

Summary
Many hypotheses have been proposed to explain how a glutamate to valine substitution in sickle haemoglobin (HbS) can cause sickle cell disease (SCD). We propose and document a new mechanism in which elevated tyrosine phosphorylation of Band 3 initiates sequelae that cause vaso-occlusion and the symptoms of SCD. In this mechanism, denaturation of HbS and release of heme generate intracellular oxidants which cause inhibition of erythrocyte tyrosine phosphatases, thus permitting constitutive tyrosine phosphorylation of Band 3. This phosphorylation in turn induces dissociation of the spectrin-actin cytoskeleton from the membrane, leading to membrane weakening, discharge of membrane-derived microparticles (which initiate the coagulation cascade) and release of cell-free HbS (which consumes nitric oxide) and activates the endothelium to express adhesion receptors). These processes promote vaso-occlusive events which cause SCD. We further show that inhibitors of Syk tyrosine kinase block Band 3 tyrosine phosphorylation, prevent release of cell-free Hb, inhibit discharge of membrane-derived microparticles, increase sickle cell deformability, reduce sickle cell adhesion to human endothelial cells, and enhance sickle cell flow through microcapillaries. In view of reports that imatinib (a Syk inhibitor) successfully treats symptoms of sickle cell disease, we suggest that Syk tyrosine kinase inhibitors warrant repurposing as potential treatments for SCD.
Keywords: anion exchanger 1, erythrocyte membrane, haemoglobinopathy, sickle cell disease, tyrosine phosphorylation
Introduction
Although most symptoms of SCD are thought to be caused by occlusion of the microvasculature,1-4 the mechanistic steps underpinning these vaso-occlusive events are still debated. The most cited mechanisms include: i) loss of erythrocyte viscoelastic properties deriving from sickle haemoglobin (HbS) polymerisation, red blood cell (RBC) dehydration, and membrane rigidification,1,3,5-9 ii) activation of adhesion receptors on the vascular endothelium and/or erythrocyte membrane,10,13 and iii) initiation of thrombosis by RBC-derived microparticles (MPs) and the subsequent activation of platelets by thrombin and other coagulation factors.12-17 Vaso-occlusive processes may result in tissue hypoxia, ischaemia-reperfusion injury, organ damage and associated morbidities, and debilitating pain which results in significant suffering and may require medical treatment/hospitalization.18,19 Sickle cell haemolysis, reduced sickle red cell lifespan, and anaemia can further aggravate clinical symptoms.20,21
While RBC dehydration,3,5,6,22 loss of membrane deformability3,23 and increased RBC/endothelial cell adhesion10-12 undoubtedly contribute to SCD, an increasing number of researchers now propose that additional pathologic sequelae may arise from the weakening of the erythrocyte membrane, leading to discharge of MPs12,14,15,17 and free haemoglobin (Hb).15,24 In this hypothesis, accelerated denaturation of HbS25 hemichrome formation26,27 and release of heme may collectively induce oxidative stress within the RBC.26,28,29 Increased oxidative stress can then cause inhibition of RBC tyrosine phosphatases which normally prevent constitutive Band 3 tyrosine phosphorylation.30-33 Upon inhibition of these phosphatases, over-phosphorylation of Band 3 then induces global destabilisation of the erythrocyte membrane,34,35 accelerating intravascular haemolysis and MP release. The increased plasma haemoglobin and heme can ‘activate’ the vascular endothelium, causing expression of adhesion receptors (e.g., p-selectin, E-selectin and von Willebrand factor),10-12 as well as sequestration of the vasodilator (NO),11,36-38 while the release of MPs can trigger intravascular thrombosis via activation of prothrombin.12,14,15,17 When initiated concomitantly with loss of RBC deformability and enhanced vaso-adhesion, the sequelae associated with membrane weakening can aggravate an already compromised blood flow, leading to micro-emboli and progressive tissue damage.
In this paper, we explore the role of Band 3 tyrosine phosphorylation and consequent membrane weakening in the development of the symptoms in SCD. We first show that the extent of tyrosine phosphorylation of Band 3 correlates with the percentage of HbS in sickle erythrocytes, the concentration of cell-free Hb in the patient’s plasma, and the number of RBC-derived MPs in a patients’ peripheral blood. We next document that the blockade of tyrosine phosphorylation of Band 3 with tyrosine kinase inhibitors prevents the release of cell-free Hb and the discharge of RBC-derived MPs, while concomitantly enhancing sickle cell deformability, reducing sickle cell sickling at lower O2 pressures, and enhancing sickle cell flow through microcapillaries. Finally, we also establish that imatinib treatment suppresses adhesion of erythrocytes to heme-activated endothelium. Based on evidence from several labs that MPs,12,14-17 cell-free Hb,36,39,40 reduced sickle cell deformability, and enhanced sickle cell adhesion to the endothelium10-12,23,41 contribute to the pathology of SCD, we argue that Band 3 tyrosine phosphorylation inhibitors could constitute a potent therapy for treatment for SCD.
Methods
Processing of SCD blood samples
All sickle cell blood samples were obtained following informed consent using procedures approved by the local institutional review boards (IRBs). Venous blood was collected from patients (genotype SS or Sβ0) and healthy volunteers in EDTA-containing vacutainer tubes and maintained at 4°C until use. Samples were centrifuged at 800 rcf (relative centrifugal force) for ten minutes and plasma was removed for analysis of MPs and cell-free Hb. RBC pellets were washed three times with phosphate buffered saline, pH 7·4, containing 5 mM glucose (PBS-G) and the buffy coat was aspirated after each wash cycle.
Quantitation of Band 3 tyrosine phosphorylation
Washed RBCs were suspended at 30% haematocrit (Hct) in PBS-G and treated with either 5 μM of a drug (imatinib, PRT062607 or R406) or vehicle (control) for 4 h at 37°C, shaking at 50 rpm. The 5 μM drug concentration was determined from the minimum concentration of imatinib required to completely reverse Band 3 tyrosine phosphorylation within 4 h. RBC membranes were prepared and processed for western blotting as described in supplemental information. Band 3 tyrosine phosphorylation intensity was quantitated using image J software. To induce Band 3 tyrosine phosphorylation and membrane fragmentation in healthy erythrocytes, blood from healthy donors was washed and cells were suspended at 30% Hct in PBS-G, containing either 5 μM imatinib or a vehicle of dimethyl sulfoxide (DMSO); ≤0·5% v/v to minimise impact of the vehicle on cells (see Fig S1), at 37°C for 1 h. After the 1h incubation, 2 mM sodium orthovanadate (OV) was added to both imatinib-treated and untreated cells, and cells were incubated for 4 h at 37°C, while shaking at 1400 rpm.
