

Abstract
The in vitro formation of stable G-quadruplexes (G4s) in human rRNA was recently reported. However, their formation in cells and their cellular roles were not resolved. Here, by taking a chemical biology approach that integrates results from immunofluorescence, G4 ligands, heme-affinity reagents, and a genetically encoded fluorescent heme sensor, we report that human ribosomes can form G4s in vivo that regulate heme bioavailability. Immunofluorescence experiments indicate that the vast majority of extra-nuclear G4s are associated with rRNA. Moreover, titrating human cells with a G4 ligand alters the ability of ribosomes to bind heme and disrupts cellular heme bioavailability as measured by a genetically encoded fluorescent heme sensor. Overall, these results suggest that ribosomes play a role in regulating heme homeostasis.
Keywords: RNA, hemin, tentacle, expansion segments, G-tract, BG4, G4, heme, ribosome, ribosomal ribonucleic acid (rRNA) (ribosomal RNA), metal homeostasis, G-quadruplex
Heme (iron protoporphyrin IX) is an essential but potentially cytotoxic metallocofactor and signaling molecule required for much of life on Earth. All heme-requiring cells and organisms must tightly regulate heme concentration and bioavailability to mitigate toxicity (1–4). Proteins that synthesize and degrade heme are relatively well-understood; structures and mechanisms of all eight heme biosynthetic enzymes and heme-degrading heme oxygenases are known (2–4). However, regulation of heme bioavailability, including its intracellular trafficking from sites of synthesis in the mitochondrial matrix or uptake at the plasma membrane, is poorly understood. Current paradigms for heme trafficking and mobilization involve heme transfer by unknown proteinaceous factors and largely ignore contributions from nucleic acids. Given that the first opportunity for protein hemylation occurs during or just after translation, rRNA or ribosomal proteins (rProteins) may be critical for shepherding labile heme to newly synthesized proteins.
We hypothesize that heme bioavailability is regulated in part by ribosomes via rRNA G-tracts, which are continuous runs of guanines. G-tracts are confined primarily to ribosomes of birds and mammals (5) and are focused in rRNA tentacles, which are seen to extend for hundreds of Å from ribosomal surfaces in these species (6). Tentacles are elaborations of rRNA expansion segments, which help form the secondary shell around the common core of eukaryotic ribosomes (7).
Tandem G-tracts can form G-quadruplexes (G4s), which are nucleic acid secondary structures composed of four guanine columns surrounding a central cavity that sequesters monovalent cations. Our rRNAG4-heme hypothesis is based in part on our observation of stable rRNA G4s in vitro (5, 8) and the extraordinary abundance of rRNA in vivo (9). Our rRNAG4-heme hypothesis is also based on work by Sen and co-workers (10–12), who has demonstrated high affinity of heme for G4s (KD ∼ nm) and proposed that RNA and DNA G4s sequester heme in vivo (13).
DNA G4s are thought to help regulate replication (14), transcription (15), and genomic stability (16). In mRNA, G4s are associated with untranslated regions and have been proposed to regulate translation (17–19). However, the in vivo folding state and functional roles of G4s are under debate. It has been proposed that eukaryotic RNA G4s are unfolded by helicases (20), although some investigators are not convinced (21, 22). The density of G4 sequences on surfaces of the human ribosome, which is extremely abundant, is high, with 17 G4 sequences in the 28S rRNA and three in 18S rRNA (Fig. 1A). Previous to this report, it was not known whether human ribosomes form G4s in vivo or what their functions might be.
Figure 1.

A, secondary structures of the human LSU rRNAs (5.8S and 28S) and SSU rRNA (18S). G4 sequences are highlighted in green. rRNA-based oligomers from the LSU (GQES7-a and GQES7-b) and from the SSU (GQes3) are indicated. B, schematic representation of a heme-G4 complex.
Here we present evidence that human rRNA tentacles form G4s in vivo that regulate cellular heme homeostasis. Results of immunofluorescence experiments with a G4 antibody, RNA pulldowns, and experiments with well-characterized G4 ligands provide strong support for in vivo formation of surface-exposed G4s on rRNA tentacles. We find that G4s on ribosomes bind heme in vitro (Fig. 1B) and that perturbation of G4s in vivo with G4 ligands affects heme interactions and bioavailability, as measured by heme-affinity reagents and genetically encoded heme sensors. The effects of in vivo G4-heme perturbations are predicted by in vitro experiments. Taken together, the results here indicate that rRNA G4s interact with heme in cells and suggest that ribosomal G4s play roles in intracellular heme metabolism.
Results
rRNA forms G4s in vivo
Confocal microscopy and G4-pulldowns were used to determine whether human ribosomes form G4s in vivo. For confocal microscopy, we used the BG4 antibody, which selectively targets G4s (23, 24) and has been broadly used for visualizing DNA G4s and non-rRNA G4s in cells (24–27). Our method of permeabilizing cells for antibody treatment does not permeabilize the nuclei (28). Therefore, DNA G4s were not anticipated or observed. To identify ribosome-associated G4s, we determined the extent to which antibodies to rProtein L19 (eL19) and to G4s colocalize and how colocalization is altered when cells are subjected to RNase or G4 ligand PhenDC3. Prior to antibody addition, the cells were cross-linked with paraformaldehyde, which has been shown to lock G4s in situ and reduce induction of G4s by small ligands (22). The extent of L19 and G4 antibody colocalization suggests that a fraction of ribosomes form G4s (Fig. 2, A and C) and that most G4s are associated with ribosomes. Specifically, we find that ∼83% of BG4 colocalizes with L19, indicating that the vast majority of RNA G4s in vivo are associated with ribosomes (Fig. 2C, green bar) and are therefore rRNA G4s. Conversely, only 5% of L19 colocalizes with BG4 (Fig. 2C, WT red bar), indicating that only a specialized fraction of ribosomes contain G4s. Similar results were obtained using an antibody against rProtein uL4 instead of eL19 (not shown). It is possible that the polymorphic nature of rRNA G4s attenuates binding to BG4, contributing to low colocalization ratios.
Figure 2.

rRNA G4s in HEK293 cells. Colocalization of (A) ribosomal protein L19 or (B) endoplasmic reticulum (red) with RNA G4s (green). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). C, extent of colocalization is quantitated as the ratio of colocalized pixels over total L19 pixels (red bars) or as the ratio of colocalized pixels over total BG4 pixels (green bar). The same analysis was performed for ER-BG4 colocalization (D). The statistical significance relative to WT is indicated by asterisks using an ordinary one-way analysis of variance with Dunnett's post-hoc test. Each dot represents a biological replicate. Images of cells treated without primary antibodies or with RNase A or PhenDC3 are shown in Fig. S6 and Fig. S7. E, the G4 ligand BioTASQ binds to 28S and 18S rRNAs in vitro. In the presence of BioTASQ and streptavidin beads, human rRNAs do not enter the native agarose gel. F, schematic representation of the BioTASQ pulldown protocol. G, RT-qPCR analysis of rRNAs pulled down by BioTASQ. The statistical significance relative to a fold-enrichment value of 1 is indicated by asterisks using a one-sample t test and a Wilcoxon test. Each dot represents a biological replicate. Data in (G) are represented as RNA enrichment under BioTASQ + streptavidin beads conditions relative to control streptavidin beads. *p < 0.05. n.s., not significant.
PhenDC3 (3,3′-[1,10–phenanthroline-2,9-diylbis(carbonylimino)]bis[1-methyl quinolinium] 1,1,1-trifluoromethanesulfonate) is a bisquinolinium phenanthroline derivative known to induce and stabilize G4s (29–32). Here, PhenDC3 appears to increase ribosomal G4 formation in vivo; treating cells with PhenDC3 increases L19-BG4 colocalization from 5 to ∼24% (Fig. 2C). The increase in colocalization upon PhenDC3 treatment supports formation of G4s by ribosomes. By contrast, treating cells with RNase A abolishes the L19-BG4 colocalization signal (Fig. 2C). Together, these results indicate that the colocalized BG4 signal is from a G4-forming RNA in close proximity to L19.
