

Abstract
To better understand the molecular basis of cancer, the NCI’s Clinical Proteomics Tumor Analysis Consortium (CPTAC) has been performing comprehensive large-scale proteogenomic characterizations of multiple cancer types. Gene and protein regulatory networks are subsequently being derived based on these proteogenomic profiles, which serve as tools to gain systems-level understanding of the molecular regulatory factories underlying these diseases. On the other hand, it remains a challenge to effectively visualize and navigate the resulting network models, which capture higher order structures in the proteogenomic profiles. There is a pressing need to have a new open community resource tool for intuitive visual exploration, interpretation and communication of these gene/protein regulatory networks by the cancer research community. In this work, we introduce ProNetView-ccRCC (http://ccrcc.cptac-network-view.org/), an interactive web-based network exploration portal for investigating phosphopeptide co-expression network inferred based on the CPTAC clear cell renal cell carcinoma (ccRCC) phosphoproteomics data. ProNetView-ccRCC enables quick, user-intuitive visual interactions with the ccRCC tumor phosphoprotein co-expression network comprised of 3,614 genes, as well as 30 functional pathway-enriched network modules. Users can interact with the network portal and can conveniently query for association between abundance of each phosphopeptide in the network and clinical variables such as tumor grade.
Keywords: co-expression network, interactive network exploration, mass spectrometry, phosphoproteomics, proteomics
Recent advances in molecular profiling technologies[1,2] enable the large-scale integrative proteomic and genomic (proteogenomic) studies of cancers. For example, the National Cancer Institute’s Clinical Proteomics Tumor Analysis Consortium (CPTAC) has recently performed comprehensive proteogenomic characterizations of tumor samples from breast,[3,4] colon,[5] ovarian,[6] and kidney[7] cancer patients. Integrative analyses based on rich proteogenomic profiles from these studies have greatly advanced our understanding of the molecular mechanisms underlying these cancers. Specifically, the co-expression networks[8–10] derived from proteomics and phosphoproteomics data revealed the regulatory relationships between proteins and post-transcriptional modifications (PTMs), bringing new insights on complicated pathway interactions and dysfunctions that drive tumor initiation and progression[3] [7].
While these protein/PTM co-expression networks contain rich information for identifying active pathways and oncogenic drivers, they are challenging to visually communicate[11,12] due to their large dimensions and complicated topologies. Furthermore, these networks are best visually explored together with associated information between gene/protein activities and biological pathway annotations as well as clinical outcomes. To better understand and interpret the multiple layers of data provided in these networks, effective visual exploration tools that address these challenges are needed.
Current network data visual exploration tools[13,14] are mainly used for visually communicating networks as static 2D images in print publications. While these provide advanced capabilities for visual customizations, they are not well suited for user-initiated interactive network explorations. Furthermore, as incorporating and exploring the meta-data associated with the CPTAC co-expression networks will make them readily available to the cancer research community, a centralized platform that is customized for CPTAC co-expression networks and associated meta-data is necessary. There is thus a pressing need to have a new open community resource tool for intuitive visual exploration, interpretation and communication of these gene/protein regulatory networks by the cancer research community.
To address these needs, we have developed a web-based interactive portal, ProNetView-ccRCC, which provides an intuitive and customized exploration environment for phosphopeptide co-expression network and other analysis results from CPTAC ccRCC study[7]. Its 3D network exploration interface enables quick and easy interactions with the ccRCC phosphopeptide co-expression network as well as 30 functional network modules and their associated meta-data. Furthermore, users can easily query for their proteins of interest, neighbors they are connected to, mapping peptides or pathways, at user-defined filters on clinical variables (stage, grade, age, gender, FDR cut-off). The portal is an open-access freely available resource at http://ccrcc.cptac-network-view.org/.
Methods
ProNetView-ccRCC enables visual explorations of two network types: i) the phosphopeptide co-expression network of ccRCC tumors and ii) 30 different network modules derived based on the topology of the overall network. The network characterizes the co-expression patterns among 20,976 phosphopeptides in ccRCC and was derived using a random-forest based network construction algorithm[10] based on phospho-peptide level data of 103 ccRCC samples. The network displays association among genes based on phosphopeptide level data with two genes being connected if at least two phosphopeptides mapping to the two genes are associated to each other. Based on the overall network topology, we identified sets of genes (i.e., network modules) tightly connected in the network. Specifically, network modules were derived by using Glay[15] clustering algorithm. 30 modules containing at least 20 genes were identified and each was presented as a different module network. For each module network, pathway enrichment analysis was performed via Fisher Exact test and those overrepresented pathways were incorporated within the portal as associated meta-data[7].
