

Abstract
Multipotent neural stem cells (NSCs) are widely applied in pre-clinical and clinical trials as a cell source to promote tissue regeneration in neurodegenerative diseases. Frequently delivered as dissociated cells, aggregates or self-organized rosettes, it is unknown whether disruption of the NSC rosette morphology or method of formation affect signaling profiles of these cells that may impact uniformity of outcomes in cell therapies. Here we generate a neural cell-cell interaction microchip (NCCIM) as an in vitro platform to simultaneously track an informed panel of cytokines and co-evaluate cell morphology and biomarker expression coupled to a sandwich ELISA platform. We apply multiplex in situ tagging technology (MIST) to evaluate ten cytokines (PDGF-AA, GDNF, BDNF, IGF-1, FGF-2, IL-6, BMP-4, CNTF, β-NGF, NT-3) on microchips for EB-derived rosettes, single cell dissociated rosettes and reformed rosette neurospheres. Of the cytokines evaluated, EB-derived rosettes secrete PDGF-AA, GDNF and FGF-2 prominently, whereas this profile is temporarily lost upon dissociation to single cells and in reformed neurospheres two additional cytokines, BDNF and β-NGF, are also secreted. This study on NSC rosettes demonstrates the development, versatility and utility of the NCCIM as a sensitive multiplex detector of cytokine signaling in a high throughput and controlled microenvironment. The NCCIM is expected to provide important new information to refine cell source choices in therapies as well as to support development of informative 2D or 3D in vitro models including areas of neurodegeneration or neuroplasticity.
Table of contents
A neural cell-cell interaction microchip evaluates 10 cytokines released by neural stem cells and their morphology and biomarker expression.
Graphical Abstract
INTRODUCTION
Neural stem cells (NSCs) present expanded opportunities for neural cell therapy due to their multipotency and ability to respond to diverse cell signaling microenvironments to differentiate and functionally integrate in vivo1,2. The frequent choice of NSCs for a variety of central nervous system (CNS) therapies3–11 is further facilitated by the ability to readily generate NSCs in culture and identify them through biomarkers and distinct rosette morphology12. Dissociated NSCs or cell aggregates have been tested for treatment of neurodegeneration12,13. However, neuroblastoma formation can occur when NSCs are used in CNS therapies, which are presumably due to interrupted lineage commitment. Cytokines that participate in autocrine and paracrine signaling are critical for regulation of NSC growth and function. How effectively cells communicate in their microenvironment has been demonstrated to be important for improving functional integration of transplanted cells in vivo. For example, the homotypic choice of CNS NSCs regionally specified for spinal applications or forebrain specified NSCs, has significantly improved cell functional integration14,15. With the awareness that cell communication plays a key role in therapeutic success of transplanted cells, tools that expand our capability to evaluate the complex relationships between cell-cell signaling in microenvironments are needed. Such strategies in regard to NSCs are expected to benefit reproducibility in cell sourcing with more predictable outcomes in vivo and provide future platforms to advance strategies that manipulate cell signaling parameters1.
Medium to high throughput formats have been introduced recently, leveraging lab-on-a-chip technologies that integrate microarrays as miniaturized multiplexed sensors. Most of those sensors rely on enzyme-linked immunosorbent assay (ELISA) for protein detection where one or more antibodies are anchored on the surface. A novel extension of this approach is the use of antibody or DNA/RNA aptamer-based arrays in high throughput microfluidic platforms that allows analysis of complex cell-cell microenvironments to look at T-cell activation16 and liver cell signaling17. Further technical optimization that advances throughput has included on-chip regeneration of aptasensors following cytokine detection for successive cycles18, decoding DNA arrays using hybridization19, rapid, automated quantification20, and multiple cytokine detection from a single aptamer-functionalized electrode21. Use of microbeads is enabling ultrahigh throughput assays, including work from this group on multiplexed single-cell protein profiling22. In earlier studies, microfluidic droplets combined with antibody-coated microbeads were used in high throughput evaluation of three specific cytokines to probe cell heterogeneity in activated T cells23 as well as evaluating growth factors in primary hepatocytes24. An important new area of interest for cell therapies is understanding cytokine signaling potential of transplanted cells that may affect their behavior in vivo. There is a continuing need to improve high throughput and miniaturization platforms that will guide cell therapies and aid development of in vitro disease models.
The NSCs remain a high priority cell resource in therapeutic neurophysiology applications. Two typically used methods to generate and expand pluripotent stem cell-derived NSC rosettes for downstream applications include formation of rosettes through embryoid body (EB)-derived intermediates25–27 or by direct differentiation of stem cells in monolayer cultures28–30. Neural rosettes obtained by these approaches in two comparative studies express early ectodermal lineage and radial glial biomarkers and have the ability to self-renew and also differentiate to neuronal and glial cell types based on the biological cues in their environment31,32. Gene expression in NSC rosettes and during neuronal development have also been examined33,34. However, the effect of two- or three-dimensional derivation strategies as well as aggregate or single-cell rosette morphology on cytokine secretion have not been explored. It has been shown that cell shape can impact cell signaling and two- or three-dimensional morphology of cells can alter their fate. The change in cell-matrix interactions is associated with varied adhesion signaling that could alter cells ability to differentiate35. It is unknown whether disruption of the NSC rosette morphology or method of formation affect signaling profiles of these cells that may impact uniformity of outcomes in animal models or clinical cell therapies. In the current study we specifically address cytokine signaling of NSCs following different derivation strategies as well as state of cells as dissociated and/or as aggregates. The strength of our approach is the ability to track changes in ten cytokines of interest simultaneously while also monitoring cell microenvironment in a high throughput multiplex array.
We report development of a neural cell-cell interaction microchip (NCCIM) for high-throughput multiplex analysis of NSCs generated by two methods, EB intermediates and monolayer cultures. We evaluate cytokine signaling using EB-formed rosettes, dissociated rosettes and three-dimensional rosette neurospheres. We integrate a multiplex in situ tagging (MIST) microarray to simultaneously evaluate 10 known neurotropic factors and growth factors important in regard to NSC/NPCs maintenance, proliferation and differentiation36–38 (Table S1). Evaluation required development of on-microchip culture conditions to form and evaluate rosettes. The delicate architecture of rosettes prevents their transplantation from culture dishes to the PDMS-fabricated microchambers array (PFMA) that forms the cell platform for the NCCIM technology. The NCCIM elegantly captures the cytokine signaling relationship between rosette populations including self-organized rosettes, dissociated rosette cells and rosette neurospheres. This study demonstrates the versatility of the NCCIM as a new high-throughput, multiplexed approach to evaluate architecture-dependent cytokine signaling in NSCs in vitro. The NCCIM technology is expected to benefit future in vivo studies in transplanted NSCs as well as provide a robust experimental platform for continued in vitro studies of 2D or 3D tissues with applications for neuronal development and diseases.
RESULTS
Development of a Neural Cell-Cell Interaction Microchip, NCCIM
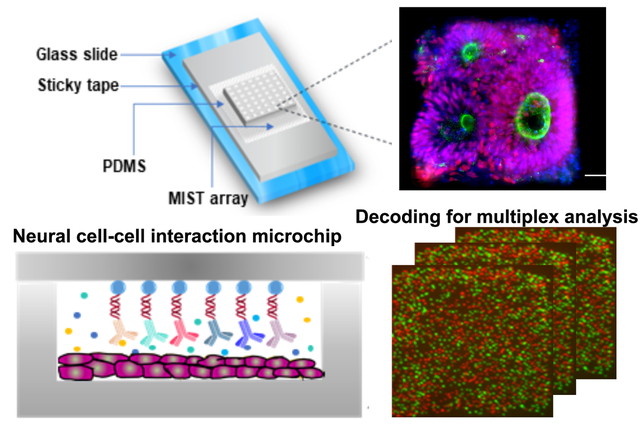
NCCIM was developed as an integrated multiplex platform that allows cytokine detection simultaneously or sequentially with visualization of rosette cell morphology and biomarker analysis. The device combines a PDMS-fabricated microchambers array (PFMA) that is customized for number and size of microchambers and integrated with a multiplexed in situ tagging (MIST) sensor array based on antibody detection and immobilized onto a non-fluorescence, pressure-sensitive tape. The PFMA and MIST arrays combine to form the NCCIM, with each component mounted onto separate glass slides. NSCs (whole or dissociated) are seeded within the PFMA in culture dishes then combined with the MIST sensor array in a custom clamp (Figure 1a). The cells can be pre-seeded and incubated in the PFMA for hours to days. Once sealed, the cells in the physically isolated microchambers release cytokines to the microenvironment, and thereafter these proteins are selectively captured and detected on the MIST array only pertinent to that microchamber. Thus, each microchamber relays unique information that is captured in MIST antibody analysis. Multiple identical or unique cell-cell interaction microenvironments can be identified and evaluated simultaneously. The MIST array is based on 3.1 μm microbeads, with ~8460 microbeads per microchamber (300μm x 300μm), each saturated with antibodies to provide a multiplex readout. Each microbead carries one of 10 types of orthogonal oligonucleotide DNA at the beginning which are permanently linked to the bead surface. Such oligonucleotides serve as the base for conversion to antibody array through hybridization with antibody-complementary DNA (cDNA) as well as for decoding to identify the type of detected proteins. To obtain a multiplex protein readout, a series of detection and decode steps are performed, followed by assignment of output readings (Figure 1b).
