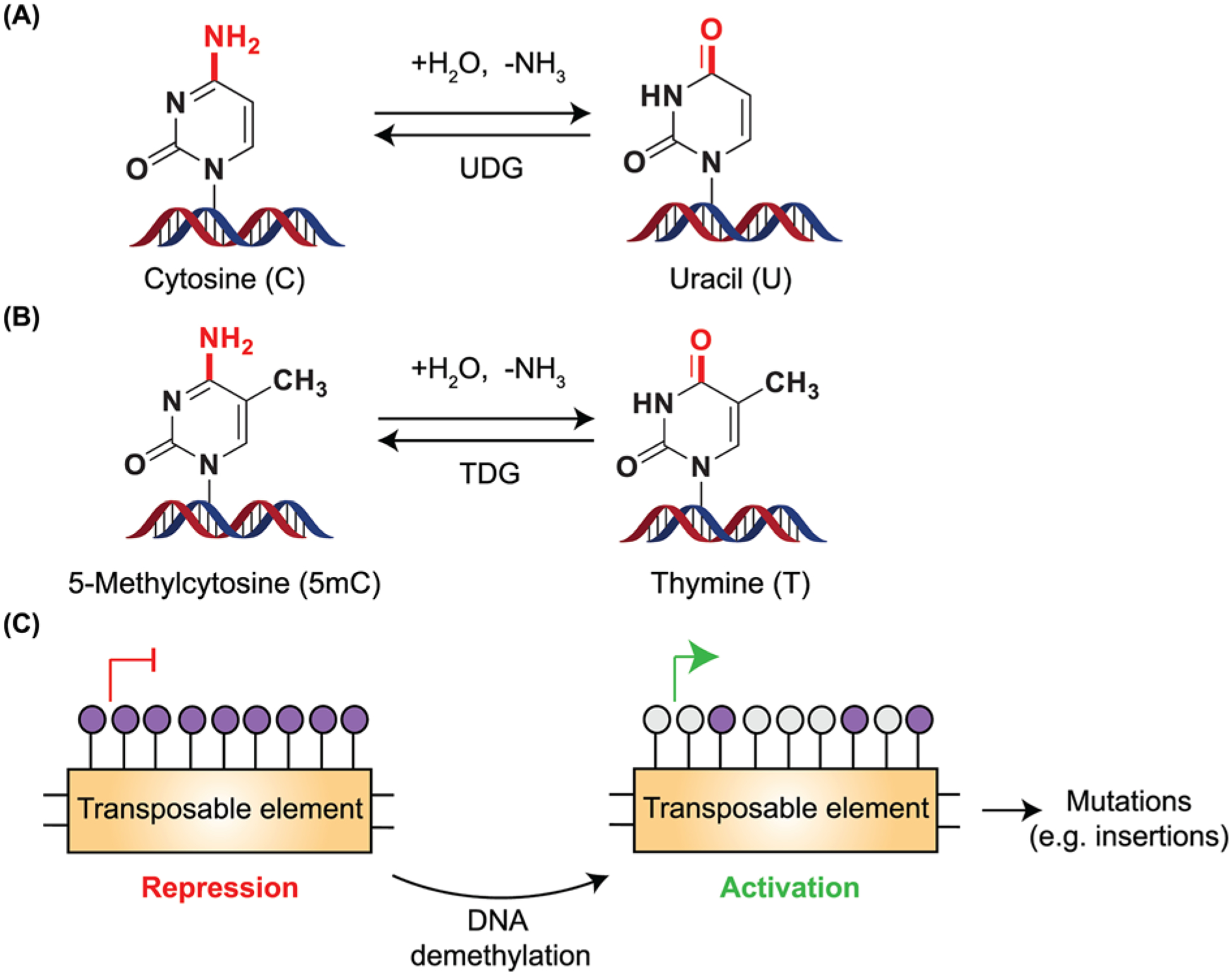
(A,B) Cytosine and 5mC undergo deamination either spontaneously or when exposed to agents that are hydrolytic or alkylating. Hydrolytic deamination (removal of -NH2 group as ammonia) from cytosine leads to the formation of uracil. Since uracil is not present in DNA, it is readily recognized by UDG, resulting in the substitution of uracil to cytosine. However, when 5mC undergoes deamination, it leads to the formation of thymine, a base normally present within DNA. TDG recognizes and corrects these misincorporated bases but inappropriate repair causes C→T transition mutations within the genome. (C) Transposable elements present in the genome are generally silenced by hypermethylation. DNA demethylation leads to the activation of these transposable elements, with the potential to increase mutations including insertions.
Transposable elements (TEs) present in mammalian genomes also rely on DNA methylation for silencing and repressing transposition. Oncogenesis is characterized by global hypomethylation, which promotes TE reactivation resulting in increased DNA breaks, insertional mutagenesis and genome instability [54] (Figure 4C). Nearly half of all human cancers have been found to express long interspersed element-1 (LINE-1), which are associated with p53 deficiency [55]. Thus, these studies highlight the diverse ways that 5mC can contribute to various types of mutations and alterations in the genome with the potential to cause genome instability and cancer.
DNA replication and DNA methylation
Maintaining and faithfully copying genetic information are an essential requirement for life. During DNA replication, DNMT1 localizes to the replication fork via its interaction with Proliferating Cell Nuclear Antigen (PCNA), the replisome clamp [56]. This interaction allows the maintenance of parental methylation on to newly synthesized daughter DNA strands during replication [9]. Although our cells have developed sophisticated mechanisms to replicate DNA with accuracy, replication is still subject to errors and interruptions. When cells are damaged during S-phase, it often gives rise to intermediates that causes the polymerases at the fork to temporarily cease their activity, known as ‘fork stalling’. Repair mechanisms are usually initiated to allow the fork to continue, but this event can also result in ‘fork collapse’, which ultimately leads to the formation of DSBs that trigger the DDR [57]. ‘Fork stalling’ can occur when the replication fork encounters transcription-replication conflicts such as R-loops, or non-canonical DNA structures like G-quadruples, Z-DNA etc [58]. Engagement of DDR and repair pathways help to alleviate replication stress in cells. There seems to be an intimate link between cell cycle regulation, DDR and DNA methylation. It is conceivable that upon activation of the DDR, the cell cycle regulator p21 or CDKN1A is activated by p53, which disrupts the interaction between DNMT1 and PCNA, suggesting a negative role for p21 in regulating DNA methylation [59]. In addition, the retinoblastoma gene product Rb can also bind to DNMT1 and inhibit its DNMT activities during DNA replication [60]. Moreover, these pathways are frequently dysregulated in cancer, which in turn could impact DNA methylation. The pathways controlling these proteins are relieved when the damage has been repaired, and this temporary stalling of DNMT1-coupled PCNA might alter DNA methylation maintenance. Thus, it is not well understood whether epigenetic patterns are faithfully maintained after DDR initiation during DNA replication.
Apart from the methylation-dependent activities of DNMT1, a recent study identified the PCNA-binding domain (PBD) of DNMT1 to be associated with increased endogenous DNA damage and mono-ubiquitination of PCNA, a known marker of replication stress, in a methylation-independent manner [61]. This observation suggests the potential for a non-canonical role for DNMT1 that is not related to DNA methylation per se in cancer initiation and progression when DNMT1 is overexpressed. While DNA methylation involving DNMT1 is targeted in cancer, the PBD of DNMT1 may provide an additional therapeutic option, which will require additional information on how DNMT1 and its mis-expression can impact genome stability. Another domain of DNMT1, namely the Replication Foci Targeting Sequence (RFTS) has recently been identified to be crucial for maintaining global DNA methylation and genome stability [62]. Here, a direct interaction between the histone marks H3K9me3 and H3 ubiquitylation with the RFTS domain was established through structural, biochemical and cellular analyses. Mutations in this domain led to decreased CpG methylation and increased sensitivity to IR. Therefore, studies directed toward understanding the functionality of the various domains of DNMT1 will be helpful in fully appreciating its multifunctional roles in maintaining genome stability.
