

Abstract
Myeloproliferative neoplasms (MPNs) are blood cancers characterized by excessive production of mature myeloid cells, which result from the acquisition of somatic driver mutations in hematopoietic stem cells (HSCs). Epidemiologic studies indicate a substantial disease heritability that is among the highest known for cancers1. However, only a limited set of genetic risk loci have been identified, and the underlying biological mechanisms leading to MPN acquisition remain unexplained. Here, we conducted a large-scale genome-wide association study (3,797 cases and 1,152,977 controls) to identify 17 MPN risk loci (p < 5.0 × 10−8), seven of which have not been previously reported. We find a shared genetic architecture between MPN risk and several hematopoietic traits spanning distinct lineages, an enrichment for risk variants mapping to accessible chromatin in HSCs, and associations of increased MPN risk with longer leukocyte telomere length and other clonal hematopoietic states, collectively implicating HSC function and self-renewal. Gene mapping identifies modulators of HSC biology and targeted variant-to-function assays suggest likely roles for CHEK2 and GFI1B in altering HSC function to confer disease risk. Overall, we demonstrate the power of human genetic studies to illuminate a previously unappreciated mechanism for inherited MPN risk through modulation of HSC function.
Myeloproliferative neoplasms (MPNs) comprise a group of blood cancers characterized by excessive production of mature myeloid cells. Although MPNs arise from somatic driver mutations, recent work has uncovered a substantial heritable component to the disease. There is a 5–7-fold increased risk of MPN acquisition in first-degree relatives of individuals with the disease1,2, amounting to a familial risk greater than that observed in breast (relative risk (RR) = 3.5)3, prostate (RR = 2.46)4, and colorectal cancer (RR=2.25)5. Genetic studies to date have identified a limited set of loci associated with MPN predisposition6,7. However, many key aspects of inherited MPN risk remain uncharacterized, including the causal genes, cell types, and underlying biological mechanisms involved.
Genetic discovery of inherited MPN risk
To characterize the germline genetic architecture that confers risk for MPNs, we conducted a genome-wide association study (GWAS) meta-analysis using three population-based cohorts (UK Biobank (UKBB), 23andMe, and FinnGen), comprising a combined sample size of 2,949 MPN cases and 835,554 controls (Supplementary Table 1, Extended Data Fig. 1). We tested 7,343,617 well-represented variants passing central and study-specific quality control measures. Linkage disequilibrium score regression (LDSC)8 showed negligible inflation in test statistics due to population structure (λ = 1.035, intercept = 1.01).
Approximate conditional analysis of GWAS signals9 revealed 15 linkage disequilibrium (LD)-independent loci at genome-wide significance (p < 5 × 10−8) and an additional 10 loci with suggestive associations (p < 1 × 10−6) (Fig. 1a, Supplementary Table 2). To replicate these associations in an independent cohort, we assessed genotypes at 24 of 25 lead variants (p < 1 × 10−6) among 848 carriers of the somatic JAK2 V617F mutation, the most common driver mutation for MPNs, and up to 317,423 controls within the Million Veteran Program. While JAK2 V617F is not an exact phenotypic proxy for MPN, previous studies have shown that JAK2 V617F strongly replicates MPN risk associations7. Because our study focuses on inherited risk prior to MPN driver mutation acquisition, we reasoned that JAK2 V617F could model a pre-MPN state. A perfect replication for direction of effects was observed (24 of 24 variants, binomial p = 5.96 × 10−8), and 17 variants were significant at the 5% level (binomial p < 2.2 × 10−16) (Extended Data Fig. 2, Supplementary Table 3). We then combined data from MVP with the discovery GWAS to reach a total of 3,797 cases and 1,152,977 controls. This combined analysis revealed 17 independent risk associations exceeding genome-wide significance, seven of which have not been previously reported (Extended Data Table 1, Supplementary Note). All 10 previously reported loci for MPN risk6,7 remained genome-wide significant in our analysis. We estimate that the 17 loci explain 18.4% of the ~5-fold familial relative risk for MPN acquisition1.
Figure 1. Genetic architecture of inherited MPN risk.
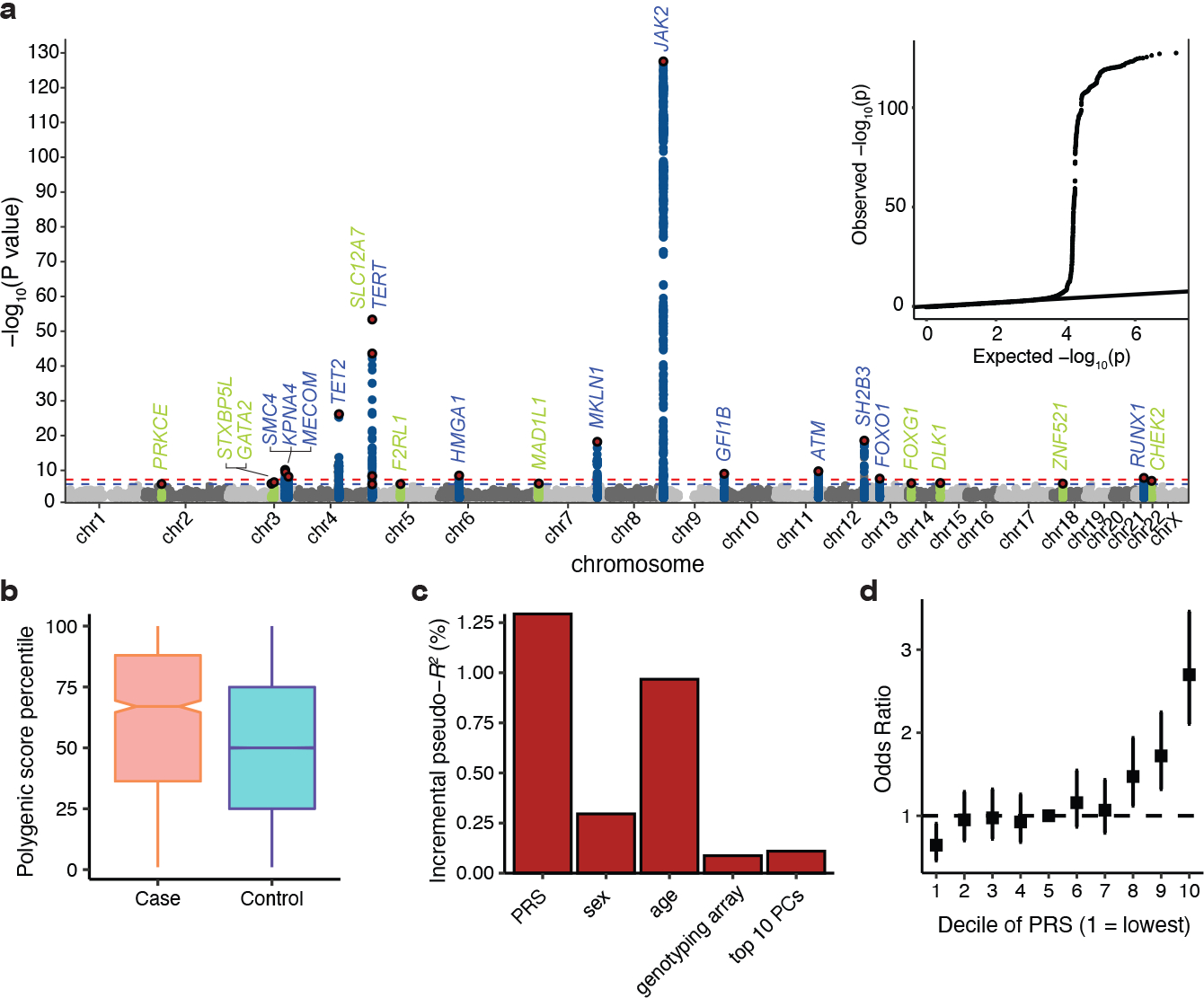
a, Manhattan plot and quantile-quantile (QQ) plot illustrating results of the genome-wide association study (GWAS) meta-analysis for MPNs (n = 2,949 cases and 835,554 controls). X axis is chromosomal position, and y axis is the −log10(P) value of association (two-tailed, logistic regression). Association signals reaching genome-wide significance (P < 5 × 10−8) and suggestive significance (P < 1 × 10−6) are shown in blue and green, respectively. Red points represent conditionally independent lead variants within each locus. Labels correspond to target gene if present (Fig. 3a), or otherwise the nearest gene at each association locus (+/− 500 kb). The QQ plot illustrates the deviation of association test statistics (points) from the distribution expected under the null hypothesis (line). b, Polygenic risk score (PRS) percentile among MPN cases (n = 1,086) versus controls (n = 407,155) in the UK Biobank test set. Box plots show the median as the line in the notch, with the top and bottom of the box indicating the interquartile range. Whiskers extend to the maximum or minimum value. Notches indicate the 95% confidence interval of the medians. c, Additional variance in MPN risk (n = 1,086 cases and 407,155 controls) explained by PRS compared to age, sex, genotyping array, and top 10 principal components of genetic relatedness. d, Odds ratio for MPN acquisition (n = 1,086 cases and 407,155 controls) stratified by deciles of the PRS, with the 5th decile as the reference. Data represent odds ratios and 95% confidence intervals.
To further characterize the polygenic architecture of MPN risk, we generated a 104-variant polygenic risk score (PRS) using a pruning and thresholding method (R2 < 0.2, P < 1×10−5) on summary statistics from 23andMe and FinnGen (Supplementary Table 4), and then tested this PRS in the out-of-sample UKBB cohort. Given the rarity of MPNs and the relatively common frequency of these risk variants, a PRS for MPNs is unlikely to have utility for population-based screening. Nonetheless, PRS analysis is useful for evaluating the distribution of genetic risk and comparing the predictive capacity of germline variants with other risk factors. Individuals with MPN in the UKBB had a median PRS percentile of 67 compared to 50 for those without MPN (Fig. 1b, Extended Data Fig. 3), and the PRS explained more variance than any other covariate in our model (Fig. 1c). Individuals in the 20th to 70th PRS percentiles had similar disease risk, but significant stratification occurred at the top and bottom quintiles of the distribution (Fig. 1d). For example, the 40,824 individuals above the 90th PRS percentile had 4.20-fold (95% CI: [3.14, 5.63]) higher odds of MPN compared to those in the lowest decile, and 2.70-fold (95% CI: [2.11, 3.46]) higher odds compared to those with average genetic risk (5th PRS decile). Finally, given that the JAK2 46/1 haplotype is one of the largest contributors to germline MPN risk10, we sought to determine the extent to which a broader set of risk loci, as represented by our PRS, could modify the penetrance of the JAK2 46/1 haplotype. When we stratified individuals by both PRS and JAK2 46/1 carrier status, MPN risk varied substantially, even among JAK2 46/1 carriers (Extended Data Fig. 3). Compared to non-carriers of JAK2 46/1, the risk among carriers with intermediate PRS risk (middle three quintiles) was 0.87-fold (95% CI: [0.72, 1.07]), but 1.87-fold (95% CI: [1.61, 2.16]) for those with high PRS (top quintile). Validation of this PRS in external cohorts is necessary to further assess the generalizability of these risk variants in different populations. Nevertheless, these results indicate that polygenic profiles can refine stratification of MPN risk.
Next, we performed Bayesian fine-mapping to prioritize putative causal variants at each MPN risk association (Methods)11. To improve detection power for downstream functional enrichments, we included all 25 suggestive loci in this analysis. Briefly, for each LD-independent signal, we extracted all variants within a 2 Mb window, calculated posterior probabilities of causality (PP) for each variant based on effect size estimates, ranked variants by decreasing PP, and added variants to credible sets until a cumulative PP > 95% was reached. Of the 25 credible sets, two mapped to a single variant, and six contained five or fewer variants. In 14 regions, the top fine-mapped variant had a PP ≥ 0.25 (Extended Data Fig. 3). To further resolve loci containing highly correlated variants, we annotated fine-mapped variants with functional criteria including regulatory features, evolutionary conservation, target genes, and transcription factor motif disruption potential (Supplementary Table 5).
Enriched cell types underlying MPN risk
We next attempted to identify relevant cell states underlying MPN predisposition. Because MPNs arise in the hematopoietic compartment, we examined genetic correlations between MPN risk and 19 blood cell traits in 408,241 European ancestry individuals from the UKBB. MPN risk demonstrated positive genetic correlations with counts of red blood cells, platelets, total white blood cells, monocytes, neutrophils, and eosinophils – all of which derive from multipotential hematopoietic stem and progenitor cells (HSPCs) (Fig. 2a, Extended Data Fig. 4). This led us to wonder whether MPN risk variants may be pleiotropically influencing distinct blood lineages from a common progenitor population. We previously showed that variants associated with multiple blood lineages preferentially co-localize with accessible chromatin of more primitive HSPCs12. Similarly here, a greater proportion of well fine-mapped MPN risk variants (PP > 0.10) exhibited pleiotropic blood trait associations compared to more weakly fine-mapped variants (PP < 0.10) (Fig. 2b, 21.4% vs. 1.6%, Fisher’s exact test p = 8.48 × 10−7), supporting the concept that MPN risk variants may act in hematopoietic progenitors to influence multiple lineages.
Figure 2. Functional enrichments in MPN risk.
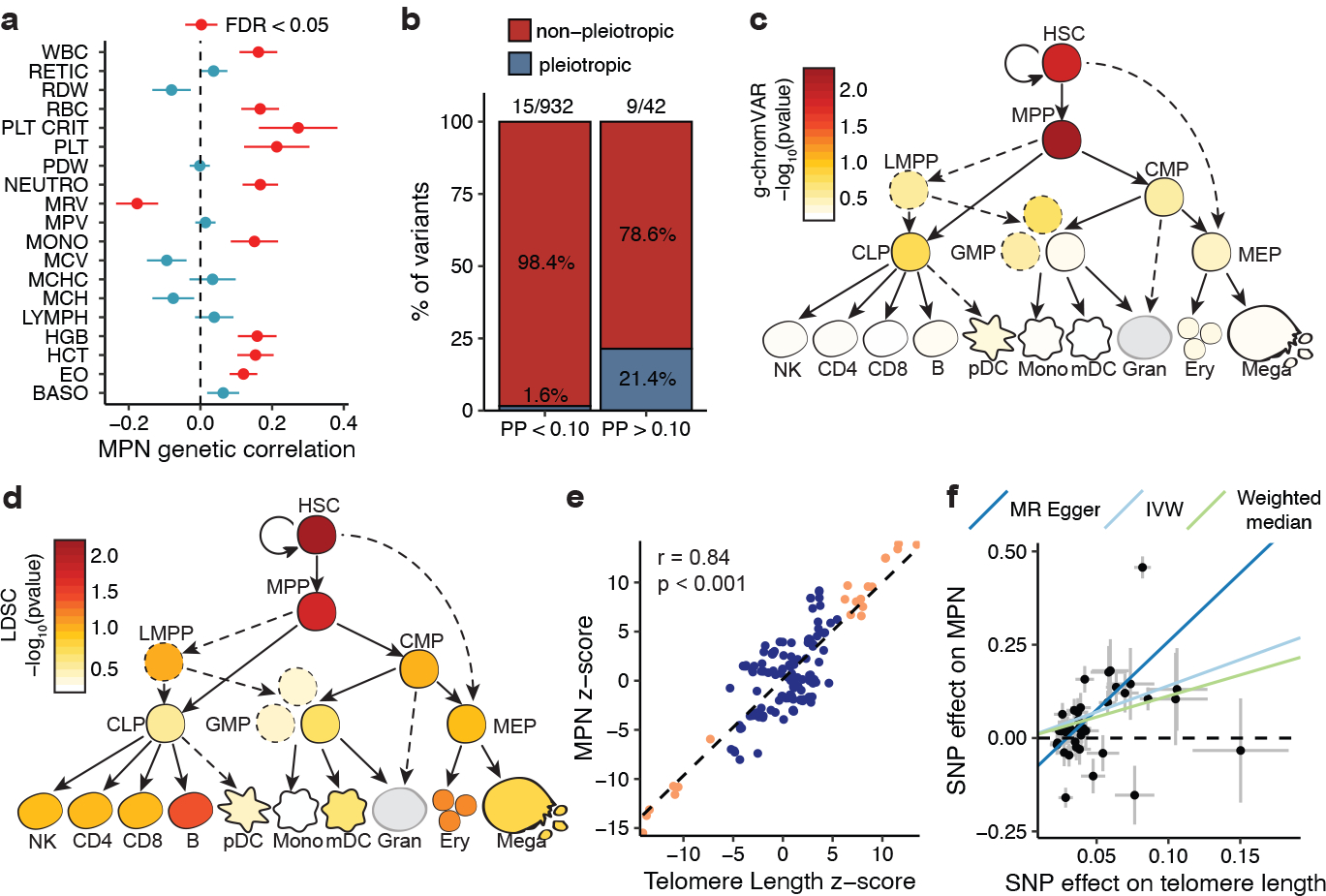
a, Genetic correlations (± standard errors) between MPN risk (n = 2,949 cases and 835,554 controls) and 19 blood traits (n = 408,241), estimated by LDSC. WBC, white blood cell count; RETIC, reticulocyte count; RDW, red cell distribution width; RBC, red blood cell count; PLT CRIT, platelet crit; PLT, platelet count; PDW, platelet distribution width; NEUTRO, neutrophil count; MRV, mean reticulocyte volume; MPV, mean platelet volume; MONO, monocyte count; MCV, mean corpuscular volume; MCHC, mean corpuscular hemoglobin concentration; MCH, mean corpuscular hemoglobin; LYMPH, lymphocyte count; HGB, hemoglobin; HCT, hematocrit; EO, eosinophil count; BASO, basophil count. Red color indicates false discovery rate-adjusted p < 0.05. b, Proportion of MPN risk variants with fine-mapped posterior probability (PP) > 0.10 vs. PP < 0.10 that exhibit pleiotropic associations with traits from two or more hematopoietic lineages (basophil, eosinophil, neutrophil, red blood cell, platelet, monocyte, lymphocyte). c-d, g-chromVAR and LD score regression results for the enrichment of MPN risk variants across 18 hematopoietic chromatin accessibility profiles. e, Correlation of GWAS effect sizes (z-scores) of variants in the TERT locus for MPN vs. telomere length (p < 2.2 × 10−16, two-tailed Pearson correlation). The dashed line is the line of best fit. Orange color indicates variants with association P < 5 × 10−8 in both the MPN and telomere length GWAS. f, Mendelian randomization (MR) plot showing LD-independent telomere length GWAS variants (p < 1 × 10−5, r2 < 0.001) and their effects on MPN risk (outcome) versus telomere length (exposure). Lines represent slopes of three regression tests: MR-Egger (p = 4.83 × 10−5), inverse-variance weighted (p = 1.36 × 10−4), and weighted median (p = 1.15 × 10−5). Data represent MR effect sizes ± standard error.