The high density of ribosomes on the surface of the endoplasmic reticulum (ER) and the lower abundance of mRNA in this location as compared with the cytosol (33) motivated us to investigate whether G4s colocalize with the ER. mRNAs in the cytosol, in the unlikely event that they form G4s at high frequency (20), may confound our ability to selectively detect rRNA G4s. Toward this end, we determined the extent to which BG4 colocalizes with an antibody against an ER membrane protein (calnexin) (Fig. 2B). Indeed, we find that ∼45% of the BG4 signal colocalizes with the ER surface (Fig. 2D, green bar), indicating a significant presence of RNA G4s near the ER membrane. As with L19, the fraction of the ER signal that colocalizes with G4s is completely abolished by RNase A and enhanced by PhenDC3 (increasing from 2 to 12%) (Fig. 2D). Thus, the data are consistent with formation of rRNA G4s by ER-bound ribosomes.
In an orthogonal approach, we pulled down RNA with BioTASQ (22, 34), a G4 ligand linked to biotin that captures G4s. We previously used BioTASQ to demonstrate that human rRNA forms G4s in vitro (Fig. 2E) (8). Here, we captured rRNA G4s from cross-linked HEK293 cells by methods summarized in Fig. 2F. BioTASQ captures 28S rRNA from cell lysates (Fig. 2G), in agreement with our previous in vitro BioTASQ data and with observations of G4-L19 colocalization above. BioTASQ also captures 18S rRNA, although the signal is significantly weaker. This observation is in agreement with the greater abundance of G-tracts in human 28S rRNA (17 G4 regions) than in 18S rRNA (three G4 regions). Taken together, our immunofluorescence and BioTASQ experiments provide strong evidence that human ribosomes form G4s in vivo.
Human ribosomes bind hemin in vitro
It has been suggested that G4s might associate with heme in vivo (10, 11, 35). In vitro, heme binds with high affinity to G4s by end-stacking (10–12, 36–38) (Fig. 1B). Here, we used UV-visible spectroscopy to assay the binding of hemin to human rRNA. rRNA oligomers GQES7-a (Fig. 3A), GQES7-b (Fig. S1A), or GQes3 (Fig. S1B) were titrated into fixed amount of hemin. GQES7-a and GQES7-b are fragments of expansion segment 7 of human large subunit (LSU) rRNA (5). GQes3 is a fragment of expansion segment 3 of human small subunit (SSU) rRNA (8). Each of these oligonucleotides is known to form G4s, and each caused a pronounced increase in the Soret band of hemin at 400 nm. The binding is specific for G4s because a mutant oligonucleotide, mutes3, that lacks G-tracts does not induce a change in the hemin Soret band (Fig. S1C). Larger human ribosomal components also bind heme. Intact 28S and 18S rRNAs extracted from human cells (Fig. S1, D and E), assembled LSUs (Fig. 3B) and SSUs (Fig. S1F), and polysomes (Fig. 3C) all induce changes in the hemin Soret bands, which is indicative of heme-rRNA interactions. Fitting of the UV-visible data in Fig. 3A, using a one-site binding model yielded an apparent limiting KD value of <100 nm (Fig. 3H), which is similar to that of other G4s (10–12, 39). The combined data are consistent with a model in which rRNA tentacles of human ribosomes bind to hemin in vitro.
Figure 3.
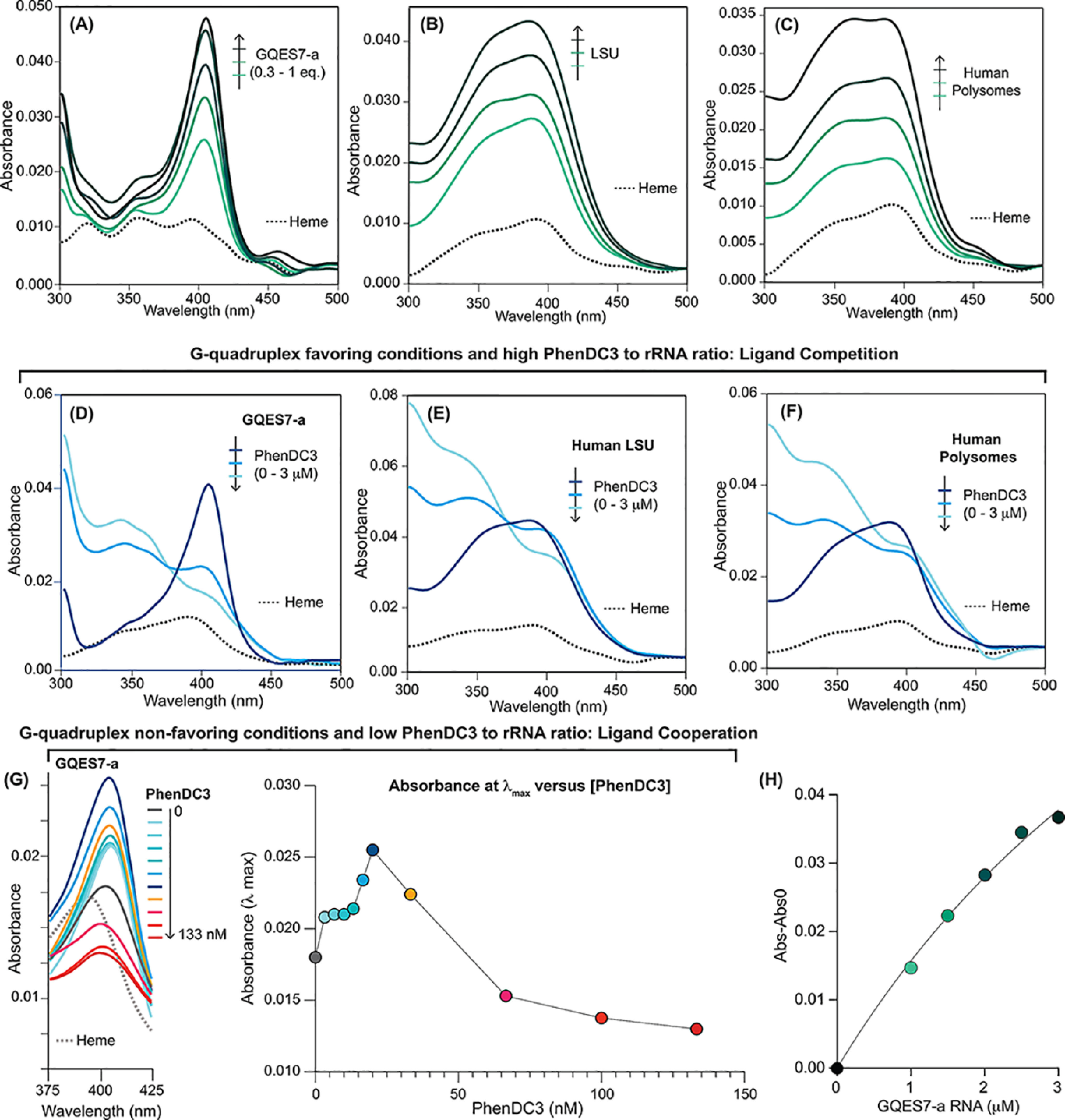
Human rRNA G4s bind to heme in vitro. UV-visible spectra (the heme Soret band) during heme titration under initial conditions that favor G4 formation with (A) GQES7-a, (B) the assembled LSU, or (C) polysomes. UV-visible spectra during titration with PhenDC3 of (D) constant [heme] and [GQES7-a], (E) constant [heme] and [LSU], and (F) constant [heme] and [polysomes]. G, UV-visible spectra during titration of constant [heme] and [GQES7-a] under initial conditions that favor G4 unfolding. The absorbance at λmax versus [PhenDC3] is plotted on the panel on the right. H, plot of absorbance at 400 nm versus [GQES7-a]. Data in (A) were fit to a one-site binding model (black line) giving an apparent limiting KD of <100 nm. The experiments in panels D–F were performed using initial conditions that favored G4 formation by G-tracts (50 mm K+ with titration of PhenDC3 in the µm range). The experiment in panel G was performed using initial conditions that favored unfolded G-tracts (10 mm Li+, 0 K+ with titration of PhenDC3 nm range).
