

Abstract
β2 integrins mediate neutrophil-endothelial adhesion and recruitment of neutrophils to sites of inflammation. The diminished expression of β2 integrins in patients with mutations in the ITGB2 (CD18) gene (leukocyte adhesion deficiency-Type 1; LAD1) results in few or no neutrophils in peripheral tissues. In the periodontium, neutrophil paucity is associated with upregulation of interleukin (IL)-23 and IL-17, which drive inflammatory bone loss. Using a relevant mouse model, we investigated whether diminished efferocytosis (owing to neutrophil scarcity) is associated with LAD1 periodontitis pathogenesis and aimed to develop approaches to restore the missing efferocytosis signals. We first showed that CD18−/− mice phenocopied human LAD1 in terms of IL-23/IL-17-driven inflammatory bone loss. Antibody-mediated blockade of c-Mer tyrosine kinase (Mer), a major efferocytic receptor, mimicked LAD1-associated upregulation of gingival IL-23 and IL-17 mRNA expression in wild-type (WT) mice. Consistently, soluble Mer-Fc reversed the inhibitory effect of efferocytosis on IL-23 expression in LPS-activated macrophages. Adoptive transfer of WT neutrophils to CD18−/− mice downregulated IL-23 and IL-17 expression to normal levels, but not when CD18−/− mice were treated with blocking anti-Mer antibody. Synthetic agonist-induced activation of liver X receptors (LXR) and peroxisome proliferator-activated receptors (PPAR), which link efferocytosis to generation of homeostatic signals, inhibited the expression of IL-23 and IL-17 and favorably affected the bone levels of CD18−/− mice. Therefore, our data link diminished efferocytosis-associated signaling due to impaired neutrophil recruitment to dysregulation of the IL-23–IL-17 axis and, moreover, suggest LXR and PPAR as potential therapeutic targets for treating LAD1 periodontitis.
Keywords: β2-integrins, leukocyte adhesion deficiency, neutrophils, interleukin-17, liver X receptors, peroxisome proliferator-activated receptors
Summary sentence:
Synthetic agonist-induced activation of metabolic nuclear receptors mimicking efferocytosis-related signaling restored periodontal tissue homeostasis in a mouse model of leukocyte adhesion deficiency type 1.
Graphical Abstract
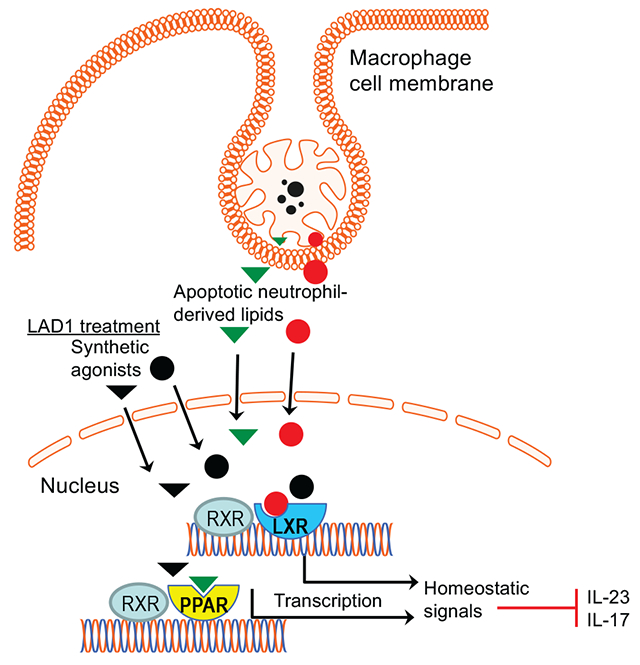
1. INTRODUCTION
Neutrophils are terminally differentiated white blood cells forming the first line of defense against pathogenic insults, although they have also been implicated in the pathogenesis of inflammatory diseases [1–4]. Huge numbers of neutrophils (about 109 cells per kg bodyweight) are produced daily in the bone marrow (BM) but they are relatively short-lived, undergoing programmed cell death by apoptosis in peripheral tissues or the BM [5]. As there is a delicate balance between the protective and potentially destructive effects of neutrophils, their production, trafficking, and clearance are tightly regulated by several homeostatic mechanisms operating in the BM or the periphery [2, 5]. In mice and humans, the granulocyte colony-stimulating factor (G-CSF) is the primary regulator of both granulopoiesis and neutrophil release from the BM [6, 7]. Interleukin (IL)-17 (also known as IL-17A) promotes granulopoiesis and induces the recruitment, activation, and survival of neutrophils by acting mainly through the upregulation of G-CSF and neutrophil-related chemokines [5, 8].
Upon their mobilization from the BM, neutrophils circulate in the blood and can migrate to extravascular sites (e.g., in the skin, gut, lungs, or gingiva) in response to infection or inflammation. The process of neutrophil extravasation involves a cascade of low- and high-affinity adhesive interactions between the neutrophils and the endothelium [1, 2, 9, 10]. The first step involves transient rolling interactions, mediated by the binding of endothelial cell surface molecules (P- or E-selectin) to their glycoprotein ligands on neutrophils. This rolling-dependent deceleration of neutrophils is followed by their firm adhesion and subsequent crawling on the endothelium, during which neutrophils seek appropriate sites for transmigration. Firm adhesion and crawling are primarily mediated by heterodimeric β2-integrins, each with a distinct CD11 subunit and a common CD18 subunit. The LFA-1 integrin (CD11a/CD18) plays a crucial role in firm adhesion, which involves interactions with endothelial counter-receptors (e.g., intercellular adhesion molecule-1). The LFA-1–dependent firm adhesion to the endothelium is required for extravasation of the neutrophils to peripheral tissues [1, 2, 11]
Neutrophil homeostasis is important for periodontal health as evident from human clinical observations and the phenotype of relevant mouse models. Indeed, neutrophil disorders affecting their numbers or function can readily precipitate periodontitis [12–18]. Periodontitis, a dysbiotic inflammatory disease that affects the tissues that surround and support the dentition (‘periodontium’, e.g., gingiva and alveolar bone), typically affects adults [19, 20]. However, individuals with disorders affecting neutrophil recruitment to the periodontium (and other peripheral tissues), such as Type 1 leukocyte adhesion deficiency (LAD1) rapidly develop severe periodontitis early in life [14, 15, 18, 21, 22]. LAD1 is an autosomal recessive primary immunodeficiency caused by mutations in the CD18-encoding ITGB2 gene that result in defective neutrophil adhesion to the endothelium (since β2-integrins such as LFA-1 are critical for this function) and hence impaired extravasation [15, 17, 23]. LAD1 patients thus have few or no neutrophils in the periodontium and other peripheral tissues and typically have recurrent bacterial infections and pathological inflammation in the skin and mucosal surfaces, as well as display profound alveolar bone loss early in life, followed by premature loss of primary and permanent teeth [14, 15, 18, 21, 22]. Rare diseases, such as LAD1, constitute an important medical burden cumulatively affecting 25 million patients in North America alone [24]. Moreover, rare monogenic diseases represent real-life models to gain insights into human biology and (patho)physiological mechanisms, thereby contributing to a better understanding of the pathogenesis of common diseases [16, 25].
LAD1–associated periodontitis (hereafter ‘LAD1 periodontitis’) has been historically attributed to lack of neutrophil surveillance of the periodontal infection; yet, this form of periodontitis has proven unresponsive to antibiotics and/or mechanical removal of the tooth-associated biofilm [14, 22, 26]. We have recently challenged this notion, however, by showing that the underlying etiology of LAD1 periodontitis involves a dysregulated host response that leads to overexpression of the proinflammatory and bone-resorptive cytokines IL-23 and IL-17 [21]. Local antibody-mediated neutralization of IL-23 or IL-17 in LFA-1-deficient mice that mimic the LAD1 phenotype inhibited periodontal inflammation and bone loss [21]. Consistently and importantly, systemic administration of an antibody that blocks the common p40 subunit of IL-23 and IL-12 (ustekinumab) in a human LAD1 patient resulted in inhibition of gingival expression of IL-17 and resolved inflammatory periodontal lesions [27].
Although the precise mechanism(s) for the dysregulated IL-23–IL-17 axis in LAD1 periodontitis is uncertain, one possibility that, in part, may explain the phenotype is related to the disruption of ‘neutrostat’, a homeostatic mechanism that coordinates the recruitment and efferocytosis of neutrophils with their production [28]: Transmigrated neutrophils become apoptotic and undergo phagocytosis (efferocytosis) by tissue phagocytes, primarily macrophages [29]. The efferocytosis of apoptotic neutrophils fulfils more than a mechanism of ‘waste disposal’ that can prevent secondary necrosis and the leakage of cytotoxic or pro-inflammatory molecules [28, 30–33]. Indeed, upon efferocytosis, macrophages are transcriptionally re-programmed to downregulate the expression of IL-23 and other proinflammatory cytokines and up-regulate the expression of pro-resolving cytokines or lipid mediators, such as TGFβ and resolvins, respectively [28, 30–33]. Liver X receptors (LXR; comprising two isoforms, LXRα and LXRβ) and peroxisome proliferator-activated receptors (PPAR; present in distinct isoforms α, β/δ, and γ) are ligand-activated transcription factors of the nuclear receptor superfamily that link efferocytosis to inflammation resolution [33–35]. In addition to its immunomodulatory effects, LXR signaling during efferocytosis also enhances the expression of a major efferocytic receptor, c-Mer tyrosine kinase (Mer), thereby further potentiating efferocytosis [30, 35, 36]. LXRs appear to be activated by sterol lipids and PPARs by polyunsaturated fatty acids, derived from the apoptotic cell plasma membrane [37].
