

SUMMARY
Differentiated cells can re-enter the cell cycle to repair tissue damage via a series of discrete morphological and molecular stages coordinated by the cellular energetics regulator mTORC1. We previously proposed the term paligenosis to describe this conserved cellular regeneration program. Here we detail a molecular network regulating mTORC1 during paligenosis in both mouse pancreatic acinar and gastric chief cells. DDIT4 initially suppresses mTORC1 to induce autodegradation of differentiated cell components and damaged organelles. Later in paligenosis, IFRD1 suppresses p53 accumulation. Ifrd1−/− cells do not complete paligenosis because persistent p53 prevents mTORC1 reactivation and cell proliferation. Ddit4−/− cells never suppress mTORC1 and bypass the IFRD1 checkpoint on proliferation. Previous reports and our current data implicate DDIT4/IFRD1 in governing paligenosis in multiple organs and species. Thus, we propose that an evolutionarily conserved, dedicated molecular network has evolved to allow differentiated cells to re-enter the cell cycle (ie undergo paligenosis) after tissue injury.
Keywords: Regeneration, Spasmolytic Polypeptide Expressing Metaplasia (SPEM), Acinar-Ductal Metaplasia (ADM), Drosophila, Schizosaccharomyces pombe
Graphical Abstract

Blurb
During regeneration or tumorigenesis, differentiated cells use an evolutionarily conserved program (paligenosis) to reprogram their metabolism and readopt a progenitor state. Miao et al. identify dedicated paligenosis genes: DDIT4 suppresses mTORC1 to induce massive autophagy that dismantles differentiated cell architecture; then IFRD1 derepresses mTORC1, licensing the dedifferentiated cell to proliferate.
INTRODUCTION
As early as 1900, pathologists observed that adult tissue damage was often repaired by mature cells recruited back into the cell cycle(Adami, 1900). They speculated that this cellular “reprogramming” must involve cells switching from using energy for physiological function (like secretion) to fueling proliferation. We now know that the reversion to a proliferative, regenerative state can occur in various contexts, such as during regeneration of limbs in amphibians or when tissue transforms into the precancerous state known as metaplasia(Burclaff and Mills, 2018a). Metaplasia itself can be regenerative, fully restoring cellular architecture, or it can persist chronically, increasing risk for cancer(Saenz and Mills, 2018). In stomach, for example, Spasmolytic Polypeptide-Expressing Metaplasia (SPEM, aka pseudopyloric metaplasia) can be induced in a synchronous, reversible fashion in mice by drugs like high-dose tamoxifen(Huh et al., 2012; Keeley et al., 2019). In humans infected with the bacterium H pylori, SPEM can be chronic and increase risk for progression to cancer(Saenz and Mills, 2018). Similarly, the drug cerulein can induce metaplasia of mature digestive-enzyme-secreting pancreatic acinar cells in mice to model Acinar-to-Ductal Metaplasia (ADM). In humans, ADM can progress to pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer(Burclaff and Mills, 2018a).
We previously showed that injury-induced regeneration in various contexts and organs can occur via the same discrete series of cellular and molecular changes(Willet et al., 2018). We reasoned that – just as dying cells, independent of species and tissue type, can access the same apoptotic cellular program – differentiated cells that need to reenter the cell cycle to regenerate lost tissue would also have access to a conserved molecular-cellular program. We termed this program paligenosis and showed that it required biphasic regulation of the metabolic sensor and protein translation effector complex mTORC1(Willet et al., 2018; Mills et al., 2019). We showed that paligenosis proceeds by discrete, sequential stages: 1) mTORC1 is quenched as cells massively upregulate lysosomes and autophagy; 2) cells induce a cohort of embryonic/fetal progenitor genes (like Sox9); 3) mTORC1 is reactivated to power cell cycle reentry(Willet et al., 2018). Cells require lysosome activity to clear Stage 1. Without mTORC1, they are blocked in G1→S-phase and don’t enter Stage 3. We proposed that multicellular organisms evolved paligenosis to allow the vast majority of cells to be harnessed for physiological function during homeostasis without sacrificing them as a potential reservoir of potential progenitors to repair large-scale tissue damage.
Here we sought to identify a conserved molecular machinery that might govern mTORC1 during paligenosis. We expect paligenosis genes to be: a) conserved across species, b) induced upon paligenosis-causing injury in multiple tissues, and c) largely dispensable for normal development or stem cell homeostasis. We identify two highly conserved genes, DDIT4 and IFRD1, that are increased specifically following injury that induces paligenosis. We show in gastric chief and pancreatic acinar cells that DDIT4 causes the initial suppression of mTORC1 observed in paligenosis. p53 continues mTORC1 suppression as DDIT4 expression is lost. IFRD1 then suppresses p53 to relieve the mTORC1 blockade and allow cells to re-enter the cell cycle. Previously published data, coupled with our findings here, indicate that IFRD1 and DDIT4 function similarly in multiple organs and across animal phyla. The data are consistent with a model wherein differentiated cells have access to specific genes largely dedicated to governing cell cycle re-entry, conceptually similar to how they have genes (eg caspases, BCL-2 family) dedicated to executing cell death (eg caspases in apoptosis).
RESULTS
Induction of DDIT4 and IFRD1 in paligenosis and requirement for IFRD1 in paligenosis are conserved across tissue and phyla
To decode the molecular network regulating mTORC1 in paligenosis, we determined the intersection of a published set of mTOR-associated genes with sets of transcripts upregulated in mice in 4 organs we and others previously have shown undergo a paligenotic response(Willet et al., 2018) (Fig.1A; Table S1). Only two transcripts, Ifrd1 (encoding Interferon related developmental regulator 1) and Ddit4 (also called Redd1, encoding DNA Damage induced transcript 4) were at the intersection of all 5 gene sets. We next analyzed publicly available data from a published single-cell RNA-Seq analysis of the regenerative injury induced by amputation in the amphibian axolotl (Fig.1B)(Gerber et al., 2018). Just as in mouse paligenosis, the fraction of cells in the regenerating wound bed expressing DDIT4 and IFRD1 transcripts increased. DDIT4 increased slightly immediately after amputation and then decreased. The fraction of cells expressing IFRD1 increased after DDIT4 and continued to rise until the stages of regeneration characterized by maximal recruitment of mature cells back into the cell cycle (marked by expression of the cell cycle transcript AURKA).
Fig. 1. IFRD1 and DDIT4 are mTORC1-associated genes induced during paligenosis, and IFRD1 is required for paligenosis in multiple tissues and species.
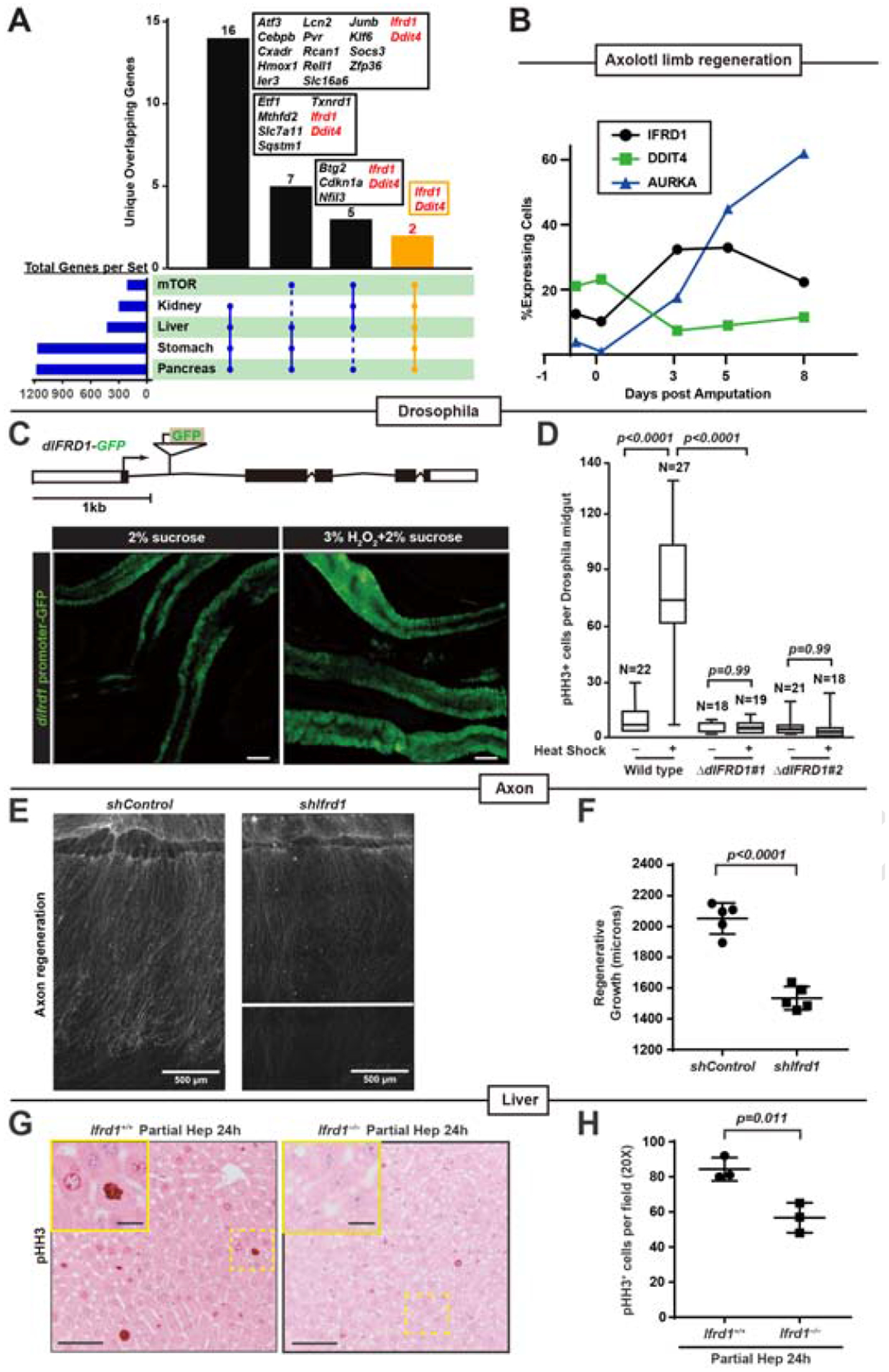
A. Intersection of transcripts upregulated by paligenotic injury in 4 organs with genes in the HALLMARK_MTORC1_SIGNALING Gene Set from Broad GSEA. Only intersections of gene sets ≥4 and transcripts unique for that intersection intersections are depicted.
B. Fraction of cells expressing DDIT4 and IFRD1 in previously published single-cell RNA-sequencing of regenerating cells after axolotl limb amputation. AURKA is representative of a cohort of cell cycle transcripts upregulated at ≈ paligenosis Stage 3.
C. GFP under regulation of difrd1 promoter in Drosophila melanogaster intestine at baseline and after stem-cell recruiting oxidative stress. Scale bar, 100μm. Above: genomic map of GFP P-element insertion in difrd1 locus.
D. Total phospho-Histone H3+ (pHH3+) cells of Wild type untreated (n=22), Wild type with heat shock (n=27), ΔdIFRD1 #1 untreated (n=18), ΔdIFRD1 #1 with heat shock (n=19), ΔdIFRD1 #2 untreated (n=21) and ΔdIFRD1 #2 with heat shock (n=18) Drosophila mid-gut. ΔdIFRD1 #1 and #2 are difrd1 hypomorphic alleles. Variance plotted as mean±SD with significance estimated by one-way ANOVA with Dunnett post-hoc test to the Ifrd+/+ control.
E. Regenerating mouse dorsal root ganglion neurites visualized by anti-SCG10 following axotomy ex vivo of control and shRNA knockdown of Ifrd1 (shIfrd1). Panels are divided at maximal extension of neurite regeneration. Scale bar, 500μm.
F. Axon regeneration quantified following axotomy from Ifrd1+/+ (n=5) and Ifrd1–/– (n=5) mice. Each datapoint: Mean±SEM maximal axon regeneration length for an individual culture from an individual mouse with significance estimated by unpaired, two-tailed t-test.
G. Immunohistochemical staining for mitotic marker pHH3 in regenerating liver 24 hours following partial resection (“Partial Hep”). Scale bar, 50μm; boxed area insert, 25μm.
H. Quantification of data as for panel (G) from Ifrd1+/+ (n=3) and Ifrd1–/– mice (n=3). Each datapoint: Mean±SEM counts of ≥10 fields in a single mouse with significance estimated by unpaired, two-tailed t-test.
DDIT4 has been previously characterized as an injury-activated scaffold that suppresses mTORC1 activity via induction of the TSC1/TSC2 complex from Drosophila to humans.(DeYoung et al., 2008; Kimball et al., 2008) However, IFRD1 conservation has not been reported. We found that nearly the entire IFRD1 protein is conserved from the fission yeast Schizosacchromyces pombe to humans (Fig.S1A,B) with similar likely secondary structure involving extensive alpha-helical character into an armadillo (or alpha-solenoid) fold (Fig.S1A,C), similar to the non-injury-induced paralog IFRD2(Brown et al., 2018). The yeast orthologue of IFRD1 has not been specifically characterized, but various transcriptomic and mutant screens indicate it is injury-induced and not required for vegetative growth(Lock et al., 2019). A recent paper similarly identified the dictyostelium Ifrd1 ortholog as mediating dedifferentiation from the multicellular slime mold to unicellular state (Nichols et al., 2020).