PhenDC3 was used to confirm binding of hemin to ribosomal G4s under initial conditions that favor G4 formation. PhenDC3, like hemin, end-stacks on G4s (30, 35). In vitro, under conditions favoring G4s (50 mm K+), essentially all rRNA G-tracts form stable G4s prior to PhenDC3 addition (5, 8). Under these conditions, PhenDC3 competed with heme for binding to rRNA G4s (Fig. 3, D–F). With fixed concentrations of GQES7-a and hemin, addition of PhenDC3 decreased the intensity of the hemin Soret peak (Fig. 3D) because of dissociation of heme. The same phenomenon was observed with assembled ribosomal particles (LSU: Fig. 3E, SSU: Fig. S2A) and with polysomes (Fig. 3F). Hemin associated with purified 28S and 18S rRNAs is dissociated by addition of PhenDC3 (Fig. S2, B and C). Solutions of hemin with control RNA mutes3 do not show a change in the Soret peak intensity upon the addition of PhenDC3 (Fig. S2D). PhenDC3 absorbs at 350 nm (Fig. S2E), causing a shoulder on the heme Soret band (Fig. 3, D–F). The results here provide strong support for association of heme with G4s of human ribosomes in vitro.
Unlike the in vitro experiments with K+, it seems probable that most rRNA G-tracts are unfolded in cells. This inference is based on our observation that only 5% of ribosomes bind to the BG4 antibody in vivo until the addition of PhenDC3, upon which BG4 binding increases to 24% of ribosomes (Fig. 2, C and D). This inefficient folding of G-tracts into G4s in our in vivo experiments is in agreement with previous studies (20).
We mimicked the in vivo environment using initial conditions that favor unfolded G4s (Li+, low PhenDC3) by GQES7-a rRNA. Under these conditions, we observed that PhenDC3 caused an increase in the binding of heme to rRNA G4s at concentrations below 25 nm PhenDC3, as inferred from an increase in the absorbance of the heme Soret peak (Fig. 3G). However, at PhenDC3 concentrations above 25 nm, we observed a decrease in the binding of heme to rRNA G4s, as indicated by a reduction in the absorbance of the heme Soret band (Fig. 3G). Thus, under initial conditions that favor unfolded G-tracts (Li+), low PhenDC3 enhanced heme-binding to GQES7-a. The cooperative relationship between PhenDC3 and heme under some conditions is expected because multiple ligand binding sites are formed by a single binding event (30). On the other hand, under initial conditions that favor folding of G-tracts to G4s (K+), PhenDC3 acted as a competitor of heme-binding to GQES7-a. It seems possible that formation of G4s by the extended arrays of G-tracts in rRNAs might be cooperative, although to our knowledge this has not been demonstrated.
Human ribosomes bind heme in vivo
We developed an assay that exploits differential interactions with hemin-agarose, an agarose resin covalently linked to heme, to report in vivo heme-binding to ribosomes and rRNA. The degree to which any biomolecule interacts with heme in cells is inversely correlated with the extent to which it interacts with hemin-agarose upon lysis because of competition between endogenous heme and hemin-agarose. Therefore, the effects of heme-binding factors in vivo can be monitored by determining whether their interaction with hemin-agarose changes upon depletion of intracellular heme.
Accordingly, HEK293 cells were grown with or without succinylacetone (SA) (40), an inhibitor of heme biosynthesis. Lysates of these cells were incubated with hemin-agarose, and hemin-agarose–interacting rRNA was quantified by RT-qPCR. Consistent with previous work (41), treatment with 0.5 mm SA for 24 h caused a 7-fold decrease in total cellular heme in HEK293 cells (results not shown). The results reveal that rRNA binding to hemin-agarose relative to control agarose lacking heme increases by ∼4-fold in cells depleted of heme (Fig. 4A). This result suggests that under heme-depleted conditions, a greater fraction of rRNA-heme–binding sites are free and available to bind hemin-agarose. In short, the data are consistent with a model in which ribosomal RNAs associate with endogenous heme.
Figure 4.

Ribosomes appropriate heme in vivo through rRNA G4s. A, RT-qPCR analysis from untreated (WT) and SA-treated human cells. Statistical significance relative to WT is represented by asterisks using Student's t test. Each dot represents a biological replicate. B, RT-qPCR analysis from PhenDC3-treated HEK293 cells. Statistical significance relative to no-treatment conditions is represented by asterisks using ordinary one-way analysis of variance with Dunnett's post-hoc test. Each dot represents a technical replicate coming from individual biological replicates. The experiment was performed two times with similar dose-dependent trends (Fig. S3A). Data in (A) and (B) are represented as RNA enrichment in hemin-agarose beads relative to control Sepharose beads. C, single-cell analysis of HS1-transfected HEK293 cells grown in HD+SA, regular media containing 5-aminolevulinic acid (R+ALA), or regular media (regular) with the indicated concentrations of PhenDC3. Statistical significance relative to regular conditions is represented by asterisks using the Kruskal-Wallis analysis of variance with Dunn's post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n.s., not significant; n ≅ 1500 cells. D, median HS1 sensor ratios obtained in (C) as a function to PhenDC3 concentration.
PhenDC3 treatment of cells increases binding of ribosomes to hemin-agarose
To probe rRNAG4-heme–binding in vivo, we determined whether rRNA from HEK293 cells treated with the G4 ligand PhenDC3 (48 h at 37 °C) would bind more extensively to hemin-agarose. RT-qPCR reveals that PhenDC3 treatment of HEK293 cells causes a dose-dependent increase in binding of the LSU to hemin-agarose (Fig. 4B). A corresponding but weaker signal is seen for the SSU, in agreement with the higher abundance of G4 regions in the LSU than in the SSU (Fig. 1A). These data are consistent with our observations that PhenDC3 promotes rRNA G4 formation in cells (Fig. 2, C–D), providing additional heme-binding sites that can interact with hemin-agarose. Control experiments show that PhenDC3 as used here does not alter rRNA levels (Fig. S4) and that carrier DMSO does not affect the results (Fig. S3, B and C).
rRNA G4s regulate heme bioavailability in vivo
To determine whether rRNA G4s regulate heme homeostasis, we deployed a previously described genetically encoded ratiometric fluorescent heme sensor, HS1. HS1 is a tri-domain fusion protein consisting of heme-binding domain cytochrome b562 fused to two fluorescent proteins. The fluorescence of eGFP is quenched by heme, and the fluorescence of mKate2 is unaltered by heme. Thus, the ratio of eGFP:mKate2 fluorescence is inversely correlated with bioavailable heme, as measured by HS1. HS1 was previously used to characterize heme homeostasis in yeast, bacteria, and mammalian cells and was instrumental in identifying new heme-trafficking factors and signals that alter heme biodistribution and dynamics (40, 42–44). We asked if cytosolic heme bioavailability is altered in response to G4 ligand PhenDC3 (40). As indicated in Fig. 4C, single-cell analysis of a population of ∼1500 HEK293 cells/condition indicated that the median HS1 eGFP/mKate2 ratio increased upon heme depletion in heme-deficient media containing SA (HD+SA) and decreased upon increasing intracellular heme when cells were conditioned with the heme biosynthetic precursor 5-aminolevulinic acid (ALA) to drive heme synthesis. Upon exposure to PhenDC3 for 24 h, which is the minimal amount of time needed to observe an effect on labile heme (Fig. S5), the HS1 sensor ratio increased in response to increasing PhenDC3 dose, indicating a decrease in heme bioavailability. This 24-h treatment is similar to treatment times used previously for small-molecule induction of G4s in cells (23, 24, 35). The difference in the time required for PhenDC3 to affect heme in vivo (24 h) versus in vitro (15 min) is likely due to the kinetics of PhenDC3 cellular import, its partitioning into rRNA, and its induction of G4 formation, followed by the consequent reequilibration of cellular heme into G4 heme-binding sites. Regardless, these observations are in agreement with our data in Fig. 3G. PhenDC3 induced G4 formation in vivo, increasing the heme-binding sites on the rRNA that can bind heme. Consequently, there was a decrease in the bioavailable heme as detected by HS1. The fractional heme saturation of HS1 decreased by ∼15% (Fig. 4D), which is on the order of what one would expect based on an equilibrium competition model between HS1 and intracellular rRNA G4s that takes into account the relative abundance of these species and their Fe(III)-heme affinities (Fig. S8). Our data indicate that rRNA G4s bind heme and regulate intracellular heme bioavailability.
