

Abstract
Gemcitabine is an intravenously administered anti-cancer nucleoside analogue. Systemic exposure following oral administration of gemcitabine is limited by extensive first-pass metabolism via cytidine deaminase (CDA) and potentially by saturation of nucleoside transporter-mediated intestinal uptake. An amino acid ester prodrug of gemcitabine, 5’-l-valylgemcitabine (V-Gem), was previously shown to be a substrate of the intestinally expressed peptide transporter 1 (PEPT1) and stable against CDA-mediated metabolism. However, preliminary studies did not evaluate the in vivo oral performance of V-Gem as compared to parent drug. In the present study, we evaluated the pharmacokinetics and in vivo oral absorption of gemcitabine and V-Gem following intravenous and oral administrations in mice. These studies revealed that V-Gem undergoes rapid systemic elimination (half-life < 1 min) and has a low oral bioavailability (< 1%). Most importantly, the systemic exposure of gemcitabine was not different following oral administration of equimolar doses of gemcitabine (gemcitabine bioavailability of 18.3%) and V-Gem (gemcitabine bioavailability of 16.7%). Single-pass intestinal perfusions with portal blood sampling in mice revealed that V-Gem undergoes extensive activation in intestinal epithelial cells and that gemcitabine undergoes first-pass metabolism in intestinal epithelial cells. Thus, formulation of gemcitabine as the prodrug V-Gem does not increase systemic gemcitabine exposure following oral dosing, due, in part, to the instability of V-Gem in intestinal epithelial cells.
Keywords: Bioavailability, Gemcitabine, PEPT1, Prodrug
Graphical Abstract
1. Introduction
Gemcitabine (2’,2’-difluoro-2’-deoxycytidine; dFdC) is a nucleoside analogue approved for use in the treatment of pancreatic, non-small cell lung, ovarian, and breast cancer [1]. It is also used off-label for treatment of other cancer types such as biliary tract and bladder cancer [2–4]. Gemcitabine exerts its anti-cancer activity through incorporation of gemcitabine triphosphate into growing DNA strands, inhibiting DNA synthesis [5] and leading to apoptosis [6]. Various self-potentiating mechanisms, including gemcitabine diphosphate inhibition of ribonucleotide reductase [7], have been reported to augment gemcitabine cytotoxicity [8]. Gemcitabine is rapidly cleared from plasma (half-life = 42 – 94 min), mainly via cytidine deaminase (CDA)-mediated deamination of gemcitabine to 2’,2’-difluoro-2’-deoxyuridine (dFdU) [9]. The in vivo activity of dFdU is currently unclear, although some work has suggested it may contribute to gemcitabine cytotoxicity [10, 11] and radiosensitization [12].
Gemcitabine has a low oral bioavailability of about 10% [13] and is, thus, administered via intravenous infusion, typically once per week at a dose of 1000 – 1250 mg/m2 [1]. However, oral administration is generally preferred as it is more patient friendly, less invasive, and reduces the costs and complications associated with intravenous drug administration. Furthermore, oral gemcitabine administration would allow for greater flexibility in designing dosing schedules, enabling both metronomic gemcitabine dosing (i.e., frequent low dose administration) and dosing which replicates gemcitabine pharmacokinetics following a prolonged intravenous infusion. There is evidence that such dosing schedules may lead to improvements in efficacy and/or reductions in toxicity [14–17].
Given the advantages of oral gemcitabine administration, much work has been dedicated to understanding the mechanistic basis of gemcitabine’s low oral bioavailability. Recent work using in situ intestinal perfusions in mice demonstrated that gemcitabine intestinal uptake is saturable and driven almost exclusively by nucleoside transporters (NT), and that gemcitabine has high effective permeability in the intestine, implying that first-pass metabolism drives gemcitabine’s low oral bioavailability [18]. This conclusion is further supported by the observation that following oral administration, gemcitabine undergoes extensive first-pass metabolism via CDA, forming dFdU [13]. Moreover, gemcitabine intestinal uptake is rapidly saturated with increasing concentration, which may further limit gemcitabine bioavailability following oral administration of larger and perhaps more clinically relevant doses [18].
In hopes of both increasing gemcitabine oral bioavailability by decreasing first-pass metabolism and enabling the oral administration of larger gemcitabine doses through reducing the saturability of gemcitabine intestinal absorption, several peptide transporter 1 (PEPT1)-targeted gemcitabine prodrugs were previously synthesized [19]. PEPT1 (SLC15A1) is a transmembrane transporter that is extensively expressed on the apical membrane of intestinal enterocytes [20]. Given that PEPT1 generally functions as a high-capacity, low-affinity intestinal transporter, it is frequently targeted to increase intestinal drug uptake via administration of a PEPT1-targeted prodrug. These prodrugs, often formed by addition of an amino acid to the parent molecule via an ester bond, undergo PEPT1-mediated uptake and are subsequently activated, releasing the active parent compound [21]. One such PEPT1-targeted gemcitabine prodrug, 5’-l-valyl-gemcitabine (V-Gem), was generated by linking l-valine to gemcitabine via an ester bond [19]. Previous in vitro work confirmed that V-Gem was stable against CDA-mediated deamination [19, 22] and was a PEPT1 substrate [19, 23].
With this in mind, the primary objective of this study was to characterize the in vivo pharmacokinetics of gemcitabine and V-Gem following intravenous and oral administrations in mice. The secondary objective, using in situ intestinal perfusions in mice, was to evaluate the ability of V-Gem to reduce first-pass metabolism and increase intestinal drug absorption, relative to gemcitabine.
2. Materials and methods
2.1. Chemicals
Gemcitabine (Gem), high-performance liquid chromatography (HPLC) grade acetonitrile, and HPLC grade trifluoroacetic acid (TFA) were purchased from Thermo Fisher Scientific (Waltham, MA). Tetrahydrouridine (THU) and the deaminated gemcitabine metabolite 2’,2’-difluorodeoxyuridine (dFdU) were purchased from Sigma-Aldrich (St. Louis, MO). 13C,15N2– Gem and 13C,15N2-dFdU internal standards were purchased from Toronto Research Chemicals (Toronto, Canada). The 5’-l-valyl-gemcitabine prodrug (V-Gem) was synthesized by AAPharmaSyn, LLC (Ann Arbor, MI). All other chemicals were obtained from standard commercial sources. See Figure 1 for the chemical structures of Gem, dFdU, and V-Gem.
Figure 1.
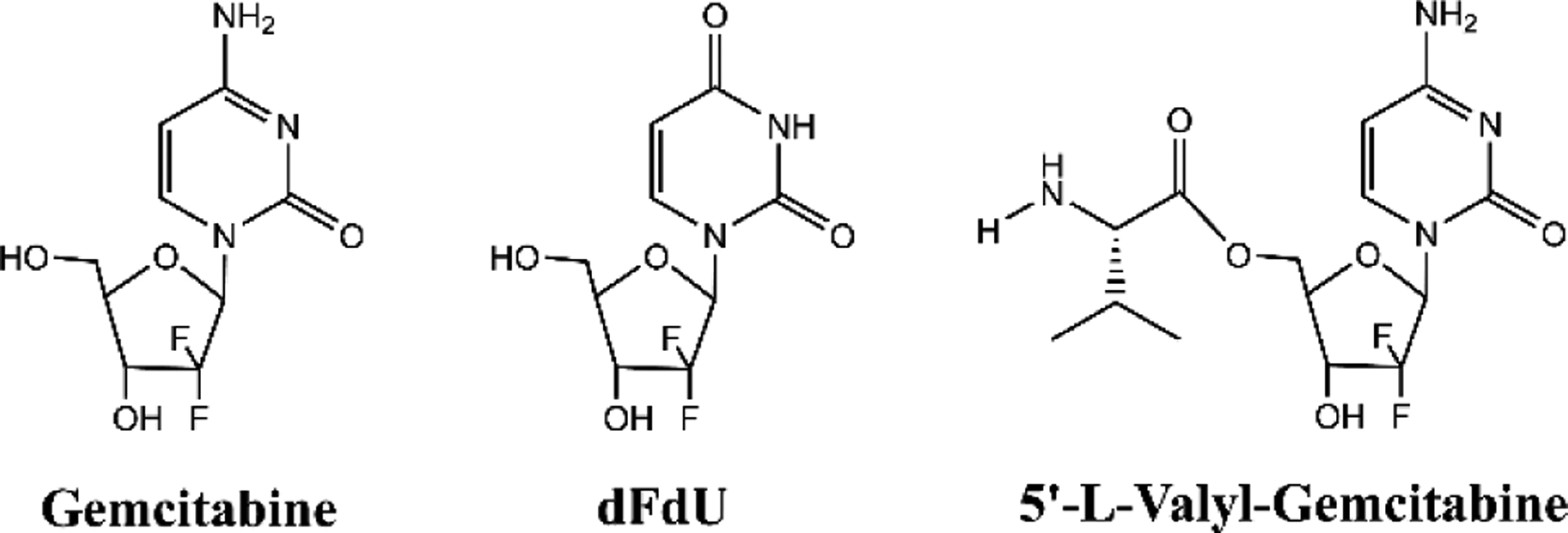
Structures of gemcitabine, the deaminated gemcitabine metabolite dFdU, and the gemcitabine prodrug 5’-l-valyl-gemcitabine
2.2. Animals
Studies were performed on 8- to 12-week old gender-matched C57BL/6 mice purchased from Charles River Laboratories (Wilmington, MA). The mice were housed in a temperature-controlled room with 12-hour light/dark cycles and were provided a standard diet with ad libitum access to water (Unit for Laboratory Animal Medicine, University of Michigan, Ann Arbor, MI). All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
2.3. Intravenous and oral pharmacokinetic studies of Gem and V-Gem
For intravenous studies, mice were administered 76 nmol/g body weight of Gem or V-Gem (in 0.1 mL saline per 25 g body weight) via bolus tail vein injection (n=4). For oral studies, mice were fasted overnight (~ 16 hr) and subsequently administered 228 nmol/g body weight of Gem or V-Gem (in 0.2 mL water per 25 g body weight) via oral gavage (n=4). Following both intravenous and oral dosing, blood samples were collected via distal tail transection at 2, 5, 15, 30, 45, 60, 90, 120, 180, 240, 300, and 360 min. At each sampling time point, approximately 20 μL of blood was collected with a pipette, added to a 0.2 mL PCR tube containing EDTA-K3 and THU (the latter included to prevent ex vivo Gem deamination by CDA), and centrifuged at 3000 rpm for 4 min at 4C. A 5 μL plasma aliquot was then mixed with 200 μL ice-cold acetonitrile, containing 0.1 μM 13C,15N2-Gem and 0.25 μM 13C,15N2-dFdU as internal standards (IS), and placed in the −20°C freezer until all samples for the given mouse were collected. Samples were then centrifuged at 17,000 g for 10 min at 4 °C and a 60 μL aliquot of the supernatant stored at −80°C until analysis.