DNA methylation and DNA damage responses
There is strong evidence that DNA methylation is associated with the DDR (Figure 5). These observations include; (1) aberrant DNA methylation patterns cause genome instability like mitotic catastrophe in cell lines [63,64]; (2) mice lacking Dnmt1 are genetically unstable [65]; (3) DNMT1 has been shown to be recruited to sites of DNA damage [66–70] and (4) interaction of DNMTs with PCNA aids DNA replication and repair [59,66,67,70].
Figure 5. Roles of DNMT1 in genome integrity.
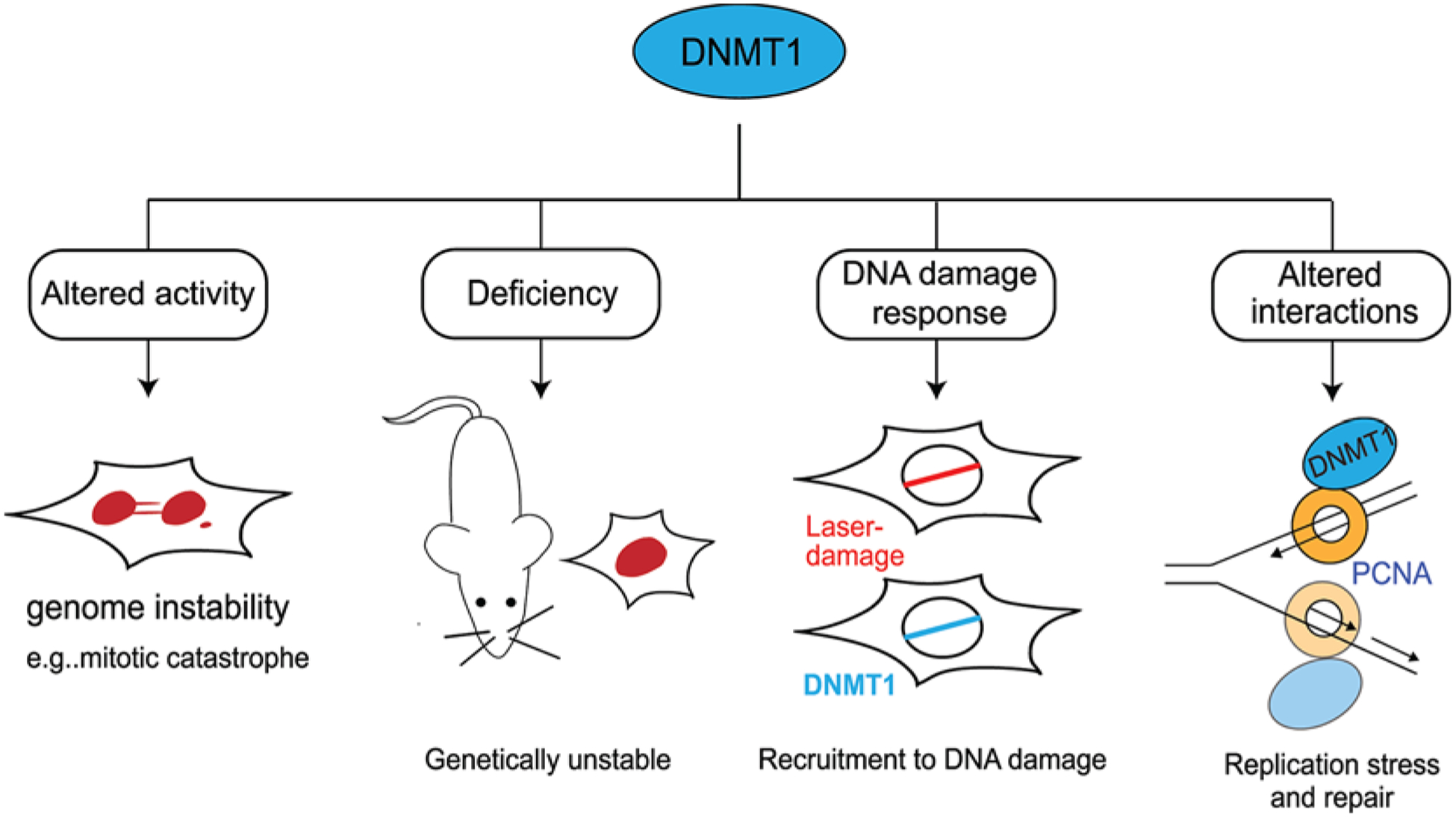
DNMT1 plays several critical roles in maintaining genome stability. These include: altered activity of DNA methylation can result in mitotic catastrophe; deficiency of DNMT1 has been found to be genetically unstable; DNMT1 is recruited to sites of laser damage; DNMT1 interacts with the replisome clamp PCNA during DNA replication and repair processes.
In more detail, the essential role of DNA methylation was contributed to p53-mediated apoptotic responses in DNMT1-deficient mouse embryonic fibroblasts [71]. Furthermore, inactivation of p53 in DNMT1 KO mice was able to rescue this lethal phenotype. This suggests that loss of DNA methylation leads to activation of p53 responses and/or the DDR. A study on the role of DNMT1 in pancreatic organogenesis observed increased p53 activation on a transcriptional level upon loss of DNMT1 [72]. On the other hand, deletion of DNMT1 in hTERT-immortalized normal human fibroblasts showed hallmarks of mismatch repair (MMR) deficiency [73]. In this case, loss of DNMT1 led to increased resistance to the nucleoside analog 6-thioguanine (6TG), and enhanced mutation rates at microsatellites. Additionally, these cells indicated loss of MMR proteins such as MLH1, PMS2 and MBD4, with increased levels of phosphorylated H2AX (γH2AX), a marker of DNA damage. Interestingly, inhibition of PARP was able to relieve the effects of DNMT1 deletion [73]. Therefore, these studies indicate the significance of DNMT1 in maintaining genomic stability, including through promoting DNA repair.
Another study on DSB repair revealed that upon oxidative stress, the chromatin remodeling complex NuRD mediates transcriptional repression by engaging with DNMT1, DNMT3A and DNMT3B causing abnormal de novo methylation [74]. This resulted in silencing of tumor-suppressive genes leading to increased proliferation, invasion and metastasis of colorectal cancer cells upon intrasplenic injection into immune-compromised mice. DNMT1 is also recruited to sites of DNA damage via its PBD, independent of its catalytic activity [66]. In addition to PCNA, DNMT1 interacts with other components of the DNA damage machinery including CHK1 and the 9-1-1 complex [66]. After recruitment to DNA lesions, DNMT1 modulates Ataxia telangiectasia and Rad3-related (ATR) signaling and suppresses abnormal activation of the DDR machinery. DNMT1 is required to repair DSBs as its loss results in delayed kinetics of IR-induced DSBs [63]. However, DNMT1 is only transiently recruited to damage sites, raising the question of whether or not methylation of DNA is its only function, especially during early stages of the DDR. Since PCNA is bound to DNMT1, it is tempting to speculate that DNMT1 restores epigenetic information on the newly synthesized DNA strand at repaired DNA lesion sites.