Discussion
Over the last two decades, whereas a handful of proteins have been implicated in regulating heme homeostasis on the basis of binding heme in vitro, there has been very little evidence of such roles from in-cell or in vivo studies (2–4, 45). Two notable exceptions include GAPDH and progesterone receptor membrane component 1 and 2. GAPDH, a key glycolytic enzyme, was found to bind and buffer heme, regulating its bioavailability (40, 43), and to deliver heme to nuclear heme-dependent transcription factors (40) and heme enzymes such as nitric oxide synthase (43, 46) and guanylate cyclase (47). Progesterone receptor membrane components, which interact with the terminal heme synthetic enzyme, ferrochelatase (48), have long been known to bind heme and affect the activity of P450 enzymes, raising the specter that they are heme chaperones (1, 49–53). More recently, they were found to enable the delivery of heme to the nucleus to control metabolism in adipocytes (54). However, current paradigms for heme trafficking and mobilization are heavily protein-centric and often ignore contributions from nucleic acids, which are highly abundant in cells.
The results here provide strong evidence that tentacles of human ribosomes form G4s in vivo and that these G4s are involved in appropriating heme. Immunofluorescence experiments with BG4 and L19 antibodies suggest that a specialized fraction of cytosolic ribosomes (∼5%) form G4s and that most extra-nuclear G4s (∼83%) are ribosomal. The small fraction of ribosomes observed to form G4s in vivo contrasts with the high stability of ribosomal G4s in vitro (5, 8). This difference supports Guo and Bartel (20), who suggest that eukaryotic cells have machinery that tends to unfold G4s. Our data show that only a small fraction of potential G4s form in vivo and that the fraction can be increased by G4-stabilizing ligands (Fig. 2). The high concentration of rRNA acts in opposition to the low frequency per ribosome so the RNA G4s are abundant. Moreover, the RNA G-quadruplexome appears to be ribosome-centric.
We previously reported that surfaces of both the SSU and the LSU contain G4 sequences (5, 8). A broad variety of data are consistent with more extensive formation of G4s on the LSU than on the SSU. These data include
more abundant and more expansive G-tracts on the LSU than the SSU (8),
greater conservation over phylogeny of LSU G-tracts than SSU G-tracts (5, 8),
higher thermodynamic stability of LSU G4s than SSU G4s (5, 8),
greater heme-binding to G4 oligomers from the LSU than those from the SSU (Fig. 3A and Fig. S1B),
greater enrichment of LSU than SSU particles in BioTASQ pulldowns (Fig. 2G),
greater enrichment of LSU than SSU particles in hemin-agarose pulldowns (Fig. 4A), and
greater effect of in vivo PhenDC3 treatment on LSU than on SSU rRNA in hemin-agarose pulldowns (Fig. 4B).
Our finding that rRNA G4s associate with heme in vivo has major implications for the physiology of G4-heme interactions. Decades of in vitro biophysical and chemical characterizations indicate that G4 and heme interact with high affinity (KD ∼ nm) and are potent redox catalysts, facilitating peroxidase and peroxygenase reactions (10–12). However, it remained unclear if heme-G4 complexes form in vivo and if heme-G4 catalyzed reactions were physiologically relevant.
The results of three orthogonal approaches from three groups appear to be self-consistent. Maizels and co-workers (35) recently proposed that heme binds to G4s in vivo, based on the transcriptional response of cells to PhenDC3. PhenDC3 causes up-regulation of heme-degrading enzymes such as heme oxygenase and other iron and heme homeostatic factors. These responses were interpreted to support a model in which G4s sequester and detoxify heme in cells. Work by Sen and co-workers (13), concurrent with ours, established G4-heme interactions in vivo by exploiting the peroxidase activity of G4-heme complexes to self-biotinylate G4s in RNA and DNA using a phenolic-biotin derivative. Here, we demonstrate in vivo heme-rRNAG4 interactions by several methods. The KD values for heme-rRNAG4 complexes (∼nm) are on the order of concentration of labile heme (25–300 nm) (40, 55, 56). Therefore, rRNA appears to be poised to buffer labile heme. We propose that heme-rRNAG4 interactions may be important for protein hemylation reactions and/or for buffering cytosolic heme, mitigating its potential toxicity. It remains to be determined how endogenous factors and processes may modulate G4 formation to regulate heme availability and homeostatic mechanisms.
Summary
The results here, building on previous in vitro work, provide the first demonstration of in vivo formation of G4s by rRNA. The levels of rRNA G4 formation observed in vivo support a model in which most G-tracts are unfolded in eukaryotes (20). Even so, the extreme density of ribosomes in vivo and the large number of G-tracts/ribosome implies a large absolute number of rRNA G4s in vivo. We provide evidence that rRNA G4s are inducible by small molecules and can bind heme in vivo. The ribosome may help regulate heme bioavailability in cells and may be directly involved in protein hemylation. These results provide new insights into the molecules and mechanisms underlying intracellular heme trafficking and bioavailability, which are currently poorly understood (1–4). Our results suggest that ribosomes, G4-containing rRNAs in particular, may regulate heme metabolism, acting to buffer intracellular heme and possibly regulate heme trafficking and cotranslational hemylation. The ribosome as a potential heme buffer is consistent with its role as a general and versatile sink for ions and small molecules, including antibiotics (57), platinum-based drugs (58–60), metabolites (61), and metal cations Mg2+, Ca2+, Mn2+, Fe2+, and K+ (62–67).
Experimental procedures
Cell culture
HEK293 cells were cultured in DMEM containing 4.5 g/liter glucose without sodium pyruvate and l-glutamine (Corning) supplemented with 10% FBS (Corning) and 2% penicillin-streptomycin solution (Gibco) in a humidified incubator kept at 37 °C with a 5% carbon dioxide atmosphere.
RNAs
GQES7-a and GQES7-b were synthesized in vitro by transcription (HiScribeTM T7 High Yield RNA Synthesis Kit, New England Biolabs). GQes3 and mutes3 were purchased from Integrated DNA Technologies. Human 28S and 18S rRNAs were extracted from HEK293 cells with TRIzol (Invitrogen). Intact rRNAs were isolated by pipetting from a native agarose gel after running the rRNA into wells in the center of the gel. The rRNA was then precipitated with 5 m ammonium acetate-acetic acid (pH 7.5) with excess ethanol. RNA sequences are listed in Table S1.
RNA annealing
RNAs were annealed by heating at 95 °C for 5 min and cooled to 25 °C at 1 °C/min and incubated for 10 min at 4 °C.
UV-visible absorbance heme-RNA binding
Stock solutions of heme chloride (1 mm) were prepared in DMSO. Prior to use, the heme chloride solution was sonicated for 10 min. RNAs (GQES7-a, GQES7-b, GQes3, and mutes3) were annealed as described above in 50 mm KCl and 10 mm Tris-HCl, pH 7.5 in increasing RNA concentrations (for rRNA oligomers: from 0.3 to 1 equivalent of heme). The annealing buffer for intact 28S and 18S rRNAs and assembled ribosomal subunits and polysomes was the same as that of the rRNA oligomers except for the inclusion of 10 mm MgCl2. After RNA annealing, heme was added to a final concentration of 3 µm. Samples (20 µl) were allowed to stand at room temperature for 30 min, then loaded onto a Corning 384-well flat clear bottom microplate. Absorbances were recorded from 300 to 700 nm on a BioTek SynergyTM H4 Hybrid plate reader.