Afterwards, the supernatants were dried in a SpeedVac concentrator for two hours (with heating at 45 °C during the first hour) and reconstituted in 90 μL water (plus 0.1% formic acid). The reconstituted samples were centrifuged at 17,000 g for 10 min at 4°C and the supernatants were analyzed via liquid chromatography-tandem mass spectrometry (LC-MS/MS), as described below.
2.4. LC-MS/MS assay conditions for pharmacokinetic study samples
The concentrations of Gem, dFdU, and V-Gem were determined in mouse plasma samples using a novel LC-MS/MS method utilizing a Shimadzu HPLC system (Shimazu, Kyoto, Japan) coupled with an Applied Biosystems API 4000 triple quadrupole/linear ion trap (QTRAP) mass spectrometer (Foster City, CA). Following plasma sample collection and preparation (see above), 8 μL of supernatant was injected onto an Agilent Poroshell 120 EC-C18 column (2.7 μm, 2.1 × 50 mm, Santa Clara, CA). Analyte separation was achieved using a gradient elution method combining water (plus 0.1% formic acid) and acetonitrile (plus 0.1% formic acid) at a flow rate of 0.35 mL/min. The gradient was initiated and held at 1% acetonitrile for 0.5 min, increased to 90% acetonitrile linearly from 0.5 – 1.0 min, held at 90% acetonitrile from 1.0 – 3.0 min, decreased to 1% acetonitrile linearly from 3.0 – 3.1 min, and held at 1% acetonitrile until the end of the run (5.1 min). To minimize and monitor for carryover, injections of blank water were performed between all sample injections.
The MS was operated in a positive multiple reaction monitoring (MRM) mode using turbo electrospray ionization. The source-dependent parameters were set as follows: curtain gas 30 psi, ionspray voltage 5500 V, temperature 500 °C, gas1 50 psi and gas2 50 psi. The analyte-specific MS parameters are summarized in Table 1.
Table 1.
Summary of analyte-specific mass spectrometry parameters.
| Compound | Parent Ion (m/z) | Product Ion (m/z) | Declustering Potential (V) | Entrance Potential (V) | Collision Energy (V) | Collision Cell Exit Potential (V) |
|---|---|---|---|---|---|---|
| Gem | 264.0 | 112.0 | 60 | 12 | 26 | 13 |
| dFdU | 265.0 | 113.0 | 60 | 12 | 22 | 13 |
| V-Gem | 363.1 | 264.0 | 6 | 8 | 22 | 14 |
| 13C,15N2-Gem (IS) | 267.0 | 115.0 | 60 | 12 | 26 | 13 |
| 13C,15N2-dFdU (IS) | 268.0 | 116.0 | 60 | 12 | 22 | 13 |
Gem, gemcitabine; dFdU, 2’,2’-difluorodeoxyuridine; V-Gem, 5’-l-valyl-gemcitabine; IS, internal standard.
2.5. Validation of LC-MS/MS assay for pharmacokinetic study samples
Calibrator, quality control (QC), and stability samples were prepared by performing a 10x dilution of aqueous drug solutions into blank mouse plasma. For all three analytes (i.e., Gem, dFdU, and V-Gem), calibrator samples were prepared at final concentrations of 0.05, 0.1, 0.25, 1, 5, 20, 50, and 100 μM, QC samples at 0.1, 5, and 50 μM, and stability samples at 0.1, 2, 5, 50, and 100 μM. The calibrator, QC, and stability samples were processed for analysis identically with plasma samples collected from the pharmacokinetic study (i.e., plasma quenched in acetonitrile containing IS, supernatant dried, dried sample reconstituted in water, supernatant analyzed).
Selectivity of the assay was evaluated by injecting blank plasma samples prepared from multiple mice, blank plasma spiked with IS, and blank plasma spiked with the three analytes at 0.05 μM to assess potential interferences. Linearity was evaluated by developing standard curves ranging from 0.05 – 100 μM for all three analytes. 13C,15N2-dFdU was used as the IS for dFdU and 13C,15N2-Gem was used as the IS for both Gem and V-Gem. Assay accuracy and precision were determined by analyzing QC samples in triplicate in three independent runs. Finally, sample stability was assessed during long-term storage by analyzing stability samples (2, 100 μM) after storage at −80 °C for 30 days. Autosampler stability was assessed by analyzing stability samples (0.1, 5, and 50 μM) after storage in the autosampler (4°C) for 24 hr.
2.6. In situ single-pass intestinal perfusions of Gem and V-Gem
Intestinal perfusions in mice were performed as previously described [18, 24]. In brief, mice were fasted overnight (~ 16 hr) with free access to water and subsequently anesthetized with sodium pentobarbital (40 – 60 mg/kg intraperitoneal). Their abdominal cavity was then exposed and an 8 cm long jejunal segment, beginning 2 cm distal from the Ligament of Treitz, was isolated and both ends cannulated. The proximal canula was attached to a syringe containing pH 6.0 perfusion solution and positioned in a perfusion pump (PHD Ultra, Harvard Apparatus, South Natick, MA). The distal canula was connected to a collection vial.
The perfusion solution contained 145 mM NaCl, 5 mM morpholinoethanesulfonic acid (MES), 5 mM glucose, 3 mM KCl, 1 mM CaCl2, 1 mM NaH2PO4, 0.5 mM MgCl2, and either 100 μM Gem or 100 μM V-Gem. To assess drug stability in the intestinal lumen, the perfusion solution containing Gem or V-Gem was perfused through the cannulated segment at 0.1 ml/min for 30 min to achieve steady-state, and then for an additional 60 min with outlet sample collections every 10 min. The concentrations of Gem, dFdU, and V-Gem were determined in inlet and outlet samples using a previously described UPLC assay [18], revalidated for the quantification of an additional analyte, V-Gem (validation data not shown here).
Perfusion studies to explore drug stability in intestinal epithelial cells were also performed by perfusing perfusion solution, containing 10 mM Gem or 10 mM V-Gem, through the cannulated jejunal segment at 0.1 mL/min for 5 min and then immediately collecting a portal blood sample (~ 200 ul). The portal blood sample was collected in a 1.5 mL microcentrifuge tube containing EDTA-K3 and THU, and immediately centrifuged at 3000 g for 4 minutes at 4 °C. A 50 μl aliquot of plasma was then mixed with 200 μl of ice-cold acetonitrile (containing caffeine as an IS) and stored at −20 °C until all samples were collected. Samples were then centrifuged at 17,000 g for 10 min at 4 °C and 60 μL of the supernatant collected. The supernatants were then dried in a SpeedVac concentrator for two hours (with heating at 45 °C during the first hour) and reconstituted in 80 μL water (plus 0.1% TFA). The reconstituted samples were centrifuged at 17,000 g for 10 min at 4°C and the supernatants analyzed via UPLC, as described below.
2.7. UPLC assay for analysis of portal blood samples
The concentrations of Gem, dFdU, and V-Gem were determined in portal plasma samples using a Waters Acquity H-Class UPLC system (Milford, MA) coupled with a photodiode array detector. Following portal plasma collection and preparation (see above), 15 μl of supernatant was injected onto a 40°C Acquity HSS T3 column (2.1 × 100 mm), fitted with an HSS T3 VanGuard precolumn (2.1 × 5 mm). Analyte separation was achieved using a gradient elution method combining water (plus 0.1% TFA) and acetonitrile (plus 0.1% TFA) at a flow rate of 0.4 ml/min. The gradient was initiated at 0% acetonitrile, increased to 6% acetonitrile linearly from 0 – 3 min, increased to 15% acetonitrile linearly from 3 – 5 min, increased to 80% acetonitrile linearly from 5 – 7 min, held at 80% acetonitrile from 7 – 8 min, returned to 0% acetonitrile linearly from 8 – 9 min, and held at 0% acetonitrile until the end of the run (12 min). Calibration curves ranging from the lower limit of quantiation to the upper limit of quantitation were generated for V-Gem (0.025 – 5 μM, detection wavelength = 275 nm), Gem (0.1 – 50 μM, detection wavelength = 284 nm), and dFdU (0.1 – 50 μM, detection wavelength = 260 nm) using caffeine (detection wavelength = 275 nm) as the IS. The method was validated with respect to selectivity, showing no endogenous compound interference with analyte detection, linearity (r2 > 0.991), accuracy (average bias of triplicated QC samples < 12%), and precision (relative standard deviation of triplicated QC samples < 7%).
2.8. Data analysis
Noncompartmental analysis (NCA) of plasma-concentration time profiles after oral and intravenous dosing was performed using Phoenix WinNonlin 8.2 (Certara, St. Louis, MO). All pharmacokinetic parameters were reported as geometric mean (geometric CV%) except for Tmax, which was reported as median (min – max). The bioavailability of Gem following oral Gem and V-Gem administrations was calculated as and , respectively. When performing calculations with AUC values, AUCinf values were used unless the percent extrapolated was > 25%, in which case AUC0–6 hr values were used. All other data were reported as arithmetic mean ± SE, unless otherwise noted. When comparing two groups, statistical differences were evaluated using an unpaired t-test (Prism version 7, GraphPad Software, La Jolla, California). A p value ≤ 0.05 was considered significant.