Likewise, the Drosophila melanogaster orthologue of Ifrd1, CG31694, had not been specifically characterized but had been shown to be rapidly induced following injuries that induce proliferation in the gut(Vodovar et al., 2005; Bou Sleiman et al., 2015). We confirmed that difrd1 increased following intestinal stem-cell-recruiting stress using GFP driven by the difrd1 promoter (Fig.1C). Various injuries induce Drosophila enterocytes to reenter the cell cycle to help fuel proliferative repair. Two hypomorphic difrd1 Drosophila pedigrees showed no homeostatic abnormalities but complete loss of proliferation in response to injury (Fig.1D), consistent with requirement for dIFRD1 in paligenosis.
We also surveyed the requirement for IFRD1 in regeneration after injury in various other mouse tissues. In nerves following axotomy, a model of regeneration that resembles paligenosis in that neurons dedifferentiate to regenerate axons(Vercellino et al., 2007; Stierli et al., 2018), regeneration was markedly retarded by knockdown of Ifrd1 (Fig.1E,F). Similarly, Ifrd1−/− livers showed nearly two-fold reduction in paligenotic proliferation post-partial hepatectomy (Fig.1G,H). Thus, evidence points to DDIT4 and IFRD1 as injury-induced genes that may have an evolutionarily conserved role in paligenosis.
IFRD1 and DDIT4 are increased in gastric and pancreatic paligenosis, with IFRD1 dispensable for early stages
Of the 4 organs in Fig.1A, the stomach is unique because injury can cause both increased proliferation of the constitutively active adult progenitor cells in the isthmus and neck of the gland (nearer the gastric lumen), while also recruiting the normally mitotically quiescent digestive-enzyme-secreting chief cells in the base of the gland as progenitors via paligenosis(Stange et al., 2013; Radyk et al., 2018; Saenz and Mills, 2018; Willet et al., 2018). Consistent with a role specifically in paligenosis, after injury with regeneration-inducing doses (250 mg/kg) of tamoxifen (hereafter, “TAM”)(Huh et al., 2012; Burclaff et al., 2017; Keeley et al., 2019), Ifrd1 was increased in the chief cells but not other cells in the epithelium (Fig.2A,B). Induction of Ifrd1 specifically within chief cells was substantial enough to be detectable via western blot and qRT-PCR by 12 and 48h. DDIT4/Ddit4 increased more modestly, peaking at 12h and dropping below baseline by 24h onward (Fig.2C,D). The pattern of earlier, modest Ddit4 increase with later, more dramatic Ifrd1 induction, was similar to that seen during axolotl regeneration.
Fig. 2. IFRD1 and DDIT4 are induced in murine gastric and pancreatic paligenosis; IFRD1 is required cell autonomously.

A. Expression of lacZ knocked into the null allele of Ifrd1+/− mice after TAM showing induction of expression in basal chief cells early in paligenosis. Scale bar, 50μm.
B. RNAScope™ shows Ifrd1 mRNA in chief cells after TAM. Scale bar; inset highlights two red puncta from boxed area; arrows indicate other puncta, Scale bar 50μm.
C. Western blot for DDIT4 and IFRD1 and mTORC1 proxy pS6 240/244, tubulin as loading control.
D. qRT-PCR for Ifrd1 and Ddit4. Each datapoint represents mean±SEM from n=3 mice cohorts.
E. Proliferation measured by BrdU 72h after TAM. Scale bar, 50μm.
F. Data as in panel (E) quantified, focusing on the basal 100 μm gastric of Ifrd1+/+ (n=6) and Ifrd1–/– (n=6) mice gastric unit. Datapoint: Mean±SEM of ≥40 glands quantified per mouse with significance estimated by unpaired, two-tailed t-test.
G. As per panel (F) but for SOX9+ progenitor cells of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice.
H. Organoids from stomach body at various days after isolation. Scale bar, 100μm. Initial outgrowth of wildtype organoids occurs throughout the gastric unit, whereas the bases (arrow) of Ifrd1−/− mice do not expand, slowing overall organoid growth
I. Data as in panel (H), quantified for organoid diameter after 5 day in culture of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice. Datapoint: Mean±SEM of ≥100 gastroids per mouse with significance estimated by unpaired, two-tailed t-test.
J. BrdU (green) identifying proliferating acinar cells 5 days post cerulein-injury-induced paligenosis (acinar cells labeled by digestive enzyme amylase, red). Yellow arrows=representative paligenotic cells; white arrows=non-epithelial cells. Scale bar, 50μm
K. Paligenotic (ie amylase-positive) proliferative (ie BrdU+) cells of Ifrd1+/+ (n=3) and Ifrd1–/– (n=3) mice from panel (J) are quantified. Datapoint: Mean±SEM of ≥10 random fields per mouse with significance estimated by unpaired, two-tailed Student’s t-test.
L. As per panel (K) but with quantification of SOX9+ progenitor cells of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice.
We first focus on IFRD1 following gastric and pancreatic injury. Ifrd1−/− stomachs were defective in paligenosis, with markedly decreased proliferation induction (Fig.2E,F; Fig.S2A). To confirm paligenosis-specificity of IFRD1, we killed acid-secreting parietal cells in HK-iDTR;Ifrd1−/− mice with diphtheria toxin, an injury that induces proliferation of the existing stem and progenitor cells in the neck of the gastric unit but not paligenosis in the chief cells in the base of the unit(Burclaff et al., 2017). Activation of the constitutive progenitors was not affected by loss of IFRD1 (Fig.S2B,C), furthering the interpretation that IFRD1 is required only for chief cells to proliferate by paligenotic induction of proliferation.
We have previously shown that paligenosis occurs via 3 Stages: 1) massive activation of autophagy and lysosomal activity as mTORC1 is extinguished; 2) re-expression of progenitor or embryonic markers (eg SOX9 or the epitope for the lectin GS-II, normally found exclusively in progenitor cells of the isthmus and neck zones); 3) induction of high-level mTORC1 with cell cycle entry. Chief cells not only return to the cell cycle during paligenosis but return to a progenitor-like phenotype(Nam et al., 2010; Stange et al., 2013; Weis et al., 2017; Willet et al., 2018; Burclaff et al., 2019). Induction of lysosomal activity still occurred in Ifrd1−/− mice at 24h post-TAM (Fig.S2D,E), and SOX9 was still induced at later timepoints (Fig.S2F), though overall census of SOX9+, paligenotic cells was reduced by 72h (Fig.2G). The reduction of SOX9+ cells was roughly equivalent to the overall loss of paligenotic cells in Ifrd1−/− mice (eg, Fig.S2A), so we interpret the results as indicating IFRD1 is largely dispensable for Stages 1 and 2.
We next determined if IFRD1 was required in a cell-autonomous way within chief cells by growing organoids from stomach bodies, which are established from the epithelium, initially from both normal epithelial progenitors and proliferation of paligenotic chief cells(Stange et al., 2013; Moore et al., 2015). As expected, organoids generated from Ifrd1−/− mice were delayed in their outgrowth (Fig.2H,I), consistent with IFRD1 being specifically required for paligenotic growth intrinsically within chief cells. Thus, our results collectively showed: chief-cell-specific Ifrd1 expression (Fig.2A,B) induced during paligenosis; lack of phenotype in injured HK-iDTR;Ifrd1−/− mice in which chief cell paligenosis does not occur (Fig.S2B,C); and slower outgrowth from epithelial organoids that depend in part on proliferation from paligenotic chief cells.
In pancreas, as in stomach, proliferating, paligenotic acinar cells induced by cerulein injury were greatly reduced in Ifrd1−/− mice, while proliferating cells in the stroma were largely unaffected (Fig.2J,K, Fig.S3A). Likewise, Stage 1 autophagic induction and Stage 2 SOX9 induction still occurred (Fig.S3B,C). As in stomach, SOX9+ cells overall were reduced (Fig.2L; Fig.S3C). However, in Ifrd1−/− mice at peak paligenosis (72h), 35±2% of gastric units showed cell loss in gland bases vs. only 7.0±2.6% of control units (n=3–4 mice per genotype, p=0.0004 by two-tailed student’s t-test; see also Fig.S2A), so loss of SOX9+ cells correlates with overall cell death in the absence of IFRD1 (as we observed previously in stomach). Thus, overall, in pancreas and stomach, IFRD1 is largely required for the final, proliferative/regenerative stage of paligenosis.
IFRD1 loss causes p53 stabilization and failed mTORC1 reinduction in Stage 3 of paligenosis
The proliferative/regenerative Stage 3 of paligenosis requires the reinduction of mTORC1 activity to drive paligenotic cells into S-phase of the cell cycle(Willet et al., 2018). Ifrd1−/− stomachs showed the usual induction of mTORC1 activity in non-paligenotic isthmal/neck progenitor cells (Fig.3A), as measured by expression of phosphorylated S6 ribosomal protein (pS6), a critical downstream target of mTORC1 (which we have previously vetted as a proxy for mTORC1 activity in tissue)(Willet et al., 2018). In contrast, even in regions of more limited cell loss at d3 post TAM, there was marked reduction in paligenotic reinduction of mTORC1 in the absence of IFRD1 (Fig.3A). p53 suppresses mTORC1 activity by various routes(Hasty et al., 2013; Akeno et al., 2015; Deng et al., 2016; Liu et al., 2019a) and thus could be a key mechanism for mTORC1 suppression and failed paligenotic proliferation as well as potentially contribute to the increased cell loss seen in Ifrd1−/− mice. Accordingly, we observed increased p53 in the base of Ifrd1−/− gastric units (Fig.3B,C).
Fig. 3. Loss of IFRD1 causes p53 stabilization and failed mTORC1 reinduction in Stage 3 of paligenosis.
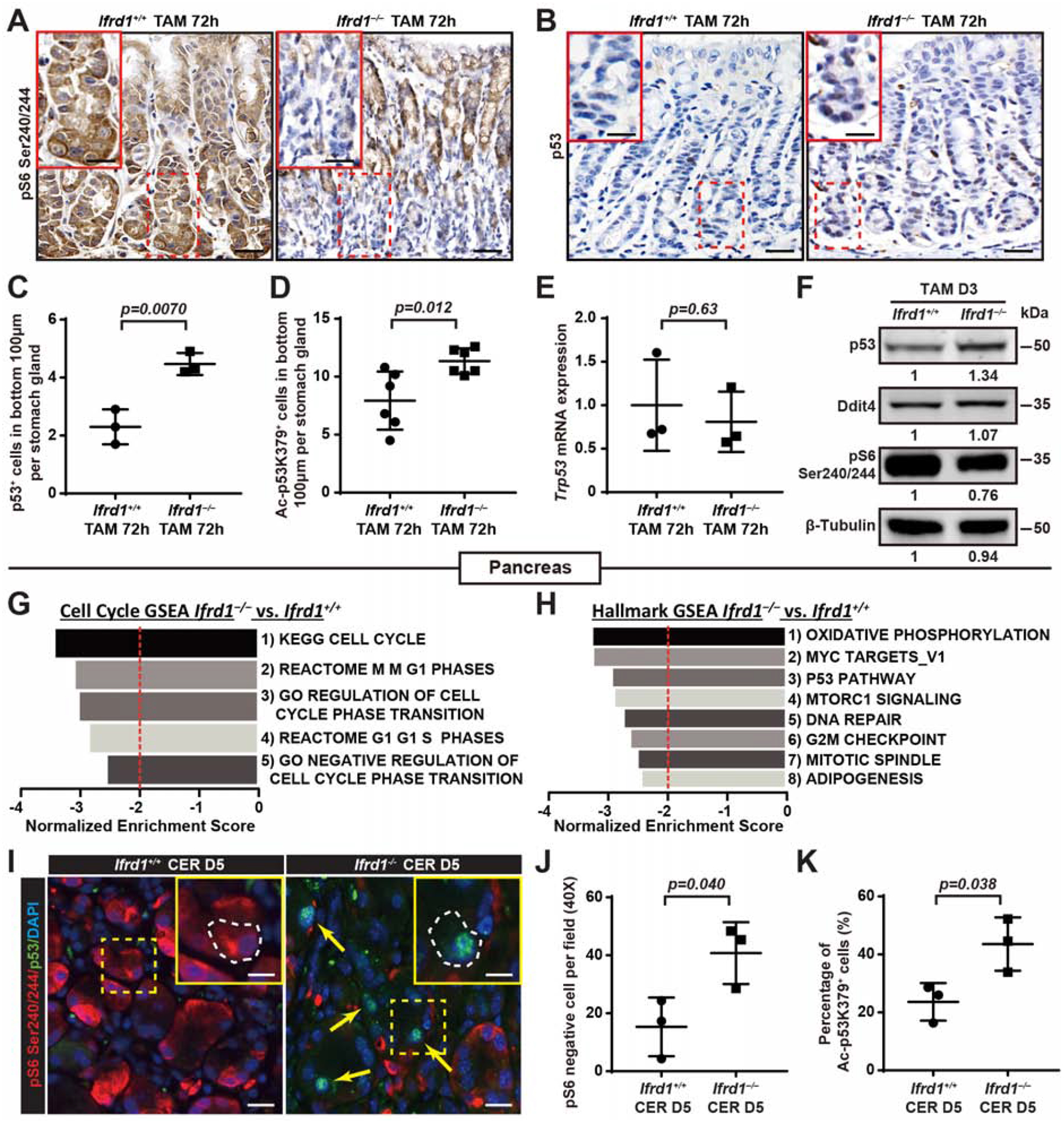
A. Anti-S6 phosphorylated on serines 240/244 (mTORC1 activity proxy) showing most surviving, paligenotic (ie basal) Ifrd1−/− cells are pS6-negative. Non-paligenotic cells higher in the unit are unaffected by IFRD1 loss. Scale bar, 50 μm; boxed area insert, 25 μm.