For intuitive exploration of these dense and complex networks, ProNetView-ccRCC enables interactive explorations in 3D. We implemented this capability by building on the Three.js framework, which provides an interface for WebGL. We calculated the network layouts using 3D layout algorithms within iCAVE [16,17], which is a desktop-based tool we have developed earlier for the stereoscopic and immersive exploration of networks. To handle various user interaction events in real time, we have utilized multiple client-side Javascript libraries (e.g. D3.js, dataTables.js, JQuery, etc.). For the styling of web interface elements, we have primarily utilized Bootstrap v3.3.7, integrated with some styling of our own. Since ProNetView-ccRCC utilizes only standard libraries and does not require the use of any additional plug-ins, the portal runs on all modern web browsers.
Use Protocols
ProNetView-ccRCC landing page (http://ccrcc.cptac-network-view.org/) provides the global ccRCC tumor phosphoproteomics co-expression network (Figure 1A) and links it to functional network modules (Figure 1B). Users can interactively explore the large and dense topology of the whole ccRCC phosphopeptide co-expression network in 3D by rotating and zooming in/out the view. Six modules within this network (Figure 1B) were significantly associated with biological pathways[7], as highlighted within the network using different coloring of nodes. Users can easily query for genes of their interest in the network. For example, one of the genes highlighted in the CPTAC ccRCC paper[7] was ERG, a gene associated with VEGF response. By searching for the oncogene ERG (Figure 1C), the corresponding node is highlighted in red in the network exploration panel (Figure 1A) and it can easily be identified as member of the Angiogenesis enriched module network (Figure 1B). The query further returns the list of genes whose phosphopeptides were directly connected to ERG (i.e. co-expressed phosphopeptides) and provides the association of its corresponding phosphoprotein with tumor grade (p-value: 0.003, FDR: 0.118) and other phenotypes of interest (Figure 1C). Overall, these help understand the phosphopeptide network in the context of markers of user interest to help guide hypothesis generation for future studies.