3. Results
3.1. LC-MS/MS assay validation for use in pharmacokinetic studies
Selectivity of the assay was demonstrated by injecting blank plasma samples from multiple mice, blank plasma spiked with IS, and blank plasma spiked with the analytes and IS. No significant interference of co-eluting peaks on analysis of the three analytes (i.e., Gem, dFdU, and V-Gem) and two IS (i.e., 13C,15N2– Gem and 13C,15N2– dFdU) was observed. Next, calibration curves ranging from 0.05 – 100 μM were developed for all analytes and shown to be linear (r2 > 0.990). As shown in Table 2, the method showed excellent accuracy and precision in quantification of all analytes at low, medium, and high concentrations. Finally, sample stability was demonstrated for all analytes during long-term (30 days) storage at −80°C (recovery range: 102 – 111 %) and during short-term (24 hr) storage on the autosampler at 4°C (recovery range: 96.2 – 113 %).
Table 2.
Summary of intra-day and inter-day accuracy and precision of the LC-MS/MS assay for quantification of Gem, dFdU, and V-Gem at low (0.1 μM), medium (5 μM), and high (50 μM) concentrations in mouse plasma.
| Intra-day (n=3) | Inter-day (n=9) | ||||
|---|---|---|---|---|---|
| Analyte | Concentration (μM) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| Gem | 0.1 / 5 / 50 | 104 / 108 / 102 | 3.2 / 2.6 / 6.7 | 94 / 109 / 103 | 9.3 / 8.4 / 9.4 |
| dFdU | 0.1 / 5 / 50 | 104 / 108 / 112 | 4.1 / 3.7 / 4.5 | 114 / 106 / 102 | 6.1 / 4.5 / 4.0 |
| V-Gem | 0.1 / 5 / 50 | 107 / 91.1 / 102 | 2.9 / 12.3 / 6.4 | 112 / 90 / 108 | 11.3 / 10.4 / 4.0 |
Gem, gemcitabine; dFdU, 2’,2’-difluorodeoxyuridine; V-Gem, 5’-l-valyl-gemcitabine.
3.2. Pharmacokinetics following intravenous and oral Gem administrations
The mean plasma concentration-time profiles of Gem and dFdU are shown following intravenous administration of 76 nmol Gem/g body weight (Figure 2) and oral administration of 228 nmol Gem/g body weight (Figure 3). Pharmacokinetic parameters for Gem and dFdU are summarized in Table 3. Following intravenous administration, Gem reached an initial concentration (C0 = Cmax) of 67.2 μM and was converted to dFdU, with the Tmax of dFdU occurring at 30 min. Following oral administration, Gem was rapidly absorbed reaching a maximum concentration (Cmax) of 9.8 μM at a Tmax of 30 min. The terminal half-life (T1/2) of Gem following intravenous Gem administration (42.5 min) was not different than the T1/2 after oral Gem administration (29.8 min) (p = 0.258). The T1/2 of dFdU was shorter following intravenous Gem administration (106 min), as compared to the T1/2 after oral Gem administration (210 min) (p = 0.050). Route-dependent differences were observed in the systemic exposure (i.e., AUC) ratios for dFdU/Gem, where the ratio was 1.5 after intravenous Gem dosing but 6.9 after oral Gem dosing.
Figure 2.
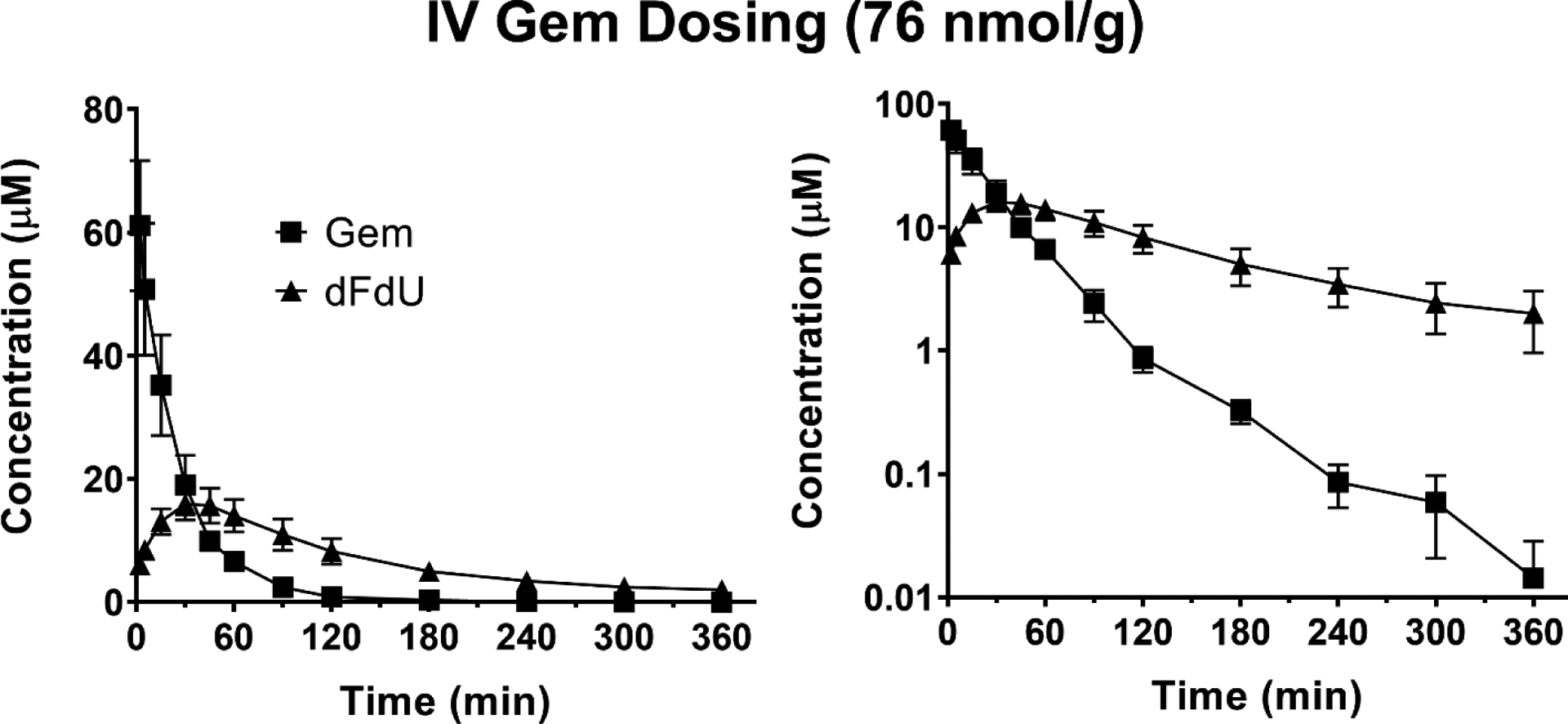
Mean plasma concentration-time profiles of Gem and dFdU following intravenous (IV) administration of 76 nmol Gem/g body weight in mice. Data are expressed as mean ± SE (n=4) with the y-axis displayed on linear (left panel) and logarithmic (right panel) scales.
Figure 3.
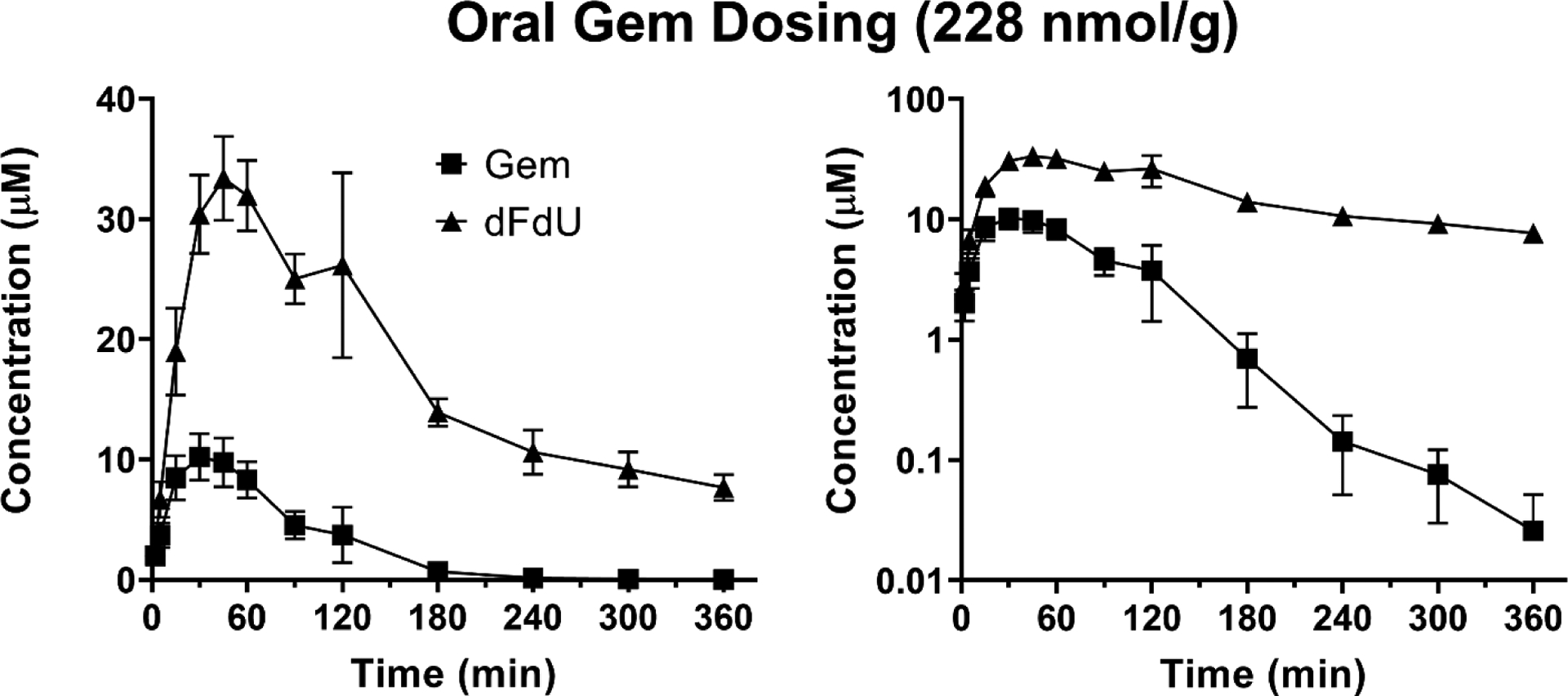
Mean plasma concentration-time profiles of Gem and dFdU following oral administration of 228 nmol Gem/g body weight in mice. Data are expressed as mean ± SE (n=4) with the y-axis displayed on linear (left panel) and logarithmic (right panel) scales.