UV-visible absorbance, heme-PhenDC3 competition/cooperation assay
For heme-PhenDC3 competition assays, RNAs were annealed and allowed to bind to heme as above. Final heme concentration was 3 µm. Final RNA concentrations were GQES7-a (3 µm), intact human 18S rRNA (65 nm), and intact human 28S rRNA (22 nm). After solutions were incubated for 30 min at room temperature, PhenDC3 or carrier DMSO was added to final concentrations consisting of 1.5 µm, 3 µm, and 6 µm. Samples (20 µl) were allowed to stand at room temperature for 15 min and were loaded onto a Corning 384-well flat clear bottom microplate. Absorbance was recorded from 300 to 700 nm. For PhenDC3-heme cooperation assay (data on Fig. 3G), GQES7-a RNA (1 µm) was added to 3 µm heme in 10 mm LiCl and 10 mm Tris-HCl, pH 7.5 with no RNA-annealing step. After RNA-heme solutions were incubated for 30 min at room temperature, PhenDC3 was added to a final concentration range consisting of 1.33–133 nm and allowed to mix for 15 min. The remainder of the experiment was performed as the competition assay described above in this section.
Heme-rRNA dissociation constants were determined from the one-site binding model (68) depicted in the below equations using nonlinear least squares regression analysis software KaleidaGraph 4.5 (Synergy Software, Reading, PA).
Where KD is the rRNA-heme dissociation constant, HmT is the concentration of heme, rRNA-Hm is the concentration of the rRNA-heme complex, rRNAT is the concentration of rRNA that is being titrated, Abs is the absorbance at any given concentration of rRNAT, Abso is the initial absorbance of heme in the absence of rRNA, and ΔAbs is the change in fluorescence due to the formation of the rRNA-heme complex.
For data fitting, Abso, ΔAbs, rRNAT, and HmT were treated as fixed parameters derived from experiments, and the KD was a “floating” parameter that was derived from regression analysis. The absorbance signals utilized to determine KD were from the heme Soret band at 400 nm.
Total heme quantification of untreated and SA-treated HEK293 cells
Heme was quantified as described (69). Briefly, HEK293 cells were seeded in complete DMEM media at an initial confluency of 10% and incubated at 37 °C for 48 h. Media for SA-treated cells was replaced by DMEM supplemented with 10% heme-depleted FBS and 0.5 mm SA. Heme depletion of serum was performed as described (30). Media for untreated cells was replaced by complete media (supplemented with 10% regular FBS) and allowed to seed at 37 °C for 24 h. The cells were harvested by scrapping and counted using an automated TC10 cell counter (Bio-Rad). Then, 2.5 × 104 cells/condition were treated with 20 mm oxalic acid and incubated at 4 °C overnight in the dark. An equal volume of 2 m oxalic acid was added to the cell suspensions. The samples were split, with half incubated at 95 °C for 30 min and half incubated at room temperature for 30 min. The samples were centrifuged at 21,000 × g for 2 min, 200 µl of each was transferred to a black Greiner Bio-One flat bottom fluorescence plate, and porphyrin fluorescence (excitation (ex): 400 nm, emission (em): 620 nm) was recorded on a Synergy Mx multi-modal plate reader. Heme concentration was calculated from a standard curve prepared by diluting a 0.1 µm hemin chloride stock solution in DMSO and treated in the same way as the cell suspensions above. To calculate heme concentration, the fluorescence of the unboiled samples was taken as the background level of protoporphyrin IX and subtracted from the fluorescence of the boiled sample, which is used as the free base porphyrin produced upon the release of the heme iron. Using this method, our data suggest SA-treatment of HEK293 cells results in a 7-fold decrease in the total cellular heme concentration.
Hemin-agarose binding
HEK293 cells were seeded onto a 6-well plate at an initial confluency of 20% in DMEM with 10% FBS and allowed to seed for 48 h at 37 °C. Media was then replaced for DMEM with 10% heme-depleted FBS supplemented with 0.5 mm succinyl acetone (for SA-treated cells). For untreated cells, media was changed for DMEM in 10% regular FBS. Both treated and untreated samples were allowed to incubate at 37 °C for 24 h. The cells were then collected by scrapping and lysed using 1.5-mm zirconium beads (Benchmark Scientific). Lysates were quantified by Bradford assay. In the meantime, hemin-agarose beads and Sepharose beads were equilibrated three times by centrifugation with lysis buffer (0.1% Triton X-100, 10 mm sodium phosphate, 50 mm KCl, 5 mm EDTA, pH 7.5, 1 × protease arrest, and RNasin RNase Inhibitor (Promega)). 100 µl of beads (50-µl bed volume) were used per biological replicate. After bead equilibration, each lysate was divided into two and 10 µg were loaded to hemin-agarose and 10 µg to Sepharose beads. The mixtures were allowed to bind for 60 min, rotating at 20 rpm at room temperature. Then, three washes were performed using lysis buffer and supernatants were discarded. Each wash consisted of 10 min of incubation at room temperature with 20 rpm rotation followed by centrifugation at 700 × g for 5 min. Bead-bound fractions were eluted by a 15-min incubation at room temperature with 20-rpm rotation in 50 µl of 1 m imidazole in lysis buffer, then centrifuged at maximum speed for 2 min, and supernatants were collected. RNA was then extracted from eluted fractions with TRIzol using the manufacturer's protocol. For the PhenDC3 titration in the HEK293 cells experiment, the same protocol was followed, with the difference that the cells were allowed to seed for 24 h (20% initial confluency), and then PhenDC3 was added in increasing concentrations (5 µm, 10µm, and 20 µm). DMSO carrier treatment was performed the same way but with equivalent DMSO volumes. The cells were left at 37 °C for 48 h and collected and lysed as described above.
RT-qPCR
The sets of primers used can be found in Table S2. Luna Universal One-Step RT-qPCR kit (New England Biolabs) was used following the manufacturer's protocol. Fold enrichments were calculated by comparing the C(t) values obtained from RNAs extracted from hemin-agarose to RNAs extracted from Sepharose beads. Three biological replicates were performed for all the RT-qPCR experiments. For BioTASQ experiments, fold enrichments were calculated by comparing the C(t) values obtained from the lysates containing BioTASQ + beads with those containing beads only.
Heme bioavailability assay using the HS1 sensor
HEK293 cells were plated and transfected in polystyrene-coated sterile 6-well plates (Greiner) for flow cytometry. The cells were plated in basal growth medium DMEM containing 10% FBS. At 30% confluency, the cells were transfected with the heme sensor plasmid pEF52α-hHS1 using Lipofectamine LTX according to the manufacturer's protocols. After 48 h of treatment with transfection reagents, the cells were treated with PhenDC3 (1 mm stock) in fresh DMEM 10% FBS for 24 h prior to harvesting. Heme-depleted cells were treated with 500 µm SA in DMEM containing 10% heme-depleted FBS for 72 h prior to harvesting. Heme-sufficient cells were treated with 350 µm ALA in DMEM 10% FBS for 24 h. The cells were harvested in 1× PBS for flow analysis. Flow cytometric measurements were performed using a BD FACSAria III Cell Sorter equipped with an argon laser (ex 488 nm) and a yellow-green laser (ex 561 nm). Enhanced GFP (eGFP) was excited using the argon laser and was measured using a 530/30-nm bandpass filter, and mKate2 was excited using the yellow-green laser and was measured using a 610/20-nm bandpass filter. Data evaluation was conducted using FlowJo v10.4.2 software. Single cells used in the analysis were selected for by first gating for forward scatter and side scatter, consistent with intact cells, and then fluorescence intensities above background were selected by gating for cells with mKate2. The fraction of sensor bound to heme may be quantified according to the following equation (40):
where R is the median eGFP/mKate2 fluorescence ratio in regular media and Rmin and Rmax are the median sensor ratios when the sensor is depleted of heme or saturated with heme. Rmin and Rmax values are derived from cells cultured in HD+SA or in media conditioned with ALA (40). The plot in Fig. 4D was obtained by fitting the median sensor ratios in Fig. 4C to the following one-site binding model (40, 68):
where x is the independent variable, PhenDC3.