Quantitation of erythrocyte-derived microparticles and free plasma haemoglobin
Plasma from sickle cell samples was centrifuged two times at 2500 rcf for 15 min to remove platelets, and 100 μl supernatant, containing MPs, was incubated for 20 min on ice with 0·5 μl mouse anti-human glycophorin A antibody (BD Biosciences #562938). To test if the MPs are CD71 positive, 1·0 μl of CD71 antibody (BD biosciences #12-0711-82) was added alongside the glycophorin A antibody. Samples were diluted with 1 ml stain buffer (BD Biosciences #554656), transferred to BD Trucount™ tubes (BD Biosciences #340334) and analysed on Attune NxT Flow Cytometer, utilising a violet fluorescent trigger channel.42 The absolute number of MPs was calculated as follows:
For evaluation of the effect of tyrosine kinase inhibitors on release of MPs, 500 μL sickle cells, suspended at 30% Hct in PBS-G, were treated for 1 h with the desired tyrosine kinase inhibitor or vehicle control, and shaken at 1400 rpm for 4 h. Newly released microparticles were separated from residual RBCs by centrifuging at 800 rcf for 10 min, collecting the supernatant, and quantitating the MPs as described above.
Cayman’s haemoglobin colorimetric assay was used to quantitate cell-free Hb according to manufacturer’s instructions.
Measurement of red blood cell deformability
Erythrocyte deformability was measured using Technicon™ Ektacytometer and plotted as elongation index versus shear stress. Data were acquired while accelerating the ektacytometer from 0 to 250 rpm and shear stress was calculated:
Measurements of RBC flow through microcapillaries
The effect of imatinib on the rate of sickle cell flow through 5 μm × 6·5 μm microcapillaries at different oxygen pressures was measured using a high speed camera mounted onto an inverted microscope, focused on a microfluidic device through which sickle blood was flowed at constant pressure (1·6 psi), as described in Supplement Information and reference.43
Oxygen gradient ektacytometry (oxygenscan)
To determine the point of sickling (PoS) upon deoxygenation, oxygenscans were performed using a Laser Optical Rotational Red Cell Analyzer, as described.44 Patient samples were washed and resuspended at 20% haematocrit in HBSS, modified with 10 mM HEPES and 10 mM MgCl2, and incubated with 5 μM imatinib or DMSO for 4 h at 37 °C. RBCs of 300 × 106 were added to 5 ml Oxylso solution (RR Mechatronics, Zwaag, the Netherlands) and loaded into the Lorrca where they were subjected to constant shear stress (30 Pa). Deformability was measured while partial pressure of oxygen (pO2) was gradually decreased from 150 mmHg to < 20 mmHg before reoxygenation with ambient air. The PoS was determined as the pO2 at which samples reached 95% of their initial deformability and began to sickle.
Measurements of RBC adhesion to endothelial cell functionalised microchannels
Microfluidic channels were fabricated and incubated with fibronectin prior to coating with a monolayer of human umbilical vein endothelial cells (HUVEC) and human pulmonary microvascular endothelial cells (HPMECs), as described earlier.16,45 Two hours prior to analysis of RBC adhesion, adherent HUVECs and HPMECs were activated with 40 μM heme to induce expression of adhesion receptors.16 Freshly isolated sickle cells were simultaneously incubated in basal medium for 4 h and perfused through the microchannels at a physiological shear stress level of 1 dyne/cm2 in precisely controlled physiological hypoxia (SpO2 of 83%).16 The SpO2 level was chosen to be pathologically relevant to SCD, based on clinical studies.46 Non-adherent RBCs were washed away and adherent erythrocytes were counted (see details in Supplemental Information).
Statistical analysis
Data are reported as mean ± standard error of the mean (SEM) and F-test on linear regression. Statistical significance was set at a 95% confidence level for all tests (P < 0·05).
Results
To test the hypothesis that sickle cells are distinguished by increased tyrosine phosphorylation of Band 3, membrane weakening, and release of both cell-free Hb and MPs, we focused studies on RBCs from non-transfused children (age range 3–20 years, mean 9·3 years; n = 48) with SCD – all undergoing hydroxycarbamide treatment. As shown in Fig. 1A, the concentration of cell-free Hb in patient plasma was more than twice that of healthy volunteers, i.e. in agreement with previous studies.12,15,24,47 Moreover, the number of MPs, identified as glycophorin A positive particles of 0·1–1·0 μm diameter (Figure S2), were also more than twice as abundant in patients than in healthy volunteers (panel B), and therefore also consistent with previous observations14 by12,15,48 It is worth noting that the glycophorin A positive microparticles identified herein are CD71 negative (Figure S3), which indicates that the MPs observed are mainly derived from mature erythrocytes. Because release of MPs would be expected to render the membrane-depleted erythrocytes less deformable (due to their smaller surface to volume ratios and higher Hb concentrations2,3,5,23,41), the reduced deformability of SCD blood versus normal controls (panel C) was expected. Importantly, the nearly linear correlation between MP count and cell-free Hb in each patient’s blood sample (panel D) suggested a possible relationship between cell-free Hb and RBC-derived MPs. The weakening of the membrane by tyrosine phosphorylation of Band 3 could account for this correlation.
Fig 1.

Quantitation of cell-free Hb and erythrocyte membrane-derived MPs in blood from patients with SCD. The concentration of cell-free Hb in the plasma (A) and numbers of erythrocyte membrane-derived MPs (B) were evaluated in both patients with SCD and healthy controls (n = 48). Erythrocyte elongation index as a function of increasing shear stress (i.e. deformability) was also compared for SCD and control blood samples (C, representative sample of three different healthy controls and five different SCD patients). A plot of free plasma Hb versus MP count was constructed to explore a possible common mechanism leading to their production (D). (Error bars are expressed as SEM, *** denotes P ≤ 0·005 computed from one-way ANOVA or student t-test; Pearson’s r = 0·65, and P = 0·0001 in panel D). (Hb, haemoglobin; MP, microparticle; SCD, sickle cell disease; SEM, standard error of the mean).