Figure 1.
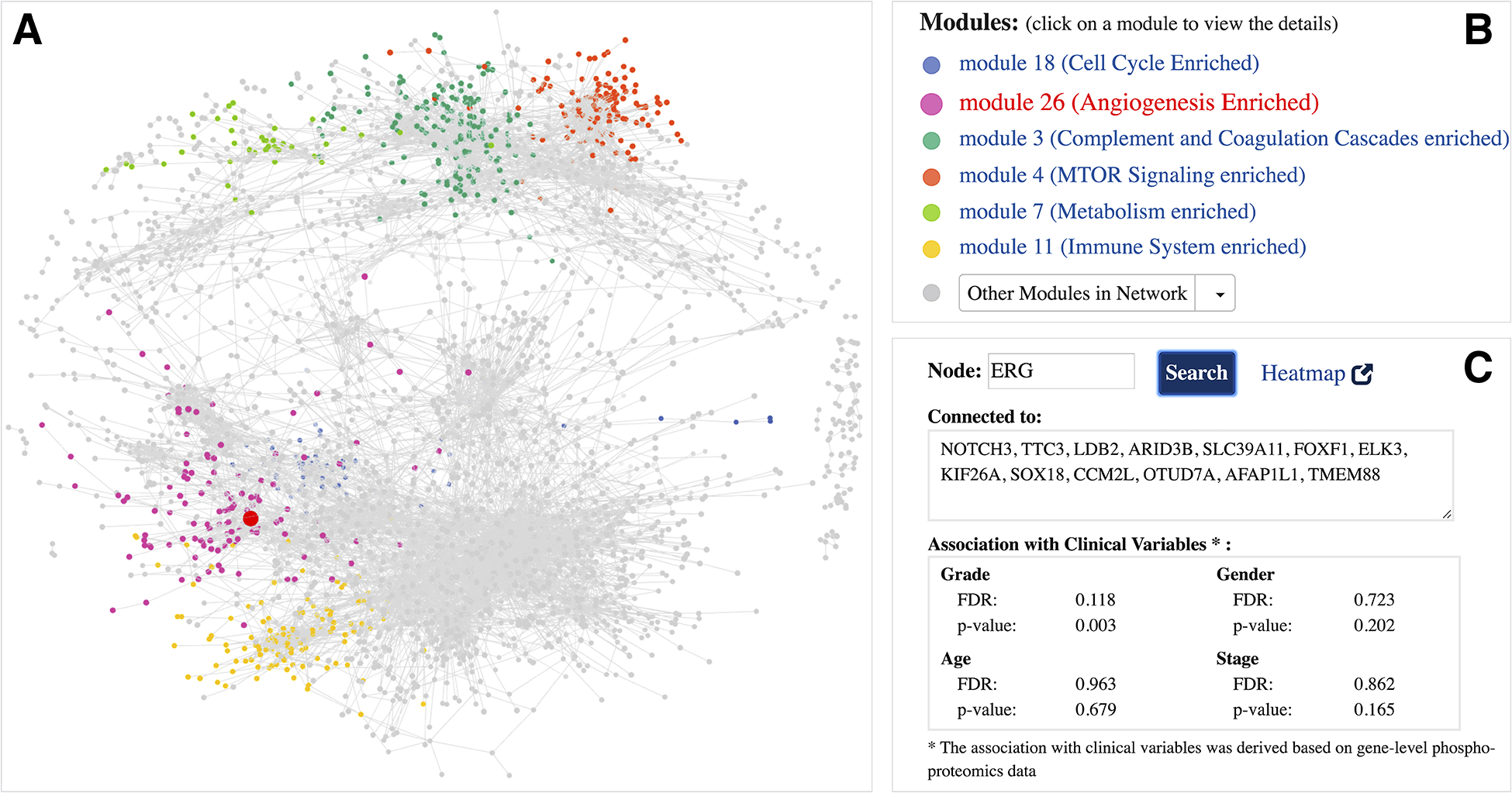
Global ccRCC tumor phosphoproteomics co-expression network A interactive 3D network exploration panel. Subsets of genes are color-coded based on their association with modules that are part of significant biological pathways. Large, red-colored node highlights the ERG gene; B list of module networks. 6 modules that are part of significant biological pathways are color-coded, the remaining 24 module networks are listed under the Other Modules in Network dropdown menu; C meta-data information associated with ERG gene. Gene-specific information includes the list of connections (co-expressed genes) and its association with clinical variables.
To explore a module network of interest and its associated meta-data in detail, users can click on a module name (Figure 1B). For example, Figure 2A displays a snapshot of the Angiogenesis enriched module network (module 26) within the ProNetView-ccRCC panel. Node sizes are proportional to the number of connections to let users easily identify the genes that are more central than others. To interactively explore the network, users can hover their mouse over a node, which will display the gene name of the phosphopeptide, and click on the node, which will display the associated gene-level meta-data in a separate panel (Figure 2B). As shown in Figure 2B, using ProNetView-ccRCC, users can quickly visualize the location of ERG in the network and display the connected nodes. As shown, ERG is directly connected to 11 genes within the same module and maps to 5 different peptides. In addition, users can click on the Heatmap link (Figure 2B), which will direct them to ProTrack tool,[18] which provides a web-based interface to explore ccRCC multi-omics data through interactive heatmaps. To query whether other genes have similar association to tumor grade as ERG, users can easily construct a simple query for those genes (Figure 2C) by specifying the phenotype of interest (grade) and the FDR cutoff value (FDR= 20%). Genes that satisfy these metrics are listed in the text box and are highlighted in the network exploration panel in red (Figure 2D). Users can click the Reset button to return to the original network display.
Figure 2.