BG4 purification
pSANG10-3F-BG4 was a gift from Shankar Balasubramanian (Addgene plasmid 55756; RRID:Addgene_55756). BL21 cells transformed with this plasmid were grown in room temperature and induced overnight with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were pelleted, then resuspended in xTractor (Takara Bio) supplemented with Protease Arrest (G-Biosciences), lysozyme, and DNase I. Sonicated cell lysate was combined with nickel-nitrilotriacetic acid resin (Invitrogen) and purified via the His-tag. BG4 was further purified by FPLC using a Superdex75 size exclusion column (GE Healthcare).
Immunofluorescence
Immunofluorescence was performed by standard protocols. HEK293 cells were seeded onto poly-l-lysine–coated cover glass 2 days before the experiment and fixed in 4% formaldehyde for 15 min. The cells were permeabilized with 0.1% Triton X-100 for 3 min and blocked with 5% donkey serum (Jackson ImmunoResearch Laboratories), followed by incubation with antibodies for 1 h at room temperature or overnight at 4 °C. Antibodies used here are BG4, rabbit anti-FLAG (Cell Signaling Technology, 14793S), mouse anti-L19 (Santa Cruz Biotechnology, sc-100830), mouse anti-rRNA (Santa Cruz Biotechnology, sc-33678), mouse anti-Calnexin (Santa Cruz Biotechnology, sc-23954), Alexa Fluor 488 conjugated donkey anti-rabbit (Jackson ImmunoResearch Laboratories, 711-545-152), and Rhodamine Red-X conjugated donkey anti-mouse (Jackson ImmunoResearch Laboratories, 715-295150). After staining, the cells were carefully washed with Dulbecco's PBS supplemented with 0.1% Tween 20. Nuclear DNA was stained with 4′,6-diamidino-2-phenylindole. Images were acquired with a Zeiss 700 Laser Scanning Confocal Microscope. PhenDC3 treatment consisted of incubation at 37 °C overnight at 10 µm. PhenDC3 treatment was done prior to cell fixation. Determination of colocalization ratios was performed as described in Zen software (Zeiss). No-primary-antibody controls and RNase A– and PhenDC3-treated images are reported in Fig. S6 and Fig. S7. The colocalization image in Fig. 2, A and B shows the G4 signal that colocalizes with L19 and with the ER (yellow pixels) and the one that does not colocalize (green pixels). L19, ER, and BG4 images only present their respective fluorescence signals.
BioTASQ capture of cellular RNAs
BioTASQ experiments followed published protocols in vitro (8) and in vivo (22). Briefly, HEK293 cells were seeded onto a 6-well plate at 20% confluency and allowed to incubate at 37 °C for 48 h. The cells were then cross-linked with 1% paraformaldehyde/PBS for 5 min at room temperature. Cross-linking was stopped by incubating cells with 0.125 m glycine for 5 min at room temperature. The cells were harvested by scrapping and resuspended in lysis buffer (200 mm KCl, 25 mm Tris-HCl, pH 7.5, 5 mm EDTA, 0.5 mm DTT, 1% Triton X-100, RNasin RNase Inhibitor, and 1× protease arrest). The cells were lysed by sonication (30% amplitude, 10 s on and off intervals, and 2 min sonication time). The lysate was then split: BioTASQ was added at a final concentration of 100 µm to one of the samples, and the other sample was left untreated. The lysates were incubated at 4 °C overnight with gentle rotation. Sera-Mag magnetic streptavidin-coated beads (GE Healthcare) were washed three times with wash buffer (5 mm Tris-HCl, pH 7.5, 0.5 mm EDTA, and 1 m KCl). Each wash was followed by centrifugation at 3,500 rpm for 5 min at 4 °C. The beads were then treated with buffer 1 (0.1 m NaOH, and 0.05 m KCl in RNase/DNase-free water) two times at room temperature for 2 min and then centrifuged at 3,500 rpm at 4 °C for 5 min and washed with buffer 2 (0.1 m KCl in RNase/DNase-free water). Lastly, to block, the beads were treated with 1 µg/ml BSA and 1 µg/ml yeast tRNA and allowed to incubate at 4 °C overnight with gentle rotation.
After incubation overnight with BioTASQ, the cell lysates were treated with 1% BSA for 1 h at 4 °C. Washed magnetic beads were added to the lysates (20 µg beads/sample) and allowed to mix with gentle rotation at 4 °C for 1 h. The beads were then washed three times with lysis buffer for 5 min, and then cross-linking was reversed by incubating the beads at 70 °C for 1 h. Finally, TRIzol was used to extract RNAs for analysis by RT-qPCR.
Data availability
All data are contained within this article and in the supporting information.
Supplementary Material
Acknowledgments
We thank Drs. Rebecca Donegan, David A. Hanna, Jonathan B. Chaires, Aaron Engelhart, David Monchaud, and Judy Wong, and Claudia Montllor-Albalate for helpful discussions. We acknowledge Andrew Shaw and the core facilities at the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology for expert advice and the use of equipment. Purified human ribosomes and polysomes were a gift from Immagina BioTechnology. BioTASQ was a gift from Dr. David Monchaud.
This article contains supporting information.
Author contributions—S. M.-F., A. R. R., and L. D. W. conceptualization; S. M.-F., C. I., C. M. M., and A. R. R. data curation; S. M.-F., and C. I. formal analysis; S. M.-F. and C. I. validation; S. M.-F., C. I., and C. M. M. investigation; S. M.-F. and C. I. visualization; S. M.-F. methodology; S. M.-F., A. R. R., and L. D. W. writing-original draft; A. R. R. and L. D. W. resources; A. R. R. and L. D. W. supervision; A. R. R. and L. D. W. funding acquisition; A. R. R. and L. D. W. project administration.
Funding and additional information—This work was supported by NASA Grants 80NSSC17K0295 and 80NSSC18K1139 (Center for the Origin of Life) (to L. D. W.), the National Institutes of Health Grant ES025661 (to A. R. R.), and the National Science Foundation Grant MCB-1552791 (to A. R. R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflict of interest with the contents of this article.
- G4
- G-quadruplex
- rProtein
- ribosomal protein
- ER
- endoplasmic reticulum
- SSU
- small subunit
- LSU
- large subunit
- SA
- succinylacetone
- HS
- heme sensor
- HD+SA
- heme-deficient media containing SA
- ALA
- 5-aminolevulinic acid
- eGFP
- enhanced GFP
- ex
- excitation
- em
- emission.