B. As for panel (A) with anti-p53. Scale bar, 50μm; boxed area insert, 25μm.
C. Data as for panel (B) quantified but focusing on the basal 100 μm of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice, where paligenosis occurs. Datapoint: Mean±SEM of ≥40 glands quantified per mouse with significance estimated by unpaired, two-tailed t-test.
D. As per panel (C) but quantifying anti-p53 acetylated on lysine residue 379 (Ac-p53K379) of Ifrd1+/+ (n=6) and Ifrd1–/–(n=6) mice.
E. Quantitative real-time PCR of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice stomach bodies 72h after TAM injury. Datapoint: Mean±SEM of 3 technical replicates per mouse, significance by unpaired, two-tailed t-test.
F. Western blot for mouse stomach body extracts 72h after TAM. Tubulin as loading control. Numbers under bands indicate relative expression of null to control.
G. Gene Set Enrichment Analysis (GSEA) of global mRNA expression profiling from whole pancreas using cell cycle-related gene sets from Broad Institute. All statistically significant cell cycle gene sets (FDR and p-value <0.001) exhibiting a Normalized Enrichment Score (NES) ≤−2.0 (dashed red line) in Ifrd1−/− vs. Ifrd+/+ mice are depicted.
H. Unbiased GSEA using all gene sets in Broad “Hallmark” compilation. All statistically significant gene sets with NES ≤−2.0 Ifrd1−/− vs. Ifrd+/+ mice depicted.
I. phospho-S6 (red) and p53 (green) after 5 days of cerulein. Arrows highlight representative p53-positive/pS6-negative cells. Scale bar, 50μm; boxed area insert, 25μm. Note Ifrd1−/− mice have increased nuclear p53 and decreased pS6, individual p53+ cells lack pS6.
J. pS6-negative cells of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice from experiments such as in panel (I) quantified by HALO™ automated cell counting and image-normalizing software. Datapoint: Mean±SEM of ≥10 random fields quantified per mouse with significance estimated by unpaired, two-tailed t-test.
K. As per panel (J) but with Acetylated-p53 K379+ cells of Ifrd1+/+ (n=3) and Ifrd1–/–(n=3) mice as a fraction of total paligenotic cells.
IFRD1 is thought to act as a scaffold that helps coordinate deacetylation of lysines of various proteins(Micheli et al., 2011; Tummers et al., 2015). IFRD1 effects on p53 have not been previously shown, yet it is known that acetylation promotes p53 transcriptional activity and blocks its degradation(Tang et al., 2008; Wang et al., 2016; Yun et al., 2016; Liu et al., 2019b). Ifrd1−/− mice exhibited increased expression of p53 acetylated on lysine 379 in paligenotic cells (Fig.3D, Fig.S2G). The effects of IFRD1 were, as expected, at the post-translational level, as Trp53 mRNA levels were not affected by loss of IFRD1 (Fig.3E). The increased p53 protein in Ifrd1−/− mice correlated in western blots with the immunohistochemical results, with decreased phosphorylation of S6 kinase target: serine residues 240 and 244 of S6 ribosomal protein (Fig.3F) (Hutchinson et al., 2011). Note that p53 and pS6 are all also expressed in non-paligenotic cells (ie in cells that don’t express IFRD1), so differences on western blot of the entire body of the stomach are not as dramatic as changes in expression detected by immunohistochemistry specifically in paligenotic cells. DDIT4 protein was slightly increased by loss of IFRD1 at d3 (Fig.3F; Fig.S2H).
Results in the pancreas were similar. Gene Set Enrichment Analysis (GSEA) of Ifrd1−/− mice ±cerulein at d5 showed substantial de-enrichment of multiple proliferation-associated gene sets in Ifrd1−/− mice (Fig.3G Fig.S3G), consistent with IFRD1 requirement for Stage 3 of paligenosis. Unbiased analysis of expression patterns using the entire Broad Institute “Hallmark” collection of GSEA datasets (a compendium of gene sets designed for unbiased screens(Subramanian et al., 2005)) further highlighted de-enrichment of cell cycle-related gene sets and highlighted p53 and mTORC1 alterations (Fig.3H; Fig.S3H). Moreover, as in stomach, paligenotic acinar cells at d5 showed increased nuclear p53 and decreased pS6, and we noted that individual p53+ cells generally lacked pS6 expression, indicating p53 and mTORC1 activity were mutually exclusive during paligenosis as predicted (Fig.3I,J; Fig.S3D,E). As in stomach, K379-acetylated p53 increased in paligenotic acinar cells (Fig.3K; Fig.S3F).
We further confirmed the role of IFRD1 in p53 stability by inducing IFRD1 via tunicamycin stress(Zhao et al., 2010) in p53-competent, intestinal L174 cells in tissue culture. As IFRD1 was induced, p53 acetylated on K382 (the human equivalent of mouse K379) decreased. When IFRD1 was stably knocked down, acetylated-p53 was elevated at both baseline and under stress (Fig.S3I). Thus, overall the results are consistent with IFRD1 acting to destabilize p53.
p53 is required for suppression of mTORC1 after Stage 1 of paligenosis
To further dissect the relationship between p53 and mTORC1, we induced paligenosis in Trp53−/− mice with TAM (Fig.S4A). Induction of autophagy in Stage 1 could still be seen at 24h (Fig.S4B,C) in Trp53−/− mice; however, as predicted based on the known role of p53 as a suppressor of mTORC1(Hasty et al., 2013; Akeno et al., 2015; Deng et al., 2016), the dynamics of mTORC1 suppression were aberrant (Fig.4A,B). mTORC1 was extinguished in both Trp53−/− and control paligenotic cells at 6h; however, while it remained diminished in control cells at 12h, it was already increasing in the absence of p53 (Fig.4A–C). The more rapid reinduction of mTORC1 correlated predictably with an earlier and more robust increase in paligenotic proliferation, starting by 24h and continuing through peak paligenosis at 72h (Fig.4D; Fig.S4D). Loss of p53 stimulated re-entry into all stages of the cell cycle, as the number of chief cells (labeled via gastric intrinsic factor, GIF) remaining quiescent (ie remaining negative for the cell-cycle marker Ki-67) was dramatically reduced in Trp53−/− mice (Fig.4E,F). Trp53−/− stomachs showed the expected decrease in the p53-induced, cell-cycle suppressor p21 and increase in the cell cycle proteins Cyclins D1 (Fig.4G). Gastroids generated from Trp53−/− mice also showed increased outgrowth (Fig.4H,I), consistent with an epithelial cell-autonomous mechanism.
Fig. 4. p53 is required to delay mTORC1 reactivation during Stages 2–3 of paligenosis.

A. Western blot showing pS6 240/244 (ie mTORC1 activity) is restored earlier (at 12h) in Trp53−/− mouse vs. control.
B. Quantification of pS6 240/244 vs. tubulin intensity from the blot in panel (A) from n=1 mouse cohort.
C. Anti-pS6 240/244 with insets highlighting paligenotic cells in the base showing that pS6 is lost in basal cells in both genotypes but reappears prematurely in Trp53−/− cells by12h. Because non-paligenotic cells also express pS6, pS6 western blots in panel (A) do not show complete loss of pS6. Scale bar, 50 μm; boxed area insert, 25 μm.
D. Quantification of BrdU-positive (ie proliferative) cells from Trp53+/+ (TAM 24h, n=3; TAM 48h, n=3; TAM 72h, n=3) and Trp53−/− (TAM 24h, n=3; TAM 48h, n=3; TAM 72h, n=3) mice shows cell proliferation increases in Trp53−/− mice as early as 24h. Datapoint: Mean±SEM of ≥40 glands quantified per mouse with significance estimated by unpaired, two-tailed t-test.
E. Cell cycle marker Ki67 (red) and chief cell lineage marker GIF (green) shows decreased Ki67−/GIF+ cells (ie chief cells that are non-paligenotic in Trp53−/− mice. Scale bar, 50 μm.
F. Non-paligenotic cells from Trp53+/+ (n=3) and Trp53−/− (n=3) mice as in panel (E) were quantified. Datapoint: Mean±SEM of ≥40 glands quantified per mouse with significance estimated by unpaired, two-tailed t-test.
G. Western blots from stomach body for p53 activated target proliferation-suppressing p21 and p53 repressed target proliferation-promoting Cyclin D1.
H. Organoids at day 6 after culturing from Trp53−/− and Trp53+/+ mice. Scale bar, 100μm.
I. Data as in panel (H) from Trp53+/+ (n=3) and Trp53−/− (n=3) mice, quantified for organoid diameter. Datapoint: Mean±SEM of ≥100 gastroids per mouse with significance estimated by unpaired, two-tailed t-test.
J. Anti-BrdU pancreas 5d after cerulein injury showing markedly increased proliferation of Trp53−/− acinar cell paligenosis. Scale bar, 50 μm; boxed area insert, 25 μm.
K. BrdU-positive cells as in panel (J) from Trp53+/+ (n=3) and Trp53−/− (n=3) mice were quantified. Datapoint: Mean±SEM of ≥10 random fields quantified per mouse with significance estimated by unpaired, two-tailed Student’s t-test.
L. As per panel (K) but quantifying p21+ cell-cycle-arrested cells of Trp53+/+ (n=3) and Trp53−/− (n=3) mice.
Results in pancreas were equivalent with significantly increased BrdU+ and reduced p21+ cells by d5 after cerulein (Fig.4J–L; Fig.S4E,F). Overall, the results show that loss of p53 leads to briefer mTORC1 diminishment that did not substantially affect induction of autodegradation of mature cell architecture but did lead to premature mTORC1 reinduction causing aberrant mTORC1-reactivation-mediated transition from Stage 2 to Stage 3 of paligenosis.
Previously, we showed mTORC1 inhibition with rapamycin during TAM treatment specifically blocks Stage 3 cell cycle entry(Willet et al., 2018). If the principal reason for the increased paligenotic proliferation in Trp53−/− mice were due to dysregulated mTORC1 at that stage, then rapamycin should render the Trp53−/−phenotype equivalent to that of rapamycin-treated wildtype mice (Fig.S5A). Accordingly, rapamycin caused near complete loss of pS6 in Trp53−/− and wildtype stomachs following TAM (Fig.S5B). Consistent with our hypothesis, it also blocked not only paligenotic S-phase entry in wildtype mice (confirming previous results) but also in Trp53−/− mice (Fig.S5C,D). We previously showed that rapamycin inhibits progression specifically from G1- to S-phase(Willet et al., 2018). Consistent with that observation, rapamycin did not significantly affect Ki-67-positive cells (which include G1-phase cells) in wildtype mice (Fig.S5E,F). Trp53−/− mice, however, showed increased Ki-67+ cell census (Fig.S5E,F), consistent with the increased early mTORC1 in Trp53−/− mice affecting not just S-phase entry in Stage 3 but also causing more efficient paligenotic recruitment of cells overall into all phases of the cell cycle. This increased efficiency of paligenosis was also mTORC1-dependent, as it was corrected by rapamycin.
Because IFRD1 works to destabilize p53 in the Stage 2 to 3 transition, the Ifrd1−/− phenotype should be corrected by loss of p53. Accordingly, Ifrd1−/−;Trp53−/− stomachs (Fig.S6A–C) showed a phenotype statistically indistinguishable from that of wildtype in terms of: paligenotic induction of proliferating progenitors (Fig.5A–F; Fig.S6A) and mTORC1 activity at 72h (Fig.S6B). p21 expression was even lower than in wildtype (Fig.5G; Fig.S6D). The pancreas showed similar rescue of mTORC1 activity, and suppression of p21 (Fig.5H–K; Fig.S6E–H). Summing the above results: p53 works to maintain mTORC1 suppression after the initial decrease in Stage 1 of paligenosis, so when IFRD1 destabilizes p53, the mTORC1 blockade is removed so that cells can enter the cell cycle in Stage 3.
Fig. 5. Loss of p53 rescues the Ifrd1−/− phenotype, indicating IFRD1 destabilizes p53.