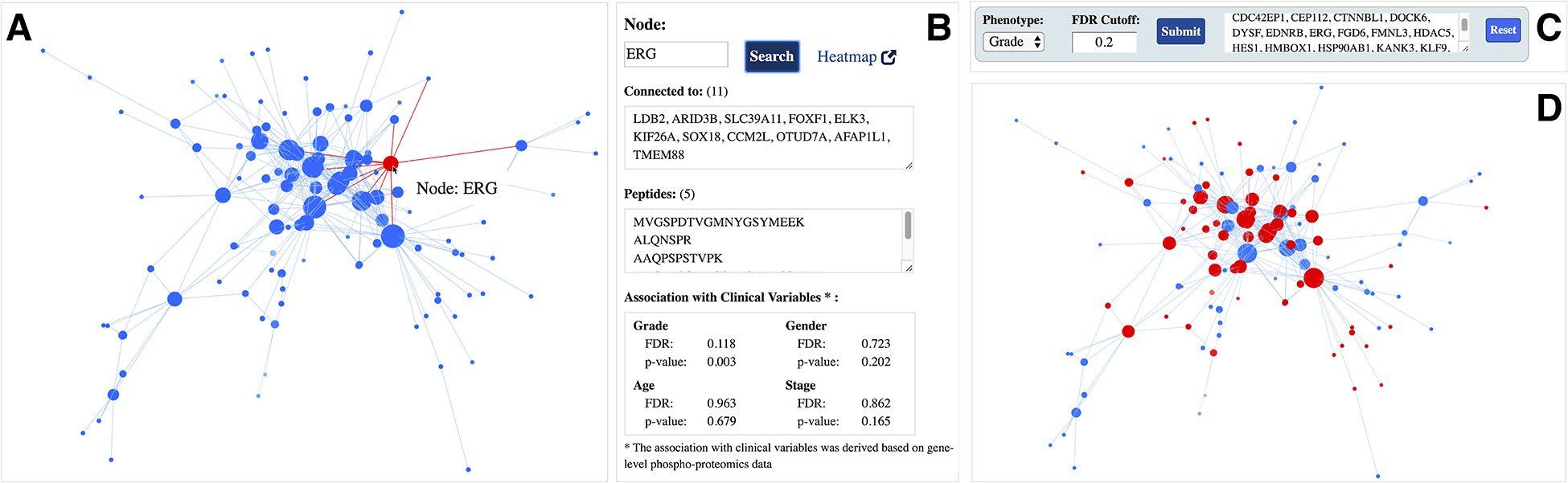
Angiogenesis enriched module network A interactive 3D network exploration panel. Hovering over gene ERG displays its name, highlights it in red, and clicking on the node highlights the connections in red as well; B meta-data information associated with ERG gene. Gene-specific information includes the list of connections (co-expressed genes), mapping peptides, and its association with clinical variables; C phenotype-based search panel. User selects the phenotype of interest and enters the FDR cutoff value. After clicking the Submit button, genes satisfying the criteria are listed in the text-box; D genes satisfying the phenotype-based search are highlighted in red in the interactive network exploration panel.
ProNetView-ccRCC also displays and provides interactive exploration capabilities for any pathway enriched in a network module. For example, Figure 3A displays the interactive table listing the enriched pathways, sorted by their p-values, associated with module 18. To identify the list of genes associated with a certain pathway (e.g. Cell cycle), users can click on the pathway name, which returns the list in a textbox (e.g. BRCA1, CDC20, CDK1, etc.) as well as highlights in the network exploration panel in red (Figure 3B). Clicking on the Reset button returns to the original network display.
Figure 3.
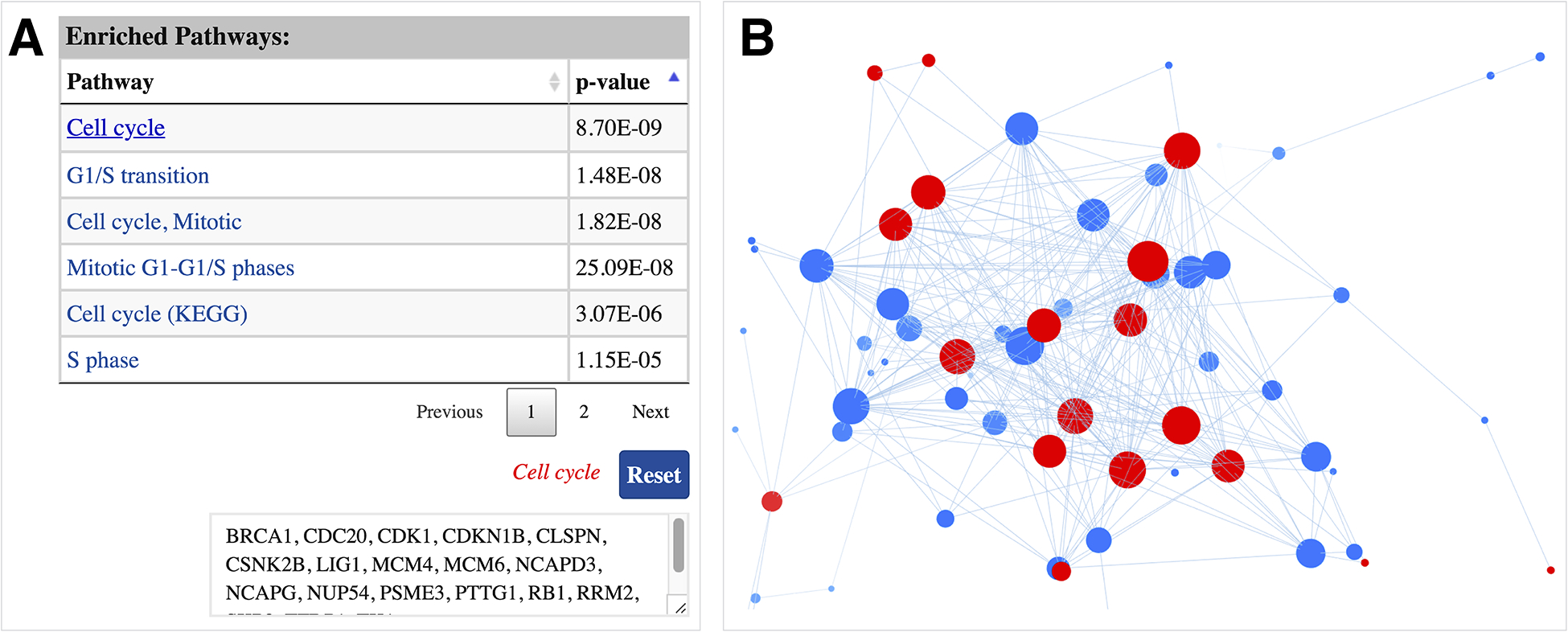
Cell cycle enriched module network A interactive table displaying the list of enriched pathways, sorted by their p-values, associated with the network. List of genes associated with a pathway can be displayed by clicking on the pathway name; B genes associated with the pathway of interest are highlighted in red in the interactive network exploration panel.
In conclusion, ProNetView-ccRCC, provides a free, open access and custom environment for the interactive exploration of CPTAC ccRCC networks and their associated meta-data. The tool runs on any modern web browser without the need for installing any specific plugins or libraries. We anticipate that ProNetView-ccRCC will facilitate researchers from a wide-spectrum computational skill levels to conduct their own analyses on the rich CPTAC ccRCC network data, and share and communicate their results. To this end, users can easily export network visuals as PNG images, as well as download network input and/or clinical variable association data files as text files.