Figure 1. Principle of the NCCIM assay for evaluating neural progenitor cells.

(a) layout of the fully assembled NCCIM. The MIST array is fabricated on the surface of a sticky tape which is placed on a glass slide. A PDMS replica carrying microchambers is mated with a MIST array with a mechanical clamp (not shown). Cells cultured in the microchambers release cytokines which will be captured by the antibodies on the MIST array for ELISA detection. The biotinylated detection antibodies are labeled with streptavidin-horseradish (HRP) for tyramide-Cy3 assisted signal amplification. (b) Procedure to identify the type of secreted proteins detected on individual microbeads. After protein detection, the fluorescence intensity of each microbead is quantified as the cytokine expression level. 0.5M NaOH is added to the array to dissociate the hybridized DNAs and release the antibodies and the captured proteins. Only single-stranded oligo DNAs are left on the MIST array. In each of 4 cycles of the decoding process, a cocktail of fluorophore tagged oligo DNAs is applied, and the same array is imaged by a fluorescence microscope. Then the hybridized DNAs are dissociated before the next cycle. Once all the images are overlapped, the sequence of fluorescent colors on each microbead is obtained, which corresponds to a specific type of protein. Such reiterative hybridization and dissociation enable 10-plex detection of cytokines.
The MIST arrays use a fluorescence sandwich ELISA procedure to quantify released cytokines. In its generation, the MIST array is convertible between an antibody-tethered array and DNA-only tethered bead array by hybridization or dissociation of antibody-complementary DNA conjugates onto DNA-beads. Tyramide-Cy3 is used to enhance signal strength so the fluorescence signals on microbeads can be easily detected by a common fluorescence microscope. Multiplex detection is done here with two fluorescent tags, Cy3 and Cy5, by processing and decoding over successive cDNA hybridization labeling and dissociation steps performed over a few cycles. The DNA dissociation after protein detection and after each cDNA labeling is highly efficient as no residual fluorescence is observed (Figure S1). Multi-color images acquired from each microarray cycle together with the protein detection image are aligned perfectly so that the same microbead at a given location generate a particular sequence of color changes, a color barcode, over the successive labeling/dissociation steps, corresponding to a given cytokine protein and its conjugated antibody-cDNA signal. The predesigned codes used are indexed (Table S2). The number of color tags applied and cycle number together determine the multiplexity which is the number of barcodes and exponentially increases with each cycle. With 2 fluorescent color tags and 4 cycles, the maximum multiplexity is 24=16 types of cytokines achievable. Each cycle needs ~ 1 h operation time, and thus 4 h is needed to decode for analyzing 10 cytokines in this work. This decoding procedure can be done either before or after the protein detection. Such design is cost effective and overcomes the limit of available fluorescent tags on an imaging system with emission/excitation overlap that often limits evaluation to a maximum of four cytokines.
Direct formation of NSC rosettes on microchips for NCCIM analysis
When cultured in a dish, NSCs spontaneously form radial structures commonly referred to as rosettes; the importance of this structure for self-regulation of multipotency and proliferation is not well known. The distinct but delicate morphology of rosettes prevents consistent transplantation of these structures by lifting from dishes to the PFMA of the NCCIM. Instead rosettes were formed directly in the PFMA (Figure 2) either by dissociating rosettes from 2D adherent cultures in dishes and reformation in the PFMA or by direct seeding of custom-sized 300 μm embryoid body (EB) intermediates. We previously described the use of custom microwells to generate uniformly sized EBs39 and generation and characterization of the human induced pluripotent stem cell line F3.5.2 used in these studies40,41. EBs were loaded into the PFMA and cultured 4 days by SMAD dual inhibition method42 in neural induction medium. Before loading, the PFMA is plasma-treated and coated with 1% Matrigel to retain cells. We determined that the microchamber dimension of 300 μm x 300 μm x 130 μm in the PFMA ensured the highest retention efficiency of EBs or seeded. This was done by comparing rosette formation from two different size EBs (300 and 500 μm) by immunocytochemistry and testing two different chamber depths to choose the optimum chamber dimensions for rosette formation (Figure S2). For dissociated neural rosettes, hiPSC line F3.5.2 was used to generate a monolayer in a culture dish with NSCs forming after 7 days27. NSCs were dissociated into single cells and equally distributed into the PFMA. Seeded cells spontaneously formed into uniform neurospheres containing rosettes after 48 hours (Figure 2). This is different from seeded EBs in the PFMA that when induced neurally form planar rosette structures.
Figure 2. Analysis of neural rosettes by microplatform seeding onto the NCCIM.

(a) Neural induction strategies using either custom sized EB intermediates or preformed rosettes in 2D cultures, dissociated and reseeded into the PDMS-fabricated microchamber array (PFMA). Rosette formation in the PFMA from neural-induced EBs generates 3 size categories (1 large rosette, or 3–4 medium rosettes or >5 small rosettes), whereas dissociated NSC rosettes spontaneously form uniformly sized rosette neurospheres in the PFMA (b) Phase images of rosette and rosette neurosphere formation on the PFMA. Scale bar is 100 μm.
PFMA-formed NSC rosettes that were derived from EBs settle into the typical distinct flower shape of rosettes in the PFMA chambers similar to what is previously observed on the dish (Figure 3a). However, when dissociated cells are seeded into the PFMA they form neurospheres within 48 hours (Figure 3b). Within these neurospheres one can clearly see rosettes and appropriate biomarkers for rosettes. When we compare newly formed neurospheres from monolayer rosette and EB-derived rosettes on PFMA to rosette formed on the dish, we observed similar quantitively expression of early neuronal and glial biomarkers (Figure S3). PFMA-formed NSC rosettes display biomarker expression patterns as discussed below. Immunocytology of six proteins expressed in rosettes were similar to rosettes formed in culture dishes (Figure 3a and 3b). This includes paired box (Pax) 6, as one of the earliest transcription factors expressed in NSCs43, Nestin that is an intermediate filament protein expressed in NSCs prior to differentiation and impacts self-renewal44, MKI67 (ki67) that is a nuclear protein and is expressed in dividing cells and Sox 1 and Sox 2 that are members of Sox B family of transcription factors that regulate NPCs by maintaining the progenitor state and preventing neuronal differentiation45. The tight junction protein ZO-1 was also evaluated since it is initially expressed evenly on the surface of undifferentiated cells, but after neural induction and polarization of rosette cells, ZO-1 relocalizes centrally to the apical membrane of rosettes and is a key feature in formed rosettes26. For on-microchip analysis with the NCCIM, we focused on Sox1 (neural commitment) and ZO-1 (rosette polarization) for optimizing NCCIM conditions. The localization of ZO-1 at the center of radial neuroepithelial cells in rosettes is particularly useful to count rosettes by identifying the central ring of the polarized cells. Although uniformly sized EBs are seeded into microwells of the PFMA, we observe that the planar rosettes that form are a mixture of various sizes and numbers (Figure 2, 3), and they are defined as three categories for analysis (Figure 3c): Category 1, greater than five small rosettes, with diameters of the centers ranging from 5 μm to 25 μm; Category 2, less frequent, with one single large rosette with occasionally small scattered rosettes surrounding; Category 3, the least frequent group, with two or three rosettes of 40 μm to 80 μm center size per microchamber. All rosettes were visible initiating from day two of culture in the PFMA.