Since efferocytosis inhibits IL-23 [28, 30–33], which is key to the induction and amplifies the expression of IL-17 in both innate and adaptive immune cells [38], the production of IL-17 is also suppressed, in turn leading to decreased production of G-CSF and thereby limiting the stimulus for neutrophil production to maintain steady-state neutrophil counts [28]. However, in LAD1, neutrophils cannot transmigrate to the periodontium. Therefore, the regulatory (inhibitory) signals for the expression of IL-23 and IL-17 are absent, or diminished, whereas the local microbial/inflammatory challenge remains; as a consequence, the local expression of IL-23 and IL-17 should be unrestrained. Consistent with this notion, in this paper we showed that antibody-mediated blockade of a major efferocytic receptor, c-Mer tyrosine kinase (Mer), resulted in upregulation of gingival IL-23 and IL-17 mRNA expression in wild-type (WT) mice, thus mimicking the LAD1 phenotype of CD18−/− mice. Adoptive transfer of WT neutrophils to CD18−/− mice restored normal expression of IL-23 and IL-17, although the expression of these cytokines remained at high levels if the CD18−/− mice were additionally treated with blocking anti-Mer antibody. We then reasoned that, if defective efferocytosis in CD18−/− mice contributes to the upregulation of the IL-23–IL-17 axis in the periodontium, then pharmacologic induction of signals that are normally produced by efferocytosis should restore homeostasis in the LAD1 periodontium. In line with this hypothesis, CD18−/− mice locally treated with a combination of synthetic agonists of LXRα/LXRβ and PPARβ/δ, which link efferocytosis to induction of homeostatic signals [33–35], exhibited decreased expression of IL-23 and IL-17 and improved bone levels. We conclude that the pharmacologic induction of signals that mimic the anti-inflammatory action of apoptotic neutrophil efferocytosis provides a novel and effective option to treat LAD1 periodontitis.
2. MATERIALS AND METHODS
2.1. Mice
Mice genetically deficient in all β2 integrins (CD18−/−; B6.129S7-Itgb2tm2Bay/J) [39] or only in the LFA-1 β2-integrin (LFA-1−/−; B6.129S7-Itgaltm1Bll/J) [40] were purchased from the Jackson Laboratories. These mice were crossed with wild-type C57BL/6J mice (Jackson Laboratories) to generate gene-deficient (homozygotes and heterozygotes) mice and wild-type littermate controls for use in experiments. Groups of mice within the same experiment were sex- and age-matched. When euthanized, the mice were 18-week old, an age in which substantial periodontal bone loss has occurred associated with the LAD1 phenotype. As there were no significant differences in the results obtained with males and females (e.g., CD18 deficiency resulted in similar bone loss regardless of sex), their respective data were pooled per treatment group. In experiments comparing CD18−/− and LFA-1−/− mice, WT controls included both CD18+/+ and LFA-1+/+ mice at equal numbers; as the results obtained with CD18+/+ and LFA-1+/+ mice were indistinguishable, the data were pooled and presented as WT group data. Mice were used as early as 4 weeks of age for cytokine blocking experiments (section 2.3). Mice at 6 weeks of age were used for bone marrow transplantation studies. Chimeric mice were generated by adoptive transfer of donor bone marrow (BM) cells into lethally irradiated recipient mice (9.5 Gy of total-body irradiation). BM cells, harvested by flushing both femurs and tibias of donor mice, were injected at a dose of 5×106 into each recipient mouse. The following combinations (donor BM ➜lethally irradiated recipient) were generated: WT➜CD18−/−, CD18−/−➜CD18−/−, WT+CD18−/−(1:1)➜CD18−/−, WT➜WT, CD18−/−➜WT and WT+CD18−/−(1:1)➜WT. Recipient mice were analyzed 12 weeks after BM reconstitution, i.e. when they were 18 weeks of age. All animal procedures were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania, in compliance with established federal and state policies.
2.2. Periodontal bone measurements
Periodontal bone heights (i.e., the distances from the cement–enamel junction [CEJ] to the alveolar bone crest [ABC]) were measured in defleshed maxillae under a Nikon SMZ800 microscope using a 40x objective. Briefly, images of the maxillae were captured using a Nikon Digital Sight DS-U3 camera controller and CEJ–ABC distances were measured at 14 predetermined sites [41] or, in the case of split-mouth experiments, at 6 predetermined sites [42] using NIS-Elements software (Nikon Instruments). For calculating change in bone levels (e.g., relative bone loss in CD18−/− mice versus WT controls), the 14-site (or 6-site) total CEJ-ABC distance for each mouse was subtracted from the mean CEJ-ABC distance of control mice. The results are presented in mm; negative values indicate bone loss and positive values indicate bone gain relative to controls. In experiments involving split-mouth design (treatments with drug vs. control in opposite halves of the maxilla), the data shown involve measurements for each hemi-maxilla.
2.3. Cytokine-blocking experiments in mice
To determine cytokine involvement in naturally occurring bone loss in CD18−/− mice, anti-cytokine blocking antibodies, or their controls, were microinjected locally into the palatal gingiva (5 μg per site), three times weekly from the age of 4 to 18 weeks, using a 28.5-gauge MicroFine needle (BD Biosciences). Microinjections were performed on the mesial of the first molar and in the papillae between first and second and third molars on both sides of the maxilla [43]. The following anti-mouse antibodies were used: anti-IL-17A (clone 17F3) and IgG1 isotype control, both from BioXcell; anti-IL-12p35 (clone C18.2; eBioscience) and IgG2a isotype control (R&D Systems); anti-IL-12/IL-23p40 (clone C17.8) and IgG2a isotype control, both from R&D Systems; polyclonal anti-IL-23p19 and non-immune IgG control, both from R&D Systems; anti-mouse IL-6 (clone MP5-20F3), anti-TNF (clone MP6-XT22) and IgG1 isotype control, all three reagents from R&D Systems.
2.4. Blocking of efferocytic receptors in vivo
Antibodies to the following mouse efferocytic receptors (or isotype controls) were locally microinjected (each at 5 μg) to the gingiva of WT mice, which were sacrificed 72h later to dissect gingival tissue for measuring cytokine mRNA expression by quantitative real-time PCR (section 2.6): TIM-1 (affinity-purified polyclonal IgG; R&D Systems); TIM-4 (clone RMT4-53) and IgG2b isotype control (BioXcell); CD11b (clone M1/70) and IgG2b isotype control (both from R&D Systems); CD14 (clone Sa14-2, IgG2a; eBioscience/Thermo-Fisher); CD36 (clone MF3, IgG2a; eBioscience/Thermo-Fisher); Mer (clone 108921) and IgG1 isotype control (both from R&D Systems).
2.5. Split-mouth experiments with nuclear receptor agonists
18-week-old CD18+/− and CD18−/− mice were locally microinjected with a combination of GW3965 and GW0742 (both at 8.1 nmol; Selleckchem) or dimethyl sulfoxide (DMSO) vehicle control. GW3965 is a selective ligand for LXRs although it cannot distinguish between LXRα and LXRβ (EC50s of 190 and 30 nM, respectively) [44]. GW0742 is a selective agonist of PPARβ/δ with EC50 value of 2 nM; although it can exhibit agonistic activity also for PPARγ, the corresponding EC50 value is much higher (2.4 μM) [45]. These agonists were administered using a split-mouth experimental design; i.e., one side was locally injected in the palatal gingiva of the 2nd molar with the drug combination and the contralateral side with vehicle control. After 72h, the mice were euthanized. Dissected gingiva were processed for quantitative real-time PCR (section 2.6) and defleshed maxilla were used to measure bone heights (section 2.2).
2.6. Quantitative real-time PCR
Total RNA was extracted from cultured cells or from gingival tissue using Trizol (ThermoFisher) or NucleoSpin RNA/Protein (Masherey-Nagel) and quantified by spectrometry at 260 and 280 nm. The RNA was reverse-transcribed using the High Capacity RNA-to-cDNA Kit (ThermoFisher) and real-time PCR with cDNA was performed using the Applied Biosystems 7500 Fast Real-Time PCR System, according to the manufacturer’s protocol (ThermoFisher). Data were analyzed using the comparative (ΔΔCt) method. TaqMan probes, sense primers, and antisense primers for detection and quantification of genes investigated in this paper were purchased from ThermoFisher. Gapdh was included as an internal control.
2.7. Immunoblotting
Excised gingival tissue was used to extract total protein using NucleoSpin RNA/Protein. The concentration of protein was quantified using Protein Quantification Assay (Masherey-Nagel). Samples with equal protein content were separated by NuPAGE Bis-Tris Protein Gels (Invitrogen) and transferred to polyvinylidene difluoride membrane (Bio-Rad) by electroblotting. The membranes were incubated in Blotting-Grade Blocker (Bio-Rad) followed by probing with rabbit anti-phospho-Stat3 (Tyr705) or rabbit anti-Stat3 antibody (Cell Signaling Technology) and visualization with HRP-conjugated anti-rabbit IgG (Cell Signaling Technology) and chemiluminescence using Immobilon Crescendo Western HRP substrate (MilliporeSigma). The immunoblots were stripped and reprobed with HRP-conjugated anti-β-actin antibody (Cell Signaling Technology) as control for sample loading. Images were captured using a FluorChem M imaging system (ProteinSimple). The density of bands was analyzed using AlphaView analysis software (ProteinSimple).
2.8. Neutrophil detection by flow cytometry
Periodontal tissue was harvested from CD18+/− and CD18−/− and single-cell suspensions were prepared as previously described [46]. The cells were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Invitrogen) to gate out dead cells and then with anti-CD45 (Clone 30-F11; Biolegend) and anti-Ly6G (Clone 1A8, Biolegend) antibodies to detect neutrophils [47]. Flow cytometry data were acquired on a NovoCyte flow cytometer (ACEA Biosciences) and analyzed with NovoExpress software (ACEA Biosciences).
2.9. Mouse BM-derived macrophages and neutrophils
BM cells were obtained after flushing femurs and tibiae of wild-type and CD18−/− mice with RPMI 1640 containing 10% FBS. Upon lysis of erythrocytes using red blood cell lysis buffer (eBioscience), the cells were plated and cultured in the presence of recombinant mouse granulocyte macrophage colony-stimulating factor (20 ng/ml; eBioscience). Culture medium was replaced every 2 days, and on day 7 differentiated BM-derived macrophages were used in experiments [33]. To isolate neutrophils from the BM, upon erythrocyte lysis as described above, BM cells were resuspended in 45% Percoll. The cell suspension was then overlaid onto a four-layer Percoll gradient (50%, 55%, 62% and 81%) and centrifuged at 1,200 g for 30 min at 4°C. Mature neutrophils were collected at the 81% interface and washed twice in PBS [48]. To induce apoptosis, the neutrophils were cultured overnight in Hank’s balanced salt solution containing 1% FBS. Apoptosis was confirmed by annexin V staining [33].