An additional important question to consider is whether DDR activation and processing of DSBs alters DNA methylation patterns. A study conducted by Cuozzo et al. showed that upon introduction of a DSB using the endonuclease I-Sce1 and gene conversion by HR, there was a concomitant change in methylation patterns pre- and post-DSB repair [75]. They propose that DNMT1 could act as a de novo methyltransferase that is recruited to DNA breaks by PCNA to methylate one of the newly synthesized strands causing differential methylation patterns, resulting in a hemi-methylated DNA segment. Upon replication of this region, cells containing both hypomethylated and methylated regions would occur, resulting in differential expression of the GFP reporter. These data suggest that DNA damage could cause alterations in DNA methylation and transcriptional status around the break site. Using a similar system with I-Sce1 and GFP+ selection of HR-repaired breaks, both ‘high’ and ‘low’ GFP expressing cells were isolated and analyzed for de novo methylation [70]. Interestingly, DNMT1, UHRF and DNMT3A were found to be recruited to DSB sites and required for methylating DNA at I-Sce1 induced DNA breaks, with DNA methylation patterns being regulated by transcription. Thus, there is strong evidence linking DNA methylation and DNA repair processes, although the molecular function of DNA methylation in DNA repair remains poorly understood. With this picture remaining unclear, further investigations are warranted. Given the prevalence of DNA repair deficiency, genome instability and alterations in DNA methylation in cancer, understanding these links may aid in dissecting their contribution to tumor development and their potential involvement in anti-cancer therapies.
TET enzymes have been shown to be critical for promoting genome integrity during replication stress [76]. Depletion of TET in mouse embryonic stem cells (mESCs) led to mitotic abnormalities upon aphidicolin treatment. Furthermore, 5hmC induction at sites of aphidicolin- and laser micro-irradiation-induced DNA damage occurred in a TET-dependent manner. Another report identified TET3 as an ATR target leading to DNA demethylation with increased 5hmC levels upon UV and camptothecin (CPT) exposure [77]. Depletion of TET3 resulted in defects in repair of UV and CPT lesions as well as survival from these DNA damaging agents. These results suggest DNA demethylation by TET3 as a requisite step in repairing these types of DNA lesions. TET1 has been found to be a target of Ataxia Telangiectasia Mutated (ATM). While irradiation increased 5hmC in neurons and fibroblasts, this response was lost in ATM-deficient cells [78]. It has been proposed that 5hmC loss due to ATM deficiency may preferentially affect cerebellar Purkinje cells, linking defects in TET1 and 5hmC to ataxia-telangiectasia disorder that results from ATM defects and resulting in neurodegeneration. Thus, TET proteins and 5hmC play an important role in regulating the DDR through DNA damage signaling and DNA repair.
DNA methylation and carcinogenesis
DNA repair machinery has evolved to maintain genomic integrity by suppressing the formation of mutations. Epigenetic silencing of DNA repair proteins can result in cells deficient for these repair pathways, resulting in mutations that promote carcinogenesis [79]. Aberrant methylation at the promoter CGIs within the promoter of key genes can lead to alterations in gene expression and defects in cellular pathways. Similarly, mutations in driver genes can result in downstream changes in DNA methylation that contribute toward oncogenesis. For example, mutations in the gene Isocitrate dehydrogenase 1 (IDH1) in glioblastoma patients result in abnormal production of 2-hydroxyglutarate. This leads to a CGI methylator phenotype (CIMP) that remodels the methylome and transcriptome due to inactivation of TET-mediated demethylation pathway [80]. The aberrant regulation by DNA methylation on the p53 gene remains controversial due to a lack of direct methylation over the p53 core promoter. Multiple investigations have been conducted to identify relationships between the mutation status of p53 and tumor grade with promoter DNA methylation in cancers [81,82]. However, no clear correlations were recognized, indicating that the primary mechanism of transcriptional silencing of the p53 promoter does not seem to depend on DNA methylation. In the case of BRCA1, methylation of CpG sites close to the transcriptional start site (TSS) is associated with reduced mRNA and protein levels [83]. Additionally, functional loss of BRCA1 involves methylation of a single copy of BRCA1, followed by loss of heterozygosity (LOH) event. This results in loss of HR activity with a pattern of genome-wide mutations and genome instability [84].
Aside from gene silencing by methylation, mutations or loss of methylation writers or erasers can also contribute to mutagenesis. Defects in DNMT1 have been reported to have a significant impact on microsatellite instability (MSI), a hallmark of MMR deficiency [85]. Deficiency of DNMT1 triggers defects in MMR through reduced levels of repair proteins like MLH1, PMS2 and MBD4 [73]. Somatic missense mutations in DNMT3A have been reported in ~20% of Acute Myeloid Leukemia (AML) patients and mutations are also observed in other hematological malignancies [86–89]. These mutations have been associated with poor overall survival in AML patients [90]. Although the downstream effects of DNMT3A mutations in AML are not well understood, a recent report observed association between DNMT3A and the leukemogenic HOX cofactor MEIS1, in the absence of Mixed Lineage Leukemia (MLL) fusions [91,92]. These findings may suggest a connection between altered DNA methylation through DNMT3A mutations and other transcriptional regulators, including MEIS1. Mutations in methylation erasers like TET2 are frequent in a wide spectrum of myeloid malignancies, causing aberrant DNA methylation patterns [93]. These mutations impair the catalytic activity of TET2 in vitro and in vivo [94]. Since TET2 converts 5mC into 5hmC, inactivating TET2 mutations would contribute to increased 5mC in the genome. Contrasting roles for TET1 have been reported. On one hand, the loss of Tet1 in mice leads to the development of B-cell lymphoma, suggesting a tumor suppressive role [95]. On the other hand however, TET1 can act as an oncogene since it is also a transcriptional target of MLL fusion proteins that activate the expression of downstream oncogenic targets to promote leukemogenesis [96]. Additional work is required to further establish how alterations in DNA methylation and demethylation perturb normal cellular functions, including those involved in the DDR, which could impact tumorigenesis and genome stability. Given that increased DNA damage is prevalent in cancer, it cannot be ruled out that DNA methylation at breaks sites, if impaired, could also directly be involved in cancer promoting events including mutagenesis.
Targeting DNA methylation
Inhibition of DNMTs can lead to increased expression of tumor suppressor genes along with a reduction in tumorigenicity in some settings [97]. These findings have made DNMTs a valuable target for cancer therapy. Indeed, the first Federal Drug Administration (FDA) approved drugs targeting an epigenetic pathway were DNMT inhibitors [98]. Commonly used catalytic inhibitors of DNMTs are nucleoside analogs consisting of 5-azacytidine (azacitidine), 5-aza-2′-deoxycytidine (decitabine) and zebularine. Currently, both azacitidine and decitabine are FDA approved for use against myelodysplastic syndrome (MDS) and decitabine for AML. Azacitidine and decitabine are nucleoside inhibitors that are incorporated into replicating DNA [99]. When DNA is methylated, the presence of azacytidine prevents the resolution of a covalent reaction intermediate, leading to the formation of protein-DNA adducts [100]. This results in decreased DNMT activity while also increasing the presence of protein-DNA adducts, which can lead to DNA damage and impair DNA replication [101,102]. Despite some poor outcomes against tumors upon using catalytic inhibitors of DNMTs, these compounds are able to demethylate and reactivate tumor suppressor gene expression in solid tumors [103]. Zebularine (1-(β-D-ribofuranosyl)-2(1H)-pyrimidinone) is an orally bioavailable DNMT inhibitor [104]. Treatment in cell systems indicates complete demethylation by depleting DNMT1 and partially depleting DNMT3A and DNMT3B [105,106]. These compounds exemplify the active area of research in drug discovery and cancer therapeutics focused on targeting DNA methylation.