Figure 3. Immunoctyology of neural rosettes formed on the NCCIM.

(a) EB-derived rosettes in the dish and in the NCCIM. Cells are stained with DAPI, tight junction protein ZO1, NSC multipotency marker Nestin, neural ectoderm (Pax6), proliferation marker ki67, and neural progenitor (Sox1, Sox2) biomarkers as indicated. (b) Monolayer rosettes in the dish are dissociated and seeded into the NCCIM and form uniformly sized NSC rosette neurospheres by Day 2. Cells are evaluated with similar biomarkers as (a). (c) Phase and fluorescence microscopy images of EB-derived rosettes in the PDMS microchambers of the NCCIM. Spontaneously formed rosettes in microwells results in three main size distribution categories: one large rosette, 3–4 medium size rosettes, and >5 small size rosettes. Cells are stained with ZO-1 (green) and Sox1 (red) antibodies. Scale bar is 50 μm.
Validation of cytokines for NCCIM MIST array multiplex protein detection
The performance and quality of the MIST array were first fully validated as follows. Each well on a 300 μm X 300 μm microarray contains 8460 ± 646 microbeads on average. Each type of the DNA coated microbeads has 305 ± 44 copies on a microarray (Table S2). After labeling with the same concentrations of complementary DNAs tagged with Cy3, the fluorescence intensities of all 10 types of DNA coated microbeads are The quantified and compared (Figure S4), with <5% variation being observed. <2% variation of batch-to-batch DNA coating is found by the similar evaluation method. Half of the microbeads are blank (without carrying DNA), to improve the spatial separation of the signals detected and sensitivity by lowering the likelihood of a missed recognition event by computational algorithms. The MIST array validation check revealed no crosstalk in protein detection, which is achieved by comparing the locations of the microbeads with Cy3-cDNA labeling versus fluorescent microbeads with protein detection. For protein detection, all the DNA carrying microbeads are anchored with antibodies using antibody-cDNA conjugates, while only a single recombinant protein is applied to each array before applying all the ten detection antibodies. We observed that the DNA and antibody-cDNA hybrid pairs were highly orthogonal with less than 0.5% crosstalk by comparing the fluorescence intensities of bright microbeads that correctly detect the DNA/proteins and the DNA encoded microbeads that should not detect the molecules. If cross reactivity were present there would be more microbeads with detected signal than anticipated, and the distribution of fluorescent microbeads would be different from Cy3-cDNA labeled microbeads. By contrast, if no crosstalk occurs, the distribution should be the same and should completely overlap. The destructive overlap between images shows the difference of location of each microbeads in two images. The percentage of non-overlapped microbeads over total microbeads is quantified as the rate of bead cross-reactivity. As shown in Figure 4, the images of destructive overlap indicate less than 2% bead cross-reactivity with confidence indicating that our multiplexed assay of 10 proteins gives unique detection of cytokine signals. The cDNA labeling in decoding achieves a signal-to-noise (S/N) ratio at 37 with 3% fluorescence variation (standard deviation), and no interference of non-specific binding to the cDNA labeling is observed (Figure S5). The high S/N ratio and low fluorescence variation indicate high accuracy and efficiency of the decoding process. Variation of protein detection was measured using recombinant protein BDNF at 1 ng/mL. Although the variation for protein detection at 17% is higher than cDNA labeling, such variation is still negligible compared with biological variations which are normally a few times higher.
Figure 4. Crosstalk check and sensitivity of the MIST arrays for multiplexed protein detection.

(a) Validation of no crosstalk for analyzing the cytokines and growth factors in the 10-protein panel. Images of the 1st row are fluorescence signals on the MIST arrays via ELISA protein detection. Images of the 2nd row are fluorescence signals through Cy3-cDNA labeling. Each column corresponds to the protein in the multiplex panel that is coupled with a particular cDNA sequence. Subtracting fluorescence signal of the 1st row images by that of the 2nd row images results in destructive overlap images on the 3rd row. Low signal in the destructive overlap images corresponds to low crosstalk. (b) Calibration curves for immunoassays on the MIST arrays using recombinant proteins with the concentrations ranging from 33 pg/mL to 10 ng/ml. Scale bars are 100 μm. The experiments were repeated for 3 times.
A panel of ten cytokines was selected (Table S1) that includes glial cell-derived neurotrophic factor (GDNF), platelet-derived growth factor-AA (PDGF-AA), interleukin 6 (IL-6), ciliary neurotrophic factor (CNTF), Insulin-like growth factor (IGF-1), brain-derived neurotrophic factor (BDNF), basic fibroblast growth factor (FGF-2), neurotrophin-3 (NT-3), bone morphogenetic protein-4 (BMP-4), and nerve growth factor (β-NGF). Recombinant proteins of the 10 cytokines were validated at various concentrations (10, 3.3, 1, 0.33, 0.1 and 0.03 ng/mL; Figure 4a) when applied to the MIST array for the sensitivity test (Figure 4b). Calibration curves were generated and plotted for each protein. The detection limit is calculated by the background plus 3 standard deviation of the fluorescence intensities at 0 pg/mL for each calibration curve46. The detection limit is comparable, though slightly higher than a non-microchip conventional ELISA method. Detection was estimated as 20.4 pg/mL for GDNF, 18.0 pg/mL for PDGF-AA, 2.7 pg/mL for IL-6, 8.5 pg/mL for CNTF, 14.9 pg/mL for IGF-1, 23.3 pg/mL for BDNF, 12.0 pg/mL for FGF-2, 57.2 pg/mL for NT-3, 11.2 pg/mL for BMP-4, and 39.1 pg/mL for β-NGF. In our initial tests we observed that tyramide enhancement provided only an increased 5 to 10 times additional improvement of signal over the one without enhancement (Figure S6), and we did not observe the reported 100 times achievable on immunohistochemistry and microarray47. Since tyramide enhancement is based on the available tyrosine on proteins in proximity, the low density of proteins on microbeads might be the limiting factor. The NCCIM additionally enables interrogation of other parameters, such as morphological information and biomarkers beyond simply cytokine release information.
NCCIM analysis of NSC rosettes enclosed with a MIST sensor array is performed over an 8-hour time period before multiplexed analysis of the 10 cytokines (Figure 5). The time period was chosen based on previous experience with enclosed chambers22 and after testing various incubation durations within a limited timeframe (4 h, 8 h and 14 h) to optimize cell viability and protein detection of low expression cytokines (Figure S7). Long incubation results in more accumulation of released cytokines and hence low expression cytokines becomes detectable, while the cell viability decreases with time due to limited nutrition in a chamber. Some loss of NSC viability occurs in this timeframe, but substantial viability is retained based on cell physiology, strong biomarker expression and by use of propidium iodide (PI) staining (Figure 5b, Table S3). The NSC rosettes maintained polarized expression of ZO-1 at rosette centers and expression of the neuroprogenitor biomarker SOX1 after 8 hours (Figure 5c). By PI staining (Table S3), we observed 60–100% viable cells for 84% of chambers with only 6% of microchambers exhibiting significant cell death. Thus the 8-hour incubation time was applied in all analysis
Figure 5. Viability and multipotency of rosettes on the NCCIM.

(a) Flow diagram from seeding of cells into the PFMA that is combined with the MIST array to generate the assembled NCCIM. Cells were maintained within the NCCIM for 8 hours then evaluated by(b) propidium iodide (PI) staining of EB-derived rosettes and rosette neurospheres to visualize dead cells at hour 0 and hour 8. In (c) neural progenitor marker SOX1 is shown with adherens junction protein ZO-1 that highlights the center of polarized rosette cells. Cells maintain their multipotent state after NCCIM incubation for 8 hours. Scale bar is 50 μm.