2.10. Adoptive transfer of neutrophils
Purified neutrophils (5×106 cells) from the BM of WT or CD18−/− mice were i.v. injected into CD18−/− mice. In some experiments, just prior to adoptive neutrophil transfer, CD18−/− mice were microinjected once in the gingiva with 5 μg anti-Mer antibody (clone 108921) or IgG1 isotype control (both from R&D Systems). 72h after neutrophil adoptive transfer, the mice were euthanized and cytokine mRNA expression in the gingival tissue was measured by quantitative real-time PCR (section 2.6).
2.11. In vitro activation of macrophages in the presence of apoptotic neutrophils
Mouse BM-derived macrophages from WT or CD18−/− mice were allowed to adhere to 24-well culture plates and then incubated for 2h with apoptotic neutrophils at a ratio of 3:1 apoptotic neutrophils/macrophages [28]. The wells were then washed with media to remove non-phagocytosed neutrophils, and LPS (100 ng/ml; Invivogen) was added for an additional 6-h incubation. In some experiments, the macrophages were pretreated for 30 min with Mer-Fc or Fc control (both at 10 μg/ml; R&D Systems), or with cytochalasin D (5 μM; Sigma-Aldrich), prior to exposure to apoptotic neutrophils. In other experiments, the macrophages were pretreated for 2h with 5 μM GSK2033 (Axon Medchem), or DMSO as vehicle control, before they were exposed to apoptotic neutrophils. GSK2033 is an antagonist of LXRα and LXRβ (IC50s of 52 nM and 11 nM, respectively); although GSK2033 may display off-target effects in in vivo models, it performs as expected in cell-based assays (as used in the current study) [49]. The inhibitors used and their controls remained for the entire incubation period, i.e., the cell cultures were not washed before adding the apoptotic neutrophils. In certain experiments, in lieu of apoptotic cells, the LXR agonist GW3965 (1 μM; Selleckchem) or DMSO vehicle control was used to pretreat BM-derived macrophages for 18h [36, 50, 51]. Without intermediate washing, the cell cultures were then challenged with LPS as above. In all experiments, cells were assayed for IL-23p19 mRNA by quantitative real-time PCR (section 2.6).
2.12. Statistical analysis
Data were evaluated by one-way analysis of variance (ANOVA) and the Dunnett’ or Tukey’s multiple-comparison test using the Prism Software (version 8.2.1; GraphPad). Where appropriate (comparison of two groups only from split-mouth experiments), paired Student t-tests were performed. P values < 0.05 were considered to be statistically significant.
3. RESULTS
3.1. CD18−/− mice phenocopy human LAD1 periodontitis
Mice deficient in CD18 or CD11a (LFA-1) display blood neutrophilia and defective neutrophil adhesion and thus reproduce important aspects of the human LAD1 phenotype [28, 40]. However, CD18-deficient (CD18−/−) mice, which are deficient in all β2-integrins as human LAD1 patients, exhibit more severe phenotypes than LFA-1-deficient (LFA-1−/−) mice, including susceptibility to colitis and skin ulcerations [28, 39, 40, 47] and may thus be a more appropriate model of human LAD1. In the current study, therefore, we used CD18−/− mice. As these mice were not previously used in periodontitis studies, we first confirmed their expected periodontal phenotype. CD18−/− mice had very few neutrophils in the gingival tissue as compared to heterozygous (CD18+/−) littermate controls (Fig. 1A), which are periodontally healthy and indistinguishable from WT (CD18+/+) mice in terms of their bone levels (data not shown). Moreover, 18-week-old CD18−/− mice displayed significantly more alveolar bone loss than age-matched LFA-1−/− mice (Fig. 1B). Consistently, CD18−/− mice showed even higher mRNA expression of IL-23p19 and IL-12/IL-23p40 and of downstream cytokines of the granulopoietic cytokine cascade (IL-17 and G-CSF) than LFA-1−/− mice did (Fig. 1C). Interestingly, in contrast to IL-23p19 and the IL-12/IL-23-shared p40 subunit, the IL-12p35 subunit– which is unique to IL-12– was not upregulated in CD18−/− or LFA-1−/− mice relative to WT controls (Fig. 1C). In our previous publication, LFA-1−/− mice treated locally in the gingiva with anti-IL-23p19 or anti-IL-17 antibody (three times weekly from the age of 4 to 18 weeks) were protected from bone loss [21]. Using the same protocol, we have now shown that the same treatments inhibited bone loss in CD18−/− mice, while untreated or control-treated CD18−/− mice displayed progressive bone loss relative to WT mice (Fig. 1D). Importantly, moreover, treatments with antibodies against other proinflammatory cytokines (TNF, IL-6, and IL-12) had no significant effect (Fig. 1D). These data underscore the specificity of the IL-23–IL-17 axis in driving the LAD1 periodontal phenotype.
Figure 1. CD18−/− mice develop IL-23/IL-17-driven periodontal bone loss that is reversible by bone marrow transplantation.
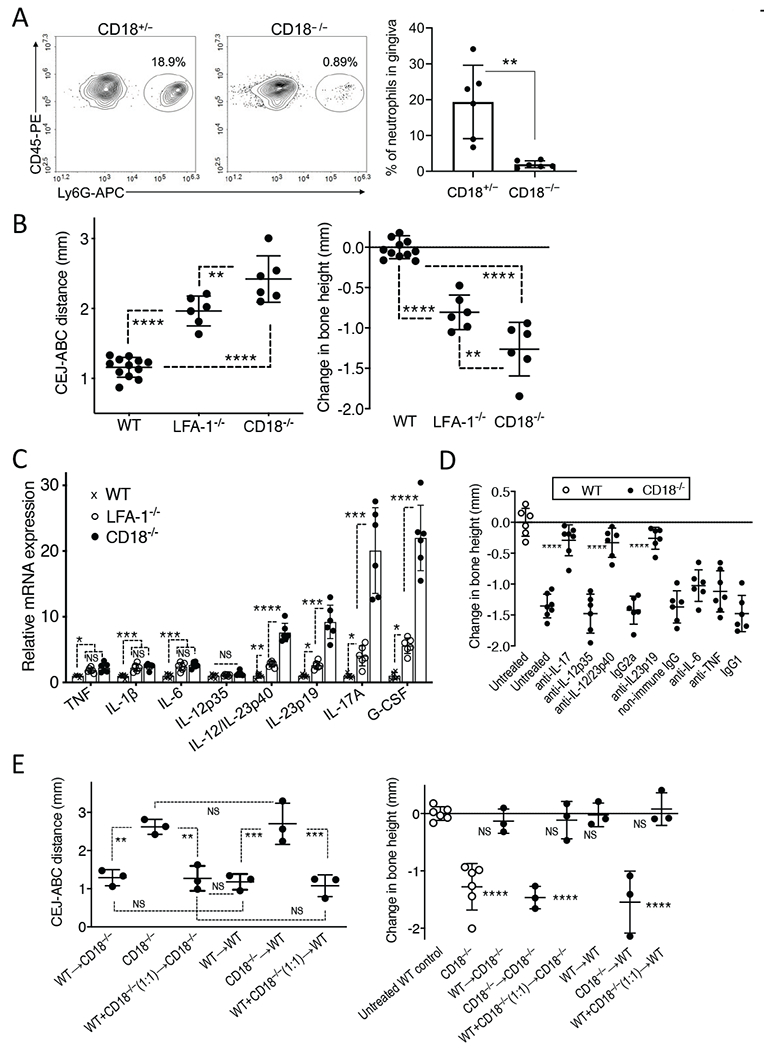
(A) Representative FACS plot of neutrophils in the gingival tissue of CD18+/− and CD18−/− mice (left panel) and bar graph showing percentage of neutrophils gated on live, single CD45+ cells (right panel). (B,C) 18-week-old WT mice and age-matched LFA-1−/− and CD18−/− mice were assessed for the following parameters: (B) Periodontal bone heights (distance between the cementoenamel junction [CEJ] and alveolar bone crest [ABC]) in the indicated mice (left panel) and bone loss (right panel), which was calculated as bone height in 18-week-old WT control mice (“0” baseline) minus bone height in experimental (LFA-1−/− and CD18−/−) mice. (C) The mRNA expression of the indicated cytokines was determined by quantitative real-time PCR. Data were normalized against Gapdh mRNA and expressed as fold induction relative to the transcript levels of 18-week-old WT mice, assigned an average value of 1. (D) CD18−/− mice were either left untreated or were microinjected locally in the gingiva with blocking mAbs to IL-17A, IL-12p35, IL-12/IL-23p40 (or IgG2a isotype control), polyclonal anti-IL-23p19 (or non-immune IgG) antibody, blocking mAbs to IL-6, or TNF (or IgG1 isotype control) three times weekly from the age of 4 to 18 weeks. Bone loss was calculated as described in B with 18-week-old WT control mice serving as the “0” baseline. (E) Lethally irradiated 6-week-old CD18−/− or WT mice underwent transplantation with WT, CD18−/−, or a 1:1 mixture of WT and CD18−/− BM cells (5×106 cells per recipient mouse). Transplanted mice were euthanized 12 weeks after BM reconstitution and bone heights were measured (left panel). Bone loss in the transplanted mice as compared to untreated WT and CD18−/− mice (data from panel B) was calculated as mean bone height in 18-week-old WT control mice (“0” baseline) minus bone height in the other indicated groups of mice. Data are means ± SD and each dot represents the value of an individual mouse (A n = 6-12 mice/group; B,C n = 6 mice/group; D n = 6-7 mice/group; E n = 3-6 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 between indicated groups or as compared with corresponding isotype control (panel D) or untreated WT control (panel E right). Unpaired t test (A), one-way ANOVA with Tukey’s (B,C, E left) or Dunnet’s (D,E right) multiple-comparison tests.
In the absence of bone marrow transplantation, LAD1 patients invariably develop severe periodontitis; however, successful bone marrow transplantation reverses the periodontal disease phenotype in these patients [52]. Similarly, lethally irradiated six-week-old CD18−/− mice transplanted with either WT BM cells or a 1:1 mixture of WT and CD18−/− BM cells exhibited, 12 weeks later, normal bone heights (distance between the cementoenamel junction [CEJ] and alveolar bone crest [ABC]), i.e., similar to those of WT mice receiving WT BM cells (Fig. 1E, left panel). In stark contrast, mice receiving exclusively CD18−/− BM cells, regardless of whether the recipients were WT or CD18−/−, had significantly higher CEJ–ABC distance values (Fig. 1E, left panel). When the CEJ-ABC data from the transplanted mice were transformed to directly show bone loss relative to age-matched (18-week-old) untreated WT mice (data from Fig. 1B), the following conclusion was made: Regardless of their genetic background (WT or CD18−/−), mice receiving exclusively CD18−/− BM cells developed bone loss similar to that of age-matched untreated CD18−/− mice; conversely, WT or CD18−/− mice receiving WT BM cells (even in a 1:1 mixture with CD18−/− cells) were protected from bone loss (Fig. 1E, right panel). Taken together, the findings from figure 1 underscore important similarities between the human LAD1 and mouse CD18−/− phenotypes, rendering the CD18−/− mice an ideal study model of LAD1 periodontitis.