We next leveraged chromatin accessibility (ATAC-seq) data across 18 human hematopoietic populations to elucidate causal cell types underlying MPN predisposition. 11.6% (34/294) of MPN risk variants with PP > 0.01 fell within accessible chromatin of one or more hematopoietic populations, compared to 4.82% (686/14,235) of variants with PP < 0.01 (χ2 p = 2.13 × 10−6), suggesting that stronger fine-mapped variants are enriched for regulatory function in hematopoietic populations. We next used g-chromVAR12 to compute enrichments of fine-mapped variants across these 18 hematopoietic chromatin accessibility profiles. Strikingly, in contrast to variants associated with peripheral blood cell traits, which are maximally enriched in terminally differentiated hematopoietic populations, MPN risk variants showed the strongest enrichments in multipotent progenitors (MPPs) and HSCs (p = 5.59 × 10−3 and 1.42 × 10−2, respectively) (Fig. 2c). Because g-chromVAR only considers variants from significant loci, we applied LDSC to assess for enrichments in cell type-specific accessible chromatin on a genome-wide level. We again observed that HSCs and MPPs showed the highest enrichment relative to all other hematopoietic populations (p = 7.10 × 10−3 and 1.89 × 10−2, respectively) (Fig. 2d).
Linking genetic MPN risk to other traits
To gain additional insights into mechanisms of MPN predisposition, we examined the relationship between MPN risk and leukocyte telomere length (LTL). LTL is correlated with telomere length of earlier hematopoietic progenitors13, telomere length is associated with HSC self-renewal potential14, and individuals with telomerase loss-of-function associated-diseases have fewer HSCs15. Motivated by the robust MPN risk associations at the telomerase reverse transcriptase (TERT) locus (Extended Data Table 1), we assessed the genetic overlap between MPN risk and LTL from a study involving 78,592 individuals16. At the TERT locus, the top two independent variants for increased telomere length, rs7705526 (p = 5.34 × 10−45) and rs2853677 (p = 3.35 × 10−31), were also lead variants for MPN risk (p = 4.78 × 10−54 and p = 2.79 × 10−44, respectively; PP = 1 for both variants) (Extended Data Fig. 4). We observed a positive genetic correlation between LTL and MPN risk both within the TERT locus (Pearson r = 0.84, p < 0.001) (Fig. 2e) and genome-wide (LDSC rg = 0.19, s.e.m. = 0.093, p = 0.037). We then tested whether increased telomere length may be causally linked to MPN risk by performing two-sample Mendelian randomization (MR), using independent genetic predictors of telomere length as instruments. Applying four related MR algorithms with varying assumptions regarding horizontal pleiotropy, we found that increased telomere length was consistently associated with increased MPN risk at a conservative multiple-testing-adjusted threshold of p < 6.25 × 10−3 (Fig. 2f). The reverse association was significant in only one out of the four tests (see Methods). These results support a positive risk effect of increased telomere length on MPN acquisition. Importantly, however, other causal pathways may be mediating these effects. For instance, given the known association between HSC self-renewal and telomere length, a causal effect of HSC self-renewal on MPN risk could lead to similar MR estimates.
Because variants affecting telomere length have been implicated in many other cancers16, we performed a phenome-wide association study (pheWAS) to measure the effects of MPN risk variants on 1,130 well-represented case-control phenotypes from the UKBB17 (Supplementary Table 6). Consistent with previous studies, the two lead variants at TERT (rs7705526 and rs2853677), as well as others near ATM, SH2B3, and MRPS3, demonstrated associations with several other cancers (Extended Data Fig. 4). In addition, we noted a positive correlation between genetic risk for MPN and clonal hematopoiesis of indeterminate potential (CHIP) (rg = 0.51, s.e.m. = 0.25, p = 0.04)18. Finally, we identified a greater than expected replication of MPN risk loci with acute myeloid leukemia (AML) risk – 5/17 lead MPN variants were significant for AML risk at the 5% level with concordant direction of effect (binomial p = 1.17 × 10−3, Supplementary Table 7). These findings suggest that MPN risk loci may promote risk not only for MPNs, but also somatic mutation acquisition in HSCs leading to other clonal disorders.
Genes underlying inherited MPN risk
Next, we sought to gain further biological insights into germline MPN predisposition by using an integrative approach to map risk variants to target genes. We first nominated three genes from risk loci containing variants at PP > 0.10 with missense consequences: rs1800057 causes the P1054R substitution in ATM, rs3184504 causes the R262W substitution in SH2B3, and rs17879961 causes the I157T substitution in CHEK2. For the remaining 22 non-coding loci, we nominated target genes based on five markers of biological evidence for regulatory function: 1) gene body colocalization, 2) mapping to hematopoietic promoter capture Hi-C (PCHi-C) interactions, 3) correlation between chromatin accessibility and nearby gene expression across hematopoietic cell types, 4) MAGMA gene-wise association signal, and 5) overlap with genes harboring recurrent somatic MPN driver mutations. Genes were selected if they scored in two or more non-coding criteria and were the highest scoring gene in the locus. Using this approach, combined with the three genes harboring coding variants, we identified a single high-confidence target gene in 15/25 MPN risk loci (Extended Data Fig. 5). The nominated genes displayed a much stronger set of functional interactions than expected by chance (STRING database19, 17 vs. 4 expected interactions, p = 3.1 × 10−7) (Extended Data Fig. 5). Remarkably, 11 of the 15 genes have known roles as modulators of HSC self-renewal and function, including ZNF52120, GATA221, MECOM22, RUNX123, HMGA124, ATM25, FOXO126, TET227, JAK228, SH2B329,30 and TERT31 (Fig. 3a). The most enriched biological annotations included replicative senescence, cell cycle, and hematopoietic and lymphoid development (Extended Data Fig. 5, Supplementary Table 8). Analysis of bulk RNA sequencing of 16 primary hematopoietic populations showed a maximal enrichment of target genes for expression in HSCs, MPPs, and myeloid committed progenitors (Fig. 3a). To extend these observations to single cell resolution, we examined the expression of MPN target genes in 278,978 human bone marrow cells. We observed an MPN gene signature that was higher in HSC-enriched cell clusters compared to all other hematopoietic cells. Following gene imputation, we also noted co-localization between MPN and HSC signatures across all cells (Spearman rs = 0.13, p < 2.2 × 10−16) (Fig. 3b–c, Supplementary Figs. 1–2). Together, these results indicate that target genes implicated in MPN risk are enriched for HSC function and expression.
Figure 3. Target genes for MPN risk.
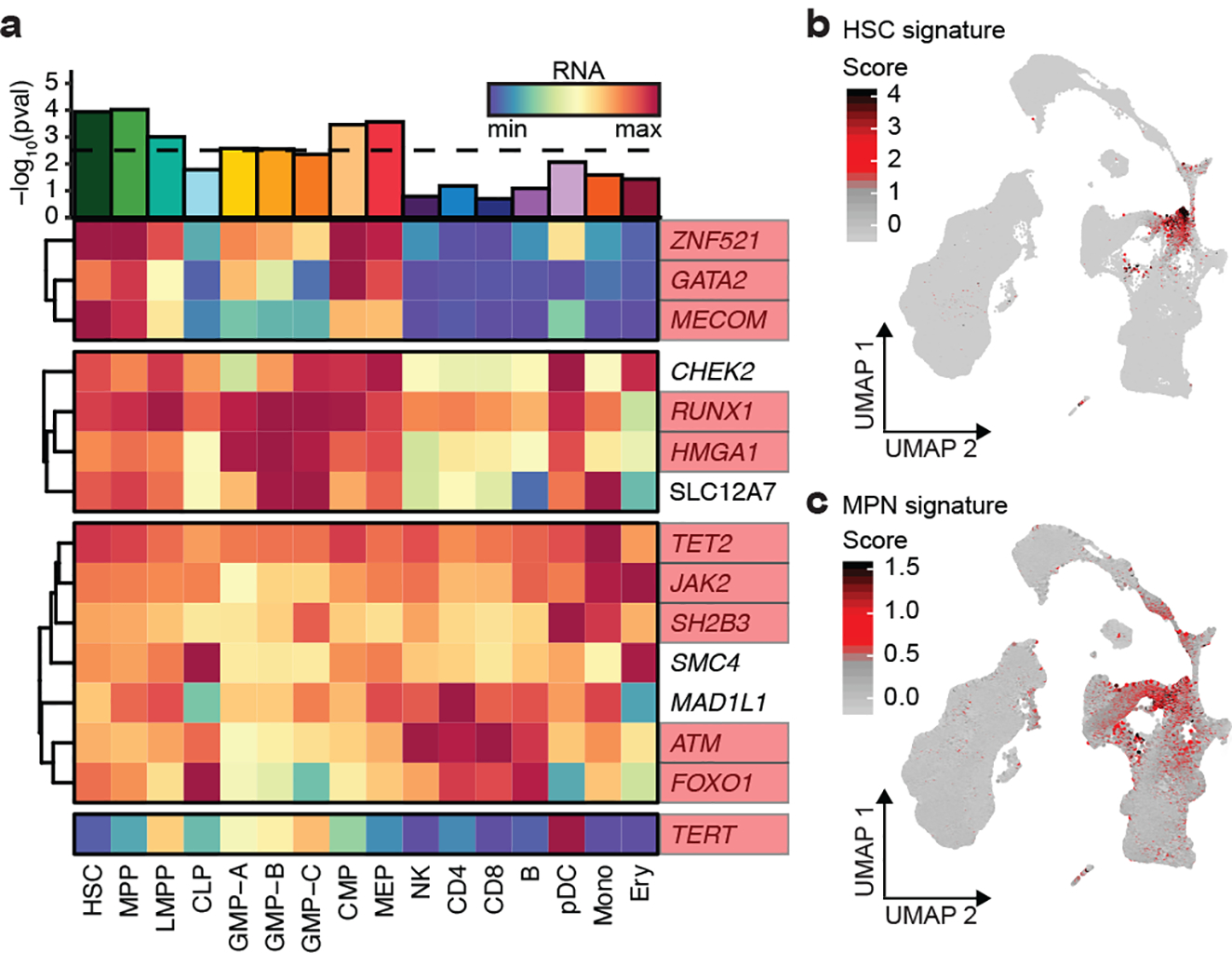
a, Heatmap of 15 MPN target genes, visualizing RNA expression across 16 hematopoietic populations. Bar plot depicts the enrichment of target gene expression in each cell type (two-tailed rank-sum permutation test). Genes with known involvement in hematopoietic stem cell function are boxed in red. b-c, UMAP projections of 278,978 single cells from human bone marrow, colored according to (b) HSC and (c) MPN target gene signatures.
Characterizing mechanisms of MPN risk
In two cases, we performed targeted variant-to-function analyses to gain mechanistic insights into MPN predisposition, starting with the I157T missense variant (rs17879961) in CHEK2. Importantly, this variant is known to be a hypomorphic allele with functionally impaired activation of downstream effectors32,33, and has been previously linked to increased risk for several cancers. Structural analysis of the CHEK2 protein revealed that the I157T variant would diminish hydrophobic interfaces and destabilize the CHEK2 dimer (Extended Data Fig. 6), consistent with prior reports examining the biochemical consequences of this loss-of-function mutation32. We found that CHEK2 inhibition34 reduced cell death after irradiation of primitive human Lin−CD34+CD38− cells, as compared to more differentiated progenitors (Extended Data Fig. 7). Remarkably, while we did not observe skewed lineage commitment in colony assays by CHEK2 inhibition (Extended Data Fig. 7), suppression of CHEK2 through RNA interference increased expansion of human cord blood Lin−CD34+ in long-term cultures in the absence of genotoxic stress (Fig. 4a). These results suggest that CHEK2 ordinarily constrains HSPC expansion, and the I157T variant may reduce CHEK2 function to promote self-renewal and thereby increase MPN risk.
Figure 4. Characterizing the mechanisms of two MPN risk variants.
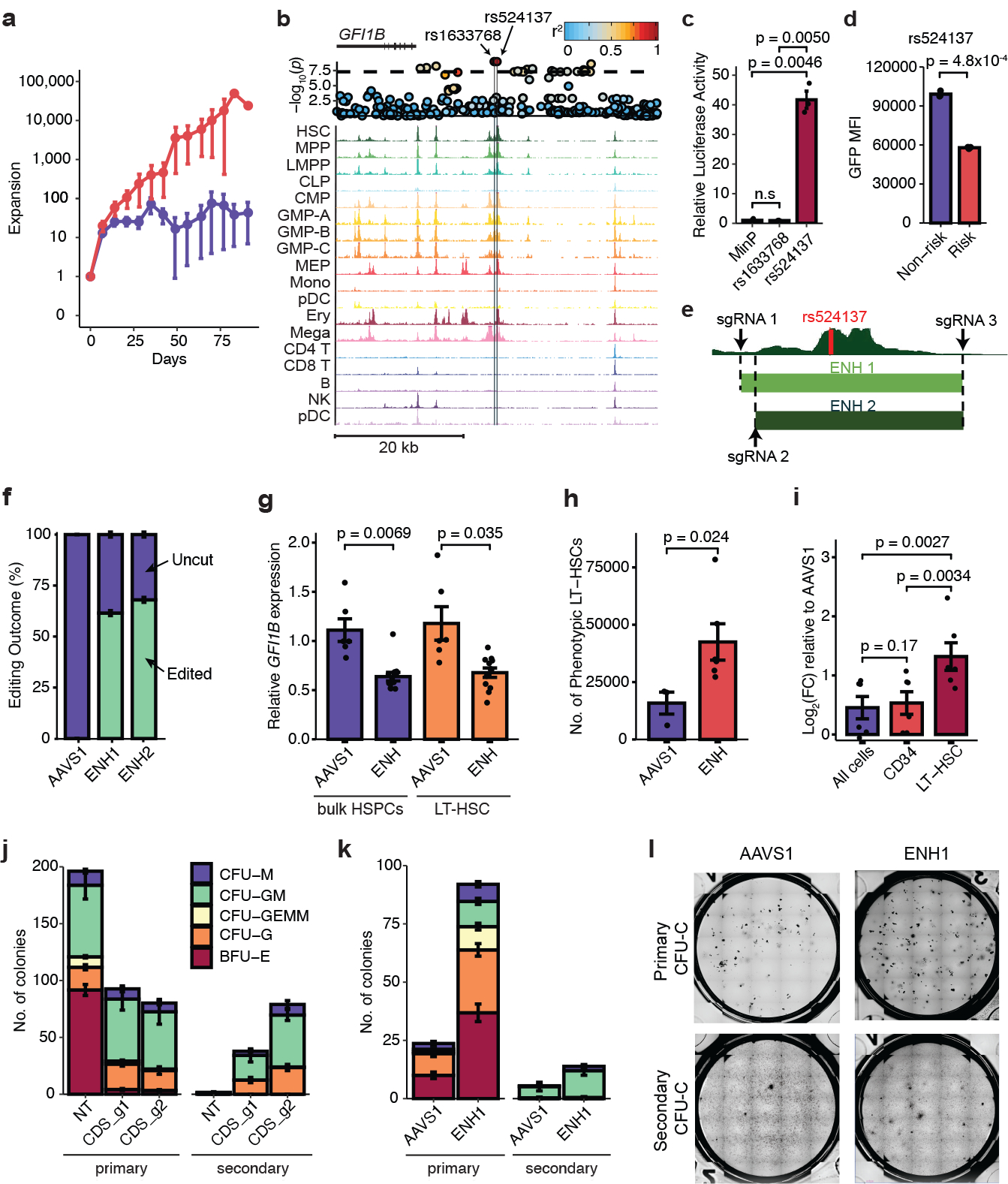
a, Expansion of Lin-CD34+ derived hematopoietic stem and progenitor cells (HSPCs) after short hairpin RNA knockdown of CHEK2 vs. control (CHEK2, n = 4; control, n = 9). b, rs1633768 and rs524137 fall in a region of hematopoietic accessible chromatin downstream of GFI1B. A locus plot is shown above, plotting -log10(p) of MPN association; color reflects linkage disequilibrium to rs524137. c, Luciferase reporter assay testing regulatory activity of genomic regions containing rs1622768 and rs524137 in hematopoietic cells compared to a minimal promoter (MinP) construct (n = 3). d, Lentiviral reporter assays testing allele-specific activity of rs524137 in hematopoietic cells (n = 3). e, HSC chromatin accessibility around rs524137 and the two CRISPR-Cas9 guide RNA pairs (ENH1 and ENH2) used to delete this region. f, Frequency of uncut and edited alleles after editing of GFI1B enhancer or control AAVS1 site in human CD34+ HSPCs (n = 6). g, GFI1B expression in bulk HSPCs and sorted phenotypic long-term HSCs (LT-HSCs) following GFI1B enhancer deletion (n = 12) compared to AAVS1 editing (n = 6). Due to similar editing outcomes, ENH1 and ENH2 were combined as ENH in this and subsequent experiments. h, Total number of phenotypic LT-HSCs in HSC maintenance culture, 6 days after editing of GFI1B enhancer (n = 6) or AAVS1 (n = 3). i, Relative expansion of cell numbers in various compartments (All cells, CD34+, and LT-HSCs) upon GFI1B enhancer deletion (n = 6) compared to AAVS1 controls (n = 3). j, GFI1B coding disruption (CDS_g1 and CDS_g2, n = 3 each) leads to reduced erythroid primary colony formation compared to non-targeting (NT) control (n = 3), but increases secondary colony formation. CFU-M, colony forming unit-macrophage; CFU-GM, granulocyte macrophage; CFU-GEMM, granulocyte erythrocyte macrophage megakaryocyte; CFU-G, granulocyte; BFU-E, burst forming unit-erythroid. k, GFI1B enhancer deletion increases secondary colony formation without affecting erythroid colony formation (n = 3). l, Representative images of primary (top) and secondary (bottom) CFU-C colonies. Data from a, c-d, f-k are means ± s.e.m. n denotes the number of biologically independent replicates. Statistical methods used were two-tailed unpaired t-test (c, d, g, h) and two-tailed paired t-test (i).