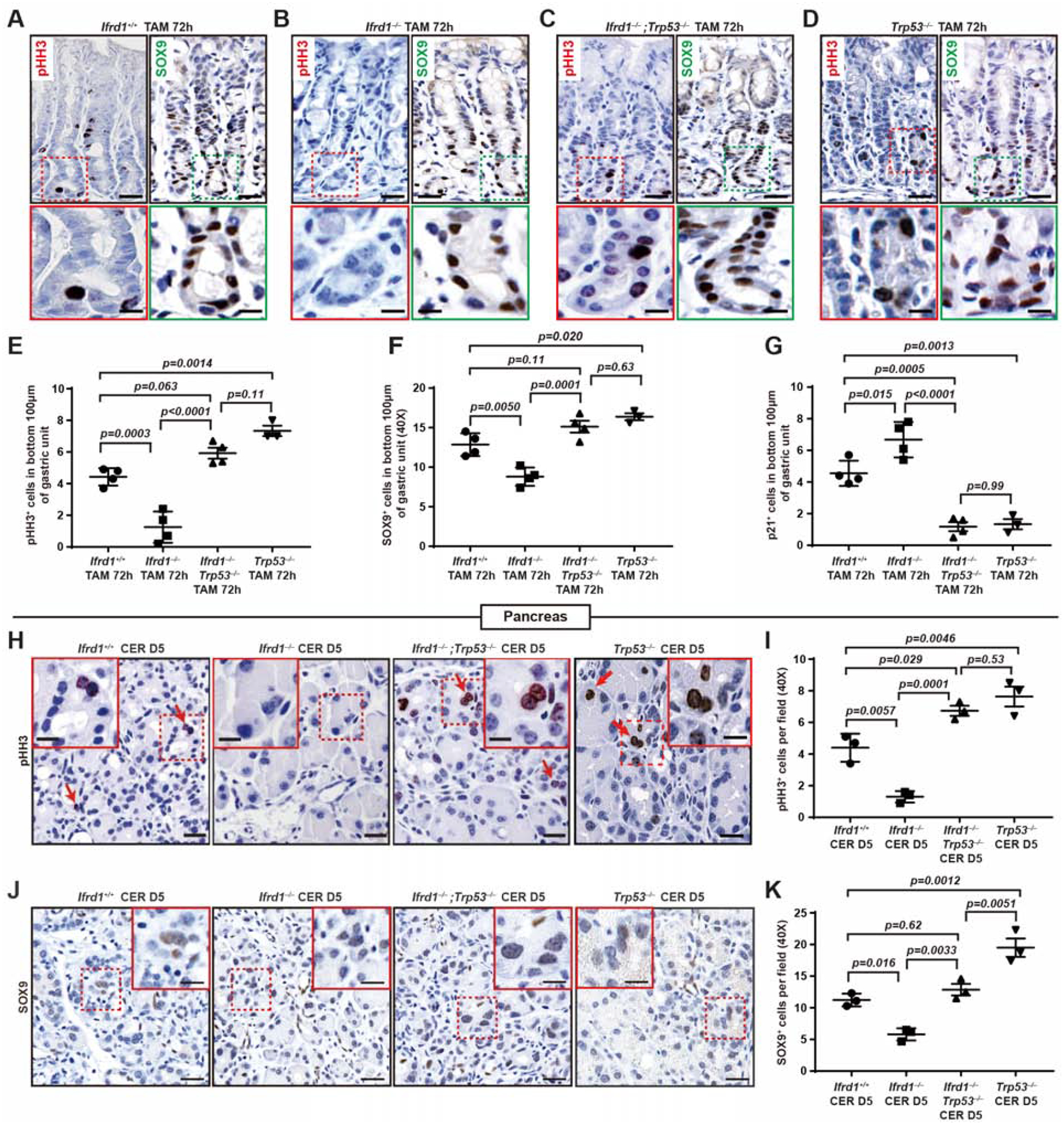
A. Anti-phospho-histone H3 (pHH3), marking mitotic cells and SOX9 (progenitor cells) in control mice, TAM day 3; bottom: higher magnification of bases where paligenosis occurs. Scale bar, 30 μm; boxed area insert, 15 μm
B. As for panel (A) but with Ifrd1–/– mice.
C. As for panel (A) but with Ifrd1−/−;Trp53−/− mice.
D. As for panel (A) but with Trp53−/− mice
E. pHH3 quantified from paligenotic bases of Ifrd1+/+ (n=4), Ifrd1–/– (n=4), Ifrd1−/−;Trp53−/− (n=4) and Trp53−/− (n=3) mice as in bottoms of panels (A-D). Datapoint: Mean±SEM of ≥40 glands quantified per mouse with significance estimated by one-way ANOVA with multiple comparisons to Ifrd+/+
control (Dunnett post-hoc test).
F. As per panel (E) but quantifying SOX9+ progenitor cells of Ifrd1+/+ (n=4), Ifrd1–/– (n=4), Ifrd1−/−;Trp53−/− (n=4) and Trp53−/− (n=3) mice induced in the base.
G. As per panel (E) but quantifying p21+ cell-cycle-arrested cells of Ifrd1+/+ (n=4), Ifrd1–/– (n=4), Ifrd1−/−;Trp53−/− (n=4) and Trp53−/− (n=3) mice induced in the base.
H. Anti-pHH3 in Ifrd+/+, Ifrd1−/−, Ifrd1−/−;Trp53−/− and Trp53−/− mice pancreases 5 days after cerulein. Scale bar, 50 μm; boxed area insert, 25 μm.
I. pHH3 quantified as from panel (H) of Ifrd1+/+ (n=3), Ifrd1–/– (n=3), Ifrd1−/−;Trp53−/− (n=3) and Trp53−/− (n=3) mice. Datapoint: Mean±SEM of ≥10 random fields quantified per mouse with significance estimated by one-way ANOVA with multiple comparisons to the Ifrd+/+ control (Dunnett post-hoc test).
J. Anti-SOX9 in Ifrd+/+, Ifrd1−/−, Ifrd1−/−;Trp53−/− and Trp53−/− mice pancreases 5 days after cerulein. Scale bar, 50 μm; boxed area insert, 25 μm.
K. As per panel (I) but for SOX9+ progenitor cells of Ifrd1+/+ (n=3), Ifrd1–/– (n=3), Ifrd1−/−;Trp53−/− (n=3) and Trp53−/− (n=3) mice.
The initial suppression of mTORC1 in paligenosis Stage 1 depends on DDIT4
The results so far explain the molecular machinery dictating the transition from Stage 2 through Stage 3. However, neither loss of IFRD1 nor p53 prevented the initial decrease in mTORC1 and the massive induction of autophagy and lysosomes seen in Stage 1. We reasoned DDIT4, as a known suppressor of mTORC1 and the other gene identified by our initial screen (Fig.1A), could be the key gene to initiate Stage 1.
In accordance with DDIT4 being required for mTORC1 suppression early in paligenosis, DDIT4 was expressed only in chief cells and only early in paligenosis, coinciding with the nadir (4–12h, Stage 1) of mTORC1 in TAM-treated mouse stomachs (Fig.6A,B). Conversely, DDIT4 was lost as paligenosis progressed to the proliferative Stage 3 (48h-72h) when mTORC1 was reinduced to suprabasal levels (Fig.2C; Fig.6A,B). The loss of mTORC1 that occurs specifically in paligenotic chief cells by 12h after TAM was substantial enough to result in consistent decrease of 57±7% in western blots of the entire stomach body (Fig.6C,D). In contrast, mTORC1 in Ddit4−/− mice was nearly unchanged or increased at all timepoints by western blot (Fig.6C,D), and immunohistochemistry showed that mTORC1 at 12h persisted aberrantly specifically in the paligenotic cells in Ddit4−/− stomachs and in pancreas at the 24h timepoint after cerulein, when mTORC1 is lowest in pancreas (Willet et al., 2018) (Fig.6B,L). Thus, the results indicate DDIT4 is activated early in paligenosis to perform the initial suppression of mTORC1 that is later maintained by p53.
Fig. 6. The initial suppression of mTORC1 and autophagic/lysosomal recycling of cell architecture in paligenosis Stage 1 depends on DDIT4.

A. DDIT4 (green) is confined to gastric chief cells, increasing in Stage 1 (4–24h) then lost later. GSII (red, a progenitor marker) labels Stage 2–3 paligenotic chief cells at base of gland and normal progenitor cells higher in the gland. As paligenotic cells become GSII+, they lose DDIT4, indicating DDIT4 is in homeostatic chief cells, increased in Stage 1, and lost thereafter. Blue=nuclei. Scale bar, 30 μm; boxed area insert highlighting base, 15 μm.
B. Anti-pS6 240/244 showing that at mid-Stage 1 (12h), Ddit4−/− paligenotic cells fail to extinguish mTORC1. Scale bar, 30 μm; boxed area insert highlighting base, 15 μm.
C. Representative western blot for pS6 240/244 showing decreased mTORC1 in Stage 1 (12h) only in Ddit4+/+ mice. Note pS6 is also in non-paligenotic cells, so a blot of whole stomach body registers only partial decrease. This blot is one of those quantified in panel (D).
D. Western blots quantified from n=3 independent mouse cohorts represented as mean±SEM, significance estimated by unpaired, two-tailed Student’s t-test at the 12h timepoint.
E. H&E staining of stomachs in Ddit4+/+ and Ddit4−/− mice at 72h after tamoxifen injury (TAM 72h). Red arrows show representative glands with dropout (ie the cells at the base of units are lost) in Ddit4+/+ control. Note that cells are mostly cuboidal/columnar. There is no dropout apparent in Ddit4−/− mice; black arrows show representative bases with most chief cells preserving their cytoplasm 72h after tamoxifen injury. Scale bar, 30 μm; boxed area insert, 15 μm.
F. Western blot of GIF and β-tubulin control expression from whole corpus protein extracts of Ddit4+/+ and Ddit4−/− mice at various time points after tamoxifen injury.
G. GIF (red, chief cells), GSII (green, progenitor or paligenotic cells), BrdU (white), DAPI (blue). Note paligenotic cells Ddit4−/− mice are both more proliferative (BrdU+) and have more GIF+ granules, indicating failed Stage 1 downscaling. Scale bar, 30 μm; boxed area insert, 15 μm.
H. Proliferating (BrdU+) cells of Ddit4+/+ (n=3) and Ddit4−/− (n=3) mice quantified by cell type in Stage 3, proliferative paligenosis (72h TAM). GIF-negative cells lack chief cell markers so are non-paligenotic: ie derived from GSII+ progenitors from neck (green) or progenitors even higher in the unit (gray); GIF+GSII− cells aberrantly retain chief cell features without inducing progenitor markers, indicating failed Stage 1. Ddit4−/− mice have significantly increased paligenotic (GIF+GSII+) and aberrant paligenotic cells (GIF+) but no difference in non-paligenotic proliferating cells. Datapoint: mean±SEM across ≥40 fields, significance estimated between paired cell types by unpaired, two-tailed t-test.
I. Total BrdU+ (proliferating) cells from Ddit4+/+ (TAM 24h, n=3; TAM 48h, n=3; TAM 72h, n=8) and Ddit4−/− (TAM 24h, n=3; TAM 48h, n=3; TAM 72h, n=8) mice per unit were quantified, Datapoint: Mean±SEM of ≥40 glands quantified per mouse, significance estimated by unpaired, two-tailed t-test at each timepoint.
J. Organoids at day 6 after culturing from Ddit4−/− and Ddit4+/+ mice. Scale bar, 100μm.
K. Data as in panel (J), quantifying organoid diameter from Ddit4+/+ (n=3) and Ddit4−/− (n=3) mice. Datapoint: Mean±SEM of ≥100 gastroids per mouse with significance estimated by unpaired, two-tailed t-test.
L. Anti-pS6 240/244 showing normal, homeostatic mTORC1 in Ddit4+/+ and Ddit4−/− pancreas, with Ddit4−/− acinar cells failing to extinguish mTORC1 at cerulein injury day 1 (mid-Stage 1 in pancreas); inset in Ddit4+/+ highlights a rare pS6+ cell, whereas most cells are positive in the absence of DDIT4. Scale bar, 30 μm; boxed area insert, 15 μm.
M. H&E staining of Ddit4+/+ and Ddit4−/− pancreas after 5 days of cerulein treatment (CER D5). Most acinar cells preserve their cytoplasm in Ddit4−/− mice, consistent with failure of Stage 1 turnover of mature cell architecture. Scale bar, 30 μm; boxed area insert, 15 μm.
N. Anti-BrdU of pancreas 5d after CER injury. Note increased BrdU+ cells in Ddit4−/− mice and failed downscaling of cell architecture. Scale bar, 30 μm; boxed area insert, 15 μm.
O. BrdU-positive cells from Ddit4+/+ (n=6) and Ddit4−/− (n=6) mice as in panel (N) quantified. Datapoint: Mean±SEM of ≥10 random fields quantified per mouse with significance estimated by unpaired, two-tailed Student’s t-test.