3.2. Reconstitution of CD18−/− mice with transmigration-competent neutrophils downregulates IL-23 and IL-17 expression to normal levels
Neutrophil apoptosis has been documented in gingival biopsies and the majority of apoptotic cells in gingival tissue sections are neutrophils [53, 54]. Thus, the lack or paucity of neutrophils in the LAD1 periodontium is likely associated with diminished efferocytic activity, which is of paramount importance for inflammation resolution and maintenance of tissue homeostasis [32, 55, 56]. We hypothesized that the increased mRNA levels of IL-23 and IL-17 in the gingiva of CD18−/− mice are due to diminished efferocytosis as a result of defective neutrophil recruitment. If the hypothesis is correct, then reconstitution of these mice with transmigration-competent neutrophils should lead to reduction of the gingival mRNA expression of IL-23 and IL-17 to normal levels (i.e., similar to those of WT mice). To test this notion, we i.v. injected transmigration-competent neutrophils (purified from the BM of WT mice) or transmigration-incompetent neutrophils (purified from the BM of CD18−/− mice) into CD18−/− mice. Adoptive transfer of WT (but not CD18−/−) neutrophils resulted, 72h later, in significantly decreased expression of IL-12/IL-23p40, IL-23p19, and IL-17A mRNA in the gingiva of recipient CD18−/− mice, indistinguishable from WT levels of expression (Fig. 2). These findings not only confirm the importance of neutrophils for periodontal tissue homeostasis, but also establish an adoptive transfer model for obtaining further mechanistic insights into the pathogenesis of LAD1 periodontitis.
Figure 2. Adoptive transfer of WT neutrophils to CD18−/− mice restores normal expression of gingival IL-23 and IL-17.
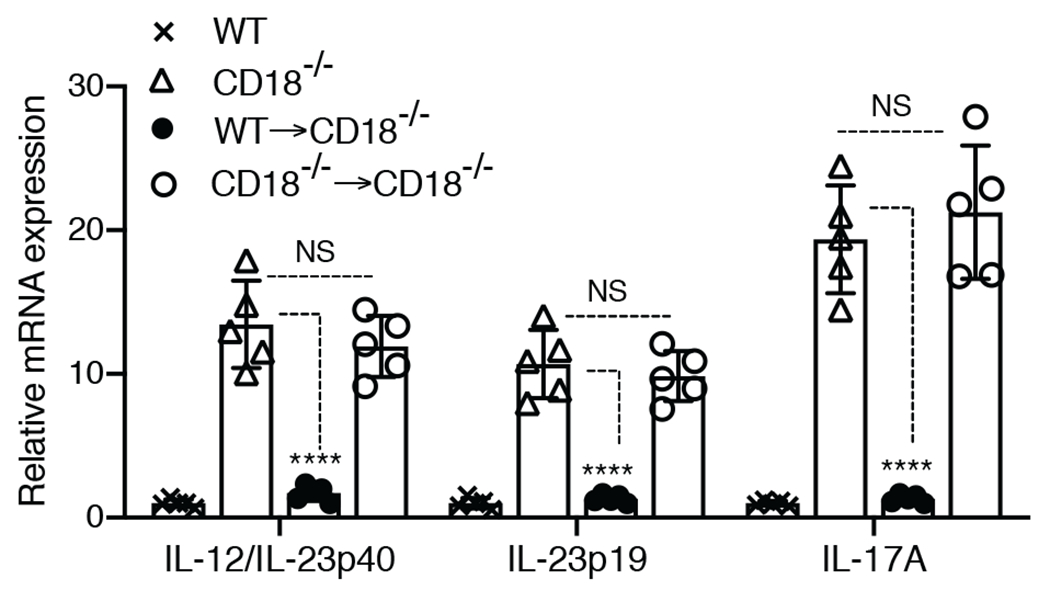
Purified neutrophils (5×106 cells) from WT or CD18−/− mice were i.v. injected into CD18−/− mice. 72h after adoptive transfer the mice were euthanized and expression of the indicated cytokines in the gingival tissue was measured by quantitative real-time PCR. Cytokine mRNA expression was normalized against Gapdh mRNA and expressed as fold induction relative to the transcript levels of WT mice, assigned an average value of 1. Data are means ± SD and each dot represents the value of an individual mouse (n = 5 mice/group). ****P < 0.01 compared to untreated CD18−/− mice. NS, non-significant.
3.3. Blockade of a major efferocytic receptor mimics the LAD1-associated upregulation of IL-23 and IL-17
The efferocytosis of apoptotic cells by tissue phagocytes involves a number of receptors that bind phosphatidylserine on the surface of apoptotic cells, often through the help of opsonins acting as bridging molecules [30, 55]. Apoptotic cell receptors include the T-cell immunoglobulin- and mucin-domain-containing molecule (TIM)-1 and TIM-4, CD14, CD36, complement receptor 3 (CR3; CD11b/CD18), and c-Mer tyrosine kinase receptor (MerTK; briefly known as Mer); however, not all receptors are expressed by a given phagocyte and the associated clearance mechanisms may occur in a tissue-specific manner [30, 31, 55]. We reasoned that if defective efferocytosis contributes to upregulation of IL-23 and hence IL-17, then blockade of receptors involved in efferocytosis in WT mice should increase the mRNA expression of IL-23 (IL-23p19 and IL-12/IL-23p40) and IL-17. To this end, using WT mice, we performed local gingival microinjections with blocking monoclonal antibodies (mAbs) to the aforementioned receptors and 72h later we examined the expression of these cytokines, as compared to untreated controls. Antibody-mediated blockade of most of the receptors investigated had no significant effect on cytokine expression with a notable exception (Fig. 3A). Indeed, significant upregulation of IL-23p19, IL-12/IL-23p40 and IL-17, but not of IL-12p35 (negative control), was observed only in mice treated with anti-Mer (Fig. 3A). These data show that the efferocytic receptor Mer regulates the expression of IL-23 and IL-17 in the gingival tissue.
Figure 3. Mer regulates IL-23 in vivo and in vitro.
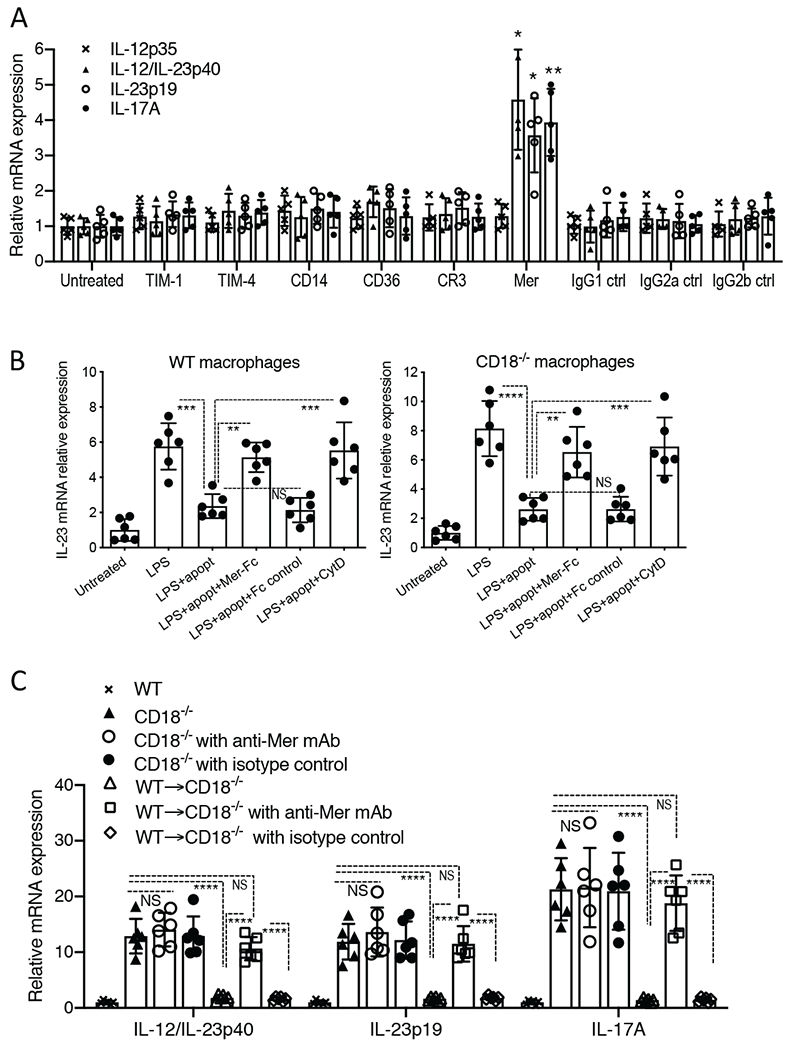
(A) Antibodies (5 μg) to each of the indicated receptors (or isotype controls) were locally microinjected once to the gingiva and the mice were sacrificed 72h later to dissect gingival tissue for cytokine mRNA expression by quantitative real-time PCR. The mRNA expression levels for the indicated cytokines were normalized against Gapdh mRNA and expressed as fold induction relative to the transcript levels of untreated mice, assigned an average value of 1. (B) Mouse BM-derived macrophages from WT (left panel) or CD18−/− (right panel) mice were allowed to adhere to 24-well culture plates and then incubated for 2h with apoptotic neutrophils (‘apopt’) at a ratio of 3:1 apoptotic neutrophils/macrophages. The wells were then washed with media to remove non-phagocytosed neutrophils, and LPS was added at 100 ng/ml for a 6-h incubation. IL-23p19 mRNA expression was measured by quantitative real-time PCR. Results were normalized against Gapdh mRNA and expressed as fold induction relative to the transcript levels of the untreated group, assigned an average value of 1. In certain groups, Mer-Fc or Fc control (both at 10 μg/ml) were added to the macrophages 30 min prior to adding apoptotic neutrophils. In other groups, prior to adding apoptotic neutrophils, macrophages were incubated for 30 min with cytochalasin D (5 μM) to block phagocytosis. (C) Purified neutrophils (5 × 106 cells) from WT mice were i.v. injected or not into groups of CD18−/− mice, which were earlier treated or not with anti-Mer or isotype control (local intragingival injection with 5 μg). 72h after adoptive transfer, the mice were euthanized and expression of the indicated cytokines in the gingival tissue was assayed by quantitative real-time PCR, as outlined in panel A. Data are means ± SD and each dot represents the value of an individual mouse or cell culture (A n = 5 mice/group; B n = 6 cultures/group; C n = 6 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 between indicated groups or as compared with untreated control (A). One-way ANOVA with Dunnet’s (A) or Tukey’s (B,C) multiple comparison tests.