Table 3.
Pharmacokinetic parameters of Gem, dFdU, and V-Gem following IV (76 nmol/g) and PO (228 nmol/g) administrations of Gem and V-Gem in mice (n = 4).
| Treatment | Parameter | Analyte | ||
|---|---|---|---|---|
| Gemcitabine | dFdU | Prodrug | ||
| Gem IV (76 nmol/g) | AUC0_6 hr (min × μM) | 1621 (38%) | 2218 (54%) | - |
| AUCinf (min × μM) | 1628 (38%) | 2484 (65%) | - | |
| % Extrapolated | 0.4 (40%) | 8.2 (88%) | - | |
| T1/2 (min) | 42.5 (44%) | 106 (32%) | - | |
| Tmax (min) | - | 30 (30 – 45) | - | |
| Cmax (μM) | 67.2 (30%)† | 15.5 (34%) | - | |
| CL (mL/hr/g) | 2.8 (38%) | - | - | |
| Vss (mL/g) | 1.4 (44%) | - | - | |
| Gem PO (228 nmol/g) | AUC0–6 hr (min × μM) | 887 (55%) | 6091 (15%) | - |
| AUCinf (min × μM) | 893 (55%) | 8613 (22%) | - | |
| % Extrapolated | 0.6 (55%) | 26.2 (52%) | - | |
| T1/2 (min) | 29.8 (41%) | 210 (41%) | - | |
| Tmax (min) | 30 (15 – 45) | 45 (30 – 120) | - | |
| Cmax (μM) | 9.8 (42%) | 35.3 (30%) | - | |
| Foral | 18.3% | - | - | |
| V-Gem IV (76 nmol/g) | AUC0–6 hr (min × μM) | 794 (23%) | 2017 (49%) | 103 (101%) |
| AUCinf (min × μM) | 801 (22%) | 2345 (61%) | 106 (98%) | |
| % Extrapolated T1/2 (min) | 0.8 (92%) 46.2 (34%) | 11.3 (84%) 128 (38%) | 1.1 (253%) 3.7 (33%) | |
| Tmax (min) | 2 (2 – 2) | 22.5 (15 – 30) | - | |
| Cmax (μM) | 41.6 (26%) | 13.9 (31%) | 32.7 (139%)† | |
| CL (mL/hr/g) | - | - | 43.1 (98%) | |
| Vss (mL/g) | - | - | 2.4 (145%) | |
| V-Gem PO (228 nmol/g) | AUC0–6 hr (min × μM) | 806 (27%) | 7732 (19%) | 1.9 (113%) |
| AUCinf (min × μM) | 814 (27%) 0.9 | 11,125 (24%) 30.2 | CND | |
| % Extrapolated | (17%) | (13%) | CND | |
| T1/2 (min) | 39.1 (25%) | 216 (9%) | CND | |
| Tmax (min) | 15 (5 – 30) | 30 (15 – 30) | 3.5 (2 – 5) | |
| Cmax (μM) | 11.1 (24%) | 47.9 (6%) | 0.2 (101%) | |
| Foral | 16.7% | - | 0.6% | |
Parameters are reported as geometric mean (geometric CV%), except for Tmax, which is reported as median (min – max), and oral bioavailability (Foral), which is reported as the dose normalized ratio (oral/IV) of the geometric mean AUCinf.
C0 reported for Cmax. CND, could not be determined; IV, intravenous; PO, oral; Gem, gemcitabine; dFdU, 2’,2’-difluorodeoxyuridine; V-Gem, 5’-l-valyl-gemcitabine.
3.3. Pharmacokinetics following intravenous and oral V-Gem administrations
The mean plasma concentration-time profiles of Gem, dFdU, and V-Gem are shown following intravenous administration of 76 nmol V-Gem/g body weight (Figure 4) and oral administration of 228 nmol V-Gem/g body weight (Figure 5). Pharmacokinetic parameters for Gem, dFdU, and V-Gem are summarized in Table 3. Following intravenous V-Gem administration, V-Gem reached an initial concentration (C0 = Cmax) of 32.7 μM and was rapidly eliminated with a T1/2 of 3.7 min. Gem was rapidly formed with a Tmax occurring at the first sampling time point (2 min) in all mice. Systemic exposure of prodrug following intravenous V-Gem administration was quite small relative to Gem (V-Gem to Gem AUC ratio = 0.13) and dFdU (V-Gem to dFdU AUC ratio = 0.05). Following oral V-Gem administration, the systemic exposure of prodrug was negligible (V-Gem oral to intravenous dose adjusted AUC ratio = 0.006) and Gem was rapidly formed, reaching a Cmax of 11.1 μM with a Tmax of 15 min. Again, the T1/2 of Gem following intravenous V-Gem administration (46.2 min) was not different than the T1/2 following oral V-Gem administration (39.1 min) (p = 0.406). However, the T1/2 of dFdU was shorter following intravenous V-Gem administration (128 min) as compared to oral V-Gem administration (216 min) (p < 0.001). Similar to Gem dosing, the average dFdU to Gem exposure ratio increased from 2.9 to 9.6 following intravenous and oral V-Gem dosing, respectively.
Figure 4.
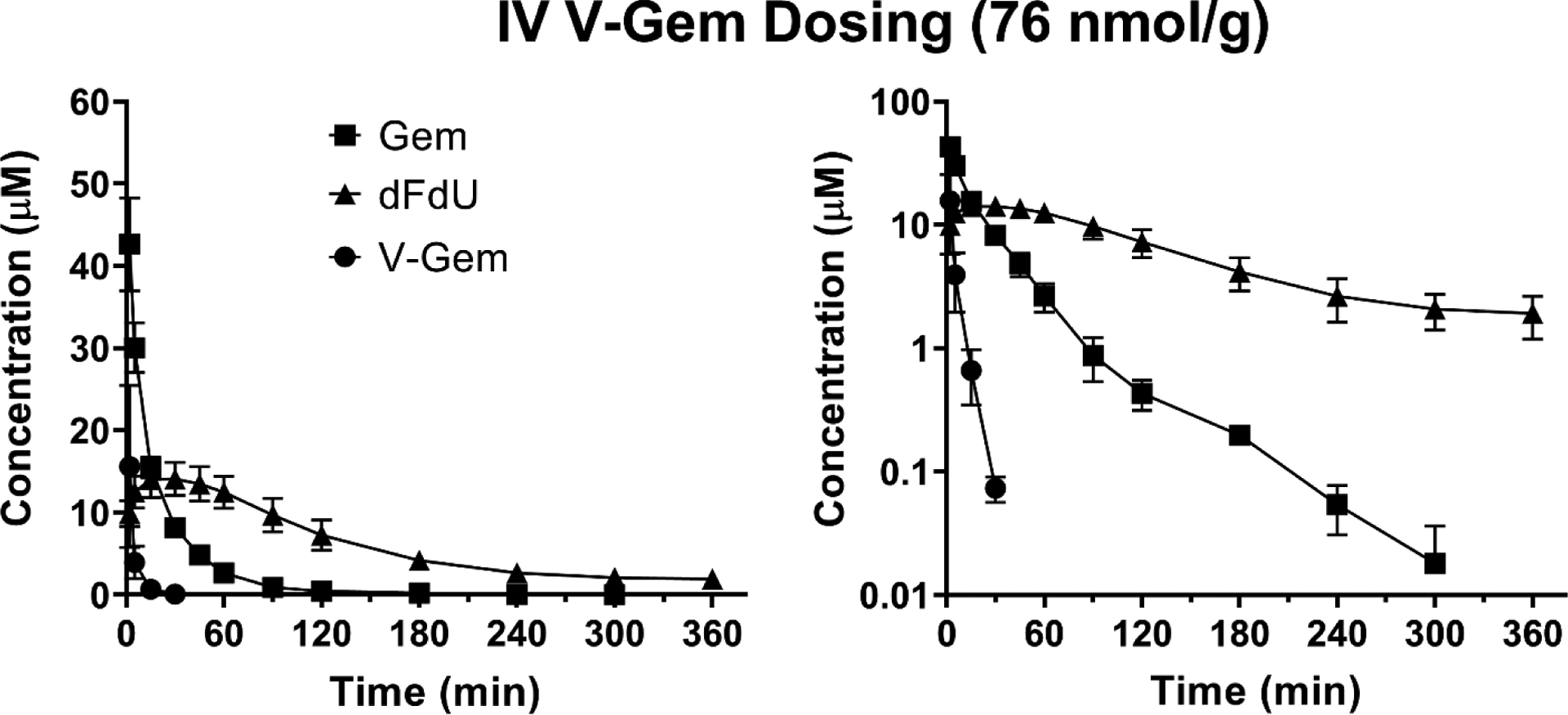
Mean plasma concentration-time profiles of Gem, dFdU, and V-Gem following intravenous (IV) administration of 76 nmol V-Gem/g body weight in mice. Data are expressed as mean ± SE (n=4) with the y-axis displayed on linear (left panel) and logarithmic (right panel) scales.