It has been frequently reported that oxidative stress leads to inactivation of erythrocyte tyrosine phosphatases,30,31,49 which in turn allow unimpeded tyrosine phosphorylation of Band 3 by constitutively active tyrosine kinases.30,35,50 Because this tyrosine phosphorylation induces an intramolecular interaction in Band 3, which causes dissociation of the spectrin-actin cortical cytoskeleton from the membrane,34,35 we hypothesised that oxidative stress deriving from premature HbS denaturation25,28,29 might initiate a phosphorylation cascade, which would lead to dissociation of the spectrin-based cytoskeleton from the membrane, causing membrane destabilisation and fragmentation. To test this hypothesis, we compared tyrosine phosphorylation of Band 3 in sickle cells and healthy controls. As shown in Fig. 2A, tyrosine phosphorylation of Band 3 in healthy cells was almost undetectable, whereas phosphorylation in sickle cells was prominent. Evidence that phosphorylation in sickle cells was dependent on their levels of HbS is provided in panel B, where a positive correlation (Pearsons r = 0·70) and a significant linear ‘relationship (P = 0·008) was observed between Band 3 tyrosine phosphorylation and the percentage of HbS in each patient’s sample.
Fig 2.
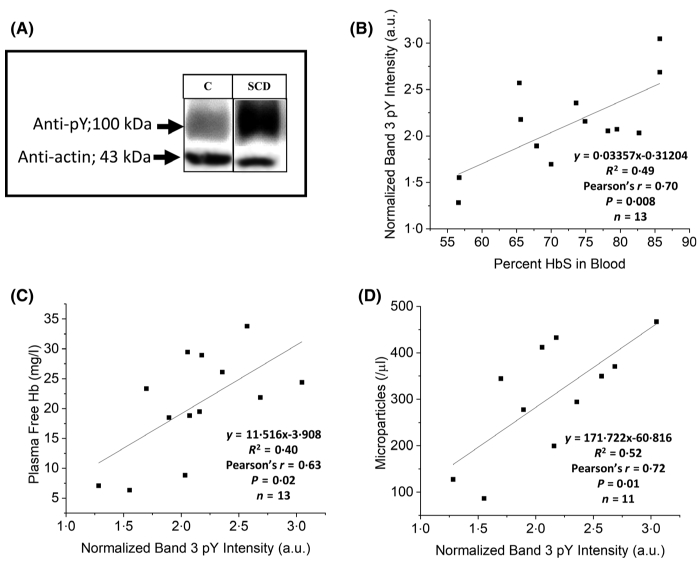
Analysis of the relationship between Band 3 tyrosine phosphorylation, cell-free Hb and MP count in sickle cell blood samples. A) Representative anti-phosphotyrosine immunoblot of Band 3 in erythrocyte membranes from a healthy control (C) and a sickle cell patient (SCD) treated with hydroxycarbamide. The correlation between Band 3 tyrosine phosphorylation and percentage of HbS in the blood sample (B), cell-free Hb in the plasma (C), and RBC membrane-derived MPs (D), is also plotted. (Statistical data from F-tests are as indicated on the respective figures). (Hb, haemoglobin; MP, microparticle; HbS, sickle haemoglobin; RBC, red blood cell).
Documentation that tyrosine phosphorylation of Band 3 was likely also related to release of Hb into plasma was demonstrated by a significant correlation (P = 0·02, Pearson’s r = 0·63) between these two parameters (panel C). Furthermore, an indication that Band 3 tyrosine phosphorylation was related to the release of MPs is shown in panel D (Pearson’s r = 0·72, P = 0·01). These data suggest that elevated tyrosine phosphorylation of Band 3 in sickle cells is related to the membrane destabilisation that causes release of MPs and free Hb.
To further test the hypothesis that Band 3 tyrosine phosphorylation might promote release of Hb and MPs from sickle erythrocytes, we explored the effect of imatinib on tyrosine phosphorylation of Band 3. As shown in Fig. 3A, healthy erythrocytes displayed low levels of Band 3 phosphorylation, whereas sickle erythrocytes exhibited higher levels of phosphorylation. Moreover, treatment of sickle erythrocytes with 5 μM imatinib lowered their Band 3 tyrosine phosphorylation to levels similar to control cells, demonstrating that imatinib can inhibit the natural tyrosine phosphorylation of Band 3 in sickle cells. As documented in panels B and C, imatinib treatment also reduces the release of cell-free Hb and MPs, suggesting that Band 3 phosphorylation is directly related to both characteristics of sickle blood. Importantly, although the tyrosine phosphorylation of Band 3 and accompanying Band 3 conformational changes are readily reversible, Hb and MP release are not reversible.33-35
Fig 3.

Effect of treatment of sickle cells with imatinib or other tyrosine kinase inhibitors on: A) tyrosine phosphorylation of Band 3 in healthy erythrocytes (control), erythrocytes from a SCD-T patient, and erythrocytes from non-SCD-T patients treated with hydroxycarbamide, B) release of cell-free Hb (n = 5), and C) discharge of RBC membrane-derived MPs from non-transfused sickle cell blood samples in vitro (n = 6). Panels D and E illustrate the effect of two Syk-specific inhibitors on tyrosine phosphorylation of Band 3 in erythrocytes from healthy controls, and non-SCD-T patients on hydroxycarbamide. Whole blood samples from healthy controls or SCD patients were washed three times in PBS-G and incubated for 4 h at 37°C in PBS-G, containing or lacking 5 μM imatinib, or 5 μM PRT062607, or 5 μM R406 while shaking at 50 (A), or 1400 (B-E) rpm prior to analysis. Note that the sickle cell patient receiving transfusions (SCD-T) exhibits no increase in Band 3 tyrosine phosphorylation. (Error bars are expressed as SEM, *denotes P ≤ 0·05, computed from one-way ANOVA). (SCD-T, transfused sickle cell disease; Hb, haemoglobin; RBC, red blood cell; MP, microparticle; PBS-G, phosphate-buffered saline with glucose; SEM, standard error of the mean).
Because imatinib inhibits several kinases besides Syk,51 the question arose whether other more Syk-specific inhibitors might similarly suppress Band 3 tyrosine phosphorylation in sickle cells. As shown in Fig. 3, incubation of sickle cells with either 5 μM PRT062607 (panel D) or R406 (panel E) resulted in an analogous diminution of Band 3 tyrosine phosphorylation. Moreover, the same Syk-specific inhibitors also suppressed MP and Hb release from both sickle cells (Figure S4) and o-vanadate-treated healthy cells (Figure S5). These data demonstrate that Syk-specific inhibitors also suppress the tyrosine kinase that phosphorylates Band 3 in SCD, suggesting that at least one of the kinases that phosphorylates Band 3 in SCD is Syk.