To determine whether Mer can mediate a similar effect in the context of efferocytosis, we employed an in vitro assay and soluble Mer-Fc fusion protein, which was previously shown to inhibit Mer-dependent efferocytosis [57]. As expected, LPS-stimulated expression of IL-23p19 mRNA in WT macrophages was inhibited by apoptotic neutrophils; however, this inhibitory effect was reversed in the presence of Mer-Fc but not Fc control (Fig. 3B, left panel). This finding was reproduced using CD18−/− macrophages in the same assay (Fig. 3B, right panel), suggesting that CD18−/− macrophages can potentially regulate their IL-23 expression if they are exposed to apoptotic neutrophils, a notion that is consistent with our in vivo findings (Fig. 2). Cytochalasin D, which blocks phagocytosis, also prevented the downregulation of IL-23p19 mRNA expression in WT or CD18−/− macrophages (Fig. 3B) further linking the phagocytosis of apoptotic neutrophils to IL-23 regulation.
We next determined whether Mer is involved in the in vivo regulation of gingival IL-23 by adoptively transferred neutrophils. To this end, we performed a neutrophil adoptive transfer experiment similar to the one in figure 2 with the exception that the gingiva were locally injected with anti-Mer antibody or isotype control. Consistent with the earlier experiment (Fig. 2), adoptive transfer of WT neutrophils to CD18−/− mice (WT-CD18−/−) resulted in potent inhibition of IL-23 (IL-23p19 and IL-12/IL-23p40) and IL-17 mRNA expression relative to untreated CD18−/− mice (Fig. 3C). However, the ability of adoptively transferred WT neutrophils to reverse the upregulation of IL-23 and IL-17 was abrogated by local injection of anti-Mer, though not by isotype control (Fig. 3C). Interestingly, in the same experiment, anti-Mer did not significantly affect the expression of IL-23 and IL-17 in CD18−/− mice that did not receive WT neutrophils (Fig. 3C). Taken together, the restoration of IL-23 and IL-17 expression to normal levels following neutrophil adoptive transfer to CD18−/− mice likely involves neutrophil-derived efferocytic signals.
3.4. Liver X receptor (LXR) activation compensates for the lack of apoptotic neutrophils and downregulates IL-23 expression in activated macrophages
The ability of efferocytosis to downregulate the expression of IL-23 by activated tissue macrophages was shown to be mediated by liver X receptor α (LXRα) and LXRβ signaling (hereafter LXR signaling) [36]. Consistent with this report, we showed that the apoptotic neutrophil-induced downregulation of IL-23 in LPS-stimulated macrophages (from WT or CD18−/− mice) was reversed by GSK2033 (Fig. 4A), a selective and potent LXRα/LXRβ antagonist in both humans and mice [58, 59]. Importantly, moreover, GW3965, a selective LXRα/LXRβ agonist in both humans and mice [44], reproduced the regulatory effect of apoptotic neutrophils. Indeed, GW3965 diminished IL-23p19 expression by LPS-stimulated WT or CD18−/− macrophages (Fig. 4B). This finding suggested that GW3965 may be used as a therapeutic agent to downregulate IL-23 and hence IL-17 in the gingiva of CD18−/− mice, restoring the expression of these cytokines to normal levels as seen in WT mice. Interestingly, LPS-stimulated CD18−/− macrophages released higher levels of IL-23 than their WT counterparts (Supplementary Fig. 1), suggesting that macrophage-intrinsic CD18 deficiency might contribute to the increased inflammation associated with LAD1 periodontitis. However, when exposed to apoptotic neutrophils, LPS-stimulated WT and CD18−/− macrophages released similar amounts of IL-23 (Supplementary Fig. 1).
Figure 4. Downregulation of IL-23 in LPS-stimulated macrophages by apoptotic neutrophils involves LXR signaling.
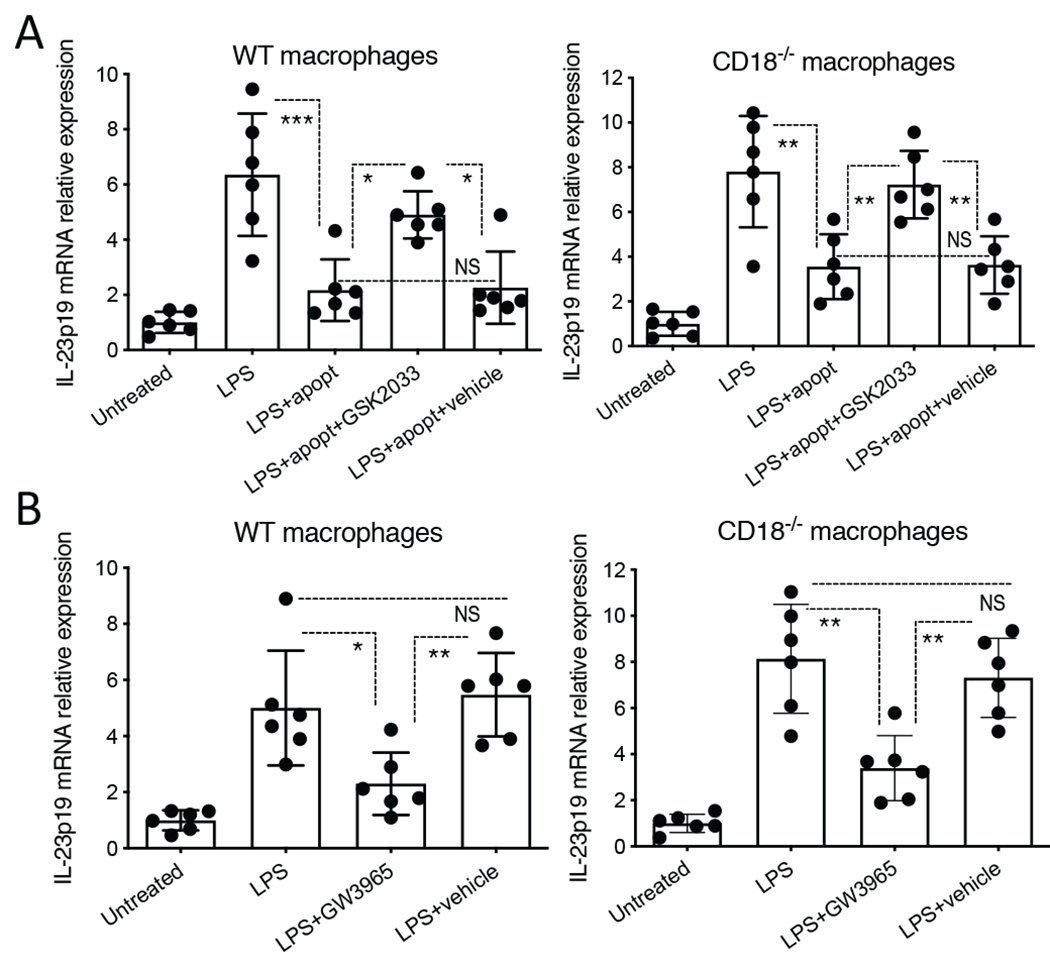
(A) Mouse BM-derived macrophages from WT or CD18−/− mice were incubated for 2h in the absence or presence of apoptotic neutrophils (‘apopt’), with or without GSK2033 (5 μM; LXR antagonist), which was added 1h earlier than the apoptotic cells and remained for the entire incubation period (3h). The wells were then washed with media to remove non-phagocytosed neutrophils, and LPS was added at 100 ng/ml for a 6-h incubation. (B) Mouse BM-derived macrophages from WT or CD18−/− mice were pretreated for 18h with GW3965 (1 μM; LXR agonist) or DMSO (vehicle control) and, without intermediate washing, stimulated with LPS (100 ng/ml) for 6h. In both A and B, IL-23p19 mRNA expression was measured by quantitative real-time PCR. Results were normalized against Gapdh mRNA and expressed as fold induction relative to the transcript levels of the untreated group, assigned an average value of 1. Data are means ± SD (n = 6 cultures/group). *P < 0.05, **P < 0.01 and ***P < 0.001 between indicated groups. NS, not significant. One-way ANOVA with Tukey’s multiple comparison test.