Autophagic/lysosomal recycling of cell architecture in Stage 1 depends on DDIT4 suppression of mTORC1 and rapamycin rescues the Ddit4−/− phenotype
We have shown that decreased mTORC1 in Stage 1 is required for the induction of the massive autophagic and lysosomal activity that governs eventual turnover of mature cell architecture(Willet et al., 2018). Accordingly, in Ddit4−/− mice, the failure to suppress mTORC1 in Stage 1 correlated with greatly decreased induction of lysosomes and autophagosomes (Fig.S7A–C), indicating aberrant autodegradation in Stage 1. The failure of Stage 1 correlated subsequently with aberrant cells at 72h (ie Stage 3, peak metaplasia): many cells still preserved key aspects of chief cell morphology, including large size, abundant apical cytoplasm, well-formed secretory granules containing GIF (Fig.6E–G; Fig.S7D)(Willet et al., 2018). Loss of mTORC1 downregulation in Stage 1 also led to earlier and more dramatic increase in the number of paligenotic cells entering the cell cycle in Stage 3 (Fig.6G–I). Furthermore, organoids derived from Ddit4−/− stomach bodies showed significantly increased growth, consistent with a paligenotic-cell-autonomous effect on proliferation due to sustained mTORC1 activation (Fig.6J,K).
The results so far indicated that, in the absence of DDIT4, mTORC1 in Stage 2 paligenotic cells is already elevated because it does not get suppressed in Stage 1. Thus, Ddit4−/− cells can complete paligenosis and progress to Stage 3 regardless of the IFRD1-regulated p53 checkpoint. That is because p53 works to suppress only reactivation of mTORC1 in Stage 2 to 3 progression, so this checkpoint is irrelevant if mTORC1 never decreases in Stage 1. Accordingly, IFRD1 abundance was not affected by loss of DDIT4. Corroborating the gastric results, Ddit4−/− acinar cells also preserved mature cell features during peak Stage 3 paligenosis and showed increased proliferation (Fig.6M–O).
To confirm that the increased proliferation and persistent mature cell architecture in Ddit4−/− mice were due to failure to suppress mTORC1, we treated Ddit4−/− mice with rapamycin to force mTORC1 inactivation. As expected, rapamycin suppressed mTORC1 in Ddit4−/− mice as measured by decreased phosphorylation of S6 and the mTORC1 direct target S6 Kinase (Fig.7A,B). Similarly, the mTORC1 suppressive protein TSC2 – which, as expected, was decreased in Ddit4−/− mice – was increased by rapamycin (Fig.7B). Reducing mTORC1 with rapamycin administration in Ddit4−/− mice corrected the Stage 3 hyperproliferation and mature-architecture-retention phenotypes in both stomach (Fig.7A,C,D; Fig.S7E,F) and pancreas (Fig.7G,H; Fig.S7G). Consistent with the key role of DDIT4 in paligenosis being to suppress mTORC1, rapamycin administration in Ddit4−/− mice did not affect SOX9 expression (Stage 2–3 marker) in either stomach or pancreas (Fig.7E,F; Fig.S7H,I). Overall, thus, DDIT4 is required for the autophagic/lysosomal recycling of cell architecture that characterizes Stage 1, and, because rapamycin completely rescues the Stage 1 defects caused by Ddit4−/−, DDIT4 regulates Stage 1 via mTORC1 suppression.
Fig. 7. mTORC1 inhibition rescues Ddit4−/− Stage 1 phenotype.

A. Rapamycin blocks pS6 (mTORC1) in Ddit4−/− and Ddit4+/+ mice. Scale bar, 30 μm; boxed area highlighting bases of units where paligenosis occurs; inset, 15 μm.
B. Western blot of whole corpus ±rapamycin. Note IFRD1 is less abundant at 72h, so exposure was longer than for Fig. 2c.
C. As in panel (A) but with anti-BrdU. Paligenotic proliferation (BrdU) in Ddit4−/− stomachs is corrected by rapamycin, such that both genotypes have the usual loss of proliferation upon mTORC1 reactivation. Scale bar, 30 μm; boxed area insert, 15 μm.
D. Data from Ddit4+/+ (n=5), Ddit4−/− (n=5), Ddit4+/++ rapamycin (n=5) and Ddit4−/−+ rapamycin (n=5) mice as for panel (C) were quantified. Note rapamycin does not significantly affect non-paligenotic proliferation, so only paligenotic proliferation is inhibited. Datapoint: Mean±SEM of ≥40 glands quantified per mouse, significance by one-way ANOVA, Dunnett post-hoc to Ddit4+/+ control.
E. Immunohistochemistry images of SOX9 staining after 5 days cerulein injury (CER D5) ± rapamycin (RAP).
F. Quantification of immunostaining depicted in panel (E) from Ddit4+/+ (n=5), Ddit4−/− (n=5), Ddit4+/++ rapamycin (n=5) and Ddit4−/−+ rapamycin (n=5) mice. Each data point is the mean of ≥10 random field quantified per mouse. Variance is plotted as standard error of the mean with significance estimated by one-way ANOVA with multiple comparisons to the Ifrd+/+ control (Dunnett post-hoc test).
G. BrdU in pancreas. Boxes: individual proliferating cells. Proliferating cells after rapamycin are stromal, not paligenotic acinar cells. Scale bar, 30 μm; boxed area inset, 15 μm.
H. Data from Ddit4+/+ (n=5), Ddit4−/− (n=5), Ddit4+/++ rapamycin (n=5) and Ddit4−/−+ rapamycin (n=5) mice as in panel (G) quantified. Datapoint: Mean±SEM of ≥10 random fields quantified per mouse, significance as per panel (D).
DISCUSSION
We summarize the molecular logic of DDIT4 and IFRD1 action in the conserved cellular dedifferentiation program we have termed paligenosis as follows: A stressor that may eventually cause differentiated cells to return to the cell cycle causes acute activation of DDIT4 along with a more gradual accumulation of IFRD1 and p53. The DDIT4 activation is required for the initial suppression of mTORC1 activity during Stage 1, as Ddit4−/− mice never substantially decrease mTORC1 and largely do not activate the autodegradative machinery to turnover mature cell features. Passage into Stage 3 depends on mTORC1 reactivation as we show here and in prior studies(Willet et al., 2018). Thus, Ddit4−/− cells, which maintain constitutively high mTORC1, do not need to wait for mTORC1 reactivation, because mTORC1 is never deactivated, and they become proliferative more rapidly. DDIT4 protein decreases dramatically by the end of Stage 1, so its direct role is limited to the initial mTORC1 decline.
As Stage 1 progresses, the massive activation of lysosomes would be expected to begin to reactivate mTORC1 via liberation of amino acids and other products of lysosome digestion. During most of Stage 1, DDIT4 suppresses mTORC1 despite this reactivation pressure. But as DDIT4 decreases, mTORC1 still does not reactivate because p53 peaks during this time period. We know p53 is critical for suppressing mTORC1 reactivation and not its initial deactivation, because Trp53−/− cells still undergo Stage 1 autodegradation and mTORC1 deactivation. Loss of p53 only makes that phase more transient with earlier mTORC1 reactivation. Eventually, late in Stage 2, p53 begins to decrease, allowing full mTORC1 reactivation with progression through the cell cycle. The p53 decrease is abetted by IFRD1 which promotes the less active, deacetylated form of p53. Ifrd1−/− cells have hyperactive p53 in Stage 2 and 3, and they largely fail to upregulate mTORC1, with many simply dying.
Thus, initial loss of mTORC1 is via DDIT4 while reactivation is via IFRD1 suppressing p53-mediated inhibition of mTORC1. The two phases of mTORC1 regulation appear relatively autonomous. Loss of DDIT4 does not affect IFRD1 abundance. Loss of IFRD1 does cause increased DDIT4 but does not affect Stage 1, indicating it has no effect on DDIT4 function. The mTORC1 reactivation in Stage 2 to 3 appears to be a critical checkpoint, as rapamycin leads to a dramatic blockade in paligenosis, with cells all expressing progenitor markers like SOX9 but unable to divide(Willet et al., 2018). Thus, the actions of IFRD1 and p53 may help ensure fidelity of cells before they undertake the drastic final step of going from a quiescent, mature state (which they might have been in for weeks to even years) back into the cell cycle. We saw some evidence of the importance of this step because loss of DDIT4 maintained high levels of mTORC1, which correlated with cells carrying DNA damage into Stage 3. Thus, loss of either DDIT4 or p53 may promote tumorigenesis, because both mutants would bypass the cell-cycle fidelity check in Stage 3. Accordingly, it has been reported in other tissues that loss of DDIT4 mimics loss of p53 in increasing tumorigenesis risk, because mice lacking either gene fail to respond appropriately to DNA damage(Janic et al., 2018).
The mechanisms DDIT4 and IFRD1 use to regulate mTORC1 appear to be confined to injury-induced proliferation of mature or quiescent cells (ie paligenosis) and do not apply to constitutively active stem and progenitor cells or to proliferation during development. DDIT4 and IFRD1 are typically injury-induced in all organisms and tissues where they have so far been studied, and mice null for Ddit4 and Ifrd1 develop normally and have normal adult stem cell activity. Previous reports have implicated DDIT4 in recruiting hematopoietic stem cells for regeneration after 5-FU with DDIT4 suppressing mTORC1 and helping to mediate scavenging of reactive oxygen species until cells are ready to renter the cell cycle, potentially an analogous process to what we observe in the current study(Basu, 2014). IFRD1 has also been shown to be required for skeletal muscle regeneration(Micheli et al., 2011) and for recruitment of quiescent myogenic stem cells after muscle crush injury(Vadivelu et al., 2004), and is induced during dictyostelium dedifferentiation (Nichols et al., 2020). We previously showed that Ifrd1−/− mice do not efficiently recruit stem cells for intestinal repair after resection(Garcia et al., 2014). In all these earlier studies, the cells that get recruited to eventually become stem cells must first emerge from mitotic quiescence. Moreover, in some cases, their emergence is known to require first suppression and then reactivation of mTORC1(Basu, 2014). In our studies and in the extant literature, the mechanism of action of IFRD1 and DDIT4 in regulating injury-induced proliferation is cell-autonomous (eg, both genes are induced in chief cells and govern paligenosis in chief cells).
We are only just beginning to understand the dynamic, cell biological differences between constitutively dividing stem and progenitor cells (like LGR5+ crypt-base intestinal cells or isthmal gastric epithelial cells (Miao et al., 2020) and so-called reserve or quiescent “stem cells”. It has been proposed that many of the so-called reserve stem cells recruited to proliferate in the intestine after injury to the constitutively dividing stem cells (eg via doxorubicin or radiation) may be differentiated cells like Paneth cells(Mills and Sansom, 2015; Burclaff and Mills, 2018b; Yu et al., 2018; Jones et al., 2019). Thus, the role for mTORC1 in nutritional and metabolic regulation of stem cell activity and tumorigenesis (Faller et al., 2015; Beyaz et al., 2016; Mihaylova et al., 2018; Miao et al., 2020) may involve effects on paligenotic recruitment of new stem cells from quiescent or mature cells.
Overall, we argue that DDIT4 and IFRD1 are conserved elements of the paligenotic program that mature cells (and perhaps mitotically quiescent cells in general) use after injury to become regenerative. Their role seems principally to be to legislate the biphasic mTORC1 changes. It is too early to argue whether they are universal elements, as there may be different forms of injury-induced reprogramming (besides paligenosis), just as apoptosis is only one form of programmed cell death. We also cannot argue yet that they have evolved exclusively to regulate paligenosis, as evolution constantly repurposes genes, just as genes that are core elements of mitosis, apoptosis, and autophagy are also used by cells in other contexts. However, we expect that the elucidation of this core machinery involving DDIT4, IFRD1, p53, and mTORC1 will play out in numerous cellular and tissue contexts.
Multicellular organisms can dedicate the vast majority of cells to specific functions; however, paligenosis also allows those cells to act as a large reservoir of potential progenitors to repair damage of organs throughout life. In the absence of tissue stem cells, the inability to undergo paligenosis, can be catastrophic, because regeneration cannot occur. For example, if we continue cerulein injury for 2 weeks in Ifrd1─/─ mice, there is near complete loss of exocrine pancreatic tissue (unpublished observations). There are risks, however, in allowing long-lived cells to cycle between replicative and differentiated states, because mutations could potentially accumulate over time increasing risk for accumulation of oncogenic mutations with each paligenotic event(Burclaff and Mills, 2018a, b). Accordingly, the pathologists and cell biologists of over a century ago who first recognized that mature cells could be recruited back into the cell cycle also noted that this process likely fueled lesions like metaplasia that increase risk for tumorigenesis(Adami, 1900). The IFRD1 and p53 cell-cycle licensing we show here that occurs in Stage 3 of paligenosis may be one reason why p53 evolved in multicellular organisms and not yeast(Wahl and Carr, 2001; Wylie et al., 2014): multicellular organisms can afford to waste damaged cells via apoptosis to avoid cancer risk.