In a second instance, we identified a compelling risk locus in a region of hematopoietic accessible chromatin located ~12 kb downstream of GFI1B, a master transcription factor necessary for maintaining HSC quiescence35 and promoting megakaryocytic/erythroid differentiation36 (Fig. 4b). The top fine-mapped variants at this locus were rs1633768 (PP = 0.28) and rs524137 (PP = 0.27). While rs1633768 lacked a target gene, rs524137 mapped to GFI1B by ATAC-RNA correlation (Pearson r = 0.88, p = 2.0 × 10−14) and was predicted to be deleterious (scaled CADD score: 12.73) (Supplementary Table 5). In an exogenous reporter assay in hematopoietic cells, the regulatory region containing rs524137 conferred a 40-fold increase in transcriptional activity compared to a minimal promoter construct, whereas rs1633768 did not change transcriptional activity (Fig. 4c). In otherwise matched genetic constructs, rs524137-T (risk allele) resulted in a 1.7-fold decrease in enhancer activity compared to the non-risk allele when using integrating lentiviral reporters in the presence of the endogenous GFI1B promoter (Fig. 4d, Extended Data Fig. 8). Deletion of a 1.6 kb region encompassing this putative enhancer in human CD34+ HSPCs using two largely overlapping pairs of CRISPR guide RNAs resulted in 61.5% (ENH1) and 68% (ENH2) editing efficiency and up to 36.3% reduction in GFI1B expression, nominating GFI1B as the causal gene (Fig. 4e–g). Enrichment of LT-HSCs based on surface marker phenotypes revealed a 32.3% decrease in GFI1B expression (Fig. 4g, Extended Data Fig. 8). Previous studies have found that knockout of Gfi1b in mice results in expansion of hematopoietic stem cells in vivo35. To test whether GFI1B enhancer deletion also expands human LT-HSCs, we quantified total numbers of LT-HSCs in vitro at the end of a 7-day culture. We observed a 2.7-fold increase in LT-HSCs in the GFI1B enhancer-edited group compared to control AAVS1-edited cells (Fig. 4h), a higher degree of expansion compared with total cells and CD34+ progenitors (Fig. 4i). To assess the effects of HSPC expansion using an independent approach, we performed methylcellulose colony re-plating assays after GFI1B gene disruption and enhancer deletion. GFI1B coding disruption greatly increased self-renewal of progenitors as demonstrated through the presence of increased secondary colonies (Fig. 4j, Extended Data Fig. 8, Supplementary Fig. 3). Loss of GFI1B also reduced formation of primary erythroid colonies, a phenotype consistent with its role in erythroid differentiation37. Deletion of the GFI1B enhancer similarly increased formation of secondary colonies, but did not impact erythroid colony formation (Fig. 4k–l), supporting a mechanism by which rs524137 affects an HSPC-selective enhancer of GFI1B. Analogous to the results obtained for CHEK2, these results show that this MPN risk variant reduces GFI1B expression in HSPCs and thereby increases self-renewal (Extended Data Fig. 9).
Discussion
In summary, we have elucidated the germline genetic architecture of MPN risk through a large-scale GWAS and implicate HSC function as an important driver through a number of functional assays. These results support a model in which MPN risk alleles expand the baseline pool of HSCs, which increases the risk of any one HSC acquiring an MPN somatic driver mutation, consistent with previous studies showing that MPNs arise in only a small subset of HSCs38,39.
MPN risk variants likely exert effects both before and after acquisition of somatic driver mutations (e.g., JAK2 V617F). In this study, we focused on how these variants act before driver mutation acquisition in order to better understand underlying mechanisms of germline risk, which is more commonly observed than the rare occurrence of overt MPNs. Work in other cancers has demonstrated that germline susceptibility can also cooperate with pathogenic driver mutations40, and testing for the presence of this mechanism in MPNs is an important area for future research.
Our discoveries of genetic variation underlying MPN risk complement other studies identifying the genetic determinants of clonal mosaicism in blood cells41–43. Understanding the fundamental mechanisms of MPN predisposition may inform the development of novel interventions for the disease, analogous to the implementation of human papillomavirus testing and colonoscopic surveillance to reduce cervical and colorectal cancer incidence44,45. In this context, improved surveillance or modulation of HSC function may enable therapies to prevent progression to clonal hematopoietic disorders like MPNs46–48. More broadly, our findings serve as a model for dissecting the underlying basis of cancer predisposition alleles.
Methods
UK Biobank GWAS
We performed a GWAS on MPNs using the UK Biobank (UKBB)49. Written consent was obtained for all participants. The study was conducted with the approval of the North-West Multi-centre Research Ethics Committee, in accordance with the principles of the Declaration of Helsinki. Individuals were genotyped using the UK Biobank Axiom Array. Variant imputation was performed as previously described using a combined 1000 Genomes Phase 3-UK10K panel and Haplotype Reference Consortium (http://biobank.ctsu.ox.ac.uk/crystal/label.cgi?id=263)49. The following sample-level quality control filters (previously generated by UKBB) were applied to subset our samples: self-reported British ancestry with similar genetic ancestry based on a principal components analysis of the genotypes, included in kinship inference, no excess (>10) of putative third-degree relatives inferred from kinship, no outlier in heterozygosity and missing rates, and no putative sex chromosome aneuploidy. Samples were filtered from UKBB .bgen files using bgenix version 1.0.1 and qctools v2.0-rc7. 408,241 individuals satisfied all of these criteria and were included for genetic association studies.
We curated a definition for the MPN phenotype within UKBB using the following codes: polycythemia (ICD10 D45; ICD9 2384), essential thrombocythemia (ICD10 D47.3, D75.2), osteomyelofibrosis (ICD10 D47.4, D75.81), chronic myeloid leukemia (ICD10 C921, C922, C931; ICD9 2051), and chronic myeloproliferative disease (ICD10 D47.1). Individuals were also classified as cases if they had a self-reported cancer, self-reported illness code, or histology of cancer tumor code for polycythemia vera, essential thrombocythemia, myelofibrosis, chronic myeloid leukemia, or malignant mastocytosis.
We used SAIGE version 0.29.450 to perform the GWAS using a generalized linear mixed model that controls test statistic inflation and improves power in studies with unbalanced case:control ratios. To select for variants used to estimate the genetic relatedness matrix (GRM), genotyped variants were filtered using the following criteria: minor allele frequency (MAF) > 0.01, LD pruned with r2 threshold of 0.2, Hardy-Weinberg p-value > 1 × 10−6, and genotype missingness < 0.01. Variants were filtered from UKBB .bgen files using bgenix version 1.0.1 and qctools v2.0-rc7. Principal components of ancestry were calculated with these variants using PLINK2 (--pca approx). Age, sex, genotyping array, and the top 10 principal components were included as covariates when fitting the logistic mixed model. For association testing, 26,942,478 autosomal and 1,039,234 X chromosome variants (excluding the pseudo-autosomal region) with MAF > 0.0001 and Info > 0.6 were included.
We also performed GWAS on 19 continuous blood traits within the same 408,241 individuals and same Info and MAF-filtered variants from the UKBB. These associations were performed using BOLT-LMM v2.3.251, with the same covariates of age, sex, genotyping array, and top 10 principal components.
23andMe GWAS
GWAS summary statistics on MPNs from the cohort collected by the personal genetics company 23andMe, Inc. were obtained from a previous study, whose analysis has been described in-depth elsewhere52. A list of 23andMe contributors is presented in the Supplementary Note. Ethical approval was obtained by Ethical & Independent Review Services, an independent external AAHRPP-accredited Institutional Review Board (IRB). Written consent was obtained for all participants. Individuals were genotyped using a custom 23andMe version of the Illumina HumanOmniExpress + BeadChip. Imputation was performed using the August 2010 release of 1000 Genomes reference haplotypes. The MPN phenotype was defined using participant self-report for the following diseases: polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), post-PV/ET myelofibrosis [MF], systemic mastocytosis, chronic myelogenous leukemia, chronic eosinophilic leukemia, and hypereosinophilic syndromes. For our meta-analysis, we used the revised phenotype in their study which also included carriers of the somatic JAK2 V617F mutation. There was a total of 1,223 cases and 252,140 controls in this population. The GWAS was performed using a logistic regression model with covariates of age, gender, and the top five principal components.
FinnGen GWAS
FinnGen is a public–private partnership project combining genotype data from Finnish biobanks and digital health record data from Finnish health registries (https://www.finngen.fi/en), and has been approved by the Ethics Review Board of the Hospital District of Helsinki and Uusimaa. A list of FinnGen contributors is presented in the Supplementary Note. The GWAS on MPNs in the FinnGen cohort was performed using SAIGE version 0.29.4, modified to handle missing genotypes and complete separation in covariates. We used the FinnGen release 4 data in this project, which comprised of 640 cases of MPN and 176,259 controls. Written consent was obtained for all participants. The MPN phenotype was defined by individuals with one or more clinical codes in nationwide hospital discharge or cause-of-death registries for polycythemia vera (ICD10 D45; ICD9 238.4; ICD8 208), chronic myeloproliferative disease (ICD10 D47.1), thrombocythemia (ICD10 D47.3; ICD9 238.7B; ICD8 287.2), myelofibrosis (ICD10 C9.45; ICD9 238.7A; ICD8 209), and chronic myeloid leukemia (ICD10 C92.1; ICD9 205.1). The majority of individuals were genotyped using a custom-designed Affymetrix array for the FinnGen project, with a subset of legacy samples genotyped using various generations of Illumina GWAS arrays. Variant imputation was performed by beagle4.1 software using a reference panel of 3,775 whole-genome sequenced individuals from Finland. The GRM was calculated with 49,811 variants that were imputed in every cohort, and which satisfied the following quality control filters: INFO > 0.95, MAF > 0.05, genotype missingness < 0.05, LD pruned with r2 threshold of 0.1. Age, sex, the top 10 principal components, and genotyping batch/cohort were applied as covariates to the SAIGE logistic regression model. For association testing, 16,289,692 autosomal and X chromosome variants with Info > 0.6 were included.
GWAS meta-analysis
We aggregated association summary statistics from the UKBB, 23andMe, and FinnGen GWAS used a fixed effects model with inverse-variance weighting of log(odds ratios), as implemented in the METAL software53. We meta-analyzed 7,343,617 variants which had association statistics in at least the two largest cohorts (UKBB and 23andMe). Linkage disequilibrium score regression (LDSC) of the meta-analysis showed an LDSC intercept of 1.01 and genomic control factor of 1.035, indicating negligible inflation in test statistics due to population structure. Thus, we did not adjust test statistics using genomic control.
Approximate conditional association analysis
GCTA was used to perform approximate conditional and joint association analyses (COJO) to identify independent MPN risk loci54. In brief, this method performs a stepwise model selection (--cojo-slct) to identify all conditionally independent risk signals at a given p-value of association, using GWAS summary statistics and estimated LD from a reference panel. For estimation of LD, we used a reference sample of 6,000 unrelated individuals of white British origin, randomly selected from the UKBB. After excluding variants with low imputation quality (INFO < 0.4) or deviation from Hardy-Weinberg equilibrium (p < 1 × 10−6), this reference panel included ~36 million variants. The reference panel was converted from BGEN files to hard-called PLINK files using PLINK2. When running GCTA-COJO, we set the threshold p-value to p < 1 × 10−6 and the distance for assuming complete linkage equilibrium (--cojo-wind) at 10000 kb (i.e. 10 Mb).
Million Veteran Program replication
We performed replication of our discovery meta-analysis results in up to 318,271 individuals of European descent from the Million Veteran Program (MVP)55. Written consent was obtained for all participants. The MVP received ethical and study protocol approval from the VA Central Institutional Review Board in accordance with the principles outlined in the Declaration of Helsinki. Individuals were genotyped using the MVP 1.0 custom Axiom array. We attempted to replicate lead variants at 24 / 25 suggestive loci identified from our discovery analysis which also had genotype information within MVP (Supplementary Table 2, Extended Data Fig. 2); the one exception was rs75405916 (p = 7.4 × 10−7, RAF = 0.0004), which was not detected in MVP. Within the MVP cohort, cases (n = 848) were defined as individuals carrying substantial JAK2 V617F mutation burden, determined by specifying a threshold variant mutant allele intensity for rs77375493, the genotyping probe for the V617F mutation. To set the threshold, we utilized a previously reported odds ratio of 2.04 between rs7868130 (a JAK2 46/1 haplotype variant) and JAK2 pV617F, determined from an out-of-sample association study of 446 V617F carriers vs. 169,021 non-carriers52, as an estimate of the true association between JAK2 46/1 and V617F cases. Setting the rs77375493 allele intensity threshold to achieve this odds ratio resulted in a V617F case prevalence of 0.27% within MVP, which was comparable to the previously reported population prevalence of ~0.2%52. The slightly higher point estimate may reflect the fact that the MVP cohort is predominantly male and older than other population studies, both of which have been associated with increased rates of V617F56.
We also attempted to replicate within MVP using an MPN definition based on Phecodes (https://phewascatalog.org/phecodes) and ICD9 codes: polycythemia vera (Phecode 200.1), chronic myeloid leukemia (Phecode 204.22), essential thrombocythemia (ICD9 238.71), and myelofibrosis (Phecode 289.1, ICD9 238.76). The replication rate using this definition was substantially lower. One reason for this could be winner’s curse bias. However, even odds ratios for known MPN risk loci showed consistently weaker effect sizes compared to all three discovery cohorts, whereas the JAK2 V617F phenotype produced effect sizes that were closer to the three discovery cohorts (Extended Data Fig. 2). To quantify this discrepancy, we calculated the M statistic, a measure of between-cohort heterogeneity that combines information across independent lead variants to reveal systematic patterns of heterogeneity57. Across the 15 genome-wide significant lead variants from the discovery analysis, the MVP cohort exhibited significant heterogeneity using the ICD-coded MPN definition (M = −1.26, Bonferroni p = 2.81 × 10−9), but not when using the JAK2 V617F definition (M = −0.45, Bonferroni p = 0.32). This heterogeneity is unlikely to be explained by differences in cohort characteristics, as all cohorts were restricted to European ancestry and controlled for age, sex, and ancestry-informed principal components. Moreover, the heterogeneity was resolved upon changing the phenotype to a more objective measure of a pre-MPN state (JAK2 V617F). These reasons led us to suspect the presence of spurious ICD-based phenotype designations for MPN within the MVP cohort. Thus, we chose to use the JAK2 V617F carrier as a proxy for a “pre-MPN” state for final replication. Notably, the use of JAK2 V617F for replication of MPN phenotypes has also been done in a previous GWAS52. Logistic regression for each replication variant was performed using the PLINK2–glm function, with age, sex, and the top 5 principal components included as covariates. Inverse-variance weighted meta-analysis was used to compute joint p values combining the discovery meta-analysis and MVP replication associations.