3.5. Combined treatment with LXR and PPAR agonists restores tissue homeostasis in CD18−/− mice
We next set out to investigate whether local treatment with the LXR agonist GW3965 can ameliorate the periodontal status of 18-week-old CD18−/− mice (CD18+/− mice were used for comparative reasons). To this end, we used a split-mouth design; i.e., one side of the maxilla was locally microinjected in the gingiva (specifically on the palatal side of the 2nd molar tooth) with 5 μg GW3965 and the contralateral side was injected with DMSO (vehicle control). The mice were euthanized after 72h for gingival cytokine mRNA analysis. GW3965 failed to reduce the gingival mRNA expression of IL-17A relative to the expression of this pro-inflammatory/pro-osteoclastogenic cytokine in the control-treated sites; (Supplementary Fig. 2A); moreover, GW3965 did not significantly affect the bone levels of either CD18−/− or CD18+/− mice as compared to the control treatment (Supplementary Fig. 2B). In addition to LXRs, other ligand-activated transcription factors/nuclear receptors that link efferocytosis to induction of anti-inflammatory/pro-resolution signals are the peroxisome proliferator-activated receptors (PPARs), which may cross-talk with LXRs [34, 35, 60, 61]. PPARβ/δ-deficient macrophages have decreased ability to phagocytose apoptotic cells and to switch to an anti-inflammatory/pro-resolving phenotype [62]. We thus examined whether activation of LXR and PPAR signaling by combined administration of GW3965 and GW0742 (a selective PPARβ/δ agonist [63]) can inhibit periodontal inflammation in 18-week-old CD18−/− mice as well as age-matched CD18+/− littermates, using the same approach as above. The two agonists were used at equal molar amounts (GW3965, 5 μg or 8.1 nmol; GW0742, 3.8 μg or 8.1 nmol). 72h after the microinjections, the mice were euthanized and the mRNA expression of Th17-related and/or IL-17-dependent cytokines in the gingival tissue was assayed by quantitative real-time PCR. The combination of GW3965 and GW0742 significantly inhibited the mRNA expression of IL-23p19, IL-6, IL-17, and IL-17 downstream cytokines (G-CSF, CXCL1, RANKL) relative to their expression in the control-treated contralateral sides in CD18−/− mice (Fig. 5A). On the other hand, IL-1β and TNF mRNA expression was not significantly inhibited by the GW3965/GW0742 combination (Fig. 5A). None of the cytokines tested were affected by the combined injection of GW3965 and GW0742 in CD18+/− mice (Fig. 5A). Consistent with these data, the levels of activated (phosphorylated) Stat3, an important transcription factor for Th17 development and hence IL-17 secretion [64, 65], in the gingival tissue of CD18−/− mice were decreased in the GW3965/GW0742-treated side as compared to the DMSO control-treated side (Fig. 5B). Consistent with their periodontal health (modest/homeostatic level of proinflammatory cytokine expression), CD18+/− mice exhibited lower levels of phosphorylated Stat3 in their control-treated sides relative to the control-treated sides of CD18−/− mice (Fig. 5B). Intriguingly, the treated sides of CD18−/− mice (but not of CD18+/− mice) exhibited significantly reduced CEJ-ABC distances relative to the control-treated sides (Fig. 5C left panel). When the data were transformed to show change in bone levels relative to the control sides serving as baseline, the GW3965/GW0742-treated sides in CD18−/− (but not CD18+/−) mice displayed positive values suggestive of bone regeneration (Fig. 5C right panel). To determine whether the effect required both GW3965 and GW0742 or whether GW072 would be sufficient, we performed a similar experiment using GW0742 alone; however, microinjection of GW0742 in the absence of GW3965 failed to regulate periodontal inflammation (as assessed by IL-17A mRNA expression) or the bone levels of CD18−/− mice (Supplementary Fig. 2 C and D, respectively). Overall, the concomitant pharmacological activation of LXR and PPAR signaling appears to promote periodontal tissue homeostasis in CD18−/− mice, perhaps by reinstating the absent (or diminished) efferocytosis-associated regulatory signals.
Figure 5. Combined treatment with LXR agonist GW3965 and the PPARβ/δ agonist GW0742 regulates periodontal inflammation and bone levels in CD18−/− mice.
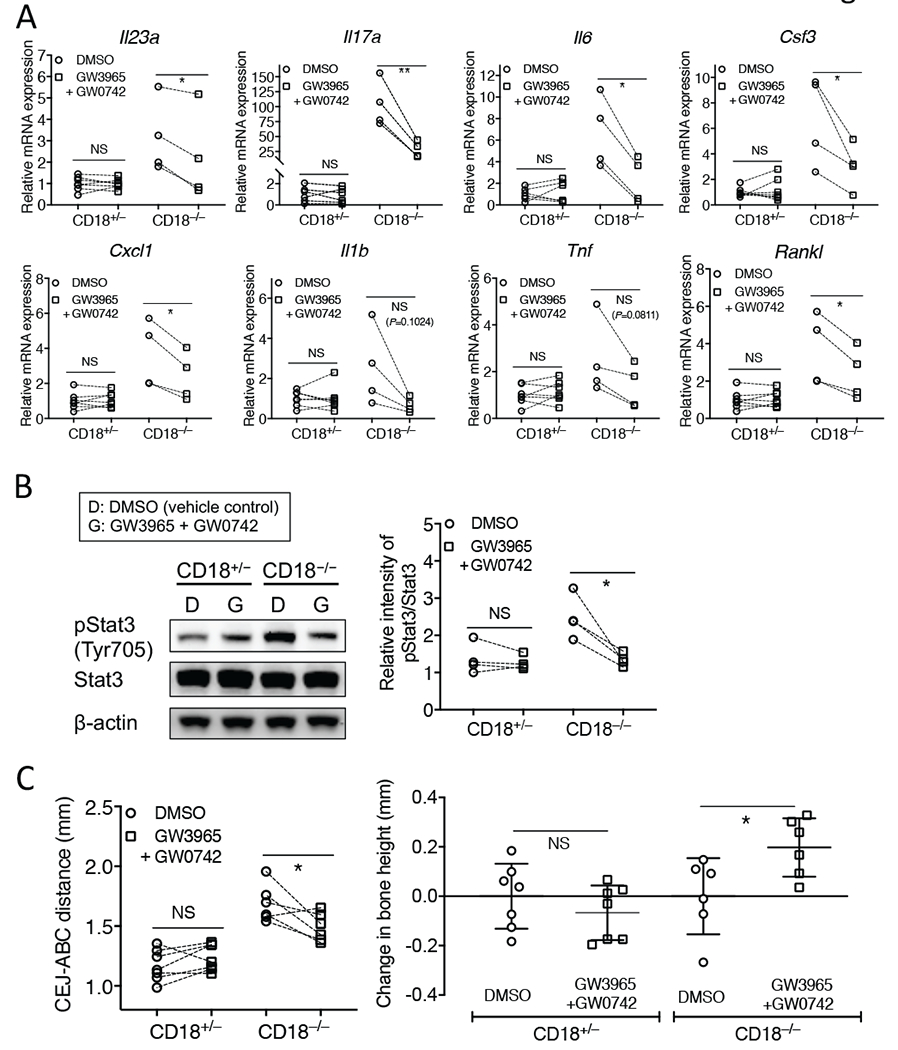
18-week-old CD18+/− and CD18−/− mice were locally microinjected in the palatal gingiva of the 2nd molar with a combination of GW3965 and GW0742 (each at 8.1 nmol; left side) or with DMSO vehicle control (right side). After 72h, the mice were euthanized. (A) Dissected gingiva were processed for quantitative real-time PCR to determine mRNA expression of the indicated cytokines. Results were normalized to Gapdh mRNA and presented as fold change in the transcript levels relative to DMSO injection sites in CD18+/− mice (assigned an average value of 1). (B) Gingiva were processed for immunoblotting to detect phosphorylated Stat3 (pStat3) (left panel). The relative intensity of immunoblotting bands of pStat3 and total Stat3 was quantified by densitometry (right panel). The pStat3/Stat3 ratio values were then normalized to the smallest pStat3/Stat3 ratio value in the CD18+/− DMSO control group, which was set as 1. (C) Defleshed maxillae were used to measure the bone heights (distance between the cementoenamel junction [CEJ] and alveolar bone crest [ABC]) (left panel). The change in bone heights was calculated relative to the corresponding vehicle control group (right panel). In A, C (left panel), and B (right panel), the pair of data for each mouse is connected by a line (n = 4-7 mice/group). In C (right panel), data are means ± SD (n = 6-7 mice/group). *P < 0.05 and **P < 0.01 (Student’s paired t test). NS, non-significant.
4. DISCUSSION
Our earlier study on LAD1 has shown that the presence of neutrophils is required for the homeostatic regulation of the IL-23/IL-17 axis and periodontal tissue health [21]. It should be noted that IL-17 has also been implicated in the pathogenesis of the common or adult-type chronic periodontitis in humans [65]. Interestingly, the extent of periodontal tissue destruction in LAD1 patients is inversely correlated with the remaining CD18 expression on their peripheral blood neutrophils, hence directly linking the patients’ defective neutrophil phenotype to periodontal disease severity [21]. Thus, although neutrophils have been traditionally implicated in inflammatory conditions (including adult-type chronic periodontitis) through their role in bystander injury [18, 66], in LAD1 periodontitis, the inflammatory pathology is associated with the absence (or paucity) of neutrophils [14, 21].
The precise mechanism whereby the absence of neutrophils from the periodontal tissue leads to increased expression of IL-23 and hence IL-17 is uncertain. However, key findings from the present study suggest that the overactivation of the IL-23/IL-17 axis is, at least in part, due to diminished efferocytosis. Consistent with this notion, we showed that antibody-mediated blockade of a major efferocytic receptor (Mer), mimicked LAD1-associated upregulation of gingival IL-23 and IL-17 mRNA expression in WT mice. Moreover, soluble Mer-Fc, an antagonist of Mer-dependent efferocytosis [57], reversed the inhibitory effect of apoptotic neutrophil efferocytosis on IL-23 expression in macrophages. Importantly, adoptive transfer of WT neutrophils to CD18−/− mice could downregulate IL-23 and IL-17 expression to normal levels as long as the CD18−/− mice were not treated locally with blocking anti-Mer antibody. In other words, the ability of transmigration-competent neutrophils to restore normal tissue levels of IL-23 and IL-17 depends on functional Mer, and perhaps intact neutrophil-associated efferocytic signals. In this regard, Mer is not only a major apoptotic cell receptor in macrophages but its phagocytic function is restricted to apoptotic cells [30, 55], therefore it is unlikely that its blockade exerts effects unrelated to efferocytosis. These findings also indicate that, in the presence of neutrophils, the lack of CD18 from macrophages does not play a major role in the regulation of the periodontal IL-23/IL-17 responses, since the hyper-responsive phenotype of CD18−/− mice was reversed by transmigration-competent neutrophils. This notion is also supported by our in vitro findings that CD18−/− and WT macrophages exposed to apoptotic neutrophils released similar levels of IL-23 in response to LPS challenge. On the other hand, in the absence of neutrophils (as is the case of LAD1), CD18−/− macrophages released more IL-23 in response to LPS stimulation than their WT counterparts did. Together, these findings suggest that, besides the lack of efferocytosis-associated pro-resolution signals, macrophage-intrinsic CD18 deficiency per se might contribute to the increased inflammation associated with LAD1 periodontitis. The mechanism accounting for the increased pro-inflammatory potential of CD18−/− macrophages is uncertain, although it may be related to the fact that these macrophages do not express the CD11b/CD18 integrin, which can restrain proinflammatory Toll-like receptor signaling in macrophages [67].