NCCIM multiplex cytokine analysis of EB-derived rosettes
NSC rosettes were formed spontaneously in PFMA after four days of culturing EBs in N2/B27 neural induction media and were then evaluated by a ten cytokine MIST sensor array. The NCCIM cytokine profiles were quantified and our analysis showed the same trend in detected cytokines in both fluorescence and quantified data (Figure 6). To exclude the assay noise and facilitate data analysis, the signal detection limit (background fluorescence intensity + 3×SD) is set as the threshold for each protein assay, and the raw NCCIM fluorescence intensities are subtracted with the thresholds to obtain the fluorescence signals which represent the detectable signals (Figure 6c). Thus the assay noise is largely reduced through this processing. Based on the calibration curves (Figure 4b) and the signal detection limit, the fluorescence intensities are converted to protein concentrations.
Figure 6. NCCIM cytokine detection assay of EB-derived neural rosettes.

Evaluation of EB-derived rosettes by NCCIM. (a) Plot of quantified ten proteins assayed by NCCIM in EB-derived rosettes. Plot shows the range of the data points for each cytokine. A horizontal line on each box shows the mean expression of each cytokine. The significant differences of mean values between cytokines for each category are shown in Figure S8. (b) Heatmap of quantified proteins assayed by NCCIM. (c) Plot of fluorescence signals of proteins assayed by NCCIM. Protein signal is obtained by subtracting the raw fluorescence intensity by the background + 3×SD for each protein. (d) Heatmap of fluorescence signals of proteins assayed by NCCIM. (e) Dot plots of quantify proteins produced by 3 categories of rosettes in individual microchambers. (f) Dot plots of detected fluorescence signals of proteins in individual microchambers. Red dash lines indicate the level of background + 3×SD as the cutoff for signal vs noise. P values: *, P<0.05; **, P<0.01; ***, P<0.01; ****, P<0.0001.
Detection results of EB-generated rosettes were based on 25 microchambers from each rosette category (Figure 2 & 3). Category 1 contains five or more small rosettes in each microchamber, Category 2 with one large rosette in each microchamber and Category 3 with three to four medium rosettes in each microchamber. Based on quantified data, one cytokine, PDGF-AA is significantly (P < 0.0001) produced in all three categories (Figure 6a–d). GDNF was also significantly expressed in category 2 (P < 0.01). Fluorescence data showed FGF-2 was also significantly produced in all three categories with different p-values (Cat 1: P < 0.0001, Cat 2: P: 0.0005, Cat 3: P: 0.0006) and IL-6 was produced significantly in category 1 (P: 0.01) (see Table S4 and Figure S8). Category 1, Category 2 and Category 3 rosettes, which are distinctive in rosette size and number, secreted high amount of PDGF-AA, 931.5 pg/ml, 722.6, pg/ml and 586.1 pg/ml PDGF-AA, respectively. Little impact of rosette size for categories 1, 2, 3 on individual cytokines with some outliers was observed (Table S5). We observed that PDGF-AA is detected frequently and regardless of distributions in rosette size (and number). PDGF-AA dominates cytokine profiles and is detected in all 25 microchambers for Category 1 and 23 microchambers for Category 2 and 3 rosettes. Dot plot of detected cytokines is shown in Figure 6e and 6f. To further evaluate possible differences between the three categories of rosettes, we have applied principal component analysis (PCA) to the data and are able to determine that multiple small rosettes (Category 1) and one large rosette (Category 3) in a microwell behave similarly. However, Category 3 rosettes that are in medium in size are now seen as more distinct (Figure S9). Such an intrinsic difference can be distinguished only by the analysis of the highly multiplexed data generated by the NCCIM assay. ELISA assay on supernatants when neural rosettes were cultured in petri dishes showed predominant secretion of PDGF-AA as well (Figure 7a).
Figure 7. NCCIM cytokine detection of neural rosettes and cytokine levels in supernatants.

(a) Quantified cytokine concentrations and fluorescence signals of ELISA assay on supernatant of EB-derived rosettes in a monolayer in the dish. (b) Quantified cytokine concentrations and fluorescence signals of ELISA assay on supernatant of rosette neurospheres in suspension. (c) Plots of quantified cytokine concentrations and fluorescence signals of ten proteins assayed by NCCIM on rosette neurospheres. (d) Variation of size of rosette neurospheres in a culture dish after two days versus rosette neurospheres in PFMA. Spheres suspended in the dish range in size from 50 microns to 300 microns while the spheres in chambers of NCCIM are 150 microns. P values: *, P<0.05; **, P<0.01; ***, P<0.01; ****, P<0.0001.
NCCIM multiplex cytokine analysis of dissociated NSCs and rosette neurospheres
We hypothesized that disrupted rosette structure may alter cytokine secretion patterns due to lost polarization and changes in cell-cell communication. We tested this hypothesis by two experimental strategies. In the first approach, monolayer NSC rosette cells cultured in a petri dish are dissociated into single cells and are seeded into PFMA and secreted cytokines of 100 microchambers examined at 24 hours. No signal was detected on the NCCIM platform, which was confirmed by conventional ELISA (data not shown). In the second approach, we transferred dissociated monolayer rosettes to microchambers, and evaluated secreted proteins at 48 hours when highly uniform neurospheres were formed. ELISA assay was also done on supernatant of rosette spheres formed in suspension (Figure 7b). Decoded information from five microchambers is shown, revealing uniformly low expression across microchambers. The 3D structure of rosette neurospheres is morphologically different from EB-derived rosettes that are planar in the wells and the cytokine secretion profile reflects differences. Figure 7c shows PDGF-AA and GDNF cytokines are highly released by the rosette neurospheres. However, many of other cytokines including BDNF, FGF-2, and β-NGF are also significantly produced by the neurospheres (P value < 0.05). Thus, the neurosphere format of NSCs favors release of a much wider spectrum of neurotropic factors. Table S6 shows cytokine quantification in individual chambers, including PDGF-AA, GDNF, FGF2 and BDNF are detected in chambers at various level.
DISCUSSION
Cytokine signaling in cell-cell communication plays a critical role in differentiation and maintenance of tissues and in guiding organotypic use of stem cell derived cells in vitro and in vivo. The power of the NCCIM, demonstrated in this study, is its ability to couple detailed changes in signaling by multiple cytokines to developmental and morphological changes of NSCs impacting cell-cell communication. NSC rosettes are a common cell source for therapeutic intervention, either as rosettes or as a dissociated cell source. We analyzed NSC cytokine signaling between the morphologically distinct populations by developing a strategy for direct formation in the PFMA and analysis. The NCCIM PFMA wells were loaded with pluripotent custom sized EBs and induced neurally to generate rosettes directly in the NCCIM microenvironment. Dissociated cultured NSC rosettes were seeded directly onto the NCCIM for analysis along with later forming rosette neurospheres. Our findings reveal that EB-derived NSC rosettes have a distinct cytokine signaling profile that is lost in dissociated rosettes and rosette neurospheres that reform from dissociated cells.
In this study we co-evaluate ten cytokines with important roles in NSC survival, maintenance, proliferation, neurogenesis and gliogenesis. The panel includes PDGF-AA, GDNF, BDNF, IGF-1, FGF-2, IL-6, BMP-4, CNTF, β-NGF and NT-3. The EB-derived rosettes express three prominent cytokines in all categories from this panel that are PDGF-AA, FGF-2 and GDNF while IL-6 is expressed in category 1 and 2. These cytokines play known roles in multiple signaling pathways acting through mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), protein kinase B (PKB or Akt) and phospholipase C-γ (PLC-γ)48–52. PDGF-AA is responsible for proliferation of NSCs and maintaining pluripotency by delaying neuronal differentiation53. It is also involved in the CREB (cAMP response element-binding protein) signaling pathway through activation of PI3K/Akt regulators. This pathway can trigger continued signaling for growth and contribute to formation of tumors54. GDNF is a member of transforming growth factor-β (TGF-β) family and acts through MAPK and other regulators to affect neuronal survival, proliferation, differentiation and migration. Previous studies reveal that the level of GDNF impacts downstream signaling pathways necessary for maintaining NPCs in an immature state51. IL-6 has a determining role in glial versus neuronal commitment and differentiation. It’s activation of the JAK/STAT signaling pathway promotes glial differentiation, whereas activation of the MAPK/CREB signaling pathway mediates neuronal differentiation53,55. The multipotent state of rosettes supports mixed potential for neuronal and glial differentiation via IL-6. In a previous study, cooperation of IL-6 with β-NGF or FGF-2 was shown to lead to neuronal differentiation56 FGF-2 signaling also has a role in self-renewal and proliferation of NSCs33,57,58. Thus FGF-2 has contrasting roles in proliferation versus neuronal differentiation that must be balanced with other cytokines. In our studies we observed FGF-2 expressed in all categories but expressed more strongly in Category 1 and 2.