Polygenic risk score analysis
We trained a polygenic risk score (PRS) on a meta-analysis of the 23andMe and FinnGen cohorts, which included 1,863 cases of MPN and 428,399 controls. To determine the risk variants to be used in the PRS, we performed a pruning and thresholding analysis using the LD clumping method in PLINK version 1.9058 (--clump) with an r2 threshold of 0.2 and a p-value threshold of 1 × 10−5. In brief, this algorithm forms clumps around variants with association P values less than the specified threshold, in which each clump contains all variants within 250 kilobases of the lead variant that are correlated with the lead variant (r2 > 0.2), based on an LD reference panel of 6,000 randomly selected European individuals from the UKBB. The final output was a PRS containing 104 independent (r2 < 0.2), MPN-associated (P < 1×10−5) variants representing the strongest risk variants for each LD clump across the genome. We performed a sensitivity analysis by testing different p-value thresholds for variant inclusion into the PRS. Across a broad range of thresholds, the area under the receiver operating characteristic (AUROC) for the PRS was similar (Supplementary Table 4).
We applied the PRS to the UKBB, an out-of-sample test set containing 1,086 cases of MPN and 401,155 controls. The PRS was computed for each individual by multiplying the genotype dosage of each risk allele for each variant by its association estimate beta (log-odds) from the UKBB summary statistics as a weight. This was performed using the PLINK2 --score function.
We modeled the PRS using a logistic regression with MPN case-control status as the phenotype and PRS, age, sex, top 10 principal components of ancestry, and genotyping array as covariates. We calculated the area under the receiver-operator curve (AUROC) for the model using the pROC R package. The proportion of variance explained was calculated by using the Nagelkerke’s pseudo-R2 metric. We used this metric to calculate the incremental R2, which quantifies the gain in R2 when a variable is added to a logistic regression of MPN case-control status on a set of other covariates (sex, age, genotyping array, 10 principal components of ancestry). To use PRS to stratify individual risk for MPNs, we divided individuals in the UKBB cohort into deciles based on their PRS. For each non-reference decile, logistic models were fitted with the same covariates as used above, comparing MPN risk for members of the given decile compared to those in the 5th decile (i.e., those with average risk) and lowest decile (i.e., those with the lowest 10% of PRS). We also performed a logistic regression for MPN risk while stratifying on individual carrier status for JAK2 46/1, a major risk haplotype for MPNs, and PRS category (low = bottom quintile of PRS, intermediate = quintiles 2–4, high = top quintile).
Genetic fine-mapping
For each distinct association signal, we calculated approximate Bayes’ factors (ABFs)59 for all variants within 1-Mb of the lead variant. ABFs were calculated as:
where r = ω/(s.e.2+ω) and z = β/s.e. The parameter ω denotes the prior variance in allelic effects, estimated here as 0.035 based on formula (8) of the original publication of Wakefield’s formula and the 95% interval of variant effect sizes in the GWAS. For loci with multiple distinct signals, association statistics were based on GCTA approximate conditional analysis adjusting for all other index variants in the region. We then calculated the posterior probability of being causal (PP) by dividing the ABF of each variant by the sum of ABF values over all variants in the locus. The 95% credible set for each locus was constructed by 1) ranking all variants in descending order of PP and 2) including ordered variants until the cumulative PP reached 95%.
Contribution of variants to overall familial relative risk
We estimated the proportion of the familial risk of MPNs that can be explained by variants identified in our GWAS under a log-additive model, as previously described60. We applied the formula , where λg is the proportion of familial risk explained, pi is the MAF for variant i, βi is the log(odds ratio) estimate for variant i, τi is the standard error of βi, and λ is the overall familial relative risk. We assumed the overall familial relative risk for MPNs to be 4.93 based on a recent epidemiological study61.
Definition of known loci
We compiled a list of 10 previously reported genome-wide significant MPN association signals from literature52,62. Loci were only included if they were identified using a similar MPN phenotype (i.e., combination of JAK2 V617F carriers and all MPN subtypes, without any sub-stratification of MPN subtypes). All loci reaching genome-wide significance (p < 5 × 10−8) before or after replication steps were included.
g-chromVAR cell type enrichment analysis
Bias-corrected enrichment of MPN risk variants for chromatin accessibility of 18 hematopoietic populations was performed using g-chromVAR (https://github.com/caleblareau/gchromVAR), whose methodology has been previously described63. In brief, this method weights chromatin peaks by fine-mapped variant posterior probabilities and computes the enrichment for each cell type versus an empirical background matched for GC content and feature intensity. For the chromatin accessibility component, we used a consensus peak set for all 18 hematopoietic cell types with a uniform width of 250 bp centered at the summit. For the variant scores, we used the fine-mapped PP for all MPN risk variants with fine-mapped PP > 0.001 across suggestive loci.
Linkage disequilibrium score regression
We used LD score regression (LDSC) to estimate the narrow-sense heritability estimate of MPN risk and compute genetic correlations between MPN risk and other phenotypes64. Reference LD scores were computed with a subset of European individuals combined from the 1000 Genomes Phase 3 (1000GP3) and UK10K cohorts. Variants were filtered by MAF > 1%, and 5,653,963 variants were used as input to LDSC. We estimated the liability scale heritability of MPN to be ~6.5% (s.e. = 2.98%) based on common genetic variation (MAF > 1%). For this estimation, the sample prevalence of MPNs was 0.00352 in our GWAS, and the population prevalence of MPNs was estimated to be 0.000328 based on previous reports65,66.
To calculate genetic correlations with blood traits, we first used BOLT-LMM to perform GWAS on 19 blood traits in 408,241 European ancestry individuals from the UKBB, the same samples used for the MPN GWAS. Imputation and variant quality control filters were the same as those applied in the MPN GWAS. To calculate cross-trait correlations, we used the same 1000GP3-UK10K reference panel used to estimate LDSC heritability. We constrained the intercept by accounting for the known sample overlap between the MPN and blood trait GWAS, as well as adjusting for phenotypic correlations between MPN case-control status and each of the 19 blood traits. To calculate genetic correlations with telomere length and CHIP, we obtained previously generated summary statistics from recent studies67,68 and calculated genetic correlations as described above, but without constraining the intercept.
To calculate cell type enrichments, we generated LD scores for 18 primary hematopoietic ATAC peak sets. We also generated a “pan-heme” peak set representing the union of peaks across all 18 populations. Adopting the approach previously used for LDSC tissue-specific enrichments69, we jointly modeled the annotation for each cell type of interest, the “pan-heme” annotation for all hematopoietic peaks, as well as the 52 annotations in the baseline model.
Blood trait pleiotropy analysis
We tested whether fine-mapped MPN risk variants were more likely to demonstrate pleiotropic associations with common blood traits from distinct lineages. To do this, we performed fine-mapping on all genome-wide significant regions for each of 18 lineage-specific blood traits (all except white blood cell count) using FINEMAP v1.3.170. We then assigned each blood trait to a major hematopoietic lineage: basophil count to basophils; eosinophil count to eosinophils; neutrophil count to neutrophils; red blood cell count, hematocrit, hemoglobin, RDW, MCH, MCHC, MCV, reticulocyte count, and mean reticulocyte volume to red blood cells; platelet count, MPV, platelet crit, platelet distribution width to platelets; monocyte count to monocytes; and lymphocyte count to lymphocytes. We considered a variant to be pleiotropic if it had a fine-mapped PP > 0.10 for blood traits from multiple lineages. Then, we constructed a contingency table for the proportion of MPN fine-mapped variants (PP > 0.10 vs. PP < 0.10) which were classified as pleiotropic vs. not pleiotropic, and calculated an enrichment using a two-sided Fisher’s exact test.
Phenome-wide association study
To identify associations between MPN risk variants and other clinical phenotypes, we conducted a phenome-wide association study (PheWAS) using summary statistics of 1,403 ICD-based clinical phenotypes analyzed from the UK Biobank (see URLs). As input, we included all fine-mapped MPN risk variants with PP > 0.001. To reduce noise from phenotypes with low case numbers, we restricted our analysis to only variant-phenotype combinations with an expected case minor allele count > 50 (calculated as 2 * variant MAF * number of cases), resulting in 1,130 unique remaining phenotypes. Variants in the HLA region were excluded. We used a Bonferroni-corrected p-value threshold to define significant pheWAS associations (0.05 / 1130 phenotypes = 4.42 × 10−5).
Comparison with acute myeloid leukemia risk
We examined the effects of the 17 lead variants (p < 5 × 10−8) from the MPN GWAS on inherited risk for acute myeloid leukemia (AML). To do this, we obtained AML association statistics for these 17 variants from three independent cohorts: UK Biobank, FinnGen, and Alliance for Clinical Trials in Oncology (Alliance). The AML GWAS for UK Biobank and FinnGen were conducted using the same quality control metrics and covariates as were used for the MPN analysis. Association statistics for the 17 variants from Alliance were obtained from a previous study, whose analysis has been described in-depth elsewhere71. We then meta-analyzed these statistics used a fixed effects model with inverse-variance weighting of log(odds ratios), as implemented in the METAL software53.
Transcription factor motif analysis
Prediction of the effects of fine-mapped variants on transcription factor binding sites (TFBS) was performed using the motifbreakR R package72 and a comprehensive collection of 426 human TFBS models (HOCOMOCO73). For all fine-mapped variants with PP > 0.001, we applied the ‘information content’ scoring algorithm and used a P-value cutoff of 1 × 10−3 for TFBS matches; all other parameters were kept at default settings.
Mendelian randomization
Mendelian randomization (MR) analysis was performed using the R packages TwoSampleMR74 and MRPRESSO75. MR consists of two steps: (i) identification of proper instrumental variables or genetic predictors, i.e., variants independently associated with the exposure factor, and (ii) calculation of causal estimates76. To achieve step 1, we first obtained GWAS summary statistics for leukocyte telomere length. We filtered for variants associated with leukocyte telomere length at a minimum p < 1 × 10−5, and then clumped these variants with an LD threshold of r2 < 0.001 to obtain 38 independent genetic instruments.
Next, we extracted the MPN risk effect sizes of these genetic instruments from our MPN GWAS and harmonized the data to ensure the variant statistics for telomere length and MPN were oriented to the same allele. To calculate causal estimates of telomere length on MPN risk, we implemented four non-independent methods with varying assumptions regarding horizontal pleiotropy: MR-Egger regression with bootstrap77, the IVW method, the weighted median test, and MR-PRESSO. We adopted a conservative multiple-testing-adjusted threshold of p < 6.25 × 10−3 (0.05/(4*2)) to account for the use of four tests and testing bidirectionally. All tests found a significant causal relationship between telomere length and MPN risk: IVW, p = 1.36 × 10−4; outlier-corrected MR-Presso, p = 1.05 × 10−5; MR-Egger, p = 4.83 × 10−5; weighted-median, p = 1.15 × 10−5.
We also tested the reverse association to assess causal estimates of MPN risk on telomere length. The same parameters were used, except this time we first clumped MPN risk variants (p < 1 × 10−5), and then extracted the corresponding effect sizes for telomere length. The MR test statistics for the reverse association were the following: IVW, p = 5.78 × 10−3; outlier-corrected MR-Presso, p = 0.057; MR-Egger, p = 0.058; weighted-median, p = 0.070.
Target gene identification
We implemented a two-stage process to identify high-confidence target genes at MPN risk loci. In all stages, the HLA locus (chromosome 6:28866528–33775446) was excluded due to its complex linkage structure. In the first stage, we checked whether any fine-mapped risk variants (PP > 0.10) resulted in coding consequences or splice alterations. To check for splice variants, we annotated variants with spliceAI78, a neural net prediction tool for splice altering variants. No variants had a delta score > 0.2, indicating a low probability for any variant to cause splicing changes. We used Variant Effect Predictor79 to screen for coding variants, which identified three variants in distinct loci with PP > 0.10 causing missense mutations: rs17879961 for CHEK2, rs1800057 for ATM, and rs3184504 for SH2B3. These three regions were mapped to these respective target genes and were not analyzed further using non-coding variant approaches, described below.
The second stage consisted of mapping noncoding regions to target genes. Because non-coding gene regulation is more difficult to pinpoint, we applied a lower variant inclusion threshold of PP > 0.01 as a high-sensitivity screen for target gene interactions. To increase specificity for high-confidence target genes, we incorporated five different functional annotations for evidence of noncoding gene regulation: 1) gene body colocalization, 2) mapping to hematopoietic promoter capture Hi-C (PCHi-C)80, 3) correlation between chromatin accessibility and nearby gene expression across hematopoietic cell types, 4) MAGMA gene-wise association signal, and 5) overlap with a recurrent somatically mutated gene in MPNs.
To map variants to gene bodies, we used gene annotation coordinates from GENCODE release 3381 for the GRCh37 genome build. We removed all ribosomal protein genes by excluding any genes with names starting with “RP”. We then identified the nearest genes to risk variants using the nearest command in the GenomicRanges R package.
For nominating target genes by enhancer promoter interactions, we used a published PCHi-C dataset spanning 15 hematopoietic cell types80. We filtered for looping interactions with a CHiCAGO score >5. If multiple gene targets were nominated for one variant, only the gene with the top CHiCAGO score was kept.
ATAC-RNA correlations were generated by computing Pearson correlations between hematopoietic ATAC peaks and RNA counts of genes within a 1-Mb window of the ATAC peak, as previously described63.
We performed gene-based associations for 18,987 protein-coding genes using MAGMA82, implemented through FUMA83. In brief, this test provides aggregate association statistics based on all variants located in a gene, adjusting for LD. Default MAGMA parameters were used, which mapped variants to genes with no window around genes (window size = 0). A gene was considered positive by MAGMA if it passed the genome-wide significance of p = 0.05/18987 = 2.63 × 10−6.
For overlap with genes with known somatic mutations in individuals with MPN, we used all genes identified with driver mutations in at least 5 patients from a recent large-scale study which performed targeted sequencing on a cohort of 2,035 patients with MPN84.
Collectively, non-coding target genes were selected if they scored in two or more criteria and were also the highest scoring gene in the risk locus. Finally, target genes from the coding and non-coding annotations were combined and filtered for protein-coding genes, as annotated by Ensembl using the annotables R package.
Target gene cell type enrichments
To measure the enrichment of MPN target genes in bulk RNA-seq data in 16 hematopoietic populations, we performed a rank-sum permutation test. First, we summed the ranks of all target genes ordered by expression (log2 counts per million) amongst all assayed protein-coding genes with non-zero expression in each cell type. Next, we randomly sampled 10,000 equally sized gene sets and obtained their rank sums within each cell type. We calculated the target gene enrichment z-score as the difference between the mean rank-sum of the permuted sets and the target gene rank sum, divided by the standard deviation of the permuted gene set rank-sums. The z-score was then converted into a two-sided p-value for each cell type.
Gene set enrichments
We used g:Profiler85 to map putative target genes to enriched gene sets. All protein-coding genes with one or more annotations were used as the background.
Single-cell RNA sequencing analysis
Single-cell RNA-seq of 378,000 cells from human bone marrow were generated as part of the Human Cell Atlas project using 10X Genomics sequencing technology and aligned to the GRCh38 reference genome using the Cell Ranger pipeline as previously described (https://preview.data.humancellatlas.org/). Downstream analyses including normalization, scaling, and cell clustering were performed using the R software package Seurat86 version 2 (http://satijalab.org/seurat/). We filtered out low-expressed genes expressed in fewer than 50 cells and low-quality cells with fewer than 500 detected genes, leaving 19,156 genes and 278,978 cells for downstream analysis. Raw gene expression counts of each cell were normalized over total counts and log transformed, and gene expression was then scaled to have a mean of 0 and variance of 1 across cells. We performed dimensionality reduction using PCA, with the top 1000 most variable genes as input, and computed the top 50 principal components (PCs). To identify clusters of cells, we used the ‘FindClusters’ function from Seurat, which applies a shared nearest neighbor modularity optimization-based clustering algorithm to identify clusters based on their PCs (in this case, top 50). To infer the HSC population, we used a marker gene signature similar to one recently applied in a different scRNA-seq human hematopoiesis dataset87 – CD34, HLF, and CRHBP. All 15 MPN target genes were detected in at least 50 cells and thus included in the aggregate MPN signature. To calculate scores based on specific gene sets (e.g., HSC marker genes, MPN target genes) for each cell, we calculated the average of the Z-normalized expression (across all cells) of each gene in the list. To adjust for dropout in single-cell data when estimating the correlation between MPN and HSC signatures, we applied a gene imputation approach called MAGIC88 to infer missing transcripts in cells. To do so, gene expression values in all cells were normalized, dimensionally reduced and transformed by internal algorithms in MAGIC with the parameters: n_pca_components = 100, t = 6, k = 10, alpha = 15, rescale_percent = 99. Following imputation, marker gene expression was again used to calculate an MPN and HSC signature per cell, as described above, and a Spearman correlation of these scores was calculated.