Figure 5.
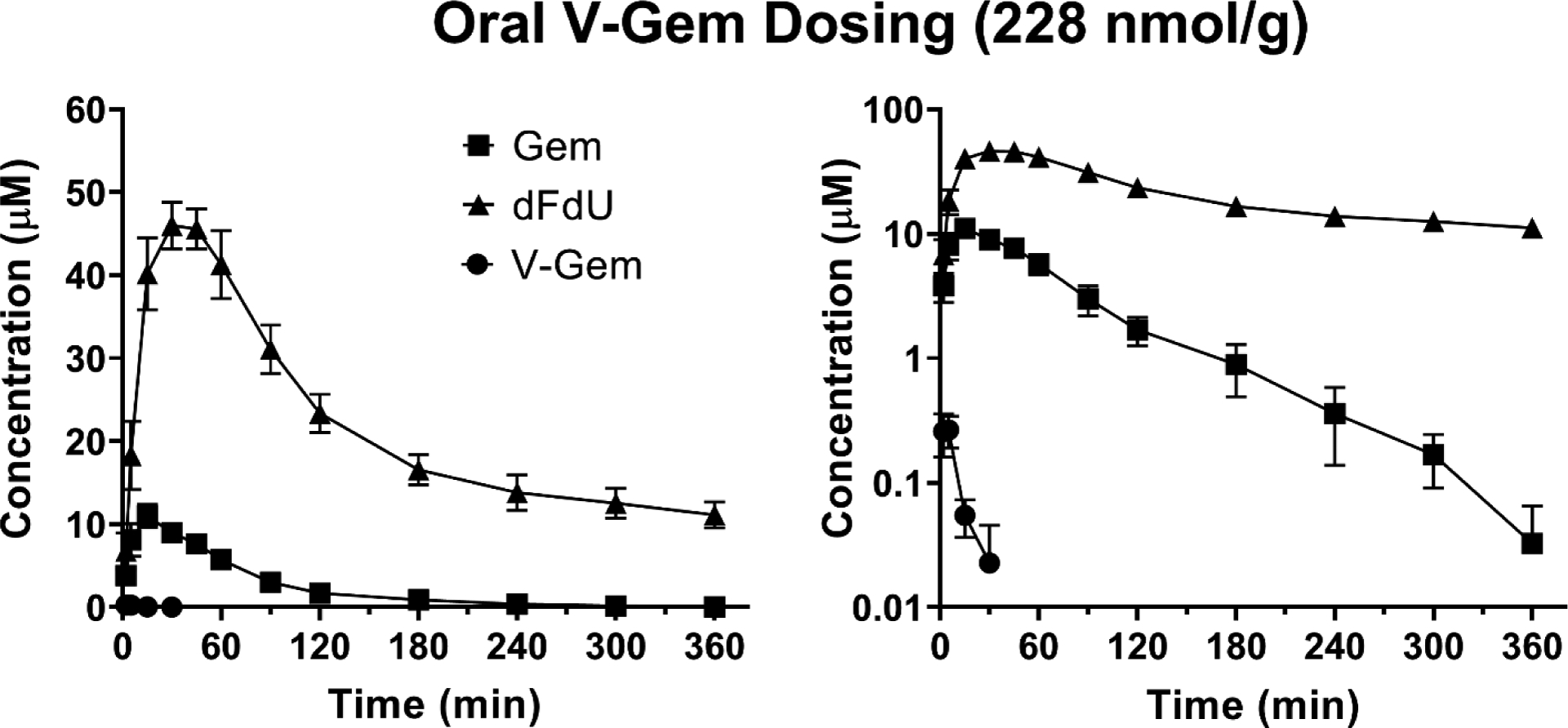
Mean plasma concentration-time profiles of Gem, dFdU, and V-Gem following oral administration of 228 nmol V-Gem/g body weight in mice. Data are expressed as mean ± SE (n=4) with the y-axis displayed on linear (left panel) and logarithmic (right panel) scales.
3.4. V-Gem activation following intravenous V-Gem administration
As observed in Table 3, mean systemic Gem exposure was ≈ 50% lower following V-Gem intravenous administration (801 min × μM) relative to Gem intravenous administration (1628 min × μM) (p < 0.05). In contrast, mean systemic dFdU exposure was not different following V-Gem intravenous administration (2345 min × μM) and Gem intravenous administration (2484 min × μM) (p = 0.848).
3.5. Comparing systemic Gem and dFdU exposure following oral Gem and V-Gem administrations
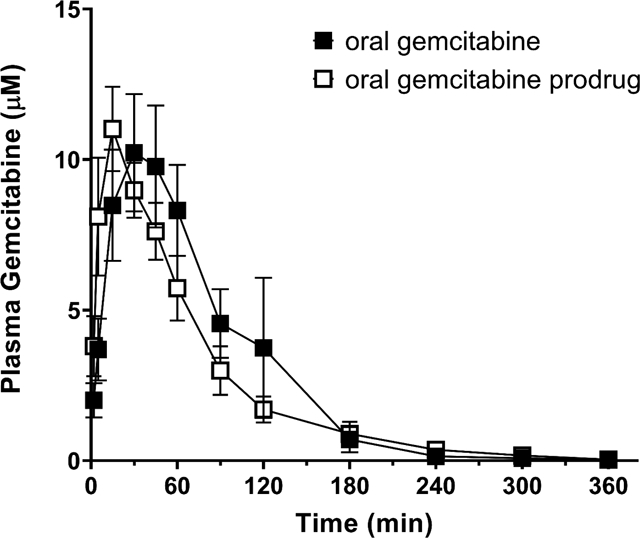
As observed in Figure 6A, the mean plasma concentration-time profiles of Gem following oral Gem and V-Gem administrations are very similar. In fact, no statistically significant difference in Gem exposure exists following oral Gem administration (893 min × μM) and oral V-Gem administration (814 min × μM) (p = 0.594). Moreover, the bioavailability of Gem following oral Gem and V-Gem administrations are 18.3% and 16.7%, respectively. As shown in Figure 6B, the concentration-time profiles of dFdU are also similar following oral Gem and V-Gem administrations. Additionally, there is no statistically significant difference in dFdU exposure (p = 0.084).
Figure 6.
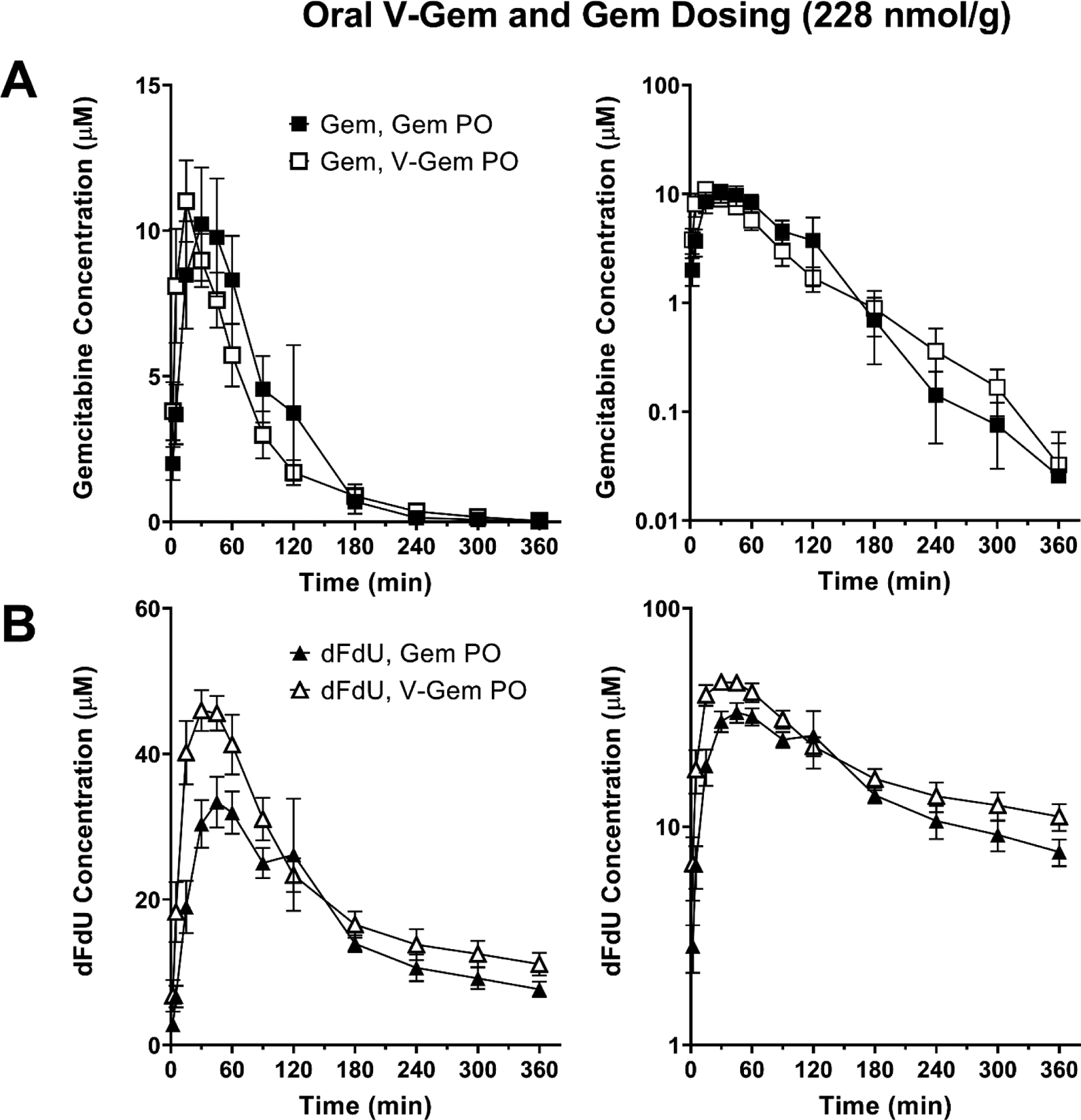
Mean plasma concentration-time profiles following oral (PO) administration of 228 nmol/g body weight Gem and V-Gem for (A) Gem and (B) dFdU. Data are expressed as mean ± SE (n=4) with the y-axis displayed on linear (left panel) and logarithmic (right panel) scales.