References
- 1. Swenson S. A., Moore C. M., Marcero J. R., Medlock A. E., Reddi A. R., and Khalimonchuk O. (2020) From synthesis to utilization: the ins and outs of mitochondrial heme. Cells 9, 579 10.3390/cells9030579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Donegan R. K., Moore C. M., Hanna D. A., and Reddi A. R. (2019) Handling heme: the mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 133, 88–100 10.1016/j.freeradbiomed.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanna D. A., Martinez-Guzman O., and Reddi A. R. (2017) Heme gazing: illuminating eukaryotic heme trafficking, dynamics, and signaling with fluorescent heme sensors. Biochemistry 56, 1815–1823 10.1021/acs.biochem.7b00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reddi A. R., and Hamza I. (2016) Heme mobilization in animals: a metallolipid's journey. Acc. Chem. Res. 49, 1104–1110 10.1021/acs.accounts.5b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mestre-Fos S., Penev P. I., Suttapitugsakul S., Hu M., Ito C., Petrov A. S., Wartell R. M., Wu R., and Williams L. D. (2019) G-quadruplexes in human ribosomal RNA. J. Mol. Biol. 431, 1940–1955 10.1016/j.jmb.2019.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman J. C., Petrov A. S., Frenkel-Pinter M., Penev P. I., and Williams L. D. (2020) Root of the tree: the significance, evolution, and origins of the ribosome. Chem. Rev. 120, 4848–4878 10.1021/acs.chemrev.9b00742 [DOI] [PubMed] [Google Scholar]
- 7. Melnikov S., Ben-Shem A., De Loubresse N. G., Jenner L., Yusupova G., and Yusupov M. (2012) One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 19, 560–567 10.1038/nsmb.2313 [DOI] [PubMed] [Google Scholar]
- 8. Mestre-Fos S., Penev P. I., Richards J. C., Dean W. L., Gray R. D., Chaires J. B., and Williams L. D. (2019) Profusion of G-quadruplexes on both subunits of metazoan ribosomes. PLoS ONE 14, e0226177 10.1371/journal.pone.0226177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Milo R., and Phillips R. (2015) Cell biology by the numbers. Garland Science; 10.1201/9780429258770 [DOI] [Google Scholar]
- 10. Poon L. C.-H., Methot S. P., Morabi-Pazooki W., Pio F., Bennet A. J., and Sen D. (2011) Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 133, 1877–1884 10.1021/ja108571a [DOI] [PubMed] [Google Scholar]
- 11. Sen D., and Poon L. C. (2011) RNA and DNA complexes with hemin [Fe (III) heme] are efficient peroxidases and peroxygenases: how do they do it and what does it mean? Crit. Rev. Biochem. Mol. Biol. 46, 478–492 10.3109/10409238.2011.618220 [DOI] [PubMed] [Google Scholar]
- 12. Li Y., and Sen D. (1996) A catalytic DNA for porphyrin metallation. Nat. Struct. Biol. 3, 743–747 10.1038/nsb0996-743 [DOI] [PubMed] [Google Scholar]
- 13. Lat P. K., Liu K., Kumar D. N., Wong K. K. L., Verheyen E. M., and Sen D. (2020) High specificity and tight spatial restriction of self-biotinylation by DNA and RNA G-quadruplexes complexed in vitro and in vivo with heme. Nucleic Acids Res. 48, 5254–5267 10.1093/nar/gkaa281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paeschke K., Capra J. A., and Zakian V. A. (2011) DNA replication through G-quadruplex motifs is promoted by the saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691 10.1016/j.cell.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siddiqui-Jain A., Grand C. L., Bearss D. J., and Hurley L. H. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 99, 11593–11598 10.1073/pnas.182256799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribeyre C., Lopes J., Boulé J.-B., Piazza A., Guédin A., Zakian V. A., Mergny J.-L., and Nicolas A. (2009) The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 5, e1000475 10.1371/journal.pgen.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beaudoin J.-D., and Perreault J.-P. (2010) 5′-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res. 38, 7022–7036 10.1093/nar/gkq557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arora A., Dutkiewicz M., Scaria V., Hariharan M., Maiti S., and Kurreck J. (2008) Inhibition of translation in living eukaryotic cells by an RNA G-quadruplex motif. RNA 14, 1290–1296 10.1261/rna.1001708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumari S., Bugaut A., Huppert J. L., and Balasubramanian S. (2007) An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 3, 218–221 10.1038/nchembio864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo J. U., and Bartel D. P. (2016) RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353, aaf5371 10.1126/science.aaf5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fay M. M., Lyons S. M., and Ivanov P. (2017) RNA G-quadruplexes in biology: principles and molecular mechanisms. J. Mol. Biol. 429, 2127–2147 10.1016/j.jmb.2017.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang S. Y., Lejault P., Chevrier S., Boidot R., Robertson A. G., Wong J. M., and Monchaud D. (2018) Transcriptome-wide identification of transient RNA G-quadruplexes in human cells. Nat. Commun. 9, 4730 10.1038/s41467-018-07224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biffi G., Di Antonio M., Tannahill D., and Balasubramanian S. (2014) Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat Chem 6, 75–80 10.1038/nchem.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biffi G., Tannahill D., McCafferty J., and Balasubramanian S. (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 5, 182–186 10.1038/nchem.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moye A. L., Porter K. C., Cohen S. B., Phan T., Zyner K. G., Sasaki N., Lovrecz G. O., Beck J. L., and Bryan T. M. (2015) Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 6, 7643 10.1038/ncomms8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conlon E. G., Lu L., Sharma A., Yamazaki T., Tang T., Shneider N. A., and Manley J. L. (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in als brains. eLife 5, e17820 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hänsel-Hertsch R., Beraldi D., Lensing S. V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M., Tannahill D., and Balasubramanian S. (2016) G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 48, 1267–1272 10.1038/ng.3662 [DOI] [PubMed] [Google Scholar]
- 28. Laguerre A., Wong J. M., and Monchaud D. (2016) Direct visualization of both DNA and RNA quadruplexes in human cells via an uncommon spectroscopic method. Sci. Rep. 6, 32141 10.1038/srep32141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monchaud D., Allain C., Bertrand H., Smargiasso N., Rosu F., Gabelica V., De Cian A., Mergny J.-L., and Teulade-Fichou M.-P. (2008) Ligands playing musical chairs with G-quadruplex DNA: a rapid and simple displacement assay for identifying selective G-quadruplex binders. Biochimie 90, 1207–1223 10.1016/j.biochi.2008.02.019 [DOI] [PubMed] [Google Scholar]
- 30. De Cian A., Delemos E., Mergny J.-L., Teulade-Fichou M.-P., and Monchaud D. (2007) Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 129, 1856–1857 10.1021/ja067352b [DOI] [PubMed] [Google Scholar]
- 31. Piazza A., Boulé J.-B., Lopes J., Mingo K., Largy E., Teulade-Fichou M.-P., and Nicolas A. (2010) Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 38, 4337–4348 10.1093/nar/gkq136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonnat L., Bar L., Génnaro B., Bonnet H., Jarjayes O., Thomas F., Dejeu J., Defrancq E., and Lavergne T. (2017) Template-mediated stabilization of a DNA G-quadruplex formed in the HIV-1 promoter and comparative binding studies. Chem. Eur. J. 23, 5602–5613 10.1002/chem.201700417 [DOI] [PubMed] [Google Scholar]
- 33. Reid D. W., and Nicchitta C. V. (2012) Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 287, 5518–5527 10.1074/jbc.M111.312280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Renard I., Grandmougin M., Roux A., Yang S. Y., Lejault P., Pirrotta M., Wong J. M., and Monchaud D. (2019) Small-molecule affinity capture of DNA/RNA quadruplexes and their identification in vitro and in vivo through the G4RP protocol. Nucleic Acids Res. 47, 5502–5510 10.1093/nar/gkz215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gray L. T., Lombardi E. P., Verga D., Nicolas A., Teulade-Fichou M.-P., Londoño-Vallejo A., and Maizels N. (2019) G-quadruplexes sequester free heme in living cells. Cell Chem. Biol. 26, 1681–1691.e5 10.1016/j.chembiol.2019.10.003 [DOI] [PubMed] [Google Scholar]
- 36. Shimizu H., Tai H., Saito K., Shibata T., Kinoshita M., and Yamamoto Y. (2015) Characterization of the interaction between heme and a parallel G-quadruplex DNA formed from d(TTAGGGT). Bull. Chem. Soc. Jpn. 88, 644–652 10.1246/bcsj.20140374 [DOI] [Google Scholar]
- 37. Saito K., Tai H., Hemmi H., Kobayashi N., and Yamamoto Y. (2012) Interaction between the heme and a G-quartet in a heme–DNA complex. Inorg. Chem. 51, 8168–8176 10.1021/ic3005739 [DOI] [PubMed] [Google Scholar]