Structural analysis
The X-ray crystallographic structure of the human CHEK2 protein was used to create a CHEK2 structural model. Structural superposition, analyses, and figures were rendered using PyMOL Molecular Graphics System v2.3.289.
Irradiation experiments
Umbilical cord blood Lineage negative (Lin-) and CD34+ cells were obtained using StemSep system according to the manufacturer’s protocol (Stem Cell Technologies, Canada). Lin−CD34+ cells were sorted to obtain HSC (CD34+38−/lowCD45RA−CD90+), CMP (CD34+38+CD45RA−CD135+), GMP (CD34+38+CD45RA+CD135+), and MEP (CD34+38+CD45RA−CD135−) fractions. Then cells were resuspended in X-VIVO 10 (BioWhittaker, Waldersville, MD) medium supplemented with 1% BSA, SCF (100 ng/ml), FLT3L (100 ng/ml), TPO (15 ng/ml), G-CSF (10 ng/ml), and IL-6 (10 ng/ml) and incubated for 72–96 hours followed by irradiation with 3Gy. When indicated, cells were pre-treated with CHEK2i (CHEK2 Inhibitor II,10uM final, Sigma 220486) or DMSO for 1hr prior to irradiation. Assessment of IR-induced cell death in the indicated populations 18hr post IR relied on double staining with Annexin and Sytox. IR-induced cell death was calculated to reduce the variability between CD34+ batches by subtracting the fraction of AnnexinV+Sytox+ cells scored in the untreated sample from the same fraction in the IR sample.
Viral constructs, human CD34+ transduction and long-term expansion assays
The following pLKO-puro lentiviral shRNA constructs from the RNAi consortium shRNA library (TRC, https://portals.broadinstitute.org/gpp/public/) were used: shCHEK2 (TRCN0000039946) and shControl (TRCN0000231746). This shRNA has been shown to induce knockdown of total and phosphorylated CHEK2 protein90. Viral particles pseudotyped with VSV-G were prepared using transient transfection of 293T cells as described elsewhere91.
Lin−CD34+ cells were incubated with the indicated lentiviruses, at multiplicity of infection 50–100, in the X-VIVO 10 medium supplemented with 1% BSA, SCF (100 ng/ml), FLT3L (100 ng/ml), TPO (15 ng/ml), G-CSF (10 ng/ml), and IL-6 (10 ng/ml) for 16 hours followed by cell wash and medium replacement. Puromycin (500 ng/ml) was added to the infected cells two days post infection for the additional two days. At the end of puromycin selection CD34+ cells were seeded in the ex vivo expansion cultures as previously described92. Briefly, CD34+ cells were plated in IMDM, 10% FCS (Sigma) supplemented with FLT3L (50 ng/ml), TPO (20 ng/ml), SCF (50 ng/ml), and IL-6 (10 ng/ml) at the density of 1 × 105 cells/ml. Every seven days, cells were counted, washed, and resuspended at the density of 1 × 105 cells/ml in fresh medium and cytokines.
Luciferase reporter assays
The genomic region containing risk and non-risk alleles of the variants rs524137 (~501 bp) and rs1633768 (~294 bp) were synthesized as gblocks (IDT Technologies) and cloned into the Firefly luciferase reporter constructs (pGL4.24) using NheI and EcoRV sites. Each allele-specific construct differed by only the single nucleotide of the variant of interest (i.e., all other nucleotides in the construct are the same). The Firefly constructs (500 ng) were co-transfected with pRL-SV40 Renilla luciferase constructs (50 ng) into 100,000 K562 cells using Lipofectamine LTX (Invitrogen) according to manufacturer’s protocols. Cells were harvested after 48 hours and the luciferase activity measured by Dual-Glo Luciferase Assay system (Promega). For each sample, the ratio of firefly to Renilla luminescence was measured and normalized to the minimal promoter construct. Location of rs524137 and rs1633768 gblocks in hg19 are chr9:135879384–135879677 and chr9:135878924–135879424 respectively.
Lentiviral reporter assays
The lentiviral reporter constructs were designed to deliver enhancer elements containing risk and non-risk alleles of rs524137 positioned upstream of human GFI1B promoter (~400 bp) driving a reporter GFP. The constructs were generated from a pLKO based lentiviral construct overexpressing GFP from a PGK promoter. Fragments containing human GFI1B promoter and enhancer elements were synthesized as gblocks (IDT technologies) and cloned using Kfl1 and BamHI sites upstream of the GFP start site replacing the PGK promoter. Lentiviral supernatants produced by transient transfection in 293Ts were used to transduce 50,000 K562 cells by spinfection at 2000 rpm for 90 min. K562s infected with the lentiviral reporters were cultured for 6 days and reporter GFP expression was measured by flow cytometry. Location of GFI1B promoter gblock in hg19 is chr9:135853723–135854128.
Ribonucleoprotein (RNP) electroporation of CD34+ human HSPCs
Electroporation was performed using the Lonza 4D Nucleofector with 20 μl Nucleocuvette Strips. For the GFI1B enhancer element deletion experiment, CD34+ HSPCs were thawed 24 hrs before electroporation. The RNP complex was made by mixing Cas9 (50 pmol) and modified sgRNAs from Synthego (100 pmol in total), including two pairs of guides ENH1 (sgRNA1: TAAGTCTGGGGTCTACAAAG & sgRNA2: ATGACTTGCTTAGAGCACCA) and ENH2 (sgRNA3: ATAGAAGACCACTTCTCGCA & sgRNA2: ATGACTTGCTTAGAGCACCA) targeting the five and three prime end of the enhancer element (hg19 chr9:135878960–135880490). For negative control, a guide targeting AAVS1 site was used (GGGGCCACTAGGGACAGGAT). GFI1B gene expression and editing outcome of the electroporation were measured at 6 days post-electroporation. Quantitative PCR was performed using SYBR green (Bio-Rad) to access the editing efficiency of the target sequences in bulk and sorted primitive LT-HSC cell populations, with primers designed to detect the percentage of unedited wild-type cells (forward: TCTACCACTCCCAGCAGCT; reverse: CGTCTCCTCTCCTGGGTCTT). GFI1B RNA expression was measured using quantitative PCR with specific primers (forward: GCAGGAAGATGAACCGCTCT; reverse CCAGGCACTGGTTTGGGAA).
For the coding deletion experiment, CD34+ HSPCs were thawed 48 hrs before electroporation. The RNP complex was prepared by mixing Cas9 (50 pmol) and modified sgRNA from Synthego (100 pmol) and incubating for 15 min at room temperature immediately before electroporation. HSPCs (3.75 × 105) resuspended in 20 μl P3 solution were mixed with RNP and transferred to a cuvette for electroporation with program DZ-100. The electroporated cells were resuspended with Stemspan II media with CC100 cytokine cocktail (Stem Cell Technologies). Two guides targeting GFI1B coding regions (sgRNA4: GGGGTCGGGACAGCACAATG; sgRNA5: CCTTGTTGCACTTCACACAG) and control non-targeting guide (NT) was used in these experiments. Gfi-1b protein expression was measured at 5 days post-electroporation using anti-GFI1B antibody (Santa Cruz Biotechnology, sc-28356; 1:3000 dilution) and a loading control anti-Lamin B1 antibody (Santa Cruz Biotechnology, sc-374015; 1:1000 dilution).
HSC maintenance cultures
Enhancer edited HSPCs were cultured for 6 days post-editing in HSC maintenance conditions using serum free StemSpan II media (Stem Cell technologies) supplemented with CC100 cytokine cocktail (Stem Cell technologies), 50ng/ml TPO (Peprotech), and a small molecule UM171 (35 nM) shown to help maintain and potentially expand HSCs in culture93,94. At the end of the culture, total cells were enumerated and stained with a panel of antibodies to quantify and enrich for LT-HSCs. The antibody panel includes anti-CD45RA-Alexa Fluor® 488 (Biolegend, #304114), anti-CD34-PerCP-CY5.5 (Biolegend, #343612), anti-CD90-PECy7 (BD, #561558), anti-CD133-superbright 436 (Ebioscience, #62-1388-42), anti-EPCR-PE (Biolegend, #351904), and anti-ITGA3-APC (Biolegend, #343808). 3 μl of anti-CD34-PerCP-CY5.5, anti-CD90-PECy7, anti-EPCR-PE, anti-ITGA3-APC, and 6 μl of anti-CD45RA-Alexa Fluor® 488, anti-CD133-superbright 436 were used per one hundred thousand cells in 100 μl staining volume. LT-HSCs were enriched by FACS sorting using previously published methods95. Both genomic DNA and RNA were extracted from sorted LT-HSCs and bulk HSPCs using AllPrep DNA/RNA Mini Kit (Qiagen) according manufacturer’s recommendations. Total LT-HSC numbers were calculated as a product of frequency of LT-HSC by FACS and total cell numbers counted at the end of culture. Flow cytometry data were analyzed on FlowJo™ software (v10.6.1).
Colony-forming unit cell assays
3 days RNP post-electroporation, 500 CD34+ HSPCs were plated in 1ml methylcellulose media (#H4034, Stem Cell Technologies). Primary CFU-C colonies were counted after 14 days. For the colony replating experiments, 2 weeks after the primary plating, the colonies from two plates were pooled, washed with PBS, and the cells were plated in new methylcellulose media at 25000 cells/ml for an additional 14 days. Images of primary and secondary colonies were taken using StemVision (Stem Cell technologies) using manufacturer’s recommendations.
URLs
1000 Genomes Phase 3: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/; UK10K: https://www.uk10k.org/data_access.html; Haplotype Reference Consortium: http://www.haplotype-reference-consortium.org/; HOCOMOCO: https://hocomoco11.autosome.ru/; Protein Data Bank: https://www.rcsb.org/pdb/static.do?p=general_information/about_pdb/index.html; g-chromVAR: https://github.com/caleblareau/gchromVAR; UK Biobank phenome-wide association study: https://www.leelabsg.org/resources; Immune cell atlas: https://preview.data.humancellatlas.org/.
Extended Data
Extended Data Figure 1.
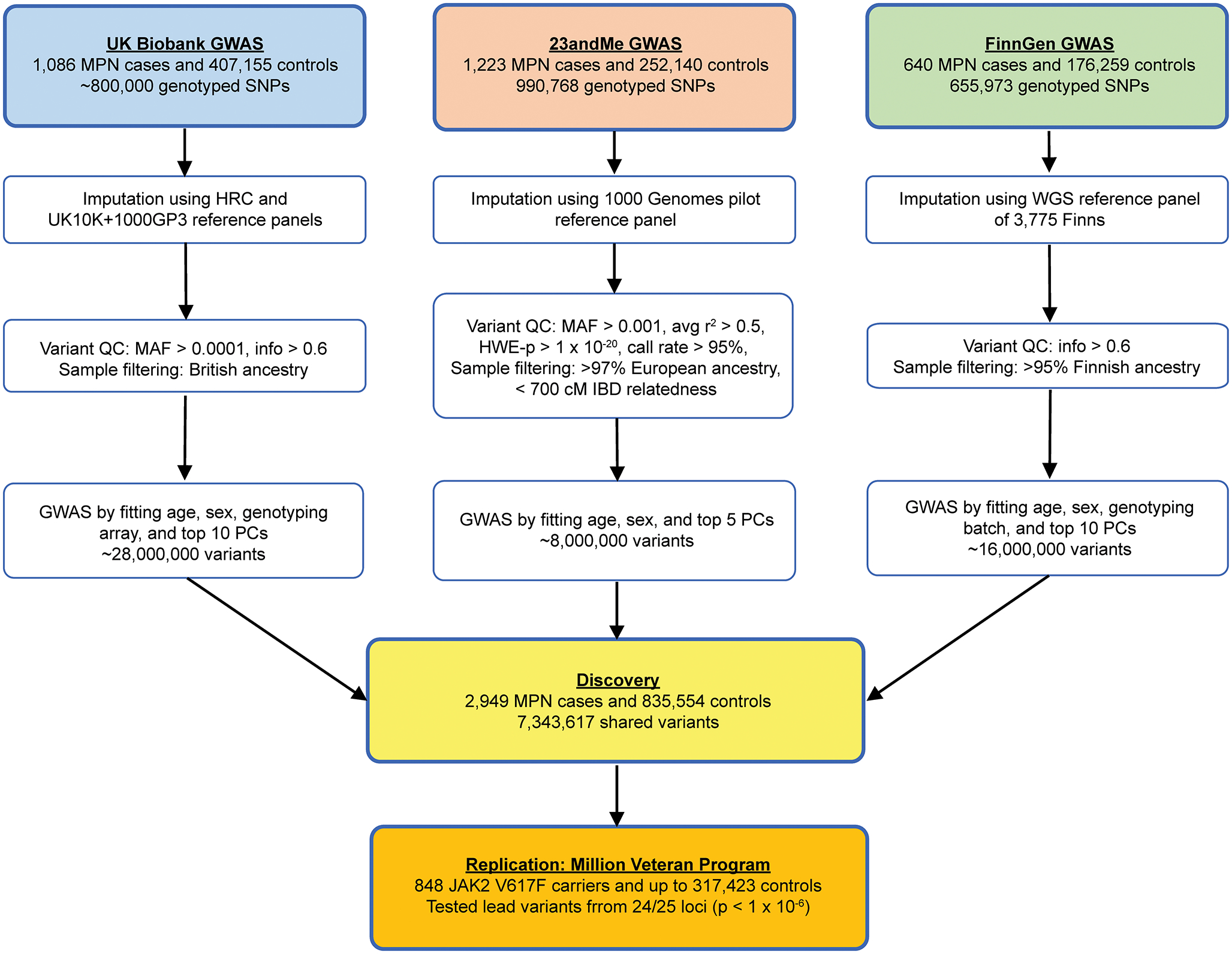
Flowchart of genetic association analyses. Flowchart of the quality control steps and analysis methods for the three discovery-phase genome-wide association studies (GWAS) in the UK Biobank, 23andMe, and FinnGen, followed by replication in the Million Veteran Program.
Extended Data Figure 2.

MPN GWAS cohort-specific effect sizes. a, Forest plot displaying cohort-specific odds ratios for lead variants of the 17 loci reaching genome-wide significance after replication. Sample sizes are: UKBB, n = 1,086 cases and 407,155 controls; 23andMe, n = 1,223 cases and 252,140 controls; FinnGen, n = 640 cases and 176,259 controls; MVP, n = 848 cases and 317,423 controls. Data represent odds ratios and 95% confidence intervals. b, Overall correlation of effect sizes between MVP cohort and combined discovery cohort (UKBB + 23andMe + FinnGen) for all 24 variants reaching suggestive significance (p < 1×10−6) which underwent replication (p = 3.76 × 10−5, two-tailed Pearson correlation). c, Forest plot displaying cohort-specific odds ratios for lead variants of the three most significant loci in the meta-analysis: the JAK2 46/1 haplotype and two independent signals at the TERT locus. MVP_jak2 = JAK2 V617F phenotype in MVP, MVP_jak2_or_mpn = JAK2 V617F or ICD-based MPN definition in MVP. Data are odds ratios and 95% confidence intervals. Sample sizes are: UKBB, n = 1,086 cases and 407,155 controls; 23andMe, n = 1,223 cases and 252,140 controls; FinnGen, n = 640 cases and 176,259 controls; MVP_jak2, n = 848 cases and 317,423 controls; MVP_jak2_or_mpn, n = 2,203 cases and 218,607 controls.
Extended Data Figure 3.