A challenge to working with rosettes is their delicate nature that prevented generating these cells in culture and transferring the rosettes to the PFMA. By generating uniformly sized 300 microns EBs and seeding these into the PFMA we formed rosettes directly. Nonetheless we observed that when rosette formation was induced, the size and number of rosettes in each microchamber showed differences that were sorted as three categories. This provided an opportunity to examine cytokine signaling for multiple smaller rosettes (category 1), a single large rosette (category 2) and several medium-sized rosettes (category 3). The sensitivity of the NCCIM reveals subtle differences between the rosette microenvironments. We did not fully convert the fluorescence signals into concentrations using calibration curves for each microwell, because the background levels are slightly fluctuating between cell types and between experimental conditions. Thus the quantitative comparisons are limited within each cell type. In this regard, it is more reliable to draw conclusions based on raw fluorescence data and appropriate statistics within a given quantification range.
Development of central nervous system stems from the neural tube that is generated from a polarized epithelium surrounding that tube in vivo59. The precise developmental equivalent of the neural rosettes has not been determined but the structure in the neural stem cell field is universally seen as the precursor for CNS development of multiple neural cell types based on early multipotent neural markers26, neural induction in vitro typically results in spontaneous formation of multiple sized rosettes in a dish that are polarized around a central ring or oval shape. This is reflected in the PFMA chambers as well. Since no study has specifically looked at rosette size in regard to cytokine signaling, ours is the first to do so. The categories done by size and number helped us to independently compare cytokine profiles. We find that certain cytokine levels are independent of categorization while others are impacted (Table S4 and S5). Future studies may relate these changes to maturation or define threshold signaling pathways related to rosette size. Overall, the medium-sized rosettes secreted lower amounts of PDGF-AA and GDNF. Therefore, the NCCIM has the ability to distinguish subtleties in cytokine signaling that can be related to size and number of rosettes. It is interesting to note that dissociated rosette cells when seeded onto the PFMA prefer to reform as 150 microns diameter neurospheres in the 48 hours timeframe. Even though we detect biomarkers of rosettes in these neurospheres, they lack the distinct cytokine signaling profile observed with rosettes that have more of a 2D morphology. The rosette neurospheres on NCCIM, produce BDNF and β-NGF at higher levels than EB-derived rosettes and it’s shown that combination of BDNF and β-NGF improves neuronal differentiation of neural stem cells60. Further, when we form the rosette neurospheres by suspension culture in the dish they have a broader range in size from 50 microns to 300 microns (Figure 7d) and show the emergence of PDGF-AA and GDNF (Figure 7b). Thus, size of reformed rosette neurospheres and/or chamber geometry may contribute to defining cytokine signaling.59
An original goal of this new technology applied to rosettes, was to obtain a cytokine signature of a rosette and to also characterize any changes in the cytokine signaling when the cells of a rosette structure are disrupted. As is often the case when these cells are used therapeutically. This would allow for an interesting nuanced discussion on the role of cytokines in cell-cell communication for this structure. We defined three categories of rosettes based on size and number in our chambers. The category 2 that is a single rosette within the PFMA is the first cytokine signature for a single whole rosette structure. By fully dissociating rosettes and comparing the cytokine signaling of dissociated cells to that of a rosette we were looking for information on how different cytokines responded to this disruption of morphology. However, for our cytokine panel, a fully dissociated rosette gives uniformly low cytokine signaling. It is possible to use our system to look at interactions of subsets within the rosette by partially dissecting rosettes manually or by light chemical dissociation. This was not done in this study but may be an interesting application in the future to evaluate the heterogeneity between cell positioning in the rosette and cell-cell communication. We take a step towards this point when we investigated the heterogeneity of different categories of rosette structure in relation to size. This has been described in the text for our 3 categories (Figure 3c). We analyzed the cytokine profiles for each of these categories in scatter plots (Figure 6a–d). The multiple wells for each category were clustered before comparison and there is heterogeneity within each category as the data in figure 6c indicate. Since the NCCIM microwell design does not need to be rectangular, one can apply this technic to a variety of interesting morphological tissue studies.
CONCLUSION
This study on NSC rosettes demonstrates the development, versatility and utility of a novel neural cell-cell interaction microchip, NCCIM, as a sensitive multiplex detector of cytokine signaling for neural applications. The NCCIM provides a unique approach to link cell morphology and tissue architectures to cytokine signaling and has provided new information on NSC rosettes to benefit in vitro studies and in vivo applications. In future studies, in addition to evaluating cell sources, the NCCIM is expected to be a valuable tool for modeling neurodegeneration or neuroplasticity. For example, evaluation of stretch induced response to mimic traumatic brain injury or opioid drug response and screening of relevant neuronal models and contribution of genetic mutations to neurodegeneration.
MATERIALS AND METHODS
Expansion of hiPSCs
The human induced pluripotent stem cell line F3.5.239 was cultured on Matrigel coated 6-well plates in mTeSR™ Plus media (STEMCELL Technologies, Inc., Cambridge UK) and passaged every five days after reaching 70% confluency using Gentle Cell Dissociation Reagent (STEMCELL Technologies, Inc., Cambridge UK). Each well of a 6-well plate was expanded 6 times, and cells were supplemented with fresh mTeSR™ Plus media every other day.
Rosette Formation
EB-derived rosettes
Rosettes were generated from hiPSC embryoid bodies (EBs). After hiPSC culture reached 70% confluency, 1 ml Accutase (STEMCELL Technologies, Inc., Cambridge UK) was added to five well of a 6-well plate and incubated for five minutes at 37° C. The cell-enzyme mixture from all wells, was centrifuged at 300 x g for five minutes, supernatant removed, and the dissociated cells resuspended in 100 μl mTeSR™ Plus media on custom-made microarrays of polydimethylsiloxane (PDMS) with 300 μm wells according to published protocols37 and dish was supplemented with ROCK inhibitor Y-27632 (STEMCELL Technologies). Media was changed to fresh mTeSR™ Plus media every other day. After four days incubation in 37° C, uniform EBs formed in the wells. The EBs were unloaded and plated onto a Matrigel coated dish and supplemented with neural induction media (DMEM/F12, N2, NEAA, B27, Glutamax, p/s) and refreshed every other day. Two selective inhibitors, brain morphogenic protein (BMP) signaling inhibitor, LDN-193189 (STEMCELL Technologies) and the TGFβ type 1 receptor kinase (ALK5) inhibitor SB-431542 (STEMCELL Technologies) were added fresh to the media each time the media was exchanged.
Monolayer rosettes
To generate monolayer neural rosettes, cultured hiPSCs were dissociated with Accutase and 1.5 × 106 single cells were plated on each Matrigel coated 35 mm dish and supplemented with neural induction medium and ROCK inhibitor Y-27632 the first day. Media was refreshed every other day to neural induction media supplemented with SB-431542 and LDN-193189 for 7 days until NSCs were generated.
Seeding Cells on the NCCIM
EB-derived rosettes
The PFMA was UV sterilized and plasma treated for 3 minutes then coated with 1% Matrigel overnight at 37˚C. Sized EBs were unloaded from custom microarrays and filtered by a cell strainer (40 μm) to remove single cells before being seeded onto the Matrigel coated PFMA. The PFMA is maintained in a 35 mm diameter dish and neural induction media is changed every other day. Viability of rosettes in chambers was studied on day 4 of neural induction on PFMA using live/dead assay (Thermofisher) according to manufacturer’s protocol at hour 0 and after 8 hours of incubation in custom made clamp.
Dissociated rosettes
To seed dissociated neural rosettes on the PFMA, Accutase was added to generated monolayer rosettes on the dish for 5 minutes at 37° C and single cells suspension was centrifuged at 300 x g for 5 minutes. Supernatant was removed and cell pellet was resuspended in 200 μl neural induction media. Cell suspension was added onto Matrigel coated PFMA in 35 mm dish and was placed on shaker (brand) at 40 rpm. After 15 minutes, 2 ml of neural induction media was added to the dish and incubated at 37° C for one day.