Because paligenosis is at the heart of regeneration and tumorigenesis – but not a part of normal homeostasis – delineating the paligenosis machinery may lead to druggable targets that can inhibit cancer progression or spur regeneration without affecting homeostatic processes like the proliferation of constitutively active tissue stem cells.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should directed to, and will be fulfilled by the Lead Contact, Jason Mills (jmills@wustl.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the microarray data files reported in this paper is NCBI GEO: GSE71580, GSE3644, GSE44925, GSE6998 and GSE121925
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cancer cell
LS174T Cells (ATCC) grown under standard conditions (DMEM, 10% FBS, and Primocin at 37°C and 5% CO2) were cotransfected with a transposon expressing shRNA against IFRD1 (TRCN0000151595) under a U6 promoter as well as a plasmid expressing Hyperactive piggyBac transposase to stably incorporate the shRNA vector into the genome. After selection with puromycin, passaged cells were treated with tunicamycin (4 ug/mL; Sigma T7765) for 15–18 hours, lysed with RIPA buffer, and analyzed via western blot. Effect of IFRD1 knockdown was compared to LS174T cells stably transfected with an empty vector and treated in an identical manner in parallel.
Mice
All experiments using vertebrate animals followed protocols approved by the Washington University School of Medicine or China Medical University Animal Studies Committees. WT C57BL/6 and Trp53tm1Tyj/J (Trp53−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Ifrd1tm1Lah (Ifrd1−/−) mice were previously described(Vadivelu et al., 2004). Ddit4tm1.1 (KOMP)Vlcg/J (Ddit4−/−) mice were the property of Quark Pharmaceuticals Inc and were generated by Lexicon Inc by Quark’s request. Atp4b-Cre;LSL-DTR mice were generated as described previously(Burclaff et al., 2017). All strains of mice in these studies were maintained in a specified pathogen-free barrier facility under a 12-hour light cycle with free access to standard chow pellets and water. Each mouse used was genotyped by PCR amplification. For full list of genotyping primers, see Table S2.
Experimental and control groups each contained at least three mice, with representatives from both genders. There were no differences in results observed between male and female mice. All mice used were between 8–12 weeks old. When possible, mice of equivalent age and gender (littermate controls) were used for experimental and control groups otherwise, mice were randomly chosen and sorted into experimental groups.
Drosophila
The mammalian ifrd1 orthologue is CG31694 (difrd1 here). Drosophila stocks were obtained from the Bloomington Drosophila stock center. yw; P(EPgy2)EY11632 (BL#20811; designated as difrd1 #1), yw; P(PTT-GA)CA07748 (BL#52520; designated as difrd1 #2 or ifrd1-GFP). w1118 or Canton-S are used as wild types(Rudolf et al., 2012). All Drosophila were cultured on yeast-molasses based on food at the room temperature.
Gastroid
Gastric organoids (gastroids) collected from the gastric glands in the corpus of the stomach were isolated from Wildtype, Ifrd1−/−, Trp53−/− and Ddit4−/− mice. After dissecting the stomach, flush stomachs with PBS and cut the forestomach and the antrum off. Wash the corpus with cold chelating solution, remove the muscle layer and cut the mucosa into small pieces. Immerse the small pieces in chelating solution + 10 mM EDTA for 3hours with gently shaking as described previously(Barker et al., 2010; Osaki et al., 2019). Approximately 100 gastric glands were mixed with 50 μL of Matrigel (Corning), plated in 24-well plates and cultured in Advanced DMEM/F12 medium (Invitrogen), 50% Wnt3a conditioned medium, 10% R-Spondin1 and Noggin conditioned medium(Osaki et al., 2019) supplemented with 10mM HEPES, 1X N-2, 1X B27, 1X glutamax (Invitrogen), 1.25 mM N-Acetylcysteine (Sigma-Aldrich), 50 ng/mL EGF, 100 ng/mL FGF10 (Peprotech), 10 nM gastrin (Sigma-Aldrich) and 0.1% Primocin (Invivogen). 10 μM ROCK inhibitor (Y-27632, Sigma-Aldrich) was provided for the first generation gastroids to prevent anoikis. Conditioned medium was changed every 3 days.
METHODS DETAILS
Drug Treatments
Stomach SPEM was induced by Tamoxifen (5 mg/20 g body weight; Toronto Research Chemicals) dissolved in 10% ethanol and 90% sunflower oil, injected intraperitoneally (IP) daily. Mice were euthanized at 6–72h post Tamoxifen treatment as described previously(Huh et al., 2012). Pancreatic ADM was induced by 6 hourly IP injections of 50 μg/kg cerulein (Sigma-Aldrich) given every other day for 1–14 days as described previously(Willet et al., 2018). Rapamycin (60 μg/20 g body weight; LC Laboratories) was injected IP in 0.25% Tween-20, 0.25% polyethylene glycol in PBS daily for 3–7 days pre-treatment and throughout the injury time course. Partial hepatectomy was performed as previously described(Blanc et al., 2010). Mice were given an intraperitoneal injection containing 5-bromo-2’-deoxyuridine (BrdU; 120 mg/kg) and 5-fluoro-2’-deoxyuridine (12 mg/kg) in sterile water 90 min before sacrifice for all BrdU labeling experiments. Mice were anesthetized with isothesia (Henry Schein Animal Health) and sacrificed via cervical dislocation. Mouse stomachs were immediately excised and flushed with PBS after sacrifice and fixed in freshly prepared 10% formalin overnight and moved to 70% ethanol before preparation for embedding. Stomach were cut into rings or strips, embedded in 3% agar in a tissue cassette, and underwent routine paraffin processing. Tissue was sectioned 5μm thick and mounted on glass slides.
For Drosophila heat shock stress, female Drosophila were incubated at 37°C for 90 minutes and recovered for more than 2 days at room temperature. For Drosophila oxidative stress, female Drosophila were raised in vials containing 1 mL of 2% sucrose solution with or without 3% hydrogen peroxide for overnight. Drosophila guts were dissected in saline and fixed (7% picric acid/4% paraformaldehyde, 1X PBS) for 60 min.
Imaging and tissue analysis
For immunohistochemistry staining, sections were deparaffinized in Histo-Clear (National Diagnostics) and rehydrated in a gradient of ethanol, then blocked with 5% normal serum in 0.15% triton X-100 and incubated overnight with primary antibodies. The following day, sections were washed in PBS and incubated for 1 h with biotin conjugated secondary antibodies then incubated with avidin/biotin complex (ABC, Vector) reagent for 30 min at room temperature. Sections were developed with DAB for desired time then counterstained in hematoxylin prior to mounting. For antibodies used in this study, see Table S3. For immunofluorescence staining, sections were deparaffinized in xylene and rehydrated, then blocked with 1% BSA in 0.15% triton X-100 and incubated overnight with primary antibodies. Primary antibodies were detected with Alexa-fluor (Invitrogen) secondary antibodies and 4’,6-Diamidino-2-phenylindole (DAPI) was used to detect nuclei in immunofluorescence images. For X-gal staining, fresh mouse tissue was embedded in OCT and sectioned using a frozen microtome (Leica). Tissue frozen sections were immersed in 1mg/ml X-gal staining solution overnight at 37°C in the dark. The following day, slides were post-fixed in 4% PFA for 10 min, then rinsed in PBS for 10 min and counter-stained with Nuclear Fast Red(Nagy et al., 2007). In Situ hybridization was used to detect RNA expression of IFRD1 using RNAscope 2.5 HD-Brown kit according to manufacturer’s instructions (Advanced Cell Diagnostics). To reduce background staining in parietal cell, slides were pretreated with 0.3M hydrochloric acid for 30 minutes. Negative controls were used according to manufacturer’s instructions.
Drosophila midguts were dissected in Drosophila saline (182mM KCl, 46mM NaCl, 10mM Tris Base, 3mM CaCl2, pH adjusted to 7.2 with 1N HCl) and transferred to fixation solution (4% formaldehyde, 7% picric acid in 1X PBS) for 1h at room temperature while shaking. All samples were washed by Washing buffer (1X PBT: 1x PBS and 0.3% Triton X-100) several times for 1h and blocked in the pre-incubation buffer (1X PBT containing 1% BSA and 1% normal goat serum, 0.01% Sodium Azide). Primary antibodies were diluted in the same buffer. Midguts were incubated in antisera overnight at 4°C with shaking. After intensive washing, midguts were incubated with secondary antibodies for 1h at room temperature. After washing twice for 15 minutes, midguts were stained with 1 μg/mL DAPI in 1x PBT for 15 min, followed twice more washing and mounted in Vectashield medium (Vector Laboratories).
Quantitative PCR
Total RNA was extracted from the corpus of the stomach and isolated using RNeasy Mini Kit (Qiagen) per the manufacturer’s protocol. Quality of mRNA was verified with a BioTek Take3 spectrophotometer. RNA was treated with DNase I (Invitrogen), and 1 μg RNA was reverse-transcribed to make cDNA with SuperScript III (Invitrogen) following the manufacturer’s protocol. qRT-PCR was performed using PowerUp SYBR Green Master Mix (ThermoFisher) and gene specific primers on a QuantStudio 3 PCR System (ThermoFisher) and data analyzed using QuantStudio Design & Analysis Software. Every run was standardized to TATA Box Binding Protein (TBP) primers and all samples were run in triplicate. All primers were exon-spanning when possible, (i.e. for genes having multiple exons of sufficient length). For full list of qPCR primers, see Table S4.
Western Blots
Approximately 100 mg of mouse stomach corpus tissue or LS174T Cells were lysed in T-PER Tissue Protein Extraction Reagent (Thermo) or RIPA Lysis and Extraction Buffer (Thermo) with 1× protease/phosphatase inhibitor cocktail (Thermo). Protein concentrations were measured using the DC protein assay (BioRad) or Pierce™ BCA Protein Assay Kit (Thermo). 20–50 μg protein was separated using a 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Thermo), then transferred to PVDF membranes (Millipore). Membranes were blocked in TBS/Tween containing 3% BSA, and subsequently incubated with primary antibody overnight at 4°C then with either infrared fluorescent dyeconjugated secondary antibodies (LI-COR Biosciences) or horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch) for one hour. Protein signal intensities were normalized against a tubulin loading control for each sample. Fluorescent intensity values were determined and quantified using Image Studio Lite Ver 5.2 software (LI-COR Biosciences). When HRP secondary antibody was used, immunoreactive bands were visualized with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo). Antibody sources and dilutions are listed in Table S3.
Axon Regeneration
Embryonic DRG neurons were cultured as previously described(Cho and Cavalli, 2012). Briefly, e13.5 DRG neurons were dissected from wildtype mice, trypsinized (.05%) for 25 minutes, and triturated 60x to dissociate the cells. Neurons were resuspended in neuronal media consisting of Neurobasal, 1x B27, 1x Glutamax, FDU, and pen/strep, and were plated in spots of 10,000 neurons on plates coated with poly-d-lysine and laminin. Lentivirus containing FCIV-Bclxl was added at DIV2 and shIFRD1 at DIV4. At DIV 9, spots were axotomized with an 8mm long microtome blade and fixed 48 hours after injury. Spots were immunostained for SCG10 and regenerative growth was measured from the blade mark to the axon tips. The experiments were completed in technical triplicate with 8 biological replicates.
Electron Microscopy
Stomach corpus tissue was collected as described above, fixed overnight at 4˚C in modified Karnovsky’s fixative, and sectioned into rings. Tissue rings were processed for EM by the Washington University in St. Louis Department of Pathology and Immunology Electron Microscopy Facility.
QUANTIFICATION AND STATISTICAL ANALYSIS
Imaging and Quantifications
Fluorescence microscopy was performed using a Zeiss Axiovert 200 microscope with an Axiocam MRM camera and Apotome II instrument for grid-based optical sectioning. Immunofluorescence images were taken on a Zeiss Apotome (Zeiss). Bright field images were taken on a 2.0 HT Nanozoomer (Hamamatsu) whole slide scanner or Olympus BX43 light microscope. For imaging of RNAscope samples, individual images were taken at 40x and stitched together digitally. Images were analyzed and post-imaging adjustments were performed with Adobe Photoshop CS6. All time points and treatments were quantified with at least three mice, with representatives from both genders.
Human sample immunohistochemstry staining was scored on the following scale: 0, no staining; 1, minimal staining in less than 20% of cells; 2, moderate to strong staining in at least 20% of cells; 3, strong staining in at least 50% of cells. The scoring system was designed and independently verified by two human pathologists.
Quantification of pancreatic proliferation and SOX9+ cells was done by counting at least 10 randomly sampled whole 40× fields per condition, representing 500 μm in maximal dimension. Tissue preparation and imaging for electron microscopy were as previously described(Ramsey et al., 2007). For counts of paligenotic cells in immunohistochemically stained tissue, we used a method previously described(Willet et al., 2018), in which we counted cells only in the bottom 100 μM of each well-oriented gastric unit. For all genotypes in the current study, we have calculated from n=3 mice of 10 fields each (as well as years of previous experience) that this method is 99.9% sensitive and 80% specific for paligenotic cells (meaning almost no paligenotic cells are uncounted, but 20% might be miscounted as paligenotic, though there is no systemic bias that would affect comparisons across genotypes).