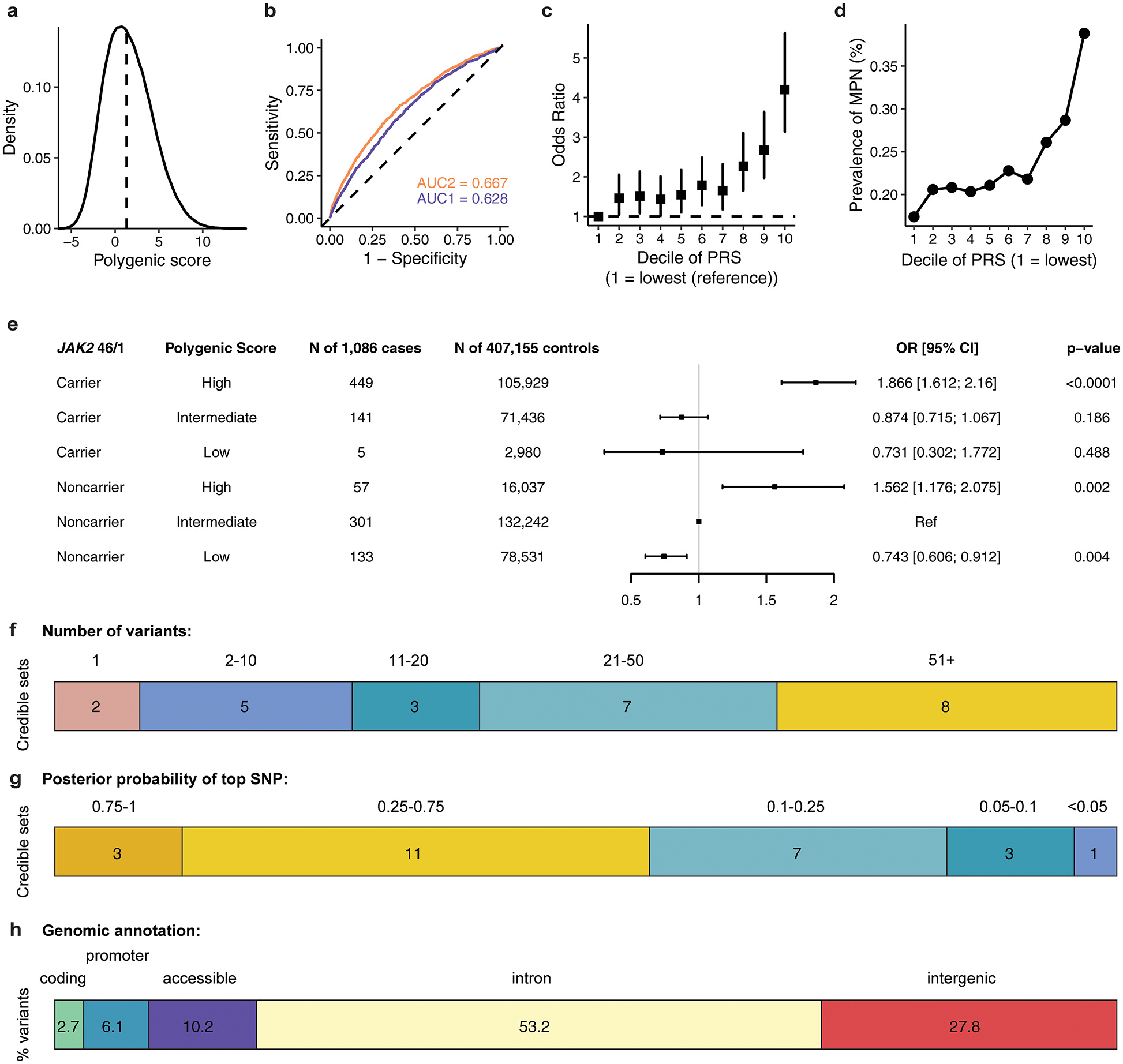
Assessing the distribution and prevalence of MPN polygenic risk score in UK Biobank. a, Density distribution of the MPN polygenic risk score (PRS) within the UK Biobank. b, Receiver operating characteristic curves for MPN predictions (n = 1,086 cases and 407,155 controls), using information from age, sex, genotyping array, and ancestry-informed principal components (AUC2, blue) alone, or with the addition of PRS (AUC1, orange). c, Odds ratio (mean and 95% confidence interval) for MPN acquisition according to deciles of the PRS (n = 1,086 cases and 407,155 controls), with decile 1 (10% of individuals with lowest PRS) as the reference group. d, Prevalence of MPN within each decile of the PRS in the UK Biobank population (n = 1,086 MPN cases, 407,155 controls). e, MPN cases and controls in the UK Biobank were stratified into three groups according to their PRS – low, intermediate, or high defined as the lowest quintile, the middle three quintiles, and the highest quintile of the PRS distribution respectively. For carriers and noncarriers of the JAK2 46/1 haplotype, the odds ratio for MPN was calculated in a logistic regression model with PRS group, age, sex, and the top ten principal components of ancestry as covariates. Non-carriers with intermediate PRS served as the reference group. Data are odds ratios and 95% confidence intervals. f, Fine-mapped 95% credible sets for all 25 MPN risk loci reaching suggestive significance, stratified by the number of variants comprising each credible set. g, The fine-mapped posterior probability of causality for the highest fine-mapped variant in each locus credible set. h, Variants within the 95% credible sets and posterior probability (PP) > 0.001 across all regions, grouped by genomic annotation.
Extended Data Figure 4.
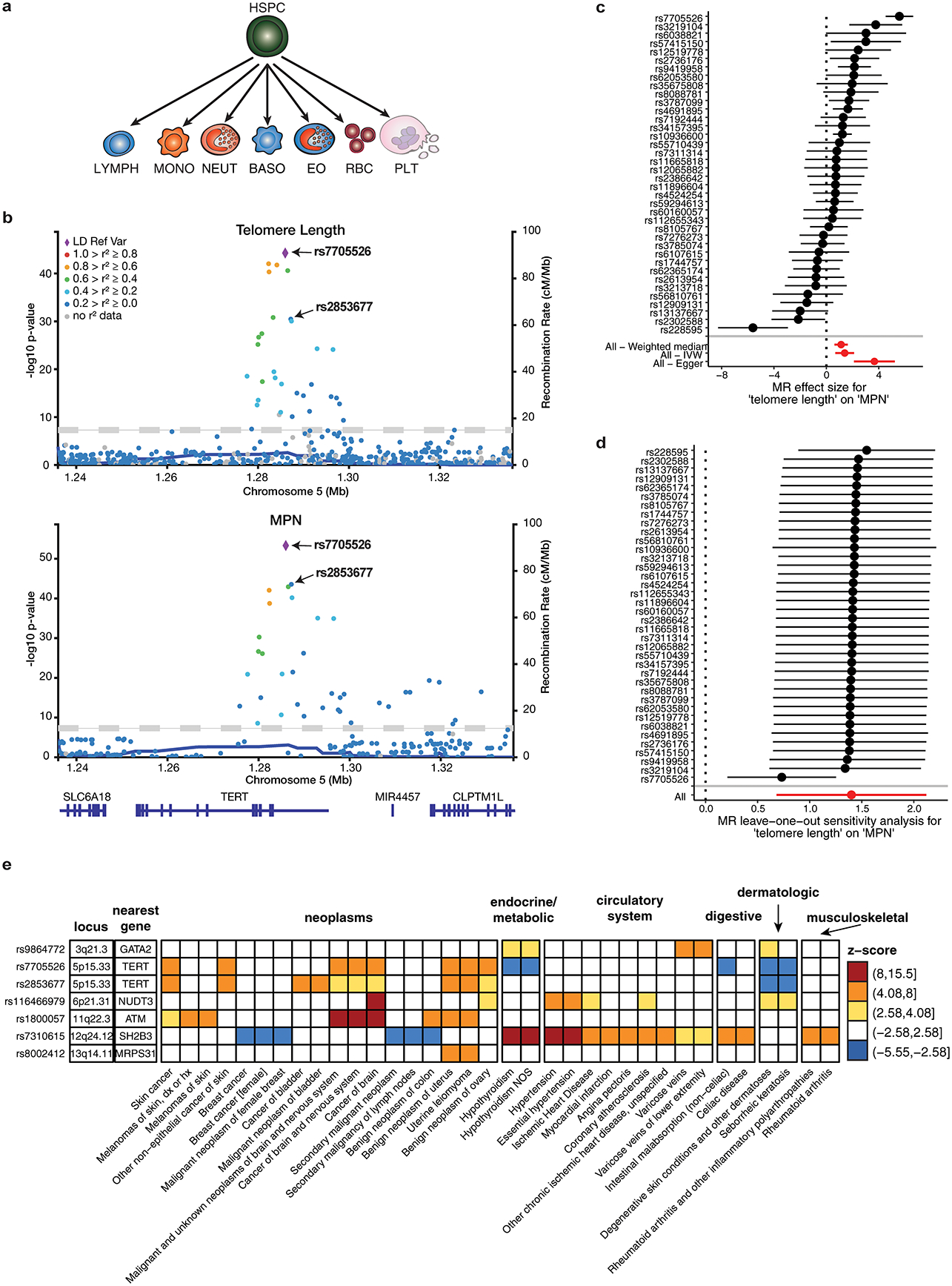
Shared genetic associations between MPN risk and other phenotypes. a, Schematic depicting the trajectory of undifferentiated hematopoietic stem and progenitor cells (HSPCs) into various committed cell types: lymphocytes (LYMPH), monocytes (MONO), neutrophils (NEUT), basophils (BASO), eosinophils (EO), red blood cells (RBC), and platelets (PLT). b, Regional association plots at the TERT locus (+/− 50kb from lead variant), showing the associations of variants with leukocyte telomere length and MPN. The colors of the points depict pairwise linkage disequilibrium (r2) to sentinel variant rs7705526. The two conditionally independent lead variants for both traits, rs7705526 and rs2853677, are labeled. c, Individual SNPs associated with telomere length and their effect sizes on MPN risk (n = 2,949 cases and 835,554 controls), calculated using the fixed effects meta-analysis method. Aggregate mendelian randomization (MR) effects, calculated from three different methods (weighted median, inverse-variance weighted, and Egger regression), are shown at the bottom. Data are MR effect sizes and standard errors. Red color indicates significance. d, MR leave-one-out sensitivity analysis, showing MR effect estimates using the inverse variance weighted approach after excluding each individual SNP from the analysis (n = 2,949 cases and 835,554 controls). Data are MR effect sizes and standard errors. e, Phenome-wide association study (pheWAS) of MPN risk variants. We tested fine-mapped MPN risk variants (PP > 0.10 or lead variant) for associations with 1,130 well-represented case-control phenotypes from the UK Biobank, calculated by two-tailed logistic mixed model association test. Shown in this heatmap are the top MPN-associated variants at each locus with one or more associations reaching Bonferroni-corrected significance (p = 0.05 / 1130 phenotypes = 4.4 × 10−5, or abs(z-score) = 4.08). Heatmap color indicates association z-score. All variant effects are oriented with respect to the risk-increasing MPN allele. Phenotypes are divided into major clinical categories, as listed in the annotations above the heatmap.
Extended Data Figure 5.
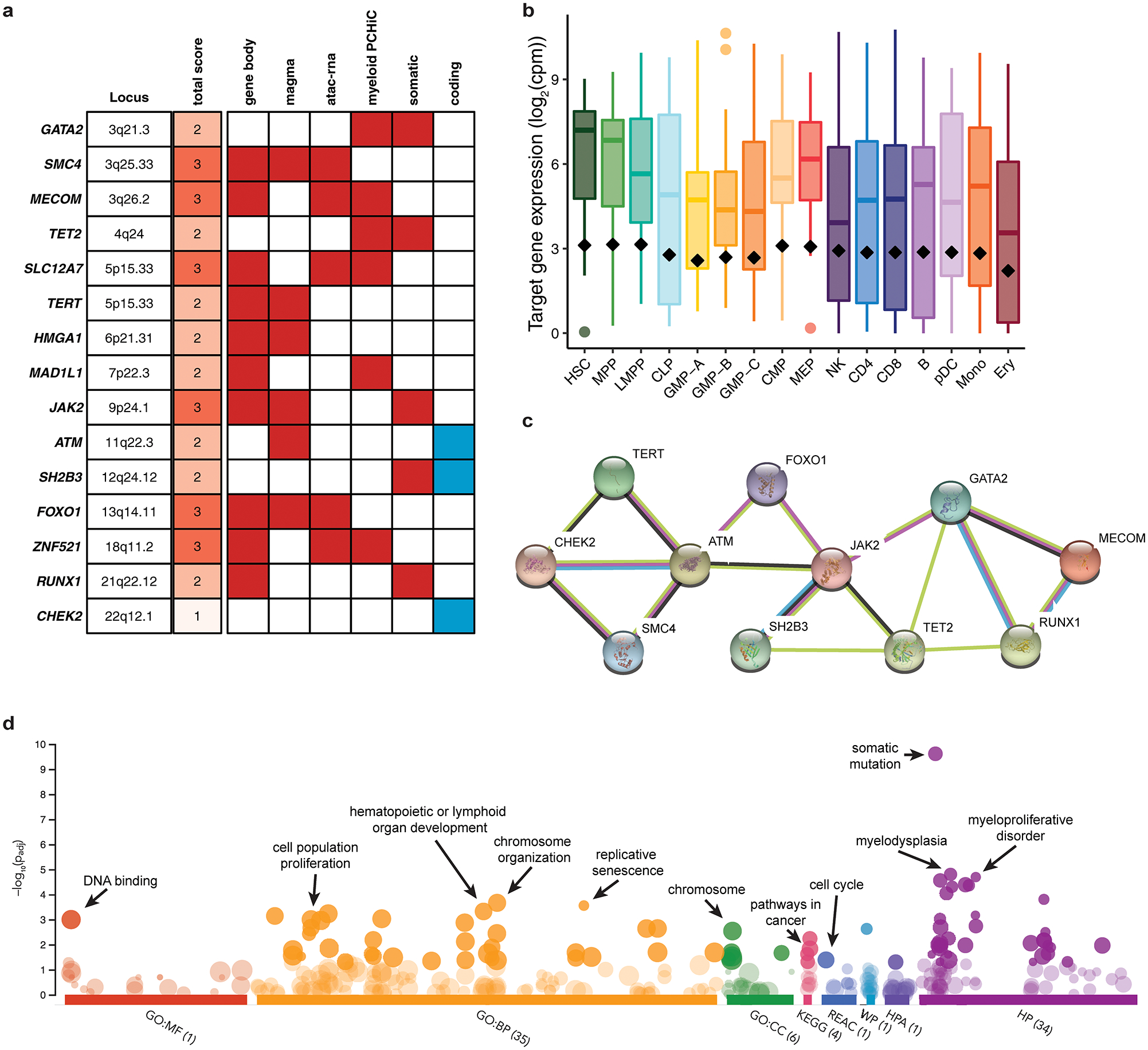
Characterizing MPN target genes. a, Target genes prioritized based on non-coding criteria (red boxes) and coding consequences (blue boxes) and scored based on the number of criteria met. Only the highest scoring gene per locus is reported, and for non-coding loci, only genes with a score of 2 or more are reported. b, Average expression (log2 counts per million) of MPN target genes (n = 15) across 16 primary hematopoietic cell types. Black diamonds indicate the mean expression of all non-zero expressed protein-coding genes in each cell type. Box plots show the median at the center, with the top and bottom of the box indicating the interquartile range. Whiskers extend to either the maximum/minimum value or 1.5x the interquartile range. c, Protein-protein interaction network showing known and predicted associations between the protein products of MPN target genes, generated with STRING database. d, Top-enriched biological annotations for MPN target genes identify key pathways associated with hematopoiesis and oncogenesis.
Extended Data Figure 6.