Because defects in molecular bridges connecting the erythrocyte membrane to its cortical spectrin-actin cytoskeleton have been shown to compromise erythrocyte deformability,34,35 we next examined whether inhibition of Band 3 tyrosine phosphorylation might restore the disrupted bridges and thereby improve sickle cell deformability. Firstly, sickle blood samples were incubated for 4 h in the presence or absence of 5 μM imatinib and then examined by ektacytometry for changes in cell deformability. As shown in Fig. 4A, although the deformability of sickle cells was lower than that of healthy controls, incubation with imatinib improved their deformability. Secondly, the deformability of sickle erythrocytes under constant shear stress was examined during deoxygenation and re-oxygenation of the sickle cells.44 As shown in Fig. 4B, sickle RBCs pre-incubated with imatinib exhibited higher baseline deformability, initiated sickling only when exposed to lower pO2 (point of sickling 5%), and displayed improved minimal deformability (EImin, panel B) compared to untreated cells from the same patient. Thirdly, because pO2-regulated RBC capillary flow velocity relates mechanistically to cell deformability,43 we studied the flow of sickle erythrocytes through microcapillaries at controlled pO2 (panel C). Relative to untreated cells, imatinib-treated cells were found to experience a significant increase in capillary velocity, which improved as the concentration of imatinib was increased (panel C). Since similar observations were obtained at all other O2 pressures examined, we conclude that imatinib improves the flow of sickle cells through microcapillaries.
Fig 4.

Effect of imatinib on the deformability and rheology of sickle cells at different oxygen pressures. A) The deformability of healthy erythrocytes and washed sickle cells is determined by ektacytometry in the presence and/or absence of 5 μM imatinib. B). A representative scan of inhibition of sickling after treatment of blood from SCD patients with imatinib or DMSO (vehicle control) as detected by Oxygenscan. Elmax, point of sickling (PoS5%) and Elmin are recorded as the sample is deoxygenated and then reoxygenated after 4 h incubation with 5 μM imatinib or an equal volume of DMSO carrier. Note both the left-shift in PoS5% and higher Elmin in sickle RBCs following incubation with imatinib, which are indicative of improved deformability of the cells. C) Washed sickle or healthy cells were incubated for 4 h at 37°C in the indicated concentrations of imatinib and then equilibrated at the indicated partial pressures of oxygen, prior to analysis of flow rates. [***denotes P ≤ 0·05, figures are representative of three (panel A) and five (panels B and C) different patient samples]. (SCD, sickle cell disease; DMSO, dimethyl sulfoxide; Elmax or Elmin, Eadie-Hofstee linearisation maximum or minimum deformability).
To directly demonstrate that tyrosine phosphorylation of Band 3 promotes membrane weakening and release of cell-free Hb and RBC membrane-derived MPs, we induced tyrosine phosphorylation of Band 3 in healthy erythrocytes by treatment with the tyrosine phosphatase inhibitor, orthovanadate, and then examined release of cell-free Hb and MPs in the presence and absence of imatinib. As shown in Fig. 5, treatment of control RBCs with orthovanadate induced tyrosine phosphorylation of Band 3 (panel A), as well as the release of cell-free Hb (panel B) and the discharge of MPs (panel C) in a manner that could be inhibited by imatinib. These data demonstrate that tyrosine phosphorylation of Band 3 constitutes the cause of Hb and MP release and that imatinib prevents these pro-embolic processes by inhibiting Band 3 tyrosine phosphorylation.
Fig 5.
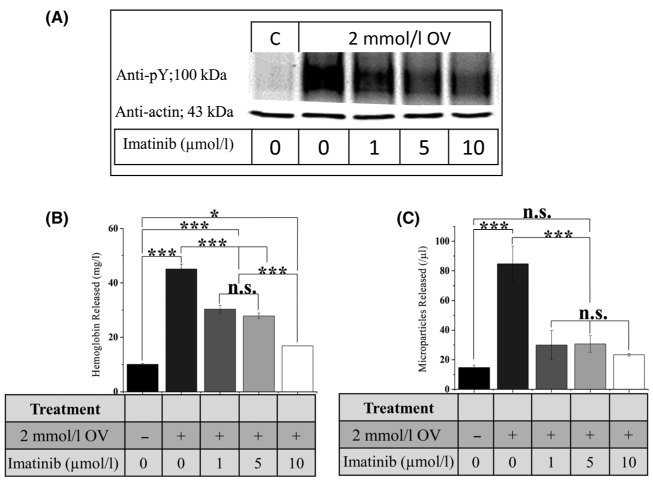
Effect of imatinib on release of cell-free Hb and membrane-derived MPs from healthy RBCs following treatment with the tyrosine phosphatase inhibitor OV. Effect of sodium OV and imatinib on (A) Band 3 tyrosine phosphorylation, (B) release of cell-free Hb, and (C) discharge of membrane-derived MPs from healthy HbAA RBCs. Cells were incubated with the indicated concentrations of imatinib and OV for 4 h with shaking at 1400 rpm prior to analysis. (Error bars are expressed as SEM, * denotes P ≤ 0·05, **denotes P ≤ 0·025, ***denotes P ≤ 0·01 computed from one-way ANOVA) (OV, orthovanadate; Hb, haemoglobin; MPs, microparticles; RBC, red blood cell; SEM, standard error of the mean).
Finally, we examined the effect of imatinib on the adhesion of flowing sickle cells to heme-activated endothelial cells. As seen in representative images of adherent RBCs in Fig. 6, untreated sickle cells (panels A and B) are more adherent to heme-activated HUVECs and HPMECs under hypoxia than imatinib-treated sickle cells (panels C and D). The mean adhesion of naive sickle cells was 383 ± 57 (control), compared to 171 ± 30 for imatinib-exposed sickle cells (panel E, n = 13 patients; P < 0·001, paired t-test). These data suggest that imatinib can further reduce vaso-occlusive events by suppressing adhesion of sickle cells to activated endothelial cells.
Fig 6.