In vitro stimulation of LXR with the synthetic agonist GW3965 compensated for the lack of apoptotic neutrophils and downregulated IL-23 expression in activated macrophages to an extend comparable to that of apoptotic neutrophils. However, GW3965 failed to modulate IL-23 expression when locally microinjected into the gingiva of CD18−/− mice, suggesting additional requirements for the in vivo regulation of this cytokine. Importantly, nevertheless, the combined activation of LXR and PPARβ/δ, by GW3965 and GW0742, respectively, downregulated the expression of IL-23, IL-17 and downstream cytokines and favorably affected the bone levels of CD18−/− mice. The mechanism(s) for the observed cooperation between GW3965 and GW0742 is uncertain. However, cross-talk between LXRs and PPARs and formation of LXR/PPAR heterodimers have been described in the literature [60, 61]. Systemic administration of agonists of distinct PPARs (WY14643, PPARα; GW0742, PPARβ/δ; or rosiglitazone, PPARγ) resulted in protection of rats from ligature-induced periodontal inflammation and tissue damage, although efferocytosis-related mechanisms have not been addressed [68–70]. Consistent with a homeostatic role for LXRs, specific-pathogen-free LXRαβ-deficient mice have increased naturally occurring bone as compared to WT controls [71].
Our finding that the GW3965/GW0742-treated sites of CD18−/− mice had reduced CEJ-ABC distances (as compared to the control-treated sites) 3 days after the treatment was quite surprising and might be explained in two different ways. Given that IL-17 induces RANKL expression and stimulates osteoclastogenesis [72], the inhibition of IL-17 by the GW3965/GW0742 combined treatment might have slowed down on-going bone resorptive activity in the treated sites. In this regard, RANKL expression was indeed inhibited in the treated sites relative to the untreated sites. However, naturally occurring bone loss CD18−/− mice is slow (relative to induced models of periodontitis, such as the ligature-induced periodontitis model [42]) and, therefore, it is unlikely that osteoclastogenesis inhibition could have resulted in clinically noticeable effects in bone levels after only 3 days. Another possibility is that the treatment stimulated new bone formation which, at least in mice, can be observed within a few days even in the absence of drug treatment (i.e., as occurs after ligature removal that promotes inflammation resolution). In the case of our treatment, if there was indeed bone regeneration, this might not be only the result of inflammation resolution but also a contribution by a potential specific effect of the nuclear receptor agonists used. In this regard, it was shown that activation of PPARβ/δ signaling (by the specific agonist GW501516) enhances Wnt- and β-catenin-dependent gene expression in osteoblasts and mesenchymal stem cells, including the master transcription factor of osteogenesis, Runt-related transcription factor 2 [73]. In fact, activation of PPARβ/δ signaling not only promoted osteoblast differentiation but also inhibited osteoclastogenesis in a manner dependent on osteoblast-derived osteoprotegerin, a natural inhibitor of RANKL [73]. Whereas the activation of LXR and PPAR signaling by the synthetic agonists used mimics efferocytic signals, it is conceivable that LXR and PPAR signaling pathways may promote periodontal tissue homeostasis also by additional mechanisms.
Mice that are deficient in the protein designated developmental endothelial locus-1 (DEL-1) display the opposite phenotype from that of CD18−/− mice, in terms of neutrophil infiltration in the periodontium. This is because endothelial cell-secreted DEL-1 binds LFA-1 on neutrophils and inhibits their adhesion to the endothelium, thus restraining neutrophil recruitment to the periodontium; consequently, DEL-1 deficiency in mice results in pronounced neutrophil infiltration [12, 74]. Strikingly, in common with CD18−/− mice, Del-1−/− mice also exhibit an overactive IL-23–IL-17 axis (IL-12 is similarly not upregulated) and IL-17-driven bone loss. This paradox may be reconciled as follows. When secreted by macrophages, DEL-1 promotes apoptotic neutrophil efferocytosis and LXR-dependent reprogramming of the efferocytic macrophage, which in turn contributes to the resolution of inflammation [33, 56]. Therefore, both Del-1−/− and CD18−/− mice cannot generate efferocytosis-associated anti-inflammatory signals, the former because they lack DEL-1 mediating efferocytosis and the latter because they lack apoptotic neutrophils. DEL-1 binds the ‘eat-me’ signal phosphatidylserine on apoptotic neutrophils and bridges them to macrophages by binding the αvβ3 integrin, rather than Mer (which uses growth arrest specific factor-6 as bridging molecule) [33]. However, efferocytic integrins (αvβ3/β5) cross-talk with other efferocytic receptor pathways including Mer, which synergizes with integrins in apoptotic cell phagocytosis and downstream signaling [75, 76].
In our present study, antibodies to IL-23p19 or to the IL-12/IL-23-shared p40 subunit inhibited bone loss in CD18−/− mice, whereas antibody to the IL-12p35 subunit, which is unique to IL-12, did not. Based on these findings, we may conclude that the protective effect in human LAD1 periodontitis of ustekinumab (which blocks the common p40 subunit of IL-23 and IL-12) [27] can be more likely attributed to inhibition of IL-23 rather than of IL-12.
Nuclear metabolic receptors are ideal targets for therapeutic approaches, in great part attributed to their inherent features, i.e., their capacity to selectively bind “drug-like” small molecules [77]. Whereas the cost of antibody–based biologic drugs is high and constantly increasing, small-molecule therapeutics, such as agonists of nuclear receptors, entail considerably more affordable costs [78–80]. In addition, small-molecule compounds have increased stability in biological fluids and low or no antigenicity [78, 79]. Our study strongly suggests that small-molecule therapeutics that activate efferocytosis-associated signaling, such as GW3965 and GW0742, have the potential to provide affordable and effective treatment options in LAD1 periodontitis by inhibiting the IL-23–IL-17 axis.
Supplementary Material
ACKNOWLEDGEMENTS
Supported by grants from the NIH (DE024153, DE024716 to GH and DE026152 to GH and TC), the Intramural Research Program of the NIDCR (NM) and the German Research Foundation (SFB1181 and SFB-TR127) to TC.
ABBREVIATIONS
- ABC
alveolar bone crest
- BM
bone marrow
- CEJ
cement–enamel junction
- DEL-1
developmental endothelial locus-1
- DMSO
dimethyl sulfoxide
- G-CSF
granulocyte colony-stimulating factor
- IL
interleukin
- LAD1
leukocyte adhesion deficiency-Type 1
- LXR
liver X receptor
- mAb
monoclonal antibody
- Mer
c-Mer tyrosine kinase
- PPAR
peroxisome proliferator-activated receptor
- TIM
T-cell immunoglobulin- and mucin-domain-containing molecule
- WT
wild-type
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no commercial or financial conflict of interest.
REFERENCES
- 1.Phillipson M Kubes P (2011) The neutrophil in vascular inflammation. Nat Med 17, 1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD (2018) Neutrophils: New insights and open questions. Sci Immunol 3, eaat4579. [DOI] [PubMed] [Google Scholar]
- 3.Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD (2015) Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol 98, 539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uriarte SM, Edmisson JS, Jimenez-Flores E (2016) Human neutrophils and oral microbiota: a constant tug-of-war between a harmonious and a discordant coexistence. Immunol Rev 273, 282–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Vietinghoff S Ley K (2008) Homeostatic regulation of blood neutrophil counts. J Immunol 181, 5183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR (2005) Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood 105, 584–91. [DOI] [PubMed] [Google Scholar]
- 7.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR (1994) Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84, 1737–46. [PubMed] [Google Scholar]
- 8.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194, 519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramanian P, Mitroulis I, Hajishengallis G, Chavakis T (2016) Regulation of tissue infiltration by neutrophils: role of integrin alpha3beta1 and other factors. Curr Opin Hematol 23, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitroulis I, Alexaki VI, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T (2015) Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther 147, 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajishengallis G Chavakis T (2013) Endogenous modulators of inflammatory cell recruitment. Trends Immunol 34, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, Ciero PA, Krauss JL, Li F, Rauner M, Hofbauer LC, Choi EY, Chung KJ, Hashim A, Curtis MA, Chavakis T, Hajishengallis G (2012) The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol 13, 465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niederman R, Westernoff T, Lee C, Mark LL, Kawashima N, Ullman-Culler M, Dewhirst FE, Paster BJ, Wagner DD, Mayadas T, Hynes RO, Stashenko P (2001) Infection-mediated early-onset periodontal disease in P/E-selectin-deficient mice. J Clin Periodontol 28, 569–75. [DOI] [PubMed] [Google Scholar]
- 14.Waldrop TC, Anderson DC, Hallmon WW, Schmalstieg FC, Jacobs RL (1987) Periodontal manifestations of the heritable Mac-1, LFA-1, deficiency syndrome. Clinical, histopathologic and molecular characteristics. J Periodontol 58, 400–16. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt S, Moser M, Sperandio M (2012) The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol 55, 49–58. [DOI] [PubMed] [Google Scholar]
- 16.Moutsopoulos NM, Lionakis MS, Hajishengallis G (2015) Inborn errors in immunity: unique natural models to dissect oral immunity. J Dent Res 94, 753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva LM, Brenchley L, Moutsopoulos NM (2019) Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol Rev 287, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hajishengallis E Hajishengallis G (2014) Neutrophil homeostasis and periodontal health in children and adults. J Dent Res 93, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinane DF, Stathopoulou PG, Papapanou PN (2017) Periodontal diseases. Nat Rev Dis Primers 3, 17038. [DOI] [PubMed] [Google Scholar]
- 20.Hajishengallis G (2015) Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, Uzel G, Chen W, Chavakis T, Holland SM, Hajishengallis G (2014) Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med 6, 229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanna S Etzioni A (2012) Leukocyte adhesion deficiencies. Ann N Y Acad Sci 1250, 50–5. [DOI] [PubMed] [Google Scholar]
- 23.Anderson DC Springer TA (1987) Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med 38, 175–94. [DOI] [PubMed] [Google Scholar]
- 24.Schieppati A, Henter JI, Daina E, Aperia A (2008) Why rare diseases are an important medical and social issue. Lancet 371, 2039–41. [DOI] [PubMed] [Google Scholar]