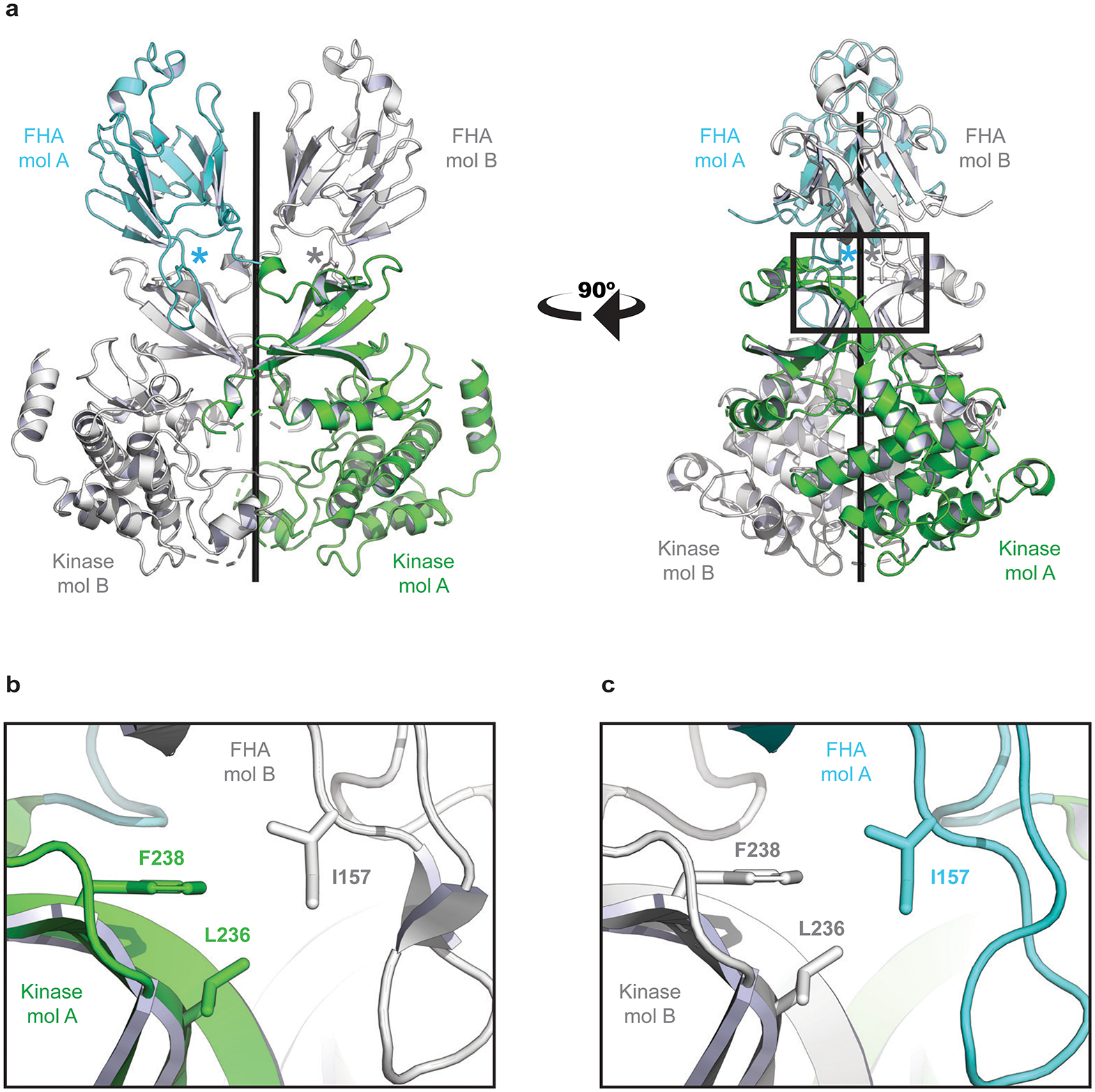
Structural basis for CHK2 homodimer disruption by Isoleucine 157 mutation. a, The crystal structure of the CHK2 (FHA-Kinase) homodimer (PDB: 3I6U). The FHA domain of molecule A (mol A) is shown in cyan and the kinase domain is colored green. A second CHK2 (mol B) has both domains colored white. The two CHK2 molecules are nearly symmetric – coiling around the central axis (black rod). The location of each Isoleucine 157 residue is marked with an asterisk. b, A zoomed window showing details of the interactions. I157 links the FHA of one CHK2 molecule (white) to the kinase domain of a second (green). The side chain of I157 mediates an FHA-Kinase hydrophobic interface, interacting with Phenylalanine 238 (F238) and Leucine 236 (L236) on the kinase domain. c, The second interface of the CHK2 dimer (180° rotation from panel b) is nearly identical. A Threonine at position 157 would diminish these hydrophobic interfaces and destabilize the CHK2 dimer, as has been previously reported.
Extended Data Figure 7.
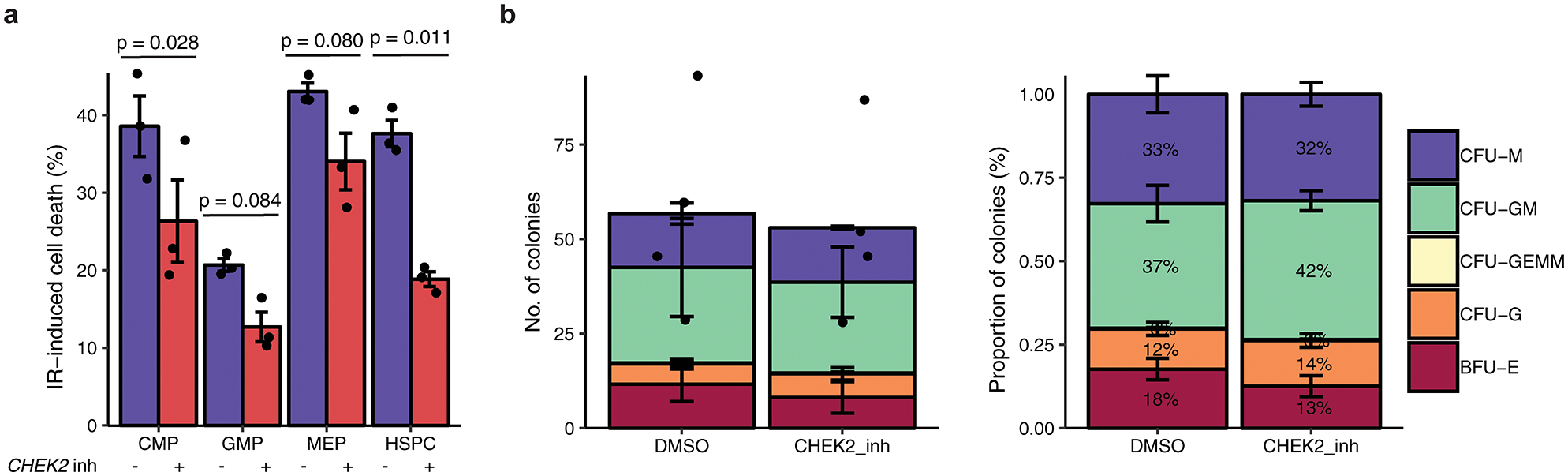
CHEK2 is required for apoptosis of cycling HSPCs, but not for lineage commitment. a, Assessment of IR-induced cell death of cycling HSPCs and myeloid progenitors (CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; MEP, megakaryocyte-erythroid progenitor) following sublethal irradiation, after treatment with CHEK2 inhibitor (n = 3) or dimethylsulfoxide control (n = 3) (two-sided paired t-test). n is the number of biologically independent experiments. Data are mean ± s.e.m. b, Numbers (left) and percent (right) of HSPC colonies formed following CHEK2 inhibition (CHEK2 inhibitor II, Sigma 220486) (n = 4) vs. dimethylsulfoxide (DMSO) control (n = 4). n is the number of biologically independent experiments. Data are mean ± s.e.m. CFU-M, colony forming unit-macrophage; CFU-GM, granulocyte macrophage; CFU-GEMM, granulocyte erythrocyte macrophage megakaryocyte; CFU-G, granulocyte; BFU-E, burst forming unit-erythroid.
Extended Data Figure 8.
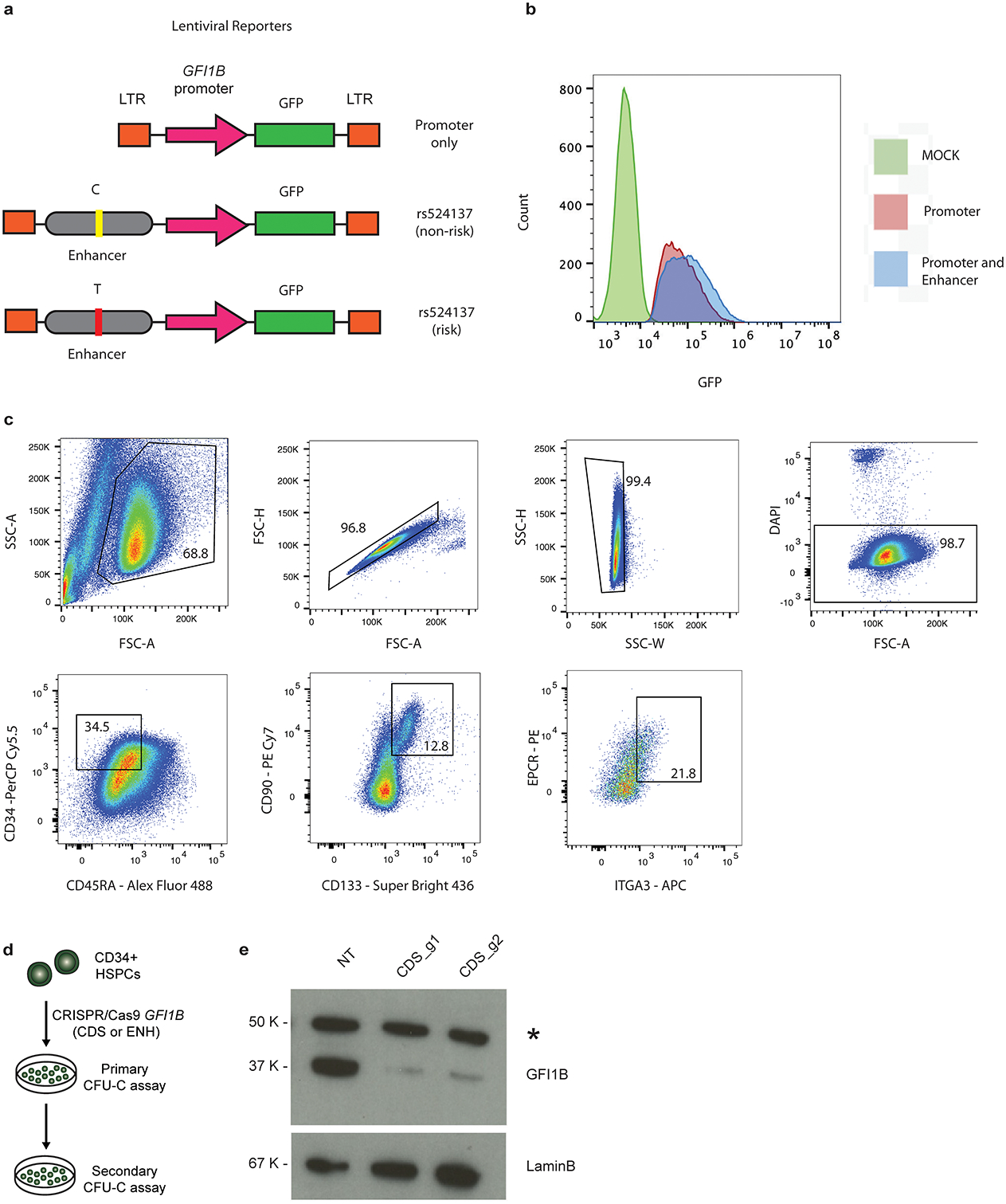
Supplementary data for variant-to-function studies at GFI1B locus. a, Map of the lentiviral constructs designed to assess enhancer activity at rs524137. b, Histogram displays GFP mean fluorescence intensity (MFI) of hematopoietic K562 cells infected with Promoter only vs Promoter and Enhancer lentiviral constructs. Compared to mock uninfected control cells, cells infected with the construct carrying both GFI1B promoter and enhancer show greater GFP intensity. c, FACS gating for sorting and identifying the primitive CD34+CD45RA−CD90+CD133+EPCR+ITGA3+ LT-HSC population in day 7 CD34+ HSPCs presented in Fig. 4g, h, i. d, Schematic of colony-replating assays using human HSPCs edited with GFI1B coding (CDS) and enhancer guides (ENH). e, Representative western blot measuring GFI1B protein expression 5 days following CRISPR/Cas9 targeting with non-targeting control (NT), or coding regions of GFI1B (g1, g2). LaminB expression used as loading control. LaminB controls was probed on the same blot as the GFI1B. Similar results were obtained in 3 independent experiments. For gel source data, see Supplementary Fig. 3.
Extended Data Figure 9.
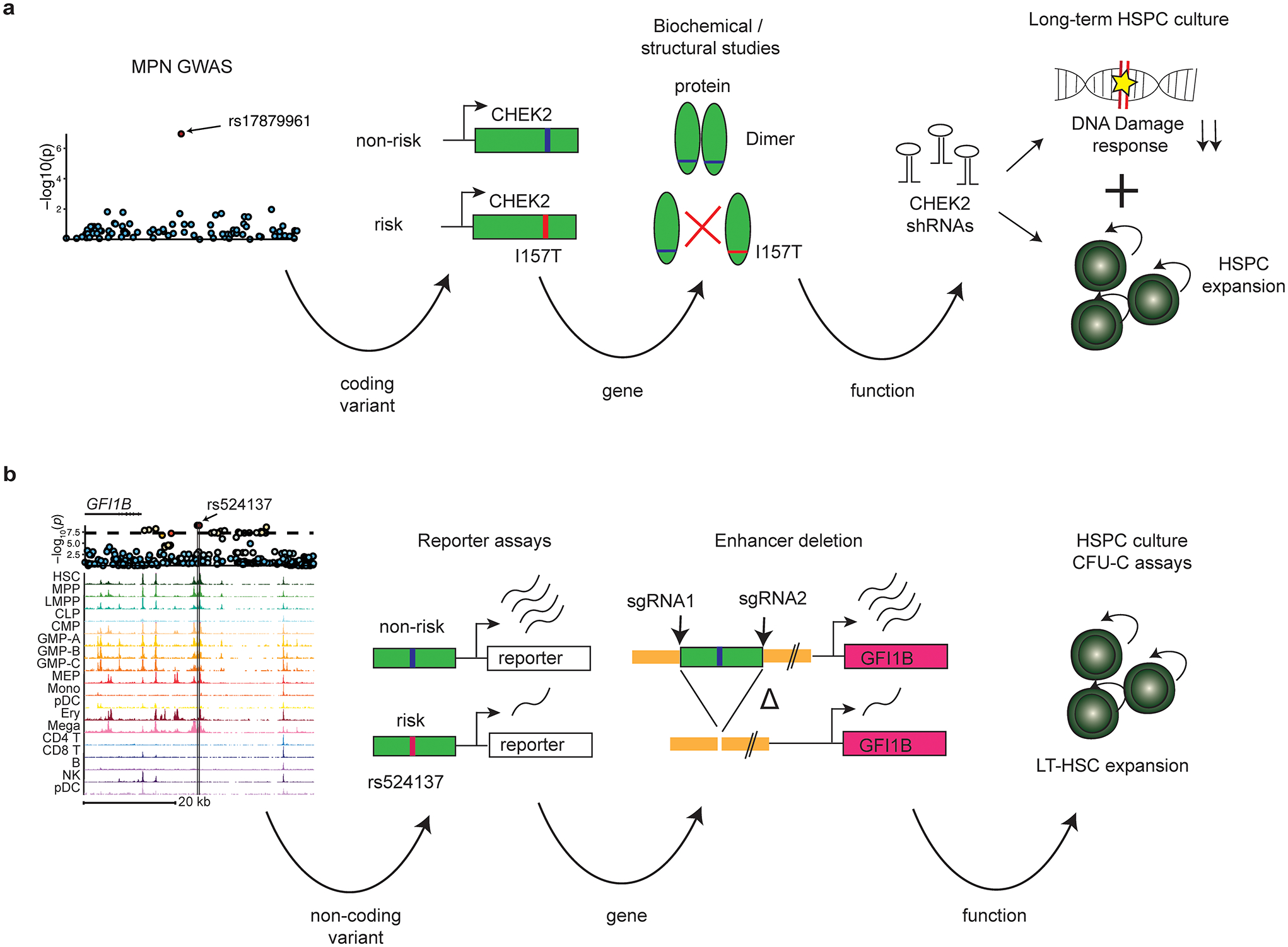
Schematics illustrating the variant-to-function arcs for MPN risk loci at (a) CHEK2 and (b) GFI1B demonstrated in this study.
Extended Data Table 1.