ACKNOWLEDGEMENT
This work was supported in part by the NIH, National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) grant U24CA210993.
Abbreviations:
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- ccRCC
clear cell renal cell carcinoma
- iCAVE
interactome-CAVE
Footnotes
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
REFERENCES
- [1].Moulder R, Bhosale SD, Goodlett DR, Lahesmaa R, Mass Spectrom. Rev 2018, DOI 10.1002/mas.21550. [DOI] [PubMed] [Google Scholar]
- [2].Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI, Nat. Methods 2017, DOI 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL, Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyö D, Ellis MJ, Carr SA, Nature 2016, DOI 10.1038/nature18003. [DOI] [Google Scholar]
- [4].Petralia F, Song WM, Tu Z, Wang P, J. Proteome Res 2016, DOI 10.1021/acs.jproteome.5b00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, Davies SR, Wang S, Wang P, Kinsinger CR, Rivers RC, Rodriguez H, Townsend RR, Ellis MJC, Carr SA, Tabb DL, Coffey RJ, Slebos RJC, Liebler DC, Gillette MA, Klauser KR, Kuhn E, Mani DR, Mertins P, Ketchum KA, Paulovich AG, Whiteaker JR, Edwards NJ, McGarvey PB, Madhavan S, Chan D, Pandey A, Shih IM, Zhang H, Zhang Z, Zhu H, Whiteley GA, Skates SJ, White FM, Levine DA, Boja ES, Hiltke T, Mesri M, Shaw KM, Stein SE, Fenyo D, Liu T, McDermott JE, Payne SH, Rodland KD, Smith RD, Rudnick P, Snyder M, Zhao Y, Chen X, Ransohoff DF, Hoofnagle AN, Sanders ME, Wang Y, Ding L, Nature 2014, DOI 10.1038/nature13438. [DOI] [Google Scholar]
- [6].Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, Sun S, Yang F, Chen L, Wang J, Shah P, Cha SW, Aiyetan P, Woo S, Tian Y, Gritsenko MA, Clauss TR, Choi C, Monroe ME, Thomas S, Nie S, Wu C, Moore RJ, Yu KH, Tabb DL, Fenyö D, Vineet V, Wang Y, Rodriguez H, Boja ES, Hiltke T, Rivers RC, Sokoll L, Zhu H, Shih IM, Cope L, Pandey A, Zhang B, Snyder MP, Levine DA, Smith RD, Chan DW, Rodland KD, Carr SA, Gillette MA, Klauser KR, Kuhn E, Mani DR, Mertins P, Ketchum KA, Thangudu R, Cai S, Oberti M, Paulovich AG, Whiteaker JR, Edwards NJ, McGarvey PB, Madhavan S, Wang P, Whiteley GA, Skates SJ, White FM, Kinsinger CR, Mesri M, Shaw KM, Stein SE, Fenyo D, Rudnick P, Snyder M, Zhao Y, Chen X, Ransohoff DF, Hoofnagle AN, Liebler DC, Sanders ME, Shi Z, Slebos RJC, Zimmerman LJ, Davies SR, Ding L, Ellis MJC, Townsend RR, Cell 2016, DOI 10.1016/j.cell.2016.05.069. [DOI] [Google Scholar]