Combinations of azacytidine or decitabine with standard chemotherapy have shown increased clinical activity. For example, co-treatment of cisplatin and 5-azacytidine treatments revealed an increase in DNA lesions that triggered the activation of DDR pathways [107]. Cisplatin and decitabine co-treatment resulted in partial response in one patient with cervical cancer and two minor responses - in one patient with non-small cell lung cancer and the other with cervical cancer [108]. However, it has to be noted that this combination led to significant hematological toxicity. Treatment with decitabine rescued cisplatin resistance in head and neck squamous cell carcinoma, leading to reduced tumor growth and reduced dosage of cisplatin in a xenograft model [109]. Further analysis revealed differences in methylation patterns between cisplatin-sensitive and cisplatin-resistant patient tumors, suggesting a role for gene methylation arrangements as possible biomarkers for cisplatin resistance. Apart from methylation-dependent effects on drug combinations, activation of signaling pathways can result in drug sensitivity. Cytotoxicity mediated by cisplatin or doxorubicin was found to be augmented by decitabine addition in bladder cancer cells by activation of Hippo pathway through RASSF1A [110]. An ongoing clinical trial (NCT03467178) is studying the combination of decitabine and carboplatin in platinum-resistant ovarian cancers [111]. While some promising synergistic tumor suppressive phenotypes have been observed when combining DNA damaging agents and DNA methylation inhibitors, mechanisms explaining these connections remain incomplete.
PARP1, a poly (ADP-ribose) polymerase involved in gene expression and the DDR, and DNMT1 have been found to interact, which may provide a direct link between the DDR and DNA methylation [112]. Combination of low doses of PARP and DNMT1 inhibitors have shown increased retention of PARP1 and DNMT1 at laser-damaged sites with increased binding of PARP1 to chromatin. This combination of inhibitors led to increased frequency of DSBs and synergistic cell death in AML cell lines, primary cells and mouse xenografts. In addition to PARP trapping on to chromatin by PARP inhibitors, PARP1 has also been shown to antagonize DNA end-resection in DSB repair [113] and also promote NHEJ [114]. Together, the function of PARP in the DDR is likely to impact cell death and sensitivity to other inhibitors, including DNMT inhibitors, in several ways that future work is needed to decipher. Interestingly, a recent investigation disclosed treatment with DNMT1 inhibitors led to hypermethylation of certain CGIs corresponding to genes differentially expressed in cancer tissue such as NFAT, LEF1 and MAZ-regulated [115]. This suggests that these inhibitors possess a complex mechanism of action and a deeper understanding of the response to DNMT1 inhibitors at the gene level is necessary to understand both their effects on the DDR and how combination of therapies can result in therapeutic benefit. Apart from the canonical DNA methylation inhibitors, targeting the ability of DNMT1 to interact with PCNA may provide another avenue to inhibit pro-tumorigenic functions of DNMT1 [61]. In this study, DNMT1 overproduction led to increased endogenous DNA damage in a methylation-independent and PBD-dependent manner, which also resulted in increased mutations, a cancer promoting process. The PBD of DNMT1 may represent an actionable drug target that could be pursued in future studies. The development and use of these drugs may also alleviate potential side effects of catalytic DNA methylation inhibitors.
Concluding remarks
Epigenetic patterns like DNA methylation serve as important regulatory roles in DNA-based processes, including transcription, replication and DNA repair. DNA methylation serves as a conduit between gene expression and important regulatory mechanisms that govern genome integrity. Aberrant DNA methylation patterns like global hypomethylation and promoter hypermethylation are observed in tumors and are linked with cancer progression. This makes it unsurprising that their activities are frequently modulated during tumorigenesis, making them potential ‘druggable’ targets. Although DNA methylation and its associated regulatory factors have been widely studied, it remains unclear their precise role in genome integrity pathways, including the potential link between DNA methylation and genome instability in cancer.
Does DNA methylation play a specific role during DNA repair and how does this contribute to epigenetic stability? A study has linked the recruitment of DNMT1 and MMR proteins MSH2 and MSH6 to the chromatin in response to oxidative damage [116]. Interestingly, they observed a reduction in nascent transcription after H2O2 treatment, which was abrogated upon knockdown of DNMT1 and/or MSH6. Furthermore, catalytically inactive DNMT1 was also recruited to chromatin, and could interact with MSH2/MSH6 upon oxidative damage. This suggests that the methylation activity of DNMT1 is likely not required, at least at the level of DNA lesion recognition. Additionally, reduction in transcription at sites of damage prevents interference between transcription and repair processes [117,118]. Is it possible that in addition to repair of the break, epigenetic modifications including DNA methylation marks are restored at repaired lesions? It has been noted that breaks occurring at gene promoters are most often repaired with no promoter hypermethylation and removal of silencing factors, including through the actions of the deacetylase SIRT1, but they are occasionally retained, which results in sustained gene silencing [68]. This suggests that repair of DNA breaks may lead to heritable silencing of CGI-containing promoters. Methylation of promoter CGIs is frequently associated with gene silencing and cancer.
Another question is whether DNA methylation in cancer cells directly affects repair processes or indirectly through gene expression alterations, or both. Reported data that could address this point is complex. The deficiency of multiple repair genes is often observed in cancers, including MLH1, O6-methylguanine-DNA methyltransferase (MGMT) and MSH2 in gastric cancers due to aberrant methylation patterns [119]. This can occur directly by promoter hypermethylation of repair proteins, e.g. MLH1 or other indirect mechanisms including overexpression of miRNAs that control the expression of many cellular proteins, e.g. miR-155 that targets MLH1 gene for repression [120]. Faulty DNA repair leads to increased mutations and epigenetic alterations, central to causing genome instability. Investigations on the role of chromatin regulator ATRX in DNA repair identified the role of its PCNA-interacting protein (PIP) motif for efficient HR-repair in G2 phase of the cell cycle [121]. This could suggest a plausible role for DNMT1 in mediating HR since it also binds to PCNA and conversely, DNMT1 overexpression may affect DNA synthesis during HR. Another repair pathway mediated by PCNA is MMR, which requires interaction between the PIP-motifs of MSH2 and MSH6 for identification of the mismatch and loading of RFC1 that acts as a processivity factor for DNA polymerases DNA pol δ and pol [122]. Thus, DNMT1 overexpression may impact other PCNA-dependent repair pathways, in addition to replication.
At the genomic level, DNA methylation within gene bodies is positively correlated with expression levels. Although the function of intergenic DNA methylation is not clearly understood, recent studies suggest that a loss of gene body methylation could result in activation of unscheduled intragenic transcription [123], and alternative promoter activation [124]. In addition, a casual relationship between gene body methylation and transcription has been identified. Treatment with azacytidine led to demethylation of gene bodies, resulting in reduced transcription of oncogenes, including c-MYC [125]. Thus, demethylation inhibitors seem to not only cause promoter demethylation, but also gene body demethylation. Alterations in these pathways could increase transcriptional noise, and possibly interfere with the DDR and genome stability.
Connections between DNA methylation and R-loops, a structure that can affect DNA template processes, have been reported. R-loops are enriched at promoter CGIs, and how these are recognized and modulated by epigenetic readers is not well known. Recently, it was shown that GADD45A could bind directly to R-loops formed by long non-coding RNAs (lncRNAs) and mediate DNA demethylation by recruiting TET1 [126]. This opens other questions such as whether GADD45A specifically recognizes R-loops at lncRNAs and whether other DNA methylation readers exist which can identify R-loops formed due to transcription-replication conflicts. Aside from R-loops, single-stranded guanine-rich DNA sequences can form G-quadruplexes, which are four-stranded DNA structures [127]. They are usually found at active chromatin and were found to sequester DNMT1 leading to a local inhibition of DNMT1 function at CGIs [128]. This suggests that G-quadruplex structures protect certain CGIs by sequestering DNMT1, and may play an important role in establishing the epigenome at these loci, although connections between DNA methylation, G-quadruplexes and genome stability have not been firmly established to date.