3.6. Intestinal stability and absorption of Gem and V-Gem
Single-pass intestinal perfusions of Gem and V-Gem, with analysis of perfusion outlet samples, showed that < 1% of perfused Gem was found in outlet samples as dFdU and < 10% of perfused V-Gem was found in outlet samples as Gem. Furthermore, as shown in Figure 7, portal plasma concentrations of Gem, dFdU, and V-Gem were determined following 5 min perfusions of Gem and V-Gem. Following Gem perfusion, dFdU accounted for about 30% of the total drug found in portal plasma. Following V-Gem perfusion, V-Gem accounted for < 12% of the total drug found in portal plasma. Furthermore, total drug concentrations (i.e., Gem + dFdU + V-Gem) in portal plasma were no different following perfusions of either Gem or V-Gem (p = 0.608).
Figure 7.
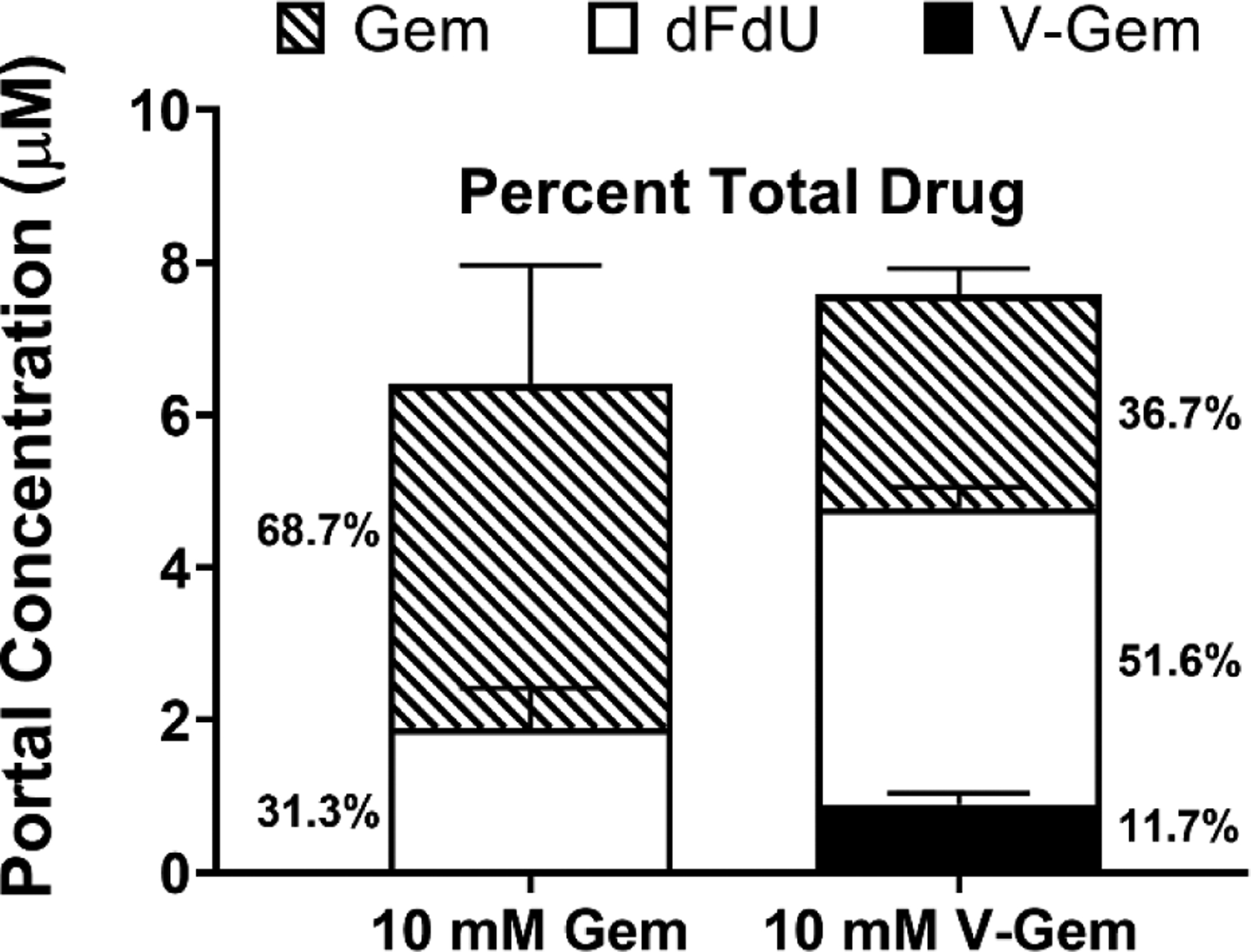
Portal plasma concentrations of Gem, dFdU, and V-Gem following 5 min jejunal perfusions of 10 mM Gem and V-Gem. The percent of total drug found as each analyte is also presented. Data are expressed as mean ± SE (n=4). Total drug concentrations were not significantly different following Gem and V-Gem perfusions, as determined by unpaired t-test.
4. Discussion
With an oral bioavailability in humans of only 10%, gemcitabine’s therapeutic application is currently hindered by a reliance on intravenous administration [13]. Recent work using in situ intestinal perfusions in mice showed that the intestinal effective permeability of gemcitabine is high at low drug concentrations and rapidly decreases with increasing drug concentration as uptake via high-affinity nucleoside transporters becomes saturated [18]. Importantly, gemcitabine’s low systemic exposure following oral dosing was reported when gemcitabine was administered at low doses (≤ 8 mg) unlikely to saturate intestinal uptake [13], implying that first-pass metabolism of gemcitabine via cytidine deaminase (CDA) drives its low bioavailability. This conclusion is further supported by data in human and mouse showing that gemcitabine undergoes extensive presystemic deamination following oral administration, forming the deaminated gemcitabine metabolite dFdU [13, 25]. To decrease first-pass metabolism, and thus increase gemcitabine bioavailability, gemcitabine may be formulated as a prodrug which reduces the ability of CDA to bind and metabolize gemcitabine. For example, an l-valine ester gemcitabine prodrug, 5’-l-valyl-gemcitabine (V-Gem) (Figure 1), was previously synthesized and shown to be stable against CDA mediated deamination, relative to gemcitabine, via incubations with recombinant human CDA [19, 22]. Furthermore, V-Gem was shown to be transported by peptide transporter 1 (PEPT1) [19, 23], a low-affinity, high-capacity transporter found on the apical membrane of intestinal enterocytes [21].
By formulating gemcitabine as a prodrug that confers stability against CDA-mediated first-pass metabolism and is targeted to a high-capacity, low-affinity intestinal uptake transporter (i.e., PEPT1), V-Gem may both reduce first-pass gemcitabine metabolism and mitigate the potential for saturation of intestinal gemcitabine uptake. As a result, we decided to characterize the in vivo pharmacokinetic performance of gemcitabine and V-Gem following intravenous and oral administrations in mice as well as the ability of V-Gem to reduce first-pass metabolism and increase drug absorption, relative to gemcitabine. In doing so, our studies revealed for the first time that: 1) V-Gem prodrug undergoes rapid systemic elimination (T1/2 < 4 min) and has very low oral bioavailability (<1%), 2) oral administration of V-Gem does not increase systemic exposure to gemcitabine, relative to oral gemcitabine administration, 3) V-Gem undergoes extensive first-pass activation in intestinal epithelial cells, and 4) gemcitabine undergoes first-pass metabolism in intestinal epithelial cells.
Using a novel and validated LC-MS/MS assay, the concentration-time profiles of gemcitabine, dFdU, and V-Gem (following V-Gem administration) were determined in mice following intravenous administration of 76 nmol gemcitabine/g body weight, oral administration of 228 nmol gemcitabine/g body weight, intravenous administration of 76 nmol V-Gem/g body weight, and oral administration of 228 nmol V-Gem/g body weight. As shown in Table 3, the pharmacokinetic parameters describing gemcitabine disposition following intravenous and oral gemcitabine administrations are in agreement with previously reported values in mice [25]. Additionally, the initial gemcitabine concentration (C0 = Cmax) following intravenous administration of 76 nmol gemcitabine/g body weight (i.e., 20 mg gemcitabine/kg body weight) in mice was 67.2 μM, replicating the gemcitabine maximum concentration (Cmax) of 50 – 70 μM following intravenous infusion of 1,250 mg gemcitabine/m2 in human [26]. It was observed that the metabolite (dFdU) to parent (gemcitabine) exposure ratio was >1 following both intravenous (ratio = 1.5) and oral (ratio = 6.9) gemcitabine administrations, reflecting the extensive conversion of gemcitabine to dFdU and the slow elimination of dFdU, relative to gemcitabine. This ratio was much higher following oral administration due to extensive presystemic conversion of gemcitabine to dFdU, in accordance with previously published work [25, 27]. Interestingly, in previous studies reporting T1/2 values of dFdU in mice, there is substantial variability in both the calculated T1/2 and the length of time over which plasma samples were collected [25, 27–29]. Examining the mean concentration-time profile of dFdU in studies where plasma samples were collected for ≥ 24 hr following gemcitabine administration [25, 29] suggests that the sampling scheme employed in the current study, which was designed to ensure adequate characterization of the concentration-time profile of gemcitabine, may bias downward estimates of dFdU T1/2. Thus, the statistically significant difference in dFdU T1/2 following oral gemcitabine administration (210 min) and intravenous gemcitabine administration (106 min) may be due to our sampling schedule being limited to six hours.