- [7].Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TSM, Chang HY, Ma W, Huang C, Ricketts CJ, Chen L, Krek A, Li Y, Rykunov D, Li QK, Chen LS, Ozbek U, Vasaikar S, Wu Y, Yoo S, Chowdhury S, Wyczalkowski MA, Ji J, Schnaubelt M, Kong A, Sethuraman S, Avtonomov DM, Ao M, Colaprico A, Cao S, Cho KC, Kalayci S, Ma S, Liu W, Ruggles K, Calinawan A, Gümüş ZH, Geizler D, Kawaler E, Teo GC, Wen B, Zhang Y, Keegan S, Li K, Chen F, Edwards N, Pierorazio PM, Chen XS, Pavlovich CP, Hakimi AA, Brominski G, Hsieh JJ, Antczak A, Omelchenko T, Lubinski J, Wiznerowicz M, Linehan WM, Kinsinger CR, Thiagarajan M, Boja ES, Mesri M, Hiltke T, Robles AI, Rodriguez H, Qian J, Fenyö D, Zhang B, Ding L, Schadt E, Chinnaiyan AM, Zhang Z, Omenn GS, Cieslik M, Chan DW, Nesvizhskii AI, Wang P, Zhang H, Cell 2019, DOI 10.1016/j.cell.2019.10.007. [DOI] [Google Scholar]
- [8].Langfelder P, Horvath S, BMC Bioinformatics 2008, DOI 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Song WM, Zhang B, PLoS Comput. Biol 2015, DOI 10.1371/journal.pcbi.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Petralia F, Wang P, Yang J, Tu Z, in Bioinformatics, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kerren A, Kucher K, Li YF, Schreiber F, PLoS One 2017, DOI 10.1371/journal.pone.0187341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kalaycı S, Demiralp Ç, Gümüş ZH, arXiv 2018, arXiv:1810.02391. [Google Scholar]
- [13].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Genome Res. 2003, 13, 2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bastian M, Heymann S, Jacomy M, Third Int. AAAI Conf. Weblogs Soc. Media 2009, 361. [Google Scholar]
- [15].Su G, Kuchinsky A, Morris JH, States DJ, Meng F, Bioinformatics 2010, DOI 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liluashvili V, Kalayci S, Fluder E, Wilson M, Gabow A, Gümüş ZH, Gigascience 2017, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kalayci S, Gümüş ZH, Curr. Protoc. Bioinforma 2018, 61, 8.27.1. [DOI] [PubMed] [Google Scholar]
- [18].Calinawan AP, Song X, Ji J, Dhanasekaran SM, Petralia F, Wang P, Reva B, bioRxiv 2020, 2020 02.05.935650. [DOI] [PMC free article] [PubMed] [Google Scholar]