Immunocytochemistry
For analysis of biomarkers, the NSCs on chamber slides or PFMA were fixed by 10% formalin for 20 minutes at room temperature, washed three times with PBS and permeabilization solution (PBS, 0.5% Triton X-100) added for 10 minutes at room temperature, then washed three times and incubated with blocking buffer (PBS, 1% BSA and 0.1% Triton X-100) for one hour at room temperature. The antibodies, including Sox1 (R&D systems AF3360), Sox2 (R&D systems MAB2018), ZO-1 (Thermofisher Scientific 33–9100), Ki67 (Abcam 15580), Nestin (R&D systems MAB1259, Pax6 (DSHB PAX6, Pax6 was deposited to the DSHB by Kawakami, A.) were added (1:1000) and incubated at 4˚C overnight. Cells were subsequently washed three times with PBS and secondary antibodies added (Alexa Fluor 488 donkey anti mouse A32766, Alexa Fluor 594 donkey anti goat A32758, Alexa Fluor 594 donkey anti rabbit A32754, Thermofisher Scientific) for one hour at room temperature, and washed 3 times with PBS. Coverslips were mounted with anti-fade diamond mountant (Thermofisher Scientific) and imaged with Zeiss Axio Observer Z1 fluorescence microscope. Post detection immunocytochemistry on NCCIM was done following the same method after 8 hours’ incubation of seeded NCCIM in custom made clamp. This experiment was repeated for 4 times.
Conjugation of microbeads with single-stranded DNAs (ss-DNA)
The aldehydic polystyrene microbeads (Life Technologies) were coated with poly-L-lysine (PLL; Ted Pella) first to increase the DNA load to the microbeads then crosslinked to the amino ssDNA. 100 μL of microbeads (3 μm, 4% w/v) is mixed with 1.5 mL of aqueous solution of PLL (0.1% w/v) with the addition of 5 μL of 0.5 M NaOH to adjust the PH at (8.5). Then, the PLL coating quality is validated with the nonspecific binding of the PLL coat with Cy3 conjugated ssDNA. After that, the microbeads were washed and centrifuged three times with 0.05% Tween 20 in PBS. 10 μL of 1mM amino ssDNA is mixed with the PLL coated beads in the presence of 0.85 mM of bis(sulfosuccinimidyl)suberate (BS3; Life Technologies) for 4 hours followed by washing and centrifuging for three times using distilled water.
Conjugation of cDNA to antibodies
40 μL amino complimentary DNAs (c-DNA, 400 μm) were reacted with succinimidyl-4-formylbenzamide (S-4FB; Trilink Biotechnologies) in 200 times excess for 4 h with addition of 10 μL DMF to prevent precipitation of the S-4FB. At the same time, every capture antibody (50 μg) were reacted with succinimidyl-6-hydrazinonicotinamide (S-HyNic; TrilinkBiotechnologies) in 10 times excess for 4 h. Both conjugated c-DNAs and antibodies were purified by centrifugation using 7K zeba spin column (ThermoFisher Scientific) at 3800 rpm for two minutes. They were mixed together and shaken overnight at pH 6 which is essential for the cross-linking reaction between S-4FB and S-HyNic. In order to ensure high sensitivity, the antibody-DNA conjugates were purified by fast protein liquid chromatography purification (FPLC) at 0.45 ml/min. The 10 captured antibodies including GDNF, PDGF-AA, IL6, CNTF, IGF, BDNF, FGF, NT-3, BMP-4 and β-NGF were tagged with D′, E′, F′, G′, H′, J’, K’ L’, M’ and O’ oligo DNAs (Integrated DNA Technologies;), respectively.
Chip fabrication
The PDMS mold that is used to form the PFMA with feature size of (300 μm x 300 μm x 130 μm) was fabricated by soft-lithography method using SU-8 2100 (Microchem) on a 4-inch silicon wafer. Preparation of the PDMS poly(dimethylsiloxane) chip (PFMA) was started with a prepolymer by mixing Silicone Elastomer Curing Agent with Silicone Elastomer Base (SYLGARD 184; Dow Silicones) in the ratio of 1:10. The air bubbles of the mixture were removed by keeping it under vacuum for 20–30 min before the mixture was poured into a mold on a silicon wafer followed by baking for 1 hour at 70 °C. The solidified PDMS chip that becomes the PFMA was peeled off from the mold and cut into square shapes before use.
Patterning the MIST array and characterization
All the ten ssDNA-conjugated microbeads were mixed together (20 μL each) with 200 μL of non-conjugated blank beads to lower the signal overcrowding during detection. The mixture was centrifuged and concentrated to 50% w/v in Milli-Q water. Then the microbeads were spread into a confined area on a PDMS slab (the PDFM) and kept for 15–20 mins until dry. The dried microbeads were spread on a pressure sensitive tape (TPA cleanroom adhesive tape; Texwipe) which is attached to a plain glass slide using another sticky tape (Scotch magic tape). The whole array was sonicated to remove excess layers of microbeads and make only a monolayer of microbeads. The PFMA was added over the microbeads monolayer to act as a big well to hold the working solutions of the experiment. The microbeads array is validated by using a cocktail of c-DNA conjugated with Cy3 (200 nM), which was applied to the array and incubated for 1 hour at room temperature. The intensity of the microbeads was measured by a fluorescence microscope. The number of microbeads for each array is counted by a Matlab program developed in the lab.
The panel of 10 oligo DNAs has been well validated without crosstalk22,61. We use destructive images for validation of antibody cross-reactivities. For this experiment, a Cy3 tagged cDNA was added to the MIST array, and positions of bright microbeads were recorded. Then the cDNA was removed by 0.5 M NaOH, followed with incubation of a cocktail of 10 antibody-cDNA conjugates at 0.03 mg/mL for each for 1 h. A single type of recombinant protein at 10 ng/mL corresponding to the selected cy3-cDNA was applied to the array and was incubated for 1 h, before washing and incubation for 1 h with a cocktail of 10 detection antibodies at various concentrations according to their product descriptions. Then streptavidin conjugated horseradish peroxidase (HRP 1:200; R&D systems) was incubated with the MIST array for 30 minutes. Finally, the tyramide conjugated Cy3 (Akoya Biosciences) were applied to the array for 15 minutes before imaging. Each of the steps above needs washing for three times with 3% BSA in PBS at the end. The protein detection image is aligned and overlapped with Cy3-cDNA image on photoshop (Adobe) where the “blend” mode of difference is selected to show the “destructive” image. The same procedure is repeated for each of Cy3 tagged cDNAs and the corresponding protein detection on the same array.
On chip cytokine detection procedure
A cocktail of the ten capture antibodies conjugates were mixed together in 3% BSA in PBS (final concentration for each conjugate = 0.03 mg/mL) and incubated for 1 hour at room temperature to convert the DNA array to antibody array followed by washing the array with 3% BSA for three times. The microchamber side of the PDMS with cells on it (the PFMA) was clamped with the array and incubated for 8 hours at 37 °C in a 5% CO2 incubator. Then the biotinylated detection antibodies were added and incubated for 2 hours at room temperature. After washing, the streptavidin conjugated HRP was incubated with the MIST array for 30 minutes followed by washing three times by 3% BSA in PBS. The tyramide conjugated Cy3 (Akoya Biosciences) were applied to the array for 15 minutes followed by washing several times. The detected protein images were taken and the ELISA components were quenched using 0.5 M NaOH for 5 mins followed by decoding procedure to identify the detected protein signal. This experiment was repeated twice for EB-derived rosettes and dissociated rosettes and once for neurosphere rosette condition.
Decoding procedure
To identify 10 proteins, a cocktail of Cy3 c-DNAs and Cy5 c-DNAs is mixed together in 4 different cycles (24 = 16 proteins; see Figure 1 and Table S2) and incubated for 1 hour at room temperature followed by taking images by the fluorescent microscope. The fluorescent microbeads were quenched by 0.5 M NaOH for 5 mins and the same steps were repeated with the following cycles.