Sickle RBC adhesion to heme-activated endothelial cells under physiologic hypoxia in microfluidic channels in vitro. Representative images of adherent RBCs to heme-activated endothelial cells are also shown in the control group (A, B on HUVECs and HPMECs) and in the imatinib-treated group (C, D on HUVECs and HPMECs). Arrows indicate RBCs adherent to endothelium. (E) Sickle RBC adhesion to heme-activated endothelial cells is significantly reduced by imatinib (5 μM) treatment, compared with control (vehicle, DMSO) treatment (N = 13 subjects, mean adhesion of untreated vs. imatinib-treated sickle cells ± SEM = 383 ± 57 vs. 171 ± 30, P < 0·001, paired t-test).). (RBC, red blood cell; HUVEC, human umbilical vein endothelial cells; HPMEC, human pulmonary microvascular endothelial cells).
Discussion
Multiple publications have reported that oxidative stress is elevated in sickle cells,11,28,29 that this oxidative stress inhibits erythrocyte tyrosine phosphatases,26,30,31,50 and that inhibition of erythrocyte tyrosine phosphatases leads to elevated tyrosine phosphorylation of Band 3.34,34,35 We document here that elevated tyrosine phosphorylation of Band 3 causes destabilisation of the membrane, promoting the release of both MPs and cell-free Hb.34,35,52 Recognising that erythrocyte-derived MPs14 (as previously observed by 12,15,48) and cell-free Hb12,15,47 are pro-embolic, we formulated the hypothesis that elevated oxidative stress in sickle cells should sequentially induce: i) heightened tyrosine phosphorylation of Band 3,30,32,34,53,54 ii) destabilisation of the sickle cell membrane,34,35 iii) release of MPs and cell-free Hb,4,12,14,15,47 iv) activation of adhesion receptors on the vascular endothelium by the released cell-free Hb and heme,10,11,14,16 v) stimulation of micro-embolisms by the prothrombotic MPs,4,12,14,15 vi) enhancement of adhesive properties of sickle cells,10-13,39 and vii) induction of vaso-occlusive events due to concurrent activation of the above processes. The data presented here provide strong evidence that these sequelae do in fact occur in SCD and that Band 3 tyrosine phosphorylation is critical for their occurrence.
In addition to the effects of inhibitors of Band 3 tyrosine phosphorylation on SCD symptoms outlined above, we also envision that these inhibitors may exert other unanticipated positive effects on SCD. Although inhibition of MP and HbS/heme release can be predicted to reduce micro-embolic events, the concomitant reduced blebbing/loss of the erythrocyte membrane area should also improve sickle cell deformability by maintaining a higher cell surface to volume ratio, thereby improving the flow of sickle erythrocytes (Fig. 4A, C). This maintenance of sickle cell volume should also suppress the cell’s tendency to sickle, since the delay in sickling is related to the 30th power of HbS concentration (i.e. a change in only 8% in RBC volume will cause a 10-fold change in the lag time before sickling8,9), and prevention of membrane loss will prevent the associated increase in cytoplasmic HbS concentration (Fig. 4B). The observed decline in sickle cell adhesiveness upon treatment with imatinib (which must derive from an effect on the erythrocyte membrane since the endothelial cells were not exposed to imatinib) should also improve sickle cell flow through the vasculature, and this improved flow should reduce the time each sickle cell remains deoxygenated, thereby further decreasing the tendency to sickle (Fig. 6).8,9 Furthermore, while our studies did not examine sickle cell lifespan, it’s predictable that inhibition of Band 3 phosphorylation should also improve sickle cell survival, since prevention of membrane loss should prolong maintenance of RBC flexibility and thereby reduce its susceptibility to phagocytosis by macrophages55,56 and hence improve SCD-associated anaemia.
Erythrocyte deformability is thought to depend on three parameters: i) the cell’s surface to volume ratio, ii) the viscosity of the cell’s cytoplasm (which is determined by the concentration of Hb), and iii) the intrinsic deformability of the RBC plasma membrane3,5 As shown in Fig. 4, both sickle cell deformability and sickle blood rheology are improved within 4 h of exposure to imatinib. Because significant changes in either RBC volume or surface to volume ratio did not occur over this short time span, the rapid improvement in RBC rheology must have derived from an enhancement in membrane deformability. These data therefore suggest that restoration of the disrupted bridges between Band 3 and the spectrin-actin cytoskeleton by imatinib can improve membrane deformability. The fact that exposure of the isolated sickle cells to imatinib also reduced their tendency to bind heme-activated human endothelial cells (Fig. 6) also suggests that imatinib has a positive effect on sickle cell membrane properties.
With more potent kinase inhibitors readily available,34 the question naturally arises why we selected imatinib to test involvement of Band 3 phosphorylation in SCD. Following initial observations that inhibition of Band 3 phosphorylation by tyrosine kinase inhibitors suppressed release of cell-free Hb and MPs, it seemed logical to explore whether any SCD patients might have fortuitously been treated for another disease with such inhibitors. Upon screening FDA-approved tyrosine kinase inhibitors for inhibition of Band 3 tyrosine phosphorylation, we found that imatinib was an effective inhibitor at clinically relevant concentrations. We then looked for reports in the literature where chronic myelogenous leukaemia (CML) patients who coincidentally suffered from SCD might have been treated with imatinib. We found two anecdotal publications that essentially reported the same observation, namely that administration of imatinib successfully treated the symptoms of SCD in their CML patient.57,58 Although neither author linked his/her findings to any RBC property, their results nevertheless suggest that an inhibitor of Band 3 tyrosine phosphorylation could constitute a therapy for SCD. While chronic use of imatinib should not be considered for the treatment of SCD in children because it can stunt a child’s growth,59,60 a well-designed short term clinical evaluation of imatinib in a more mature population could provide a proof-of-concept test which would inform whether a search for a more selective inhibitor of Band 3 tyrosine phosphorylation might be worthwhile.
Supplementary Material
Fig S1. Effect of DMSO on release of Hb (panel A) and MPs (panel B). Erythrocytes suspended at 30% Hct were treated with 0.5% v/v of DMSO and/or a Band 3 tyrosine phosphorylation stimulant, OV, prior to analysis of the amount of Hb and MPs released. (DMSO, dimethyl sulfoxide; Hb, haemoglobin; MPs, microparticles; Hct, haematocrit; OV, sodium orthovanadate.)