Finally, the question arises on whether there is an association between tumors exhibiting genome instability and defective DNA methylation. Genome instability can arise from many types of damage, including to telomeres, centromeres, replication stress and DSBs. A meta-analysis study identified a positive correlation between cancer incidence and DNA methylation at the promoter regions of genes involved in regulating telomere maintenance and regulation [129]. This suggests that cancer cells can alter telomere homeostasis through DNA methylation. Centrosomal defects are observed in breast cancer and result in aneuploidy due to chromosomal instability (CIN) [130]. Since p53 signaling axis is often compromised, p21, its bonafide target gene, can no longer inhibit the PCNA-DNMT1 complex from methylating DNA [59]. This results in aberrant DNA methylation, which could contribute to the CIN-phenotype. Thus, these studies highlight increasing evidence connecting defective DNA methylation and genome instability in cancer.
Although much of what we know about DNA methylation and genome stability relies on studies involving DNMT1, it is exciting to consider that this epigenetic mark is reversible. While, this makes DNA methylating inhibitors an attractive target in cancer therapeutics, very little is known about DNA demethylation in the context of the DDR and genome stability. Given that DNA methylation is read by a host of reader proteins (Figure 4), the potential impact of DNA methylation on the binding and function of these proteins in genome integrity pathways is clear but has not yet been studied comprehensively. Our current limited knowledge on DNA methylation and its biological effects on genome maintenance needs to be extended to allow additional forays into therapeutic targeting of these pathways to be made. It is conceivable that DNA methylation inhibitors in combination with other drugs, including those targeting the DDR, or in mutational backgrounds that could provide genetic vulnerabilities to these compounds can offer promising possibilities for cancer treatment (Figure 6). Future studies should focus on understanding canonical and non-canonical roles of DNA methylation in not only gene expression, but also their roles in maintaining genome integrity. This information has the potential to be leveraged to better identify and treat cancer.
Figure 6. Future perspectives.
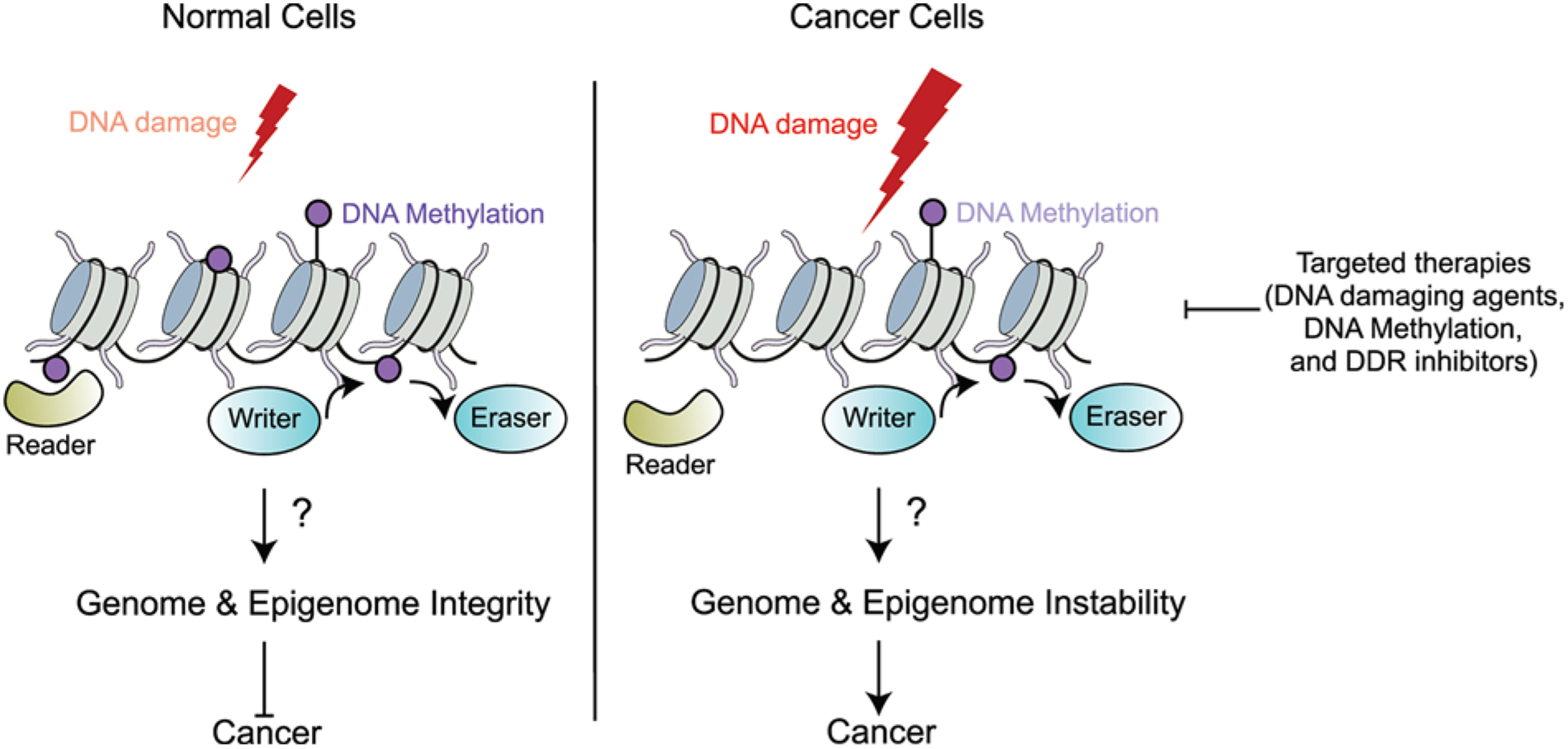
When normal cells encounter DNA damage, readers, writers and erasers of DNA methylation may contribute to the cellular response to DNA damage via gene regulation, DDRs and repair processes to ensure the maintenance of genome and epigenome integrity. However, in cancer cells, the function of readers, writers and erasers of DNA methylation may be altered. The changes in the methylation landscape could result in genomic and epigenomic instability due to differential gene expression, mutations and endogenous DNA damage, resulting in genome instability, a hallmark of cancer. Increased understanding of the mechanisms surrounding DNA methylation upon DNA damage and maintenance of genome integrity is necessary to extend current therapeutic strategies. Combinatorial treatments of inhibitors of DNA methylation along with DNA damaging agents and drugs targeting the DDR (i.e. PARP inhibitors) could offer promising drug treatment options to target cancer cells with altered DNA methylation patterns.
Summary.
DNA methylation is a reversible epigenetic mark that plays an important role in gene expression and DDRs.
Exposure to DNA damaging agents can affect DNA methylation patterns, causing mutations like deamination and increased transposon activation. However, it still remains to be determined if loss of DNA methylation patterns impacts the DDR directly, which could result in additional genomic and epigenomic instabilities.
DNMT1 interacts with the replisome clamp PCNA and its dysregulation results in replication stress and mutations. Testing for roles of DNA methylation directly in replication and repair fidelity is warranted.
Epigenetic inactivation of tumor suppressor genes due to aberrant methylation contributes toward increased mutations and genome instability.
DNMT targeting drugs beyond those targeting DNA methylation catalytic activities should be considered.
Many questions remain about the molecular mechanisms that govern DNA methylation and genome integrity, including the role of DNA demethylases, adenine methylation and readers of methylated DNA in the DDR.
Acknowledgements
We thank Dr. Bethany A. Buck-Koehntop (University of Utah) for careful reading and helpful comments in the review. We also thank the handling editor and reviewers for their comments and contributions to this work. We apologize to colleagues whose work could not be cited due to space limitations.
Funding
K.M.M.’s lab was supported in part by the National Cancer Institute, the National Institute of Health (N.I.H.) [grant numbers CA198279, CA201268]; and the American Cancer Society [grant number RSG-16-042-01]. B.X. lab was supported by the N.I.H. [grant number GM127802 (to B.X.)].