Autophagosome area was calculated by outlining the full regions of autophagosomes in each zymogenic cell cytoplasm and calculating that as a fraction of total cytoplasmic area using ImageJ (NIH). HALO image analysis platform (Indica Labs) was used in some cases to quantify intensity of fluorescent staining (eg for phospho-S6 in CER pancreases). We selected 10 random pancreas images per Ifrd1−/− and control animals that exhibited pathological ADM. Exposure times were kept constant across all samples, and images were analyzed based on the intensity of fluorescence per cell. The staining threshold was set based on control tissue for positive staining.
Bioinformatics, microarray and in silico screening
Microarray data from this manuscript have previously been deposited in the National Center for Biotechnology Gene Expression Omnibus (GEO): Stomach (TAM-treated, 12h, GSE71580)(Moore et al., 2015); Pancreas (Cerulein, 24h, GSE3644)(Kowalik et al., 2007); Kidney (Glycerol, 24h, GSE44925)(Fahling et al., 2013), Liver (Hepatectomy, 6h, GSE6998)(Otu et al., 2007). Genes whose expression was increased 1.5-fold (stomach sample) or 2-fold (other organs) with unadjusted p-value of <0.05, were selected, and those with unique, official Gene Symbols were analyzed for intersections with the Gene Set called “mTORC1-associated Hallmark” gene set from the Broad GSEA(Liberzon et al., 2015). Analysis of intersection among the 5 gene sets was performed in R (version 3.6.1) using the UpSetR(Conway et al., 2017) and ComplexHeatMap(Gu et al., 2016) implementations of the UpSet(Lex et al., 2014) approach to data visualization. Intersections were depicted in “distinct” mode, meaning lists of intersecting transcripts represent those that are exclusive to that intersection of gene sets (eg, Ifrd1, and Ddit4 are the only transcripts in all 5 gene sets and are also, consequently, members of the intersection of “Stomach”, “Pancreas”, “Liver”, “Kidney”; however, because they are classified in the 5 set intersection, they are not also listed as part of the intersection of “Stomach”, “Pancreas”, “Liver”, “Kidney”). Intersections that contained no transcripts unique to that intersection were not plotted.
GSEA(Subramanian et al., 2005) was done using default 3.0 GSEA app settings, except: “Permutation type” was set to “gene_set” and “Metric for ranking genes” was set to: “Diff_of_Classes”. GMX files were made using microarray data generated de novo from Ifrd1+/+ and Ifrd1−/− pancreas tissue ± treatment with cerulein at d5 (GSE121925, publically available November 1, 2019). RNA to generate expression profiles for GSEA was isolated using the RNEasy Micro Kit (Qiagen) following the manufacturers’ instructions. Mouse Gene 2.0 ST Array (Affymetrix) Genechips were used to determine mRNA expression, and Partek Genomic Suite 6.6 was used to determine normalized expression for GMX files. Multisequence alignments and phylogenetic analysis are presented as uncurated results from Clustal Omega Webserver(Sievers and Higgins, 2018). Secondary structure prediction was computed with the JPred4 webserver(Drozdetskiy et al., 2015) using the sequence of human IFRD1.
Axolotl data were downloaded(Gerber et al., 2018) and fraction of cells expressing each gene at each timepoint calculated by formulas in Microsoft Excel.
Statistics
Statistical analyses were performed in GraphPad Prism 7 (La Jolla, CA). In cases involving more than two samples to compare, ANOVA with post-hoc correction (Tukey) was used to assess statistical significance if multiple conditions were compared; Dunnett post-hoc when comparing multiple samples to a single control. Otherwise unpaired, two tailed Student’s t-test was used unless otherwise stated. Most data (eg cell counts) were first analyzed as mean values of measurements from multiple fields across multiple sections of an organ for each animal, and then the mean of those means was plotted. Area measurement analysis was done using Student’s t-test (for data that showed presumptive normal distribution). Almost all data were expressed as mean of the means from each mouse ± standard error of mean (SEM) or, when noted, standard deviation (SD) when only a sample mean is plotted.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Ddit4 | Protein Tech | Cat#10638–1-AP RRID: AB_2245711 |
| Goat anti-Ddit4 | Santa Cruz | Cat# sc-46034 RRID: AB_2292589 |
| Rabbit anti-Ki67 | Abcam | Cat# ab15580 RRID: AB_443209 |
| Rabbit anti-pS6 240/244 | Cell Signaling | Cat#2215 RRID: AB_331682 |
| Rabbit anti-pS6k 389 | Cell Signaling | Cat#9205 RRID: AB_330944 |
| Rabbit anti-TSC2 | Cell Signaling | Cat#4308 RRID: AB_10547134 |
| Mouse anti-β-tubulin | Abcam | Cat# ab21057 RRID: AB_727043 |
| Mouse anti-BrdU | DHSB (G3G4) | Cat# G3G4 RRID:AB_2618097 |
| Rabbit anti-human gastric intrinsic factor | Gift of Dr. David Alpers, Washington University | N/A |
| Sheep anti-Pepsinogen | Abcam | Cat# ab31464 RRID: AB_2160912 |
| Rabbit anti-Sox9 | Millipore | Cat# AB5535 RRID: AB_2239761 |
| Rabbit anti-LC3B | Cell Signaling | Cat#2775 RRID: AB_915950 |
| Rabbit anti-Amylase | Sigma-Aldrich | Cat# A8273 RRID: AB_258380 |
| Rat anti-Lamp1 | DHSB | Cat#1D4B RRID: AB_528127 |
| Rabbit anti-pHH3 | Cell Signaling | Cat#9701 RRID: AB_331535 |
| Mouse anti-P53 | Cell Signaling | Cat#2524 RRID: AB_331743 |
| Rabbit anti-P53 | Proteintech | Cat#10442–1-AP RRID: AB_2206609 |
| Rabbit anti-Acetyl p53 379 | Cell Signaling | Cat#2570 RRID: AB_823591 |
| Rabbit anti-Acetyl p53 382 | Cell Signaling | Cat#2525 RRID: AB_330083 |
| Mouse anti-P21 | Santa Cruz | Cat# sc-6246 RRID: AB_628073 |
| Mouse anti-cyclin D1 | Santa Cruz | Cat# sc-8396 RRID: AB_627344 |
| Rabbit anti-IFRD1 | Novus Biologicals | Cat# NBP1–87327 RRID: AB_11052376 |
| Ifrd1 Probe-RNAscope 2.5 HD-RED | ACD | Cat#569021 |
| Bacterial and Virus Strains | ||
| XL-10 Gold | Agilent | Cat #200317 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Toronto Research Chemicals Inc | Cat#T00600 |
| 5-Bromo-2-deoxyuridine (BrdU) | Sigma-Aldrich | Cat#B5002 |
| EDTA | Thermo-fisher | Cat#AM2691 |
| DTT | Gold Biotechnology | Cat#DTT10 |
| BSA | Sigma-Aldrich | Cat#A7906 |
| Matrigel | Corning | Cat#354234 |
| Triton X-100 | Lab Chem | Cat#LC262801 |
| Advanced DMEM/F12 | Invitrogen | Cat#12634–010 |
| HEPES | Gibco | Cat#15630–080 |
| Glutamax | Gibco | Cat#35050–061 |
| N- Acetylcysteine | Sigma-Aldrich | Cat#A9165 |
| EGF | Peprotech | Cat#315–09 |
| FGF10 | Peprotech | Cat#100–26 |
| Gastrin | Sigma-Aldrich | Cat#G9020 |
| ROCK Inhibitor (y-27632) | Sigma-Aldrich | Cat#Y0503 |
| N-2 | Gibco | Cat#17502–048 |
| B27 | Gibco | Cat#17504–044 |
| Primocin | Invivogen | Cat#ant-pm-1,2 |
| trypLE Express | Gibco | Cat#12605–028 |
| DPBS | Gibco | Cat#14190–136 |
| Isothesia | Henry Schein Animal Health | Cat#NDC11695-6776-2 |
| Tween20 | Sigma-Aldrich | Cat#P5927 |
| ProLong Gold antifade mountant with DAPI | Molecular Probes | Cat#P36930 |
| Permount Mounting Medium | Fisher Chemical | Cat#SP150 |
| DNase I | Invitrogen | Cat# 18047019 |
| SuperScript III | Invitrogen | Cat# 11752050 |
| PowerUp SYBR Green Master Mix | ThermoFisher | Cat# A25742 |
| RNA Protect Reagent | Qiagen | Cat# 76526 |
| T-PER Tissue Protein Extraction Reagent | ThermoFisher | Cat#78510 |
| DC protein assay | BioRad | Cat# 5000111 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat# 23225 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher | Cat#34580 |
| 5-fluoro-2’-deoxyuridine | Sigma-Aldrich | Cat# F0503 |
| Potassium hexacyanoferrate(III) | Sigma-Aldrich | Cat# 244023 |
| Potassium hexacyanoferrate(II) trihydrate | Sigma-Aldrich | Cat# 455989 |
| N,N-Dimethylformamide | Sigma-Aldrich | Cat# 227056 |
| 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside | Sigma-Aldrich | Cat# B4252 |
| DMEM | Gibco | Cat# 11965084 |
| RIPA Lysis and Extraction Buffer | Thermo Scientific | Cat# 89900 |
| Tunicamycin | Sigma-Aldrich | Cat# T7765 |
| Cerulein | Bachem | Cat# H-3220 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail, EDTA-Free (100X) | Thermo Scientific | Cat# 78443 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Vectastain Elite ABC HRP Kit | Vector Laboratories | Cat#PK6100 |
| DAB Substrate Kit | Thermo Scientific | Cat#36000 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| GeneChip™ Mouse Gene 2.0 ST Array | Affymetrix | Cat#902119 |
| Deposited Data | ||
| Microarray data files (High dose tamoxifen induced SPEM) |
NBCI Gene Expression | GSE71580 |
| Microarray data files (Cerulein induced pancreatitis) |
NBCI Gene Expression | GSE3644 |
| Microarray data files (Acute kidney injury) |
NBCI Gene Expression | GSE44925 |
| Microarray data files (Liver regeneration) |
NBCI Gene Expression | GSE6998 |
| Microarray data files (Ifrd1+/+ and Ifrd1−/− pancreas ± Cerulein at D5) |
NBCI Gene Expression | GSE121925 |
| Experimental Models: Cell Lines | ||
| LS174T | ATCC | Cat# CL-188 RRID:CVCL_1384 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ddit4tm1.1 (KOMP)Vlcg/J | Generated by Quark Pharmaceuticals Inc, (Brafman et al., 2004) | N/A |
| Mouse: Trp53tm1Tyj/J | Jackson laboratories | RRID: IMSR_JAX:002101 |
| Mouse: Ifrd1tm1Lah | Generated by Dr. Lukas Huber Medical University of Innsbruck | RRID: MGI:3043558 |
| Mouse: Wild type C57BL/6 | Jackson laboratories | RRID: IMSR_JAX:000664 |
| Mouse: Atp4b-cre: B6.FVB-Tg(Atp4b-cre)1Jig/JcmiJ | Jackson Laboratories | RRID: IMSR_ JAX: 030656 |
| Mouse: Gt(ROSA)26Sortm1(HBEGF)Awai | Jackson Laboratories | RRID: IMSR_JAX:007900 |
| D. melanogaster, Canton -S | Bloomington Drosophila Stock Center | BDSC:64349, flybaseID: FBsn0000274 |
|
D. melanogaster, ΔIFRD1#1 yw;CG31694[EY11632] |
Bloomington Drosophila Stock Center | BDSC:20811, flybaseID: FBst0020811 |
| D. melanogaster, ΔIFRD1#2; yw;CG31694[CA07748] | Bloomington Drosophila Stock Center | BDSC:51520, flybaseID: FBst0051520 |
| D. melanogaster, IFRD1-GFP yw;CG31694[CA07748]. | Bloomington Drosophila Stock Center | BDSC:51520, flybaseID: FBst0051520 |
| D. melanogaster, w1118 | Bloomington Drosophila Stock Center | BDSC: 3605, FlybaseID: FBal0018186 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S4 | This paper | N/A |
| Primers for mouse genotyping, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
|
IFRD1 shRNA (TRCN0000151595) |
This paper | N/A |
| Software and Algorithms | ||
| QuantStudio3 Design and Analysis Software | ThermoFisher | https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/ab-quantstudio-3-and-5-real-time-pcr-system.html |
| Photoshop CS6 | Adobe | RRID:SCR_014199; https://www.adobe.com/downloads.html |
| Partek Genomic Suite 6.6 | Partek Incorporated | RRID: SCR_011860 http://www.partek.com/partek-genomics-suite/ |
| GSEA-P Software | Broad Institute | RRID:SCR_003199; http://software.broadinstitute.org/gsea/index.jsp |
| NDP.View2 | Hamamatsu Photonics | Cat#U12388–01 |
| Prism 3 | GraphPad | RRID:SCR_002798; https://www.graphpad.com/scientific-software/prism/ |
| Cytation 3 cell imaging multi-mode reader | BioTek | Cat# BTCYT3MV |
| Image J | NIH | RRID: SCR_003070 https://imagej.nih.gov/ij/ |
| AxioVision 4.9.1 | Zeiss | https://www.zeiss.com/microscopy/us/downloads/axiovision-downloads.html |
Highlights.