Fig S2. Quantitation of erythrocyte-derived microparticles by Attune NxT Flow Cytometer. Microparticles were identified as glycophorin A positive particles in the size range of 0.1-1.0 μm. The size range was determined using flow cytometry sub-micron particle size reference beads (Thermo Fisher # F13839) as illustrated in panels A, B and C for 0.5 μm, 1.0 μm and 2.0 μm diameter beads respectively, on V-SSC-H (violet side scatter; for better small-particle resolution) vs FSC-H (forward scatter) dot plot. Gate R2 (green) was set as the region for microparticles/events smaller than 1.0 μm diameter. Blue arrows in panels A-C indicate the positions of 0.5 μm, 1.0 μm and 2.0 μm diameter beads respectively, while panel D shows Trucount counting beads (~4.2 μm diameter) which are gated in R1. As can be seen in panels C and D, 2.0 μm beads falls outside gate R2 as do the Trucount beads. Trucount beads were detected in the blue fluorescence detector (488 nm) as shown in panel E, gated R3. Panel F and G show glycophorin A positive erythrocyte-derived microparticles (red arrows) in the plasma from healthy and sickle cell patients, respectively (gate R2). Panel H represents events within gate R2 where glycophorin A positive events (erythrocyte-derived microparticles) are gated in R4 (which constitute the number of erythrocytederived microparticles reported in this article).
Fig S3. Identification of surface markers of erythroid-derived MPs. MPs were stained with 0.5 μL of mouse anti-human glycophorin A antibody and 1.0 μL of CD71 antibody (BD Biosciences #12-0711-82) and incubated for 20 min on ice prior to analysis with Attune NxT flow cytometer. Glycophorin A positive MPs (circled R6) from healthy control (panel A) and from a non-transfused SCD patient (panel B) turned out to be CD71 negative (quadrant R7 and R8). (MPs, microparticles; SCD, sickle cell disease.)
Fig S4. Inhibition of haemolysis and microparticle formation by well-established Syk inhibitors R406 (panels A and B) and PRT062607 (panels C and D). Washed sickle erythrocytes suspended in PBS-G at 30% Hct were incubated with 0.5 μM of either Syk inhibitor or an equivalent volume of vehicle (DMSO) at 37°C for 4 h under 1400 rpm shaking. [n = 3; error bars are expressed as SEM; * denotes P < 0.05; n.s., not statistically significant). (PBS-G, phosphate-buffered saline with glucose; Hct, haematocrit; DMSO, dimethyl sulfoxide; SEM, standard error of the mean.)
Fig S5. Effect of treatment of sickle cells with Syk inhibitors on inhibition of: A) release of free Hb, and B) discharge of erythrocyte-derived MPs. Both Syk inhibitors significantly reduced the amount of Hb and MPs released following treatment with OV, suggesting that Syk is likely the predominant tyrosine kinase which phosphorylates Band 3. (n = 3; error bars are expressed as standard deviation; *** denotes P < 0.001, ** denotes P < 0.01 and n.s. denotes not statistically significant). (Hb, haemoglobin; MPs, microparticles; OV, sodium orthovanadate.)
Acknowledgements
This work was supported by NIH grants R01GM24417-40 (P.S.L.), 1RF1 NS110049-01 (J.W.), R01HL133574 (U.A.G. and J.A.L.), T32HL134622 (R.A.), and NSF #1552782 (U.A.G.). We thank Suzie A. Noronha for providing sickle cell samples from URMC. We thank Ruhani Sansoya for her assistance with the lab-related work.
Footnotes
Conflict of interest disclosures
The authors declare no conflict of interest.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discovery. 2019; 18(2):139–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connes P, Alexy T, Detterich J, Romana M, Hardy-Dessources M-D, Ballas SK. The role of blood rheology in sickle cell disease. Blood Rev. 2016;30:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballas SK, Mohandas N. (2004) Sickle red cell microrheology and sickle blood rheology. Microcirculation (New York, N.Y.: 1994), 11, 209–225. [DOI] [PubMed] [Google Scholar]
- 4.Camus SM, Gausseres B, Bonnin P, Loufrani L, Grimaud L, Charue D, et al. Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood. 2012;120:5050–8. [DOI] [PubMed] [Google Scholar]
- 5.Ellory JC, Robinson HC, Browning JA, Stewart GW, Gehl KA, Gibson JS. Abnormal permeability pathways in human red blood cells. Blood Cells Mol Dis. 2007;39:1–6. [DOI] [PubMed] [Google Scholar]
- 6.Joiner CH, Franco RS. The activation of KCL cotransport by deoxygenation and its role in sickle cell dehydration. Blood Cells Mol Dis. 2001;27:158–64. [DOI] [PubMed] [Google Scholar]
- 7.Wandersee NJ, Hillery CA. (2016) Red Blood Cells and the Vaso-Occlusive Process - Sickle Cell Anemia: From Basic Science to Clinical Practice. In, Costa FF & Conran N (eds). Cham: Springer International Publishing; pp 75–90 [Google Scholar]
- 8.Mozzarelli A, Hofrichter J, Eaton WA. (1987) Delay time of hemoglobin S polymerization prevents most cells from sickling in vivo. Science (New York, N.Y.), 237, 500–506. [DOI] [PubMed] [Google Scholar]
- 9.Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017;129:2719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zennadi R, Whalen EJ, Soderblom EJ, Alexander SC, Thompson JW, Dubois LG, et al. Erythrocyte plasma membrane–bound ERK1/2 activation promotes ICAM-4–mediated sickle red cell adhesion to endothelium. Blood. 2012;119(5):1217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123:377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood. 2015;125:3805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wautier J-L, Wautier M-P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin Hemorheol Micro. 2013;53:11–21. [DOI] [PubMed] [Google Scholar]
- 14.van Beers EJ, Schaap MCL, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, et al. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica. 2009;94:1513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westerman M, Pizzey A, Hirschman J, Cerino M, Weil-Weiner Y, Ramotar P, et al. Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br J Haematol. 2008;142:126–35. [DOI] [PubMed] [Google Scholar]