Imaging and data processing
A fluorescent microscope (Zeiss Axio Observer Z1) were used to take all images. The green and red fluorescence were taken using a Cy3 and Cy5 filters, respectively. The microscope objective of Plan-APOCHROMAT 20x/0.8 was used to collect light. A custom MATLAB code is developed to measure the signal intensity of the detected protein. The program also aligns six images for the same array including the bright-field image, the protein detection image and 4 decoding images. The fluorescent color combination of each microbead on the PFMA was extracted and then matched with the predesigned color map (Table S2). Each microbead on the array is analyzed through the program to identify the type of protein detected. The intensities of microbeads for detecting the same protein were averaged across the same array to obtain cytokine profile. The same method was repeated to extract the data of ∼100 arrays on a chip where each array carries information of one well.
Signal detection limit is set as background fluorescence intensity plus 3 times of its standard deviation by measuring 5 microarrays and each with 20 microbeads for each protein, while the measured intensities are quantified only on the microbeads for detection of this particular protein. To make calibration curves, various concentrations (10, 3.3, 1, 0.33, 0.1 and 0.03 ng/mL) of recombinant proteins were applied to the microarrays and the fluorescence intensities were quantified. A linear line is fitted to each calibration curve after taking log scale of both fluorescence intensities and the concentrations to obtain protein concentrations. All the fluorescence intensities lower than the threshold, or signal detection limit, are taken as noise. The “fluorescence signal” is obtained by subtracting the raw fluorescence intensity by the signal detection limit for each protein, and this unit is used to represent the detectable true protein signal on NCCIM in Figure 6 & 7.
Unpaired, two-tailed t-test was used to determine statistically significant differences using GraphPad Prism. A P value less than 0.05 is considered statistically significant and is denoted with *, while **, *** and **** represent P < 0.01, P < 0.001 and P < 0.0001, respectively. Mean value of cytokines for each condition is calculated and shown by a horizontal line on each box in the plots. Mean value of the background is shown by horizontal dotted line in the plots.
Supplementary Material
Acknowledgement
This research is supported by the New York Stem Cell Science (C32574GG) to J.W. and J. P., and the National Institute of Health (R01GM12898401) to J.W.
Footnotes
Conflict of interest
There are no conflicts to declare.
References
- 1.Zhang BB et al. Nanomaterials in neural-stem-cell-mediated regenerative medicine: imaging and treatment of neurological diseases. Adv. Mater, 2018, 30(17). [DOI] [PubMed] [Google Scholar]
- 2.Farrukh A, Zhao SF & del Campo A Microenvironments designed to support growth and function of neuronal cells. Front. Mater, 2018, 5(62). [Google Scholar]
- 3.Mazzini L, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J. Transl. Med, 2015, 13(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, Polak M, Bordeau J, Sakowski SA, & Glass JD Intraspinal neural stem cell transplantation in Amyotrophic Lateral Sclerosis: phase 1 trial outcomes. ANNALS of neurology, 2014, 75, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagoshi N, Tsuji O, Nakamura M, & Okano H Cell therapy for spinal cord injury using induced pluripotent stem cells. Regenerative Therapy, 2019, 11, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereira IM, Marote A, Salgado AJ, & Silva NA Filling the gap: neural stem cells as a promising therapy for spinal cord injury. Pharmaceuticals, 2019, 12(65). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagan M, Wennersten A, Meijer X, Holmin S, Wahlberg L, Mathiesen T Neuroprotection by human neural progenitor cells after experimental contusion in rats. Neurosci. Letters, 2003, 351, 149–152. [DOI] [PubMed] [Google Scholar]
- 8.Ma H, Yu B, Kong L, Zhang Y, & Shi Y Transplantation of neural stem cells enhances expression of synaptic protein and promotes functional recovery in a rat modelo of traumatic brain injury. Mol. Med. Rep, 2011, 4, 849–856. [DOI] [PubMed] [Google Scholar]
- 9.Skardelly M, Gaber K, Burdack S, Scheidt F, Hillbig H, Boltze J, Forschler A, Schwarz S, Schwarz J, Meixensberger J, & Schumann MU Long-term benefit of human fetal neuronal progenitor cell transplantation in a clinically adapted model after traumatic brain injury. J. Neurotrauma, 2011, 28, 401–414. [DOI] [PubMed] [Google Scholar]
- 10.Jain M, Armstrong RJE, Elneil S, & Barker RA Transplanted human neural precursor cells migrate widely but show no lesion-specific tropism in the 6-hydroxydopamine rat model of Parkinson’s disease. Cell Transplantation, 2006, 15, 579–593. [DOI] [PubMed] [Google Scholar]
- 11.Zou F, Bao X, Sun X, Wu J, Bai Q, Chen G, Li X, Zhou Q, Yang Y, Shen Q, & Wang R Transplantation of human neural stem cells in a parkinsonian model exerts neuroprotection via regulation of the host microenvironment. Int. J. Mol. Sci, 2015, 16, 26473–26492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Y, Yu P, & Chen L Current progress in the derivation and therapeutic application of neural stem cells. Cell Death and Disease, 2017, 8, e3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirahmadi M, Rezanejadbardaji H, Irfan-Maqsood M, Mokhtari MJ, & Naderi-Meshkin H Stem cell therapy for neurodegenerative diseases: Strategies for regeneration against degeneration. Cell Therapy & Regenerative Medicine, 2016, 1(1), 03–19. [Google Scholar]
- 14.Dell’Anno MT, Wang X, Onorati M, Li M, Talp F, Sekine Y, Ma S, Liu F, Cafferty WBJ, Sestan N, & Strittmatter SM Human neuroepithelial stem cell regional specificity enables spinal cord repair through a relay circuit. Nat. Comm, 2018, 9(3419). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dulin JN et al. Injured adult motor and sensory axons regenerate into appropriate organotypic domains of neural progenitor grafts. Nat. Commun, 2018, 9, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Med, 2011, 17(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matharu Z et al. Detecting transforming growth factor-beta release from liver cells using an aptasensor integrated with microfluidics. Anal. Chem, 2014, 86, 8865–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Q et al. On-chip regeneration of aptasensors for monitoring cell secretion. Lab Chip, 2014, 14, 276–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunderson KL et al. , Decoding Randomly Ordered DNA Arrays, Genome Res. 2004, 14(5), 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu ZTF et al. Rapid, automated, parallel quantitative immunoassays using highly integrated microfluidics and AlphaLISA. Sci Rep-Uk, 2015, 5: 11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Matharu Z, Rahimian A & Revzin A Detecting multiple cell-secreted cytokines from the same aptamer-functionalized electrode. Biosens. Bioelectron, 2015, 64, 43–50. [DOI] [PubMed] [Google Scholar]
- 22.Zhao P, Bhowmick S, Yu JC & Wang J Highly multiplexed single-cell protein profiling with large-scale convertible DNA-antibody barcoded arrays. Adv. Sci, 2018, 5, 1800672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chokkalingam V et al. Probing cellular heterogeneity in cytokine-secreting immune cells using droplet-based microfluidics. Lab Chip, 2013, 13, 4740–4744. [DOI] [PubMed] [Google Scholar]
- 24.Son KJ, Gheibi P, Stybayeva G, Rahimian A & Revzin A Detecting cell-secreted growth factors in microfluidic devices using bead-based biosensors. Microsyst. Nanoeng, 2017, 3, 17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Wernig M, Duncan ID, Brustle O, & Thomson JA In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nature Biotech, 2001, 19, 1129–1133. [DOI] [PubMed] [Google Scholar]
- 26.Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, & Studer L Human ES cell-derived neural rosettes reveal functionally distinct early neural stem cell stage. Genes & Development, 2008, 22, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meinhardt A, Eberle D, Tazaki A, Ranga A, Niesche M, Wilsch-Brauninger M, Stec A, Schackert G, Lutolf M, & Tanka EM 3D resconstruction of the patterned neural tube from embryonic stem cells. Stem Cell Reports, 2014, 3, 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabiec M, Hribkova H, Varecha M, Stritecka D, Hampl A, Dvorak P, & Sun Y Stage-specific roles of FGF2 signaling in human neural development. Stem Cell Research, 2016, 17, 330–341. [DOI] [PubMed] [Google Scholar]