Abbreviations
- 5-caC
5-carboxycytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5-mC
5-methylcytosine
- AID
activity-induced cytosine deaminase
- ALKBH1
alkylated DNA repair protein B homolog
- AML
acute myeloid leukemia
- AP site
apyrimidinic site
- APOBEC
apolipoprotein B mRNA editing complex
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3-related
- BER
base excision repair
- bZIP
basic leucine-zipper
- CGI
CpG island
- CPT
camptothecin
- DDR
DNA damage response
- DNMT
DNA (cytosine-5)-methyltransferase 1
- DSB
double-strand break
- FDA
Federal Drug Administration
- hm6dA
human N6-hydroxymethyladenine
- HR
homologous recombination
- IR
ionizing radiation
- lncRNA
long non-coding RNA
- MBD
methyl-CpG-binding domain
- MGMT
O6-methylguanine-DNA methyltransferase
- MLL
mixed lineage leukemia
- MMR
mismatch repair
- N6AMT1
N6-adenine-specific DNA methyltransferase 1
- NHEJ
non-homologous end joining
- PBD
PCNA-binding domain
- PCNA
proliferating cell nuclear antigen
- PIP
PCNA-interacting protein
- R-loop
RNA:DNA hybrid
- SAM
S-adenosyl-l-methionine
- TDG
thymine DNA glycosylase
- TE
transposable element
- TET
ten-eleven translocation protein
- UHRF1
Ubiquitin-like, containing PHD and RING finger domains 1
- UV
ultraviolet
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Chatterjee N and Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen 58, 235–263, 10.1002/em.22087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SP and Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078, 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tubbs A and Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656, 10.1016/j.cell.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puget N, Miller KM and Legube G (2019) Non-canonical DNA/RNA structures during transcription-coupled double-strand break repair: roadblocks or bona fide repair intermediates? DNA Repair (Amst.) 81, 102661, 10.1016/j.dnarep.2019.102661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JJ, Lee SY, Gong F, Battenhouse AM, Boutz DR, Bashyal A et al. (2019) Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 33, 1751–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeggo PA, Pearl LH and Carr AM (2016) DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 16, 35–42, 10.1038/nrc.2015.4 [DOI] [PubMed] [Google Scholar]
- 7.Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492, 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 8.Goto T and Monk M (1998) Regulation of X-chromosome inactivation in development in mice and humans. Microbiol. Mol. Biol. Rev 62, 362–378, 10.1128/MMBR.62.2.362-378.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermann A, Goyal R and Jeltsch A (2004) The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem 279, 48350–48359, 10.1074/jbc.M403427200 [DOI] [PubMed] [Google Scholar]
- 10.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S and Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764, 10.1126/science.1147939 [DOI] [PubMed] [Google Scholar]
- 11.Okano M, Bell DW, Haber DA and Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257, 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- 12.Hata K, Okano M, Lei H and Li E (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 13.Bourc’his D, Xu G-L, Lin C-S, Bollman B and Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536, 10.1126/science.1065848 [DOI] [PubMed] [Google Scholar]
- 14.Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X et al. (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398, 10.1126/science.1120976 [DOI] [PubMed] [Google Scholar]
- 15.Xhemalce B (2013) From histones to RNA: role of methylation in cancer. Brief. Funct. Genomics 12, 244–253, 10.1093/bfgp/els064 [DOI] [PubMed] [Google Scholar]
- 16.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935, 10.1126/science.1170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer LM, Tahiliani M, Rao A and Aravind L (2009) Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8, 1698–1710, 10.4161/cc.8.11.8580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q et al. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307, 10.1126/science.1210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303, 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiti A and Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem 286, 35334–35338, 10.1074/jbc.C111.284620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Gu TP, Weber AR, Shen JZ, Li BZ, Xie ZG et al. (2015) Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 43, 3986–3997, 10.1093/nar/gkv283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA and Cairns BR (2008) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135, 1201–1212, 10.1016/j.cell.2008.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu SC and Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol 11, 607–620, 10.1038/nrm2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagiwada S, Kurimoto K, Hirota T, Yamaji M and Saitou M (2013) Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 32, 340–353, 10.1038/emboj.2012.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudson NO and Buck-Koehntop BA (2018) Zinc finger readers of methylated DNA. Molecules 23, 10.3390/molecules23102555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilas CK, Emery LE, Denchi EL and Miller KM (2018) Caught with one’s zinc fingers in the genome integrity cookie jar. Trends Genet. 34, 313–325, 10.1016/j.tig.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong S, Wang D, Horton JR, Zhang X, Speck SH, Blumenthal RM et al. (2017) Methyl-dependent and spatial-specific DNA recognition by the orthologous transcription factors human AP-1 and Epstein-Barr virus Zta. Nucleic Acids Res. 45, 2503–2515, 10.1093/nar/gkx057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S et al. (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239, 10.1126/science.aaj2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginder GD and Williams DC Jr (2018) Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Ther 184, 98–111, 10.1016/j.pharmthera.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahmood N and Rabbani SA (2019) DNA methylation readers and cancer: mechanistic and therapeutic applications. Front. Oncol 9, 489, 10.3389/fonc.2019.00489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A and Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935, 10.1101/gad.13.15.1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stirzaker C, Song JZ, Ng W, Du Q, Armstrong NJ, Locke WJ et al. (2017) Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene 36, 1328–1338, 10.1038/onc.2016.297 [DOI] [PubMed] [Google Scholar]
- 33.Hendrich B and Bird A (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol 18, 6538–6547, 10.1128/MCB.18.11.6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leighton G and Williams DC Jr (2019) The Methyl-CpG-binding domain 2 and 3 proteins and formation of the nucleosome remodeling and deacetylase complex. J. Mol. Biol 432, 1624–1639, 10.1016/j.jmb.2019.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendrich B, Hardeland U, Ng HH, Jiricny J and Bird A (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401, 301–304, 10.1038/45843 [DOI] [PubMed] [Google Scholar]
- 36.Kimura H and Shiota K (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem 278, 4806–4812, 10.1074/jbc.M209923200 [DOI] [PubMed] [Google Scholar]
- 37.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA et al. (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912, 10.1038/nature06397 [DOI] [PubMed] [Google Scholar]
- 38.Zhou T, Xiong J, Wang M, Yang N, Wong J, Zhu B et al. (2014) Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell 54, 879–886, 10.1016/j.molcel.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 39.Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G et al. (2001) The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filion GJ, Zhenilo S, Salozhin S, Yamada D, Prokhortchouk E and Defossez PA (2006) A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol 26, 169–181, 10.1128/MCB.26.1.169-181.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buck-Koehntop BA, Stanfield RL, Ekiert DC, Martinez-Yamout MA, Dyson HJ, Wilson IA et al. (2012) Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc. Natl. Acad. Sci. U.S.A 109, 15229–15234, 10.1073/pnas.1213726109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodges AJ, Hudson NO and Buck-Koehntop BA (2019) Cys2His2 zinc finger methyl-CpG binding proteins: getting a handle on methylated DNA. J. Mol. Biol 432, 1640–1660, 10.1016/j.jmb.2019.09.012 [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F et al. (2012) Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 22, 1680–1688, 10.1101/gr.136101.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG and Cheng X (2017) Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell 66, 711.e3–720.e3, 10.1016/j.molcel.2017.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lang F, Li X, Zheng W, Li Z, Lu D, Chen G et al. (2017) CTCF prevents genomic instability by promoting homologous recombination-directed DNA double-strand break repair. Proc. Natl. Acad. Sci. U.S.A 114, 10912–10917, 10.1073/pnas.1704076114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hilmi K, Jangal M, Marques M, Zhao T, Saad A, Zhang C et al. (2017) CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci. Adv 3, e1601898, 10.1126/sciadv.1601898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao CL, Zhu S, He M, Chen D, Zhang Q, Chen Y et al. (2018) N(6)-Methyladenine DNA modification in the human genome. Mol. Cell 71, 306.e7–318.e7, 10.1016/j.molcel.2018.06.015 [DOI] [PubMed] [Google Scholar]