- 16.Kucukal E, Ilich A, Key NS, Little JA, Gurkan UA. Red blood cell adhesion to heme-activated endothelial cells reflects clinical phenotype in sickle cell disease. Am J Hematol. 2018;93:1050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nebor D, Bowers A, Connes P, Hardy-Dessources M-D, Knight-Madden J, Cumming V, et al. Plasma concentration of platelet-derived microparticles is related to painful vaso-occlusive phenotype severity in sickle cell anemia. PLoS ONE. 2014;9(1):e87243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148:94–101. [DOI] [PubMed] [Google Scholar]
- 19.Elmariah H, Garrett ME, De Castro LM, Jonassaint JC, Ataga KI, Eckman JR, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89:530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bensinger TA, Gillette PN. Hemolysis in sickle cell disease. Arch Intern Med. 1974;133:624–31. [PubMed] [Google Scholar]
- 21.Steinberg MH. (2016) Overview of sickle cell anemia pathophysiology In Sickle Cell Anemia: From Basic Science to Clinical Practice. Costa FF & Conran N (eds). Cham: Springer International Publishing; pp 49–73 [Google Scholar]
- 22.Merciris P, Claussen WJ, Joiner CH, Giraud F. Regulation of K-Cl cotransport by Syk and Src protein tyrosine kinases in deoxygenated sickle cells. Pflugers Arch. 2003;446:232–8. [DOI] [PubMed] [Google Scholar]
- 23.Mohandas N, Clark MR, Jacobs MS, Shohet SB. Analysis of factors regulating erythrocyte deformability. J Clin Investig. 1980;66:563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor JG, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT & Steinberg MH Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PLoS ONE. 2008;3:e2095–e2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hebbel RP, Morgan WT, Eaton JW, Hedlund BE. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Nati Acad Sci. 1988;85(1):237–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferru E, Pantaleo A, Carta F, Mannu F, Khadjavi A, Gallo V, et al. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica. 2014;99:570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shalev O, Hebbel RP. Catalysis of soluble hemoglobin oxidation by free iron on sickle red cell membranes. Blood. 1996;87:3948–52. [PubMed] [Google Scholar]
- 28.George A, Pushkaran S, Konstantinidis DG, Koochaki S, Malik P, Mohandas N, et al. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood. 2013;121:2099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nolfi-Donegan D, Pradhan-Sundd T, Pritchard KA, Hillery CA. Redox signaling in sickle cell disease. Current Opin Physiol. 2019;9:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merciris P, Hardy-Dessources M-D, Giraud F. Deoxygenation of sickle cells stimulates Syk tyrosine kinase and inhibits a membrane tyrosine phosphatase. Blood. 2001;98(10):3121–7 [DOI] [PubMed] [Google Scholar]
- 31.Zipser Y, Piade A, Kosower NS. Erythrocyte thiol status regulates band 3 phosphotyrosine level via oxidation/reduction of band 3-associated phosphotyrosine phosphatase. FEBS Lett. 1997;406(1-2):126–30. [DOI] [PubMed] [Google Scholar]
- 32.Brunati AM, Bordin L, Clari G, James P, Quadroni M, Baritono E, et al. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: identification of primary and secondary phosphorylation sites. Blood. 2000;96:1550–7. [PubMed] [Google Scholar]
- 33.Hierso R, Lemonne N, Villaescusa R, Lalanne-Mistrih M-L, Charlot K, Etienne-Julan M, et al. Exacerbation of oxidative stress during sickle vaso-occlusive crisis is associated with decreased anti-band 3 autoantibodies rate and increased red blood cell-derived microparticle level: a prospective study. Br J Haematol. 2017;176:805–13. [DOI] [PubMed] [Google Scholar]
- 34.Puchulu-Campanella E, Turrini FM, Li Y-H, Low PS. Global transformation of erythrocyte properties via engagement of an SH2-like sequence in band 3. Proc Nati Acad Sci. 2016;113(48):13732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferru E, Giger K, Pantaleo A, Campanella E, Grey J, Ritchie K, et al. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood. 2011;117:5998–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Micro. 2018;68:263–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36:707–17. [DOI] [PubMed] [Google Scholar]
- 38.Liu C, Zhao W, Christ GJ, Gladwin MT, Kim-Shapiro DB. Nitric oxide scavenging by red cell microparticles. Free Radic Biol Med. 2013;65:1164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hebbel RP. Reconstructing sickle cell disease: a data-based analysis of the ‘hyperhemolysis paradigm’ for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol. 2011;86:123–54. [DOI] [PubMed] [Google Scholar]
- 40.Almeida CB, Souza LEB, Leonardo FC, Costa FTM, Werneck CC, Covas DT, et al. Acute hemolytic vascular inflammatory processes are prevented by nitric oxide replacement or a single dose of hydroxyurea. Blood. 2015;126(6):711–20. [DOI] [PubMed] [Google Scholar]
- 41.Parrow NL, Tu H, Nichols J, Violet P-C, Pittman CA, Fitzhugh C, et al. Measurements of red cell deformability and hydration reflect HbF and HbA2 in blood from patients with sickle cell anemia. Blood Cells Mol Dis. 2017;65:41–50. [DOI] [PubMed] [Google Scholar]
- 42.Welsh JA, Holloway JA, Wilkinson JS, Englyst NA. Extracellular vesicle flow cytometry analysis and standardization. Front Cell Development Biol. 2017;5:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou S, Giannetto M, DeCourcey J, Kang H, Kang N, Li Y, et al. Oxygen tension–mediated erythrocyte membrane interactions regulate cerebral capillary hyperemia. Sci Adv. 2019;5(5):eaaw4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rab MAE, van Oirschot BA, Bos J, Merkx TH, van Wesel ACW, Abdul-malik O, et al. Rapid and reproducible characterization of sickling during automated deoxygenation in sickle cell disease patients. Am J Hematol. 2019;94:575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim M, Alapan Y, Adhikari A, Little JA, Gurkan UA. (2017) Hypoxia-enhanced adhesion of red blood cells in microscale flow. Microcirculation (New York, N.Y. : 1994), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mondal P, Stefek B, Sinharoy A, Sankoorikal B-J, Abu-Hasan M, Aluquin V. The association of nocturnal hypoxia and an echocardiographic measure of pulmonary hypertension in children with sickle cell disease. Pediatr Res. 2019;85:506–10. [DOI] [PubMed] [Google Scholar]