- 29.Koch P, Optiz T, Steinbeck JA, Ladewig J, & Brustle O A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. PNAS, 2009, 106(9), 3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lukovic D, et al. Highly efficient neural conversion of human pluripotent stem cells in adherent and animal-free conditions. Stem Cells Trans. Med, 2017, 6, 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandrasekaran A, et al. Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Research, 2017, 25, 139–151. [DOI] [PubMed] [Google Scholar]
- 32.Pauly MG, Krajka V, Stengel F, Seibler P, Klein C, & Capetian P Adherent vs. free-floating neural induction by dual SMAD inhibition for neurosphere cultures derived from human induced pluripotent stem cells. Front. Cell. Dev. Biol, 2018, 6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valensisi C, Andrus C, Buckberry S, Jayavelu ND, Lund RJ, Lister R, & Hawkins RD Epigenomic landscape of hESC-derived neural rosettes: modeling neural tube formation and disease. Cell Rep, 2017, 20(6), 1448–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fathi A, Hatami M, Hajhosseini V, Fattahi F, Kiani s., Baharvand H, & Salekdeh GH Comprehensive gene expression analysis of human embryonic stem cells during differentiation into neural cells. Plos One, 2011, 6(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilak F et al. Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell, 2009, 5, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawryluk G, W. J., Mothe A, Wang J, Wang S, Tator C, & Fehlings MG An in vivo characterization of trophic factor production following neural precursor cell or bone marrow stromal cell transplantation for spinal cord injury. Stem Cells & Development, 2012, 21(12), 2222–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drago D, Cossetti C, Iraci N, Gaude E, Musco G, Bachi A, & Pluchino S The stem cell secretome and its role in brain repair. Biochimie, 2013, 95, 2271–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quiroz EN, Quiroz RN, Ahmad M, Escorcia LG, Villarreal JL, Ponce CF, & Martinez GA Cell signaling in neural stem cells. Cells, 2018, 7, 75–101. [Google Scholar]
- 39.Tomov ML, Olmsted ZT, & Paluh JL The human embryonic body cystic core exhibits architectural complexity revealed by use of high throughput polymer microarrays. Macromol. Biosci, 2015, 15(7). [DOI] [PubMed] [Google Scholar]
- 40.Tomov ML, Olmsted ZT, Dogan H, Gongorurler E, Tsompana M, Out HH, Buck M, Chang E, Cibellu J, & Paluh JL Distinct and shared determinants of cardiomyocyte contractility in multi-lineage competent ethnically diverse human iPSCs. Sci. Rep 2016, 6 (37637). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang EA, Tomov ML, Suhr ST, Luo J, Olmsted ZT, Paluh JL, Cibelli J Derivation of Ethnically Diverse Human Induced Pluripotent Stem Cell Lines. Sci Rep, 2015, 5, 15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, & Studer L Highly effiecient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 2009, 27(3), 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell, 2010, 7(2), 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park D, Xiang AP, Mao FF, Zhang L, Di C, Liu X, Shao Y, Ma B, Lee J, Ha K, Walton N, & Lahn BT Nestin is required for the proper self-renewal of neural stem cells. Stem Cells, 2010, 28, 2162–2171. [DOI] [PubMed] [Google Scholar]
- 45.Archer TC, Jin J, & Casey ES Interaction of sox1, Sox2 and Sox3 and Oct4 during primary neurogenesis. Dev Biol., 2011, 350(2), 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desimoni E and Brunetti B About Estimating the Limit of Detection by the Signal to Noise Approach. Pharmaceutica Analytica Acta, 2015, 6:4. [Google Scholar]
- 47.Meany DL, Hackler L, Zhang L, Chan DW Tyramide Signal Amplification for Antibody-Overlay Lectin Microarray: A Strategy to Improve the Sensitivity of Targeted Glycan Profiling. J. Proteome Res. 2011, 10(3), 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Funa K, & Sasahara M The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol, 2014, 9, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoch RV, & Soriano P Roles of PDGF in animal development. Development, 2003, 130(20), 4769–4784. [DOI] [PubMed] [Google Scholar]
- 50.Sariola H, & Saarma M Novel functions and signaling pathways for GDNF. J. Cell Sci, 2003, 116, 3855–3862. [DOI] [PubMed] [Google Scholar]
- 51.Uesaka T, Nagashimada M, & Enomoto H GDNF signaling levels control migration and neuronal differentiation of enteric ganglion precursors. J. Neuroscience, 2013, 33(41), 16372–16382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Islam O, Gong X, Rose-John S, & Heese K Interleukin-6 and neural stem cells: more than gliogenesis. Mol. Biol. of the Cell, 2009, 20, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erlandsson A, Enarsson M, & Forsberg-Nilsson K Immature neurons from CNS stem cells proliferate in response to platelet-derived growth factor. J. Neuroscience, 2001, 21(10), 3483–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mantamadiotis T, Papalexis N, & Dworkin S CREB signaling in neural stem/progenitor cells: recent developments and the implications for brain tumor biology. Bioassays, 2012, 34, 293–300. [DOI] [PubMed] [Google Scholar]
- 55.Gu F, Hata R, Ma YJ, Tanaka J, Mitsudu N, Kumon Y, Hanakawa Y, Nakajima K, & Sakanaka M Suppression of STAT3 promotes neurogenesis in cultured neural stem cells. J. Neurosci. Res, 2005, 81(2). 163–171. [DOI] [PubMed] [Google Scholar]
- 56.Sterneck E, Kaplan DR, & Johnson PF Interleukin-6 induces expression of peripherin and cooperates with Trk receptor signaling to promote neuronal differentiation in PC12 cells. J. Neurochem, 1996, 67(4), 1365–1374. [DOI] [PubMed] [Google Scholar]
- 57.Stravridis MP, Lunn JS, Collins BJ, & Storey KG A discrete period of FGF-induced Erk 1/2 signaling is required for vertebrate neural specification. Development, 2007, 134, 2889–2894. [DOI] [PubMed] [Google Scholar]
- 58.Sterneckert J, et al. Neural induction intermendiates exhibit distinct roles of FGF signaling. Stem Cells, 2010, 28, 1772–1781. [DOI] [PubMed] [Google Scholar]
- 59.Knight GT et al. Engineering induction of singular neural rosette emergence within hPSC-derived tissues. eLife, 2018, 7:e37549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu F, Xuan A, Chen Y, Zhang J, Xu L, Yan Q, & Long D Combined effect of nerve growth factor and brain-derived neurotrophic factor on neuronal differentiation of neural stem cells and potential molecular mechanism. Mol. Med. Rep, 2014, 10, 1739–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdullah M, AA, and Wang J “Ultrasimple Single-Cell Detection of Multiple Cytokines by a Nanowell Chip Integrated with Encoded Microarrays.” ACS sensors, 2019, 4(9), 2296–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kowianski P, Lietzau G, Czuba E, Waskow M, Steliga A, & Morys J BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol. Neurobiol, 2018, 38, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poyhonen S, Er S, Domanskyi A, & Airavaara M Effect of neurotrophic factors in glial cells in the central nervous system: expression and properties in neurodegeneration and injury. Front. Physiol, 2019, 10:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang H, Wang R, Thrimawithana T, Little PJ, Xu J, Feng Z, & Zheng W The nerve growth factor signaling and its potential ad therapeutic target for glaucoma. Biomed Research International, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang RN, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes & Diseases, 2014, 1, 87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reichardt LF Neurotrophin-regulated signaling pathways. Phil Trans. R. Soc. B, 2006, 361, 1545–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schuster B, Kovaleva M, Sun Y, Regenhard P, Matthews V, Grotzinger J, Rose-John S, Kallen K Signaling of human ciliary neurotrophic factor (CNTF) revisited. J. Biol. Chem, 2003, 278(11), 9528–9535. [DOI] [PubMed] [Google Scholar]
- 68.Aberg MAI, Aberg ND, Palmer TD, Alborn A-M, Carlsson-Skwirut C, Bang P, Rosengren LE, Olsson T, Gage FH, & Eriksson PS IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol. & Cell. Neurosci, 2003, 24, 23–40. [DOI] [PubMed] [Google Scholar]
- 69.Aberg MAI, Aberg ND, Hedbacker H, Oscarsson J, & Eriksson PS Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J. Neurosci, 2000, 20(8), 2896–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.