Following intravenous V-Gem administration, the prodrug rapidly disappeared from plasma with a T1/2 of 3.7 min, while gemcitabine rapidly appeared in plasma, with the gemcitabine Tmax occurring in the first sample (2 min) for all mice. Two enzymes, RBBP9 and the biphenyl hydrolase like enzyme (BPHL), have previously been shown to catalyze V-Gem activation in vitro via V-Gem incubations with these recombinant human enzymes [30, 31]. These studies, however, did not rule out the potential involvement of additional esterase enzymes in V-Gem activation. Interestingly, systemic gemcitabine exposure (AUCinf) was about 50% lower following intravenous V-Gem administration, relative to intravenous administration of an equimolar gemcitabine dose (p < 0.05). Total dFdU exposure (AUCinf), however, was not different following intravenous V-Gem and gemcitabine administrations. One potential explanation for this observation is that V-Gem is not completely activated to gemcitabine following intravenous administration but, instead, a portion of the administered V-Gem is first deaminated, forming 5’-l-valyl-dFdU (V-dFdU), and V-dFdU is subsequently cleaved releasing dFdU. Given that gemcitabine’s clearance and first-pass metabolism is driven by CDA-mediated deamination [9, 13], the ability of V-Gem to reduce CDA-mediated deamination was tested and confirmed in vitro with recombinant human enzyme [19, 22]. However, it is feasible that V-Gem becomes a substrate of a different deaminase enzyme [32–35], whose activity on V-Gem has not been previously evaluated. It is also possible that V-Gem is stable against human, but not murine CDA. This species difference in CDA substrate specificity seems unlikely, however, as the amino acid residues believed to dictate CDA substrate specific are completely conserved between mouse and human CDA [36]. To confirm the hypothesized V-Gem metabolic scheme, verficiation of V-dFdU formation in vivo would be required. Regardless, the T1/2 of gemcitabine and dFdU following V-Gem intravenous administration closely mirrored the corresponding values following gemcitabine intravenous administration.
The mean oral bioavailability of V-Gem was about 0.6% and, thus, systemic exposure to V-Gem following oral administration was negligible. The mean plasma concentration-time profiles of gemcitabine following oral gemcitabine and V-Gem administrations (Figure 6A) are very similar, demonstrating that oral administrations of gemcitabine and V-Gem lead to equivalent systemic gemcitabine exposure. Quantitatively, this is evidenced by the fact that systemic gemcitabine exposure (AUCinf) following oral administration of gemcitabine (893 min × μM) and V-Gem (814 min × μM) were not statistically significantly different. In fact, the average gemcitabine exposure following oral V-Gem administration was about 9% lower. Gemcitabine oral bioavailability was 18.3% following oral gemcitabine administration, in line with previously reported values [25], and 16.7% following oral V-Gem administration. Interestingly, the concentration-time profile of dFdU appears to differ slightly following oral administrations of gemcitabine and V-Gem (Figure 6B). However, there was no statistically significant difference in dFdU exposure following oral gemcitabine and V-Gem dosing.
To further understand why oral V-Gem administration did not increase systemic gemcitabine exposure, relative to oral gemcitabine administration, additional intestinal perfusion experiments were performed in mice. Importantly, for V-Gem to undergo PEPT1-mediated uptake and confer resistance against first-pass metabolism, it must be stable in both the stomach and the intestinal lumen. Previous work demonstrated V-Gem stability in pH 1.2 simulated gastric fluid (T1/2 > 120 min) [23]. The stability of V-Gem in the intestinal lumen was explored in the current work by perfusing V-Gem through a cannulated jejunal segment in an anesthetized mouse and quantifying the concentration of activated gemcitabine in perfusion outlet samples. These experiments demonstrated that V-Gem underwent some activation in the intestinal lumen, however, < 10% of the perfused V-Gem was found in outlet samples as activated Gem.
Prodrug stability in intestinal epithelial cells was then explored by perfusing V-Gem for 5 min and quantifying the concentration of gemcitabine, dFdU, and V-Gem in portal plasma samples (Figure 7). The short perfusion time was selected to minimize the impact of recirculated drug on the estimation of prodrug activation in intestinal epithelial cells. These results show that V-Gem undergoes extensive activation in intestinal epithelial cells as intact prodrug accounted for < 12% of total drug found in portal plasma. This observation is consistent with other work reporting extensive activation of amino acid ester prodrugs in mouse and rat intestinal epithelium [30, 37–39]. Additionally, perfusion experiments were performed showing gemcitabine is quite stable in the intestinal lumen (< 1% of perfused gemcitabine found in outlet samples as dFdU) but undergoes first-pass metabolism in the intestinal enterocytes, as evidenced by the appearance of dFdU in portal plasma, accounting for about 30% of total drug. Thus, the extensive activation of V-Gem in intestinal epithelial cells diminishes the ability of V-Gem to protect against first-pass gemcitabine metabolism in both the intestine and the liver and, thus, the ability of V-Gem to increase systemic gemcitabine exposure. Importantly, interspecies differences (i.e., mouse vs human) in the activity of various esterases have been reported, suggesting that V-Gem may be more stable in the human intestinal epithelium [40]. However, this seems unlikely given in vitro work showing that V-Gem undergoes extensive (> 90%) activation during transit through a Caco-2 cell monolayer [19].
Furthermore, total drug concentrations in portal plasma samples following perfusion of 10 mM gemcitabine and V-Gem were not different, indicating that even at high intestinal concentrations expected to completely saturate nucleoside transporter mediated gemcitabine uptake, partitioning of total drug from the intestinal lumen into portal plasma was not increased by V-Gem. An important caveat to this conclusion, however, is the assumption that no other V-Gem metabolites are present in portal plasma (e.g., V-dFdU). To address this possibility, perfusions with radiolabeled V-Gem could be performed and total drug concentrations in portal blood assayed via total radioactivity.
As alluded to above, the incomplete conversion of V-Gem to gemcitabine, which was hypothesized to occur following intravenous V-Gem administration, could also contribute to the inability of oral V-Gem administration to increase systemic gemcitabine exposure, relative to oral gemcitabine administration. However, it is important to note that incomplete V-Gem activation following intravenous V-Gem administration remains speculative and that V-Gem activation may differ following oral and intravenous administrations (i.e., V-Gem may undergo complete activation following oral but not intravenous administration). Thus, additional studies would be needed to further explore V-Gem activation in vivo and the impact of potential incomplete activation on gemcitabine exposure following oral V-Gem administration.
In conclusion, the in vivo performance of a PEPT1-targeted gemcitabine prodrug, V-Gem, was evaluated following intravenous and oral administrations in mice. This work demonstrated that V-Gem is rapidly removed from plasma following intravenous administration and has very low oral bioavailability (< 1%). Furthermore, our studies demonstrate that formulation of gemcitabine as V-Gem did not lead to increased systemic gemcitabine exposure following oral dosing as gemcitabine bioavailability was no different following oral gemcitabine and V-Gem administrations. These results suggest that future prodrug strategies aimed at increasing systemic exposure of gemcitabine following oral dosing should focus on prodrugs with high intestinal effective permeability, good stability during first-pass transit through the intestinal enterocytes and liver, and complete conversion to the active gemcitabine species. Alternatively, future work aimed at enabling oral gemcitabine administration could focus on decreasing first-pass gemcitabine metabolism and, thus, increasing systemic gemcitabine exposure through co-administration of gemcitabine with a cytidine deaminase inhibitor.
Acknowledgements
The authors thank Dr. Yongjun Hu for his technical guidance on drug administrations. This work was supported, in part, by the National Institutes of Health National Institute of General Medical Sciences grants R01GM115481 (to DES) and T32GM007767 (to BRT).
Abbreviations:
- AUC0–6 hr
area under the plasma concentration-time curve from time 0 to 6 hr
- AUCinf
area under the plasma concentration-time curve from time 0 to infinity
- CDA
cytidine deaminase
- C0
initial plasma concentration at time 0
- Cmax
maximum plasma concentration
- dFdU
2’,2’-difluoro-2’-deoxyuridine
- Gem
gemcitabine
- NT
nucleoside transporters
- PEPT1
peptide transporter 1
- THU
tetrahydrouridine
- T1/2
half-life
- Tmax
time to reach the maximum plasma concentration
- V-Gem
5’-l-valyl-gemcitabine
- Vss
volume of distribution steady-state
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest, financial or otherwise.