- 48.Woodcock CB, Yu D, Hajian T, Li J, Huang Y, Dai N et al. (2019) Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 5, 63, 10.1038/s41421-019-0136-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao Z, Wu T, Cui X, Zhu P, Tan C, Dou X et al. (2020) N(6)-Deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell 78, 382–395.e8, 10.1016/j.molcel.2020.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong J, Ye TT, Ma CJ, Cheng QY, Yuan BF and Feng YQ (2019) N(6)-Hydroxymethyladenine: a hydroxylation derivative of N6-methyladenine in genomic DNA of mammals. Nucleic Acids Res. 47, 1268–1277, 10.1093/nar/gky1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greenblatt MS, Bennett WP, Hollstein M and Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878 [PubMed] [Google Scholar]
- 52.Sassa A, Kanemaru Y, Kamoshita N, Honma M and Yasui M (2016) Mutagenic consequences of cytosine alterations site-specifically embedded in the human genome. Genes Environ. 38, 17, 10.1186/s41021-016-0045-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tommasi S, Denissenko MF and Pfeifer GP (1997) Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res. 57, 4727–4730 [PubMed] [Google Scholar]
- 54.Deniz Ö, Frost JM and Branco MR (2019) Regulation of transposable elements by DNA modifications. Nat. Rev. Genet 20, 417–431, 10.1038/s41576-019-0117-3 [DOI] [PubMed] [Google Scholar]
- 55.Rodic N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS et al. (2014) Long interspersed element-1 protein expression is a hallmark of many human cancers. Am. J. Pathol 184, 1280–1286, 10.1016/j.ajpath.2014.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moldovan GL, Pfander B and Jentsch S (2007) PCNA, the maestro of the replication fork. Cell 129, 665–679, 10.1016/j.cell.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 57.Zeman MK and Cimprich KA (2014) Causes and consequences of replication stress. Nat. Cell Biol 16, 2–9, 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mirkin EV and Mirkin SM (2007) Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev 71, 13–35, 10.1128/MMBR.00030-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G and Li BF (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277, 1996–2000, 10.1126/science.277.5334.1996 [DOI] [PubMed] [Google Scholar]
- 60.Pradhan S and Kim GD (2002) The retinoblastoma gene product interacts with maintenance human DNA (cytosine-5) methyltransferase and modulates its activity. EMBO J. 21, 779–788, 10.1093/emboj/21.4.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia J, Chiu LY, Nehring RB, Bravo Nunez MA, Mei Q, Perez M et al. (2019) Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell 176, 127.e24–143.e24, 10.1016/j.cell.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren W, Fan H, Grimm SA, Guo Y, Kim JJ, Yin J et al. (2020) Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc. Natl. Acad. Sci. U.S.A 117, 18439–18447, 10.1073/pnas.2009316117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones PA and Gonzalgo ML (1997) Altered DNA methylation and genome instability: a new pathway to cancer? Proc. Natl. Acad. Sci. U.S.A 94, 2103–2105, 10.1073/pnas.94.6.2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B et al. (2007) Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet 39, 391–396, 10.1038/ng1982 [DOI] [PubMed] [Google Scholar]
- 65.Okano M and Li E (2002) Genetic analyses of DNA methyltransferase genes in mouse model system. J. Nutr 132, 2462S–2465S, 10.1093/jn/132.8.2462S [DOI] [PubMed] [Google Scholar]
- 66.Ha K, Lee GE, Palii SS, Brown KD, Takeda Y, Liu K et al. (2011) Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum. Mol. Genet 20, 126–140, 10.1093/hmg/ddq451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC and Leonhardt H (2005) Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U.S.A 102, 8905–8909, 10.1073/pnas.0501034102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Hagan HM, Mohammad HP and Baylin SB (2008) Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 4, e1000155, 10.1371/journal.pgen.1000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee GE, Kim JH, Taylor M and Muller MT (2010) DNA methyltransferase 1-associated protein (DMAP1) is a co-repressor that stimulates DNA methylation globally and locally at sites of double strand break repair. J. Biol. Chem 285, 37630–37640, 10.1074/jbc.M110.148536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morano A, Angrisano T, Russo G, Landi R, Pezone A, Bartollino S et al. (2014) Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 42, 804–821, 10.1093/nar/gkt920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G et al. (2001) Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet 27, 31–39, 10.1038/83730 [DOI] [PubMed] [Google Scholar]
- 72.Georgia S, Kanji M and Bhushan A (2013) DNMT1 represses p53 to maintain progenitor cell survival during pancreatic organogenesis. Genes Dev. 27, 372–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loughery JEP, Dunne PD, O’Neill KM, Meehan RR, McDaid JR and Walsh CP (2011) DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum. Mol. Genet 20, 3241–3255, 10.1093/hmg/ddr236 [DOI] [PubMed] [Google Scholar]
- 74.Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D et al. (2017) CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 31, 653.e7–668.e7, 10.1016/j.ccell.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A et al. (2007) DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 3, e110, 10.1371/journal.pgen.0030110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I et al. (2016) 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep. 14, 1283–1292, 10.1016/j.celrep.2016.01.035 [DOI] [PubMed] [Google Scholar]
- 77.Jiang D, Wei S, Chen F, Zhang Y and Li J (2017) TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep. 18, 781–796, 10.15252/embr.201643179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang D, Zhang Y, Hart RP, Chen J, Herrup K and Li J (2015) Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain 138, 3520–3536, 10.1093/brain/awv284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lahtz C and Pfeifer GP (2011) Epigenetic changes of DNA repair genes in cancer. J. Mol. Cell Biol 3, 51–58, 10.1093/jmcb/mjq053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483, 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang JH, Kim SJ, Noh DY, Park IA, Choe KJ, Yoo OJ et al. (2001) Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab. Invest 81, 573–579, 10.1038/labinvest.3780266 [DOI] [PubMed] [Google Scholar]
- 82.Chmelarova M, Krepinska E, Spacek J, Laco J, Beranek M and Palicka V (2013) Methylation in the p53 promoter in epithelial ovarian cancer. Clin. Transl. Oncol 15, 160–163, 10.1007/s12094-012-0894-z [DOI] [PubMed] [Google Scholar]
- 83.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E et al. (2000) Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst 92, 564–569, 10.1093/jnci/92.7.564 [DOI] [PubMed] [Google Scholar]
- 84.Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G et al. (2017) A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet 49, 1476–1486, 10.1038/ng.3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin B and Robertson KD (2013) DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol 754, 3–29, 10.1007/978-1-4419-9967-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE et al. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med 363, 2424–2433, 10.1056/NEJMoa1005143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan X-J, Xu J, Gu Z-H, Pan C-M, Lu G, Shen Y et al. (2011) Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet 43, 309–315, 10.1038/ng.788 [DOI] [PubMed] [Google Scholar]
- 88.Roller A, Grossmann V, Bacher U, Poetzinger F, Weissmann S, Nadarajah N et al. (2013) Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia 27, 1573–1578, 10.1038/leu.2013.65 [DOI] [PubMed] [Google Scholar]