Genome-wide-significant loci from MPN GWAS. Variants shown are the most associated variant at each locus.
| locus | SNP | bp | risk | non-risk | RAF | OR | OR 95CI | pvalue | meta pvalue | Nearest gene | Other genes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3q21.3 | rs9864772* | 128316939 | G | A | 0.6075 | 1.15 | 1.09–1.21 | 2.74E-07 | 2.06E-08 | GATA2 | |
| 3q25.33 | rs77249081* | 159633461 | G | C | 0.0096 | 3.7 | 2.49–5.49 | 8.04E-11 | 5.54E-10 | SCHIP1 | |
| 3q25.33 | rs74676712* | 160284736 | T | C | 0.1138 | 1.3 | 1.2–1.41 | 3.57E-10 | 3.64E-11 | KPNA4 | |
| 3q26.2 | rs9847631 | 168832107 | T | G | 0.3979 | 1.17 | 1.11–1.23 | 7.06E-09 | 4.89E-10 | MECOM | |
| 4q24 | rs62329718 | 105758059 | A | T | 0.0374 | 2.11 | 1.84–2.42 | 7.46E-27 | 2.72E-34 | TET2 | |
| 5p15.33 | rs7705526 | 1285974 | A | C | 0.3358 | 1.58 | 1.49–1.67 | 4.78E-54 | 2.42E-64 | TERT | |
| 5p15.33 | rs2853677 | 1287194 | G | A | 0.4176 | 1.46 | 1.38–1.54 | 2.79E-44 | 4.32E-54 | TERT | |
| 6p21.31 | rs116466979* | 34235378 | C | T | 0.0453 | 1.5 | 1.31–1.71 | 3.34E-09 | 1.86E-12 | NUDT3 | HMGA1 |
| 7q32.3 | rs62471615 | 130746955 | C | A | 0.3037 | 1.3 | 1.23–1.38 | 6.55E-19 | 7.20E-21 | MKLN1 | |
| 9p24.1 | rs1327494 | 4999303 | G | A | 0.2735 | 2 | 1.89–2.12 | 3.07E-128 | 1.11E-170 | JAK2 | |
| 9q34.13 | rs1633768 | 135879138 | T | C | 0.2708 | 1.2 | 1.13–1.27 | 1.03E-09 | 2.15E-12 | GFI1B | |
| 11q22.3 | rs1800057 | 108143456 | G | C | 0.0255 | 1.65 | 1.41–1.92 | 2.00E-10 | 2.94E-12 | ATM | |
| 12q24.12 | rs7310615 | 111865049 | C | G | 0.4768 | 1.27 | 1.21–1.34 | 3.05E-19 | 2.46E-18 | SH2B3 | ATXN2 |
| 13q14.11 | rs8002412* | 41331497 | C | T | 0.1803 | 1.2 | 1.13–1.29 | 2.59E-08 | 5.23E-10 | MRPS31 | |
| 18q11.2 | rs9946154* | 22810619 | T | C | 0.6436 | 1.15 | 1.09–1.21 | 6.95E-07 | 1.50E-08 | ZNF521 | |
| 21q22.12 | rs55857134* | 36347627 | C | T | 0.3353 | 1.17 | 1.11–1.24 | 1.63E-08 | 1.93E-09 | RUNX1 | |
| 22q12.1 | rs17879961 | 29121087 | G | A | 0.0221 | 2.23 | 1.66–3 | 1.12E-07 | 3.60E-08 | CHEK2 | HSCB |
Novel associations are denoted with an asterisk after the SNP. Coordinates based on hg19 genome build. Alleles are on the + strand. Locus, chromosome band and locus; SNP, variant RSID identifier; bp, base position; risk, risk-increasing allele; non-risk, other allele; RAF, risk allele frequency; OR, odds ratio estimate for risk allele; OR 95CI, 95% confidence interval for OR; pvalue, discovery GWAS association p-value (two-tailed logistic regression); meta pval, joint discovery + replication association p-value (inverse-variance weighted meta-analysis); nearest gene, nearest gene to variant; other genes, additional genes located within 25 kb of the variant. SNPs marked with
indicate a novel locus.
Supplementary Material
Acknowledgements
We thank members of the Sankaran laboratory for valuable comments. We thank Wei Zhou for technical guidance on the implementation of SAIGE. We would like to thank the research participants and employees of 23andMe, UK Biobank, FinnGen, and Million Veteran Program. This research has been conducted using the UK Biobank Resource under application 31063. ELB received support from the Howard Hughes Medical Institute Medical Research Fellowship. This work was supported by the Claudia Adams Barr Program for Innovative Cancer Research, the New York Stem Cell Foundation, the MPN Research Foundation, the Leukemia & Lymphoma Society, and National Institute of Health Grant R01 DK103794 (VGS). VGS is a New York Stem Cell Foundation-Robertson Investigator.
Footnotes
Author disclosures
P.N. reports research grants from Amgen, Apple, and Boston Scientific, and is a scientific advisor to Apple and Blackstone Life Sciences, all unrelated to the present work. A.R. is a cofounder of and equity holder in Celsius Therapeutics, and a SAB member of ThermoFisher Scientific, Neogene Therapeutics, and Syros Pharmaceuticals. S.K. is an employee of Verve Therapeutics, and holds equity in Verve Therapeutics, Maze Therapeutics, Catabasis, and San Therapeutics. He is a member of the scientific advisory boards for Regeneron Genetics Center and Corvidia Therapeutics; he has served as a consultant for Acceleron, Eli Lilly, Novartis, Merck, Novo Nordisk, Novo Ventures, Ionis, Alnylam, Aegerion, Haug Partners, Noble Insights, Leerink Partners, Bayer Healthcare, Illumina, Color Genomics, MedGenome, Quest, and Medscape. The remainder of the authors report that they have nothing to disclose.
Data availability
Summary statistics for variants with fine-mapped posterior probability >0.1% from the full GWAS meta-analysis (UK Biobank, Finngen, 23andMe) are available in Supplementary Table 5. Full summary statistics from 23andMe data cannot be reported due to a clause in the 23andMe data transfer agreement, intended to protect the privacy of the 23andMe research participants. Thus, we provide full summary statistics for the MPN meta-analysis comprising UK Biobank and Finngen cohorts on GWAS Catalog under accession code GCST90000032 (https://www.ebi.ac.uk/gwas/downloads/summary-statistics). To fully recreate our meta-analysis results for MPN: researchers can (1) obtain MPN summary statistics from 23andMe (https://research.23andme.com/dataset-access/), and (2) conduct a meta-analysis of our summary statistics with the 23andMe summary statistics. For downloads of FinnGen summary statistics, information on how to access individual level FinnGen data by application to responsible agencies (FinBB and THL), and other collaborative access inquiries, please see https://www.finngen.fi/en/access. Individual genetic and phenotypic data for the following cohorts are available by application: UK Biobank (https://www.ukbiobank.ac.uk) and Million Veteran Program (https://www.research.va.gov/MVP/research.cfm).
Code availability
Code and source data required for reproducing results and figures discussed herein are available on GitHub (https://github.com/sankaranlab/mpn-gwas).
References
- 1.Sud A et al. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 132, 973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landgren O et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 112, 2199–2204, doi: 10.1182/blood-2008-03-143602 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer HR, Jones ME, Schoemaker MJ, Ashworth A & Swerdlow AJ Family history and risk of breast cancer: an analysis accounting for family structure. Breast cancer research and treatment 165, 193–200, doi: 10.1007/s10549-017-4325-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albright F et al. Prostate cancer risk prediction based on complete prostate cancer family history. The Prostate 75, 390–398, doi: 10.1002/pros.22925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johns LE & Houlston RS A systematic review and meta-analysis of familial colorectal cancer risk. American Journal Of Gastroenterology 96, 2992, doi: 10.1111/j.1572-0241.2001.04677.x (2001). [DOI] [PubMed] [Google Scholar]
- 6.Tapper W et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun 6, 6691, doi: 10.1038/ncomms7691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinds DA et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 128, 1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature Genetics 47, 291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nature Genetics 44, 369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones AV et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nature Genetics 41, 446 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakefield J A Bayesian Measure of the Probability of False Discovery in Genetic Epidemiology Studies. The American Journal of Human Genetics 81, 208–227, doi: 10.1086/519024 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulirsch JC et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nature Genetics 51, 683–693, doi: 10.1038/s41588-019-0362-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura M et al. Synchrony of telomere length among hematopoietic cells. Experimental hematology 38, 854–859, doi: 10.1016/j.exphem.2010.06.010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison SJ, Prowse KR, Ho P & Weissman IL Telomerase Activity in Hematopoietic Cells Is Associated with Self-Renewal Potential. Immunity 5, 207–216, doi: 10.1016/S1074-7613(00)80316-7 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi H et al. Mutations in TERT, the Gene for Telomerase Reverse Transcriptase, in Aplastic Anemia. New England Journal of Medicine 352, 1413–1424, doi: 10.1056/NEJMoa042980 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Li C et al. Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length. The American Journal of Human Genetics 106, 389–404, doi: 10.1016/j.ajhg.2020.02.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics 50, 1335–1341, doi: 10.1038/s41588-018-0184-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bick AG et al. Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. bioRxiv, 782748, doi: 10.1101/782748 (2019). [DOI] [Google Scholar]
- 19.Roth A et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Research 43, D447–D452, doi: 10.1093/nar/gku1003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrison BS et al. ZFP521 regulates murine hematopoietic stem cell function and facilitates MLL-AF9 leukemogenesis in mouse and human cells. Blood 130, 619–624, doi: 10.1182/blood-2016-09-738591 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodrigues NP et al. Haploinsufficiency of GATA2 perturbs adult hematopoietic stem-cell homeostasis. Blood 106, 477, doi: 10.1182/blood-2004-08-2989 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Kataoka K et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. The Journal of Experimental Medicine 208, 2403, doi: 10.1084/jem.20110447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tober J, Yzaguirre AD, Piwarzyk E & Speck NA Distinct temporal requirements for Runx1 in hematopoietic progenitors and stem cells. Development 140, 3765, doi: 10.1242/dev.094961 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabezas-Wallscheid N et al. Identification of Regulatory Networks in HSCs and Their Immediate Progeny via Integrated Proteome, Transcriptome, and DNA Methylome Analysis. Cell Stem Cell 15, 507–522, doi: 10.1016/j.stem.2014.07.005 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ito K et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Tothova Z et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 128, 325–339, doi: 10.1016/j.cell.2007.01.003 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Moran-Crusio K et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 20, 11–24, doi: 10.1016/j.ccr.2011.06.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akada H et al. Critical Role of Jak2 in the Maintenance and Function of Adult Hematopoietic Stem Cells. STEM CELLS 32, 1878–1889, doi: 10.1002/stem.1711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buza-Vidas N et al. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes & Development 20, 2018–2023 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seita J et al. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proceedings of the National Academy of Sciences 104, 2349, doi: 10.1073/pnas.0606238104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allsopp RC, Morin GB, DePinho R, Harley CB & Weissman IL Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 102, 517, doi: 10.1182/blood-2002-07-2334 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Cai Z, Chehab NH & Pavletich NP Structure and Activation Mechanism of the CHK2 DNA Damage Checkpoint Kinase. Molecular Cell 35, 818–829, doi: 10.1016/j.molcel.2009.09.007 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Falck J, Mailand N, Syljuåsen RG, Bartek J & Lukas J The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842–847, doi: 10.1038/35071124 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Zipin-Roitman A et al. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget 8, 16712–16727, doi: 10.18632/oncotarget.15147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khandanpour C et al. Evidence that Growth factor independence 1b regulates dormancy and peripheral blood mobilization of hematopoietic stem cells. Blood 116, 5149, doi: 10.1182/blood-2010-04-280305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polfus Linda M. et al. Whole-Exome Sequencing Identifies Loci Associated with Blood Cell Traits and Reveals a Role for Alternative GFI1B Splice Variants in Human Hematopoiesis. The American Journal of Human Genetics 99, 481–488, doi: 10.1016/j.ajhg.2016.06.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vassen L et al. Growth Factor Independence 1b (Gfi1b) Is Important for the Maturation of Erythroid Cells and the Regulation of Embryonic Globin Expression. PLOS ONE 9, e96636, doi: 10.1371/journal.pone.0096636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lundberg P et al. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. The Journal of Experimental Medicine 211, 2213–2230, doi: 10.1084/jem.20131371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mansier O et al. Description of a knock-in mouse model of JAK2V617F MPN emerging from a minority of mutated hematopoietic stem cells. Blood 134, 2383–2387, doi: 10.1182/blood.2019001163 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Musa J et al. Cooperation of cancer drivers with regulatory germline variants shapes clinical outcomes. Nature Communications 10, 4128, doi: 10.1038/s41467-019-12071-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson DJ et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657, doi: 10.1038/s41586-019-1765-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh P-R, Genovese G & McCarroll SA Monogenic and polygenic inheritance become instruments for clonal selection. Nature, doi: 10.1038/s41586-020-2430-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terao C et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature, doi: 10.1038/s41586-020-2426-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naucler P et al. Human Papillomavirus and Papanicolaou Tests to Screen for Cervical Cancer. New England Journal of Medicine 357, 1589–1597, doi: 10.1056/NEJMoa073204 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Løberg M et al. Long-Term Colorectal-Cancer Mortality after Adenoma Removal. New England Journal of Medicine 371, 799–807, doi: 10.1056/NEJMoa1315870 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Cimmino L et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095.e1020, doi: 10.1016/j.cell.2017.07.032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nature Medicine 25, 103–110, doi: 10.1038/s41591-018-0267-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agathocleous M et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209, doi: 10.1038/s41586-018-0579-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou W et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics 50, 1335–1341, doi: 10.1038/s41588-018-0184-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loh P-R et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics 47, 284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinds DA et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 128, 1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191, doi: 10.1093/bioinformatics/btq340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nature Genetics 44, 369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunter-Zinck H et al. Measuring genetic variation in the multi-ethnic Million Veteran Program (MVP). bioRxiv, 2020.2001.2006.896613, doi: 10.1101/2020.01.06.896613 (2020). [DOI] [Google Scholar]
- 56.Nielsen C, Birgens HS, Nordestgaard BG & Bojesen SE Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. British Journal of Haematology 160, 70–79, doi: 10.1111/bjh.12099 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Magosi LE, Goel A, Hopewell JC, Farrall M & on behalf of the, C. D. C. Identifying systematic heterogeneity patterns in genetic association meta-analysis studies. PLOS Genetics 13, e1006755, doi: 10.1371/journal.pgen.1006755 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, doi: 10.1186/s13742-015-0047-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wakefield J A Bayesian Measure of the Probability of False Discovery in Genetic Epidemiology Studies. The American Journal of Human Genetics 81, 208–227, doi: 10.1086/519024 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michailidou K et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sud A et al. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 132, 973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tapper W et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun 6, 6691, doi: 10.1038/ncomms7691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulirsch JC et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nature Genetics 51, 683–693, doi: 10.1038/s41588-019-0362-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nature Genetics 47, 1236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roaldsnes C, Holst R, Frederiksen H & Ghanima W Myeloproliferative neoplasms: trends in incidence, prevalence and survival in Norway. European Journal of Haematology 98, 85–93, doi: 10.1111/ejh.12788 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Höglund M, Sandin F & Simonsson B Epidemiology of chronic myeloid leukaemia: an update. Annals of Hematology 94, 241–247, doi: 10.1007/s00277-015-2314-2 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Li C et al. Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length. The American Journal of Human Genetics 106, 389–404, doi: 10.1016/j.ajhg.2020.02.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bick AG et al. Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. bioRxiv, 782748, doi: 10.1101/782748 (2019). [DOI] [Google Scholar]
- 69.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nature Genetics 50, 621–629, doi: 10.1038/s41588-018-0081-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501, doi: 10.1093/bioinformatics/btw018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walker CJ et al. Genome-wide association study identifies an acute myeloid leukemia susceptibility locus near BICRA. Leukemia 33, 771–775, doi: 10.1038/s41375-018-0281-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coetzee SG, Coetzee GA & Hazelett DJ motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849, doi: 10.1093/bioinformatics/btv470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kulakovskiy IV et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Research 46, D252–D259, doi: 10.1093/nar/gkx1106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hemani G et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408, doi: 10.7554/eLife.34408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verbanck M, Chen C-Y, Neale B & Do R Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature Genetics 50, 693–698, doi: 10.1038/s41588-018-0099-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanna S et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nature Genetics, doi: 10.1038/s41588-019-0350-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bowden J, Davey Smith G & Burgess S Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. International Journal of Epidemiology 44, 512–525, doi: 10.1093/ije/dyv080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaganathan K et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548.e524, doi: 10.1016/j.cell.2018.12.015 (2019). [DOI] [PubMed] [Google Scholar]
- 79.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biology 17, 122, doi: 10.1186/s13059-016-0974-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Javierre BM et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384.e1319, doi: 10.1016/j.cell.2016.09.037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research 47, D766–D773, doi: 10.1093/nar/gky955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Computational Biology 11, e1004219, doi: 10.1371/journal.pcbi.1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nature Communications 8, 1826, doi: 10.1038/s41467-017-01261-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grinfeld J et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. New England Journal of Medicine 379, 1416–1430, doi: 10.1056/NEJMoa1716614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Research 47, W191–W198, doi: 10.1093/nar/gkz369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pellin D et al. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nature Communications 10, 2395, doi: 10.1038/s41467-019-10291-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Dijk D et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e727, doi: 10.1016/j.cell.2018.05.061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Delano WL The PyMOL Molecular Graphics System. http://www.pymol.org (2002).
- 90.Zipin-Roitman A et al. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget 8, 16712–16727, doi: 10.18632/oncotarget.15147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Milyavsky M et al. A Distinctive DNA Damage Response in Human Hematopoietic Stem Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Cell Stem Cell 7, 186–197, doi: 10.1016/j.stem.2010.05.016 (2010). [DOI] [PubMed] [Google Scholar]
- 92.Piacibello W et al. Lentiviral gene transfer and ex vivo expansion of human primitive stem cells capable of primary, secondary, and tertiary multilineage repopulation in NOD/SCID mice. Blood 100, 4391, doi: 10.1182/blood.V100.13.4391 (2002). [DOI] [PubMed] [Google Scholar]
- 93.Cohen S et al. Hematopoietic stem cell transplantation using single UM171-expanded cord blood: a single-arm, phase 1–2 safety and feasibility study. The Lancet Haematology 7, e134–e145, doi: 10.1016/S2352-3026(19)30202-9 (2020). [DOI] [PubMed] [Google Scholar]
- 94.Fares I et al. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 345, 1509, doi: 10.1126/science.1256337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tomellini E et al. Integrin-α3 Is a Functional Marker of Ex Vivo Expanded Human Long-Term Hematopoietic Stem Cells. Cell Reports 28, 1063–1073.e1065, doi: 10.1016/j.celrep.2019.06.084 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.