- 47.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO 3rd, Schechter AN, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–9. [DOI] [PubMed] [Google Scholar]
- 48.Hebbel RP, Key NS. Microparticles in sickle cell anaemia: promise and pitfalls. Br J Haematol. 2016;174(1):16–29. [DOI] [PubMed] [Google Scholar]
- 49.Metere A, Iorio E, Pietraforte D, Podo F, Minetti M. Peroxynitrite signaling in human erythrocytes: synergistic role of hemoglobin oxidation and band 3 tyrosine phosphorylation. Arch Biochem Biophys. 2009;484:173–82. [DOI] [PubMed] [Google Scholar]
- 50.Pantaleo A, Ferru E, Pau MC, et al. (2016) Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p72 Syk. Hindawi, 2016, Available at: https://www.hindawi.com/journals/omcl/2016/6051093/cta/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atwell S, Adams JM, Badger J, Buchanan MD, Feil IK, Froning KJ, et al. A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J Biolog Chem. 2004;279:55827–32. [DOI] [PubMed] [Google Scholar]
- 52.Stefanovic M, Puchulu-Campanella E, Kodippili G, Low PS. Oxygen regulates the band 3-ankyrin bridge in the human erythrocyte membrane. Biochem J. 2013;449:143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terra HT, Saad MJ, Carvalho CR, Vicentin DL, Costa FF, Saad ST. Increased tyrosine phosphorylation of band 3 in hemoglobinopathies. Am J Hematol. 1998;58:224–30. [DOI] [PubMed] [Google Scholar]
- 54.Bordin L, Brunati AM, Donella-Deana A, Baggio B, Toninello A, Clari G. Band 3 is an anchor protein and a target for SHP-2 tyrosine phosphatase in human erythrocytes. Blood. 2002;100:276–82. [DOI] [PubMed] [Google Scholar]
- 55.Buffet PA, Milon G, Brousse V, Correas J-M, Dousset B, Couvelard A, et al. Ex vivo perfusion of human spleens maintains clearing and processing functions. Blood. 2006;107:3745–52. [DOI] [PubMed] [Google Scholar]
- 56.Mohandas N Of mice and men: the voracious spleen. Blood. 2006;107(9):3426. [Google Scholar]
- 57.Stankovic Stojanovic K, Thiolière B, Garandeau E, Lecomte I, Bachmeyer C, Lionnet F. Chronic myeloid leukaemia and sickle cell disease: could imatinib prevent vaso-occlusive crisis? Br J Haematol. 2011;155(2):271–2 [DOI] [PubMed] [Google Scholar]
- 58.Murphy M, Close J, Lottenberg R, Rajasekhar A. Effectiveness of imatinib therapy for sickle cell anemia and chronic myeloid leukemia. Am J Med Sci. 2014;347:254–5. [DOI] [PubMed] [Google Scholar]
- 59.Narayanan KR, Bansal D, Walia R, Sachdeva N, Bhansali A, Varma N, et al. Growth failure in children with chronic myeloid leukemia receiving imatinib is due to disruption of GH/IGF-1 axis. Pediatr Blood Cancer. 2013;60:1148–53. [DOI] [PubMed] [Google Scholar]
- 60.Rastogi MV, Stork L, Druker B, Blasdel C, Nguyen T, Boston BA. Imatinib mesylate causes growth deceleration in pediatric patients with chronic myelogenous leukemia. Pediatr Blood Cancer. 2012;59:840–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Effect of DMSO on release of Hb (panel A) and MPs (panel B). Erythrocytes suspended at 30% Hct were treated with 0.5% v/v of DMSO and/or a Band 3 tyrosine phosphorylation stimulant, OV, prior to analysis of the amount of Hb and MPs released. (DMSO, dimethyl sulfoxide; Hb, haemoglobin; MPs, microparticles; Hct, haematocrit; OV, sodium orthovanadate.)
Fig S2. Quantitation of erythrocyte-derived microparticles by Attune NxT Flow Cytometer. Microparticles were identified as glycophorin A positive particles in the size range of 0.1-1.0 μm. The size range was determined using flow cytometry sub-micron particle size reference beads (Thermo Fisher # F13839) as illustrated in panels A, B and C for 0.5 μm, 1.0 μm and 2.0 μm diameter beads respectively, on V-SSC-H (violet side scatter; for better small-particle resolution) vs FSC-H (forward scatter) dot plot. Gate R2 (green) was set as the region for microparticles/events smaller than 1.0 μm diameter. Blue arrows in panels A-C indicate the positions of 0.5 μm, 1.0 μm and 2.0 μm diameter beads respectively, while panel D shows Trucount counting beads (~4.2 μm diameter) which are gated in R1. As can be seen in panels C and D, 2.0 μm beads falls outside gate R2 as do the Trucount beads. Trucount beads were detected in the blue fluorescence detector (488 nm) as shown in panel E, gated R3. Panel F and G show glycophorin A positive erythrocyte-derived microparticles (red arrows) in the plasma from healthy and sickle cell patients, respectively (gate R2). Panel H represents events within gate R2 where glycophorin A positive events (erythrocyte-derived microparticles) are gated in R4 (which constitute the number of erythrocytederived microparticles reported in this article).
Fig S3. Identification of surface markers of erythroid-derived MPs. MPs were stained with 0.5 μL of mouse anti-human glycophorin A antibody and 1.0 μL of CD71 antibody (BD Biosciences #12-0711-82) and incubated for 20 min on ice prior to analysis with Attune NxT flow cytometer. Glycophorin A positive MPs (circled R6) from healthy control (panel A) and from a non-transfused SCD patient (panel B) turned out to be CD71 negative (quadrant R7 and R8). (MPs, microparticles; SCD, sickle cell disease.)
Fig S4. Inhibition of haemolysis and microparticle formation by well-established Syk inhibitors R406 (panels A and B) and PRT062607 (panels C and D). Washed sickle erythrocytes suspended in PBS-G at 30% Hct were incubated with 0.5 μM of either Syk inhibitor or an equivalent volume of vehicle (DMSO) at 37°C for 4 h under 1400 rpm shaking. [n = 3; error bars are expressed as SEM; * denotes P < 0.05; n.s., not statistically significant). (PBS-G, phosphate-buffered saline with glucose; Hct, haematocrit; DMSO, dimethyl sulfoxide; SEM, standard error of the mean.)
Fig S5. Effect of treatment of sickle cells with Syk inhibitors on inhibition of: A) release of free Hb, and B) discharge of erythrocyte-derived MPs. Both Syk inhibitors significantly reduced the amount of Hb and MPs released following treatment with OV, suggesting that Syk is likely the predominant tyrosine kinase which phosphorylates Band 3. (n = 3; error bars are expressed as standard deviation; *** denotes P < 0.001, ** denotes P < 0.01 and n.s. denotes not statistically significant). (Hb, haemoglobin; MPs, microparticles; OV, sodium orthovanadate.)