- 25.Peltonen L, Perola M, Naukkarinen J, Palotie A (2006) Lessons from studying monogenic disease for common disease. Hum Mol Genet 15 Spec No 1, R67–74. [DOI] [PubMed] [Google Scholar]
- 26.Deas DE, Mackey SA, McDonnell HT (2003) Systemic disease and periodontitis: manifestations of neutrophil dysfunction. Periodontol 2000 32, 82–104. [DOI] [PubMed] [Google Scholar]
- 27.Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, Uzel G, Axelrod KC, Lisco A, Notarangelo LD, Hajishengallis G, Notarangelo LD, Holland SM (2017) Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1. N Engl J Med 376, 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K (2005) Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–94. [DOI] [PubMed] [Google Scholar]
- 29.Poon IK, Lucas CD, Rossi AG, Ravichandran KS (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14, 166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kourtzelis I, Mitroulis I, von Renesse J, Hajishengallis G, Chavakis T (2017) From leukocyte recruitment to resolution of inflammation: the cardinal role of integrins. J Leukoc Biol 102, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabas I (2010) Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortega-Gomez A, Perretti M, Soehnlein O (2013) Resolution of inflammation: an integrated view. EMBO Mol Med 5, 661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, Grzybek M, von Renesse J, Czogalla A, Troullinaki M, Ferreira A, Doreth C, Ruppova K, Chen LS, Hosur K, Lim JH, Chung KJ, Grossklaus S, Tausche AK, Joosten LAB, Moutsopoulos NM, Wielockx B, Castrillo A, Korostoff JM, Coskun U, Hajishengallis G, Chavakis T (2019) DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol 20, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon YS, Kim SY, Kim MJ, Lim JH, Cho MS, Kang JL (2015) PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol 8, 1031–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alonso-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A (2009) Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong C, Kidani Y, N AG, Phung T, Ito A, Rong X, Ericson K, Mikkola H, Beaven SW, Miller LS, Shao WH, Cohen PL, Castrillo A, Tontonoz P, Bensinger SJ (2012) Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest 122, 337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.A-González N Castrillo A (2011) Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1812, 982–994. [DOI] [PubMed] [Google Scholar]
- 38.Cua DJ Tato CM (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10, 479–89. [DOI] [PubMed] [Google Scholar]
- 39.Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL (1998) Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med 188, 119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 163, 5029–38. [PubMed] [Google Scholar]
- 41.Baker PJ, Dixon M, Roopenian DC (2000) Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect. Immun 68, 5864–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe T Hajishengallis G (2013) Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods 394, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, Hajishengallis G (2012) Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 189, 5442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA, Willson TM (2002) Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 45, 1963–6. [DOI] [PubMed] [Google Scholar]
- 45.Chehaibi K, le Maire L, Bradoni S, Escola JC, Blanco-Vaca F, Slimane MN (2017) Effect of PPAR-β/δ agonist GW0742 treatment in the acute phase response and blood–brain barrier permeability following brain injury. Translational Research 182, 27–48. [DOI] [PubMed] [Google Scholar]
- 46.Mizraji G, Segev H, Wilensky A, Hovav AH (2013) Isolation, processing and analysis of murine gingival cells. J Vis Exp, e50388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Lim JH, Kajikawa T, Li X, Vallance BA, Moutsopoulos NM, Chavakis T, Hajishengallis G (2019) Macrophage beta2-Integrins Regulate IL-22 by ILC3s and Protect from Lethal Citrobacter rodentium-Induced Colitis. Cell Rep 26, 1614–1626 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T (2007) A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J 26, 1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffett K Burris TP (2016) Promiscuous activity of the LXR antagonist GSK2033 in a mouse model of fatty liver disease. Biochemical and Biophysical Research Communications 479, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pascual-García M, Rué L, León T, Julve J, Carbó JM, Matalonga J, Auer H, Celada A, Escolà-Gil JC, Steffensen KR, Pérez-Navarro E, Valledor AF (2013) Reciprocal negative cross-talk between liver X receptors (LXRs) and STAT1: effects on IFN-γ-induced inflammatory responses and LXR-dependent gene expression. Journal of immunology (Baltimore, Md. : 1950) 190, 6520–6532. [DOI] [PubMed] [Google Scholar]
- 51.Fan A, Wang Q, Yuan Y, Cheng J, Chen L, Guo X, Li Q, Chen B, Huang X, Huang Q (2016) Liver X receptor-α and miR-130a-3p regulate expression of sphingosine 1-phosphate receptor 2 in human umbilical vein endothelial cells. Am J Physiol Cell Physiol 310, C216–26. [DOI] [PubMed] [Google Scholar]
- 52.Thomas C, Le Deist F, Cavazzana-Calvo M, Benkerrou M, Haddad E, Blanche S, Hartmann W, Friedrich W, Fischer A (1995) Results of allogeneic bone marrow transplantation in patients with leukocyte adhesion deficiency. Blood 86, 1629–35. [PubMed] [Google Scholar]
- 53.Gamonal J, Bascones A, Acevedo A, Blanco E, Silva A (2001) Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J Periodontol 72, 517–25. [DOI] [PubMed] [Google Scholar]
- 54.Gamonal J, Sanz M, O’Connor A, Acevedo A, Suarez I, Sanz A, Martinez B, Silva A (2003) Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 30, 616–23. [DOI] [PubMed] [Google Scholar]
- 55.Ravichandran KS (2011) Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 35, 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hajishengallis G Chavakis T (2019) DEL-1-Regulated Immune Plasticity and Inflammatory Disorders. Trends Mol Med 25, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK (2007) A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 109, 1026–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuercher WJ, Buckholz RG, Campobasso N, Collins JL, Galardi CM, Gampe RT, Hyatt SM, Merrihew SL, Moore JT, Oplinger JA, Reid PR, Spearing PK, Stanley TB, Stewart EL, Willson TM (2010) Discovery of tertiary sulfonamides as potent liver X receptor antagonists. J Med Chem 53, 3412–6. [DOI] [PubMed] [Google Scholar]
- 59.Solt LA, Kamenecka TM, Burris TP (2012) LXR-mediated inhibition of CD4+ T helper cells. PLoS One 7, e46615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ide T, Shimano H, Yoshikawa T, Yahagi N, Amemiya-Kudo M, Matsuzaka T, Nakakuki M, Yatoh S, Iizuka Y, Tomita S, Ohashi K, Takahashi A, Sone H, Gotoda T, Osuga J. i., Ishibashi S, Yamada N (2003) Cross-Talk between Peroxisome Proliferator-Activated Receptor (PPAR) α and Liver X Receptor (LXR) in Nutritional Regulation of Fatty Acid Metabolism. II. LXRs Suppress Lipid Degradation Gene Promoters through Inhibition of PPAR Signaling. Molecular Endocrinology 17, 1255–1267. [DOI] [PubMed] [Google Scholar]
- 61.Gao M, Bu L, Ma Y, Liu D (2013) Concurrent Activation of Liver X Receptor and Peroxisome Proliferator-Activated Receptor Alpha Exacerbates Hepatic Steatosis in High Fat Diet-Induced Obese Mice. PLOS ONE 8, e65641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chawla A (2010) Control of macrophage activation and function by PPARs. Circ Res 106, 1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galatou E, Kelly T, Lazou A (2014) The PPARβ/δ agonist GW0742 modulates signaling pathways associated with cardiac myocyte growth via a non-genomic redox mechanism. Molecular and Cellular Biochemistry 395, 145–154. [DOI] [PubMed] [Google Scholar]
- 64.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, Morell RJ, Freeman AF, Lazarevic V, Trinchieri G, Diaz PI, Holland SM, Belkaid Y, Hajishengallis G, Moutsopoulos NM (2018) A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med 10, eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scapini P Cassatella MA (2014) Social networking of human neutrophils within the immune system. Blood 124, 710–719. [DOI] [PubMed] [Google Scholar]
- 67.Han C, Jin J, Xu S, Liu H, Li N, Cao X (2010) Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 11, 734–42. [DOI] [PubMed] [Google Scholar]
- 68.Di Paola R, Briguglio F, Paterniti I, Mazzon E, Oteri G, Militi D, Cordasco G, Cuzzocrea S (2011) Emerging role of PPAR-beta/delta in inflammatory process associated to experimental periodontitis. Mediators Inflamm 2011, 787159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Briguglio E, Di Paola R, Paterniti I, Mazzon E, Oteri G, Cordasco G, Cuzzocrea S (2010) WY-14643, a Potent Peroxisome Proliferator Activator Receptor-alpha PPAR-alpha Agonist Ameliorates the Inflammatory Process Associated to Experimental Periodontitis. PPAR Res 2010, 193019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Paola R, Mazzon E, Maiere D, Zito D, Britti D, De Majo M, Genovese T, Cuzzocrea S (2006) Rosiglitazone reduces the evolution of experimental periodontitis in the rat. J Dent Res 85, 156–61. [DOI] [PubMed] [Google Scholar]
- 71.Huang N, Shaik-Dasthagirisaheb YB, LaValley MP, Gibson FC 3rd (2015) Liver X receptors contribute to periodontal pathogen-elicited inflammation and oral bone loss. Mol Oral Microbiol 30, 438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miossec P Kolls JK (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11, 763–76. [DOI] [PubMed] [Google Scholar]
- 73.Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, Zaiss MM, Stock M, Donhauser L, Bohm C, Kleyer A, Hess A, Engelke K, David JP, Djouad F, Tuckermann JP, Desvergne B, Schett G, Kronke G (2013) PPARbeta/delta governs Wnt signaling and bone turnover. Nat Med 19, 608–13. [DOI] [PubMed] [Google Scholar]
- 74.Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, Kundu RK, Orlandi A, Zheng YY, Prieto DA, Ballantyne CM, Constant SL, Aird WC, Papayannopoulou T, Gahmberg CG, Udey MC, Vajkoczy P, Quertermous T, Dimmeler S, Weber C, Chavakis T (2008) Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 322, 1101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flannagan RS, Canton J, Furuya W, Glogauer M, Grinstein S (2014) The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell 25, 1511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Y, Singh S, Georgescu MM, Birge RB (2005) A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci 118, 539–53. [DOI] [PubMed] [Google Scholar]
- 77.Moore JT, Collins JL, Pearce KH (2006) The nuclear receptor superfamily and drug discovery. ChemMedChem 1, 504–23. [DOI] [PubMed] [Google Scholar]
- 78.Morand EF, Leech M, Bernhagen J (2006) MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov 5, 399–410. [DOI] [PubMed] [Google Scholar]
- 79.Morizane A, Doi D, Kikuchi T, Nishimura K, Takahashi J (2011) Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J Neurosci Res 89, 117–126. [DOI] [PubMed] [Google Scholar]
- 80.Cheng J Feldman SR (2014) The cost of biologics for psoriasis is increasing. Drugs Context 3, 212266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.