- 38. Shinomiya R., Katahira Y., Araki H., Shibata T., Momotake A., Yanagisawa S., Ogura T., Suzuki A., Neya S., and Yamamoto Y. (2018) Characterization of catalytic activities and heme coordination structures of heme–DNA complexes composed of some chemically modified hemes and an all parallel-stranded tetrameric G-quadruplex DNA formed from d(TTAGGG). Biochemistry 57, 5930–5937 10.1021/acs.biochem.8b00793 [DOI] [PubMed] [Google Scholar]
- 39. Grigg J. C., Shumayrikh N., and Sen D. (2014) G-quadruplex structures formed by expanded hexanucleotide repeat RNA and DNA from the neurodegenerative disease-linked C9orf72 gene efficiently sequester and activate heme. PLoS ONE 9, e106449 10.1371/journal.pone.0106449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanna D. A., Harvey R. M., Martinez-Guzman O., Yuan X., Chandrasekharan B., Raju G., Outten F. W., Hamza I., and Reddi A. R. (2016) Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. U.S.A. 113, 7539–7544 10.1073/pnas.1523802113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ebert P. S., Hess R. A., Frykholm B. C., and Tschudy D. P. (1979) Succinylacetone, a potent inhibitor of heme biosynthesis: effect on cell growth, heme content and δ-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem. Biophys. Res. Commun. 88, 1382–1390 10.1016/0006-291x(79)91133-1 [DOI] [PubMed] [Google Scholar]
- 42. Hanna D. A., Hu R., Kim H., Martinez-Guzman O., Torres M. P., and Reddi A. R. (2018) Heme bioavailability and signaling in response to stress in yeast cells. J. Biol. Chem. 293, 12378–12393 10.1074/jbc.RA118.002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sweeny E. A., Singh A. B., Chakravarti R., Martinez-Guzman O., Saini A., Haque M. M., Garee G., Dans P. D., Hannibal L., Reddi A. R., and Stuehr D. J. (2018) Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 293, 14557–14568 10.1074/jbc.RA118.004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinez-Guzman O., Willoughby M. M., Saini A., Dietz J. V., Bohovych I., Medlock A. E., Khalimonchuk O., and Reddi A. R. (2020) Mitochondrial–nuclear heme trafficking in budding yeast is regulated by GTPases that control mitochondrial dynamics and ER contact sites. J. Cell Sci. 133, jcs237917 10.1242/jcs.237917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hannibal L., Collins D., Brassard J., Chakravarti R., Vempati R., Dorlet P., Santolini J. R. M., Dawson J. H., and Stuehr D. J. (2012) Heme binding properties of glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 51, 8514–8529 10.1021/bi300863a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chakravarti R., Aulak K. S., Fox P. L., and Stuehr D. J. (2010) GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 18004–18009 10.1073/pnas.1008133107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dai Y., Sweeny E. A., Schlanger S., Ghosh A., and Stuehr D. J. (2020) GAPDH delivers heme to soluble guanylyl cyclase. J. Biol. Chem. 295, 8145–8154 10.1074/jbc.RA120.013802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Piel I. I. R., Shiferaw M. T., Vashisht A. A., Marcero J. R., Praissman J. L., Phillips J. D., Wohlschlegel J. A., and Medlock A. E. (2016) A novel role for progesterone receptor membrane component 1 (PGRMC1): a partner and regulator of ferrochelatase. Biochemistry 55, 5204–5217 10.1021/acs.biochem.6b00756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Craven R. J., Mallory J. C., and Hand R. A. (2007) Regulation of iron homeostasis mediated by the heme-binding protein Dap1 (damage resistance protein 1) via the p450 protein Erg11/Cyp51. J. Biol. Chem. 282, 36543–36551 10.1074/jbc.M706770200 [DOI] [PubMed] [Google Scholar]
- 50. Mallory J. C., Crudden G., Johnson B. L., Mo C., Pierson C. A., Bard M., and Craven R. J. (2005) Dap1p, a heme-binding protein that regulates the cytochrome p450 protein Erg11p/Cyp51p in Saccharomyces cerevisiae. Mol. Cell Biol. 25, 1669–1679 10.1128/MCB.25.5.1669-1679.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghosh K., Thompson A. M., Goldbeck R. A., Shi X., Whitman S., Oh E., Zhiwu Z., Vulpe C., and Holman T. R. (2005) Spectroscopic and biochemical characterization of heme binding to yeast Dap1p and mouse PGRMC1p. Biochemistry 44, 16729–16736 10.1021/bi0511585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thompson A. M., Reddi A. R., Shi X., Goldbeck R. A., Moënne-Loccoz P., Gibney B. R., and Holman T. R. (2007) Measurement of the heme affinity for yeast dap1p, and its importance in cellular function. Biochemistry 46, 14629–14637 10.1021/bi7013739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hughes A. L., Powell D. W., Bard M., Eckstein J., Barbuch R., Link A. J., and Espenshade P. J. (2007) Dap1/PGRMC1 binds and regulates cytochrome p450 enzymes. Cell Metab. 5, 143–149 10.1016/j.cmet.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 54. Galmozzi A., Kok B. P., Kim A. S., Montenegro-Burke J. R., Lee J. Y., Spreafico R., Mosure S., Albert V., Cintron-Colon R., Godio C., Webb W. R., Conti B., Solt L. A., Kojetin D., Parker C. G., et al. (2019) PGRMC2 is an intracellular haem chaperone critical for adipocyte function. Nature 576, 138–142 10.1038/s41586-019-1774-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yuan X., Rietzschel N., Kwon H., Nuno A. B. W., Hanna D. A., Phillips J. D., Raven E. L., Reddi A. R., and Hamza I. (2016) Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc. Natl. Acad. Sci. U.S.A. 113, E5144–E5152 10.1073/pnas.1609865113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song Y., Yang M., Wegner S. V., Zhao J., Zhu R., Wu Y., He C., and Chen P. R. (2015) A genetically encoded fret sensor for intracellular heme. ACS Chem. Biol. 10, 1610–1615 10.1021/cb5009734 [DOI] [PubMed] [Google Scholar]
- 57. Wilson D. N. (2014) Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12, 35–48 10.1038/nrmicro3155 [DOI] [PubMed] [Google Scholar]
- 58. Hostetter A. A., Osborn M. F., and Derose V. J. (2012) RNA-pt adducts following cisplatin treatment of Saccharomyces cerevisiae. ACS Chem. Biol. 7, 218–225 10.1021/cb200279p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Plakos K., and Derose V. J. (2017) Mapping platinum adducts on yeast ribosomal RNA using high-throughput sequencing. Chem. Commun. 53, 12746–12749 10.1039/c7cc06708a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rijal K., and Chow C. S. (2008) A new role for cisplatin: probing ribosomal RNA structure. Chem. Commun [DOI] [PubMed] [Google Scholar]
- 61. Del Valle A. H., Seip B., Cervera-Marzal I., Sacheau G., Seefeldt A. C., and Innis C. A. (2020) Ornithine capture by a translating ribosome controls bacterial polyamine synthesis. Nat. Microbiol. 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gesteland R. F. (1966) Unfolding of Escherichia coli ribosomes by removal of magnesium. J. Mol. Biol. 18, 356–371 10.1016/s0022-2836(66)80253-x [DOI] [PubMed] [Google Scholar]
- 63. Stahli C., and Noll H. (1977) Structural dynamics of bacterial ribosomes. Mol. Gen. Genet. MGG 153, 159–168 10.1007/BF00264731 [DOI] [PubMed] [Google Scholar]
- 64. Weiss R. L., Kimes B. W., and Morris D. R. (1973) Cations and ribosome structure. III. Effects on the 30S and 50S subunits of replacing bound Mg2+ by inorganic cations. Biochemistry 12, 450–456 10.1021/bi00727a014 [DOI] [PubMed] [Google Scholar]
- 65. Gordon J., and Lipmann F. (1967) Role of divalent ions in poly U-directed phenylalanine polymerization. J. Mol. Biol. 23, 23–33 10.1016/S0022-2836(67)80064-0 [DOI] [Google Scholar]
- 66. Nierhaus K. H. (2014) Mg2+, K+, and the ribosome. J. Bacteriol. 196, 3817–3819 10.1128/JB.02297-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bray M. S., Lenz T. K., Haynes J. W., Bowman J. C., Petrov A. S., Reddi A. R., Hud N. V., Williams L. D., and Glass J. B. (2018) Multiple prebiotic metals mediate translation. Proc. Natl. Acad. Sci. U.S.A. 115, 12164–12169 10.1073/pnas.1803636115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sankar S. B., Donegan R. K., Shah K. J., Reddi A. R., and Wood L. B. (2018) Heme and hemoglobin suppress amyloid β–mediated inflammatory activation of mouse astrocytes. J. Biol. Chem. 293, 11358–11373 10.1074/jbc.RA117.001050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sinclair P. R., Gorman N., and Jacobs J. M. (1999) Measurement of heme concentration. Curr. Protoc. Toxicol. 8.3.1–8.3.7 10.1002/0471140856.tx0803s00 [DOI] [PubMed] [Google Scholar]
- 70. Zhu Y., Hon T., Ye W., and Zhang L. (2002) Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 13, 431–439 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within this article and in the supporting information.