- 89.Shah MY and Licht JD (2011) DNMT3A mutations in acute myeloid leukemia. Nat. Genet 43, 289–290, 10.1038/ng0411-289 [DOI] [PubMed] [Google Scholar]
- 90.Park DJ, Kwon A, Cho BS, Kim HJ, Hwang KA, Kim M et al. (2020) Characteristics of DNMT3A mutations in acute myeloid leukemia. Blood Res. 55, 17–26, 10.5045/br.2020.55.1.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferreira HJ, Heyn H, Vizoso M, Moutinho C, Vidal E, Gomez A et al. (2016) DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene 35, 3079–3082, 10.1038/onc.2015.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kohlmann A, Schoch C, Dugas M, Schnittger S, Hiddemann W, Kern W et al. (2005) New insights into MLL gene rearranged acute leukemias using gene expression profiling: shared pathways, lineage commitment, and partner genes. Leukemia 19, 953–964, 10.1038/sj.leu.2403746 [DOI] [PubMed] [Google Scholar]
- 93.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M et al. (2009) Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet 41, 838–842, 10.1038/ng.391 [DOI] [PubMed] [Google Scholar]
- 94.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS et al. (2010) Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843, 10.1038/nature09586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y et al. (2015) TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol 16, 653–662, 10.1038/ni.3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang H, Jiang X, Li Z, Li Y, Song CX, He C et al. (2013) TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. U.S.A 110, 11994–11999, 10.1073/pnas.1310656110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Subramaniam D, Thombre R, Dhar A and Anant S (2014) DNA methyltransferases: a novel target for prevention and therapy. Front. Oncol 4, 80, 10.3389/fonc.2014.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ganesan A, Arimondo PB, Rots MG, Jeronimo C and Berdasco M (2019) The timeline of epigenetic drug discovery: from reality to dreams. Clin. Epigenet 11, 174, 10.1186/s13148-019-0776-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Christman JK (2002) 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495, 10.1038/sj.onc.1205699 [DOI] [PubMed] [Google Scholar]
- 100.Santi DV, Norment A and Garrett CE (1984) Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U.S.A 81, 6993–6997, 10.1073/pnas.81.22.6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD and Robertson KD (2008) DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol 28, 752–771, 10.1128/MCB.01799-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Orta ML, Calderon-Montano JM, Dominguez I, Pastor N, Burgos-Moron E, Lopez-Lazaro M et al. (2013) 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 41, 5827–5836, 10.1093/nar/gkt270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zambrano P, Segura-Pacheco B, Perez-Cardenas E, Cetina L, Revilla-Vazquez A, Taja-Chayeb L et al. (2005) A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer 5, 44, 10.1186/1471-2407-5-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE et al. (2003) Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst 95, 399–409, 10.1093/jnci/95.5.399 [DOI] [PubMed] [Google Scholar]
- 105.Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG et al. (2004) Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol. Cell. Biol 24, 1270–1278, 10.1128/MCB.24.3.1270-1278.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Johnson WD, Harder JB, Naylor J, McCormick DL, Detrisac CJ, Glaze ER et al. (2006) A pharmacokinetic/pharmacodynamic approach to evaluating the safety of zebularine in non-human primates. Cancer Res. 66, 309–310 [Google Scholar]
- 107.Zacharioudakis E, Agarwal P, Bartoli A, Abell N, Kunalingam L, Bergoglio V et al. (2017) Chromatin regulates genome targeting with cisplatin. Angew. Chem. Int. Ed. Engl 56, 6483–6487, 10.1002/anie.201701144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pohlmann P, DiLeone LP, Cancella AI, Caldas AP, Dal Lago L, Campos O Jr et al. (2002) Phase II trial of cisplatin plus decitabine, a new DNA hypomethylating agent, in patients with advanced squamous cell carcinoma of the cervix. Am. J. Clin. Oncol 25, 496–501, 10.1097/00000421-200210000-00015 [DOI] [PubMed] [Google Scholar]
- 109.Viet CT, Dang D, Achdjian S, Ye Y, Katz SG and Schmidt BL (2014) Decitabine rescues cisplatin resistance in head and neck squamous cell carcinoma. PLoS ONE 9, e112880, 10.1371/journal.pone.0112880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khandelwal M, Anand V, Appunni S, Seth A, Singh P, Mathur S et al. (2018) Decitabine augments cytotoxicity of cisplatin and doxorubicin to bladder cancer cells by activating hippo pathway through RASSF1A. Mol. Cell. Biochem 446, 105–114, 10.1007/s11010-018-3278-z [DOI] [PubMed] [Google Scholar]
- 111.Benson EA, Skaar TC, Liu Y, Nephew KP and Matei D (2015) Carboplatin with decitabine therapy, in recurrent platinum resistant ovarian cancer, alters circulating miRNAs concentrations: a pilot study. PLoS ONE 10, e0141279, 10.1371/journal.pone.0141279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y et al. (2016) Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents - a potential therapy for cancer. Cancer Cell 30, 637–650, 10.1016/j.ccell.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Caron MC, Sharma AK, O’Sullivan J, Myler LR, Ferreira MT, Rodrigue A et al. (2019) Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun 10, 2954, 10.1038/s41467-019-10741-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJL et al. (2016) PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell 61, 547–562, 10.1016/j.molcel.2016.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Giri AK and Aittokallio T (2019) DNMT inhibitors increase methylation in the cancer genome. Front. Pharmacol 10, 10.3389/fphar.2019.00385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB and O’Hagan HM (2016) Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol 8, 244–254, 10.1093/jmcb/mjv050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gong F, Clouaire T, Aguirrebengoa M, Legube G and Miller KM (2017) Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol 216, 1959–1974, 10.1083/jcb.201611135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW et al. (2015) Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 29, 197–211, 10.1101/gad.252189.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bernstein C and Bernstein H (2015) Epigenetic reduction of DNA repair in progression to gastrointestinal cancer. World. J. Gastrointest. Oncol 7, 30–46, 10.4251/wjgo.v7.i5.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M et al. (2014) MicroRNAs in the DNA damage/repair network and cancer. Int. J. Genomics 2014, 820248, 10.1155/2014/820248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Juhasz S, Elbakry A, Mathes A and Lobrich M (2018) ATRX promotes DNA repair synthesis and sister chromatid exchange during homologous recombination. Mol. Cell 71, 11.e7–24.e7, 10.1016/j.molcel.2018.05.014 [DOI] [PubMed] [Google Scholar]
- 122.Hsieh P and Zhang Y (2017) The Devil is in the details for DNA mismatch repair. Proc. Natl. Acad. Sci. U.S.A 114, 3552–3554, 10.1073/pnas.1702747114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G et al. (2017) Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77, 10.1038/nature21373 [DOI] [PubMed] [Google Scholar]
- 124.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ et al. (2010) Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6, e1001134, 10.1371/journal.pgen.1001134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA and Liang G (2014) Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590, 10.1016/j.ccr.2014.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arab K, Karaulanov E, Musheev M, Trnka P, Schäfer A, Grummt I et al. (2019) GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat. Genet 51, 217–223, 10.1038/s41588-018-0306-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hansel-Hertsch R, Di Antonio M and Balasubramanian S (2017) DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol 18, 279–284, 10.1038/nrm.2017.3 [DOI] [PubMed] [Google Scholar]
- 128.Mao S-Q, Ghanbarian AT, Spiegel J, Martínez Cuesta S, Beraldi D, Di Antonio M et al. (2018) DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol 25, 951–957, 10.1038/s41594-018-0131-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Joyce BT, Zheng Y, Nannini D, Zhang Z, Liu L, Gao T et al. (2018) DNA methylation of telomere-related genes and cancer risk. Cancer Prev. Res 11, 511–522, 10.1158/1940-6207.CAPR-17-0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carroll PE, Okuda M, Horn HF, Biddinger P, Stambrook PJ, Gleich LL et al. (1999) Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene 18, 1935–1944, 10.1038/sj.onc.1202515 [DOI] [PubMed] [Google Scholar]