Dedicated genes govern paligenosis, a conserved cellular regeneration program
DDIT4 first blocks mTORC1, inducing massive autophagy to downscale the cell
p53 activation continues mTORC1 suppression to maintain cell quiescence
IFRD1 suppresses p53 to reinduce mTORC1 and license progression into the cell cycle
ACKNOWLEDGMENTS
We thank Quark Pharmaceuticals Inc for allowing the opportunity to use the Ddit4tm1.1 (KOMP)Vlcg/J (Ddit4−/−) mice. We thank Dr. Elena Feinstein (Quark Pharmaceuticals, Inc) and Prof. Rubin Tuder (University of Colorado Denver) for providing the Ddit4tm1.1 (KOMP)Vlcg/J (Ddit4−/−) mice. We thank Dr. Paul Taghert and Dr. Craig Micchelli for Drosophila food and reagents and for Drosophila, which were also obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537). We are deeply grateful to Spencer G Willet (Washington University in St Louis) for critically revising the manuscript and figures. We thank the lab of David Denardo for training and use of HALO software. We acknowledge the AITAC of the Washington University Digestive Disease Center (DDRCC: P30 DK052574) and the NIH Shared Instrumentation Grant S10 RR0227552 (for Nanozoomer slide scanning). MAL was funded by T32-DK077653 (NIH, NIDDK) by the DeNardo Education & Research Foundation Grant, and a supplement to R01-DK094989 (NIH NIDDK); MAT by T32-DK077653 (NIH, NIDDK) and R25-GM103757 (NIGMS, NIH); JWB by T32-DK007130 (NIH NIDDK); CJC by T32-CA00954731 (NIH NCI), JB by T32-GM007067 (NIGMS, NIH) and the Philip and Sima Needleman Student Fellowship in Regenerative Medicine; NOD by R01s HL38180 (NIH NHBLI), DK112378 and DK56260 (NIH NIDDK); DCR by R01-DK106382 (NIH NIDDK); ZNW by National Key R&D Program of China (MOST-2017YFC0908300), National Natural Science Foundation of China (81961128026, U1908207), Major Scientific and Technological Special Project of Liaoning Province of China (2019020176-JH1/103); JCM by DK094989, DK105129, and DK110406, by the Alvin J. Siteman Cancer Center-Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH National Cancer Institute P30 CA091842 and R01 CA246208, and the Barnard Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- Adami JG (1900). On Growth and Overgrowth In “Festschrift” in Honor of Abraham Jacobi, MD, LLD: To Commemorate the Seventieth Anniversary of His Birth, May Sixth, 1900 (Knickerbocker Press; ), pp. 422–432. [Google Scholar]
- Akeno N, Miller AL, Ma X, and Wikenheiser-Brokamp KA (2015). p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene 34, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, et al. (2010). Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25–36. [DOI] [PubMed] [Google Scholar]
- Basu S (2014). A complex interplay between PGC-1 co-activators and mTORC1 regulates hematopoietic recovery following 5-fluorouracil treatment. Stem Cell Res 12, 178–193. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Sessa KJ, Kennedy S, Luo J, and Davidson NO (2010). Apobec-1 complementation factor modulates liver regeneration by post-transcriptional regulation of interleukin-6 mRNA stability. J Biol Chem 285, 19184–19192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou Sleiman MS, Osman D, Massouras A, Hoffmann AA, Lemaitre B, and Deplancke B (2015). Genetic, molecular and physiological basis of variation in Drosophila gut immunocompetence. Nat Commun 6, 7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Baird MR, Yip MC, Murray J, and Shao S (2018). Structures of translationally inactive mammalian ribosomes. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff J, and Mills JC (2018a). Plasticity of differentiated cells in wound repair and tumorigenesis, part I: stomach and pancreas. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff J, and Mills JC (2018b). Plasticity of differentiated cells in wound repair and tumorigenesis, part II: skin and intestine. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff J, Osaki LH, Liu D, Goldenring JR, and Mills JC (2017). Targeted Apoptosis of Parietal Cells Is Insufficient to Induce Metaplasia in Stomach. Gastroenterology 152, 762–766 e767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff J, Willet S, Saenz JB, and Mills J (2019). Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells During Homeostasis and Injury-induced Metaplasia. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, and Cavalli V (2012). HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J 31, 3063–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Cha J, Yuan J, Haraguchi H, Bartos A, Leishman E, Viollet B, Bradshaw HB, Hirota Y, and Dey SK (2016). p53 coordinates decidual sestrin 2/AMPK/mTORC1 signaling to govern parturition timing. J Clin Invest 126, 2941–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, and Ellisen LW (2008). Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, and Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43, W389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahling M, Mathia S, Paliege A, Koesters R, Mrowka R, Peters H, Persson PB, Neumayer HH, Bachmann S, and Rosenberger C (2013). Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J Am Soc Nephrol 24, 1806–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB, et al. (2015). mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AM, Wakeman D, Lu J, Rowley C, Geisman T, Butler C, Bala S, Swietlicki EA, Warner BW, Levin MS, et al. (2014). Tis7 deletion reduces survival and induces intestinal anastomotic inflammation and obstruction in high-fat diet-fed mice with short bowel syndrome. Am J Physiol Gastrointest Liver Physiol 307, G642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber T, Murawala P, Knapp D, Masselink W, Schuez M, Hermann S, Gac-Santel M, Nowoshilow S, Kageyama J, Khattak S, et al. (2018). Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hasty P, Sharp ZD, Curiel TJ, and Campisi J (2013). mTORC1 and p53: clash of the gods? Cell Cycle 12, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, and Mills JC (2012). Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142, 21–24 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JA, Shanware NP, Chang H, and Tibbetts RS (2011). Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1. J Biol Chem 286, 8688–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, Yang H, Tai L, Vandenberg CJ, Kueh AJ, et al. (2018). DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 24, 947–953. [DOI] [PubMed] [Google Scholar]
- Jones JC, Brindley CD, Elder NH, Myers MG Jr., Rajala MW, Dekaney CM, McNamee EN, Frey MR, Shroyer NF, and Dempsey PJ (2019). Cellular Plasticity of Defa4(Cre)-Expressing Paneth Cells in Response to Notch Activation and Intestinal Injury. Cell Mol Gastroenterol Hepatol 7, 533–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley TM, Horita N, and Samuelson LC (2019). Tamoxifen-Induced Gastric Injury: Effects of Dose and Method of Administration. Cell Mol Gastroenterol Hepatol 8, 365–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Do AN, Kutzler L, Cavener DR, and Jefferson LS (2008). Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem 283, 3465–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik AS, Johnson CL, Chadi SA, Weston JY, Fazio EN, and Pin CL (2007). Mice lacking the transcription factor Mist1 exhibit an altered stress response and increased sensitivity to caerulein-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 292, G1123–1132. [DOI] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, and Pfister H (2014). UpSet: Visualization of Intersecting Sets. IEEE Trans Vis Comput Graph 20, 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang C, Hu W, and Feng Z (2019a). Tumor suppressor p53 and metabolism. J Mol Cell Biol 11, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tavana O, and Gu W (2019b). p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 11, 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock A, Rutherford K, Harris MA, Hayles J, Oliver SG, Bahler J, and Wood V (2019). PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res 47, D821–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao ZF, Adkins-Threats M, Burclaff JR, Osaki LH, Sun JX, Kefalov Y, He Z, Wang ZN, and Mills JC (2020). A Metformin-Responsive Metabolic Pathway Controls Distinct Steps in Gastric Progenitor Fate Decisions and Maturation. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli L, Leonardi L, Conti F, Maresca G, Colazingari S, Mattei E, Lira SA, Farioli-Vecchioli S, Caruso M, and Tirone F (2011). PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-kappaB. J Biol Chem 286, 5691–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, et al. (2018). Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 22, 769–778 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, and Sansom OJ (2015). Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 8, re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Stanger BZ, and Sander M (2019). Nomenclature for cellular plasticity: are the terms as plastic as the cells themselves? EMBO J 38, e103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BD, Jin RU, Osaki L, Romero-Gallo J, Noto J, Peek RM Jr., and Mills JC (2015). Identification of alanyl aminopeptidase (CD13) as a surface marker for isolation of mature gastric zymogenic chief cells. Am J Physiol Gastrointest Liver Physiol 309, G955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, and Behringer R (2007). Staining Frozen Mouse Embryo Sections for {beta}-Galactosidase (lacZ) Activity. CSH Protoc 2007, pdb prot4726. [DOI] [PubMed] [Google Scholar]
- Nam KT, Lee HJ, Sousa JF, Weis VG, O’Neal RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM Jr., et al. (2010). Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139, 2028–2037 e2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JM, Antolovic V, Reich JD, Brameyer S, Paschke P, and Chubb JR (2020). Cell and molecular transitions during efficient dedifferentiation. Elife 9, pii: e55435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki LH, Bockerstett KA, Wong CF, Ford EL, Madison BB, DiPaolo RJ, and Mills JC (2019). Interferon-gamma directly induces gastric epithelial cell death and is required for progression to metaplasia. J Pathol 247, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otu HH, Naxerova K, Ho K, Can H, Nesbitt N, Libermann TA, and Karp SJ (2007). Restoration of liver mass after injury requires proliferative and not embryonic transcriptional patterns. J Biol Chem 282, 11197–11204. [DOI] [PubMed] [Google Scholar]
- Radyk MD, Burclaff J, Willet SG, and Mills JC (2018). Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology 154, 839–843 e832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey VG, Doherty JM, Chen CC, Stappenbeck TS, Konieczny SF, and Mills JC (2007). The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development 134, 211–222. [DOI] [PubMed] [Google Scholar]
- Rudolf A, Buttgereit D, Rexer KH, and Renkawitz-Pohl R (2012). The syncytial visceral and somatic musculature develops independently of beta3-Tubulin during Drosophila embryogenesis, while maternally supplied beta1-Tubulin is stable until the early steps of myoblast fusion. Eur J Cell Biol 91, 192–203. [DOI] [PubMed] [Google Scholar]
- Saenz JB, and Mills JC (2018). Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol 15, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, and Higgins DG (2018). Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, et al. (2013). Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stierli S, Napoli I, White IJ, Cattin AL, Monteza Cabrejos A, Garcia Calavia N, Malong L, Ribeiro S, Nihouarn J, Williams R, et al. (2018). The regulation of the homeostasis and regeneration of peripheral nerve is distinct from the CNS and independent of a stem cell population. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, and Gu W (2008). Acetylation is indispensable for p53 activation. Cell 133, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummers B, Goedemans R, Pelascini LP, Jordanova ES, van Esch EM, Meyers C, Melief CJ, Boer JM, and van der Burg SH (2015). The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFkappaB activation. Nat Commun 6, 6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadivelu SK, Kurzbauer R, Dieplinger B, Zweyer M, Schafer R, Wernig A, Vietor I, and Huber LA (2004). Muscle regeneration and myogenic differentiation defects in mice lacking TIS7. Mol Cell Biol 24, 3514–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, Mutani R, Giordana MT, and Cavalla P (2007). Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 66, 732–739. [DOI] [PubMed] [Google Scholar]
- Vodovar N, Vinals M, Liehl P, Basset A, Degrouard J, Spellman P, Boccard F, and Lemaitre B (2005). Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proc Natl Acad Sci U S A 102, 11414–11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl GM, and Carr AM (2001). The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol 3, E277–286. [DOI] [PubMed] [Google Scholar]
- Wang D, Kon N, Lasso G, Jiang L, Leng W, Zhu WG, Qin J, Honig B, and Gu W (2016). Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 538, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis VG, Petersen CP, Weis JA, Meyer AR, Choi E, Mills JC, and Goldenring JR (2017). Maturity and age influence chief cell ability to transdifferentiate into metaplasia. Am J Physiol Gastrointest Liver Physiol 312, G67–G76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet SG, Lewis MA, Miao ZF, Liu D, Radyk MD, Cunningham RL, Burclaff J, Sibbel G, Lo HG, Blanc V, et al. (2018). Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Lu WJ, D’Brot A, Buszczak M, and Abrams JM (2014). p53 activity is selectively licensed in the Drosophila stem cell compartment. Elife 3, e01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Tong K, Zhao Y, Balasubramanian I, Yap GS, Ferraris RP, Bonder EM, Verzi MP, and Gao N (2018). Paneth Cell Multipotency Induced by Notch Activation following Injury. Cell Stem Cell 23, 46–59 e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun T, Yu K, Yang S, Cui Y, Wang Z, Ren H, Chen S, Li L, Liu X, Fang M, et al. (2016). Acetylation of p53 Protein at Lysine 120 Up-regulates Apaf-1 Protein and Sensitizes the Mitochondrial Apoptotic Pathway. J Biol Chem 291, 7386–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Datta S, Mandal P, Xu S, and Hamilton T (2010). Stress-sensitive regulation of IFRD1 mRNA decay is mediated by an upstream open reading frame. J Biol Chem 285, 8552–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the microarray data files reported in this paper is NCBI GEO: GSE71580, GSE3644, GSE44925, GSE6998 and GSE121925