References
- [1].Gemzar [package insert]. Eli Lilly and Company, Indianapolis, IN, (2014). [Google Scholar]
- [2].Conti RM, Bernstein AC, Villaflor VM, Schilsky RL, Rosenthal MB, and Bach PB, Prevalence of Off-Label Use and Spending in 2010 Among Patent-Protected Chemotherapies in a Population-Based Cohort of Medical Oncologists, J Clin Oncol. 31 (9) (2013) 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, and Bridgewater J, Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer, N Engl J Med. 362 (14) (2010) 1273–81. [DOI] [PubMed] [Google Scholar]
- [4].von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A, and Arning M, Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer, J Clin Oncol. 23 (21) (2005) 4602–8. [DOI] [PubMed] [Google Scholar]
- [5].Huang P, Chubb S, Hertel LW, Grindey GB, and Plunkett W, Action of 2’,2’-difluorodeoxycytidine on DNA synthesis, Cancer Res. 51 (22) (1991) 6110–6117. [PubMed] [Google Scholar]
- [6].Huang P and Plunkett W, Fludarabine- and gemcitabine-induced apoptosis: incorporation of analogs into DNA is a critical event, Cancer Chemother Pharmacol. 36 (3) (1995) 181–188. [DOI] [PubMed] [Google Scholar]
- [7].Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, and Plunkett W, Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2’,2’-difluorodeoxycytidine, Mol Pharmacol. 38 (4) (1990) 567–572. [PubMed] [Google Scholar]
- [8].Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, and Gandhi V, Gemcitabine: metabolism, mechanisms of action, and self-potentiation, Semin Oncol. 22 (4 Suppl 11) (1995) 3–10. [PubMed] [Google Scholar]
- [9].Zhang L, Sinha V, Forgue ST, Callies S, Ni L, Peck R, and Allerheiligen SR, Model-based drug development: the road to quantitative pharmacology, J Pharmacokinet Pharmacodyn. 33 (3) (2006) 369–393. [DOI] [PubMed] [Google Scholar]
- [10].Honeywell RJ, Ruiz van Haperen VW, Veerman G, Smid K, and Peters GJ, Inhibition of thymidylate synthase by 2’,2’-difluoro-2’-deoxycytidine (Gemcitabine) and its metabolite 2’,2’-difluoro-2’-deoxyuridine, Int J Biochem Cell Biol. 60 (2015) 73–81. [DOI] [PubMed] [Google Scholar]
- [11].Veltkamp SA, Pluim D, van Eijndhoven MA, Bolijn MJ, Ong FH, Govindarajan R, Unadkat JD, Beijnen JH, and Schellens JH, New insights into the pharmacology and cytotoxicity of gemcitabine and 2’,2’-difluorodeoxyuridine, Mol Cancer Ther. 7 (8) (2008) 2415–2425. [DOI] [PubMed] [Google Scholar]
- [12].Pauwels B, Korst AE, Lambrechts HA, Pattyn GG, de Pooter CM, Lardon F, and Vermorken JB, The radiosensitising effect of difluorodeoxyuridine, a metabolite of gemcitabine, in vitro, Cancer Chemother Pharmacol. 58 (2) (2006) 219–28. [DOI] [PubMed] [Google Scholar]
- [13].Veltkamp SA, Jansen RS, Callies S, Pluim D, Visseren-Grul CM, Rosing H, Kloeker-Rhoades S, Andre VA, Beijnen JH, Slapak CA, and Schellens JH, Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study, Clin Cancer Res. 14 (11) (2008) 3477–3486. [DOI] [PubMed] [Google Scholar]
- [14].Cham K, Baker J, Takhar K, Flexman J, Wong M, Owen D, Yung A, Kozlowski P, Reinsberg S, and Chu E, Metronomic gemcitabine suppresses tumour growth, improves perfusion, and reduces hypoxia in human pancreatic ductal adenocarcinoma, Br J Cancer. 103 (1) (2010) 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yapp DT, Wong MQ, Kyle AH, Valdez SM, Tso J, Yung A, Kozlowski P, Owen DA, Buczkowski AK, and Chung SW, The differential effects of metronomic gemcitabine and antiangiogenic treatment in patient-derived xenografts of pancreatic cancer: treatment effects on metabolism, vascular function, cell proliferation, and tumor growth, Angiogenesis. 19 (2) (2016) 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tran Cao HS, Bouvet M, Kaushal S, Keleman A, Romney E, Kim G, Fruehauf J, Imagawa DK, Hoffman RM, and Katz MH, Metronomic gemcitabine in combination with sunitinib inhibits multisite metastasis and increases survival in an orthotopic model of pancreatic cancer, Mol Cancer Ther. 9 (7) (2010) 2068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dehua Z, Mingming C, and Jisheng W, Meta-analysis of gemcitabine in brief versus prolonged low-dose infusion for advanced non-small cell lung cancer, PLoS One. 13 (3) (2018) e0193814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thompson BR, Hu Y, and Smith DE, Mechanisms of gemcitabine oral absorption as determined by in situ intestinal perfusions in mice, Biochem Pharmacol. 168 (2019) 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song XQ, Lorenzi PL, Landowski CP, Vig BS, Hilfinger JM, and Amidon GL, Amino acid ester prodrugs of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport, Mol Pharm. 2 (2) (2005) 157–167. [DOI] [PubMed] [Google Scholar]
- [20].Drozdzik M, Groer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, Prasad B, Unadkat JD, Siegmund W, and Oswald S, Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine, Mol Pharm. 11 (10) (2014) 3547–55. [DOI] [PubMed] [Google Scholar]
- [21].Smith DE, Clemencon B, and Hediger MA, Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications, Mol Aspects Med. 34 (2–3) (2013) 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tsume Y, Drelich AJ, Smith DE, and Amidon GL, Potential Development of Tumor-Targeted Oral Anti-Cancer Prodrugs: Amino Acid and Dipeptide Monoester Prodrugs of Gemcitabine, Molecules. 22 (8) (2017) 1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tsume Y, Incecayir T, Song X, Hilfinger JM, and Amidon GL, The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: enhanced membrane permeability and enzymatic stability, Eur J Pharm Biopharm. 86 (3) (2014) 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Epling D, Hu Y, and Smith DE, Evaluating the intestinal and oral absorption of the prodrug valacyclovir in wildtype and huPepT1 transgenic mice, Biochem Pharmacol. 155 (2018) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Beumer JH, Eiseman JL, Parise RA, Joseph E, Covey JM, and Egorin MJ, Modulation of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine) Pharmacokinetics, Metabolism, and Bioavailability in Mice by 3,4,5,6-Tetrahydrouridine, Clin Cancer Res. 14 (11) (2008) 3529–3535. [DOI] [PubMed] [Google Scholar]
- [26].Kuenen BC, Rosen L, Smit EF, Parson MRN, Levi M, Ruijter R, Huisman H, Kedde MA, Noordhuis P, Vijgh W.J.F.v.d., Peters GJ, Cropp GF, Scigalla P, Hoekman K, Pinedo HM, and Giaccone G, Dose-Finding and Pharmacokinetic Study of Cisplatin, Gemcitabine, and SU5416 in Patients With Solid Tumors, J Clin Oncol. 20 (6) (2002) 1657–1667. [DOI] [PubMed] [Google Scholar]
- [27].Veltkamp SA, Pluim D, van Tellingen O, Beijnen JH, and Schellens JH, Extensive metabolism and hepatic accumulation of gemcitabine after multiple oral and intravenous administration in mice, Drug Metab Dispos. 36 (8) (2008) 1606–15. [DOI] [PubMed] [Google Scholar]
- [28].Hao WH, Wang JJ, Hsueh SP, Hsu PJ, Chang LC, Hsu CS, and Hsu KY, In vitro and in vivo studies of pharmacokinetics and antitumor efficacy of D07001-F4, an oral gemcitabine formulation, Cancer Chemother Pharmacol. 71 (2) (2013) 379–388. [DOI] [PubMed] [Google Scholar]
- [29].Reddy LH, Khoury H, Paci A, Deroussent A, Ferreira H, Dubernet C, Decleves X, Besnard M, Chacun H, Lepetre-Mouelhi S, Desmaele D, Rousseau B, Laugier C, Cintrat JC, Vassal G, and Couvreur P, Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice, Drug Metab Dispos. 36 (8) (2008) 1570–7. [DOI] [PubMed] [Google Scholar]
- [30].Shenoy VM, Thompson BR, Shi J, Zhu HJ, Smith DE, and Amidon GL, Chemoproteomic Identification of Serine Hydrolase RBBP9 as a Valacyclovir-Activating Enzyme, Mol Pharm. (2020) [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- [31].Kim I, Song X, Vig BS, Mittal S, Shin H-C, Lorenzi PJ, and Amidon GL, A novel nucleoside prodrug-activating enzyme: substrate specificity of biphenyl hydrolase-like protein, Mol Pharm. 1 (2) (2004) 117–127. [DOI] [PubMed] [Google Scholar]
- [32].Yuan G, Bin JC, McKay DJ, and Snyder FF, Cloning and characterization of human guanine deaminase. Purification and partial amino acid sequence of the mouse protein, J Biol Chem. 274 (12) (1999) 8175–80. [DOI] [PubMed] [Google Scholar]
- [33].Yeung CY, Ingolia DE, Roth DB, Shoemaker C, Al-Ubaidi MR, Yen JY, Ching C, Bobonis C, Kaufman RJ, and Kellems RE, Identification of functional murine adenosine deaminase cDNA clones by complementation in Escherichia coli, J Biol Chem. 260 (18) (1985) 10299–307. [PubMed] [Google Scholar]
- [34].Austin EA and Huber BE, A first step in the development of gene therapy for colorectal carcinoma: cloning, sequencing, and expression of Escherichia coli cytosine deaminase, Mol Pharmacol. 43 (3) (1993) 380–7. [PubMed] [Google Scholar]
- [35].Weiner KX, Weiner RS, Maley F, and Maley GF, Primary structure of human deoxycytidylate deaminase and overexpression of its functional protein in Escherichia coli, J Biol Chem. 268 (17) (1993) 12983–9. [PubMed] [Google Scholar]
- [36].Frances A and Cordelier P, The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy?, Molecular Therapy. 28 (2) (2020) 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tao W, Zhao D, Sun M, Wang Z, Lin B, Bao Y, Li Y, He Z, Sun Y, and Sun J, Intestinal absorption and activation of decitabine amino acid ester prodrugs mediated by peptide transporter PEPT1 and enterocyte enzymes, Int J Pharm. 541 (1–2) (2018) 64–71. [DOI] [PubMed] [Google Scholar]
- [38].Sun Y, Sun J, Shi S, Jing Y, Yin S, Chen Y, Li G, Xu Y, and He Z, Synthesis, transport and pharmacokinetics of 5’-amino acid ester prodrugs of 1-beta-D-arabinofuranosylcytosine, Mol Pharm. 6 (1) (2009) 315–25. [DOI] [PubMed] [Google Scholar]
- [39].Hu Y, Epling D, Shi J, Song F, Tsume Y, Zhu HJ, Amidon GL, and Smith DE, Effect of biphenyl hydrolase-like (BPHL) gene disruption on the intestinal stability, permeability and absorption of valacyclovir in wildtype and Bphl knockout mice, Biochem Pharmacol. 156 (2018) 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bahar FG, Ohura K, Ogihara T, and Imai T, Species difference of esterase expression and hydrolase activity in plasma, J Pharm Sci. 101 (10) (2012) 3979–88. [DOI] [PubMed] [Google Scholar]